
Increase in Full-Length Dystrophin by Exon Skipping in Duchenne Muscular Dystrophy Patients with Single Exon Duplications: An Open-label Study
Abstract
Single exon duplications account for disease in a minority of Duchenne muscular dystrophy patients. Exon skipping in these patients has the potential to be highly therapeutic through restoration of full-length dystrophin expression. We conducted a 48-week open label study of casimersen and golodirsen in 3 subjects with an exon 45 or 53 duplication. Two subjects (aged 18 and 23 years) were non-ambulatory at baseline. Upper limb, pulmonary, and cardiac function appeared stable in the 2 subjects in whom they could be evaluated. Dystrophin expression increased from 0.94 % ±0.59% (mean±SD) of normal to 5.1% ±2.9% by western blot. Percent dystrophin positive fibers also rose from 14% ±17% at baseline to 50% ±42% . Our results provide initial evidence that the use of exon-skipping drugs may increase dystrophin levels in patients with single-exon duplications.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is the most common inherited myopathy in childhood, affecting approximately 1 in 5000 male newborns [1]. DMD is caused by mutations in the DMD gene, most often out-of-frame deletions causing a frame shift in the rod domain of the gene [2, 3]. For a subset of these mutations, the reading frame of the gene can be restored through skipping of an adjacent exon, leading to the expression of an internally deleted dystrophin isoform. Four such exon-skipping phosphorodiamidate morpholino oligomer (PMO) antisense oligonucleotides (ASO) have now received regulatory approval, targeting exons 45, 51, and 53 [4–8]. Studies leading to the approval of casimersen, eteplirsen, and golodirsen were performed in boys with out-of-frame deletions, and showed that these PMOs restore approximately 1% of dystrophin expression by western blot. By contrast, duplications of exons 45, 51, and 53 are much rarer, accounting for only 0.3% of DMD patients [3]. Although FDA-approved labeling does not preclude the treatment of patients with these mutations, no data have been reported in this population. We therefore conducted an open-label study to search for biomarker or clinical evidence of efficacy of PMOs in these patients, under the hypothesis that skipping of single duplicated exons has the potential to be significantly therapeutic, as it restores expression of full-length dystrophin.
MATERIALS AND METHODS
Subjects
Male subjects, aged 6 months or older, with DMD caused by duplication of exon 45, 51, or 53 were eligible for this study. Enrollment of up to 6 subjects was originally planned. Subjects aged ≥5 were required to receive corticosteroid treatment at a stable dose for the duration of the study. Key exclusion criteria included any current or prior treatment with ASOs, symptomatic cardiomyopathy, and anticipated pulmonary decompensation over the duration of the study.
Study procedures
Each subject was treated with casimersen or golodirsen (according to genotype) for a duration of 48 weeks. The study drug was administered weekly via intravenous infusion at a dose of 30 mg/kg, in keeping with FDA-approved dosing. A biopsy of the gastrocnemius was obtained at baseline and, on the contralateral side, at 48 weeks. Functional assessments and patient-reported outcome scales selected according to age and ambulatory status were administered at baseline and at 12, 24, 36, and 48 weeks. In non-ambulatory patients, these were defined in the protocol as the ACTIVE-seated workspace volume measurement and the PROMIS Upper Extremity Report [9, 10]. However, no ACTIVE-seated data was ultimately included in the analysis, as a review of raw data identified errors in acquisition, potentially attributable to equipment malfunction, at several examination time points. Pulmonary function tests were performed at the same time points as functional assessments. Echocardiograms were obtained at baseline, 12, and 24 weeks. Laboratory studies for safety monitoring were obtained regularly. This study was approved by the Nationwide Children’s Hospital Institutional Review Board. All subjects or their legal guardians provided informed consent for participation. This study was registered on ClinicalTrials.gov [NCT04179409].
DNA and RNA sequencing
DNA was extracted from muscle samples using the DNeasy Kit (Qiagen). In each subject, the target exon and flanking 300 bp intronic regions underwent amplification and Sanger sequencing to identify variants that may modify endogenous splicing or PMO binding (primer sequences provided in Supplementary Table 1).
RNA was extracted from homogenized muscle samples with the Trizol reagent (Invitrogen) and the RNA Clean and Concentrator-25 kit (Zymo). Exon skipping was quantified by RT-PCR using an adapted version of a previously published method [11]. For this analysis, amplicons were generated spanning the duplicated exon (Supplementary Table 1) and quantified by agarose gel electrophoresis.
Dystrophin quantification
Table 1
Baseline characteristics
| Subject 1 | Subject 2 | Subject 3 | |
| Age (years) | 23 | 6 | 18 |
| Ambulatory status | Non-ambulatory | Ambulatory | Non-ambulatory |
| DMD mutation | Exon 53 duplication | Exon 45 duplication | Exon 45 duplication |
| Study drug | Golodirsen | Casimersen | Casimersen |
| Corticosteroid treatment | Deflazacort (daily) | Prednisolone (twice weekly) | Deflazacort (daily) |
| Corticosteroid duration at enrollment (years) | 11 | 4 | 12 |
Western blot was performed using a modified version of a previously published method [4]. Two tissue blocks were analyzed for each time point. Protein was extracted using 4 M urea buffer. 20μg of protein was loaded on a 3–8% polyacrylamide tris-acetate gel (Life Technologies), run at 150 V for 75 minutes, and transferred onto a polyvinylidene difluoride membrane at 30 V for 75 minutes. After blocking, membranes were incubated overnight at 4°C with a dystrophin rod domain antibody (DYS1, Leica Biosciences, 1:20 dilution), washed, then incubated with mouse IgG horseradish peroxidase-conjugated secondary antibody (GE Healthcare). A standard curve prepared from healthy control tissue was included on each blot.
Quantitative immunofluorescence analysis was performed at Flagship Biosciences, using two tissue blocks for each time point [12]. Briefly, 10μm frozen sections were co-stained for dystrophin (Mandys106 antibody, Milipore Sigma, 1:50 dilution) and laminin (ab11576 antibody, Abcam, 5.5μg/mL). Whole slide images were acquired at 20X magnification on a Pannoramic Scan II fluorescent scanner (3DHISTECH). The images were then analyzed using Flagship’s proprietary algorithm to quantify percent dystrophin positive fibers (PDPF). After exclusion of artefacts and background correction, a threshold was calculated for each subject based on the sarcolemmal membrane mean dystrophin staining intensity of the baseline biopsy. Fibers with ≥30% pixels above this threshold along their perimeter were considered positive.
Statistics
Descriptive statistics were used to analyze the data. Hypothesis testing was not performed given the small sample size.
RESULTS
Subjects
Three subjects were screened for inclusion and all were enrolled in the study. Enrollment was halted when no further subjects with eligible mutations, and for whom participation was feasible, could be identified. Baseline characteristics are summarized in Table 1. Subject 3 missed one casimersen infusion at Week 34. All other infusions were completed as scheduled and all subjects completed the study. Subject 2 could not complete any functional assessments due to autism spectrum disorder.
Clinical outcomes
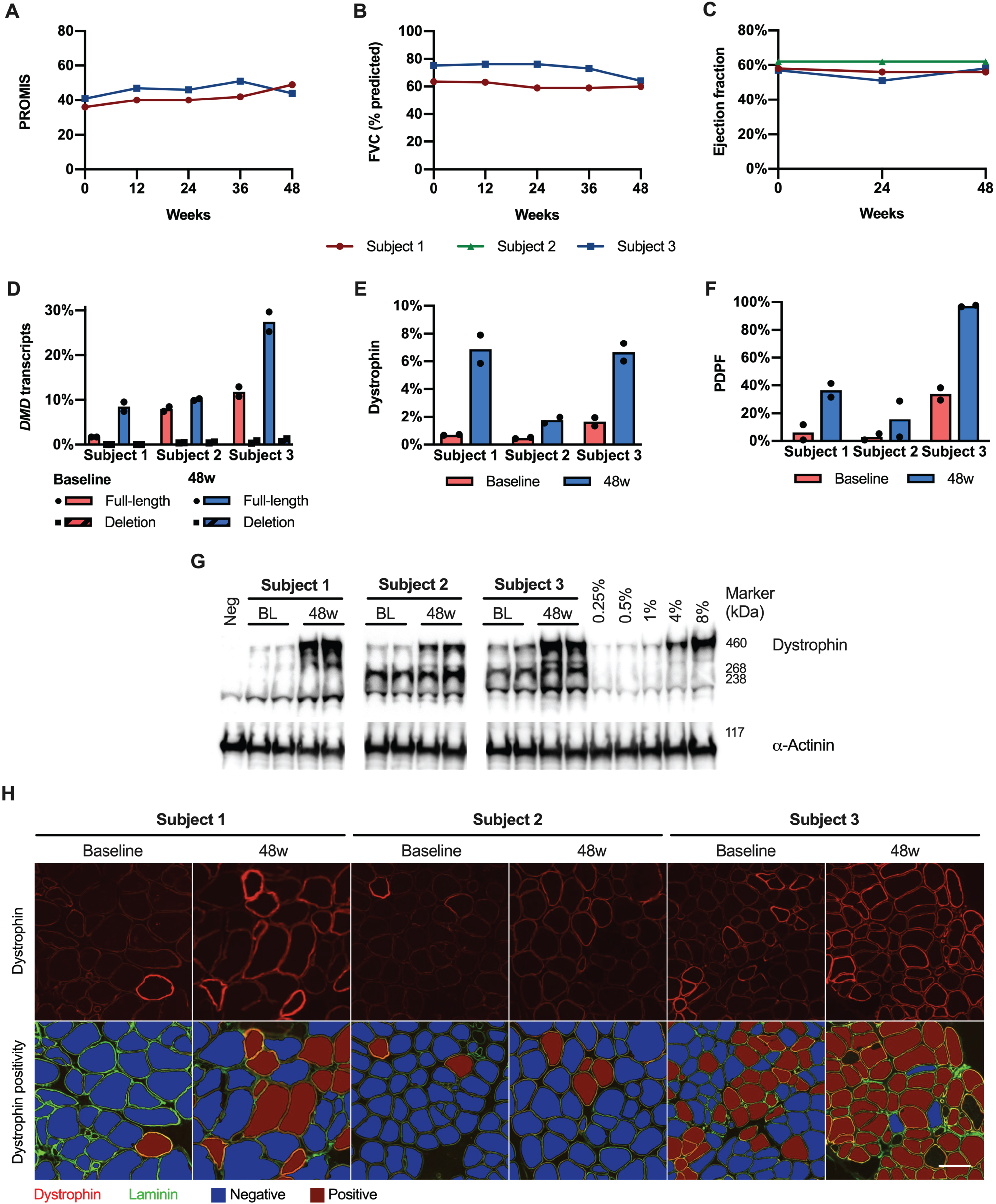
In subjects 1 and 3, the PROMIS Upper Extremity Report score and forced vital capacity remained stable throughout the duration of the study (Fig. 1 A-B). Subject 3 reported a subjective improvement in trunk control, as demonstrated by regaining the ability to remove his T-shirt, to bend down in his wheelchair to pick a water bottle from the floor, and to sit up from a semi-recumbent position (Supplementary Video). Left ventricular ejection fraction remained stable throughout the study in all subjects (Fig. 1C).
Fig.1
Exon skipping and dystrophin expression. A–C. The PROMIS score, FVC, and ejection fraction remained stable over the duration of the study. D. There was an increase in full-length DMD transcripts (relative to total DMD transcripts) in subjects 1 and 3. Deletion transcripts (i.e. skipping both copies of the target exon) were only observed at very low frequencies. E-F. Dystrophin expression increased in all subjects, as did PDPF. G. Representative western blots from each sample are shown as a composite. A 0.25% –8% standard curve prepared from healthy control tissue was included on each blot. H. Representative regions of interest stained for dystrophin are shown, along with dystrophin positivity analysis. Scale bar = 100μm. 48w, 48 weeks; BL, baseline; FVC, forced vital capacity; PDPF, percent dystrophin positive fibers.
Dystrophin expression
RT-PCR identified up to 3 transcript isoforms in each sample, and Sanger sequencing confirmed that these corresponded to skipping of none, one, or both copies of the duplicated exon (Supplementary Figure 1). There was an increase in full-length DMD transcripts in subjects 1 and 3, rising from a mean of 7.1% of all DMD transcripts at baseline, to 15% at 48 weeks (Fig. 1 D and Table 2). Transcripts skipping both copies of the target exon rose from a mean of 0.3% at baseline (likely reflecting the noise floor of our quantification) to 0.5% of all DMD transcripts at 48 weeks. Sanger sequencing did not identify any rare variants (frequency <1% in the GNOMAD v3.1.2 database) in the vicinity of the target exons that could affect skipping efficiency [13]. Dystrophin expression measured by western blot increased in all subjects over the duration of the study, from a mean of 0.94% of normal at baseline to 5.1% at 48 weeks (Supplementary Fig. 1E and G Supplementary Figure 2, and Table 2). The increase in dystrophin expression was confirmed by a second independent analysis (SupplementaryFigure 3). PDPF also increased in all subjects, from a mean of 14% at baseline to 50% at 48 weeks ( Fig. 1F and H, Supplementary Figure 4).
Table 2
Study results
| Subject 1 | Subject 2 | Subject 3 | ||||
| Baseline | 48w | Baseline | 48w | Baseline | 48w | |
| RT-PCR (% full-length transcript) | 1.7% | 8.5% | 7.8% | 10% | 12% | 27% |
| Western blot (% healthy control) | 0.70% | 6.8% | 0.48% | 1.8% | 1.6% | 6.7% |
| Percent dystrophin positive fibers | 6.2% | 37% | 3.0% | 16% | 34% | 97% |
Safety
Each subject experienced between 8 and 11 adverse events (Supplementary Table 2). No serious adverse events were recorded. Only 3 adverse events were deemed possibly related to the study drug: intermittent headache, renal calculi, and rash. No clinically significant laboratory abnormalities were detected.
DISCUSSION
Due to the rarity of the eligible genotypes, only 3 subjects could be enrolled in the present study. This study was not powered to detect a clinical benefit of the drugs due to the very limited number of subjects, clinical heterogeneity, and short follow-up. It is noteworthy that all subjects had evidence of low-level endogenous correction of the duplication at the transcript level, resulting in a baseline dystrophin expression of 0.94% of normal. This is higher than typically seen in patients with rod domain deletions, and in keeping with previous findings in cell lines derived from patients with single exon duplications [14]. Nonetheless, we found that in each subject, treatment resulted in a 1.3% to 6.1% absolute increase in dystrophin expression levels by western blot. The drug-related increase in dystrophin expression was also confirmed by immunofluorescence analysis, which showed a 13% to 63% absolute increase in PDPF. The magnitude of dystrophin restoration in these patients is greater than the approximately 1% increase typically reported in patients with rod domain deletions treated with the same drugs [4, 6]. This greater response could be attributable either to the greater stability of full-length dystrophin or to increased efficacy of ASOs in the presence of two binding sites, and argues for the continued development of therapeutic strategies aimed at restoring full-length dystrophin [15]. While skipping of both copies of the target exon can occur in vitro, we found that the in vivo incidence of skipping of both copies of a duplicated exon was very low, likely owing to the modest efficacy of current PMOs.
Subject 2 was the youngest of the 3 subjects in this study (6 years old at enrollment) and the only one who was still ambulatory. The magnitude of dystrophin restoration in this subject was smaller than in the other two. This may be related to lower dystrophin expression at baseline, increased muscle turnover, or differences in the biodistribution of the drug. Both subjects in whom clinical outcome data could be collected remained stable over the duration of the study.
This study provides initial evidence that exon skipping drugs may increase dystrophin levels in DMD patients with single exon duplications. As inferred from patients with Becker muscular dystrophy, the benefit derived from dystrophin expression depends not only on the amount, but also the function of the dystrophin isoform that is expressed [16, 17]. Low levels of expression of variant dystrophins are associated with attenuated phenotypes [18], and a level of 15% of a near-full-length dystrophin is associated with ambulation into at least the 7th decade [19]. It is reasonable to expect that expression of full-length dystrophin will result in an even larger degree of attenuation than internally-deleted isoforms at similar levels. A number of novel and more potent exon-skipping agents are currently in clinical trials, including peptide-conjugated ASOs, antibody-conjugated ASOs, and vectorized exon-skipping agents [11, 20, 21]. In treatment of patients with single-exon duplications, these agents may have advantages over alternative approaches, such as microdystrophin. Further studies will therefore be needed to identify the most effective treatment in patients amenable to more than one therapy.
ACKNOWLEDGMENTS
The authors would like to thank Ms. Lauren Bird and Ms. Kandice Roush for their assistance in conducting patient visits. This study was funded by Sarepta Therapeutics, Inc.
AUTHOR CONTRIBUTIONS
KMF, MAW, MK, and LPL conceived the study. SN, JM, PMC, EF, MK, MAI, NFR, and LPL collected and analyzed the data. SW, BJK, PW analyzed RNA-seq data. SN wrote a first draft of the manuscript.
CONFLICTS OF INTEREST
JM and PMC are employees of Sarepta Therapeutics, the manufacturer of the products used in this trial. KMF has received consulting fees from Sarepta Therapeutics for service on advisory boards. MAW has received consulting fees from Sarepta Therapeutics. LPL has received consulting fees from Sarepta Therapeutics for services as an outcomes measure consultant.
DATASETS/DATA AVAILABILITY STATEMENT
The data supporting the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy restrictions.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-230107.
REFERENCES
[1] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , Al-Dahhak R , Gastier-Foster J , et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: (3):304–13. doi: 10.1002/ana.23528. |
[2] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. (2015) ;36: (4):395–402. doi: 10.1002/humu.22758. |
[3] | Flanigan KM , Dunn DM , von Niederhausern A , Soltanzadeh P , Gappmaier E , Howard MT , et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort. Hum Mutat. (2009) ;30: (12):1657–66. doi: 10.1002/humu.21114. |
[4] | Charleston JS , Schnell FJ , Dworzak J , Donoghue C , Lewis S , Chen L , et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology. (2018) ;90: (24):e2146-54.–54. doi: 10.1212/WNL.0000000000005680. |
[5] | Wagner KR , Kuntz NL , Koenig E , East L , Upadhyay S , Han B , et al. Safety, tolerability, and pharmacokinetics of casimersen in patients with Duchenne muscular dystrophy amenable to exon 45 skipping: A randomized, double-blind, placebo-controlled, dose-titration trial. Muscle Nerve. (2021) ;64: (3):285–92. doi: 10.1002/mus.27347. |
[6] | Frank DE , Schnell FJ , Akana C , El-Husayni SH , Desjardins CA , Morgan J , et al. Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy. Neurology. (2020) ;94: (21):e2270–82. doi: 10.1212/WNL.0000000000009233. |
[7] | Clemens PR , Rao VK , Connolly AM , Harper AD , Mah JK , Smith EC , et al. Safety, Tolerability, and Efficacy of Viltolarsen in Boys With Duchenne Muscular Dystrophy Amenable to Exon 53 Skipping: A Phase 2 Randomized Clinical Trial. JAMA Neurol. (2020) ;77: (8):982–91. doi: 10.1001/jamaneurol.2020.1264 |
[8] | Komaki H , Takeshima Y , Matsumura T , Ozasa S , Funato M , Takeshita E , et al. Viltolarsen in Japanese Duchenne muscular dystrophy patients: A phase 1/2 study. Ann Clin Transl Neurol. (2020) ;7: (12):2393–408. doi: 10.1002/acn3.51235. |
[9] | Lowes LP , Alfano LN , Crawfis R , Berry K , Yin H , Dvorchik I , et al. Reliability and validity of active-seated: An outcome in dystrophinopathy. Muscle Nerve. (2015) ;52: (3):356–62. doi: 10.1002/mus.24557 |
[10] | Rose M , Bjorner JB , Becker J , Fries JF , Ware JE Evaluation of a preliminary physical function item bank supported the expected advantages of the Patient-Reported Outcomes Measurement Information System (PROMIS). J Clin Epidemiol. (2008) ;61: (1):17–33. doi: 10.1016/j.jclinepi.2006.06.025 |
[11] | Wein N , Vetter TA , Vulin A , Simmons TR , Frair EC , Bradley AJ , et al. Systemic delivery of an AAV9 exon-skipping vector significantly improves or prevents features of Duchenne muscular dystrophy in the Dup2 mouse. Mol Ther Methods Clin Dev. (2022) ;26: :279–93. doi: 10.1016/j.omtm.2022.07.005 |
[12] | Aeffner F , Faelan C , Moore SA , Moody A , Black JC , Charleston JS , et al. Validation of a Muscle-Specific Tissue Image Analysis Tool for Quantitative Assessment of Dystrophin Staining in Frozen Muscle Biopsies. Arch Pathol Lab Med. (2019) ;143: (2):197–205. doi: 10.5858/arpa.2017-0536-OA |
[13] | Chen S , Francioli LC , Goodrich JK , Collins RL , Kanai M , Wang Q , et al.A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv. 2022:2022.03.20.485034. doi: 10.1101/2022.03.20.485034 |
[14] | Wein N , Vulin A , Findlay AR , Gumienny F , Huang N , Wilton SD , et al. Efficient Skipping of Single Exon Duplications in DMD Patient-Derived Cell Lines Using an Antisense Oligonucleotide Approach. J Neuromuscul Dis. (2017) ;4: (3):199–207. doi: 10.3233/JND-170233 |
[15] | Henderson DM , Belanto JJ , Li B , Heun-Johnson H , Ervasti JM Internal deletion compromises the stability of dystrophin. Hum Mol Genet. (2011) ;20: (15):2955–63. doi: 10.1093/hmg/ddr199 |
[16] | van den Bergen JC , Wokke BH , Janson AA , van Duinen SG , Hulsker MA , Ginjaar HB , et al. Dystrophin levels and clinical severity in Becker muscular dystrophy patients. J Neurol Neurosurg Psychiatry. (2014) ;85: (7):747–53. doi: 10.1136/jnnp-2013-306350 |
[17] | Nicolas A , Raguenes-Nicol C , Ben Yaou R , Ameziane-Le Hir S , Cheron A , Vie V , et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum Mol Genet. (2015) ;24: (5):1267–79. doi: 10.1093/hmg/ddu537 |
[18] | de Feraudy Y , Ben Yaou R , Wahbi K , Stalens C , Stantzou A , Laugel V , et al. Very Low Residual Dystrophin Quantity Is Associated with Milder Dystrophinopathy. Ann Neurol. (2021) ;89: (2):280–92. doi: 10.1002/ana.25951 |
[19] | Gurvich OL , Maiti B , Weiss RB , Aggarwal G , Howard MT , Flanigan KM DMD exon 1 truncating point mutations: Amelioration of phenotype by alternative translation initiation in exon 6. Hum Mutat. (2009) ;30: (4):633–40. doi: 10.1002/humu.20913 |
[20] | Desjardins CA , Yao M , Hall J , O’Donnell E , Venkatesan R , Spring S , et al. Enhanced exon skipping and prolonged dystrophin restoration achieved by TfR1-targeted delivery of antisense oligonucleotide using FORCE conjugation in mdx mice. Nucleic Acids Res. (2020) ;50: (20):11401–14. doi: 10.1093/nar/gkac641 |
[21] | Gan L , Wu LCL , Wood JA , Yao M , Treleaven CM , Estrella NL , et al. A cell-penetrating peptide enhances delivery and efficacy of phosphorodiamidate morpholino oligomers in mdx mice. Mol Ther Nucleic Acids.17-27. (2022) ;30: :17–27. doi: 10.1016/j.omtn.2022.08.019. |


