
Bone Quality in Patients with a Congenital Myopathy: A Scoping Review
Abstract
Background:
Congenital myopathies are rare neuromuscular disorders presenting with a wide spectrum of clinical features, including long bone fractures (LBFs) that negatively influence functional prognosis, quality of life and survival. Systematic research on bone quality in these patients is lacking.
Objective:
This scoping review aims to summarize all evidence on bone quality and to deduce recommendations for bone quality management in congenital myopathies.
Methods:
Five electronic databases (Pubmed, Embase, Cochrane, Web of Science, CINAHL) were searched. All studies on bone quality in congenital myopathies were included. Decreased bone quality was defined as low bone mineral density and/or (fragility) LBFs. Study selection and data extraction were performed by three independent reviewers.
Results:
We included 244 single cases (mean: 4.1±7.6 years; median: 0 years) diagnosed with a congenital myopathy from 35 articles. Bone quality was decreased in 93 patients (37%) (mean: 2.6±6.8 years; median: 0 years). Low bone mineral density was reported in 11 patients (4.5%) (mean: 10.9±9.7; median: 11 years). Congenital LBFs were reported in 64 patients (26%). (Fragility) LBFs later at life were described in 24 patients (9.8%) (mean: 14.9±11.0; median: 14 years). Four cases (1.6%) were reported to receive vitamin D and/or calcium supplementation or diphosphonate administration.
Conclusion:
LBFs are thus frequently reported in congenital myopathies. We therefore recommend optimal bone quality management through bone mineral density assessment, vitamin D and calcium suppletion, and referral to internal medicine or pediatrics for consideration of additional therapies in order to prevent complications of low bone mineral density.
ABBREVIATIONS
ACTA1 = Actin Alpha 1
ASCC1(-RM) = Activating Signal Cointegrator 1 Complex Subunit 1 (–related myopathy)
BHI = Bone Health Index
BM = Bethlem myopathy
CCD = central core disease
CNM = centronuclear myopathy
DEXA-scan = Dual Energy X-ray absorptiometry scan
DMD = Duchenne Muscular Dystrophy
HHR = hereditary hypophosphatemic rickets
KLHL40 = Kelch Like Family Member 40
LMOD3 = Leiomodin 3
LBF = long bone fracture
NEB = Nebulin
NEM2 = nemaline myopathy type 2
NEM8 = nemaline myopathy type 8
NM = nemaline myopathy
RYR1(-RM) = Ryanodine receptor type 1(-related myopathy)
SELENON(-RM) = SELENON(-related myopathy)
SMA = spinal muscular atrophy
TNNC2 = Troponin C2
TTN = titin
UCMD = Ulrich congenital muscular dystrophy
VMA21 = Vacuolar ATPase Assembly Factor
(XL-)MTM = (X-linked) myotubular myopathy
INTRODUCTION
Congenital myopathies are a group of rare inherited myopathies that present with a wide spectrum of clinical features [1]. Frequently reported characteristics include neonatal feeding difficulties and respiratory weakness, delayed motor development and scoliosis. Due to the multi-system involvement in patients with congenital myopathies, physicians from a broad range of disciplines collaborate to provide optimal care for this patient group. The low incidence of congenital myopathies combined with the multi-system involvement might cause essential clinical aspects to remain unknown or underexposed to the health care providers. Therefore, a Consensus Statement on Standard of Care for Congenital Myopathies has been developed, in which among others neurological, cardiac, pulmonary, gastrointestinal, and orthopedic aspects of care are described [1]. Since patients with reduced mobility are at increased risk of low bone mineral density and fragility LBFs, this guideline recommends that intensive bone quality evaluation should be undertaken, and that vitamin D and calcium should be supplemented. However, systematic studies or reviews on bone quality, including bone mineral density and (fragility) LBFs, in patients with congenital myopathies are lacking.
In the adult population, osteopenia and osteoporosis are characterized by low bone mineral density (T score between –1 and –2.5 SD, and< –2.5 SD on Dual Energy X-ray absorptiometry scan (DEXA-scan), respectively), resulting in an increased risk of bone fractures after a low-impact trauma (fragility LBFs) [2, 3]. In the pediatric population, osteoporosis is defined as low bone mineral density (z-score ≤–2.0) in combination with a clinically significant fracture history (i.e. ≥2 fragility LBFs before age of 10, ≥3 fragility LBFs before age of 19, or the presence of one or more vertebral compression fractures occurring without major trauma or local disease) [4]. Primary osteoporosis occurs due to an intrinsic skeletal defect of genetic or idiopathic origin, most commonly caused by a form of Osteogenesis Imperfecta [5]. Osteoporosis associated with physiological changes during menopause and ageing (secondary osteoporosis) is the most common form in adults. Secondary osteoporosis in children and adults can also be caused by other diseases or medications, i.e. malabsorption, immobility, chronic inflammatory diseases, endocrinologic diseases or steroid use [2, 6]. A number of studies have found a link between neuromuscular diseases and a higher risk of developing low bone mineral density and fragility LBFs [7]. Indeed, it is well-known that Duchenne muscular dystrophy is associated with an increased risk of bone fragility caused by a combination of progressive muscle weakness with loss of weight-bearing activity and osteotoxicity from prolonged glucocorticoid therapy [8].
Besides systematic studies or reviews on bone qualitynatural history studies will provide more insight on bone quality that can be the basis for intervention studies [1]. Therefore, in the current LAST STRONG Study, a natural history study on patients diagnosed with LAMA2-related muscular dystrophy and SELENON-related myopathy, we included bone quality assessment by a fracture history and a DEXA-scan [9]. The first results show decreased bone mineral density and/or fragility LBFs in the majority of the patients, which was not always related to immobility alone (unpublished data).
LBFs play a role in the functional prognosis, in the quality of life and in survival of patients with neuromuscular diseases [1, 10, 11]. There are clues that limb immobilization in patients with a neuromuscular disease may cause more loss of muscle mass than in healthy subjects [12]. Patients that experience fractures of the femurs or lower legs are prone to lose ambulance permanently [11, 13]. Further, low bone quality can lead to difficulties in neuromuscular management and rehabilitation, including surgical treatments for scoliosis or foot deformities [14, 15]. Moreover, pain caused by LBFs impairs rehabilitative care resulting in decreased mobilization, pulmonary function and nutritional intake, all negatively influencing survival [1, 16]. Finally, a previous study has shown that bisphosphonate treatment in patients with Duchenne Muscular Dystrophy (DMD) with clinical bone fragility has improved bone density, backpain and even survival [17]. Preventing complications caused by low bone mineral density, including the prevention of falls, is thus of particular interest in patients with congenital myopathies.
In order to explore bone quality in congenital myopathies, to examine the correlation between bone quality and age, and to deduce recommendations for standardized bone quality assessment and/or treatment, a scoping review on bone quality in patients diagnosed with a congenital myopathy was initiated.
MATERIALS AND METHODS
This scoping review was performed according to the Preferred Reporting Items for Systematic reviews and Meta-Analyses extension for Scoping Reviews (PRISMA-Scr) guidelines, which follows a systematic approach to map evidence on a topic with emerging, heterogenous evidence and identify main concepts, theories, sources, and knowledge gaps [18]. This approach allowed us to structurally synthesize evidence and assess the scope of literature on bone quality in congenital myopathies.
Information sources and search strategy
A literature search was carried out in Pubmed (MEDLINE database) (United States National Library of Medicine, United States), Embase (Ovid) (Excerpta Medica dataBASE, Elsevier, The Netherlands), the Cochrane Library (Cochrane, England), Web of Science (Clarivate Analytics, United States) and the Cumulative Index to Nursing & Allied Health Literature (CINAHL) (EBSCO, United States) to find relevant studies, without limitations on year of publication at February 15th, 2022. Search terms included MeSH terms or Emtree terms, synonyms of congenital myopathy, bone complications (osteopenia, osteoporosis, LBF), ancillary investigations to evaluate bone mineral density (Dual Energy X-ray absorptiometry (DEXA)-scan and X-ray radiogrammetry of the hand), and possible treatment strategies (vitamin D, calcium and/or diphosphonates). It was aimed to include all known congenital myopathies subtypes, both classified on the basis of morphological features and on the genetic mutation [1, 19]. Assistance to optimize our search strategy was provided by an information specialist at the Medical Library in Nijmegen, The Netherlands. In addition, references of eligible studies were checked for any studies that were not obtained through our search strategy (forward and backward citing). The full search strategies can be found in Supplementary Table 1. Prior to the start of the study, the study protocol was registered at OSF registries (registration DOI: 10.17605/OSF.IO/HBVJP).
Inclusion strategy
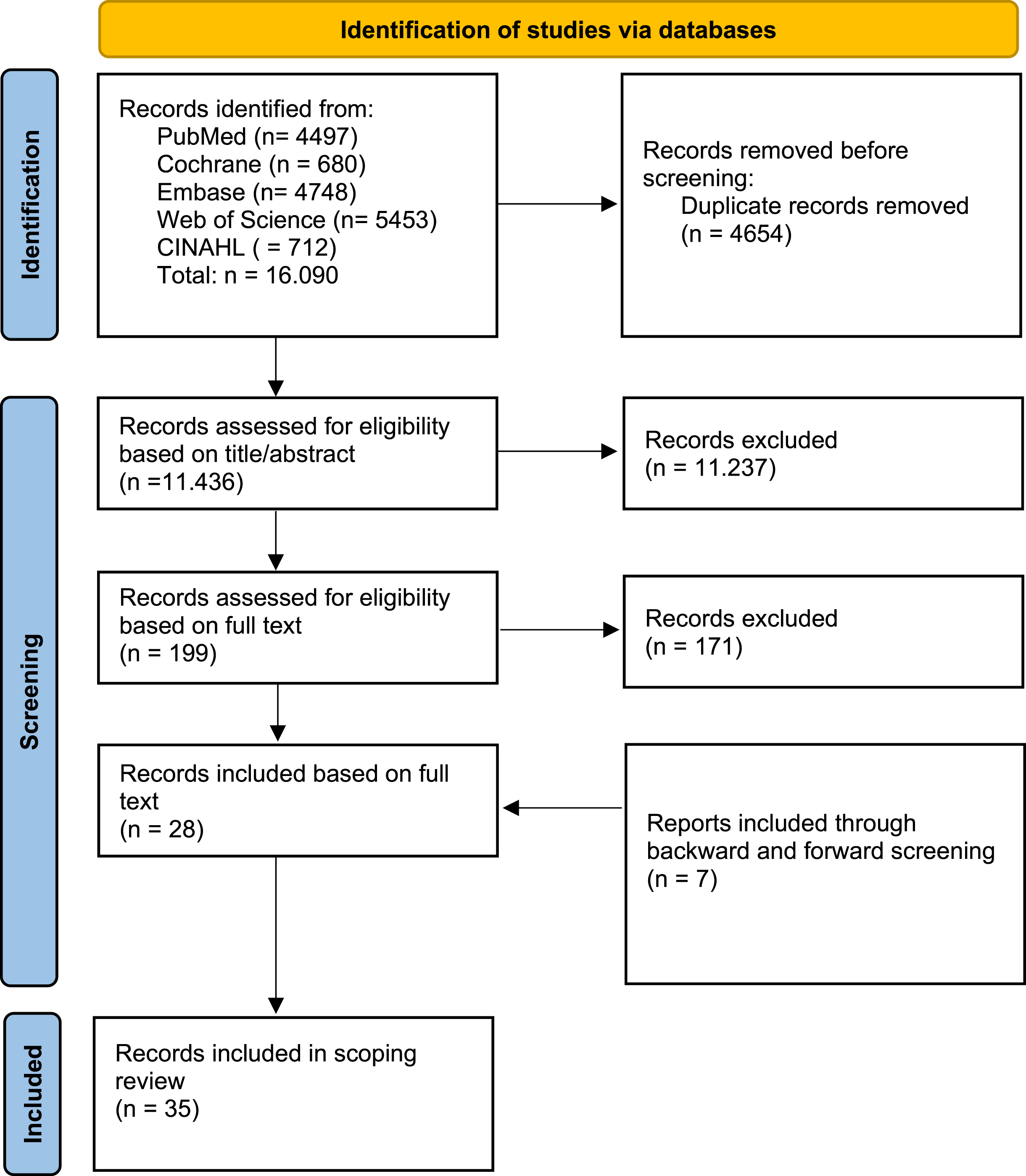
Any case report, case series, clinical study or conference abstract with information on bone quality or complications in patients diagnosed with a congenital myopathy was included, without a restriction on publication year. Decreased bone quality was defined as low bone mineral density (T < –1,0 in adults or Z≤–2,0 in pediatric population) and/or (fragility) LBFs. Articles that were not written in Dutch or English or that were not available in full text were excluded. Literature reviews were used to identify new case reports or case series through backward and forward screening, but were not themselves included in our analysis in order to prevent double inclusion of cases. All identified cases were collected in EndNote X9 and duplicates were removed. Each article was independently screened for eligibility by three authors (KB, and AD or JD) based on title and abstract by using Rayyan Systems Inc (http://rayyan.qcri.org). Full texts of all potentially relevant studies were subsequently retrieved and further examined for eligibility. Inconsistencies were discussed until consensus was reached. The PRISMA flow diagram provides more detailed information regarding the selection process of studies (See Fig. 1).
Fig. 1
PRISMA flowchart on identification of eligible studies on bone quality in congenital myopathies. The abstracts of the identified articles were screened and the full-text articles were subsequently assessed for eligibility. N = number;
Data extraction
Data was systematically extracted by two independent researchers (KB and AD) using a predesigned extraction sheet, including information on medical history (i.e. general medical history, family history, bone mineral density, LBFs, medicines used) and ancillary investigations (DEXA-scan, X-ray, blood tests on calcium and vitamin D). Since the included articles frequently lacked information on the criteria for osteopenia and osteoporosis or did not quantify the degree of decreased bone mineral density into more details, we followed the exact terminology used in the included articles to classify decreased bone mineral density into osteopenia or osteoporosis for further analysis in our scoping review. A LBF, which is defined by cortical interruption of the long bone, was either classified as congenital (present in the first four weeks of life) or as ‘later at life’ (seen after the neonatal period, after the first four weeks of life). Fragility fractures are defined as fractures after low-energy trauma as a consequence of low bone mineral density [20]. Since the included articles frequently did not make a clear distinction between fragility and non-fragility LBFs, it was decided to refer to ‘(fragility) LBFs’ in this scoping review when the origin was (partly) unknown. Additionally, demographic data (i.e. age, gender, nationality etc.) and neuromuscular disease specific information (type of congenital myopathy and specific mutation) were obtained. In more detail, the current age and the age at which the bone complication was first noted, were extracted. If the latter was not known, the current age was used for further data analysis. The data extraction sheet is added as Supplementary Table 2.
Statistical analysis
Descriptive statistics (mean±SD, range, median, percentages) were used to summarize the data on bone quality in patients diagnosed with a congenital myopathy.
RESULTS
Case demographics
We identified 244 single cases (M = 37, F = 21; unknown = 186) with information on bone quality or complications diagnosed with a congenital myopathy from 35 separate studies, with a mean age of 4.1 years (SD: 7.6 years; range 0 to 34 years; median: 0 years) [21–55]. Of all included cases, 178 patients were diagnosed with nemaline myopathy (NM) [21, 22, 25, 26, 28, 32, 34, 38, 40, 41, 45, 46, 50–54], 27 patients with Ryanodine Receptor 1-Related Myopathy (RYR1-RM) [23, 28, 35, 43, 49, 55], 13 patients with congenital titinopathy [29, 42], eight patients with X-linked myotubular myopathy (XL-MTM) [27, 28, 31, 48] and three patients with SELENON-RM [36]. Another five patients with a congenital myopathy caused by mutations in the ASCC1 gene were described [24]. Finally, ten patients were diagnosed with a congenital myopathy with a different origin or a congenital myopathy that was not further histologically or genetically specified [28, 30, 33, 37, 39, 44, 47] (See Table 1).
Table 1
Neuromuscular diagnosis of included cases
| Congenital myopathy | Number of cases (n = 244) | Genetic mutation and/or histological subdivision |
| Nemaline myopathy | 178 | |
| 4 | LMOD3 | |
| 5 | ACTA1 | |
| 20 | KLHL40 | |
| 1 | TNNC2 | |
| 1 | NEB | |
| 147 | Nemalin myopathy without specific genetic diagnosis | |
| RYR1-RM | 27 | |
| Congenital titinopathy | 13 | |
| XL-MTM | 8 | |
| SELENON-RM | 3 | |
| ASCC1-RM | 5 | |
| Remaining or unspecified congenital myopathy | 10 | ACTA1-RM, X-linked myopathy with excessive autophagy associated with VMA21 mutation, NEB-RM, Tubular aggregate myopathy, core myopathy, centronuclear myopathy, Nonspecific congenital myopathy |
Abbreviations. LMOD3 = Leiomodin 3; ACTA1 = Actin Alpha 1; KLHL40 = Kelch Like Family Member 40; TNNC2 = Troponin C2; NEB = Nebulin; RYR1-RM = Ryanodine receptor type 1-related myopathies; XL-MTM = X-linked myotubular myopathy; SELENON-RM = SELENON-related myopathies; ASCC1-RM = Activating Signal Cointegrator 1 Complex Subunit 1 – related myopathy; VMA21 = Vacuolar ATPase Assembly Factor.
Bone complications
Ninety-three of 244 patients were reported to have decreased bone quality (i.e. low bone mineral density (osteopenia or osteoporosis)) or (fragility) LBFs, with a mean age at diagnosis of 2.6 years (SD±6.8; range 0 to 34 years; median: 0 years) (See Fig. 2 and Supplementary Table 3).
Fig. 2
Bone complications in patients with a congenital myopathy. Number of cases identified with congenital LBFs, LBFs later in life, decreased bone mineral density or treatment. Treatment includes vitamin D and/or calcium supplementation or intravenous diphosphonate administration. N = number; LBF = long bone fracture.
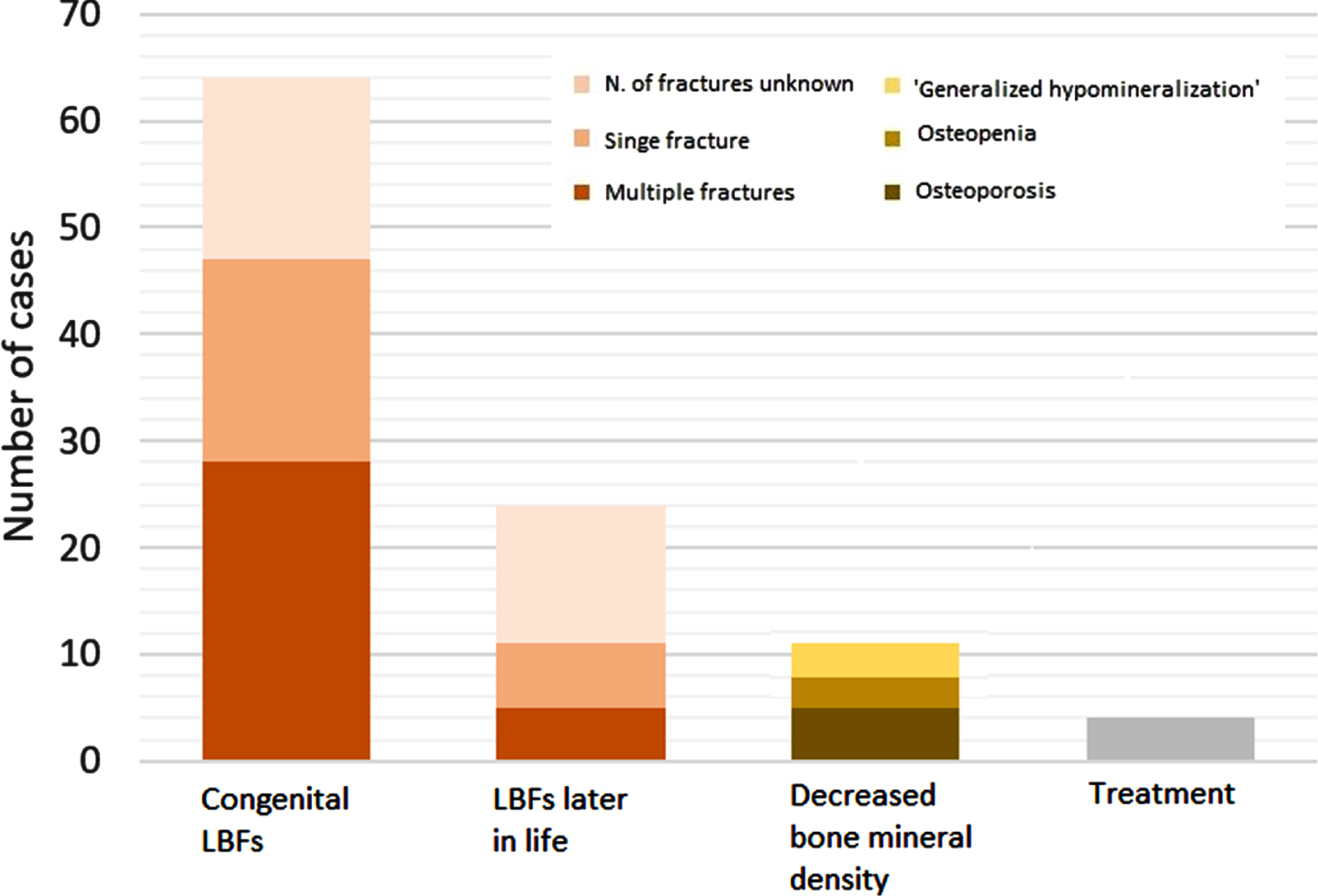
Low bone mineral density was found in 11 patients (4.5%) (mean age: 10.9±9.7 years; median: 11 years). In more detail, four patients had osteopenia (NM (LMOD3), n = 1, 8 years; NM (TNNC2), n = 1, 27 years; NM (undefined), n = 1, 26 years; CCD (RYR1), n = 1, 4 years) [25, 38, 45, 49], four patients had osteoporosis (X-linked myopathy (VMA21 mutation), n = 2, 14 years; MTM, n = 1, 11 years; CNM, n = 1, 16 years) [31, 33, 44], and three patients were reported to have ‘generalized hypomineralization’ without any further specification (undefined NM, n = 3, neonatal) [41].
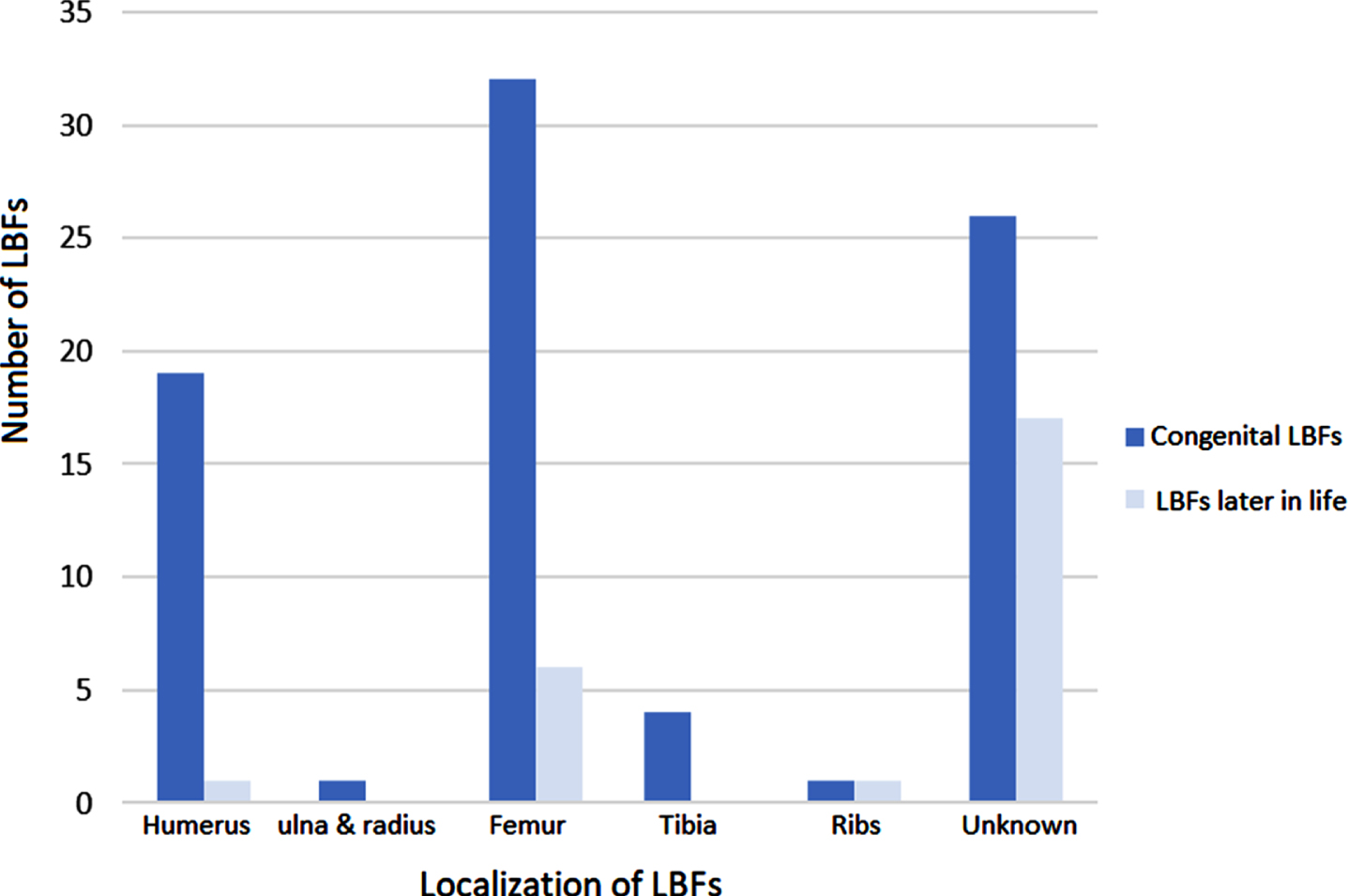
In 64 patients, one or more congenital fractures were reported (NM (LMOD3), n = 3, NM (ACTA1), n = 7; NM (KLHL40), n = 11; NM (NEB), n = 1; RYR1-RM, n = 16, ASCC1-CM, n = 5; undefined NM, n = 11; XL-MTM, n = 2; congenital titinopathy, n = 5; undefined myopathy, n = 3). Twenty-eight patients were reported to have multiple congenital fractures [21–24, 26, 28, 30, 32, 34, 35, 41, 42, 46–48, 50, 51, 54, 55], while in 19 patients only one congenital fracture was present [21, 28, 29, 43, 52, 53]. In 17 patients, the number of congenital fractures was not described [40, 41]. Congenital fractures of the humerus (n = 19 [21, 23, 24, 26, 28, 30, 32, 35, 46, 48, 51, 53, 55]) and femur (n = 32 [21–24, 26, 28, 30, 32, 34, 35, 43, 46, 47, 51, 52, 54, 55]) were most frequently reported. A congenital fracture of the tibia was reported in four patients [30, 47, 55], of the ulna and radius in one patient [34], and of the ribs also only in one patient [47]. In 26 patients, the localization of the congenital fractures was not described [24, 29, 40–42, 50] (See Fig. 3).
(Fragility) LBFs later in life (after the neonatal period) were reported in 24 patients (MTM, n = 17; RYR1-RM, n = 2; SELENON-RM, n = 1; NEB-RM, n = 1; CCD, n = 1; CNM, n = 1; undefined NM, n = 1) (See Fig. 2), with a mean age of 14.9 years (SD±11,0; range 1 to 34 years; median: 14 years). Of those 24 patients, three patients were reported to have had a fracture at ‘the pediatric age’ and 13 patients ‘later in life’, both without any further specifications on age. Of all 24 patients with a fracture later in life, five patients were reported to have had multiple fractures [27, 31, 37], while six patients were reported to have had a single fracture [25, 36, 44, 49, 56]. In 13 patients, the number of fractures was not reported [41]. A humeral fracture was described in one patient [49] and femoral fractures in six patients [25, 31, 36, 39, 44, 49]. None of the patients with a fracture later at life was reported to have had a fracture of the radius, ulna or tibia. In one patient, fractures of multiple ribs, next to fractures of both femurs, was described at age 1 year without adequate trauma mechanism [31]. In 17 patients, the localization of the fractures was not described [27, 37, 41] (See Fig. 3). We found no association between ambulation status and (fragility) LBFs later in life.
Fig. 3
Number and localization of (fragility) long bone fractures in patients with a congenital myopathy. LBF = long bone fracture.
Four patients (MTM, n = 1; NM (LMOD3), n = 1; CNM, n = 1; CCD, n = 1) were reported to receive Vitamin D and/or calcium supplementation or intravenous diphosphonate administration, which were started after low bone mineral density and/or fragility LBFs were noticed [31, 38, 44, 49]. Detailed clinical information of all affected patients can be found in Supplementary Table 3.
DISCUSSION
This scoping review shows that decreased bone quality (i.e. low bone mineral density) and (fragility) LBFs are frequently described in patients with a congenital myopathy, not only in the neonatal period, but also later in life. It underscores the need for optimal bone quality management to prevent fragility LBFs in order to improve functional prognosis, quality of life and survival in these patients. The latter is in line with the Consensus statement on standard of care for congenital myopathies and other clinical care guidelines which are based on expert opinion, that were established for specific subgroups of congenital myopathies [1, 57].
The most outstanding finding is the occurrence of low bone mineral density and (fragility) LBFs in patients with a congenital myopathy after the neonatal period. Low bone quality can be explained through multiple pathways. Firstly, impaired physical activity in patients with neuromuscular diseases is thought to be an important risk factor [10]. Physical activity has a role in stimulating osteoblast bone formation and reducing osteoclast-mediated bone resorption through biological pathways modulated by mechanical loading [58]. For example, bone protective molecules of which the release is promoted by muscle contraction, stimulate osteoblasts and inhibits osteoclast activities through the NFkB signaling pathway, increases cortical bone thickness and reduces the risk of fragility LBFs in osteoporotic woman [59]. Secondly, neuromuscular diseases generally lead to muscle atrophy. Myokines that are highly expressed in atrophic muscle, lead to osteoclast differentiation and bone loss induction, resulting in decreased bone mineral density and increased risk for fragility LBFs [60]. Thirdly, patients with a neuromuscular disorder frequently face nutritional issues, including malnutrition due to feeding disorders and swallowing problems, and decreased outdoor time and sun exposure, which could lead to a deficiency of calcium and vitamin D [61–63]. A lack of calcium and vitamin D induces bone loss, leading to low bone mineral density [64, 65]. The musculoskeletal system is significantly more complex than portrayed by traditional approaches that mainly focus on bone and skeletal muscle. The musculoskeletal system also includes tendons, ligaments, cartilage, joints, vascular and nervous tissues, and molecular and biochemical interactions within this system [66]. Patients with neuromuscular disorders have thus multiple risk factors that can contribute to an increased risk for fragility LBFs and low bone mineral density. Yet, the precise underlying pathophysiology of decreased bone quality in congenital myopathies remains unknown. Several disciplines, including internal medicine, pediatrics and neurology, should work together to reveal this pathophysiology. This scoping review is the first step to create awareness for the lacune in knowledge on bone quality in congenital myopathies.
Another important finding is the occurrence of LBFs in the neonatal period. Fetuses with reduced mobility in utero are at increased risk of decreased bone mineral density and pathological fractures, as a secondary effect of severe fetal muscular inactivity. Immobilization and decreased mechanical forces reduce cortical thickness and prevents preservation of bone structure, with the subsequent development of reduced bone mineral density, fragile bones and LBFs [67]. Further, swallowing of amniotic fluid contributes to fetal somatic growth and has been estimated to contribute approximately 10% of fetal protein intake [68]. Reduced swallowing, which is frequently observed in congenital myopathies, might thus lead to malnutrition and consequently to decreased bone quality. Moreover, we might hypothesize that mutations in congenital myopathies not only indirectly (through reduced mobility in utero or malnutrition) contribute to congenital LBFs, but also directly through decreased or altered pressure of the weakened musculoskeletal system on the bones. In short, the exact pathophysiology is unknown, and needs further investigation. Like in other neuromuscular diseases including DMD, the survival rate of children with severe congenital myopathies (for example XL-MTM, severe congenital NM) into childhood, or even adulthood, has markedly increased due to i.e. optimized respiratory and cardiac management, and rehabilitation techniques [69, 70]. Consequently, new symptoms are likely to arise and the long-term care in this patient group becomes essential, including bone quality. Caution must be paid to general underreporting of this patient group that survive to an older age, and thus to the possible underreporting on decreased bone quality.
The next striking finding was the young age at which a (fragility) LBFs took place. Since prospective, natural history data are lacking, it is unknown whether the incidence of fragility LBFs is indeed higher in the pediatric population or whether low bone quality in the adult population is excessively underreported. What we do know is that bone health and osteoporosis management are elaborately addressed in the pediatric care for boys and men with DMD [71, 72]. The focus of health care providers in the pediatric population on bone quality might thus be higher, which could explain at least partially the discrepancy in the documented incidence of low bone quality between the pediatric and adult population. Currently, a possible correlation between low bone mineral density, fragility LBFs and the age of patients with a congenital myopathy remains unknown. Yet, the frequent complications at the pediatric age, and to some lesser extent in the adult population, stresses the importance of bone health management from young age to adulthood.
It is of particular interest that both congenital LBFs and LBFs later at life were predominantly reported in the femur and humerus. Also in other neuromuscular diseases, congenital LBFs of the humerus and femur were mainly seen [73, 74]. Currently, the process of bone remodeling and the factors that can influence it, and consequently the underlying pathophysiology for the predominance for congenital LBFs in the femur and humerus, are unknown [75]. Congenital birth fractures due to traumatic birth are mostly located in the clavicula, which was not reported in congenital myopathies [76]. In severe forms of other neuromuscular diseases prenatal fractures are described [77]. We thus speculate that congenital LBFs in congenital myopathies are mostly already present prenatally, and are not caused by traumatic birth, and are therefore not seen in the clavicula. Further, due to the altered interaction between muscles and bone in congenital myopathies, we might speculate that fragility fractures are mostly expected in bones on which most force is applied, i.e. the long bones. Both in utero, due to stiffness and contractures of the uterus, and in adults, most of the force is applied on the femur and humerus, causing LBFs to mainly appear in these long bones. Fragility fractures of the vertebrae and distal radius were (almost) not reported. Vertebral fragility fractures were nevertheless frequently reported later at life in other neuromuscular diseases [78, 79]. The underreport of vertebral fragility fractures in congenital myopathies can be explained by the fact that they are often asymptomatic and routine vertebral assessment in congenital myopathies is missing [79, 80]. The low prevalence of (fragility) LBFs of the (distal) radius later at life in patients with congenital myopathies can be explained by their fall mechanism: patients have difficulty to reach out their hands in order to break the fall, and consequently fall on their hips or shoulder region [81].
Remarkably, only 11 patients were reported to have low bone mineral density (osteopenia or osteoporosis), while (fragility) LBFs were reported more frequently. Given the high number of reported (fragility) LBFs, a higher number of patients with low bone mineral density can be expected. This discrepancy can be explained by relative underreporting of low bone mineral density in the literature or the absence of routinely performed DEXA-scans in congenital myopathies.
In this scoping review, we included all articles on bone quality in a wide range of congenital myopathies. The review was preceded by the discussion on which neuromuscular diseases should be included or excluded. The subdivision of neuromuscular diseases into congenital myopathies, congenital muscular dystrophies and other neuromuscular diseases has historically been widely discussed. For example, the same genetic mutations can lead to a congenital myopathy, to a congenital muscular dystrophy or even to another neuromuscular disorder, which might cause confusion or discrepancy between different articles [24, 77, 82].
In conclusion, bone complications are seen in a broad range of congenital myopathies, both in the pediatric and adult population. Future, unselected cross-sectional studies are needed to do prevalence estimations on decreased bone quality in patients with congenital myopathies. Further, long-term follow-up studies within these patients will enable to reveal the evolution of bone quality over time. Finally, it is of major importance to determine whether the current general advices for optimizing bone mineral density are sufficient for patients with congenital myopathies, or whether specific additional recommendations are needed.
Limitations
Our scoping review includes several limitations. Firstly, retrospective studies without standardized bone quality examinations (i.e. fracture history, DEXA-scan) could cause low bone mineral density to remain unnoticed. Further, case reports could lead to publication and selection bias towards patients with abnormal bone quality. We did not find any prospective studies on bone quality (i.e. low bone mineral density or fragility LBFs) in patients with congenital myopathies. Moreover, only limited information was provided on bone quality in the majority of articles. For example, Cahill et al. 2007 described the presence of multiple (fragility) LBFs at pediatric age in three patients [27]. Unfortunately, no information on the specific trauma mechanism, bone mineral density, serum calcium or vitamin D levels, treatments or other possible causes of decreased bone quality was provided. This information is relevant to classify LBFs, to assess possible underlying pathophysiological mechanisms and to quantify bone quality. In addition, the used diagnostic criteria for osteoporosis or osteopenia are underexposed and used interchangeably in the included articles. For example, Gangfuss et al. 2021 describe the presence of a 11-year-old boy with osteoporosis. Nevertheless, the ancillary investigations used to measure bone mineral density and the degree of bone mineral density are not mentioned. Further, some articles do not follow the internationally accepted criteria of the International Society of Clinical Densitometry [83]. For example, Michael et al. 2019 describe an 8-year-old girl with osteopenia in the absence of skeletal fractures [38]. In the International Society of Clinical Densitometry, no definition for osteopenia in the pediatric population is provided. Further, for the diagnosis of osteoporosis in the pediatric population, fragility LBFs must be reported. Michael et al. 2019 thus refer to a non-existing disease entity, without further information on which criteria they used for the classification as ‘osteopenia’.
In this scoping review, we only included cases in whom information is provided on the bone quality (i.e. (fragility) LBF, low bone mineral density). All other cases are excluded from this review. This could cause an overrepresentation of bone complications in patients with congenital myopathies. Consequently, we are unable to do prevalence estimations on complications regarding bone quality. We therefore advise natural history studies on bone quality in this group of patients.
Bone Quality Assessment in congenital myopathies
Based on the findings of this scoping review combined with our clinical expertise, we advise to routinely assess bone quality in patients with congenital myopathies. Firstly, a fracture history should be included in routine visits, and pubertal assessment (Tanner staging) should be undertaken to monitor pubertal progression [1]. A dietician should be consulted to optimize dietary calcium intake [84]. Further, we advise to perform a DEXA-scan in all patients with a congenital myopathy at diagnosis and subsequently at every one to three years, depending on the severity of the osteopenia/osteoporosis. A digital X-ray radiogrammetry of the hand to assess the Bone Health Index (BHI) is a feasible alternative for DEXA-scan in case of disrupting factors (movement during measurement, metallic implants, contractures, scoliosis) or age <3 years (no normative values available) [85]. In children with a fragility LBFs or two or more traumatic fractures, bone pain, severe osteoporosis on plain radiographs or orthopedic instrumentation failure due to reduced bone strength, a DEXA-scan is indicated. A lateral vertebral assessment should be performed to look for vertebral compression fractures. Yearly blood test on calcium, phosphate, alkaline phosphatase and vitamin D, and urine tests on calcium and creatinine are advised in patients that are at risk of (fragility) LBFs [86].
Treatment options
Interventions to maintain bone quality are essential in patients with congenital myopathies. In the Consensus statement on standard of care for congenital myopathies (consortium agreement), patients are recommended to optimize physical activity, preferably through weight-bearing activities, but also sports by patients that are wheelchair bound, are beneficial. Further, vitamin D should be daily supplemented in all patients throughout the year (age <12 years: 400 IU, age ≥12 years: 800 IU). Sufficient dietary calcium intake (two to three portions of dairy products (i.e. cheese, milk, yoghurt) or other calcium containing products (calcium-set tofu, broccoli etcetera) per day) is needed. In case of insufficient calcium intake, it is advised to supplement calcium (500 mg per day). We further advise referral to internal medicine or pediatrics, so that additional therapy, for example intravenous diphosphonates, can be considered [84].
CONCLUSION
(Fragility) LBFs are frequently described in patients with a congenital myopathy, both in the pediatric and adult population. Osteopenia or osteoporosis (low bone mineral density) are reported less frequently and is possibly underreported in the literature. Decreased bone quality is predominantly described in the pediatric population and to a lesser extent in the adult population. Prospective natural history data on the prevalence of low bone mineral density and (fragility) LBFs are currently lacking and are highly needed in order to do prevalence estimations, stratified by age group. Further, the underlying pathophysiology for decreased bone mineral density in congenital myopathies should be explored. Based on generic bone quality management guidelines and this scoping review, we think that proper management on bone health is deemed essential. Hereby we aim to improve functional prognosis, quality of life and survival of patients with congenital myopathies. Further research is needed to elucidate the prevalence, the development and the pathophysiology of low bone mineral density in congenital myopathies.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable
CONSENT FOR PUBLICATION
Not applicable
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
FUNDING
The study is financially supported by a competitively awarded, peer-reviewed grant from Stichting Spieren voor Spieren, Stichting Stofwisselkracht and Stichting Voor Sara, The Netherlands. Several authors of this publication are members of the Radboudumc Center of Expertise for neuromuscular disorders (Radboud-NMD), Netherlands Neuromuscular Center (NL-NMD) and the European Reference Network for rare neuromuscular diseases (EURO-NMD).
AUTHORS’ CONTRIBUTION
KB, AD and JD contributed to the data search and data extraction. KB, AD, MJ, NV, JD, CE contributed to the study conception and design and drafted the manuscript. KB, AD, JG, BE, MJ, NV, JD and CE critically revised the manuscript. All authors have read and approved the final manuscript and agreed to be accountable for their contributions.
ACKNOWLEDGMENTS
We thank On Ying Chan (information specialist at the Medical Library in Nijmegen) for her help in optimizing the search strategy.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-221543.
REFERENCES
[1] | Wang CH , Dowling JJ , North K , Schroth MK , Sejersen T , Shapiro F , et al. Consensus statement on standard of care for congenital myopathies, J Child Neurol (2012) ;27: (3):363–82. DOI: 10.1177/0883073812436605. |
[2] | (US) OotSG. Bone Health and Osteoporosis: A Report of the Surgeon General. 2004. DOI:NBK45513. |
[3] | Siris ES , Adler R , Bilezikian J , Bolognese M , Dawson-Hughes B , Favus MJ , et al. The clinical diagnosis of osteoporosis: A positionstatement from the National Bone Health Alliance Working Group, Osteoporos Int (2014) ;25: (5):1439–43. DOI: 10.1007/s00198-014-2655-z. |
[4] | Bishop N , Arundel P , Clark E , Dimitri P , Farr J , Jones G , et al. Fracture prediction and the definition of osteoporosis in children and adolescents: The ISCD pediatric official positions, Journal of Clinical Densitometry (2014) ;17: (2):275–80. DOI: 10.1016/j.jocd.2014.01.004s. |
[5] | Bishop N . Primary osteoporosis, Endocr Dev (2009) ;16: :157–69. DOI: 10.1159/000223694. |
[6] | Mirza F , Canalis E Management of endocrine disease: Secondary osteoporosis: Pathophysiology and management, Eur J Endocrinol (2015) ;173: (3):R131–R51. DOI: 10.1530/EJE-15-0118. |
[7] | Adaikina A , Hofman PL , O’Grady GL , Gusso S . Exercise training as part of musculoskeletal management for congenital myopathy: Where are we now? Pediatr Neurol (2020) ;104: :13–8. DOI: 10.1016/j.pediatrneurol.2019.10.008. |
[8] | Ward LM , Hadjiyannakis S , McMillan HJ , Noritz G , Weber DR . Bone health and osteoporosis management of the patient with duchenne muscular dystrophy, Pediatrics (2018) ;142: (Suppl 2):S34–42. DOI: 10.1542/peds.2018-0333E. |
[9] | Bouman K , Groothuis JT , Doorduin J , van Alfen N , Udink Ten Cate FEA , van den Heuvel FMA , et al. Natural history, outcome measures and trial readiness in LAMA2-related muscular dystrophy and SELENON-related myopathy in children and adults: Protocol of the LAST STRONG study, BMC Neurol (2021) ;21: (1):313. DOI: 10.1186/s12883-021-02336-z. |
[10] | Iolascon G , Paoletta M , Liguori S , Curci C , Moretti A . Neuromuscular diseases and bone, Front Endocrinol (Lausanne) (2019) ;10: :794. DOI: 10.3389/fendo.2019.00794. |
[11] | Vestergaard P , Glerup H , Steffensen BF , Rejnmark L , Rahbek J , Moseklide L . Fracture risk in patients with muscular dystrophy and spinal muscular atrophy, J Rehabil Med (2001) ;33: (4):150–5. |
[12] | Joya JE , Kee AJ , Nair-Shalliker V , Ghoddusi M , Nguyen M-AT , Luther P , et al. Muscle weakness in a mouse model of nemaline myopathy can be reversed with exercise and reveals a novel myofiber repair mechanism, Human Molecular Genetics (2004) ;13: (21):2633–45. DOI: 10.1093/hmg/ddh285. |
[13] | HsuJD . Extremity fractures in children with neuromuscular disease, Johns Hopkins Med J (1979) ;145: (3):89–93. |
[14] | Vialle R , Thévenin-Lemoine C , Mary P . Neuromuscular scoliosis, Orthopaedics & Traumatology: Surgery & Research (2013) ;99: (1,Supplement):S124–39. DOI: 10.1016/j.otsr.2012.11.002. |
[15] | Strømsøe K . Fracture fixation problems in osteoporosis, Injury (2004) ;35: (2):107–13. DOI: 10.1016/j.injury.2003.08.019. |
[16] | Pianucci L , Sonagra M , Greenberg BA , Priestley DR , Gmuca S . Disordered eating among adolescents with chronic pain The experience of a pediatric rheumatology subspecialty pain clinic, Pediatr Rheumatol Online J (2021) ;19: (1):16. DOI: 10.1186/s12969-021-00506-4. |
[17] | Buckner JL , Bowden SA , Mahan JD . Optimizing bone health in duchenne muscular dystrophy, Int J Endocrinol (2015) ;2015: :9283. DOI: 10.1155/2015/928385. |
[18] | Tricco AC , Lillie E , Zarin W , O’Brien KK , Colquhoun H , Levac D , et al. PRISMA extension for scoping reviews (PRISMA-ScR): Checklist and explanation, Ann Intern Med (2018) ;169: (7):467–73. DOI: 10.7326/M18-0850. |
[19] | North KN , Wang CH , Clarke N , Jungbluth H , Vainzof M , Dowling JJ , et al. Approach to the diagnosis of congenital myopathies, Neuromuscular Disorders: NMD (2014) ;24: (2):97–116. DOI: 10.1016/j.nmd.2013.11.003. |
[20] | Rebolledo BJ , Unnanuntana A , Lane JM . A comprehensive approach to fragility fractures, J Orthop Trauma (2011) ;25: (9):566–73. DOI: 10.1097/BOT.0b013e3181f9b389. |
[21] | Abbott M , Jain M , Pferdehirt R , Chen Y , Tran A , Duz MB , et al. Neonatal fractures as a presenting feature of LMOD3-associated congenital myopathy, Am J Med Genet A (2017) ;173: (10):2789–94. DOI: 10.1002/ajmg.a.38383. |
[22] | Baydan F , Ozyilmaz B , Sertpoyraz F , Tuncag B , Tiftikcioglu I . Neuropathy? Myopathy? Nemaline myopathy type 2 case, Acta Myologica (2018) ;37: (2):176. |
[23] | Bharucha-Goebel DX , Santi M , Medne L , Zukosky K , Dastgir J , Shieh PB , et al. Severe congenital RYR1-associated myopathy: The expanding clinicopathologic and genetic spectrum, Neurology (2013) ;80: (17):1584–9. DOI: 10.1212/WNL.0b013e3182900380. |
[24] | Böhm J , Malfatti E , Oates E , Jones K , Brochier G , Boland A , et al. Novel ASCC1 mutations causing prenatal-onset muscle weakness with arthrogryposis and congenital bone fractures, J Med Genet (2019) ;56: (9):617–21. DOI: 10.1136/jmedgenet-2018-105390. |
[25] | Brown MJ , Song K , Vollmar M , Shelby M , Leshikar H . Flexible femoral nailing of an awake adult muscular dystrophy patient, J Am Acad Orthop Surg Glob Res Rev (2019) ;3: (12). DOI: 10.5435/JAAOSGlobal-D-19-00013. |
[26] | Buonocore G , Balestri P , Toti P , Bagnoli F . A new case of severe congenital nemaline myopathy, Acta Paediatr (1993) ;82: (12):1082–4. DOI: 10.1111/j.1651-2227.1993.tb12819.x. |
[27] | Cahill PJ , Rinella AS , Bielski RJ . Orthopaedic complications of myotubular myopathy, J Pediatr Orthop (2007) ;27: (1):98–103. DOI: 10.1097/BPO.0b013e31802b6c73. |
[28] | Castiglioni C , Diaz A , Ferrada V , Velasquez A , Erazo R , Bevilacqua J , et al. Congenital myopathies and neonatal bone fractures, Journal of Neuromuscular Diseases. (2014) ;1: (Supplement 1):S184. DOI: 10.3233/JND-149002. |
[29] | Coppens S , Deconinck N , Phadke R , Sewry C , Kadhim H , Tay C , et al. P, 241Congenital titinopathy as a cause of severe to profound congenital weakness and early death. Neuromuscular Disorders. (2019) ;29: (Supplement 1):S137. DOI: 10.1016/j.nmd.2019.06.355. |
[30] | Donaldson JS , Gilsanz V , Gonzalez G , Wittel RA , Gilles F . Tall vertebrae at birth: A radiographic finding in flaccid infants, AJR Am J Roentgenol (1985) ;145: (6):1293–5. DOI: 10.2214/ajr.145.6.1293. |
[31] | Gangfuss A , Schmitt D , Roos A , Braun F , Annoussamy M , Servais L , et al. Diagnosing X-linked myotubular myopathy - a german 20-year follow up experience, Journal of Neuromuscular Diseases (2021) ;8: (1):79–90. DOI: 10.3233/JND-200539. |
[32] | Garcia-Angarita N , Kirschner J , Heiliger M , Thirion C , Walter MC , Schnittfeld-Acarlioglu S , et al. Severe nemaline myopathy associated with consecutive mutations E74D and H75Y on a single ACTA1 allele, Neuromuscul Disord (2009) ;19: (7):481–4. DOI: 10.1016/j.nmd.2009.05.001. |
[33] | Ikeda C , Honda R , Komaki H , Sasaki M , Munteanu I , Ramachandran N , et al. Congenital form of X-linked myopathy with excessive autophagy associated with VMA21 mutation, Neuromuscular Disorders (2010) ;20: (9-10):625>. DOI: 10.1016/j.nmd.2010.07.095. |
[34] | Lacson AG , Donaldson G , Barness EG , Ranells JD , Pomerance HH . Infant with high arched palate, bell-shaped chest, joint contractures, and intrauterine fractures, Pediatr Pathol Mol Med (2002) ;21: (6):569–84. DOI: 10.1080/pd21.6.569.584. |
[35] | Laforgia N , Capozza M , De Cosmo L , Di Mauro A , Baldassarre ME , Mercadante F , et al. A rare case of severe congenital RYR1-associated myopathy, Case Rep Genet (2018) ;2018: :6184185. DOI:10.1155/2018/6184185. |
[36] | Lal D , Neubauer BA , Toliat MR , Altmüller J , Thiele H , Nürnberg P , et al. Increased probability of co-occurrence of two rare diseases in consanguineous families and resolution of a complex phenotype by next generation sequencing, PLoS One (2016) ;11: (1):e0146040. DOI: 10.1371/journal.pone.0146040. |
[37] | Lee M , Khanlou N , Shieh P . A confusing muscle biopsy, Journal of Clinical Neuromuscular Disease. (2017) ;18: (Supplement 1):S8. DOI: 10.1097/CND.0000000000000166. |
[38] | Michael E , Hedberg-Oldfors C , Wilmar P , Visuttijai K , Oldfors A , Darin N . Long-term follow-up and characteristic pathological findings in severe nemaline myopathy due to LMOD3 mutations, Neuromuscular Disorders (2019) ;29: (2):108–13. DOI: 10.1016/j.nmd.2018.12.009. |
[39] | Rao S , Wragg C , Lowe S , Majumdar A . Congenital myopathy, distinctive features and severe lymphoedema in a family-a new association? Developmental Medicine and Child Neurology, (2014) ;56: :38. DOI: 10.1111/dmcn.12350-9. |
[40] | Ravenscroft G , Miyatake S , Lehtokari VL , Todd EJ , Vornanen P , Yau KS , et al. Mutations in KLHL40 are a frequent cause of severe autosomal-recessive nemaline myopathy, Am J Hum Genet (2013) ;93: (1):6–18. DOI: 10.1016/j.ajhg.2013.05.004. |
[41] | Ryan MM , Schnell C , Strickland CD , Shield LK , Morgan G , Iannaccone ST , et al. Nemaline myopathy: A clinical study of 143 cases, Ann Neurol (2001) ;50: (3):312–20. DOI: 10.1002/ana.1080. |
[42] | Sanchez-Horvath M , Marcelino-Salas K , Llano-Rivas I , Martinez Gonzalez MJ , Garcia-Ribes A , Fernandez-Bedoya AI , et al. P87-F Severe congenital myopathy related to a novel mutation in the titin gene, A case report. Clinical Neurophysiology (2019) ;130: (7):e92–e3. DOI: 10.1016/j.clinph.2019.04.536. |
[43] | Santi M , Medne L , Bharucha-Goebel D , Bonnemann C , Dastgir J , Zukosky K , et al. Variable clinical and histological features in severe congenital RYR1 associated myopathy, Neuromuscular Disorders. (2013) ;23: (9-10):762. DOI: 10.1016/j.nmd.2013.06.447. |
[44] | Summers P , Quidgley-Nevares A , Shelton JE . Chronic pain patient with myopathy unmasked by treatment of an acute fracture: A case report, PM and R (2013) ;5: (9):S310. DOI: 10.1016/j.pmrj.2013.08.570. |
[45] | van de Locht M , de Winter J , Conijn S , Ma W , Helmes M , Irving T , et al. P, 163 Mutations in fast skeletal troponin C (TNNC2) cause contractile dysfunction. Neuromuscular Disorders. (2019) ;29: (Supplement 1):S96. DOI: 10.1016/j.nmd.2019.06.218. |
[46] | Waisayarat J , Suriyonplengsaeng C , Khongkhatithum C , Rochanawutanon M . Severe congenital nemaline myopathy with primary pulmonary lymphangiectasia: Unusual clinical presentation and review of the literature, Diagn Pathol (2015) ;10: :27. DOI:10.1186/s13000-015-0270-8. |
[47] | Rodríguez JI , Garcia-Alix A , Palacios J , Paniagua R . Changes in the long bones due to fetal immobility caused by neuromuscular disease, A radiographic and histological study. J Bone Joint Surg Am (1988) ;70: (7):1052–60. |
[48] | Han Y-M , Kwon K-A , Lee Y-J , Nam S-O , Park K-H , Byun S-Y , et al. X-linked recessive myotubular myopathy with MTM1 mutations, Korean J Pediatr (2013) ;56: (3):139–42. DOI: 10.3345/kj2013.56.3.139. |
[49] | Jean K , Mah JTJ . An overview of congenital myopathies, Continuum (2016) ;22: (6):1932–53. |
[50] | Ilkovski B , Nowak KJ , Domazetovska A , Maxwell AL , Clement S , Davies KE , et al. Evidence for a dominant-negative effect in ACTA1 nemaline myopathy caused by abnormal folding, aggregation and altered polymerization of mutant actin isoforms, Hum Mol Genet (2004) ;13: (16):1727–43. DOI: 10.1093/hmg/ddh185. |
[51] | Vendittelli F , Manciet-Labarchède C , Gilbert-Dussardier B . Nemaline myopathy in the neonate: Two case reports, European Journal of Pediatrics (1996) ;155: (6):502–5. DOI: 10.1007/BF01955190. |
[52] | Agrawal PB , Strickland CD , Midgett C , Morales A , Newburger DE , Poulos MA , et al. Heterogeneity of nemaline myopathy cases with skeletal muscle alpha-actin gene mutations, Ann Neurol (2004) ;56: (1):86–96. DOI: 10.1002/ana.20157. |
[53] | Wallefeld W , Krause S , Nowak KJ , Dye D , Horváth R , Molnár Z , et al. Severe nemaline myopathy caused by mutations of the stopcodon of the skeletal muscle alpha actin gene (ACTA1), NeuromusculDisord (2006) ;16: (9-10):541–7. DOI: 10.1016/j.nmd.2006.07.018. |
[54] | Dofash FFL , Servián-Morilla E , Rivas E , Sullivan P , Oates E , Clayton J , Taylor R , Davis M , Laing N , Cabrera-Serrano M , Ravenscroft G . Congenital myopathies –nemaline myopathies:EP34 3′UTR variant in KLHL40 causes nemaline myopathy, Journal of Neuromuscular Disorders (2021) ;31: :S60. DOI: 10.1016/j.nmd.2021.07.059. |
[55] | Catteruccia M , Fattori F , Bonetti AM , Bertini E , D’Amico A . Severe congenital RYR1-associated myopathy with multiple fractures, Acta Myologica (2019) 38: (2):124–5. |
[56] | Rao S , Majumdar A . Camurati-Englemann disease masquerading as a congenital myopathy, Developmental Medicine and Child Neurology (2014) ;56: :40 DOI: 10.1111/dmcn.12350-9. |
[57] | Rachel Bronstein M , Michael Brucker P , Brittany Coleman M , Robert Dirksen P , James Dowling M , Sarah Foye O , et al. Clinical care guidelines: What patients & families need to knowabout RYR-1. Handbook by the RYR1-Foundation. 2020. |
[58] | Takata S , Yasui N . Disuse osteoporosis, J Med Invest (2001) ;48: (3-4):147–56. |
[59] | Colaianni G , Cuscito C , Mongelli T , Pignataro P , Buccoliero C , Liu P , et al. The myokine irisin increases cortical bone mass, Proc Natl Acad Sci U S A (2015) ;112: (39):12157–62. DOI: 10.1073/pnas.1516622112. |
[60] | Qin Y , Peng Y , Zhao W , Pan J , Ksiezak-Reding H , Cardozo C , et al. Myostatin inhibits osteoblastic differentiation by suppressing osteocyte-derived exosomal microRNA- A novel mechanism in muscle-bone communication, J Biol Chem (2017) ;292: (26):11021–33. DOI: 10.1074/jbc.M116.770941. |
[61] | Chou E , Lindeback R , D’Silva AM , Sampaio H , Neville K , Farrar MA . Growth and nutrition in pediatric neuromuscular disorders, Clin Nutr (2021) ;40: (6):4341–8. DOI: 10.1016/j.clnu.2021.01.013. |
[62] | Kooi-van Es M , Erasmus CE , de Swart BJM , Voet NBM , van der Wees PJ , de Groot IJM , et al. Dysphagia and dysarthria in children with neuromuscular diseases, a prevalence study, J Neuromuscul Dis (2020) ;7: (3):287–95. DOI: 10.3233/jnd-190436. |
[63] | Bian Q , McAdam L , Grynpas M , Mitchell J , Harrington J . Increased rates of vitamin D insufficiency in boys with duchenne muscular dystrophy despite higher vitamin D(3) supplementation, Glob Pediatr Health (2019) ;6: :2333794X19835661–2333794X. DOI: 10.1177/2333794X19835661. |
[64] | Lips P , van Schoor NM . The effect of vitamin D on bone and osteoporosis, Best Pract Res Clin Endocrinol Metab (2011) ;25: (4):585–91. DOI: 10.1016/j.beem.2011.05.002. |
[65] | Zhu K , Prince RL . Calcium and bone, Clin Biochem (2012) ;45: (12):936–42. DOI: 10.1016/j.clinbiochem.2012.05.006. |
[66] | Brotto M , Bonewald L . Bone and muscle: Interactions beyond mechanical, Bone (2015) ;80: :109–14. DOI: 10.1016/j.bone.2015.02.010. |
[67] | Rodríguez JI , Palacios J , García-Alix A , Pastor I , Paniagua R . Effects of immobilization on fetal bone development, A morphometric study in newborns with congenital neuromuscular diseases with intrauterine onset. Calcif Tissue Int (1988) ;43: (6):335–9. DOI: 10.1007/bf02553275. |
[68] | El-Haddad MA , Desai M , Gayle D , Ross MG . In utero development of fetal thirst and appetite: Potential for programming, J Soc Gynecol Investig (2004) ;11: (3):123–30. DOI: 10.1016/j.jsgi.2003.12.001. |
[69] | Amburgey K , Tsuchiya E , de Chastonay S , Glueck M , Alverez R , Nguyen CT , et al. A natural history study of X-linked myotubular myopathy, Neurology (2017) ;89: (13):1355–64. DOI: 10.1212/wnl.0000000000004415. |
[70] | Houwen-van Opstal SLS , Heutinck L , Jansen M , Krom YD , Cup EHC , Hendriksen JGM , et al. Occurrence of symptoms in different stages of Duchenne muscular dystrophy and their impact on social participation, Muscle Nerve (2021) ;64: (6):701–9. DOI: 10.1002/mus.27406. |
[71] | Birnkrant DJ , Bushby K , Bann CM , Alman BA , Apkon SD , Blackwell A , et al. Diagnosis and management of Duchenne muscular dystrophy, part Respiratory, cardiac, bone health, and orthopaedic management, The Lancet Neurology (2018) ;17: (4):347–61. DOI: 10.1016/S1474-4422(18)30025-5. |
[72] | Quinlivan R , Messer B , Murphy P , Astin R , Mukherjee R , Khan J , et al. Adult north star network (ANSN): Consensus guideline for the standard of care of adults with duchenne muscular dystrophy, Journal of Neuromuscular Diseases (2021) ;8: (6):899–926. DOI: 10.3233/JND-200609. |
[73] | Borochowitz Z , Glick B , Blazer S . Infantile spinal muscular atrophy (SMA) and multiple congenital bone fractures in sibs: A lethal new syndrome, J Med Genet (1991) ;28: (5):345–8. DOI: 10.1136/jmg.28.5.345. |
[74] | Felderhoff-Mueser U , Grohmann K , Harder A , Stadelmann C , Zerres K , Bührer C , et al. Severe spinal muscular atrophy variant associated with congenital bone fractures, J Child Neurol (2002) ;17: (9):718–21. DOI: 10.1177/088307380201700915. |
[75] | Florencio-Silva R , Sasso GR , Sasso-Cerri E , Simões MJ , Cerri PS . Biology of bone tissue: Structure, function, and factors that influence bone cells, Biomed Res Int (2015) ;2015: :421746.. DOI: 10.1155/2015/421746. |
[76] | Rehm A , Promod P , Ogilvy-Stuart A . Neonatal birth fractures: A retrospective tertiary maternity hospital review, J Obstet Gynaecol (2020) ;40: (4):485–90. DOI: 10.1080/01443615.2019.1631770. |
[77] | Knierim E , Hirata H , Wolf NI , Morales-Gonzalez S , Schottmann G , Tanaka Y , et al. Mutations in subunits of the activating signal cointegrator 1 complex are associated with prenatal spinal muscular atrophy and congenital bone fractures, American Journal of Human Genetics (2016) ;98: (3):473–89. DOI: 10.1016/j.ajhg.2016.01.006. |
[78] | Singh A , Schaeffer EK , Reilly CW . Vertebral fractures in duchenne muscular dystrophy patients managed with deflazacort, J Pediatr Orthop (2018) ;38: (6):320–4. DOI: 10.1097/bpo.0000000000000817. |
[79] | Vai S , Bianchi ML , Moroni I , Mastella C , Broggi F , Morandi L , et al. Bone and spinal muscular atrophy, Bone (2015) ;79: :116–20. DOI: 10.1016/j.bone.2015.05.039. |
[80] | Karen Hertz JS-T Fragility Fracture Nursing: Holistic Care and Management of the Orthogeriatric Patient. In: Hertz K, Santy-Tomlinson J, editors. Fragility Fracture Nursing: Holistic Care and Management of the Orthogeriatric Patient. Cham (CH): Springer. Copyright 2018, The Editor(s)(if applicable) and the Author(s). This book is an open access publication.; 2018. DOI:10.1007/978-3-319-76681-2. |
[81] | Horlings CGC , Küng UM , van Engelen BGM , Voermans NC , Hengstman GJD , van der Kooi AJ , et al. Balance control in patients with distal versus proximal muscle weakness, Neuroscience (2009) ;164: (4):1876–86. DOI: https://doi.org/10.1016/j.neuroscience.2009.09.063. |
[82] | Oliveira J , Martins M , Pinto Leite R , Sousa M , Santos R . The new neuromuscular disease related with defects in the ASC-1 complex: Report of a second case confirms ASCC1 involvement, Clin Genet (2017) ;92: (4):434–9. DOI: 10.1111/cge.12997. |
[83] | Watts NB , Leslie WD , Foldes AJ , Miller PD . international society for clinical densitometry position development conference: Task force on normative databases, J Clin Densitom (2013) ;16: (4):472–81. DOI: 10.1016/j.jocd.2013.08.001. |
[84] | Compston J , Cooper A , Cooper C , Gittoes N , Gregson C , Harvey N , et al. UK clinical guideline for the prevention and treatment of osteoporosis, Archives of Osteoporosis (2017) ;12: (1):43. DOI: 10.1007/s11657-017-0324-5. |
[85] | Leijten AD , Hampsink B , Janssen M , Klein WM , Draaisma JMT . Can digital X-ray radiogrammetry be an alternative for dual-energy X-ray absorptiometry in the diagnosis of secondary low bone quality in children? European Journal of Pediatrics (2019) ;178: (9):1433–41. DOI: 10.1007/s00431-019-03425-5. |
[86] | Diab DL , Watts NB . Secondary osteoporosis: Differential diagnosisand workup, Clin Obstet Gynecol (2013) ;56: (4):686–93. DOI: 10.1097/GRF.0b013e3182a9b5f9. |


