
RNA Transcript Diversity in Neuromuscular Research
Abstract
Three decades since the Human Genome Project began, scientists have now identified more then 25,000 protein coding genes in the human genome. The vast majority of the protein coding genes (> 90%) are multi-exonic, with the coding DNA being interrupted by intronic sequences, which are removed from the pre-mRNA transcripts before being translated into proteins, a process called splicing maturation. Variations in this process, i.e. by exon skipping, intron retention, alternative 5’ splice site (5’ss), 3’ splice site (3’ss), or polyadenylation usage, lead to remarkable transcriptome and proteome diversity in human tissues. Given its critical biological importance, alternative splicing is tightly regulated in a tissue- and developmental stage-specific manner. The central nervous system and skeletal muscle are amongst the tissues with the highest number of differentially expressed alternative exons, revealing a remarkable degree of transcriptome complexity. It is therefore not surprising that splicing mis-regulation is causally associated with a myriad of neuromuscular diseases, including but not limited to amyotrophic lateral sclerosis (ALS), spinal muscular atrophy (SMA), Duchenne muscular dystrophy (DMD), and myotonic dystrophy type 1 and 2 (DM1, DM2). A gene’s transcript diversity has since become an integral and an important consideration for drug design, development and therapy. In this review, we will discuss transcript diversity in the context of neuromuscular diseases and current approaches to address splicing mis-regulation.
MECHANISMS OF TRANSCRIPT DIVERSITY REGULATION
Splicing is mediated by a dynamic small nuclear ribonucleoprotein machinery called the spliceosome [1, 2], which undergoes step-wise assembly, starting from recognition of the cis-element 5’ss and 3’ss on the pre-mRNA. Splice site choice is also modulated by additional splicing regulatory cis-elements throughout the pre-mRNA that dictate the recruitment of different trans-acting RNA binding splicing factors, known as exonic or intronic splicing enhancers and exonic or intronic splicing silencers, depending on whether they enhance or silence splicing activity. These consensus cis-element and trans-acting splicing factors act in a coordinated manner to orchestrate alternative splicing, accounting for the tissue and stage specific regulation of transcript expression. One well-studied example is the recruitment of muscleblind-like proteins (MBNL) to the “UGCU” motif to regulate alternative splicing events in muscle [3, 4]. Weyn-Vanhentenryck et al. reported temporal regulation of splice switching during neural development and maturation in different neuronal subtypes that involves coordinated regulations by RNA binding proteins including neuro-oncological ventral antigen protein (NOVA), RNA binding Fox-1 (RBFOX), MBNL and polypyrimidine tract binding protein (PTBP) [5]. The neural-specific SR-related protein of 100 kDa (nSR100/Srmm4) is also important in regulating alternative splicing events in genes involved in neural functions, as exemplified by abnormalities in the branching of motor neurons innervating the diaphragm, defects in the axonal midline crossing in the corpus callosum and altered cortical layers of the forebrain observed in nSR100 knockout mice [6]. Since the majority of splicing events occur co-transcriptionally, epigenetic factors that regulate the rate of transcription, transcription initiation and elongation can also modulate splicing. These include histone modifications and DNA methylation within the gene body or promoter regions that affect splicing factor recruitment and alter the rate of transcription elongation, thereby indirectly influencing splice site choice and recognition (Reviewed in [7, 8]). In addition, several RNA modifications have been reported to have a role in alternative RNA processing and alternative splicing, including the highly prevalent modifications pseudouridine (Ψ) and internal N6-methyladenosine (m6A) [9], through recruitment of RNA binding proteins specific to the modifications. m6A RNA methylation occurs at the N-6 position of the adenosine residue in the RRACH (R = A/G, H = A/C/U) consensus motif. Depletion of m6A writers [10, 11], erasers [12, 13] and readers [14, 15] have been shown to change the alternative splicing patterns in mammalian systems. Recently Martinez N et al. reported that pre-mRNA is pseudouridylated co-transcriptionally, with specific enrichment of Ψ near alternative splice sites, splicing regulatory elements and splicing factor binding motifs, and installation of a single Ψ is sufficient to alter the splicing outcome in vitro, suggesting regulatory potential of Ψ in alternative splicing [16]. Interestingly, similar to m6A, Ψ is also significantly enriched in 3’ UTRs of pre-mRNAs suggesting a likely role of both these modifications also in alternative polyadenylation [16–18]. Lastly, adenosine-to-inosine RNA editing mediated by Adenosine Deaminases Acting on RNA (ADAR) enzymes, also plays a role in RNA processing and splicing. This is supported by the findings that more than 95% of A-I RNA editing occurs co-transcriptionally in nascent RNAs prior to polyadenylation and splicing events. Modulation of RNA splicing events can occur via creation or elimination of splice sites and branch points by RNA editing or by altering RNA secondary structure which may affect the accessibility of splice sites or the ADAR proteins can promote or preclude binding of splicing machineries or splicing regulators to the RNA [19–22].
ALTERNATIVE SPLICING IN NEUROMUSCULAR DISEASES
Fig. 1
The DMD gene encodes several dystrophin proteins (Dp) or isoforms, which are named based on the length in kilodaltons: Dp427 (c, cortical; m, muscle; p: purkinje cells), Dp260, Dp140, Dp116, Dp71. Alternative promoters are depicted by the arrows.
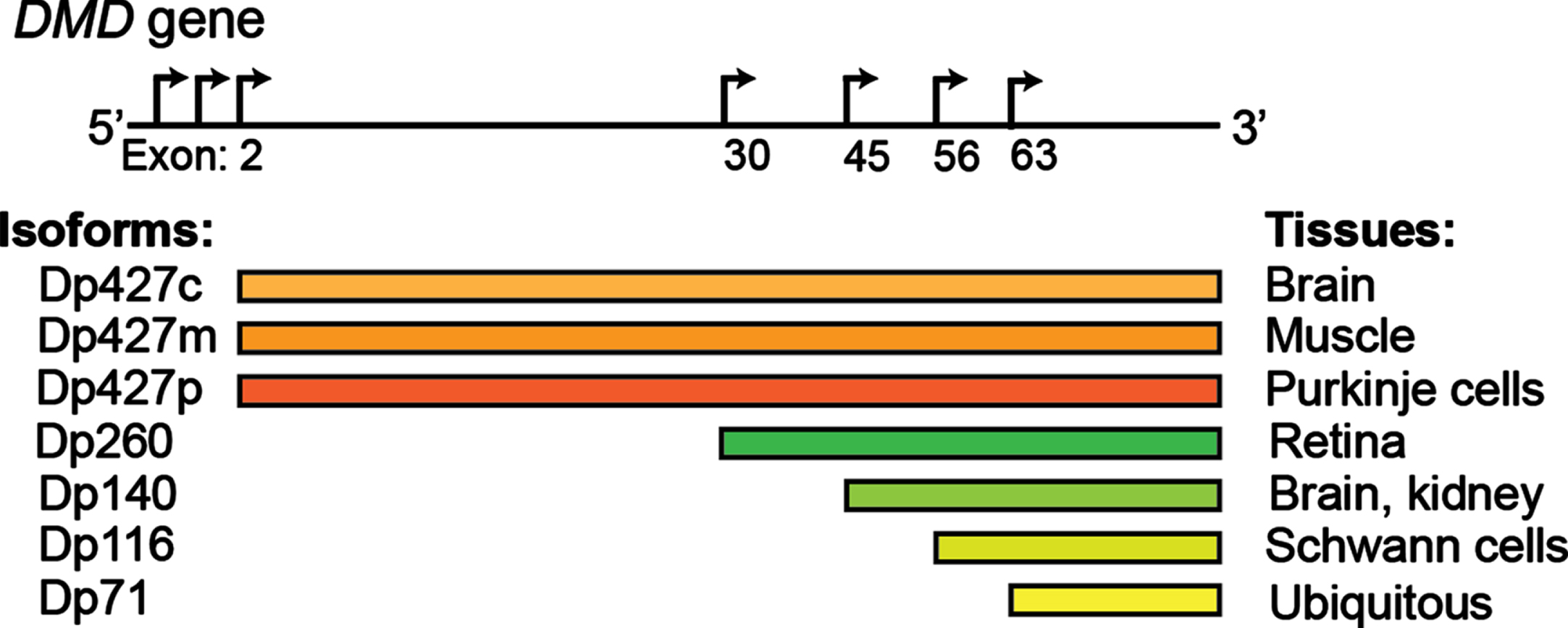
Studies indicate that 95% of human genes are alternatively spliced, representing a fundamental mechanism of spatiotemporal gene regulation [23, 24]. With the progressive maturation of the methods of detection of RNA isoforms, their contribution to human homeostasis is only starting to emerge [25]. Alternative splicing appears to be particularly prevalent in muscle and brain [26–29], where it plays a key role in numerous functions, including driving the process of development and aging [30–32]. In skeletal muscle, tropomyosin isoforms display different localization patterns along actomyosin bundles and are functionally non-redundant [33]. Transcription factor myocyte-specific enhancer factor 2D (Mef2D), a member of the Mef2 family and a key mediator of signal-dependent regulator of developmental processes such as differentiation [34], undergo a major isoform switch during myogenesis. Alternate use of mutually exclusive exons generates a muscle-specific isoform, Mef2Dα2, which is resistant to protein kinase A (PKA) phosphorylation, allowing transcriptional activation of pro-myogenic target genes in the presence of inhibitory PKA signalling [35, 36]. Alterations of tissue-specific transcript isoforms can account for the phenotypic variability observed in many neuromuscular conditions. The dystrophin (DMD) gene contains at least seven tissue-specific promoters and two alternative polyadenylation sites, producing several tissue- and developmental stage-specific transcripts, such as the Dp427 variants, Dp427c and Dp427p, which are predominantly expressed in neurons [37] and the shorter Dp140 isoform, which is predominantly expressed during foetal life stages across the brain [38]. DMD mutations resulting in absent/non-functional dystrophin protein determine a severe phenotype mainly characterised by progressive muscle wasting and weakness, and is frequently also associated with cognitive delay and autism spectrum disorders (ASD) [39, 40]. The risk of cognitive impairment has been linked with the presence of the DMD mutations post-intron 44, which affects not only the full-length isoform, but also the shorter neuronal isoforms, with a pattern of worse cognitive performances on all neuropsychological tests in these patients [41–43] (Fig. 1). Consistently, restoration of Dp140 via mRNA-mediated overexpression improves ASD-like behaviour in a dystrophic mouse model with a mutation in exon 52 [44], further confirming the importance of these neuronal isoforms in the pathophysiology of DMD. Several variants associated with Congenital Myasthenic Syndrome (CMS) fall in the exonic/intronic splicing regions of genes essential for neuromuscular synaptogenesis, such as cholinergic receptor nicotinic epsilon subunit (CHRNE), docking protein 7 (DOK7), and receptor associated protein of the synapse (RAPSN), compromising the binding affinity for trans-acting proteins [45–48], and overall highlighting the important role of splicing for neuromuscular junction formation, maintenance and function. For instance, the IVS3-8G>A change in CHRNA1, a gene encoding the muscle nicotinic acetylcholine receptor α subunit, disrupts an intronic splicing silencer and results in exclusive inclusion of the downstream P3A exon, generating an acetylcholine receptor (AChR) subunit that fails to be incorporated at the motor end plate [49]. Similarly, alterations in trans-acting factors, such as serine arginine-rich (SR) proteins and heterogeneous nuclear ribonucleoproteins (hnRNPs), chromatin landscape and RNA structure can also result in neuromuscular diseases by global perturbation of alternative splicing patterns [50–61]. In DM1 expanded microsatellite repeats in the DM1 protein kinase (DMPK1) gene lead to the formation of RNA secondary structures that sequesters RNA binding proteins, including the splicing regulator MBNL, altering the alternative splicing pattern regulation of key genes in skeletal muscles and other affected tissues [62]. ALS-causing mutations in several RNA binding proteins, including Transactive-response DNA-binding Protein, 43 kDa (TDP-43) and Fused in Sarcoma (FUS), have been causally linked with aberrant alternative splicing in diseased motor neurons [63–70]. Loss-of nuclear TDP-43 function in FTD (frontaltemporal dementia)/ALS has been reported to induce inclusion of cryptic exons, leading to nonsense-mediated delay and loss of Unc-13 Homolog A (UNC13A) and Stathmin-2 (STMN2) protein that are critical for synapse function [71, 72]. Intron retention, a dominant feature of splicing programming occurring during early motor neuron differentiation, occurs prematurely in iPS-derived motor neurons from ALS patients, with Splicing Factor Proline and Glutamine rich (SFPQ) being the most significant intron-retaining transcript across several ALS-causing mutations and representing a hallmark of both familiar and sporadic ALS [73]. Overall these examples highlight the critical importance of alternative isoform regulation in both healthy and diseased motor units.
SPLICING MODULATION AS A THERAPEUTIC TARGET FOR NEUROMUSCULAR DISEASES
Strategies to manipulate RNA splicing have gained traction in the recent years, resulting in several approved drugs and many more showing promising results at preclinical stages in the field of neuromuscular diseases and beyond [74–78]. Therapeutic modulation of RNA splicing has been proposed for two ranges of applications: 1) to correct mutation-induced abnormal splicing patterns, and 2) to selectively modulate isoform expression. One such strategy entails the use of splice switching oligonucleotides (SSOs) to promote exon inclusion or exon skipping by blocking the RNA-RNA base pairing or protein-RNA binding interactions, in order to modulate the ratio of splicing variants or correct splicing defects. To date, five SSOs have been approved for clinical use for neuromuscular diseases, including eteplirsen [79–81], golordirsen [82], viltolarsen [83, 84] and casimersen [85] for DMD and nusinersen [86, 87] for SMA. In the case of DMD, SSOs are designed to skip one or multiple mutation-containing exons to restore the reading frame of dystrophin transcripts, giving rise to truncated but functional dystrophin protein [88], while for SMA, SSOs promote inclusion of exon 7 in survival of motor neuron 2 (SMN2) transcripts, thereby rescuing the expression of full length SMN protein [87, 89–91], which is lacking in this disease. These oligonucleotides are chemically modified not only to increase their stability and affinity to target RNA and to protect them from nuclease activity, but also to prevent activation of RNase H degradation mechanisms and to allow access to the target pre-mRNAs located in the nuclei of cells [92, 93]. In addition to SSOs, other oligonucleotide therapies are also being developed to restore activity of RNA binding splicing factors. Two antisense oligonucleotide (ASO)-based strategies have been proposed to treat DM1: Steric block strategy using ASO to block the binding of MBNL1 to the hairpins, and an RNase H-active ASO that targets the CUG-expanded transcripts for degradation. Both approaches release the sequestered MBNL1 and restore splicing regulatory activity of MBNL1, having shown promise when tested in DM1 preclinical models [94–100]. ASOs have also been used to redirect the usage of alternative translation start site and alternative polyadenylation site, thereby altering the expression of transcript isoforms [101, 102]. Splicing modulation can also be achieved by small molecules, whose discovery has been greatly accelerated by the development of high-throughput screening techniques and in silico splice site prediction tools [103–106]. Risdiplam (PTC-Roche) is the first small molecule splicing modifier approved by the U.S. Food and Drug Administration in 2020 for SMA [107, 108]. While the exact mechanism of action of risdiplam is not yet completely understood, studies suggested that this class of compounds bind the exonic splicing enhancer 2 and 5’ss of SMN2 exon 7 pre-mRNA, ultimately leading to exon 7 inclusion in SMN2 transcript and increased expression of functional full length SMN protein [106, 109]. PK4C9, another RNA splicing modulator that enhances SMN2 exon 7 splicing inclusion by binding to and remodelling the stem-loop RNA structure terminal stem-loop 2 on the 5’ss of SMN2 exon 7 to improve accessibility of the 5’ss to splicing factors [110], is currently under preclinical investigations. RNA splice modulating small molecules have also been used to reduce the production of toxic proteins by inducing a pseudoexon inclusion containing a premature stop codon [111, 112]. For DMD, a strategy deploying Clustered Regularly-Interspaced Short Palindromic Repeats (CRISPR)/Cas to restore dystrophin expression levels in preclinical models have been tested with various levels of success. Olson and colleagues systemically delivered two adeno-associated virus seroptype 9, one encoding Streptococcus pyogenes Cas9 (SpCas9) driven by muscle-specific creatine kinase promoter and another expressing sgRNA targeting a region adjacent to the exon 51 splice acceptor site, to 1-month-old puppies. This strategy generated a single cut at the target site and created various INDELs, in particular, a single nucleotide adenosine (A) insertion immediately 3’ to the Cas9 cut site that resulted in restoration of the dystrophin open reading frame and dystrophin protein expression of up to 70% of wild type levels in skeletal muscles and 92% in heart [113]. In a separate study, Kupatt et al. delivered two nanoparticle coated adeno-associated virus seroptype 9 each carrying half of Cas9 fused to a split-intein moiety that self-assemble upon expression and a pair of single guide RNA targeting sequences flanking exon 51 to 10–14 year old piglets. A high dose treatment (4×1014 vp/kg) restored dystrophin protein expression, 54% and 34%, in quadriceps and diaphragm, respectively, at ∼70 days post treatment, as well as improved muscle and cardiac function [114]. CRISPR/Cas strategies relying on the repair of the double-stranded DNA break, however, may cause unwanted large deletion and in some cases, DNA rearrangement. To circumvent this, two new classes of genome editing technology, termed ‘base editor’ and ‘prime editor’[115], were developed. Base editor, through the fusion of Cas9 nickase with nucleobase deaminases (cytidine or adenine deaminase), catalyses the conversion of one base to another (adenine base editors ABEs: A-to-G or cytosine base editors CBEs: C-to-T), therefore, directly and precisely create point mutations into the DNA without making double-stranded breaks. There has been substantial interest in using the CBEs and ABEs to modulate splicing to promote skipping of exons bearing pathogenic mutations [116, 117] or to induce functional alternative splicing patterns [118]. ABEs, in particular, show great promises since nearly half of the human disease-causing point mutations are G-to-A or C-to-T. Using mouse models of DMD carrying a nonsense point mutation in DMD gene, Ryu S. M. et al. and Xu L. et al. successfully corrected the genetic mutations using ABEs and observed widespread dystrophin rescue and functional improvement in dystrophic mice [119, 120]. Base editors have also been applied to target splice sites for gene knockout. Interestingly, compared to base editor-mediated premature STOP induction, targeting splice sites, in particular the splice donors, produces more robust gene disruption, reflecting the critical role of splice sites in controlling gene splicing and gene expression [121]. While base editors are limited to transitions of A:T to G:C or C:G to T:A, prime editing that relies on a catalytically impaired Cas9 endonucleases fused to an engineered reverse transcriptase and a guide RNA specifying the target site and the desired edit, theoretically, can be used for any type of splice corrections. Using prime editors to reframe the open reading frames, Chemello F et al. restored dystrophin expression and corrected contractile abnormalities in human DMD cardiomyocytes [122]. While these new generation editors exhibit huge potential for splicing correction following a single treatment, the large size of the base editing and prime editing construct precludes single-vector adeno-associated virus packaging. Delivery strategies, longevity of the rescue and potential consequences of persistent in vivo expression of the genome editors are questions still to be addressed and investigated.
SELECTIVE TRANSCRIPT ISOFORM MODULATION
Isoforms often exhibit complementary, unique or even opposing functions to the canonical variant, representing a largely unexplored area of therapeutic opportunity. A role of isoforms has been widely demonstrated in tumorigenesis [123–129], neurological disorders [130, 131] and viral infections [132]. In several types of cancer, toxic isoforms arise as a result of aberrant proteolytic processes when selective pressure is exerted by therapy, forming a pool of escape variants. Targeted degradation of the toxic isoforms via RNA or protein targeting strategies may therefore improve treatment sensitivity and disease prognosis [129, 133, 134]. Overexpression of therapeutically beneficial isoforms have been proposed for treatment of many neuromuscular conditions. In ALS, gene therapy overexpressing trophic factor Neuregulin 1 isoform 1 (NRG1-I) in the skeletal muscles and/or Neuregulin 1 isoform 3 (NRG1-III) in the central nervous system are effective in preserving motor neuron functions [135, 136]. Recently, we showed that specific overexpression of a naturally occurring dominant negative isoform of androgen receptor (AR isoform 2) ameliorates the disease phenotype in a mouse model of spinal and bulbar muscular atrophy, by modulating the activity of the disease causing mutant androgen receptor protein [137]. Another gene therapy using a recombinant adeno-associated virus serotype 1 to deliver follistatin alternatively spliced isoform FS344, to avoid potential binding to off target sites, is currently under clinical investigation for a milder form of dystrophin deficient muscular dystrophy, Becker muscular dystrophy [138]. This strategy is also under investigation for other indications including sporadic inclusion body myositis [139], facioscapulohumeral muscular dystrophy (FSHD) and as a combinatorial therapy for DMD [140].
COMPUTATIONAL TOOLS FOR ISOFORM QUANTIFICATION
Traditional methods for transcriptome-wide identification of alternative splicing events rely on sequencing technologies producing reads ranging from 50 to 150 bp in length, which, combined with various computational tools, have already allowed the identification of thousands of transcript isoforms in human tissues [141, 142]. The methods for isoform analysis can be broadly divided based on whether they utilise a genome reference approach or a de novo assembly one [143], and whether the alternative splicing is estimated from the isoform quantification or directly from exon inclusion ratios [144], with exon-based approaches being generally more sensitive for known transcripts. Many of these tools, such as Cufflinks [145], StringTie [146], systems-level interactive data exploration (SLIDE) [147], and IsoLasso [148] are able to perform transcript discovery based on existing annotations, nevertheless the results are often inaccurate and contradictory, mainly because the detection is biased by the process itself [149, 150]. The use of de novo transcript assembly packages such as Trinity [151], Trans-AbySS [152], and Oases [153] may help mitigate the issue but such methods have failed to provide a complete assessment of isoform diversity in human tissues. Recently developed long-read sequencing technologies, such as single molecule real time sequencing from Pacific Biosciences and Oxford Nanopore Technologies, remove the challenging task of reconstructing transcript isoforms from fragmented short reads and therefore hold great potential in improving our understanding of the plethora of alternatively spliced isoforms in human and non-human tissues [154, 155]. Various computational programs have been developed to produce high-confidence isoforms from long-read sequencing data, such as full-length alternative isoform analysis of RNA (FLAIR) [156], full-length analysis of mutations and splicing (FLAMES) [157, 158], Structural and Quality Annotation of Novel Transcript Isoforms (SQANTI) [159], and Technology-Agnostic Long-Read Analysis (TALON) [160]. These methods have been employed to identify novel alternatively spliced isoforms [155, 161–164] and to characterise changes in isoform profiles upon disease state [156, 165]. Expanding on these advancements, a number of studies have successfully coupled long-read sequencing with targeted RNA capture, accelerating annotation of lowly expressed genes including long non-coding RNAs [161, 162] and allowing deep profiling of tissue-specific isoforms [155, 166, 167]. Recently, a long-read RNA sequencing approach enabled identification of differential exon usage and phasing of structural genes producing large transcripts in cardiac muscle and fast and slow skeletal muscles, that could have direct effect on the interpretation of clinical sequencing data [168]. As the throughput and accuracy of current long-read sequencing platforms improve at fast pace and more sophisticated data analysis pipelines are generated, this technology is rapidly maturing for deployment for single-cell resolution and spatially-resolved transcriptomic applications, therefore allowing deep characterization of isoform diversity in human tissues with further level of complexity and accuracy.
CONCLUSIONS
It has been more than four decades since the concept of alternative splicing was first proposed [169]. Alternative splicing is a highly regulated and sophisticated process of gene regulation that ensures proteome plasticity and diversity necessary for biological functions. However, it is prone to errors, which lead to a wide range of human diseases. The motor unit has proven to be heavily reliant on correct splicing regulation and therefore appears to be exceptionally sensitive to its perturbations. With the advent of new high throughput sequencing technologies and bioinformatics tools, our understanding of alternative splicing is rapidly advancing, with further opportunities for further characterization at single-cell and spatial resolution starting to loom. Undoubtedly, this body of knowledge is quickly showing that RNA diversity is to be accounted for when embarking in any scientific endeavours, all the way from the choice of the appropriate disease model to employ, to mechanistic understanding of physiological and pathological processes, and design and development of treatment strategies that can make a difference for patients: It is time for the modern biomedical scientists to embrace it.
REFERENCES
[1] | Yan C , et al. Structure of a yeast spliceosome at 3, 6-angstrom resolution. Science (2015) ;349: :1182–91. |
[2] | Hang J , Wan R , Yan C , Shi Y Structural basis of pre-mRNA splicing. Science (2015) ;349: :1191–8. |
[3] | Ho TH , et al. Muscleblind proteins regulate alternative splicing. EMBO J (2004) ;23: :3103–12. |
[4] | Kino Y , et al. MBNL and CELF proteins regulate alternative splicing of the skeletal muscle chloride channel CLCN1. Nucleic Acids Res (2009) ;37: :6477–90. |
[5] | Weyn-Vanhentenryck SM , et al. Precise temporal regulation of alternative splicing during neural development. Nat Commun (2018) ;9: :2189. |
[6] | Quesnel-Vallieres M , Irimia M , Cordes SP , Blencowe BJ Essential roles for the splicing regulator nSR100/SRRM4 during nervous system development. Genes Dev. (2015) ;29: :746–59. |
[7] | Naftelberg S , Schor IE , Ast G , Kornblihtt AR Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem. (2015) ;84: :165–98. |
[8] | Luco RF , Allo M , Schor IE , Kornblihtt AR , Misteli T Epigenetics in alternative pre-mRNA splicing. Cell. (2011) ;144: :16–26. |
[9] | Louloupi A , Ntini E , Conrad T , Orom UAV Transient N-6-Methyladenosine Transcriptome Sequencing Reveals a Regulatory Role of m6A in Splicing Efficiency. Cell Rep. (2018) ;23: :3429–37. |
[10] | Wei G , et al. Acute depletion of METTL3 implicates N (6)-methyladenosine in alternative intron/exon inclusion in the nascent transcriptome. Genome Res. (2021) ;31: :1395–408. |
[11] | Liu N , et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. (2015) ;518: :560–4. |
[12] | Bartosovic M , et al. , N6-methyladenosine demethylase FTO targets pre-mRNAs and regulates alternative splicing and 3’-end processing. Nucleic Acids Res. (2017) ;45: :11356–70. |
[13] | Zhao X , et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. (2014) ;24: :1403–19. |
[14] | Xiao W , et al. Nuclear m(6)A Reader YTHDC1 Regulates mRNA Splicing. Mol Cell. (2016) ;61: :507–19. |
[15] | Kasowitz SD , et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. (2018) ;14: :e1007412. |
[16] | Martinez NM , et al. Pseudouridine synthases modify human pre-mRNA co-transcriptionally and affect pre-mRNAprocessing. Mol Cell. (2022) ;82: :645–59 e649. |
[17] | Meyer KD , et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. (2012) ;149: :1635–46. |
[18] | Ke S , et al. A majority of m6A residues are in the last exons, allowing the potential for 3’ UTR regulation. Genes Dev. (2015) ;29: :2037–53. |
[19] | Hsiao YE , et al. RNA editing in nascent RNA affects pre-mRNA splicing. Genome Res. (2018) ;28: :812–23. |
[20] | Schoft VK , Schopoff S , Jantsch MF Regulation of glutamate receptor B pre-mRNA splicing by RNA editing. Nucleic Acids Res. (2007) ;35: :3723–32. |
[21] | Rieder LE , Reenan RA The intricate relationship between RNA structure, editing, and splicing. Semin Cell Dev Biol. (2012) ;23: :281–8. |
[22] | Rieder LE , Staber CJ , Hoopengardner B , Reenan RA Tertiary structural elements determine the extent and specificity of messenger RNA editing. Nat Commun. (2013) ;4: :2232. |
[23] | Pan Q , Shai O , Lee LJ , Frey BJ , Blencowe BJ Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. (2008) ;40: :1413–5. |
[24] | Wahl MC , Will CL , Luhrmann R The spliceosome: design principles of a dynamic RNP machine. Cell. (2009) ;136: :701–18. |
[25] | Wang GS , Cooper TA Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. (2007) ;8: :749–61. |
[26] | Wang ET , et al. Dysregulation of mRNA Localization and Translation in Genetic Disease. J Neurosci. (2016) ;36: :11418–26. |
[27] | Sollner JF , et al. An RNA-Seq atlas of gene expression in mouse and rat normal tissues. Sci Data. (2017) ;4: :170185. |
[28] | Merkin J , Russell C , Chen P , Burge CB Evolutionary dynamics of gene and isoform regulation in Mammalian tissues. Science. (2012) ;338: :1593–9. |
[29] | Consortium GT. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. (2015) ;348: :648–60. |
[30] | Raj B , Blencowe BJ Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron. (2015) ;87: :14–27. |
[31] | Mazin P , et al. Widespread splicing changes in human brain development and aging. Mol Syst Biol. (2013) ;9: :633. |
[32] | Tollervey JR , et al. Analysis of alternative splicing associated with aging and neurodegeneration in the human brain. Genome Res. (2011) ;21: :1572–82. |
[33] | Tojkander S , et al. A molecular pathway for myosin II recruitment to stress fibers. Curr Biol. (2011) ;21: :539–50. |
[34] | Potthoff MJ , Olson EN MEF a central regulator of diverse developmental programs. Development. (2007) ;134: :4131–40. |
[35] | Martin JF , et al. A Mef2 gene that generates a muscle-specific isoform via alternative mRNA splicing. Mol Cell Biol. (1994) ;14: :1647–56. |
[36] | Sebastian S , et al. Tissue-specific splicing of a ubiquitously expressed transcription factor is essential for muscle differentiation. Genes Dev. (2013) ;27: :1247–59. |
[37] | Nudel U , et al. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature. (1989) ;337: :76–8. |
[38] | Morris GE , Simmons C , Nguyen TM Apo-dystrophins (Dp140 and Dp71) and dystrophin splicing isoforms in developing brain. Biochem Biophys Res Commun. (1995) ;215: :361–7. |
[39] | Billard C , et al. Cognitive functions in Duchenne muscular dystrophy: a reappraisal and comparison with spinal muscular atrophy. Neuromuscul Disord. (1992) ;2: :371–8. |
[40] | Dorman C , Hurley AD , D’Avignon J Language and learning disorders of older boys with Duchenne muscular dystrophy. Dev Med Child Neurol. (1988) ;30: :316–27. |
[41] | Taylor PJ , et al. Dystrophin gene mutation location and the risk of cognitive impairment in Duchenne muscular dystrophy. PLoS One. (2010) ;5: :e8803. |
[42] | Chamova T , et al. Association between loss of dp140 and cognitive impairment in duchenne and becker dystrophies. Balkan J Med Genet. (2013) ;16: :21–30. |
[43] | Doorenweerd N , et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep. (2017) ;7: :12575. |
[44] | Hashimoto Y , et al. Brain Dp140 alters glutamatergic transmission and social behaviour in the mdx52 mouse model of Duchenne muscular dystrophy. Prog Neurobiol. (2022) ;216: :102288. |
[45] | Rahman MA , et al. SRSF1 and hnRNP H antagonistically regulate splicing of COLQ exon 16 in a congenital myasthenic syndrome. Sci Rep. (2015) ;5: :13208. |
[46] | Ohno K , Milone M , Shen XM , Engel AG A frameshifting mutation in CHRNE unmasks skipping of the preceding exon. Hum Mol Genet. (2003) ;12: :3055–66. |
[47] | Cossins J , et al. The spectrum of mutations that underlie the neuromuscular junction synaptopathy in DOK7congenital myasthenic syndrome. Hum Mol Genet. (2012) ;21: :3765–75. |
[48] | Muller JS , et al. Impaired receptor clustering in congenital myasthenic syndrome with novel RAPSN mutations. Neurology. (2006) ;67: :1159–64. |
[49] | Masuda A , et al. hnRNP H enhances skipping of a nonfunctional exon P3A in CHRNA1 and a mutation disrupting its binding causes congenital myasthenic syndrome. Hum Mol Genet. (2008) ;17: :4022–35. |
[50] | Jeong S _SR Proteins: Binders, Regulators, and Connectors of RNA. Mol Cells. (2017) ;40: :1–9. |
[51] | Zhou Z , Fu XD Regulation of splicing by_SR proteins and_SR protein-specific kinases. Chromosoma. (2013) ;122: :191–207. |
[52] | Geuens T , Bouhy D , Timmerman V The hnRNP family: insights into their role in health and disease. Hum Genet. (2016) ;135: :851–67. |
[53] | Coltri PP , Dos Santos MGP , da Silva GHG Splicing and cancer: Challenges and opportunities. Wiley Interdiscip Rev RNA. (2019) ;10: :e1527. |
[54] | Schor IE , Fiszbein A , Petrillo E , Kornblihtt AR Intragenic epigenetic changes modulate NCAM alternative splicing in neuronal differentiation. EMBO J. (2013) ;32: :2264–74. |
[55] | Schor IE , Gomez Acuna LI , Kornblihtt AR Coupling between transcription and alternative splicing. Cancer Treat Res. (2013) ;158: :1–24. |
[56] | Dujardin G , et al. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell. (2014) ;54: :683–90. |
[57] | Dujardin G , et al. Transcriptional elongation and alternative splicing. Biochim Biophys Acta. (2013) ;1829: :134–40. |
[58] | Herzel L , Ottoz DSM , Alpert T , Neugebauer KM Splicing and transcription touch base: co-transcriptional spliceosome assembly and function. Nat Rev Mol Cell Biol. (2017) ;18: :637–50. |
[59] | Zhu LY , Zhu YR , Dai DJ , Wang X , Jin HC Epigenetic regulation of alternative splicing. Am J Cancer Res. (2018) ;8: :2346–58. |
[60] | Gomez Acuna LI , Fiszbein A , Allo M , Schor IE , Kornblihtt AR Connections between chromatin signatures and splicing. Wiley Interdiscip Rev RNA. (2013) ;4: :77–91. |
[61] | Warf MB , Berglund JA Role of RNA structure in regulating pre-mRNA splicing. Trends Biochem Sci. (2010) ;35: :169–78. |
[62] | Mankodi A , et al. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science. (2000) ;289: :1769–73. |
[63] | Qiu H , et al. ALS-associated mutation FUS-R521C causes DNA damage and RNA splicing defects. J Clin Invest. (2014) ;124: :981–99. |
[64] | Kapeli K , Martinez FJ , Yeo GW Genetic mutations in RNA-binding proteins and their roles in ALS. Hum Genet. (2017) ;136: :1193–214. |
[65] | Ishigaki S , et al. Position-dependent FUS-RNA interactions regulate alternative splicing events and transcriptions. Sci Rep. (2012) ;2: :529. |
[66] | Masuda A , Takeda J , Ohno K FUS-mediated regulation of alternative RNA processing in neurons: insights from global transcriptome analysis. Wiley Interdiscip Rev RNA. (2016) ;7: :330–40. |
[67] | Deshaies JE , et al. TDP-43 regulates the alternative splicing of hnRNP A1 to yield an aggregation-prone variant in amyotrophic lateral sclerosis. Brain. (2018) ;141: :1320–33. |
[68] | Melamed Z , et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. (2019) ;22: :180–90. |
[69] | Klim JR , et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. (2019) ;22: :167–79. |
[70] | Ling JP , Pletnikova O , Troncoso JC , Wong PC TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. (2015) ;349: :650–5. |
[71] | Brown AL , et al. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. (2022) ;603: :131–7. |
[72] | Ma XR , et al. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. (2022) ;603: :124–30. |
[73] | Luisier R , et al. Intron retention and nuclear loss of SFPQ are molecular hallmarks of ALS. Nat Commun. (2018) ;9: :2010. |
[74] | Bennett CF Therapeutic Antisense Oligonucleotides Are Coming of Age. Annu Rev Med. (2019) ;70: :307–21. |
[75] | Zhu Y , Zhu L , Wang X , Jin H RNA-based therapeutics: an overview and prospectus. Cell Death Dis. (2022) ;13: :644. |
[76] | Stanley RF , Abdel-Wahab O Dysregulation and therapeutic targeting of RNA splicing in cancer. Nat Cancer. (2022) ;3: :536–46. |
[77] | El Marabti E , Abdel-Wahab O Therapeutic Modulation of RNA Splicing in Malignant and Non-Malignant Disease. Trends Mol Med. (2021) ;27: :643–59. |
[78] | Scoto M , Finkel R , Mercuri E , Muntoni F Genetic therapies for inherited neuromuscular disorders. Lancet Child Adolesc Health. (2018) ;2: :600–9. |
[79] | Mendell JR , et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol. (2016) ;79: :257–71. |
[80] | Kinali M , et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. (2009) ;8: :918–28. |
[81] | Nakamura A , Takeda S Exon-skipping therapy for Duchenne muscular dystrophy. Lancet. (2011) ;378: :546–7. |
[82] | Heo YA Golodirsen: First Approval. Drugs. (2020) ;80: :329–33. |
[83] | Dhillon S Viltolarsen: First Approval. Drugs. (2020) ;80: :1027–31. |
[84] | Clemens PR , et al. Long-Term Functional Efficacy and Safety of Viltolarsen in Patients with Duchenne Muscular Dystrophy. J Neuromuscul Dis. (2022) ;9: :493–501. |
[85] | Shirley M Casimersen: First Approval. Drugs. (2021) ;81: :875–9. |
[86] | Corey DR Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat Neurosci. (2017) ;20: :497–9. |
[87] | Hua Y , Vickers TA , Okunola HL , Bennett CF , Krainer AR Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. (2008) ;82: :834–8. |
[88] | Kole R , Krieg AM Exon skipping therapy for Duchenne muscular dystrophy. Adv Drug Deliv Rev. (2015) ;87: :104–7. |
[89] | Hua Y , Vickers TA , Baker BF , Bennett CF , Krainer AR Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. (2007) ;5: :e73. |
[90] | Singh NK , Singh NN , Androphy EJ , Singh RN Splicing of a critical exon of human Survival Motor Neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. (2006) ;26: :1333–46. |
[91] | Singh NN , Shishimorova M , Cao LC , Gangwani L , Singh RN A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. (2009) ;6: :341–50. |
[92] | Bennett CF , Swayze EE RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu Rev Pharmacol Toxicol. (2010) ;50: :259–93. |
[93] | Bennett CF , Baker BF , Pham N , Swayze E , Geary RS Pharmacology of Antisense Drugs. Annu Rev Pharmacol Toxicol. (2017) ;57: :81–105. |
[94] | Klein AF , Dastidar S , Furling D , Chuah MK Therapeutic Approaches for Dominant Muscle Diseases: Highlight onMyotonic Dystrophy. Curr Gene Ther. (2015) ;15: :329–37. |
[95] | Klein AF , et al. Peptide-conjugated oligonucleotides evoke long-lasting myotonic dystrophy correction in patient-derived cells and mice. J Clin Invest. (2019) ;129: :4739–44. |
[96] | Wheeler TM , et al. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. (2012) ;488: :111–5. |
[97] | Jauvin D , et al. Targeting DMPK with Antisense Oligonucleotide Improves Muscle Strength in Myotonic Dystrophy Type 1 Mice. Mol Ther Nucleic Acids. (2017) ;7: :465–74. |
[98] | Pandey SK , et al. Identification and characterization of modified antisense oligonucleotides targeting DMPK in mice and nonhuman primates for the treatment of myotonic dystrophy type 1. J Pharmacol Exp Ther. (2015) ;355: :329–40. |
[99] | Yadava RS , et al. Systemic therapy in an RNA toxicity mouse model with an antisense oligonucleotide therapy targeting a non-CUG sequence within the DMPK 3’UTR RNA. Hum Mol Genet. (2020) ;29: :1440–53. |
[100] | Ait Benichou S , et al. Antisense oligonucleotides as a potential treatment for brain deficits observed in myotonic dystrophy type 1. Gene Ther. ((2022) ). |
[101] | Vickers TA , Wyatt JR , Burckin T , Bennett CF , Freier SM Fully modified 2’ MOE oligonucleotides redirect polyadenylation. Nucleic Acids Res. (2001) ;29: :1293–9. |
[102] | Castanotto D , et al. A Multifunctional LNA Oligonucleotide-Based Strategy Blocks AR Expression and Transactivation Activity in PCa Cells. Mol Ther Nucleic Acids. (2021) ;23: :63–75. |
[103] | Pawellek A , et al. Identification of small molecule inhibitors of pre-mRNA splicing. J Biol Chem. (2014) ;289: :34683–98. |
[104] | Sidarovich A , et al. Identification of a small molecule inhibitor that stalls splicing at an early step of spliceosome activation. Elife. (2017) ;6. |
[105] | Zhang J , Harvey SE , Cheng C A high-throughput screen identifies small molecule modulators of alternative splicing by targeting RNA G-quadruplexes. Nucleic Acids Res. (2019) ;47: :3667–79. |
[106] | Campagne S , et al. Structural basis of a small molecule targeting RNA for a specific splicing correction. Nat Chem Biol. (2019) ;15: :1191–8. |
[107] | Ratni H , et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J Med Chem. (2018) ;61: :6501–17. |
[108] | Baranello G , et al. Risdiplam in Type 1 Spinal Muscular Atrophy. N Engl J Med. (2021) ;384: :915–23. |
[109] | Sivaramakrishnan M , et al. Binding to SMN2 pre-mRNA-protein complex elicits specificity for small molecule splicing modifiers. Nat Commun. (2017) ;8: :1476. |
[110] | Garcia-Lopez A , et al. Targeting RNA structure in SMN2 reverses spinal muscular atrophy molecular phenotypes. Nat Commun. (2018) ;9: :2032. |
[111] | Bhattacharyya A , et al. Small molecule splicing modifiers with systemic HTT-lowering activity. Nat Commun. (2021) ;12: :7299. |
[112] | Keller CG , et al. An orally available, brain penetrant, small molecule lowers huntingtin levels by enhancing pseudoexon inclusion. Nat Commun. (2022) ;13: :1150. |
[113] | Amoasii L , et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. (2018) ;362: :86–91. |
[114] | Moretti A , et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med. (2020) ;26: :207–14. |
[115] | Anzalone AV , et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. (2019) ;576: :149–57. |
[116] | Gapinske M , et al. CRISPR-SKIP: programmable gene splicing with single base editors. Genome Biol. (2018) ;19: :107. |
[117] | Winter J , et al. Targeted exon skipping with AAV-mediated split adenine base editors. Cell Discov. (2019) ;5: :41. |
[118] | Yuan J , et al. Genetic Modulation of RNA Splicing with a CRISPR-Guided Cytidine Deaminase. Mol Cell. (2018) ;72: :380–94 e387. |
[119] | Ryu SM , et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol. (2018) ;36: :536–9. |
[120] | Xu L , et al. Efficient precise in vivo base editing in adult dystrophic mice. Nat Commun. (2021) ;12: :3719. |
[121] | Kluesner MG , et al. CRISPR-Cas9 cytidine and adenosine base editing of splice-sites mediates highly-efficient disruption of proteins in primary and immortalized cells. Nat Commun. (2021) ;12: :2437. |
[122] | Chemello F , et al. Precise correction of Duchenne muscular dystrophy exon deletion mutations by base and prime editing. Sci Adv. (2021) ;7. |
[123] | Miura K , Fujibuchi W , Unno M Splice isoforms as therapeutic targets for colorectal cancer. Carcinogenesis. (2012) ;33: :2311–9. |
[124] | Bogetofte Barnkob M , Vitting-Seerup K , Ronn Olsen L Target isoforms are an overlooked challenge and opportunity in chimeric antigen receptor cell therapy. Immunother Adv. (2022) ;2: :ltac009. |
[125] | Zhao L , Sanyal S p53 Isoforms as Cancer Biomarkers and Therapeutic Targets. Cancers (Basel) (2022) ;14: . |
[126] | Meng J Distinct functions of dynamin isoforms in tumorigenesis and their potential as therapeutic targets in cancer. Oncotarget. (2017) ;8: :41701–16. |
[127] | Paur J , et al. Fibroblast growth factor receptor 3 isoforms: Novel therapeutic targets for hepatocellular carcinoma? Hepatology. (2015) ;62: :1767–78. |
[128] | Ricciuti B , et al. Comparative Analysis and Isoform-Specific Therapeutic Vulnerabilities of KRAS Mutations in Non-Small Cell Lung Cancer. Clin Cancer Res. (2022) ;28: :1640–50. |
[129] | Gupta A , et al. Isoform specific anti-TGFbeta therapy enhances antitumor efficacy in mouse models of cancer. Commun Biol. (2021) ;4: :1296. |
[130] | Boyarko B , Hook V Human Tau Isoforms and Proteolysis for Production of Toxic Tau Fragments in Neurodegeneration. Front Neurosci. (2021) ;15: :702788. |
[131] | Kerr B , et al. Transgenic complementation of MeCP2 deficiency: phenotypic rescue of Mecp2-null mice by isoform-specific transgenes. Eur J Hum Genet. (2012) ;20: :69–76. |
[132] | Ko SH , et al. Interference of DNAJB6/MRJ Isoform Switch by Morpholino Inhibits Replication of HIV-1 and RSV. Mol Ther Nucleic Acids. (2019) ;14: :251–61. |
[133] | Zhang L , et al. RNA interference: a potential strategy for isoform-specific phosphatidylinositol 3-kinasetargeted therapy in ovarian cancer. Cancer Biol Ther. (2004) ;3: :1283–9. |
[134] | Vanderborght B , et al. Effect of isoform-specific HIF-1alpha and HIF-2alpha antisense oligonucleotides ontumorigenesis, inflammation and fibrosis in a hepatocellular carcinoma mouse model. Oncotarget. (2020) ;11: :4504–20. |
[135] | Modol-Caballero G , et al. Gene Therapy Overexpressing Neuregulin 1 Type I in Combination With Neuregulin 1 TypeIII Promotes Functional Improvement in the SOD1(G93A) ALS Mice. Front Neurol. (2021) ;12: :693309. |
[136] | Modol-Caballero G , et al. Gene therapy for overexpressing Neuregulin 1 type I in skeletal muscles promotes functional improvement in the SOD1(G93A) ALS mice. Neurobiol Dis. (2020) ;137: :104793. |
[137] | Lim WF , et al. Gene therapy with AR isoform 2 rescues spinal and bulbar muscular atrophy phenotype by modulatingAR transcriptional activity. Sci Adv. (2021) ;7. |
[138] | Mendell JR , et al. A phase 1/2a follistatin gene therapy trial for becker muscular dystrophy. Mol Ther. (2015) ;23: :192–201. |
[139] | Mendell JR , et al. Follistatin Gene Therapy for Sporadic Inclusion Body Myositis Improves Functional Outcomes. Mol Ther. (2017) ;25: :870–9. |
[140] | Rodino-Klapac LR , et al. Micro-dystrophin and follistatin co-delivery restores muscle function in aged DMD model. Hum Mol Genet. (2013) ;22: :4929–37. |
[141] | Carninci P , et al. High-efficiency full-length cDNA cloning by biotinylated CAP trapper. Genomics. (1996) ;37: :327–36. |
[142] | Strausberg RL , et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc Natl Acad Sci U S A. (2002) ;99: :16899–903. |
[143] | Martin JA , Wang Z Next-generation transcriptome assembly. Nat Rev Genet. (2011) ;12: :671–82. |
[144] | Jiang W , Chen L Alternative splicing: Human disease and quantitative analysis from high-throughput sequencing. Comput Struct Biotechnol J. (2021) ;19: :183–95. |
[145] | Trapnell C , et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. (2010) ;28: :511–5. |
[146] | Pertea M , et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat Biotechnol. (2015) ;33: :290–5. |
[147] | Alamancos GP , Pages A , Trincado JL , Bellora N , Eyras E Leveraging transcript quantification for fast computation of alternative splicing profiles. RNA. (2015) ;21: :1521–31. |
[148] | Li W , Feng J , Jiang T IsoLasso: a LASSO regression approach to RNA-Seq based transcriptome assembly. J Comput Biol. (2011) ;18: :1693–707. |
[149] | Conesa A , et al. A survey of best practices for RNA-seq data analysis. Genome Biol. (2016) ;17: :13. |
[150] | Engstrom PG , et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods. (2013) ;10: :1185–91. |
[151] | Grabherr MG , et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. (2011) ;29: :644–52. |
[152] | Robertson G , et al. De novo assembly and analysis of RNA-seq data. Nat Methods. (2010) ;7: :909–12. |
[153] | Schulz MH , Zerbino DR , Vingron M , Birney E Oases: robust de novo RNA-seq assembly across the dynamic range ofexpression levels. Bioinformatics. (2012) ;28: :1086–92. |
[154] | Chen S , Qiu G , Yang M SMRT sequencing of full-length transcriptome of seagrasses Zostera japonica. Sci Rep. (2019) ;9: :14537. |
[155] | De Paoli-Iseppi R , Gleeson J , Clark MB Isoform Age - Splice Isoform Profiling Using Long-Read Technologies. Front Mol Biosci. (2021) ;8: :711733. |
[156] | Tang AD , et al. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun. (2020) ;11: :1438. |
[157] | Tian L , et al. Comprehensive characterization of single-cell full-length isoforms in human and mouse with long-read sequencing. Genome Biol. (2021) ;22: :310. |
[158] | Dong X , et al. The long and the short of it: unlocking nanopore long-read RNA sequencing data with short-read differential expression analysis tools. NAR Genom Bioinform. (2021) ;3: :lqab028. |
[159] | Tardaguila M , et al. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res, ((2018) ). |
[160] | Wyman D , et al. A technology-agnostic long-read analysis pipeline for transcriptome discovery and quantification. BioRxiv, ((2020) ). |
[161] | Lagarde J , et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nat Genet. (2017) ;49: :1731–40. |
[162] | Hardwick SA , Joglekar A , Flicek P , Frankish A , Tilgner HU Getting the Entire Message: Progress in Isoform Sequencing. Front Genet. (2019) ;10: :709. |
[163] | Workman RE , et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nat Methods. (2019) ;16: :1297–305. |
[164] | Roach NP , et al. The full-length transcriptome of C. elegans using direct RNA sequencing. Genome Res. (2020) ;30: :299–312. |
[165] | Asnani M , et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonalframeshift mutations in CD19. Leukemia. (2020) ;34: :1202–7. |
[166] | Treutlein B , Gokce O , Quake SR , Sudhof TC Cartography of neurexin alternative splicing mapped by single-molecule long-read mRNA sequencing. Proc Natl Acad Sci U S A. (2014) ;111: :E1291–1299. |
[167] | Treutlein B , et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. (2014) ;509: :371–5. |
[168] | Uapinyoying P , et al. A long-read RNA-seq approach to identify novel transcripts of very large genes. Genome Res. (2020) ;30: :885–97. |
[169] | Gilbert W , Why genes in pieces? Nature. ((1978) :271: :501. |


