
Phenotype Genotype Characterization of FKRP-related Muscular Dystrophy among Indian Patients
Abstract
Background:
The phenotypic spectrum of Fukutin-related protein (FKRP) mutations is highly variable and comprises of limb girdle muscular dystrophy (LGMD) R9 (previously LGMD 2I) and FKRP related congenital muscular dystrophies.
Objective:
To identify the distinct genotype phenotype pattern in Indian patients with FKRP gene mutations.
Methods:
We retrospectively reviewed the case files of patients with muscular dystrophy having a genetically confirmed FKRP mutation. All patients had undergone genetic testing using next-generation sequencing.
Results:
Our patients included five males and four females presenting between 1.5 years and seven years of age (median age - 3 years). The initial symptom was a delayed acquisition of gross motor developmental milestones in seven patients and recurrent falls and poor sucking in one patient each. Two patients had a language delay, with both having abnormalities on the brain MRI. Macroglossia, scapular winging, and facial weakness were noted in one, three and four patients respectively. Calf muscle hypertrophy was seen in eight patients and ankle contractures in six. At the last follow-up, three patients had lost ambulation (median age - 7 years; range 6.5–9 years) and three patients had not attained independent ambulation. Creatine kinase levels ranged between 2793 and 32,396 U/L (mean 12,120 U/L). A common mutation - c.1343C>T was noted in 5 patients in our cohort. Additionally, four novel mutations were identified. Overall, six patients had an LGMD R9 phenotype, and three had a congenital muscular dystrophy phenotype.
Conclusion:
Patients with FKRP mutations can have varied presentations. A Duchenne-like phenotype was the most commonly encountered pattern in our cohort, with c.1343C>T being the most common mutation
INTRODUCTION
Fukutin-related protein (FKRP) is a ribitol phosphate transferase involved in the glycosylation of dystroglycans on the sarcolemma, which is an important step that helps in its attachment to the laminin proteins in the extracellular matrix [1]. The gene encoding FKRP is located at chromosome 19q13.3 and spans about 12.5kb [2]. Mutations in the FKRP gene result in highly variable phenotypes and comprise of autosomal recessive LGMD 2I [3], currently renamed as LMGD R9- FKRP related [4], autosomal recessive FKRP related congenital muscular dystrophy, also called merosin-deficient congenital muscular dystrophy type 1C (MDC1C) [2], and the severe phenotype of Walker-Warburg syndrome and Muscle-Eye-Brain disease [5, 6]. Here we describe the clinical, genetic, and laboratory findings in nine patients with FKRP gene mutations from India. For this study patients who have not attained the ability to walk independently till 2 years of age and till the last available follow-up have been classified as having a congenital muscular dystrophy phenotype and others as having a LGMD phenotype in accordance to the 229th ENMC international workshop proposal for Limb girdle muscular dystrophies [4].
MATERIALS AND METHODS
This retrospective study includes all patients with a genetically confirmed FKRP mutation between 2016 and 2020. An institutional ethics committee approval was obtained for the study (NIMHANS/IEC/2020-21). Among 345 genetically confirmed cases of autosomal recessive muscular dystrophies and 73 cases of congenital muscular dystrophy (CMD), nine unrelated patients with FKRP mutations were identified and included in the current study. Clinical characteristics and laboratory results, including histopathological findings and mutational characteristics, were extracted from archived records. The data includes a pre-designed elaborate proforma maintained in each patient’s records.
Genetic testing
All patients had undergone genetic testing using next-generation sequencing. Targeted gene capture was done using Exome Research Panel (Integrated DNA technologies) or a custom capture kit. The libraries were sequenced on the Illumina sequencing platform, followed by bioinformatic analysis and variant calling. The free version of the VarSome online tool (https://www.varsome.com/) and manual application of American College of Medical Genetics and Genomics (ACMG) criteria (2015) [7] were used for the classification of individual single nucleotide variants (SNVs) and small insertion/deletions (INDELs). Larger copy number variants (CNVs) including single or multi-exon deletions and duplications when identified in recessive state were considered as ‘null’ or ‘loss of function’ variants based on ACMG guidelines [8]. The FKRP variants in this paper have been annotated as per RefSeq transcript NM_024301.5.
Histopathology
Seven patients had undergone open biopsy from either the quadriceps, biceps or tibialis anterior muscle. The biopsied muscle was subjected to snap (Flash) freezing using isopentane precooled (chilled) in liquid nitrogen, after which the frozen muscle was cryosectioned using a cryomicrotome. The transverse sections of the muscle thus obtained after cryosectioning, were subjected to non-enzyme histochemical staining that included Haematoxylin Eosin (HE) stain, modified Gomori trichrome (MGT) along with periodic stain with and without diastase stain. Also, the enzyme histochemical stains were performed on the cryosections which included succinic dehydrogenase(SDH), Nicotinamide adenine dinucleotide Tetrazolium Reductase (NADH -TR), adenosine tri phosphatase (ATPase) pH 9.4, 4.6 and succinic dehydrogenase–cytochrome C oxidase (SDH-COX). The cryosections were subjected to immunohistochemistry, the steps of which includes peroxidase blocking (NCL), protein blocking, thorough washing with tris-buffered saline (TBS), incubation with the primary, post primary (polymer) and secondary antibodies and finally application of DAB (DiaminobenzidineHCl, NCL-1:20) chromogen. Immunohistochemistry for dystrophins (Dy4/6D3, Dy8/6C5, Dy10/12B2, NovocastraTM Leica Biosystems) sarcoglycans (alpha, Beta, Gamma, Delta: Ad1/20A6, βSarc1/5B1, 35DAG/21B5, δSarc3/12C1 NovocastraTM Leica Biosystems), and dysferlin (Ham3/17B2, NovocastraTM Leica Biosystems, Germany) proteins was done in four samples (patients: 2,5,6, and 8). Immunohistochemistry for merosin (Mer3/22B2 NovocastraTM Leica Biosystems, Germany) was done in five patients (patients 2,6,7,8 and 9). Western blotting for alpha-dystroglycan (IIH6C4, Anti-α-Dystroglycan Antibody 05-593, Merck Millipore) was done in one patient (patient 7) using the anti-α-Dystroglycan antibody.
RESULTS
Demographic features
Our patients included five males and four females. The age at presentation varied from 1.5 to 7 years, with a median of 3 years.
Clinical features
The initial symptom noted was a motor developmental delay in seven patients, poor sucking and recurrent falls in one patient each. Overall eight out of nine patients had delayed acquisition of gross motor developmental milestones. Delay in language development was reported in two patients. Cranio-facial dysmorphisms were present in two patients, with one having dolicocephaly, high arched palate, and low set ears (patient 4) and one having high arched palate alone (patient 6).
Calf muscle hypertrophy was seen in eight, joint contractures in six, macroglossia in one, facial muscle weakness in four and scapular winging in three patients. None had eye movement restriction or ptosis. Most of the patients had weakness more in the proximal limb muscles than distal. Selective involvement of the quadriceps and gluteus maximus was seen in our cases where a formal assessment of power was possible. Tendon reflexes in the upper limbs and knee jerk were either just elicitable or absent in all cases. A brisk ankle reflex was noted in one and normal in two. Sensory system was normal in all and none showed any clinical evidence for pyramidal, extrapyramidal or cerebellar involvement. The distinct clinical features for individual patients are given in Table 1.
Table 1
Clinical, muscle biopsy and genetic features of the 9 patients with FKRP gene mutation
| Parameter | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 |
| Parental Consanguinity | Yes | No | Yes | Yes | Yes | Yes | Yes | No | No |
| Relevant antenatal, birth history | LSCS | Floppy infant, delayed cry | Nil | Nil | LSCS | Reduced fetal movements, LSCS | Nil | Nil | Nil |
| Initial symptom noted | Recurrent falls on walking | Delayed motor milestones | Delayed motor milestones | Delayed motor milestones | Delayed motor milestones | Poor sucking after birth | Delayed motor milestones | Delayed motor milestones | Delayed motor and language milestones |
| Motor Developmental Delay (Yes/No) | No | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Language delay (Yes/No) | No | Yes | No | No | No | No | No | No | Yes |
| Age at last follow-up(years)/ Sex | 10/M | 8/M | 7/M | 3/F | 9/F | 7/M | 3/M | 3/F | 2/F |
| Age at attaining independent ambulation (months) | 12 | N/W | 20 | 24 | 30 | 21 | N/W | 36 | N/W |
| Age of Loss of Ambulation (years) | 9.5 | N/W | NA* | NA* | 7 | 6.5 | N/W | NA* | N/W |
| Calf hypertrophy | Present | Present | Absent | Present | Present | Present | Present | Present | Present |
| Selectivity in weakness | – | Quadriceps, Gluteus maximus | – | – | – | – | Gluteus Maximus | Quadriceps, Gluteus Maximus | – |
| Other salient examination findings | Scapular winging, protruberant abdomen | Facial weakness, macroglossia, finger drop, Scapular winging, knee and ankle contractures | Lumbar lordosis, ankle contractures | Low set ears, high arched palate, dolicocephaly, hip and knee contractures | Scapular winging, ankle contractures | High arched palate, Hypotonia, Ankle contractures | Facial weakness, hypotonia, knee and ankle contractures | Facial weakness, hypotonia | High arched palate, pectus excavatum, umbilical hernia, facial weakness, hypotonia |
| CK levels (Units/litre) | 32296 | 6459 | 24000 | 2793 | 9727 | 9927 | 6978 | 13030 | 3878 |
| Muscle Biopsy findings | Not done | Dystrophic changes. IHC: Normal expression of dystrophin, sarcoglycans, dysferlin, merosin | Not done | Dystrophic changes. IHC not done | Dystrophic changes, IHC: Normal expression of dystrophin, dysferlin and sarcoglycans | Dystrophic changes, IHC: Normal expression of dystrophin, dysferlin, sarcoglycans and merosin | Dystrophy, IHC: Merosin: reduced expression, Col 6: Normal expression, Western blot: Alpha-dystroglycan: absence of 156kDa band | Dystrophy, IHC: Merosin deficient, Normal expression of dystrophin, sarcoglycans, Beta dystroglycan, dysferlin | Dystrophy, IHC: Merosin reduced expression, Col 6 –normal expression |
| Mutation | c.646C>T, homozygous | c.650C>A, homozygous | c.1343C>T, homozygous | c.1343C>T, homozygous | c.160C>T, homozygous | c.1343C>T, homozygous | c.1343C>T, homozygous | c.933_934del and c.1343C>T | c.1060_1061delGGinsCC and c.1136G>C |
| ACMG classification | Likely Pathogenic | Likely Pathogenic | Pathogenic | Pathogenic | Likely Pathogenic | Pathogenic | Pathogenic | Both mutations: Pathogenic | Both mutations: VUS |
| Phenotype | LGMDR9 | MDC1C | LGMDR9 | LGMDR9 | LGMDR9 | LGMDR9 | MDC1C | LGMDR9 | MDC1C |
LSCS: lower segment cesarean section, N/W: never walked, NA*: Not applicable, ambulant till last follow-up, ACMG: American College of Medical Genetics, IHC: immunohistochemistry, VUS: variant of uncertain significance.
At the last follow-up, three patients had lost unassisted ambulation at a median age of 7 years (range 6.5–9 years). Three patients had not attained independent ambulation until the last follow-up, between ages 2 and 7. None had symptoms of respiratory or cardiac muscle involvement till the last available follow-up; however, a 2D ECHO was available in only two cases, and it was normal.
Investigations
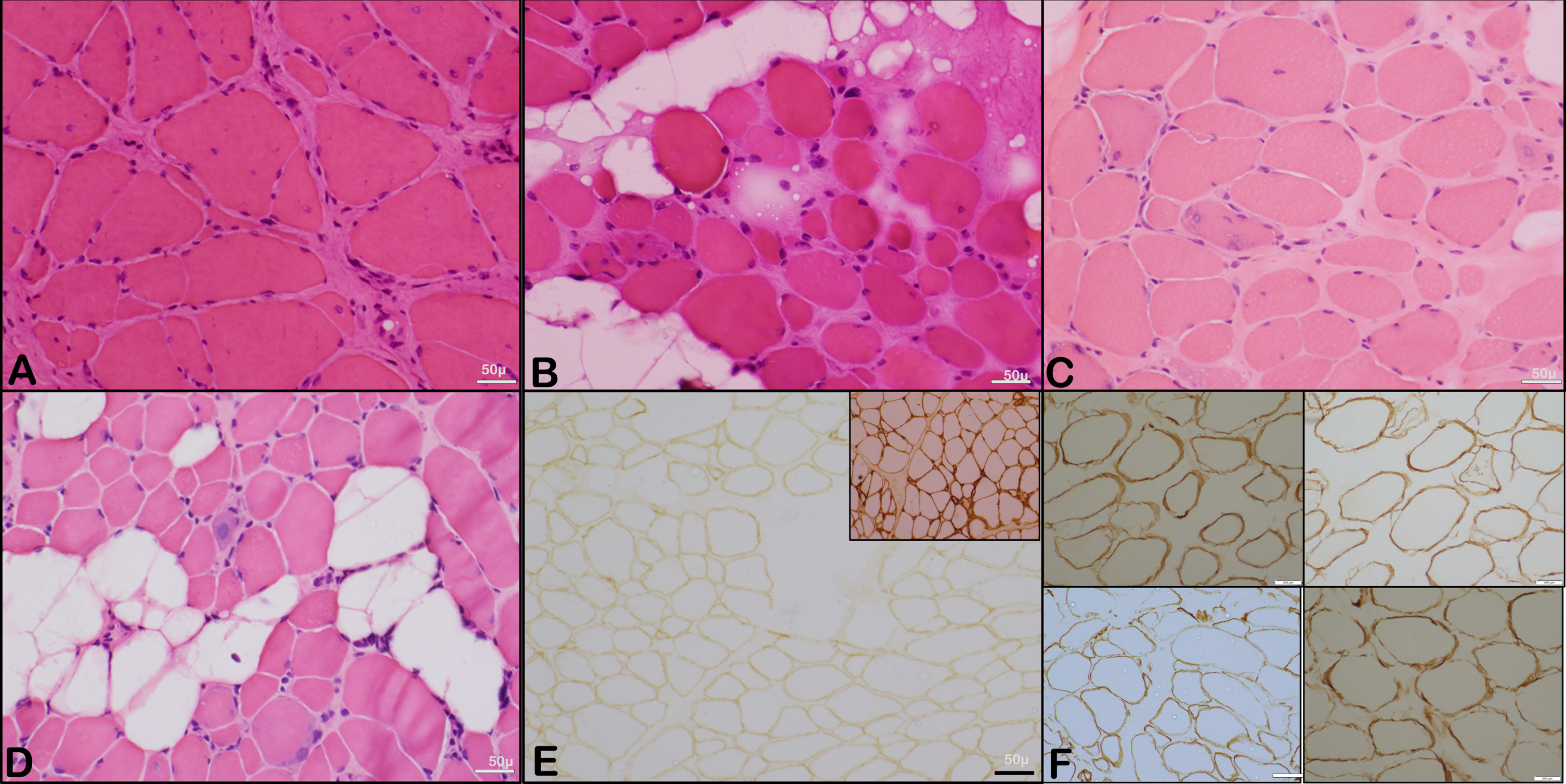
The mean creatine kinase (CK) level was 12,120 U/L (range 2793 to 32,396 U/L, lab reference value 20–200 U/L). Muscle biopsy was done in 7 patients, and histology revealed a variable degree of dystrophic changes with evolving fibrosis. Immunohistochemistry for dystrophin, sarcoglycans, and dysferlin proteins was done in four samples (patients: 2,5,6, and 8), which showed normal expression. An absent or reduced staining for merosin was noted in three patients (patients 7,8,9): and normal expression in two of them (patients 2 and 6). Western blotting for alpha-dystroglycan was done in one patient (patient 7) and showed an absence of the 156kDa band. Figure 3 shows representative microphotographs of the histopathological changes.
MRI of the brain was done on two patients (patient 2 and 9) both of whom had a delay in speech development (Fig. 1c and 1d). Both showed periventricular white matter T2 hyperintensities. CT scan of the brain done in one patient (patient 7) who had not attained ambulation till 3 years of age was normal.
Fig. 1
Clinical photographs and MRI images of patients with congenital muscular dystrophy phenotype. a and b: Calf muscle hypertrophy and scapular winging in patient 2 with congenital muscular dystrophy phenotype c and d: Brain MRI showing periventricular white matter T2 hyperintensities in patient 9 (figure c) and patient 2 (figure d) with congenital muscular dystrophy phenotype.
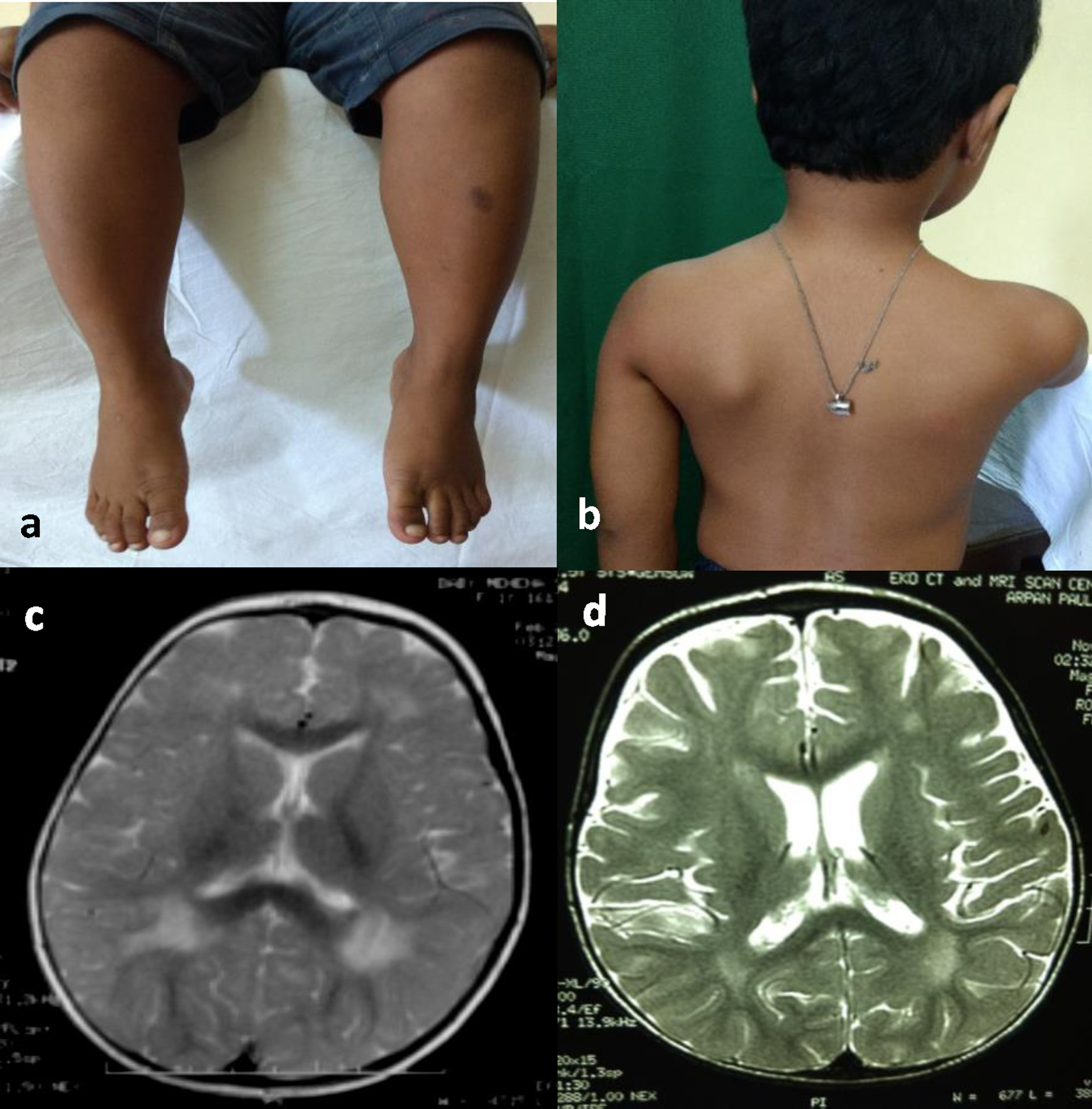
Fig. 2
Schematic representation of the FKRP gene. The exons are represented as rectangular boxes with respective exonic numbers. 5′ UTR is blackened and 3′ UTR is shown in grey. The four novel mutations identified in this study are marked in red rectangular boxes.
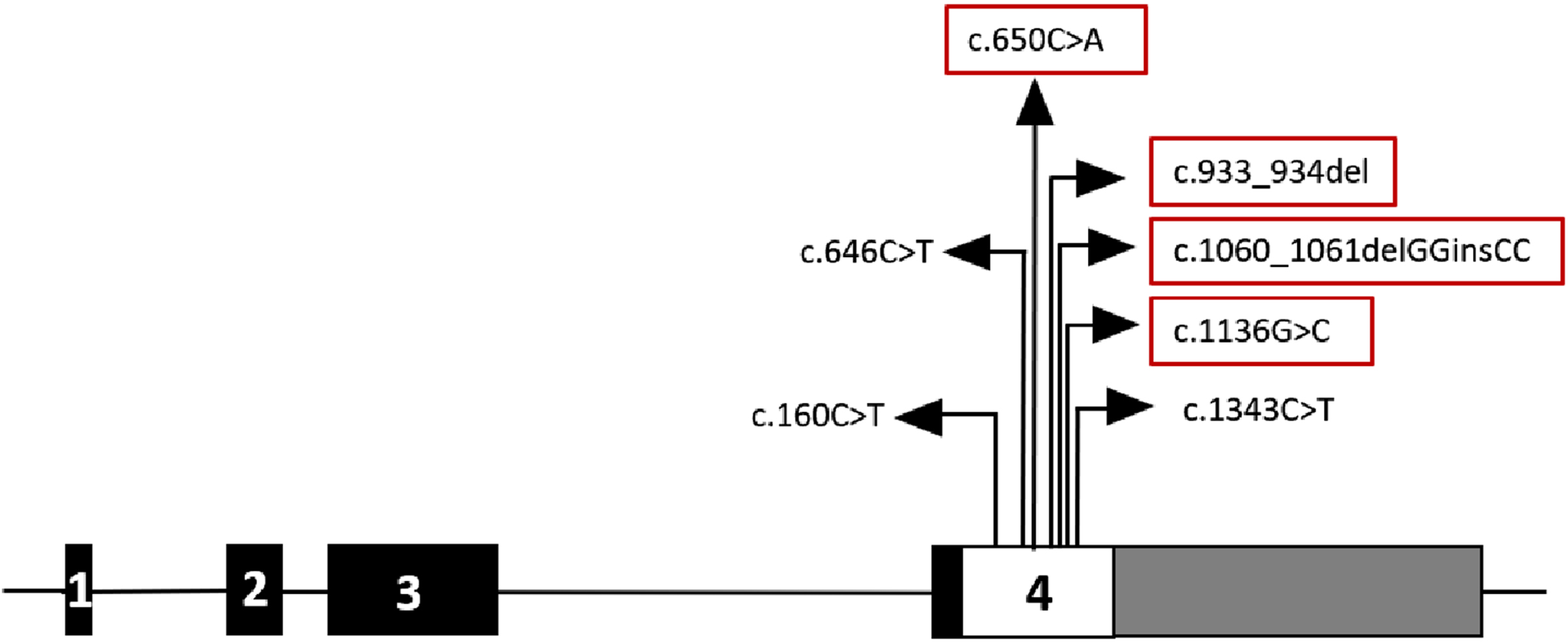
Fig. 3
Representative microphotographs showing the histopathological changes. A to D: Representative microphotographs showing variation in fibre size with variable atrophic and scattered hypertrophic fibres with regenerating fibres and evidence of interstitial (endomysial) fibrosis (Transverse section, H & E, X 200). E: Microphotograph showing muscle fibres with reduced expression of merosin (Inset shows control staining) (IHC, X 200). The perimysium is thickened and endomysial fibrosis is observed. F: Representative images of muscle immunohistochemistry (Alpha, Beta, Gamma and Delta Sarcoglycan; IHC x 200).
Family history and genetics
Six patients (patients 1,3,4,5,6 and 7) were born to consanguineous parents. Patient 4 had an elder male sibling with similar phenotype who had motor developmental delay and eventually lost ambulation by 7 years of age. Patient 9 had a history suggestive of a similar phenotype in 2 out of 3 older siblings. One sibling died at 3 years of age and the other died at 10 months. Both had delayed motor milestones and never attained ambulation. The cause of death in them was reported to be respiratory failure. Parents of patient 3 and 6 had further pregnancies where prenatal testing showed an affected fetus, following which a medical termination was done.
In total, seven different mutations were identified in the FKRP gene, all in either homozygous or compound heterozygous state. The most common mutation was c.1343C>T(P448L) in exon 4, which is a missense mutation causing the substitution of leucine for proline at codon 448. Five patients had this mutation, of which four were in a homozygous state, and one was in a compound heterozygous state. Table 1, summarizes the other mutations detected. Three mutations have previously been reported in patients with LGMD 2I, and the other four are novel variants. Of the four novel variants, c.933_934del (R312Lfs*77), a frameshift variant has been identified along with the recurrent c.1343C>T (P448L) mutation in patient 8 in a likely compound heterozygous state as noticed by the presence of only the missense change in mother, while father’s sample was not available. Among the three novel missense variants identified, c.650C>A (P217Q) was found in homozygous state in patient 2, while patient 9 had c.1060_1061delGGinsCC, (G354P) and c.1136G>C (R379P) in compound heterozygous state as confirmed by segregation in unaffected parents.
The salient clinical characteristics, muscle biopsy findings and mutations of the patients are summarized in Table 1.
Phenotypic classification
Three patients (patients 2,7,9) never attained independent ambulation, two of whom (patients 2,9) additionally had delayed language development and abnormalities on brain MRI. These three have been classified as congenital muscular dystrophy. Other six patients having attained independent ambulation are classified as LGMD R9 according to the revised LGMD classification criteria [4]. All six who had an LGMD phenotype had a Duchenne-like presentation, with the onset of symptoms in the early first decade and a tendency for early loss of ambulation.
DISCUSSION
This is the first study examining the spectrum of genetically confirmed FKRP-related muscular dystrophy from India and highlights the clinical and genetic heterogeneity of the disorder. LGMD R9 phenotype seen in 6 patients was the most common presentation in this short series, while a congenital muscular dystrophy presentation was noted in only three patients. All the patients with LGMD R9 had a Duchenne-like phenotype, however in all these patients the first symptom was noted before two years of age which is earlier than the average age of onset of Duchenne muscular dystrophy reported from India [9]. Notably the age of onset of our patients is much earlier as compared to prior reports of patients with LGMD R9 [10–13]. The mean age of onset of the Western blot confirmed LGMD2I/R9 patients from our previously published cohort of autosomal recessive LGMD was 15.4 years [14]. This wide range of presentations of the FKRP-mediated LGMD phenotype is well established. The Duchenne-like presentation was first described by Brockington et al., in 5 out of 17 families with LGMD 2I, while a later age at presentation and slower progression was seen in the other 12 families [3]. The high frequency of the c.826C>A mutation in the western population, which has a milder phenotype could explain this phenotypic difference [13]. Table 2 summarizes the age of onset and mutations reported in various case series of the FKRP related muscular dystrophy published in literature previously.
Table 2
Summary of the mutation spectrum of FKRP (NM_024301.5) gene reported in various studies
| Brockington 2001 [3] | Mercuri 2003 [16] | Poppe 2003 [10] | de Paula 2003 [18] | Boito 2005 [11] | Sveen 2006 [13] | Bourteel 2009 [15] | Liang 2013 [12] | Awano 2021 [19] | |
| Total cases: LGMD, CMD | 22,0 | 18,4 | 16,0 | 16,0 | 20,0 | 38,0 | 11,0 | 6,0 | 6,3 |
| Ethnicity of patients | NA | NA | NA | Brazilian | Italian | Danish | French | Taiwanese | Japanese –8 |
| Chinese –1 | |||||||||
| Mean age at onset years (range in years) | NA | NA | 19.2 (2–40) | NA | NA (2–50)a | 18 and 5b | 9 | 6.3±6.1 | NA |
| (<0.5 –40) | (Birth –23) | (1–40) | (NA) | (1.5–23) | (2–17) | (Birth –7) | |||
| Mutations identified | c.826C>A (p. Leu276Ile) | c.926A>G (p.Tyr309Cys) | c.826C>A (p.Leu276Ile) | c.826C>A (p.Leu276Ile) | c.826C>A (p. Leu276Ile) | c.826C>A (p.Leu276Ile) | c.826C>A (p.Leu276Ile) | c.263A>T (p. Tyr88Phe) | c.157G>A (p.Val53Met) |
| c.1016G>T (p. Arg339Leu) | c.1154C>A (p. Ser384Ter) | c.1154C>A (p. Ser384Ter) | c.545A>G (p.Tyr182Cys) | c.1384C>T (p. Pro462Ser) | c.1384C>T (p. Pro462Ser) | c.823C>G (p. Arg275Gly) | c.560C>G (p.Ala187Gly) | c.877A>G (p.Thr293Ala) | |
| cc.387_390dup (p.Asp131ThrfsTer2) | ec.1343C>T (p. Pro448Leu) | c.928G>T (p. Glu310Ter) | c.878C>T (p.Thr293Ile) | c. 427C>A (p. Arg143Ser) | c.1187dupA (p.Ala397GlyfsTer67) | c.1466T>G (p. Leu489Arg) | c.545A>G (p. Tyr182Cys) | c.778G>T (p.Glu260Ter) | |
| c.934C>T (p.Arg312Cys) | c.1016G>A (p. Arg339His) | c.1073C>T (p. Pro358Leu) | c.1073C>T (p. Pro358Leu) | c.919T>A (p.Tyr307Asn) | ic.983A>C (p.Tyr328Ser) | c.948delC (p.Cys317AlafsTer111) | c.68_69delAT (p.Tyr23CysfsTer9) | ||
| c.947C>G (p. Pro316Arg) | fc.162_165dup (p.Phe56GlyfsTer6) | c.478G>T (p. Val160Phe) | c.731G>A (p. Arg244His) | hc.469G>C (p.Ala157Pro) | c.964C>G (p. Leu322Val) | c.823C>T (p.Arg275Cys) | c.540_570dup (p.Cys191ProfsTer78) | ||
| dc.427_438del (p.Arg143_Glu146del) | c.826C>A (p. Leu276Ile) | c.235G>A (p.Val79Met) | c.478G>T (p. Val160Phe) | c.158_162dup (p.Glu55CysfsTer15) | c.341C>G (p. Ala114Gly) | c.169G>A (p.Glu57Lys) | |||
| c.427C>A (p. Arg143Ser) | cc.387_390dup (p.Asp131ThrfsTer2) | c.400C>T (p.Arg134 Trp) | c.605T>A (p.Leu202Gln) | c.1201G>A (p. Asp401Asn) | c.266C>G (p.Pro89Arg) | ||||
| c.1A>G (p.Met1Val) | c.899T>C (p.Val300 Ala) | c.501_502delinsCC (p.Arg167_Cys168delinsSerArg) | |||||||
| c.919T>A (p. Tyr307Asn) | c.898G>A (p.Val300 Met) | c.545A>G (p.Tyr182Cys) | |||||||
| gc.158_162dup (p.Glu55CysfsTer15) | c.764G>A (p.Trp255Ter) | c.692G>A (p.Trp231Ter) | |||||||
| c.946C>T (p. Pro316Ser) | c.808C>T (p.Arg270Cys) | ||||||||
| c.1384C>T (p. Pro462Ser) | c.878C>T (p.Thr293Ile) | ||||||||
| dc.427_438del (p.Arg143_Glu146del) | c.946C>T (p.Pro316Ser) | ||||||||
| c.1078G>A (p. Asp360Asn) | c.1027G>T (p.Glu343Ter) | ||||||||
| c.1170_1171delCG (p.Gly391LeufsTer72) | |||||||||
| Common mutation, No. subjects harbouring the mutationj | c.826C>A, 19 | c.826C>A, 13 | c.826C>A, 16 | c.826C>A, 5 | c.826C>A, 9 | c.826C>A, 38 | c.826C>A, 10 | c.948delC, 5 | c.169G>A, 2 |
NA: information not available. a. Series also includes asymptomatic patients. b. Mean age at onset of patients harbouring c.826C>A in homozygous and compound heterozygous states respectively. c: c.387_390dup (p.Asp131ThrfsTer2) has been previously annotated as c.390insTACC (Gly132Stop) in Brockington et al 2001. and Mercuri et al. 2003. Source: LOVD database (Variant #0000240433). d: c.427_438del (p.Arg143_Glu146del) has been previously annotated as c.426del12nt (p. 143delRMVE) in Brockington et al. 2001 and Mercuri et al 2003. Source: LOVD (Variant #0000240465). e: c.1343C>T (p.Pro448Leu) has been previously annotated as c.1378C>T in Mercuri et al. 2003. Source: LOVD (Variant #0000240364). f: c.162_165dup (p.Phe56GlyfsTer6) has been previously annotated as c.165InsGGAG (p. Asp60Ter) in Mercuri et al. 2003. Source: LOVD (Variant #0000240469). g: c.158_162dup (p.Glu55CysfsTer15) was previously annotated as c.158InsCTGGT (p. Asp60Ter) in Mercuri et al. 2003. Source: LOVD (Variant #0000240505). h: c.469G>C (p.Ala157Pro) was previously annotated as c.477G>C (p.Ala157Pro)in Sveen et al. 2006. Source: LOVD (Variant # 0000240562). i: c.983A>C (p.Tyr328Ser) was previously annotated as c.982A>C (p.Tyr328Ser) in Bourteel et al. 2009. Source:LOVD (Variant #0000240473). j. Either in heterozygous or homozygous state.
Calf muscle hypertrophy, tendo-achilles contractures, scapular winging, and macroglossia seen in our patients have previously been reported in patients with LGMD R9 [10, 15].
We found a selective weakness of the quadriceps and hip extensors in our patients where a formal assessment of the muscle power was possible. Selective weakness of the quadriceps among FKRP patients is previously described [10]. However, in contrast to observations by Poppe et al., we noted a selective weakness of the hip extensors [10]. This is in keeping with muscle MRI observation for early involvement of gluteus maximus previously reported [15].
Unassisted ambulation was lost in 3 out of 6 patients with LGMD R9 phenotype at a median age of 7 years; however, the age at last follow-up of the patients with retained ambulation was between 3 and 7 years. In their Danish cohort, Sveen et al., reported the loss of ambulation at a mean age of 44 years in patients with c.826C>A homozygous mutation and 20 years with other mutations [13]. Mercuri et al., in their study, report loss of independent ambulation in teenage with a Duchenne like phenotype and preserved ambulation beyond the second decade in the less severe group [16]. The tendency for an earlier loss of ambulation in our patients is in keeping with the Duchenne-like phenotype.
None of our patients had symptoms suggestive of cardiac dysfunction or respiratory involvement; however, none had entered into the second decade of life at last follow-up. Cardiac and respiratory involvement is well described in LGMD R9, but the age of onset is quite variable [11, 13, 15, 16]. Sveen et al., have reported a reduced ejection fraction in 10 out of 39 patients with LGMD 2I, while four in their study were on continuous ventilator support and six were using other respiratory assistive devices intermittently [13].
Three of our patients had a CMD-like presentation with language delay in two, and none achieved independent ambulation. None of our patients had the severe phenotype of Walker-Warburg syndrome or Muscle-Eye-Brain disease. The inability to walk independently after 2 years of age and till the last available follow-up was used as the criteria for classification as congenital muscular dystrophy in this study. One of the 6 patients (patient 6) classified as having LGMD R9 had a documented history of reduced fetal movements antenatally and poor sucking after birth. However, this patient has been classified as LGMD R9 as he eventually attained independent ambulation. None of the other LGMD R9 patients had a documented hypotonia at birth. Four of the seven different mutations identified were previously unreported, of which the three missense variants are currently classified as variants of uncertain significance (VUS) as per ACMG guidelines [7]. However, the c.650C>A (P217Q) in patient 2 is adjacent to the previously reported c.646C>T (R216W) [17] observed in patient 1, suggesting a possible hotspot location in exon 4. Compared to patient 2, who never attained independent ambulation and had additional language delay and MRI brain abnormalities, patient 1 had a milder phenotype having attained independent ambulation at 1 year and remaining ambulant up until 9.5 years of age. In patient 9, who like patient 2 had a CMD phenotype with language delay and brain MRI abnormalities, the two novel missense changes, c.1060_1061delGGinsCC (G354P) and c.1136G>C (R379P), were confirmed to be present in trans based on parental segregation. Based on the above findings and clinical correlation, we posit that these three FKRP missense changes can be considered disease-causing. However, further functional evidence and identification of more patients might be required to establish the pathogenicity.
The most common FKRP mutation reported from most of the studies from Europe and Brazil is the c.826C>A mutation [3, 11, 13, 15, 18] which typically has a milder presentation. In contrast, c.948delC and c.169G>A is the most common FKRP mutation reported in studies from Taiwan and Japan, respectively [12, 19]. Table 2 summarizes the mutations described in the various prior studies on FKRP.
The significant finding in our series is the recurrence of c.1343C>T(P448L) in five patients which was first reported as homozygous in a Libyan patient from a consanguineous family [2]. This patient was reported to have congenital onset hypotonia with feeding difficulties and motor delay evidenced by lack of ambulation at 16 months of age. Calf hypertrophy and facial weakness were reported with elevated CK. However, brain MRI was normal with normal intelligence suggesting a CMD phenotype without brain involvement. Few other studies have also reported the c.1343C>T(P448L) mutation in patients with LGMD phenotype and Duchenne like phenotype [20–22]. Further animal studies showed that the homozygous (P448L) mouse model exhibited a moderate to severe form of dystrophic pattern with a marked reduction in functional glycosylation of α-DG in skeletal muscles but without any brain defects consistent with severe LGMD2I (R9) or MDC1C phenotype observed in the Libyan patient [23]. In our cohort, the P448L mutation is present as homozygous in four patients (patients 3, 4, 6, 7) and as likely compound heterozygous with a novel frameshift c.935_936del(R312Lfs*77) mutation in patient 8. The P448L variant was found to be in heterozygous state in unaffected parents tested and all five families with the mutation hailed from south India. GnomAD population database reported P448L variant in 2 south Asian controls in heterozygous. Further haplotype studies can be crucial in identifying any possible founder effect. Three of the four patients with homozygous P448L had LGMD2I with a Duchenne-like phenotype. They had motor development delay, with independent ambulation attained between 20 and 24 months and loss of independent ambulation being recorded in one at the age of 6.5 years. Calf hypertrophy was seen in all 4, and scapular winging was absent. The mean CK of these patients was 10,924 U/L. One had a CMD phenotype. However, in comparison to the other two patients who had a CMD phenotype, this patient had no delay in non-motor developmental realms and had a normal brain CT scan similar to the first report of this mutation [2]. He was considered to have a CMD phenotype rather than LGMD as he was unable to stand even with support till the last follow-up, which was at three years of age [4].
One patient (patient 8) had the mutation in a compound heterozygous state. She also presented with an LGMD R9 –Duchenne like phenotype, attaining independent ambulation by 3 years, having calf hypertrophy and highly elevated CK level of 13,030 U/L. Four of the five patients harboring the P448L mutation had undergone a muscle biopsy, exhibiting a dystrophic picture of variable degree and evolving fibrosis. Among the three patients who had immunohistochemistry stains done, two of them displayed a reduced or deficient staining for merosin, including one with the P448L mutation in a homozygous state (patient 7) and one in a compound heterozygous state (patient 8). This is in contrast to the muscle biopsy findings reported previously in a patient harboring the same mutation who had normal staining for merosin [2]. Merosin deficiency in patients with FKRP-mediated muscular dystrophies is thought to be secondary to the loss of alpha-dystroglycan in the muscle membrane [2]. The reduced merosin staining in our patients with the P448L mutation could represent a more advanced stage of the dystrophic process.
CONCLUSIONS
This is the first description of the clinical and mutational spectrum of genetically confirmed cases of FKRP mutations from India. LGMD R9 with a Duchenne-like phenotype was more common than a congenital muscular dystrophy phenotype in our cohort. We report c.1343C>T(P448L) as a common recurrent mutation in Indian FKRP patients but further larger studies are required to determine the frequency of this mutation and a possible founder effect. Awareness of a Duchenne muscular dystrophy mimicker in terms of the clinical presentation, progression of symptoms, and creatine kinase levels is critical among clinicians, especially for guiding genetic testing.
CONFLICTS OF INTEREST
No potential conflicts of interest to declare.
ACKNOWLEDGMENTS
The authors received no financial support for the research, authorship, and/or publication of this article. We would like to thank the patients and their families for participating in this study.
REFERENCES
[1] | Kanagawa M , Toda T . Muscular dystrophy with ribitol-phosphate deficiency: A novel post-translational mechanism in dystroglycanopathy. J Neuromuscul Dis 4: (4):259–67. |
[2] | Brockington M , Blake DJ , Prandini P , Brown SC , Torelli S , Benson MA , et al. Mutations in the Fukutin-Related Protein Gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin α2 deficiency and abnormal glycosylation of α-dystroglycan. The American Journal of Human Genetics. (2001) ;69: (6):1198–209. |
[3] | Brockington M , Yuva Y , Prandini P , Brown SC , Torelli S , Benson MA , et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Human Molecular Genetics. (2001) ;10: (25):2851–9. |
[4] | Straub V , Murphy A , Udd B , Corrado A , Aymé S , Bönneman C , et al. 229th ENMC international workshop: Limb girdle muscular dystrophies –Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscular Disorders. (2018) ;28: (8):702–10. |
[5] | Beltran-Valero de Bernabé D , Voit T , Longman C , Steinbrecher A , Straub V , Yuva Y , et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. (2004) ;41: (5):e61. |
[6] | Reeuwijk JV , Olderode-Berends MJW , Elzen CVD , Brouwer OF , Roscioli T , Pampus MV , et al. A homozygous FKRP start codon mutation is associated with Walker–Warburg syndrome, the severe end of the clinical spectrum. Clinical Genetics. (2010) ;78: (3):275–81. |
[7] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–23. |
[8] | Brandt T , Sack LM , Arjona D , Tan D , Mei H , Cui H , et al. Adapting ACMG/AMP sequence variant classification guidelines for single-gene copy number variants. Genetics in Medicine. (2020) ;22: (2):336–44. |
[9] | Singh RJ , Manjunath M , Preethish-Kumar V , Polavarapu K , Vengalil S , Thomas PT , et al. Natural history of a cohort of Duchenne muscular dystrophy children seen between and An observational study from South India. Neurology India. (2018) ;66: (1):77. |
[10] | Poppe M , Cree L , Bourke J , Eagle M , Anderson LVB , Birchall D , et al. The phenotype of limb-girdle muscular dystrophy type 2I. Neurology. (2003) ;60: (8):1246–51. |
[11] | Boito CA , Melacini P , Vianello A , Prandini P , Gavassini BF , Bagattin A , et al. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch Neurol. (2005) ;62: (12):1894–9. |
[12] | Liang WC , Hayashi YK , Ogawa M , Wang CH , Huang WT , Nishino I , et al. Limb-girdle muscular dystrophy type 2I is not rare in Taiwan. Neuromuscular Disorders. (2013) ;23: (8):675–81. |
[13] | Sveen ML , Schwartz M , Vissing J . High prevalence and phenotype-genotype correlations of limb girdle muscular dystrophy type 2I in Denmark. Ann Neurol. (2006) ;59: (5):808–15. |
[14] | Nalini A , Polavarapu K , Sunitha B , Kulkarni S , Gayathri N , Bharath MS , et al. A prospective study on the immunophenotypic characterization of limb girdle muscular dystrophies 2 in India. Neurology India. (2015) ;63: (4):548. |
[15] | Bourteel H , Vermersch P , Cuisset JM , Maurage CA , Laforet P , Richard P , et al. Clinical and mutational spectrum of limb-girdle muscular dystrophy type 2I in 11 French patients. J Neurol Neurosurg Psychiatry. (2009) ;80: (12):1405–8. |
[16] | Mercuri E , Brockington M , Straub V , Quijano-Roy S , Yuva Y , Herrmann R , et al. Phenotypic spectrum associated with mutations in the fukutin-related protein gene. Ann Neurol. (2003) ;53: (4):537–42. |
[17] | Murphy LB , Schreiber-Katz O , Rafferty K , Robertson A , Topf A , Willis TA , et al. Global FKRP registry: Observations in more than 300 patients with limb girdle muscular dystrophy R9. Ann Clin Transl Neurol. (2020) ;7: (5):757–66. |
[18] | de Paula F , Vieira N , Starling A , Uraco Yamamoto L , Lima B , de Cássia Pavanello R , et al. Asymptomatic carriers for homozygous novel mutations in the FKRP gene: The other end of the spectrum. Eur J Hum Genet. (2003) ;11: (12):923–30. |
[19] | Awano H , Saito Y , Shimizu M , Sekiguchi K , Niijima S , Matsuo M , et al. FKRP mutations cause congenital muscular dystrophy 1C and limb-girdle muscular dystrophy 2I in Asian patients. J Clin Neurosci. (2021) ;92: :215–21. |
[20] | Nallamilli BRR , Chakravorty S , Kesari A , Tanner A , Ankala A , Schneider T , et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of patients. Ann Clin Transl Neurol. (2018) ;5: (12):1574–87. |
[21] | Tallapaka K , Ranganath P , Ramachandran A , Uppin MS , Perala S , Aggarwal S , et al. Molecular and histopathological characterization of patients presenting with the duchenne muscular dystrophy phenotype in a tertiary care center in Southern India. Indian Pediatr. (2019) ;56: (7):556–9. |
[22] | Nerakh G , Ranganath P , Murugan S . Next-generation sequencing in a cohort of Asian Indian patients with the duchenne muscular dystrophy phenotype: Diagnostic yield and mutation spectrum. J Pediatr Genet. (2021) ;10: (1):23–8. |
[23] | Blaeser A , Keramaris E , Chan YM , Sparks S , Cowley D , Xiao X , et al. Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum Genet. (2013) ;132: (8):923–34. |


