
Management of Select Adverse Events Following Delandistrogene Moxeparvovec Gene Therapy for Patients With Duchenne Muscular Dystrophy
Abstract
BACKGROUND:
Duchenne muscular dystrophy (DMD) is a rare, degenerative, recessive X-linked neuromuscular disease. Mutations in the gene encoding dystrophin lead to the absence of functional dystrophin protein. Individuals living with DMD exhibit progressive muscle weakness resulting in loss of ambulation and limb function, respiratory insufficiency, and cardiomyopathy, with multiorgan involvement. Adeno-associated virus vector-mediated gene therapy designed to enable production of functional dystrophin protein is a new therapeutic strategy. Delandistrogene moxeparvovec (Sarepta Therapeutics, Cambridge, MA) is indicated for treatment of ambulatory pediatric patients aged 4 through 5 years with DMD who have an indicated mutation in the DMD gene.
OBJECTIVE:
Evidence-based considerations for management of potential adverse events following gene therapy treatment for DMD are lacking in clinical literature. Our goal was to provide interdisciplinary consensus considerations for selected treatment-related adverse events (TRAEs) (vomiting, acute liver injury, myocarditis, and immune-mediated myositis) that may arise following gene therapy dosing with delandistrogene moxeparvovec.
METHODS:
An interdisciplinary panel of 12 specialists utilized a modified Delphi process to develop consensus considerations for the evaluation and management of TRAEs reported in delandistrogene moxeparvovec clinical studies. Panelists completed 2 Questionnaires prior to gathering for an in-person discussion. Consensus was defined as a majority (≥58% ; 7/12) of panelists either agreeing or disagreeing.
RESULTS:
Panelists agreed that the choice of baseline assessments should be informed by individual clinical indications, the treating provider’s judgment, and prescribing information. Corticosteroid dosing for treatment of TRAEs should be optimized by considering individual risk versus benefit for each indication. In all cases involving patients with a confirmed TRAE, consultations with appropriate specialists were suggested.
CONCLUSIONS:
The Delphi Panel established consensus considerations for the evaluation and management of potential TRAEs for patients receiving delandistrogene moxeparvovec, including vomiting, acute liver injury, myocarditis, and immune-mediated myositis.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is a rare, degenerative, recessive X-linked neuromuscular disease [1]. Mutations in the gene encoding dystrophin lead to the absence of functional dystrophin protein, which is expressed in skeletal and cardiac muscle, gastrointestinal vascular/smooth muscle, the retina, and the brain [1–5]. A lack of dystrophin leads to progressive muscle weakness, resulting in loss of motor function, respiratory insufficiency, and cardiomyopathy [6]. As DMD affects multiple organ systems and increases the risk of a variety of health complications, patients with DMD require an interdisciplinary care team [6].
In recent years, there have been significant advances in the use of gene therapies to treat a range of conditions [7]. Several recombinant adeno-associated virus (rAAV) vector-based gene therapies have been approved by the Food and Drug Administration (FDA) to treat various genetic disorders, including rare and life-threatening diseases, with many more in late-stage development [7–11]. Vector-mediated gene therapy is a new therapeutic strategy to treat patients living with DMD [12]. Delandistrogene moxeparvovec (Sarepta Therapeutics, Cambridge, MA) is an rAAVrh74 vector-based gene therapy designed for targeted expression of a therapeutic transgene that enables the production of functional micro-dystrophin protein, thereby addressing the direct cause of DMD [12]. In the delandistrogene moxeparvovec clinical development program (studies NCT03375164, NCT03769116, and NCT04626674), a safety dataset derived from the trial experiences of 85 patients identified 13 treatment-related adverse events (TRAEs) that required medical intervention, including vomiting, myocarditis, acute liver injury (ALI), and immune-mediated myositis (IMM) [13, 14]. The TRAEs described in this paper have also been reported in clinical trials of other systemic gene therapies [10, 11].
Due to the rapid development of rAAV vector-based gene therapies and the limited number of individuals treated, there is a dearth of published peer-reviewed evidence to inform clinical decision-making and management of patient safety events that may arise following gene therapy. As a result, there is a critical need to understand how to identify and manage adverse events (AEs) associated with rAAV vector-based gene therapies [7, 8, 15, 16]. The Delphi panel process is one method that may address clinical questions when sufficient guidance in the scientific literature is limited [17, 18]. This technique uses a structured, iterative, and scientific research methodology to build consensus among a group of experts on efficacy and safety of a particular product or health-related topic [17]. The Delphi technique is rigorous, relies on the high level of expertise and credibility of the panel, and allows for the exploration of complex clinical questions [17, 18]. A Delphi panel was convened prior to the accelerated approval granted to delandistrogene moxeparvovec by the FDA to discuss the evaluation and management of specific AEs occurring post-gene therapy in patients with DMD and was based on the clinical experience of the treating provider and clinical trial data collected as of October 17, 2022. The objective of this Delphi panel was to provide interdisciplinary consensus management considerations for TRAEs following clinical administration of delandistrogene moxeparvovec.
METHODS
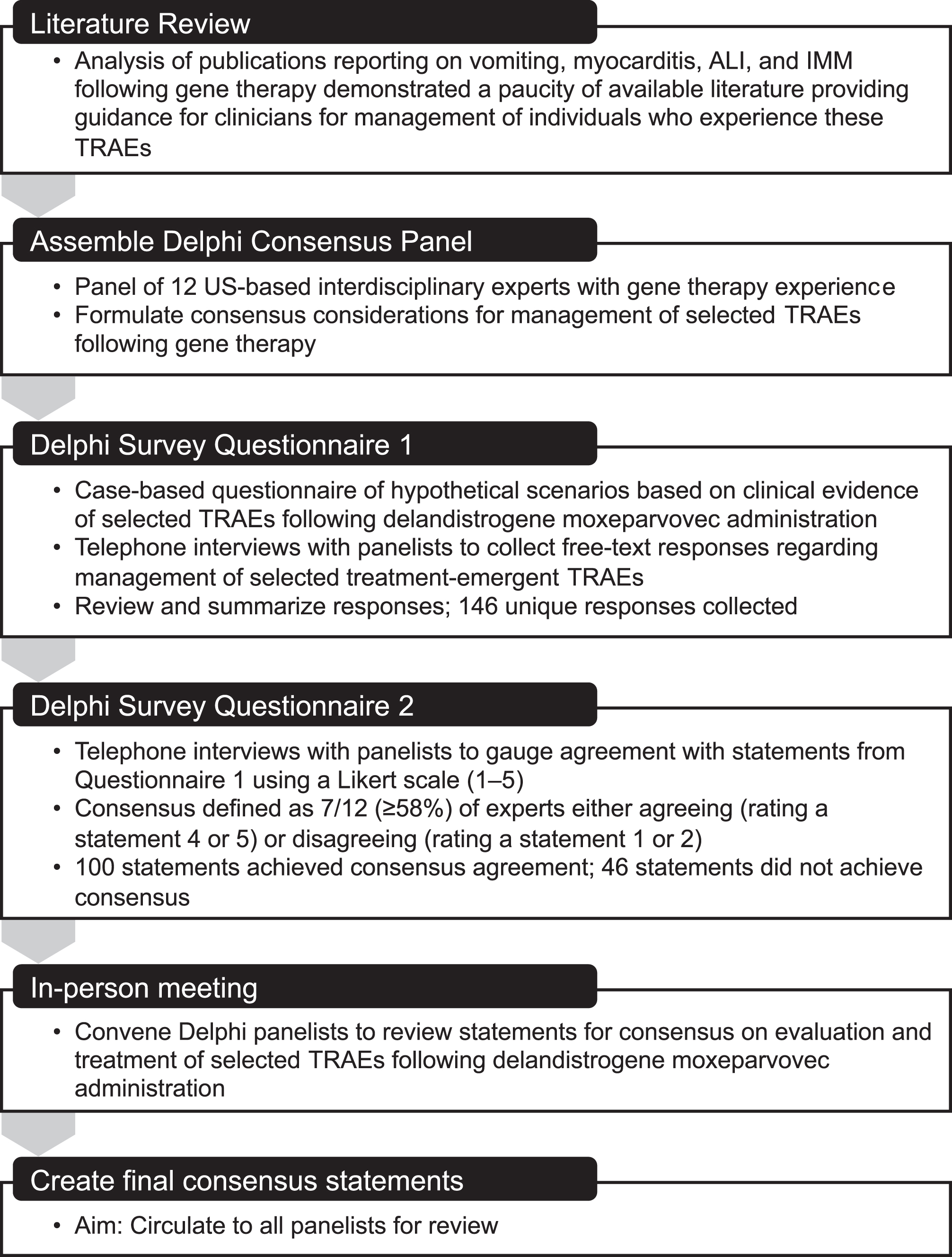
A modified Delphi process was utilized that included a literature search, two rounds of Questionnaires, and the development of consensus statements at a live meeting (Fig. 2). To ensure a diversity of expertise, the panel included neurologists, a cardiologist, a hepatologist, a pulmonologist, a gastroenterologist, and a pediatric advanced practice registered nurse. Those on the panel either administered delandistrogene moxeparvovec to patients in the context of clinical trials or had experience managing patients with DMD and/or the gene therapy-related AEs discussed here. Consistent with previously reported Delphi panel processes [19–21], a qualitative approach was adopted to establish consensus on the evaluation and management of TRAEs following treatment with delandistrogene moxeparvovec.
A search of published literature led to the identification of articles presenting clinical studies, case series, and retrospective analyses that reported strategies for monitoring and managing AEs commonly observed following gene therapy. The search results yielded few articles relevant to the evaluation and management of selected AEs following gene therapy in the DMD patient population, so the search was subsequently expanded to include all gene therapy treatments, which revealed a limited number of published manuscripts on management of TRAEs. The literature search identified 26 articles and abstracts of interest related to ALI, 25 related to myocarditis, and 6 related to IMM. A summary is presented in Supplement 1.
All panelists completed two Questionnaires and subsequently gathered for an in-person meeting to establish consensus on specific statements regarding evaluation and management of TRAEs (Supplement 2). Notably, the panel considerations for both Questionnaires and the in-person conversation were guided by the clinical trial protocols and are aligned with the current prescribing label, which stipulated that an increased dose of corticosteroids should be initiated prior to gene therapy infusion and continued for a minimum of two months to reduce the risk associated with an immune response to the vector and/or transgene. Daily corticosteroid dosing suggestions included adding 1 mg/kg/day in addition to the pre-gene delivery baseline dose, starting one day prior to the infusion, and increasing up to a maximum of 60 mg/day for a minimum of 60 days as clinically indicated, followed by a taper. The clinical trial protocol required cardiac assessment prior to study enrollment. To assess cardiac function, patients underwent baseline cardiac evaluation and imaging with echocardiogram and/or MRI. Exclusion criteria included signs of cardiomyopathy and echocardiogram with an ejection fraction below 40%.
Development of Questionnaire 1 was based on relevant information from the literature search as well as anonymized patient data from delandistrogene moxeparvovec clinical trials. Panelists provided open-ended responses to a series of questions related to managing select TRAEs observed in clinical trials of delandistrogene moxeparvovec (studies NCT03375164, NCT03769116, and NCT04626674), including vomiting, ALI, myocarditis, and IMM.
Questionnaire 2 was built upon responses from Questionnaire 1 wherein panelists were asked to rate these statements using a 5-point Likert scale (1 = strongly disagree; 5 = strongly agree). Consensus was defined as the majority (≥58% [7/12]) of panelists in concordance, either agreeing (rating a statement 4 or 5) or disagreeing (rating a statement 1 or 2).
During the in-person meeting, statements that failed to achieve majority consensus in Questionnaire 2 were discussed in the context of clinical trial experience (clinical case examples are presented in Supplement 3). While discussing management considerations, panelists were able to reword statements or debate additional suggestions to attempt to achieve consensus. After discussion, panelists were anonymously polled using an online digital platform (Slido) to determine consensus. If a statement still failed to achieve majority agreement, panelists were asked to agree that consensus was not obtained, and discussion was closed on that statement.
RESULTS
During two rounds of individual interviews and one in-person meeting, the panelists’ opinions regarding clinical assessment, laboratory studies, diagnostic studies, and treatment when specified AEs occur post-gene therapy administration were collected; these opinions were based on their clinical experience and information collected during clinical trials prior to the FDA accelerated approval of delandistrogene moxeparvovec. In total, the panelists were presented with 146 statements for consideration; of these, 17 did not achieve consensus during the in-person meeting. Full results from Questionnaire 1, Questionnaire 2, and the in-person meeting are presented in Supplement 2. Agreed-upon consensus considerations for evaluation and management of such patients are presented in Table 1.
Table 1
Consensus considerationsa for general assessment and management
| Timing of the physical exam to determine candidacy | A physical exam should be conducted one month prior to and again within 48 hours of the infusion |
| Timing of baseline lab collection | Baseline labs should be collected twice prior to gene therapy infusion: at the evaluation appointment (∼1 month prior) and again within one to three days of the procedure |
| Laboratory studies | The following tests are suggested: complement C3, complement C4, complement total CH50, CBC with differential, CMP (including ALT, AST, total bilirubin), serum IgG, aPTT, GGT, PT/INR, troponin I, and cystatin CProposed frequency of testing: |
| •Monitoring of liver enzymes prior to the infusion and weekly for the first three months following the infusion | |
| •Weekly troponin I monitoring during the first month following treatment | |
| •Weekly platelet monitoring during the first 2 weeks | |
| •Continued monitoring until results are unremarkable or if clinically indicated | |
| Treatment of gastritis | An H2 blocker is suggested to be taken as indicated by symptoms |
| Treatment of emesis | An antiemetic should be provided to be used as needed; greatest concerns are inability to tolerate oral corticosteroids and potential dehydration |
| Monitoring of patients post-infusion | Patients should be monitored for two to four hours following infusion |
| Communication strategies | Communication depends on the physician and institution, but patients/caregivers should be provided with contact options |
aThese considerations are offered based on clinical trial experience and the clinical experience of the individual panelists. aPTT, activated partial thromboplastin time; CBC, complete blood count; CMP, comprehensive metabolic panel; GGT, gamma-glutamyl transferase; H2, histamine; IgG, immunoglobulin G; PT/INR, prothrombin time and international normalized ratio.
Pretreatment considerations
Timing of exams and laboratory testing prior to infusion
Regarding the timeline for the physical exam prior to infusion, most panelists (83%) agreed that to provide the greatest level of safety for the general patient population, a physical exam should be conducted approximately one month prior to treatment and again within 48 hours of the procedure to ensure that the patient does not have any acute infections or illnesses. Panelists (75%) also agreed that baseline laboratory collection should occur twice before the infusion: one month prior to treatment and again one to three days before infusion. They emphasized that obtaining baseline labs was critical and that checking labs at two time points before the infusion could improve the utility of comparison in the future if abnormalities arise, but did not agree on timing.
Suggested laboratory tests and clinical investigations
Panelists suggested commercially available laboratory tests and clinical investigations to establish the patient’s baseline health status and to evaluate liver and heart function; these are described in Table 1.
Post-infusion monitoring
Most panelists (75%) agreed that the patient should be monitored closely for two to four hours post-infusion. Panelists acknowledged that no infusion-related reactions occurred in the clinical trials. They agreed that the care team should be readily accessible to patients and caregivers in order to discuss any concerning signs or symptoms. The method of contacting the care team and the individuals participating in an accessible care team will vary from site to site. A suggestion was made to build an interdisciplinary gene therapy team at the hospital to triage concerns and optimize patient care post-infusion, but the composition of the team was not specified because selection of representatives should be guided by the AEs associated with a specific gene therapy infusion and by the healthcare provider’s expertise and clinical experience with AEs in the context of gene therapy.
Considerations for GI events
All panelists agreed that prescribing histamine H2-receptor antagonists (H2 blockers) for the treatment of gastritis that may occur with corticosteroid treatment is appropriate; however, they did not reach consensus on whether to prophylactically administer an H2 blocker for gene therapy-associated gastritis (33%).
Vomiting
Panelists suggested that the patient/caregiver should contact the treatment provider immediately if post-infusion vomiting occurs. An antiemetic may be provided as needed. All panelists agreed that the greatest concern regarding patient emesis following gene therapy infusion is the patient’s inability to tolerate oral corticosteroids used to suppress immune responses to the capsid; if not tolerated, TRAEs such as ALI are more likely to occur. If vomiting prevents oral corticosteroid administration, parenteral administration will be necessary. Potential dehydration is another concern in patients with vomiting.
Acute liver injury
Panelists were asked to consider treatment for three different scenarios of sequentially increasing severity: 1) a patient who has mildly elevated liver function tests (aspartate aminotransferase [AST]/alanine transaminase [ALT]/-glutamyl transferase [GGT]) 1–2 times the baseline value or previous measurement); 2) a patient who has been diagnosed with ALI; and 3) a patient diagnosed with ALI who is refractory to initial interventions. The consensus opinions from the Delphi panel for ALI are summarized in Table 2.
Table 2
Consensus considerations for management of acute liver injury
| Patient presentation | Asymptomatic patient presenting with initial mild elevations of liver laboratory tests |
| Patient monitoring | Request a telehealth visit with the parent/caregiver; assess the need for a physical examination based on the clinical history Patient likely does not need to be admitted to the hospital at this time unless otherwise indicated |
| Laboratory studies | Monitor closely and repeat laboratory studies sooner than one week |
| Patient presentation | Acute liver injury is diagnosed/confirmed |
| Patient monitoring | Patient should be seen in person; assess need for hospitalization, based on laboratory and exam findings |
| Laboratory studies | If not hospitalized, monitor closely, and repeat laboratory studies sooner than one week |
| Additional diagnostic and laboratory studies | GGT, PT/INR |
| Medication &treatment | Increase oral corticosteroid dose over baseline corticosteroid dose to 2 mg/kg/day (max 120 mg/day) |
| Patient presentation | Acute liver injury that does not respond to initial treatment |
| Patient monitoring | If not previously hospitalized, patient should be admitted at this time |
| Laboratory studies | Continue to monitor laboratory studies conducted at baseline in addition to GGT, PT/INR, and direct bilirubin |
| Additional diagnostic studies | Perform viral testing (hepatitis panel, EBV, CMV) Perform a liver ultrasound |
| Medication &treatment | Begin a short-term pulse of IV methylprednisolone |
| Consultation | Consult with appropriate specialist (hepatologist), preferably one experienced with DMD and/or gene therapy |
Initial mild elevations of liver laboratory tests should be assessed with repeat laboratory studies sooner than the protocol stipulates and warrant close monitoring, with consideration of other causes of liver injury. Repeat baseline laboratory studies based on prescribing information and individual patient presentation; corticosteroid dose optimization should be based on the appropriate peri-infusion steroid dose. CMV, cytomegalovirus; DMD, Duchenne muscular dystrophy; EBV, Epstein-Barr virus; GGT, gamma-glutamyl transferase; HIV, human immunodeficiency virus; PT/INR, prothrombin time and international normalized ratio.
Scenario 1: The panelists discussed a hypothetical case of a patient with mildly elevated liver function tests. Based on laboratory abnormalities in AST/ALT/GGT (1–2 times the baseline value or previous measurement) in a hypothetical patient with suspected ALI, panelists suggested requesting a telehealth visit with the patient/caregiver (67%), repeating lab collection sooner than the protocol specifies (within one week) (75%), and maintaining the patient’s current post-infusion corticosteroid dose (92%).
Scenario 2: Upon confirmed diagnosis of ALI, most panelists (83%) agreed that a telehealth visit alone is limited or lacks physical interaction and suggested that the patient be seen in person for a physical exam. All panelists agreed to an increased frequency of laboratory monitoring, with lab collection repeated within one week and monitoring adjusted, as needed, if liver enzymes (AST, ALT, and GGT) continued to increase. Most panelists (92%) also agreed to additional testing for direct bilirubin but did not feel that viral testing for hepatitis, human immunodeficiency virus (HIV), Epstein-Barr virus (EBV), or cytomegalovirus (CMV) was necessary, unless there was a clinical indication to do so (such as failure to respond to increased corticosteroids).
In patients with ALI, most panelists (83%) would consider increasing the oral prednisone dose by adding an additional 1 mg/kg/day on top of the patient’s baseline and peri-delandistrogene moxeparvovec infusion corticosteroid dose to a total of 2 mg/kg/day with a maximum of 120 mg/day but did not think a liver ultrasound (67%) or specialist consult (58%) was indicated.
Scenario 3: For ALI that is not responding to initial interventions such as increased oral corticosteroids (the most severe hypothetical liver injury), most panelists (92%) suggested initiating a short-term pulse of intravenous (IV) methylprednisolone, expanding laboratory testing to include causes of viral hepatitis (67%), testing for direct bilirubin (92%), requesting a liver ultrasound (83%), and obtaining a consult with a hepatologist (83%). Panelists differed on the dose and duration of IV methylprednisolone treatment, and discussed a range of options including a single IV in the emergency department up to a brief hospitalization (3–5 days) for sequential IV therapy.
In summary, panelists suggested a staged approach to managing increased liver enzymes, with all patients with elevated liver enzymes requiring more frequent lab monitoring and, in moderate or severe cases, increased corticosteroids and additional laboratory and diagnostic evaluations.
Myocarditis
Panelists discussed treatment and management options in three different scenarios of increasing severity: 1) a patient with elevated troponin I; 2) a symptomatic patient with elevated troponin I; and 3) a patient with elevated troponin I who is not responding to initial interventions. Consensus opinions from the expert panel regarding management of myocarditis are shown in Table 3.
Table 3
Consensus considerations for myocarditis
| Patient presentation | Asymptomatic patient presenting with initial mild troponin I elevation |
| Patient monitoring | Request a telehealth visit with the parent/caregiver; assess the need for a physical exam based on the clinical history |
| Laboratory studies | Monitor closely and repeat baseline laboratory studies sooner than one week |
| Patient presentation | Patient presents with suspected or confirmed myocarditis |
| Patient monitoring | Patient should be seen for a physical exam; assess need for hospitalization based on laboratory and exam findings |
| Laboratory studies | Monitor closely and repeat laboratory studies sooner than one week |
| Additional diagnostic and laboratory studies | Complement C3, complement C4, complement total CH50, CK-MB, CK, urinalysis, cystatin C, CRP Perform an echocardiogram and ECG; consider cardiac MRI based on clinical scenario |
| Medication &treatment | Increase corticosteroid dose over baseline corticosteroid dosing to 2 mg/kg/day (max 120 mg/day); based on clinical scenario, consider short-term pulse of IV methylprednisolone; also consider adding IVIg |
| Consultation | Consult with cardiologist, preferably one experienced with DMD and/or gene therapy |
| Patient presentation | Myocarditis not responding to initial treatment |
| Patient monitoring | If not previously hospitalized, patient should be admitted at this time |
| Laboratory studies &diagnostics | Continue to monitor baseline laboratory studies |
| Additional diagnostic and laboratory studies | Continue to monitor complement C3, complement C4, complement total CH50, CK-MB, CK, cystatin C, CRP, urinalysis Continue to monitor echocardiogram, ECG, and cardiac MRI |
| Medication &treatment | Continue to optimize corticosteroid treatment, including IV methylprednisolone; consider adding IVIg and/or other immunosuppressant pharmacotherapy |
| Consultation | Consult with cardiologist, preferably one experienced with DMD and/or gene therapy |
Repeat baseline laboratory studies based on prescribing information and individual patient presentation; corticosteroid dose optimization should be based on the appropriate peri-infusion steroid dose. Depending on the clinical scenario, hospitalization, cardiology consultation, cardiac MRI should be considered; IVIg and/or other immunosuppressive therapy could also be considered. CK, creatine kinase; CK-MB, creatine kinase-myocardial band; CRP, C-reactive protein; DMD, Duchenne muscular dystrophy; ECG, electrocardiogram; IVIg, intravenous immunoglobulin; MRI, magnetic resonance imaging.
Scenario 1: For the asymptomatic patient with mildly elevated troponin I (defined by the group as <2.5 times the ULN or, if the baseline value of the patient is abnormal, as <2.5 times the baseline), most panelists (83%) agreed that a telehealth visit with the patient/caregiver would be appropriate and that a physical exam may be clinically indicated. Panelists would repeat baseline labs sooner than the protocol stipulated (in less than a week) (75%). All panelists agreed they would not test for complement activation (C3, C4, CH50) at this stage. Panelists would not suggest changing the dose of oral corticosteroids (67%) or obtaining a consultation with a specialist (58%).
Scenario 2: When a patient is symptomatic and has elevated troponin I, the panel suggested that the patient should be admitted to the hospital for examination (75%), a short-term pulse of IV methylprednisolone (75%) should be initiated, and a cardiology consultation should be sought (92%). They (92%) suggested repeating baseline labs within two to five days and would also request additional tests (described in Table 3). Most panelists (92%) agreed that an echocardiogram and ECG should be obtained and cardiac magnetic resonance imaging (cMRI) with gadolinium contrast should be conducted if available. Panelists recognized that experience with obtaining and interpreting cMRI for patients with DMD could vary by facility and that this should be considered when developing treatment protocols for patients undergoing gene therapy in DMD.
Scenario 3: For the symptomatic patient whose troponin I remains elevated following initial treatment with increased corticosteroids, in addition to the previous considerations, the panelists suggested a short-term pulse of IV methylprednisolone (58%) and they all agreed that IVIg should be considered (Table 3).
In summary, for an asymptomatic patient with mildly elevated troponin I, panelists suggested a clinical evaluation and increased frequency of lab monitoring. For symptomatic patients with elevated troponin I, patients should be admitted to the hospital for intravenous corticosteroids, additional diagnostic testing, a cardiology consultation, and consideration for IVIg.
Immune-mediated myositis
The panelists discussed a hypothetical case of IMM. Consensus statements developed by the panelists for management of IMM are described in Table 4. Criteria for diagnosing IMM in the context of gene therapy are lacking in the literature. In the delandistrogene moxeparvovec clinical trial program, criteria for IMM event reporting were based on patient symptoms and signs, clinical manifestations, laboratory abnormalities, and muscle biopsy results, and represented a diagnosis of exclusion with a focus on physical presentation. The group considered which clinical symptoms would be useful in determining a diagnosis of IMM following micro-dystrophin gene therapy as IMM presentation observed to date has been variable, including rapidly progressive axial, appendicular, and respiratory weakness; bulbar weakness; and swelling related to angioedema. Panelists suggested that a patient who is exhibiting physical signs indicative of possible IMM should be admitted to the hospital (67%) and baseline lab collection should be repeated within two to five days (92%). Panelists agreed that in the context of suspected IMM, patients experiencing swallowing or chewing difficulty, especially bulbar weakness, should be evaluated with a swallow study (67%) and neuromuscular strength assessment (92%). They further felt that an echocardiogram (100%) and ECG (83%) should be conducted to evaluate cardiac involvement. The group agreed that increased corticosteroids should be trialed, but that if the patient did not rapidly improve, treatment should be escalated with the consideration of adding plasmapheresis, IVIg, or other targeted immunosuppressive therapy.
Table 4
Consensus considerations for immune-mediated myositis
| Patient presentation | Patient presents with physical signs of IMM (weakness, muscle pain/tenderness, difficulty swallowing) that are progressive over days |
| Patient monitoring | Patient should be seen urgently by the prescribing physician for physical assessment (including neuromuscular strength assessment) and likely will require admission to the hospital for ongoing close observation |
| Laboratory studies | Monitor closely and repeat baseline laboratory studies sooner than one week |
| Additional diagnostic and laboratory studies | ANA, CK, CRP, aldolase, ESR, myoglobin, cystatin C, urinalysis, and urine output Echocardiogram, ECG, swallow study may be performed based on clinical scenario |
| Medication &treatment | Increase steroid therapy to either 2 mg/kg/day (max 120 mg/day) or 3-day course of high-dose IV methylprednisolone |
| Consultation | Consult with appropriate specialists (consider rheumatology, immunology, and cardiology), preferably experienced with DMD and/or gene therapy |
| Patient presentation | IMM with inadequate response to steroid optimization |
| Patient monitoring | If not already hospitalized, patient should be admitted at this time |
| Laboratory studies | Continue to monitor baseline laboratory studies |
| Additional diagnostic and laboratory studies | Continue to monitor ANA, CK, CRP, aldolase, ESR, myoglobin, cystatin C, urinalysis, and urine output Continue to assess neuromuscular strength, echocardiogram, ECG, swallow study based on clinical scenario |
| Medication &treatment | Consider escalating treatment with plasmapheresis, IVIg, and/or other immunosuppressant pharmacotherapy until response is achieved |
| Consultation | Consult with appropriate specialists (consider rheumatology, immunology, and cardiology), preferably experienced with DMD and/or gene therapy |
Experts agreed that for suspected symptomatic IMM, emergent evaluation by physical exam, laboratory studies, and additional laboratory assessments and diagnostics is required. Repeat baseline laboratory studies based on prescribing information and individual patient presentation; corticosteroid dose optimization should be based on the appropriate peri-infusion steroid dose. ANA, antinuclear antibodies; CK, creatine kinase; CRP, C-reactive protein; DMD, Duchenne muscular dystrophy; ECG, electrocardiogram; ESR, erythrocyte sedimentation rate; IMM, immune-mediated myositis; IVIg, intravenous immunoglobulin.
DISCUSSION
The Delphi Panel used a modified process to establish consensus considerations for the evaluation and treatment of vomiting, ALI, myocarditis, and IMM following delandistrogene moxeparvovec gene therapy infusion, as well as for the general assessment and management of the patient, which can be used by providers in the clinical setting given the paucity of available published data. The Delphi panel convened six months prior to the accelerated FDA approval of delandistrogene moxeparvovec, therefore, the considerations offered by this panel are based on experience with select TRAEs during the clinical trial program. Panelists agreed that the choice of baseline laboratory studies should be based on the prescribing information, individual clinical indications, and the provider’s clinical judgment. Consideration of corticosteroid dosing optimization assumes that a baseline corticosteroid dose was initiated pre-infusion to combat both innate and adaptive vector-induced immune responses, in addition to the patient’s pre-treatment daily dose for DMD. Post-infusion corticosteroid doses were dictated by the patient’s responses to treatment (ie, emergent AEs) and monitored during the post-infusion period according to the product label; patients may require additional monitoring for complications of higher dose steroids such as hypertension and hyperglycemia. In all cases involving a complex patient presentation, appropriate specialist consultations were suggested; it is preferable that specialists have experience with DMD and, whenever possible, rAAV vector-based gene therapy.
The panel was informed by and aware of results collected from the clinical trial safety database [14]. As of the clinical cutoff date of October 17, 2022, the delandistrogene moxeparvovec clinical trial safety database included data from 85 patients, with a mean (range) follow-up time of 2.2 years (0.5–4.8) [13]. Thus far, no long-term safety issues have been identified during ≥2 years of follow-up, and no evidence of late-onset or latent events has been observed [22]. In the early-phase clinical studies, 96% of patients (82/85) experienced treatment-emergent AEs, and 86% of patients (73/85) reported AEs that were subsequently deemed treatment-related [22]. The majority of AEs (98.5%) were mild to moderate in severity, generally occurred within the first 60 days following treatment, and resolved in weeks [22]. Vomiting was the most frequently occurring TRAE (61% ; 52/85) and was generally observed in the first 2 weeks following infusion [13]. Myocarditis occurred in 1/85 patients (1%) within three to four days after infusion [13]. ALI was reported in 31/85 (36%) patients and occurred 4–8 weeks after infusion, resolving spontaneously or with additional corticosteroid treatment [22]. IMM occurred in 1/85 patients (1%) approximately one month post-infusion and resolved with sequelae [22]. These SAEs in the clinical trials required admission to the hospital for increased monitoring and additional immunosuppression with either higher-dose steroids, plasmapheresis, or IVIg (see Supplement 3 for description of these cases). Thrombocytopenia was observed in 10/85 (12%) patients and all events were assessed as mild to moderate in severity. For this reason, thrombocytopenia was not selected for panel discussion. Other potential early acute/subacute AEs (such as thrombotic microangiopathy) have been reported following AAV-based gene therapy; however, these events have not been reported after delandistrogene moxeparvovec infusion [13, 22, 23].
A general timeline to AE onset is shown in Fig. 1. The timing and response to treatment of TRAEs observed during the clinical trials inform understanding of the underlying mechanisms, but more data is needed. For example, ALI occurred subacutely within 4 weeks post-infusion, responded well to increased corticosteroids, and was presumably related to an immune response to the viral vector. The episode of myocarditis that occurred four to seven days following infusion also suggests an immune response was the underlying cause (Fig. 1). In one reported case, an older, non-ambulatory patient with DMD experienced sudden cardiac decompensation and death that occurred within days of post-gene therapy [24]. The more acute TRAEs suggest a complement-mediated process favoring an innate immune response; discussion of this event led the panel to suggest monitoring complement levels. In contrast, cases of IMM have occurred following micro-dystrophin transgene expression in delandistrogene moxeparvovec studies and other DMD gene therapy products. Cases of IMM emerge four to eight weeks after infusion and likely result from adaptive immunogenicity against the newly expressed transgenic protein, possibly due to T cell immunity [13, 16, 25]. Although studies conducted to evaluate other gene therapies (including onasemnogene abeparvovec and resamirigene bilparvovec) [10, 11] have identified similar AEs, this Delphi panel was focused specifically on discussing those TRAEs arising from delandistrogene moxeparvovec infusion. The panel’s suggestions therefore may not be applicable to every gene therapy program currently approved due to the use of different viruses and transgenes. The pathophysiology of TRAEs observed following administration of delandistrogene moxeparvovec and other gene therapy products will be further elucidated following additional study and clinical experience.
Fig. 1
Observed Timeline of Adverse Events Following Treatment With Delandistrogene Moxeparvovec. ALP, alkaline phosphatase; ALT, alanine transaminase; GGT, gamma-glutamyl transferase; IMM, immune-mediated myositis; ULN, upper limit of normal.
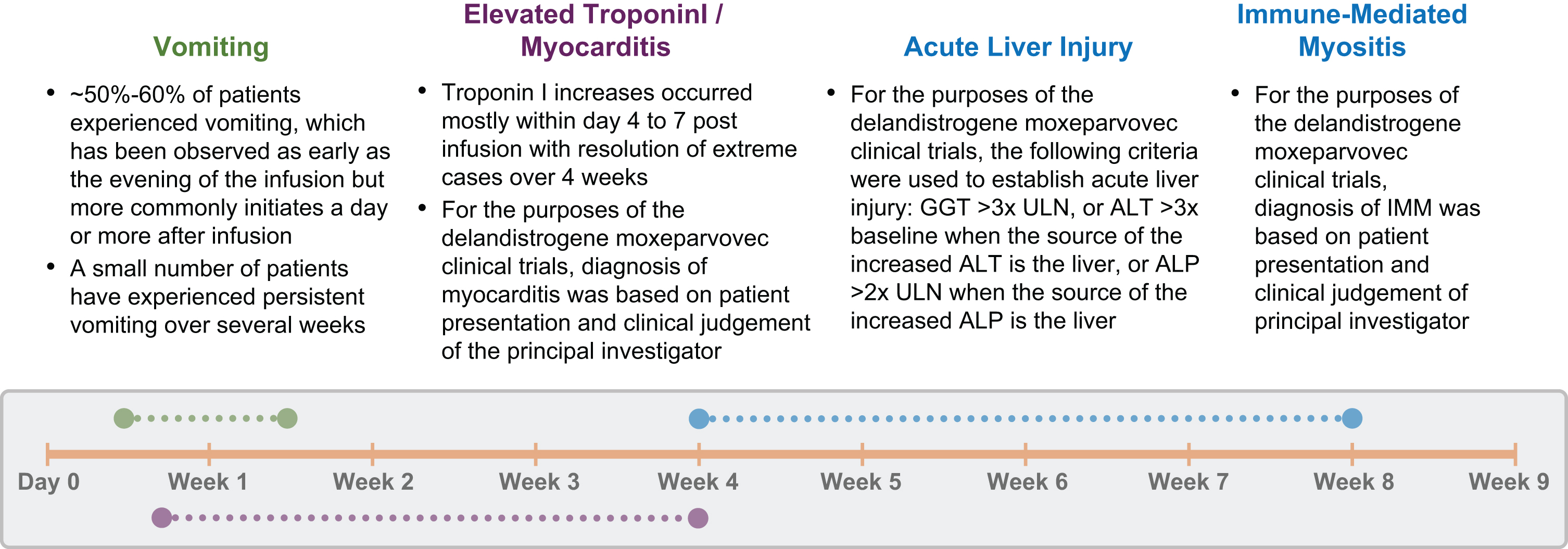
Fig. 2
Delphi Panel Modified Methodology. ALI, acute liver injury; IMM, immune-mediated myositis; TRAE, treatment-related adverse event.
Panelists were asked to consider hypothetical scenarios of these TRAEs with increasing severity and they generally responded by suggesting increasing levels of intervention, including more frequent lab monitoring and diagnostic testing, higher doses of corticosteroids, hospital admission for monitoring, and consideration of additional immunomodulation using IVIg or plasmapheresis. These recommendations pre-date the FDA-approved product label and augment the guidance now available in the prescribing information.
For the evaluation of patient candidacy and for monitoring patients who experience TRAEs, panelists suggested a battery of laboratory testing, including complement, IgG levels, and AST/ALT, among others (see Table 1). These suggestions closely parallel the study protocols used in delandistrogene moxeparvovec clinical trials and expand on the labs recommended in the FDA-approved product label, which specifies serial assessment of liver function with GGT, total bilirubin, platelets, and troponin I.
Panelists recognized certain challenges in evaluating lab abnormalities in patients with DMD; for instance, AST and ALT can derive from skeletal muscle as well as hepatocytes and these transaminases are chronically elevated in patients with DMD due to disease-related sarcolemmal fragility potentially causing muscle breakdown [2, 26]. Tracking CK levels is helpful for interpreting the source of a rise in AST/ALT, as is evaluating GGT, which derives from hepatocytes specifically [26]. Panelists also agreed that elevated GGT/AST/ALT does not inform evaluation of liver function and therefore suggested including testing for PT/INR to evaluate liver production of clotting factors. In cases of ALI not responding to treatment, viral hepatitis testing was recommended by panelists; the rationale given for this was that a patient with a prior history of viral hepatitis taking a high dose of corticosteroids could have a latent viral reactivation that should be considered. Panelists did not recommend routine screening for viral hepatitis prior to administering delandistrogene moxeparvovec; however, this should be considered if clinically indicated as a concomitant hepatitis infection has been reported as a contributing factor in a child with ALI following gene therapy [27].
Similar challenges in interpretation of abnormal lab values may arise when attempting to differentiate the cause of troponin I elevation resulting from DMD-associated cardiomyopathy versus myocarditis. Given that individuals with DMD may have baseline elevations in troponin I due to chronic myocardial injury, the delandistrogene moxeparvovec clinical program used the Brighton Collaboration myocarditis criteria which specify that myocarditis is characterized by an increase in cardiac troponin I ≥ 1 ng/mL in combination with other symptoms or abnormal test results [28]. Panelists took these criteria into consideration when developing consensus opinion and agreed that it was important to determine a threshold above which troponin I levels become clinically significant, which is challenging. One advisor suggested that the threshold should be set to the individual’s baseline and should not be tied to the reference lab range. Others suggested that a level between 2–3 times the ULN could be used as a general guide. Panelists stated that multiple tests indicating a trend of elevated troponin I levels was more significant than a single test alone. Information for determining the ideal threshold for troponin I elevation that identifies post-gene therapy myocarditis in the DMD population is lacking, largely due to the rarity of reported cases. Assay-specific variability in troponin I measurement also prevented the panel from making specific recommendations regarding ideal cutoff values to ensure high sensitivity and specificity of troponin I elevations for myocarditis.
Advantages and limitations of the Delphi process
Advantages of the Delphi process include anonymity during the polling process, which offers the opportunity to provide uncensored opinions and to vote free of peer pressure, and the capability of reaching agreement among participants in a specific area that lacks sufficient evidence-based knowledge [29]. It is also a relatively efficient, flexible, and adaptable method that can stimulate fresh ideas and provide motivation and further education for the panelists [29]. The Delphi process also has several limitations, most notably that it does not constitute empirical evidence. Results may be biased by the selection of panel members and the content of the Questionnaire, which was based on a literature search and experience in clinical trials and with gene therapy-associated AEs. In addition, consensus can be interpreted differently depending on the criteria used to define it. The panel was composed of 12 US-based participants but lacked an immunologist and other organ-specific specialists, limiting the diversity of opinion; this limitation was acknowledged by the panelists. Global perspectives were not obtained, and the patient/caregiver voice was not included. Finally, the live meeting was conducted prior to the FDA accelerated approval in the US and before the product label was finalized.
CONCLUSION
Delandistrogene moxeparvovec is a promising treatment for persons with DMD, but TRAEs arising post-infusion must be recognized and managed promptly in this patient population. A Delphi panel used a modified process to establish consensus considerations for the evaluation and treatment of vomiting, ALI, myocarditis, and IMM following gene therapy infusion, as well as for general assessment and management of the patient. These findings, based on clinical trial experience and individual health care provider experience, address the limited data available regarding management of safety issues arising post-delandistrogene moxeparvovec administration. These considerations provide additional insight and can be used by clinicians as a starting point for continued work and discussion on the management of potential TRAEs.
ACKNOWLEDGMENTS
This study was supported by Sarepta Therapeutics. The authors thank Nicole Day, PhD, MWC, and Courtney Breuel, ELS, of PharmaWrite, LLC, for medical writing and editorial assistance, which were funded by Sarepta Therapeutics. This manuscript was prepared according to the International Society for Medical Publication Professionals’ “Good Publication Practice (GPP) Guidelines for Company-Sponsored Biomedical Research: 2022 Update.”
CONFLICT OF INTEREST
CMZ: consultancy/advisory role and speaker fees with Sarepta and Optum Therapeutics; research funding from Biogen, Novartis. NLG: consultancy/advisory role with Novartis and Sarepta. AAA: consultancy/advisory role with Mirum Pharma, Albireo/Ipsen, and Sarepta Therapeutics. KDM: consultancy/advisory role with Sarepta Therapeutics, ML Bio; research funding from AMO Pharma, Capricor Therapeutics, Edgewise Therapeutics, FibroGen, Avidity, Italfarmaco, Reata, LEXEO, Biogen, Biohaven, Scholar Rock, PTC Therapeutics, Pfizer, and Sarepta Therapeutics. KNH: consultancy/advisory role with Bayer AG, Bristol-Myers Squibb, Capricor Therapeutics, Catabasis Pharmaceuticals, Daiichi Sankyo, PTC Therapeutics, Revidia Therapeutics, Sarepta Therapeutics, Inc., Stealth Biotherapeutics, Vertex Pharmaceuticals, and Wave Life Science; research funding from Sarepta Therapeutics; speakers’ bureau for NS Pharma and PTC Therapeutics; other relationship(s) with Blade Therapeutics (DSMB) and FibroGen (DSMB). RGC: equity interest in and consultancy/advisory role with LEXEO Therapeutics and XyloCor Therapeutics. AV: consultancy/advisory role with AMO Pharma, AveXis, Biogen, Edgewise Therapeutics, FibroGen, Novartis, Pfizer, PTC Therapeutics, Sarepta Therapeutics, Inc., UCB Pharma and Scholar Rock; research funding from AMO Pharma, Capricor Therapeutics, Edgewise Therapeutics, FibroGen, Muscular Dystrophy Association, Novartis, Parent Project Muscular Dystrophy, Pfizer, RegenxBio, and Sarepta Therapeutics, Inc.; other relationship(s) with MedLink Neurology for editorial services. KEG and EKS: employment with Sarepta Therapeutics, Inc. RJB: consultancy/advisory role with AavantiBio, Biogen, Reata, Sarepta Therapeutics, Inc., and Scholar Rock. AMC: consultancy/advisory role with Biohaven, Edgewise, Sarepta Therapeutics, Inc., and Scholar Rock; research funding from Biohaven, Edgewise, FibroGen, MDA, Sarepta Therapeutics, Inc., and Scholar Rock. CMP: consultancy/advisory role with AveXis/Novartis Gene Therapies, Biogen, Genentech/Roche, Sarepta Therapeutics, Inc., and Scholar Rock; research funding from AveXis/Novartis Gene Therapies, Astellas, Biogen, CSL Behring, FibroGen, PTC, Pfizer, Sarepta, and Scholar Rock; speakers’ bureau for Biogen. PBW: consultancy/advisory role with Alnylam, Astellas, Bayer, Biogen, Fore Therapeutics, RegenxBio, Roche, Sarepta, Intellia Therapeutics, Pfizer, and Verve Therapeutics. JRM: financial support from Sarepta Therapeutics, Inc., for the travel to meetings to present products sponsored by Sarepta; research funding from Sarepta Therapeutics, Inc.; patents, royalties, or other intellectual property as co-inventor of AAVrh74.MHCK; and is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer review.
DATA SHARING STATEMENT
The data supporting the findings of this study are available within the article and/or its supplementary material.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230185.
REFERENCES
[1] | Barohn RJ , Levine EJ , Olson JO , Mendell JR . Gastric hypomotility in Duchenne’s muscular dystrophy. New Engl J Med. (1988) ;319: (1):15–8. |
[2] | Kononets O , Karaiev T , Tkachenko O , Lichman L . Renal, hepatic and immune function indices in patients with Duchenne muscular dystrophy. Georgian Med News. (2020) ;(309);64–71. |
[3] | Hellebrekers DMJ , van Abeelen SAM , Catsman CE , van Kuijk SMJ , Laridon AM , Klinkenberg S , et al. Cognitive and behavioral functioning in two neurogenetic disorders; how different are these aspects in Duchenne muscular dystrophy and Neurofibromatosis type 1? PLoS One. (2022) ;17: (10):e0275803. |
[4] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol ((2010) ) 9: (1), 77–93. |
[5] | Muntoni F , Torelli S , Ferlini A . Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. (2003) ;2: (12):731–40. |
[6] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Brumbaugh D , et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. (2018) ;17: (3):251–67. |
[7] | High KA , Roncarolo MG . Gene therapy. New Engl J Med. (2019) ;381: (5):455–64. |
[8] | Manini A , Abati E , Nuredini A , Corti S , Comi GP . Adeno-associated virus (AAV)-mediated gene therapy for Duchenne muscular dystrophy: The issue of transgene persistence. Front Neurol. (2021) ;12: :814174. |
[9] | U.S. Food and Drug Administration (FDA). Approved cellular and gene therapy products 2023 [updated 05/19/2023]. Available from: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products. |
[10] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. New Engl J Med. (2017) ;377: (18):1713–22. |
[11] | Shieh PB , Kuntz NL , Dowling JJ , Müller-Felber W , Bönnemann CG , Seferian AM , et al. Safety and efficacy of gene replacement therapy for X-linked myotubular myopathy (ASPIRO): A multinational, open-label, dose-escalation trial. Lancet Neurol. (2023) ;22: (12):1125–39. |
[12] | Mendell JR , Sahenk Z , Lehman K , Nease C , Lowes LP , Miller NF , et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: A nonrandomized controlled trial. JAMA Neurol. (2020) ;77: (9):1122–31. |
[13] | Sarepta Therapeutics, Inc. SRP-9001 (delandistrogenemoxeparvovec) for Treatment of Duchenne Muscular Dystrophy. Presented at the Cellular, Tissue, and Gene Therapies Advisory Committee May 12, 2023. Available from: https://www.fda.gov/media/168095/download |
[14] | Mendell JR , Sahenk Z , Lehman KJ , Lowes LP , Reash NF , Iammarino MA , et al. Long-term safety and functional outcomes of delandistrogene moxeparvovec gene therapy in patients with Duchenne muscular dystrophy: A phase 1/2a nonrandomized trial. Muscle & Nerve. (2024) ;69: (1):93–8. |
[15] | Wang D , Tai PWL , Gao G . Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. (2019) ;18: (5):358–78. |
[16] | Bönnemann CG , Belluscio BA , Braun S , Morris C , Singh T , Muntoni F . Dystrophin immunity after gene therapy for Duchenne’s muscular dystrophy. New Engl J Med. (2023) ;388: (24):2294–6. |
[17] | Hsu C-C , Sandford BA . The Delphi technique: Making sense of consensus. Practical Assessment, Research, and Evaluation. (2007) ;12: (10):1–8. |
[18] | Okoli C , Pawlowski SD . The Delphi method as a research tool: An example, design considerations and applications. Information & Management. (2004) ;42: (1):15–29. |
[19] | Michels RE , Peters ML , Schiffers KM , Bouma PA , Hengstman GI , van Munster CE , et al. A Delphi panel on treatment of high disease activity relapsing remitting multiple sclerosis in the Netherlands. J Comp Eff Res. (2021) ;10: (2):93–100. |
[20] | Rahaghi F , Belperio JA , Fitzgerald J , Gulati M , Hallowell R , Highland KB , et al. Delphi consensus recommendations on management of dosing, adverse events, and comorbidities in the treatment of idiopathic pulmonary fibrosis with nintedanib. Clin Med Insights Circ Respir Pulm Med. (2021) ;15: :11795484211006050. |
[21] | Rivera SR , Jhamb SK , Abdel-Hamid HZ , Acsadi G , Brandsema J , Ciafaloni E , et al. Medical management of muscle weakness in Duchenne muscular dystrophy. PLoS One. (2020) ;15: (10):e0240687. |
[22] | Sarepta Therapeutics, Inc. SRP-9001 (delandistrogene moxeparvovec) for the treatment of Duchenne muscular dystrophy (DMD) – sponsor briefing document. Presented at the Cellular, Tissue, and Gene Therapies Advisory Committee May 12, 2023. Available from: https://www.fda.gov/media/168022/download |
[23] | Chand DH , Zaidman C , Arya K , Millner R , Farrar MA , Mackie FE , et al. Thrombotic microangiopathy following onasemnogene abeparvovec for spinal muscular atrophy: A case series. J Pediatr. (2021) ;231: :265–8. |
[24] | Lek A , Wong B , Keeler A , Blackwood M , Ma K , Huang S , et al. Unexpected death of a Duchenne muscular dystrophy patient in an N-of-1 trial of rAAV9-delivered CRISPR-transactivator [preprint]. MedRxiv. (2023) . |
[25] | Arjomandnejad M , Dasgupta I , Flotte TR , Keeler AM . Immunogenicity of recombinant adeno-associated virus (AAV) vectors for gene transfer. BioDrugs. (2023) ;37: (3):311–29. |
[26] | McMillan HJ , Gregas M , Darras BT , Kang PB . Serum transaminase levels in boys with Duchenne and Becker muscular dystrophy. Pediatrics. (2011) ;127: (1):e132–e6. |
[27] | Lakhotia A , Turek G , Green J , Khan M . Gene replacement therapy for spinal muscular atrophy unmasking occult hepatitis C in a pediatric patient. Muscle & Nerve. (2022) ;65: (1):E2–3. |
[28] | Sexson Tejtel SK , Munoz FM , Al-Ammouri I , Savorgnan F , Guggilla RK , Khuri-Bulos N , et al. Myocarditis and pericarditis: Case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine. (2022) ;40: (10):1499–511. |
[29] | Powell C . The Delphi technique: Myths and realities. J Adv Nurs. (2003) ;41: (4):376–82. |


