
Early-Onset Autosomal Dominant Myopathy with Vacuolated Fibers and Tubular Aggregates but No Periodic Paralysis, in a Patient with the c.1583G>A (p.R528H) mutation in the CACNA1S Gene
Abstract
Dominant mutations in CACNA1S gene mainly causes hypokalemic periodic paralysis (PP)(hypoPP). A 68-year-old male proband developed a progressive proximal weakness from the age of 35. Muscle biopsy showed atrophic fibers with vacuoles containing tubular aggregates. Exome sequencing revealed a heterozygous p.R528H (c.1583G>A) mutation in the CACNA1S gene. CACNA1S-related HypoPP evolving to persistent myopathy in late adulthood is a well-known clinical condition. However, isolated progressive myopathy (without PP) was only exceptionally reported and never with an early onset. Reporting a case of early onset CACNA1S-related myopathy in a patient with no HypoPP we intend to alert clinicians to consider it in the differential diagnosis of younger adult-onset myopathies especially when featuring vacuolar changes.
INTRODUCTION
The CACNA1S (Cav1.1) gene encodes the pore-forming subunit of the dihydropyridine receptor (DHPR) located on the skeletal muscle T-tubules. DHPR is a voltage-gated L-type Ca2 + channel responsible for the process of excitation–contraction coupling (ECC) by interacting with the ryanodine receptor (RYR1) at the triad: this interplay triggers muscle contraction. Mutations in the CACNA1S gene result in a paradoxical depolarization that reduces fiber excitability and contractility during an attack of HypoPP caused by an anomalous gating pore leakage current in mutant CaV1.1 channels [1, 2]. Dominant mutations in the CACNA1S gene are known to cause hypokalemic periodic paralysis (HypoPP) and malignant hyperthermia (MHS5) [3–6]. More recently, dominant and recessive mutations in the CACNA1S gene have been linked to a novel class of congenital myopathies and to a late-onset myopathy without HypoPP [7, 8]. In describing progressive early onset myopathy with heterozygous CACNA1S mutation we aim to expand the phenotypical spectrum of CACNA1S gene mutations beyond strict channelopathies to include other myopathies.
CASE REPORT
The proband (PII.2), a male Caucasian patient aged 68, coming from a family of 5 siblings (Fig. 1), without evidence of consanguinity, was referred to our tertiary center in 2020, for progressive weakness of the four limbs evolving since the age of 35. The patient had an asymptomatic 49-year-old daughter, a living 58-year-old brother (who was also asymptomatic), a deceased
Fig. 1
Family tree. *p.R528H (c.1583G>A).
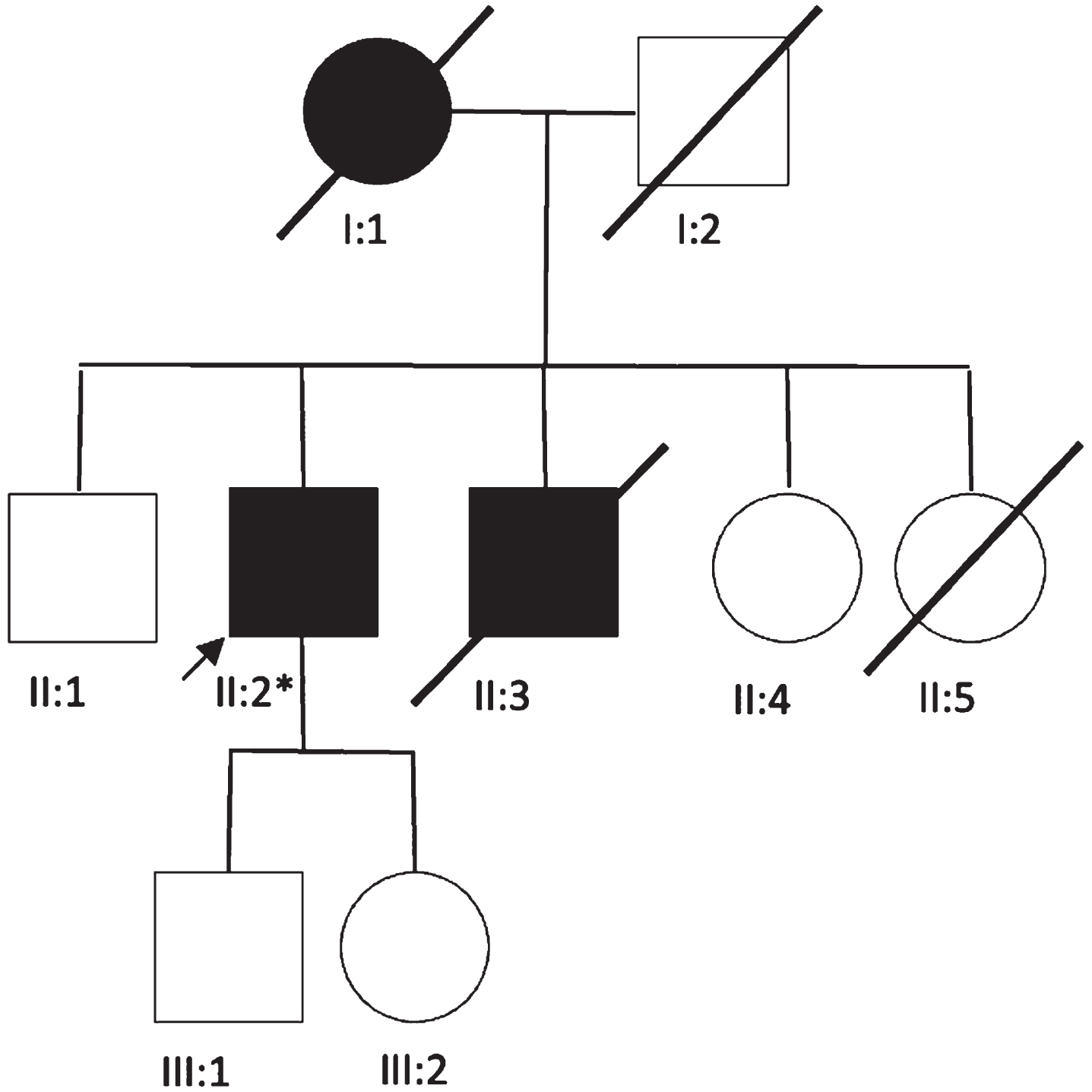
Fig. 2
Lower limb MRI. Major fatty replacement of the thigh muscles with partial sparing of the adductor muscle, the rectus femoris and the gracilis muscles (arrows).
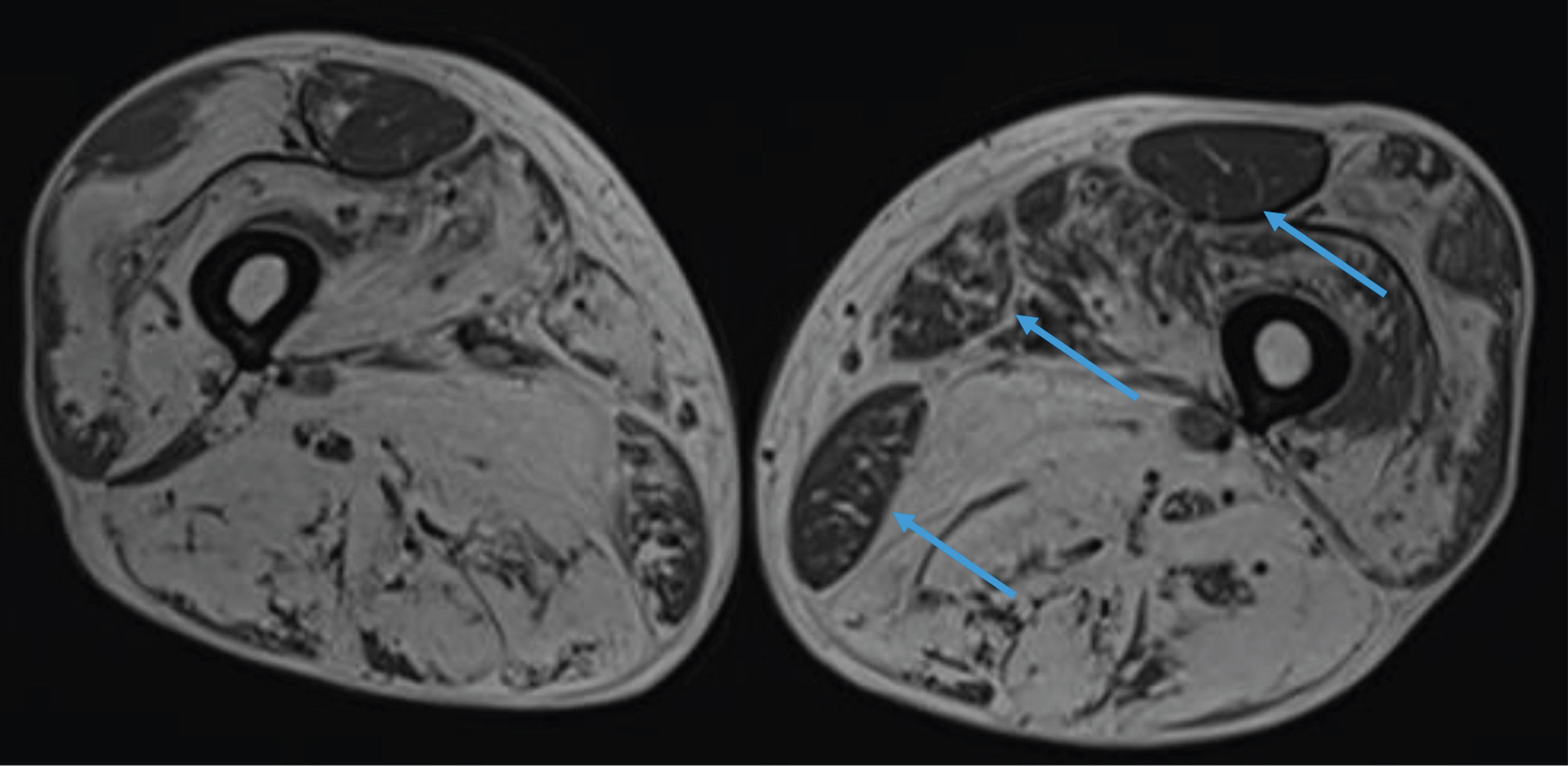
Fig. 3
MRI of the pelvis. Complete fatty replacement of the psoas muscles (arrows).
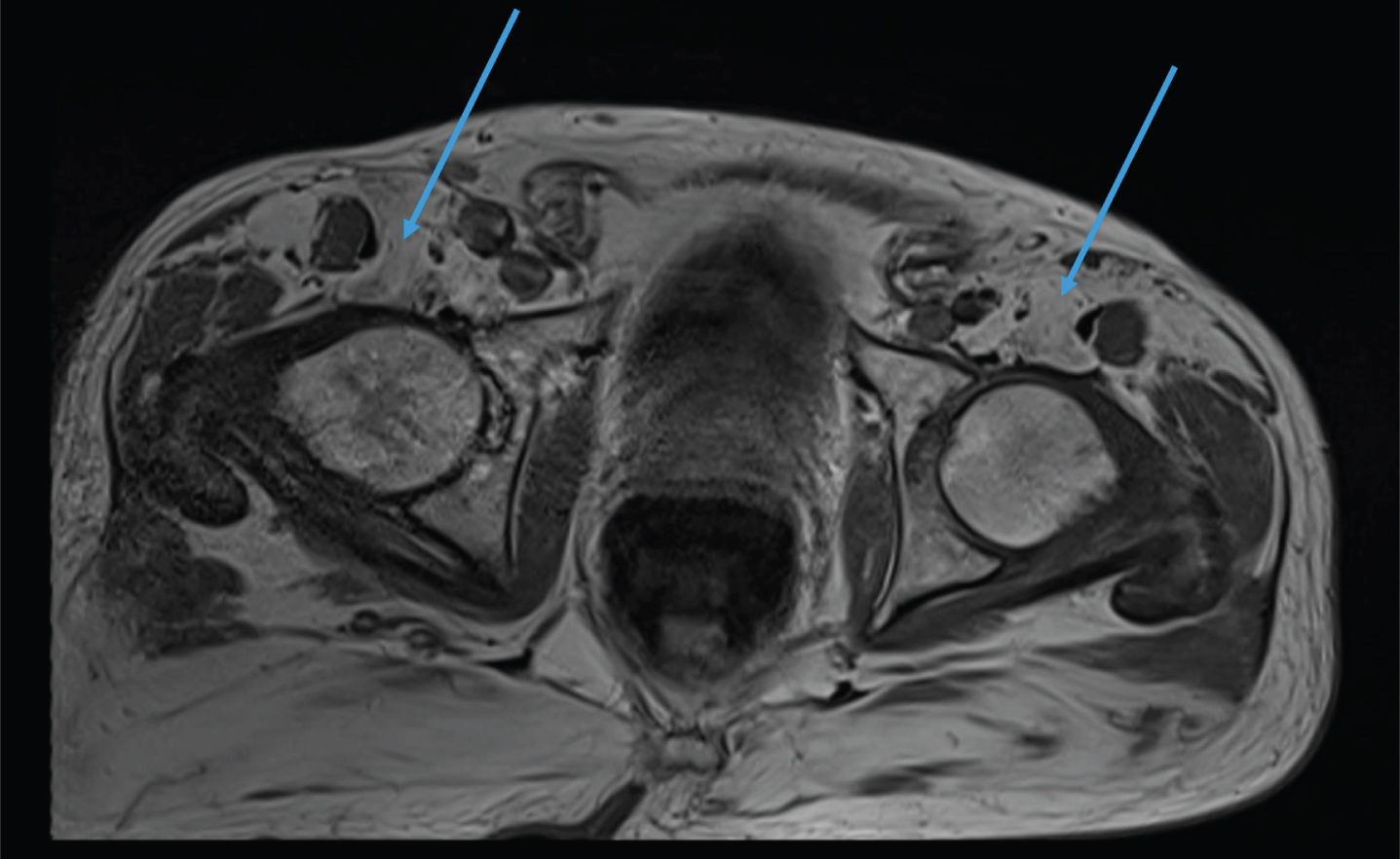
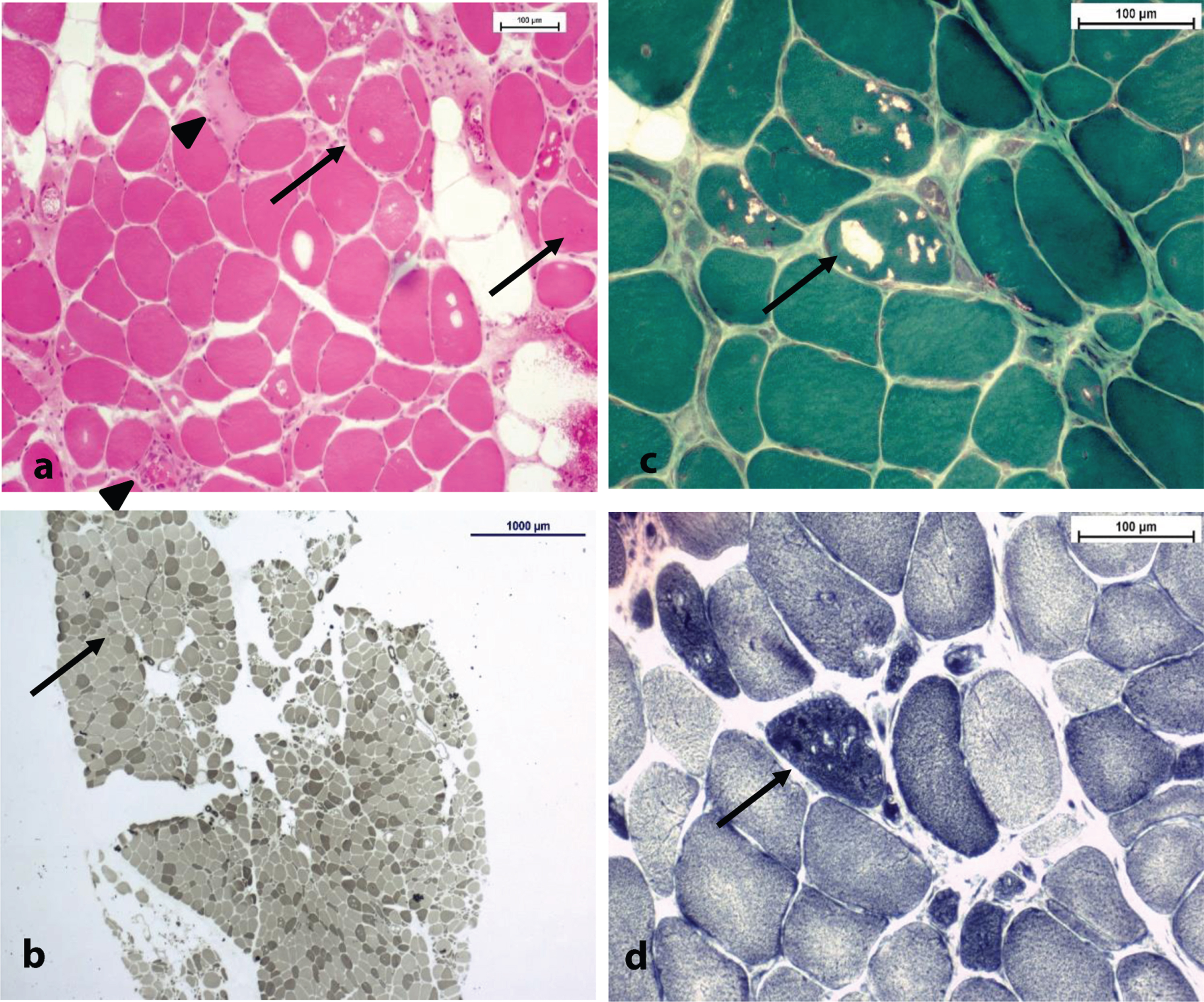
Fig. 4
(a) H& E stain x10; note the internalized nuclei (arrows), necrotic fibers (spike arrows) and the numerous atrophic fibers (non-indicated) bars = 100μm. (b) ATPase pH 9,4 stain x2; normal fiber-type distribution, as Type II atrophic fibers can be spotted (arrow); bars = 1000μm. Type I predominance. (c,d) Same spot in Gömöri trichrome stain (c) and NADH (d) stain x20, respectively; note the variable aspect of vacuoles (arrow) in each stain. Tubular aggregates have a dark staining pattern in NADH ((d) - arrow) despite the aspect of vacuoles (+/- rimmed) in Gömöri trichrome stain ((c) - arrow) bars = 100μm.
DISCUSSION
Primary hypokalemic periodic paralysis (HypoPP) typically presents before the age of 20 with transient attacks of muscle weakness. Severity and duration of the episodes vary, lasting from hours to days [9]. Paralytic attacks are usually precipitated by carbohydrate-rich meals, rest after exercise and stress-related illness; all are conditions that favor the entry of potassium into cells. The HypoPP phenotype classically involves normal muscle-strength between paralytic episodes. Some patients may however develop a permanent muscle-weakness over time, generally after the fourth decade, whereby paralytic attacks decrease in frequency [10]. The association between periodic paralysis and permanent muscle weakness has long fueled debates, since the relationship between the frequency and severity of previous attacks and the myopathy has not been well-established. It is thought that weakness might be the result of a more complex process than just the damage accumulated by repetitive attacks [11]. Pathologically, HypoPP-related myopathy is characterized by the presence in muscle fibers of vacuoles containing granular material and abnormal dilatation of the sarcoplasmic reticulum [12]. Vacuolar changes, though non-pathognomonic, are considered as the histological hallmark of HypoPP and can be found at disease-onset. Links et al. stated in 1990 that vacuolar changes were the primum movens of a myopathy unrelated to paralysis attacks [11]. Many authors nowadays agree with this statement [8]. Concerning the distribution of weakness, HypoPP related myopathy shows both a proximal and distal involvement, mainly involving the lower limbs. Our patient presented a major, symmetrical involvement of the posterior compartment of the thighs and a sparing of rectus femoris and gracilis as depicted in previous observations [8]. In the family we are reporting here, the phenotypic expression in the different family members was heterogeneous: in the proband, the myopathy was early-onset and with no PP. The two other affected family-members however seemingly presented HypoPP based on their clinical history. It is noteworthy that the p.R528H mutation was previously identified in a large family as causing HypoPP and a late-onset myopathy without paralytic attacks [13]. These findings were further confirmed in a more recent, large cohort study on the CACNA1S mutations spectrum [8]. Our case additionally highlights the intra-familial phenotypic variability of CACNA1S gene mutation and related HypoPP. The question of what determines this pronounced intra-familial variability remains open and should be investigated in future studies. In summary, the proband in this affected family presented with an early-onset myopathy evolving since the age of 35. CACNA1S-related pathogenic mutation can rarely present with a purely myopathic phenotype. To the best of our knowledge, this had been previously reported in only 7 patients, and never with a clinical onset younger than the age of 35 [8, 13, 14]. Our report on this progressive early-onset myopathy associated with a heterozygous CACNA1S mutation expands the phenotypical spectrum of CACNA1S-related muscle diseases and should imply considering it in the differential diagnosis of younger adult-onset myopathies especially when featuring myopathies with tubular aggregates.
ACKNOWLEDGMENTS
We thank the patient and his family for participating in the assessment and for agreeing to the publication of the findings.
MB, IV, LD and GR author(s) of this publication are members of the European Reference Network for Neuromuscular Diseases - Project ID N° 870177.
We thank Dr Afarine MADANI and Dr Stefano DINH for their help in editing the radiological images.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Jurkat-Rott K , Lehmann-Horn F , Elbaz A , Heine R , Gregg RG , Hogan K et al. A calcium channel mutation causing hypokalemic periodic paralysis. Hum Mol Genet. (1994) ;3: (8):1415–9. doi: 10.1093/hmg/3.8.1415. . PMID: 7987325 |
[2] | Rüdel R , Lehmann-Horn F , Ricker K , Küther G . Hypokalemic periodic paralysis: in vitro investigation of muscle fiber membrane parameters. Muscle Nerve. (1984) ;7: (2):110–20. doi: 10.1002/mus.880070205. PMID: 6325904 |
[3] | Elbaz A , Vale-Santos J , Jurkat-Rott K , Lapie P , Ophoff RA , Bady B et al. Hypokalemic periodic paralysis and the dihydropyridine receptor (CACNL1A3): genotype/phenotype correlations for two predominant mutations and evidence for the absence of a founder effect in 16 caucasian families. Am J Hum Genet. (1995) ;56: (2):374–80. PMID: 7847370; PMCID: PMC1801148. |
[4] | Jurkat-Rott K , Weber MA , Fauler M , Guo XH , Holzherr BD , Paczulla A et al. K+-dependent paradoxical membrane depolarization and Na+ overload, major and reversible contributors to weakness by ion channel leaks. Proc Natl Acad Sci U S A. (2009) ;106: (10):4036–41. doi: 10.1073/pnas.0811277106. Epub 2009 Feb 18. PMID: 19225109; PMCID: PMC2644652. |
[5] | Ptácek LJ , Tawil R , Griggs RC , Engel AG , Layzer RB , Kwieciński H , et al. Dihydropyridine receptor mutations cause hypokalemic periodic paralysis. Cell. (1994) ;77: (6):863–8. doi: 10.1016/0092-8674(94)90135-x. PMID: 8004673. |
[6] | Monnier N , Procaccio V , Stieglitz P , Lunardi J . Malignant-hyperthermia susceptibility is associated with a mutation of the alpha 1-subunit of the human dihydropyridine-sensitive L-type voltage-dependent calcium-channel receptor in skeletal muscle. Am J Hum Genet. (1997) ;60: (6):1316–25. doi: 10.1086/515454. PMID: 9199552; PMCID: PMC1716149. |
[7] | Schartner V , Romero NB , Donkervoort S , Treves S , Munot P , Pierson TM et al. Dihydropyridine receptor (DHPR, CACNA1S) congenital myopathy. Acta Neuropathol. (2017) ;133: (4):517–33. doi: 10.1007/s00401-016-1656-8.. Epub 2016 Dec 23. PMID: 28012042. |
[8] | Holm-Yildiz S , Witting N , Dahlqvist J , de Stricker Borch J , Solheim T , Fornander F et al. Permanent muscle weakness in hypokalemic periodic paralysis. Neurology. (2020) ;95: (4):e342–e352. doi: 10.1212/WNL.0000000000009828. Epub 2020 Jun 24. PMID: 32580975. |
[9] | Fouad G , Dalakas M , Servidei S , Mendell JR , Van den Bergh P , Angelini et al. Genotype-phenotype correlations of DHP receptor alpha 1-subunit gene mutations causing hypokalemic periodic paralysis. Neuromuscul Disord. (1997) ;7: (1):33–8. doi: 10.1016/s0960-8966(96)00401-4. PMID: 9132138. |
[10] | Miller TM , Dias da Silva MR , Miller HA , Kwiecinski H , Mendell JR , Tawil R et al. Correlating phenotype and genotype in the periodic paralyses. Neurology. (2004) ;63: (9):1647–55. doi: 10.1212/01.wnl.0000143383.91137.00. PMID: 15534250. |
[11] | Links TP , Zwarts MJ , Wilmink JT , Molenaar WM , Oosterhuis HJ . Permanent muscle weakness in familial hypokalaemic periodic paralysis. Clinical, radiological and pathological aspects. Brain. (1990) ;113: (6):1873–89. doi: 10.1093/brain/113.6.1873. PMID: 2276049. |
[12] | Gold R , Reichmann H . Muscle pathology correlates with permanent weakness in hypokalemic periodic paralysis: a case report. Acta Neuropathol. (1992) ;84: (2):202–6. doi: 10.1007/BF00311396. PMID: 1381862. |
[13] | Chalissery AJ , Munteanu T , Langan Y , Brett F , Redmond J . Diverse phenotype of hypokalaemic periodic paralysis within a family. Pract Neurol. (2018) ;18: (1):60–65. doi: 10.1136/practneurol-2017-001677. Epub 2017 Sep 28. PMID: 28972032. |
[14] | Sternberg D , Maisonobe T , Jurkat-Rott K , Nicole S , Launay E , Chauveau D , et al. Hypokalaemic periodic paralysis type 2 caused by mutations at codon 672 in the muscle sodium channel gene SCN4A. Brain. (2001) ;124: (Pt 6):1091–9. doi: 10.1093/brain/124.6.1091. PMID: 11353725. |


