
Brain MRI Abnormalities, Epilepsy and Intellectual Disability in LAMA2 Related Dystrophy – a Genotype/Phenotype Correlation
Abstract
Background:
LAMA2-related muscular dystrophy is a disorder that causes muscle weakness and varies in severity, from a severe, congenital type to a milder, late-onset form. However, the disease does not only affect the muscles, but has systemic involvement and can lead to alterations such as brain malformation, epilepsy and intellectual disability.
Objective:
Describe the frequency of cortical malformations, epilepsy and intellectual disability in LAMA2-RD in a Brazilian cohort and correlate the neurological findings to genetic and motor function.
Methods:
This is an observational study of 52 LAMA2-RD patients, who were divided into motor function subgroups and compared based on brain MRI findings, epilepsy, intellectual disability, and type of variants and variant domains.
Results:
44 patients (84.6%) were only able to sit, and 8 patients (15.4%) were able to walk. 10 patients (19.2%) presented with cortical malformations (polymicrogyria, lissencephaly-pachygyria, and cobblestone),10 patients (19.2%) presented with epilepsy, and 8 (15.4%) had intellectual disability. CNS manifestations correlated with a more severe motor phenotype and none of the patients able to walk presented with cortical malformation or epilepsy. There was a relation between gene variants affecting the laminin-α2 LG-domain and the presence of brain malformation (P = 0.016). There was also a relation between the presence of null variants and central nervous system involvement. A new brazilian possible founder variant was found in 11 patients (21,15%) (c.1255del; p. Ile419Leufs*4).
Conclusion:
Cortical malformations, epilepsy and intellectual disability are more frequent among LAMA2-RD patients than previously reported and correlate with motor function severity and the presence of variants affecting the laminin-α2 LG domain. This brings more insight fore phenotype-genotype correlations, shows the importance of reviewing the brain MRI of patients with LAMA2-RD and allows greater attention to the risk of brain malformation, epilepsy, and intellectual disability in those patients with variants that affect the LG domain.
INTRODUCTION
LAMA2 related dystrophies (LAMA2-RD) consists of a continuous spectrum of diseases ranging from a severe congenital muscular dystrophy) [1, 2] to a milder late-onset form [3, 4]. The congenital form is usually characterized by hypotonia, muscle weakness, delayed motor development milestones with inability to achieve independent ambulation, joint contractures, and early restrictive pulmonary disease [1, 2]. The milder form can present itself with manifestations from early childhood to adulthood and affected individuals present proximal muscle weakness, joint contractures, and respiratory insufficiency[4, 5].
LAMA2 has autosomal recessive inheritance and patients develop muscle weakness, high CK, multiple joint deformities, restrictive ventilatory disorder, and white matter abnormalities in T2-weighted imaging (T2-WI) and in fluid-attenuated inversion recovery (FLAIR) in brain magnetic resonance images (MRI). Sometimes patients present cortical malformations [6–8], epilepsy and intellectual disability[9, 10].
The LAMA2 gene codifies the laminin-α2 protein, which is part of the laminin-211 (merosin), a major constituent of the basement membrane [7]. Laminin-211 consists of three similar but non-identical polypeptide chains (α2, β1 and γ1), which assemble into a T-shaped heterotrimer with two short arms and one long arm.
Laminin-α2 has three domains: The LN domain (LN), which is responsible for protein self-assembly; the coiled coil domain (LCC); and the LG domain (LG), which binds to the glycosylated residues of α-dystroglycan and to the integrins [11]. Laminin-α2 is a widely expressed protein and is essential in skeletal muscle fibers but also important to the brain’s functions[8, 12, 13].
To date, the presence of brain malformation, epilepsy and intellectual disability in LAMA2-RD hasn’t been well characterized and the correlation between motor function and cortical malformation is not well established. Neither is the relation between type and position of variants and central nervous system findings. The aim of this study was to characterize the central nervous system (CNS) manifestations in a large cohort of patients with LAMA2-RD and determine whether they correlate with motor function and geneticfindings.
MATERIALS AND METHODS
Study design
This is an observational study conducted from March 2018 to May 2022 at the Outpatient Service of Neuromuscular Disorders at the HCFMUSP. Sixty patients evaluated in this period had a clinical/histological diagnosis of LAMA2-RD. Patients without genetic tests or a brain MRI were excluded. Therefore, 52 patients were included in this study.
Informed consent for study participation and use of clinical photographs were obtained from patients/parents/legal guardians, and the institution’s ethics committee approved the project (CAAE: 87782318.2.0000.0068).
Patient classification
For a correlation between motor function and cortical malformation, epilepsy and intellectual disability, we classified the patients, based on motor severity, into 1) not able to walk, 2) able to walk.
CNS analysis
All patients had underwent a brain MRI on either 1.5 or 3.0 Tesla machines during their clinical investigation. The three-dimensional T1-weighted, diffusion-weighted imaging, gradient-echo T2, T2-WI and FLAIR imaging were performed. The first author reevaluated all the images, and a second reevaluation was performed by an experienced neuroradiologists (SFF, LTL, AJR). In those cases where there was a divergence, there was discussion between the evaluators to reach an agreement. White matter signal alterations were classified in [1] milder: involvement of periventricular white matter with/without deep white matter, but without subcortical involvement [2] more Severe: involvement of periventricular and deep white matter, with subcortical white matter. Cortical malformation was described according to type and position and classified as extensive when more than one lobe was affected.
Intellectual disability was suspected when the patient presented speech and language delay. Mini-mental state exam was performed in all patients older than 14 years old and the modified mini-mental state exam in all patients under 14 years old. The Montreal Cognitive Assessment Scale (MOCA) were performed in all patients. Values below 31 of 37 points in the modified mini mental; 27 of 35 points in the mini mental assessment suggested mental impairment and values below 26 of 30 points in the MOCA scale were considered abnormal [14]. The epilepsy definition and classification followed the International League Against Epilepsy’s criteria [15].
Genetic analysis
Molecular studies were performed through commercial genetic panels and/or whole-exome sequencing. Variant interpretation followed the standards and guidelines for the classification of sequence variants the American College of Medical Genetics and Genomics proposed [16]. We annotated variants using ANNOVAR (http://annovar.openbioinformatics.org) and filtered them with custom scripts. We used the Clinvar database (https://www.ncbi.nlm.nih.gov) and Pubmed (https://pubmed.ncbi.nlm.nih.gob) to identify previously reported pathogenic variants. We used the UCSC Genome Browser (https://genome.ucsc.edu) and Gnomad (https://gnomad.broadinstitute.org) to determine each variant’s frequency in the general population. We used species conservation, amino acid conservation, bioinformatic predictors (Polyphen (http://genetics.bwh.harvard.edu/pph2/)), and SpliceAI (https://spliceailookup.broadinstitute.org) to predict the missense’s and splice-site variant’s effects.
To analyze if there was a correlation between protein domain and cortical malformation, the variants were divided according to the domain into LN, LCC, and LG domains.
Statistical analysis
We compared two groups of patients (ambulantory and not ambulantory), based on brain MRI findings, epilepsy, intellectual disability, and type of variants. Variant domains were compared based on cortical malformation, epilepsy, and intellectual disability. And the presence of missense variants was compared based on the patient’s ability to walk, presence of cortical malformation, epilepsy, and intellectual disability.
We presented continuous variables as mean and discrete and categorical variables as counts and percentages. We conducted univariate analysis to describe and calculate the prevalence of the patients’ characteristics. We used the Fisher’s exact test for count data with a 95% confidence interval (95% CI) and calculated odds ratios. All tests were two-sided, and we considered a P value of.05 the threshold for statistical significance. We conducted statistical analyses using R version 3.5.0.
RESULTS
The study cohort comprised 52 patients from 50 families. Of the patients, 30 were female (57.7%), and 22 were male (42.3%), and the patients were between 2 and 27 years old at last assessment (mean = 11.4; SD = 6.2).
Motor severity classification
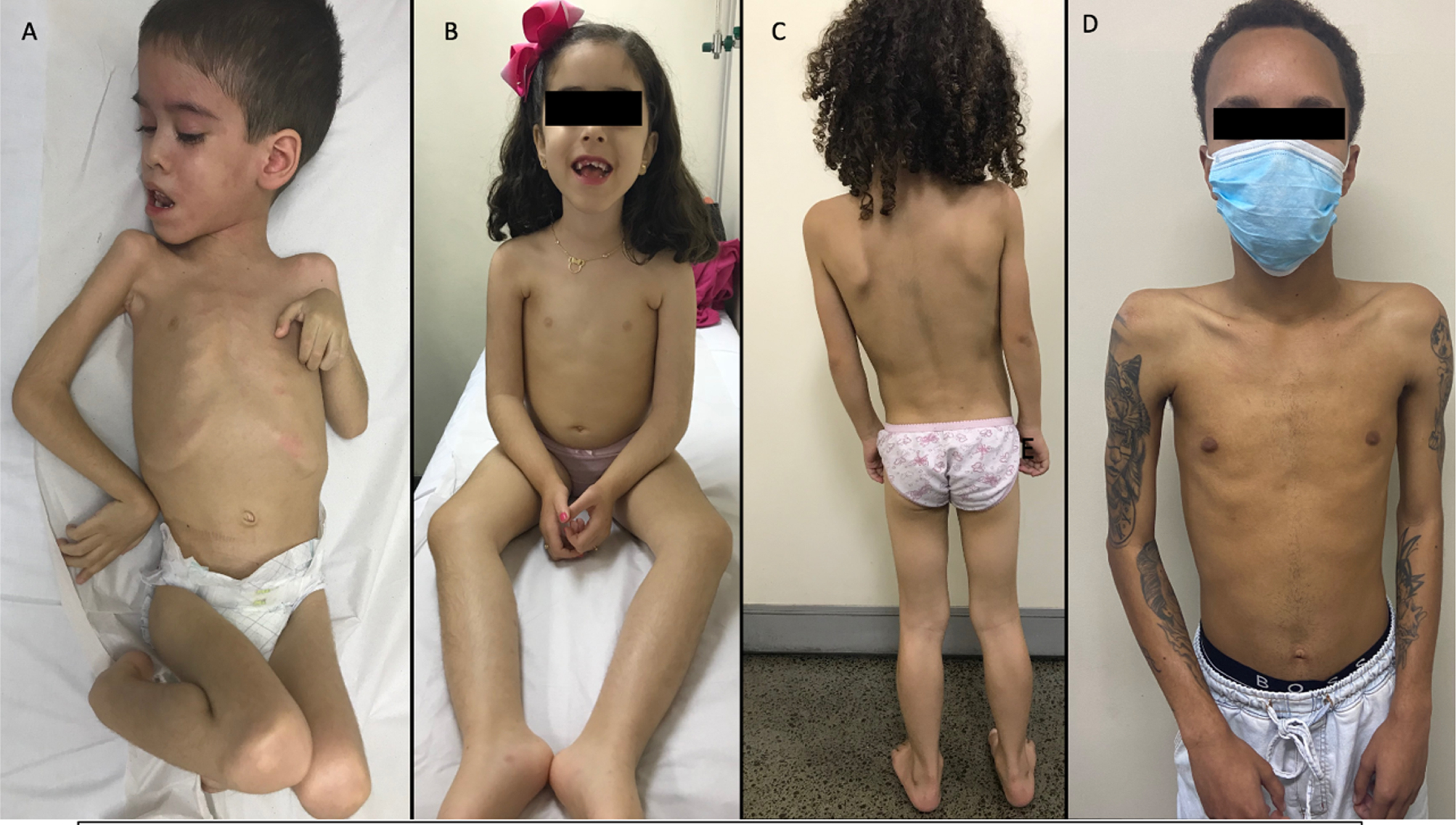
44 patients (84.6%) were unable to walk, and two of them were unable to sit (3.8%). 8 patients (15.4%) were able to walk. Of those 8 patients, 2 became wheelchair dependent at the age of 8, and 2 had the limb girdle muscular dystrophy form and acquired gait by the expected age (Fig. 1).
Fig. 1
LAMA2-RD phenotypes. (A) Patient never able to sit, (B) Patient able to sit, but unable to walk, (C) Patient able to walk, with delayed motor development, and (D) Patient able to walk with LGMD phenotype.
CNS abnormalities
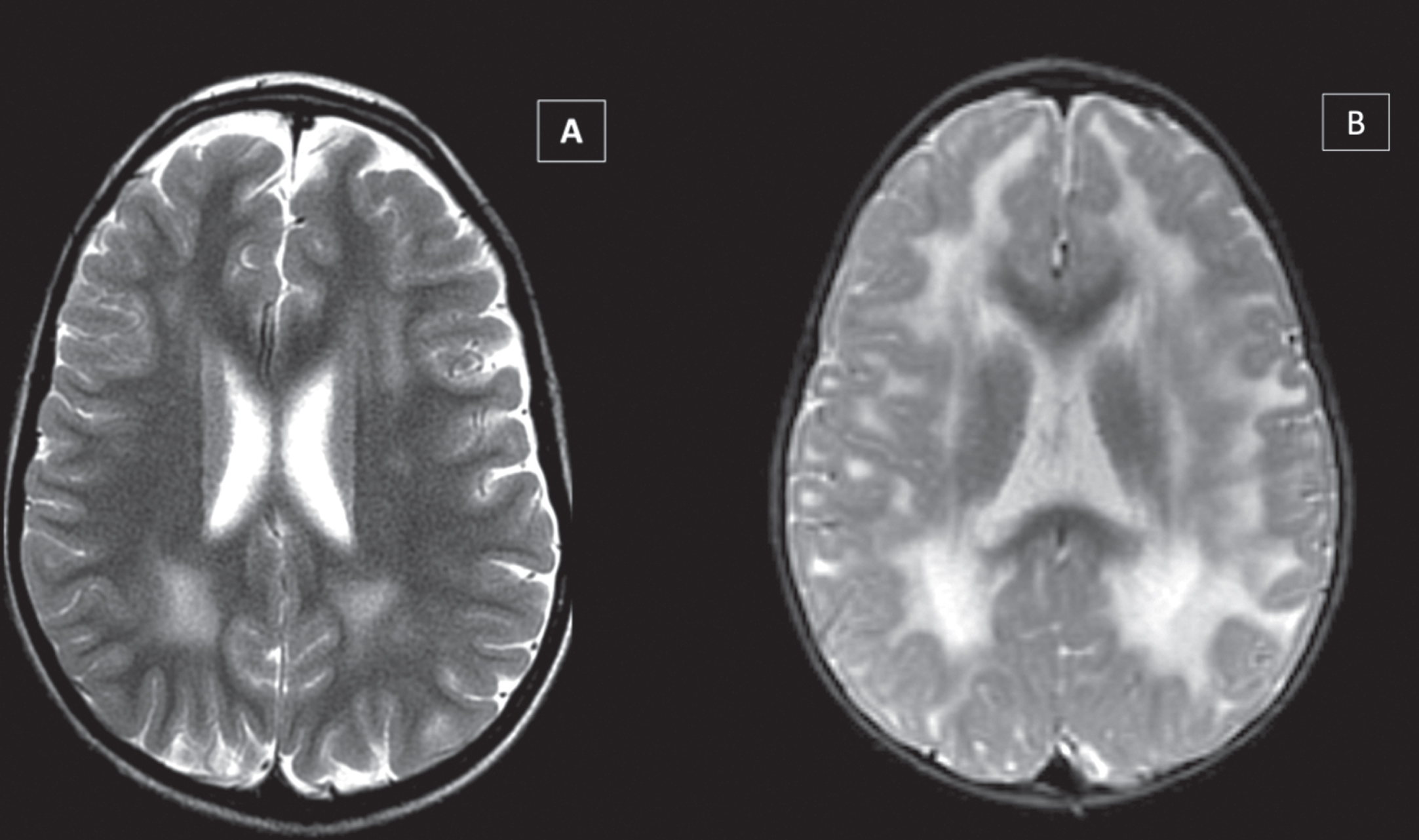
All 52 patients had some degree of white matter alteration in the brain MRI. White matter involvement varied in degree of intensity according to the patient’s clinical severity. It was milder in ambulantory patients (especially in those with a clinical picture of limb girdle muscular dystrophy) and more severe in patients unable to sit or walk (Fig. 2).
Fig. 2
White Matter abnormalities in LAMA2-RD, showing variable involvement of periventricular, central, and subcortical white matter regions on brain MRI, according to motor function - greater involvement in most severe motor phenotype. Patient able to walk (A – T2-WI), unable to walk (B – T2-WI), and unable to sit (C - FLAIR).
In this cohort, after brain MRI reevaluation, 10 (19.2%) patients presented brain cortical malformations, as follows: occipital polymicrogyria (8/10), temporal polymicrogyria (2/10), frontal polymicrogyria (1/10), occipital lissencephaly-pachygyria (2/10), and occipital cobblestone malformation (1/10). In addition, we observed temporal cysts in 3 patients (Figs. 3 and 4). Two patients had extensive cortical malformation with more than one lobe affected. Eight patients with cortical malformation had epilepsy and five had intellectual disability.
Fig. 3
Brain malformations in three different CMD-LAMA2 patients MRI’s: (A) Lissencephaly-pachygyria in occipital lobe (T2-WI), (B) Polymicrogyria and cobblestone in occipital lobe in (T1-WI), and (C) Pachygyria in occipital and temporal lobes in (T2-WI).
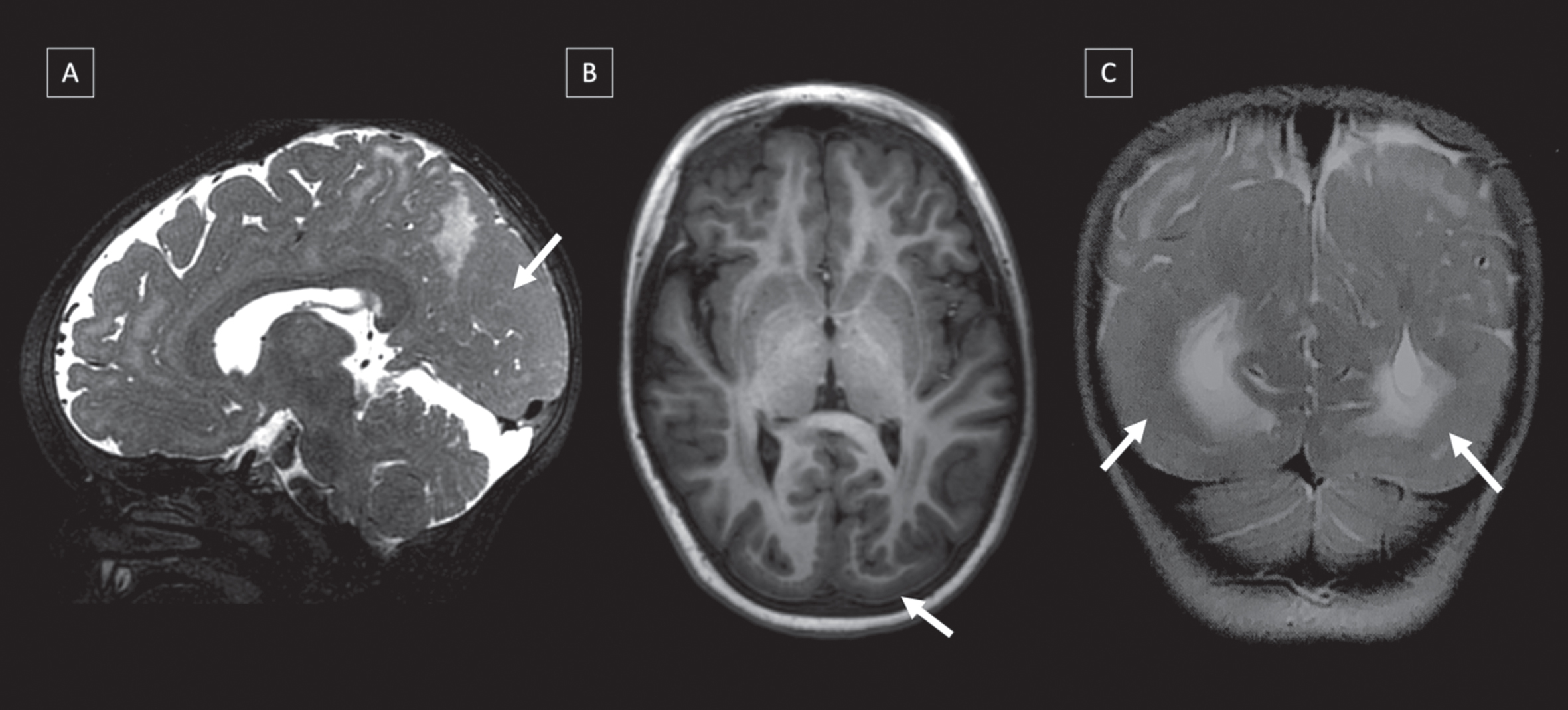
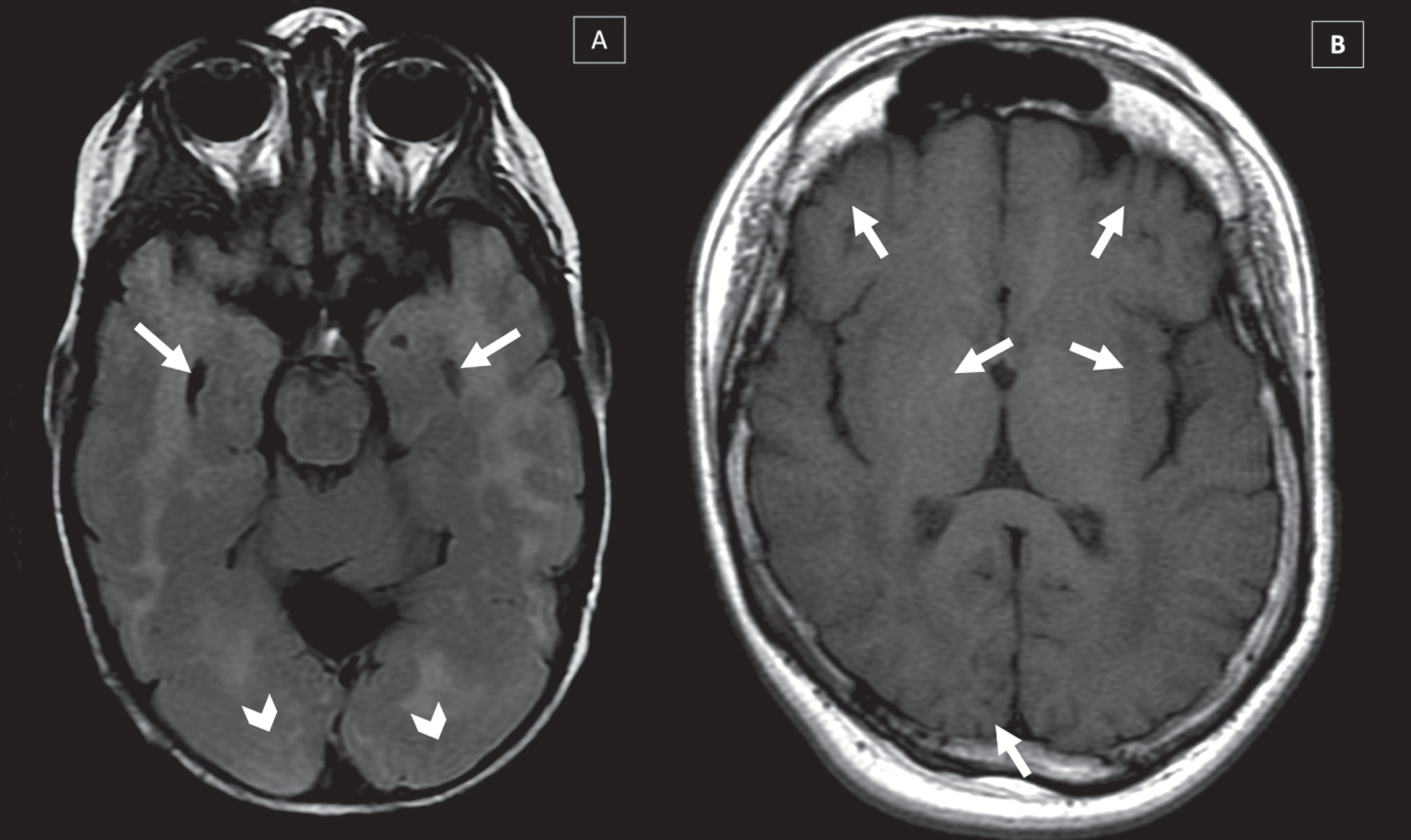
Fig. 4
Brain malformations in two different CMD-LAMA2 patients MRI’s: (A) Lissencephaly-pachygyria in occipital lobe (arrows head) and temporal cists (arrows) (T2-WI), (B) Polymicrogyria in frontal, temporal and occipital lobes in (T1-WI) (arrows).
Ten patients (19.2%) had epilepsy at a median age of 8.1 years (range: 6–13 years). All of them had focal-onset seizures with or without impaired awareness. Of those patients, 8 achieved seizure control using antiepileptic monotherapy, and both patients who needed more than one antiepileptic drug had extensive cortical malformation. Patient 36 (p36), who had the most extensive cortical malformation, had refractory epilepsy, and needed antiepileptic multi-therapy (valproate, vigabatrin, and lacosamide). Two patients with epilepsy didn’t present with cortical malformation.
Eight patients (15.4%) had intellectual disability, five of them with cortical malformation. All of them were≥ 8 years old. One ambulantory patient had intellectual disability associated with autism spectrum disorder (ASD) (Table 1).
Table 1
Phenotypic/genotypic findings in patients with and without cortical malformation
| Patients | Intellectual | Epilepsy | Ambulation | Biallelic loss-of- | At least one variant |
| disability | function variants | in the LG variant | |||
| With Cortical malformation (N = 10) | 5/10 (50%) | 8/10 (80%) | 0/10 (0%) | 7/10 (70%) | 6/10 (60%) |
| Without cortical malformation (N = 42) | 3/42 (7%) | 2/10 (20%) | 8/42 (19%) | 25/42 (60%) | 8/42 (19%) |
LAMA2 variants
In this cohort of 52 patients, we found 40 distinct variants; 80.8% were compound heterozygous and 19.2% homozygous. In two patients, we identified only 1 pathogenic variant. Twenty-three variants were predicted to be loss-of-funtion (10 frameshift, 10 nonsense, 3 splice site). Ten variants were missense, and 7 were copy number variations. Twenty-two variants were novel (65%).
Ten variants affected the LG domain, 8 the LCC domain, and 22 the LN domain.
Eleven patients (21%) presented with a recurrent novel frameshift variant (c.1255del; p. Ile419Leufs*4), 3 of them homozygous. This variant was not previously reported and is absent in Gnomad. (Fig. 5) (All variants are cited in the Table 1 in Supplementary data).
Fig. 5
LAMA2 variants found in the cohort. (A) SNV variants according to exon localization and protein domain. The number of dots represent the number of times this variant was found in the cohort. Black spots unveils the variants that originate premature termination codons (PTC): nonsense and out-of-frame. Yellow spots show splice-site variants and Green spots display missense and inframe changes. All variants are displayed in Table 1. (B) CNV variants represented as blue squares.
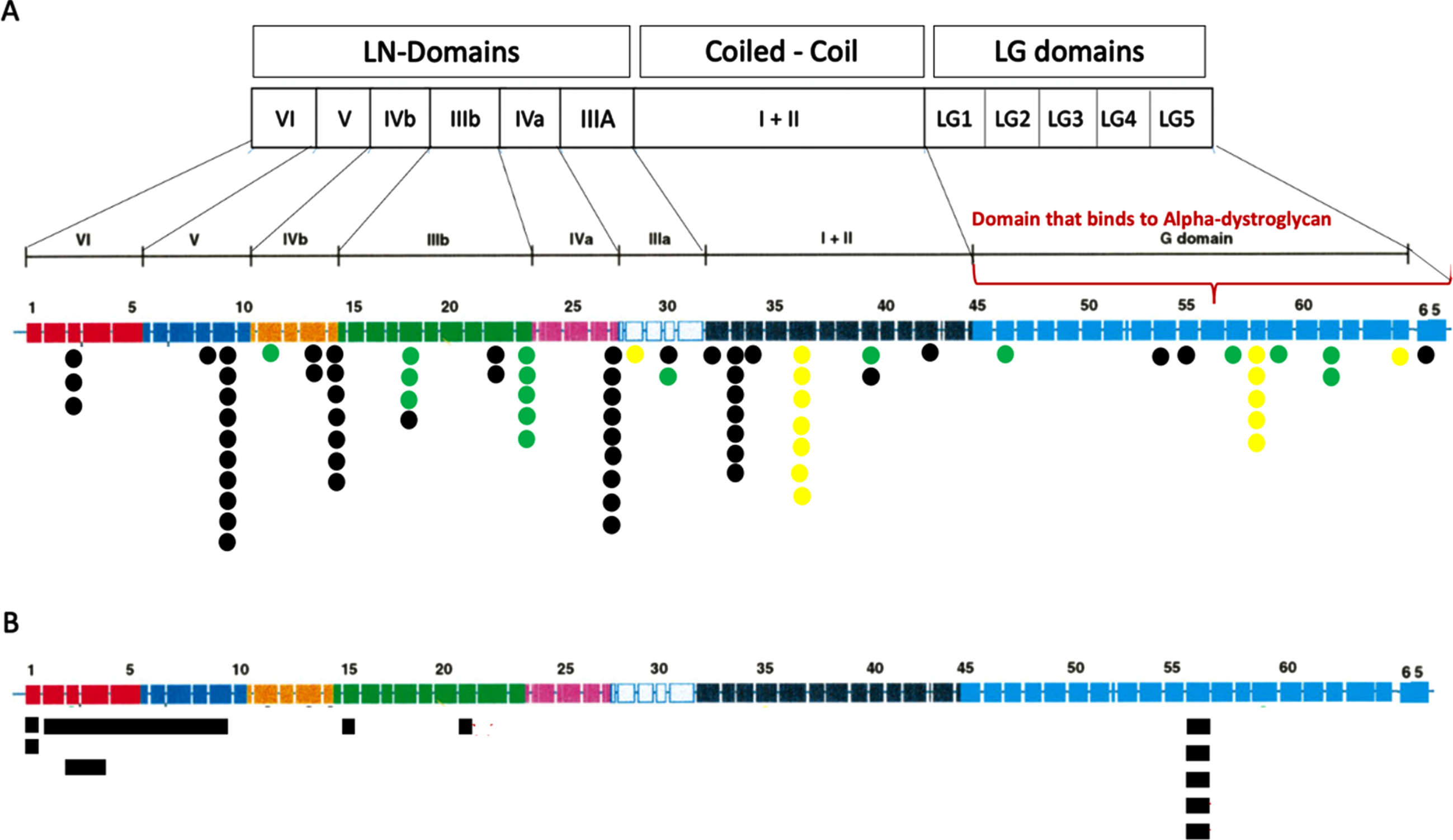
Genotype-phenotype correlations
Non-ambulantory patients
There are 44 patients in the cohort who are non-ambulantory, and only patients from the this group had cortical malformations and/or epilepsy. 10/44 (22.7%) had cortical malformations, 10/44 (22.7%) had epilepsy, and 7/44 (15.9%) had intellectual disability. Both patients unable to sit had severe intellectual disability.
From these 44 patients, 36 (81,8%) presented predicted loss-of-function variants in both alleles and 6 (13.6%) had 1 predicted loss-of-function variant with 1 missense variant in the ither allele. In two patients, only one variant was identified.
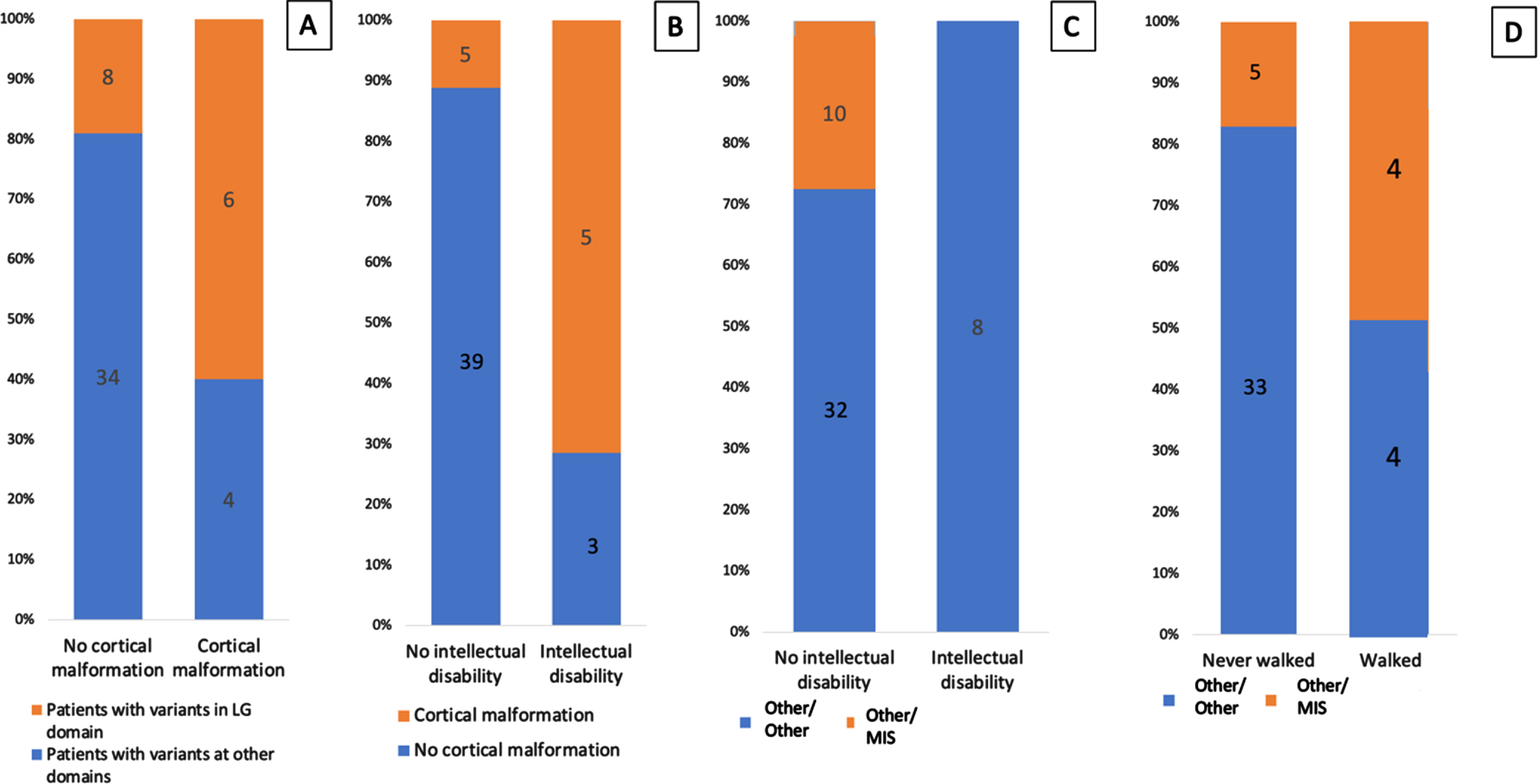
We observed cortical malformations more frequently in patients with pathogenic variants affecting the laminin-α2 LG domains (region that binds to α-dystroglycan and integrins) than in other domains (6/10 vs 8/42; p = 0.016; OR:6.48; Fig. 5), and 9 of 10 patients who presented cortical malformations had null variants in both alleles (Table 2).
Table 2
Genetic profile of patients with cortical malformation
| Patient | First Variant | Second variant | Domain | mRNA effect |
| 7 | c.4960-17C>A | c.4960-17C>A | LCC / LCC | Splicing / Splicing |
| 9 | c.3976C>T p.(Arg1326*) | c.1128_1129insG p.(Tyr2527Ter) | LN / LN | Nonsense / Frameshift |
| 17 | c.8556_8558del (p.Ile2852del) | c.8556_8558del (p.Ile2852del) | LG / LG | Inframe / Inframe |
| 21 | c.5234 + 1G>A | Exon 56 deletion | LCC / LG | Splicing / CNV |
| 30 | c.1255del p.(Ile419Leufs*4) | c.8244 + 1G>A | LN / LG | Frameshift / Splicing |
| 37 | c.4348C>T p.(Arg1450*) | c.9223C>T p.(Gln3075*) | LN / LG | Nonsense / Nonsense |
| 39 | c.8244 + 1G>A | c.8244 + 1G>A | LG / LG | Splicing / Splicing |
| 44 | c..2486_2489del p.(Leu830Serfs*57) | Exon 1 deletion | LN / LN | Frameshift / CNV |
| 51 | c.7147C>T p.(Arg2383*) | c.4523G>A p.(Arg1508Lys) | LG / LCC | Nonsense / CNV |
| 52 | c.8775del p.(His2926Metfs*35) | c.3955C>T p.(Arg1319*) | LG / LN | Frameshift / Nonsense |
The presence of variants affecting the laminin-α2 LG domains correlated with epilepsy (6/10 vs 8/42; p = 0.016; OR:6.48; Fig. 5). Epilepsy was present only in the nonambulantory group, and 8 of the 10 patients with epilepsy had cortical malformations. We confirmed a correlation between the presence of cortical malformations and intellectual disability (5/8 vs 5/44 p = 0.0017; OR:20; Fig. 6). Of the 8 patients with intellectual disability, 2 were unable to sit, 5 unable to walk, and 1 able to walk.
Fig. 6
Genotype-phenotype correlations. (A) Correlation between the presence of variants affecting the LG domain and cortical malformations. (B) Correlation between the presence of cortical malformations and intellectual disability. (C) Correlation between the type of variants and intellectual disability. (D) Correlation between the type of variants and the ability to walk.
All patients with the new variant c.1255del were able to sit and unable to walk.
AMBULANTORY PATIENTS
Of the 8 patients who were able to walk, none had cortical malformations, none had epilepsy, and 1 had intellectual disability with autism spectrum disorder. In the ambulantory group, we found a greater frequency of missense variants than of other variants (4/8 vs 5/39, p = 0.0228; OR:4.48; Fig. 6). LGMD patients had missense variants in one allele and CNV in the other. Two ambulantory patients had 2 null variants but lost ambulation during the study period.
Patients with at least 1 missense variant did not present with either epilepsy (P = 0.0891) or intellectual disability (P = 0.17; Fig. 6).
DISCUSSION
In this large brazilian cohort of LAMA2-RD we were able to evaluate cortical malformation, epilepsy and intellectual disability frequency and correlate with motor severity and genetic findings. Brain MRI was reviewed in all patients and one-fifth presented with cortical malformations, one-fifth with epilepsy, and 15% also presented with intellectual disability. Patients who presented with cortical brain malformations and epilepsy were exclusively from the non-ambulantory group. Patients with cortical malformations more frequently presented with 2 null variants and variants affecting the laminin-α2 LG-domain.
After reviewing all brain MRIs, the most common brain malformation was polymicrogyria, as previously described [9, 10, 17, 18], but we also found lissencephaly-pachygyria complex and cobblestone malformation. Although it is complex to differentiate polymicrogyria from cobblestone, it was possible to identify in one patient the cobblestone malformation. Such alterations have been described in patients of large cohorts, but without correlation to motor function or genetic findings [17, 19]. Other large cohorts of disease natural history didn’t present data on brain involvement [20].
In our study, most of the patients with cortical malformations presented lesions in the occipital lobe, but we also found alterations in the frontal and temporal lobes. In patients with LAMA2-RD, moderated or subtle changes in CNS anatomy, especially in the occipital lobe, were the most common findings [10], but gross structural abnormalities can also occur [21]. It remains unclear why malformations are more commonly found in the occipito-temporal regions; however, malformations are not easily observable and may be more extensive than believed. Jayakody and colleagues, for example, described postmortem observations of widespread, symmetric cobblestone malformation and cerebellar dysplasia in the brain of a LAMA2-RD patient, whose brain MRI showed only subtle abnormalities [21].
Laminin-211, or merosin, is a basement-membrane glycoprotein with an essential structural and signaling roles. It is a heterotrimers comprised of α, β, and γ chains combined via interaction between their LN domains and connected to α-dystroglycan and integrins through the laminin-α2 LG domain [22]. Patients with CNS manifestations had variants of LAMA2 that affect the protein’s LG domain (exons 45–65) more frequently. This finding could be explained by the fact that laminin-α2 binds with high affinity to the dystrophin-glycoprotein complex and to integrins through the LG-domain [23]. This connection appears to influence neuronal adhesion, differentiation, growth, shape, and migration [21]. Laminin-211 is expressed in various tissues, not only in muscle but also Schwann cells, glia limitans, intracerebral micro vessels, and leptomeninges [24]. The loss of laminin-α2 can result in neuronal migration defects that can result in gross cortical malformation, as happens in α-dystroglycanopathies. This suggests that LAMA2-RD and α-dystroglycanopathies may share cellular mechanisms underlying CNS pathology[6].
Ten patients manifested focal or complex partial seizures, and 9 of them presented with cortical malformations. Epilepsy was diagnosed at a median age of 8.1 years (range 6–13 years), the same median age Benito and colleagues found for the onset of seizures [9]. In their cohort of 25 patients, all 9 who presented epilepsy had polymicrogyria [9], and in our study, only 1 patient with epilepsy did not present with cortical malformation in their brain MRI. All patients in our cohort had focal-onset seizures with or without impaired awareness, which may be a more difficult type of seizure for the family to identify. Nine patients, described in this study, achieved seizure control using antiepileptic monotherapy, and only 1, who had the most extensive cortical malformation, needed multiple antiepileptic multi-therapy. Seizures in patients with LAMA2-RD are usually responsive to antiepileptic medications [10]; however, drug resistant epilepsy has been described [18], including West syndrome, a severe childhood epileptic encephalopathy [25].
Although patients with CMD-LAMA2 were previously mostly characterized with normal intelligence [1, 12, 26–29], our cohort showed that the frequency of intellectual disability among these patients is not irrelevant and should be better assessed. Profound intellectual disability, however, was found only among the patients unable to sit. As expected, we found a correlation between cortical malformation and intellectual disability. These data are in agreement with previously published ones [30–32], which confirm that these are not that homogeneous, neither clinically nor from an MRI standpoint.
Characteristic brain white matter hypointensity on T1-WI MRI and an increased T2-WI signal in the periventricular and subcortical white matter are routinely observed in CMD-LAMA2 patients older than 6 months [33] and was present in all patients of this cohort. The underlying pathogenesis of white matter changes is still unclear. One hypothesis is that white matter contains increased water content due to impaired selective filtration of blood-brain barrier caused by laminin-α2 deficiency [24, 34]. Another hypothesis is that these white matter changes indicate structural changes because interactions between laminin-α2 and integrins on developing oligodendrocytes (myelinating cells of the CNS) enhance the myelin membrane’s development [35]. In this cohort, the intensity of white matter involvement was related to motor function, with more diffuse alterations among nonambulantory patients and predominance of focal lesions in ambulantoryones.
In this study, the relation between a milder motor function phenotype, and the presence of missense variants was confirmed [12, 17, 23, 36]. Patients able to walk more frequently had at least 1 missense variant. These patients did not present with cortical malformations or epilepsy, and only 1 had an intellectual disability associated to autism spectrum disorder. Patients with epilepsy or intellectual disability did not have missense variants.
Although nonsense variants are the most common null variant in CMD-LAMA2 [12, 17, 19, 26], in our cohort, the most common variant was a novel frameshift variant (c.1255del) found in 16 allele. This variant was present in patients able to sit, unable to walk, and without cortical malformation, epilepsy or intellectual disability. Although haplotype analysis had not been performed, we speculate that c.1255del might be a founder variant in the Brazilian population.
The increased frequency of cortical malformations and epilepsy in this cohort cannot be attributed to the apparent Brazilian founder variant, as this variant was not associated with cortical malformations or epilepsy.
One limitation of this study was the small number of patients in the ambulantory group, what could provide additional information about the gravity spectrum of the brain manifestations in LAMA2-RD. Another limitation was the lack of neuropsychological evaluation and quality-of-life tests for a better clinical characterization of the patients. Last, mini mental and MOCA tests are limited tests to evaluate intellectual disability and are more reliable in patients older than 8 years and.
In conclusion, cortical malformation, epilepsy and intellectual disability may be common among LAMA2-RD patients, as it was in this cohort, and the most severe motor phenotypes correlate with these findings. A correlation also exists between the presence of variants that affect the protein’s LG domain and central nervous system involvement. This information is important to better predict the course of the disease and to improve patient care.
ACKNOWLEDGMENTS
This work didn’t have any sponsor. The content of the manuscript had the contribution and approval of all authors.
CONFLICT OF INTERESTS
The authors have no conflict of interest to report
SUPPLEMENTARY MATERIAL
[1] The supplementary table 1 is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-221638.
REFERENCES
[1] | Bonnemann CG , Wang CH , Quijano-Roy S , Deconinck N , Bertini E , Ferreiro A , et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. (2014) ;24: (4):289–311. |
[2] | Toda T , Kobayashi K , Kondo-Iida E , Sasaki J , Nakamura Y . The Fukuyama congenital muscular dystrophy story. Neuromuscul Disord. (2000) ;10: (3):153–9. |
[3] | Reed UC . Congenital muscular dystrophy. Part I: A review of phenotypical and diagnostic aspects. Arq Neuropsiquiatr. (2009) ;67: (1):144–68. |
[4] | Tome FM , Evangelista T , Leclerc A , Sunada Y , Manole E , Estournet B , et al. Congenital muscular dystrophy with merosin deficiency. C R Acad Sci III. (1994) ;317: (4):351–7. |
[5] | Løkken N , Born AP , Duno M , Vissing J . LAMA2-related myopathy: Frequency among congenital and limb-girdle muscular dystrophies. Muscle Nerve. (2015) ;52: (4):547–53. |
[6] | Arreguin AJ , Colognato H . Brain Dysfunction in LAMA2-Related Congenital Muscular Dystrophy: Lessons From Human Case Reports and Mouse Models. Front Mol Neurosci. (2020) ;13: , 118. |
[7] | Helbling-Leclerc A , Zhang X , Topaloglu H , Cruaud C , Tesson F , Weissenbach J , et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. (1995) ;11: (2):216–8. |
[8] | Oliveira J , Parente Freixo J , Santos M , Coelho T . LAMA2 Muscular Dystrophy. In: AdamMP, EvermanDB, MirzaaGM, PagonRA, WallaceSE, BeanLJH, et al., editors. GeneReviews((R)). Seattle (WA) 1993. |
[9] | Natera-de Benito D , Muchart J , Itzep D , Ortez C , Gonzalez-Quereda L , Gallano P , et al. Epilepsy in LAMA2-related muscular dystrophy: An electro-clinico-radiological characterization. Epilepsia. (2020) ;61: (5):971–83. |
[10] | Salvati A , Bonaventura E , Sesso G , Pasquariello R , Sicca F . Epilepsy in LAMA2-related muscular dystrophy: A systematic review of the literature. Seizure. (2021) ;91: :425–36. |
[11] | Smirnov SP , Barzaghi P , McKee KK , Ruegg MA , Yurchenco PD . Conjugation of LG domains of agrins and perlecan to polymerizing laminin-2 promotes acetylcholine receptor clustering. J Biol Chem. (2005) ;280: (50):41449–57. |
[12] | Oliveira J , Gruber A , Cardoso M , Taipa R , Fineza I , Goncalves A , et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-alpha2 variome and its related phenotypes. Hum Mutat. (2018) ;39: (10):1314–37. |
[13] | Sewry CA , D’Alessandro M , Wilson LA , Sorokin LM , Naom I , Bruno S , et al. Expression of laminin chains in skin in merosin-deficient congenital muscular dystrophy. Neuropediatrics. (1997) ;28: (4):217–22. |
[14] | Pike NA , Poulsen MK , Woo MA . Validity of the Montreal Cognitive Assessment Screener in Adolescents and Young Adults With and Without Congenital Heart Disease. Nurs Res. (2017) ;66: (3):222–30. |
[15] | Specchio N , Wirrell EC , Scheffer IE , Nabbout R , Riney K , Samia P , et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: Position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. (2022) ;63: (6):1398–442. |
[16] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–24. |
[17] | Geranmayeh F , Clement E , Feng LH , Sewry C , Pagan J , Mein R , et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. (2010) ;20: (4):241–50. |
[18] | Vigliano P , Dassi P , Di Blasi C , Mora M , Jarre L . LAMA2 stop-codon mutation: Merosin-deficient congenital muscular dystrophy with occipital polymicrogyria, epilepsy and psychomotor regression. Eur J Paediatr Neurol. (2009) ;13: (1):72–6. |
[19] | Tan D , Ge L , Fan Y , Chang X , Wang S , Wei C , et al. Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort. Orphanet J Rare Dis. (2021) ;16: (1):319. |
[20] | Zambon AA , Ridout D , Main M , Mein R , Phadke R , Muntoni F , et al. LAMA2-related muscular dystrophy: Natural history of a large pediatric cohort. Ann Clin Transl Neurol. (2020) ;7: (10):1870–82. |
[21] | Jayakody H , Zarei S , Nguyen H , Dalton J , Chen K , Hudgins L , et al. Cobblestone Malformation in LAMA2 Congenital Muscular Dystrophy (MDC1A). J Neuropathol Exp Neurol. (2020) ;79: (9):998–1010. |
[22] | Yoshida-Moriguchi T , Campbell KP . Matriglycan: A novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology. (2015) ;25: (7):702–13. |
[23] | Yurchenco PD , McKee KK , Reinhard JR , Rüegg MA . Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Matrix Biol. (2018) ;71–72: , 174–87. |
[24] | Menezes MJ , McClenahan FK , Leiton CV , Aranmolate A , Shan X , Colognato H . The extracellular matrix protein laminin alpha2 regulates the maturation and function of the blood-brain barrier. J Neurosci. (2014) ;34: (46):15260–80. |
[25] | Camacho A , Nunez N , Dekomien G , Hernandez-Lain A , de Aragon AM , Simon R . LAMA2-related congenital muscular dystrophy complicated by West syndrome. Eur J Paediatr Neurol. (2015) ;19: (2):243–7. |
[26] | Oliveira J , Santos R , Soares-Silva I , Jorge P , Vieira E , Oliveira ME , et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. (2008) ;74: (6):502–12. |
[27] | Darin N , Tulinius M . Neuromuscular disorders in childhood: A descriptive epidemiological study from western Sweden. Neuromuscul Disord. (2000) ;10: (1):1–9. |
[28] | Gilhuis HJ , ten Donkelaar HJ , Tanke RB , Vingerhoets DM , Zwarts MJ , Verrips A , et al. Nonmuscular involvement in merosin-negative congenital muscular dystrophy. Pediatr Neurol. (2002) ;26: (1):30–6. |
[29] | Jones KJ , Morgan G , Johnston H , Tobias V , Ouvrier RA , Wilkinson I , et al. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: Case series and review. J Med Genet. (2001) ;38: (10):649–57. |
[30] | Marques J , Duarte ST , Costa S , Jacinto S , Oliveira J , Oliveira ME , et al. Atypical phenotype in two patients with LAMA2 mutations. Neuromuscul Disord. (2014) ;24: (5):419–24. |
[31] | Mercuri E , Gruter-Andrew J , Philpot J , Sewry C , Counsell S , Henderson S , et al. Cognitive abilities in children with congenital muscular dystrophy: Correlation with brain MRI and merosin status. Neuromuscul Disord. (1999) ;9: (6-7):383–7. |
[32] | Messina S , Bruno C , Moroni I , Pegoraro E , D’Amico A , Biancheri R , et al. Congenital muscular dystrophies with cognitive impairment. A population study. Neurology. (2010) ;75: (10):898–903. |
[33] | Leite CC , Lucato LT , Martin MG , Ferreira LG , Resende MB , Carvalho MS , et al. Merosin-deficient congenital muscular dystrophy (CMD): A study of 25 Brazilian patients using MRI. Pediatr Radiol. (2005) ;35: (6):572–9. |
[34] | Saredi S , Gibertini S , Matalonga L , Farina L , Ardissone A , Moroni I , et al. Exome sequencing detects compound heterozygous nonsense LAMA2 mutations in two siblings with atypical phenotype and nearly normal brain MRI. Neuromuscul Disord. (2019) ;29: (5):376–80. |
[35] | Relucio J , Menezes MJ , Miyagoe-Suzuki Y , Takeda S , Colognato H . Laminin regulates postnatal oligodendrocyte production by promoting oligodendrocyte progenitor survival in the subventricular zone. Glia. (2012) ;60: (10):1451–67. |
[36] | Sarkozy A , Foley AR , Zambon AA , Bönnemann CG , Muntoni F . LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front Mol Neurosci. (2020) ;13: , 123. |


