
Newborn Screening for the Diagnosis and Treatment of Duchenne Muscular Dystrophy
Abstract
A pilot newborn screening (NBS) program for Duchenne muscular dystrophy (DMD) study proposes to assess the feasibility of the screening procedure, temporal course of the various steps of screening, and the public acceptability of the program. This is particularly vital to ascertain as DMD is considered a ‘non-treatable’ disease and thus does not fit the traditional criteria for newborn screening. However, modern perspectives of NBS for DMD are changing and point to possible net benefits for children and their families undertaking NBS for DMD. The aim of this workshop was to establish pathways for the successful implementation and evaluation of a pilot NBS for DMD program in Australia. Consensus was reached as to the rationale for, potential benefits, risks, barriers and facilitators of screening, alongside the establishment of screening protocols and clinical referral pathways.
ABBREVIATIONS
AAV | Adeno-associated virus |
SMA | Spinal muscular atrophy |
NPF | National Policy Framework |
GSP | Genetic screening processor |
qPCR | Quantitative polymerase chain reaction |
CNV | Copy number variant |
OHMR | Office for Health and Medical Research |
FDA | US Food and Drug Administration |
DMD | Duchenne Muscular Dystrophy |
BMD | Becker Muscular Dystrophy |
NBS | Newborn Screening |
DBS | Dried Blood Spot |
CK | Creatinine Kinase |
CK-MM | Creatine Kinase Isoenzyme (muscle specific) |
HCP | Healthcare professional |
MoH | Ministry of Health |
NATA | National Association of Testing Authority |
NMD | Neuromuscular disease |
FDA | Food and Drug Administration |
NBSTRN | Newborn Screening Translational Research Network |
NGS | Next Generation Sequencing |
NSW | New South Wales |
MPS | Massive Parallel Sequencing |
PPMD | The Parent Project Muscular Dystrophy |
PCR | Polymerase chain reaction |
SCHN | Sydney Children’s Hospital Network. |
WORKSHOP STRUCTURE
Twenty attendees including clinicians, geneticists, scientists, patient advocates, and government representatives convened on 31st May 2021 for the Newborn Screening for the Diagnosis and Treatment of Duchenne Muscular Dystrophy- Australian Health System Readiness Workshop. The stakeholder committee consisted of individuals with expertise in newborn screening, neuromuscular diseases, implementation science, practice and health policy, funding bodies and consumer facing (advocate) roles. This workshop aimed to undertake a horizon scan to evaluate the role and feasibility of a Newborn Screening (NBS) program for Duchenne muscular dystrophy (DMD) in New South Wales and the Australian Capital Territory (NSW/ACT), Australia. To aid in health system readiness for dissemination of such a program across the state, the workshop aimed to identify facilitators and address barriers of implementation.
AN OVERVIEW OF DUCHENNE MUSCULAR DYSTROPHY AND HISTORICAL PERSPECTIVE OF NEWBORN SCREENING PROGRAMS
Associate Professor Michelle Farrar, a paediatric neurologist and chair of the workshop opened the event with a rationale, the current landscape of DMD, and a historical perspective of DMD NBS programs worldwide. Duchenne muscular dystrophy (DMD) is the most common and severe type of muscular dystrophy of childhood. Predominantly affecting boys, it is characterised by progressive muscle weakness. The condition is caused by mutations in the X-linked dystrophin gene which encodes the dystrophin protein [1]. Absent or dysfunctional dystrophin leads to chronic muscle damage, inflammation, and progressive replacement with connective and adipose tissues [2, 3].
NBS is a universal public health initiative that identifies infants at high risk of having conditions that would benefit from early diagnosis and management [4]. For DMD, the benefits of a neonatal diagnosis include early access to multidisciplinary care and emerging therapies that aim to maintain muscle function and avert fibrosis and atrophy. Additionally, wider benefits of NBS for DMD include access to a diagnosis that unlocks the pathway for reproductive, economic, and social support for families. These benefits have prompted a growing number of countries since the late 1970’s to pilot the inclusion of DMD to NBS programs [5]. Centres in the USA, several European countries, the UK, Canada, and New Zealand have undertaken pilots, measuring the muscle isoform of creatine kinase (CK-MM) levels in newborn dried blood (DBS) spot samples to screen for DMD.
These historical screening approaches have involved a two step process; screening for elevated CK-MM levels in initial DBS samples soon after birth, and if elevated retesting a second DBS or venous blood at 4 to 6 weeks of age. Parents of infants with persistent CK-MM elevation are contacted, the results disclosed and discussed. Subject to parental consent, DMD gene analysis has been undertaken in blood and the diagnosis confirmed by detection of a DMD-causing mutation. The first commercial CK-MM assay kit for DMD newborn screening (PerkinElmer) was FDA-approved in 2019 [6–8]. Elevated CK levels may also be associated with birth trauma and detect several other rarer forms of muscular dystrophy, associated with false positives and necessitating extensive follow up [5].
In Australia, DMD is yet to be nominated to the National Standing Committee of Screening for inclusion into local NBS programs. A state-wide pilot screening was previously proposed in Western Australia but was not supported because of scientific limitations and ethical concerns, in particular insufficient evidence that early treatment facilitated by newborn screening resulted in long term clinical gains [5, 9, 10]. There was also controversy over the technical performance characteristics of the DMD screening tool and the level of analytical error that could be considered ethically acceptable [11]. Additionally, there was some collective concern about the psychosocial implications of establishing the diagnosis in a presymptomatic infant, particularly its potential impact on the crucial early period of parent-infant bonding [11, 12].
These remain legitimate concerns, however the ongoing advances in technologies available to NBS programs, in concert with the fast-changing current and future therapeutic landscape for DMD, make it imperative to re-evaluate the case for introducing an Australian pilot NBS program for the condition. The use of genomics as a second tier in NBS may limit false positives and be useful in establishing a pathway for DMD NBS. In the absence of NBS-facilitated early diagnosis, affected infants cannot gain early access to proven therapies or to emerging therapeutic trials [13]. While fundamental NBS principles and practices centre on the effectiveness of testing and improving health outcomes for identified newborns, the value of genomic technologies in modern screening practices may also be associated with broader ethical, social, epidemiological, and reproductive consequences, which are important to value. The workshop provided an opportunity for detailed discussion of these matters and further assessment of the need and feasibility of establishing a NSW-wide NBS pilot program for DMD.
NEWBORN SCREENING FOR DUCHENNE MUSCULAR DYSTROPHY IN THE CONTEXT OF THE AUSTRALIAN NATIONAL FRAMEWORK FOR NEWBORN SCREENING
Sarah Alland and Julia Warning from the NSW Ministry of Health (MoH) gave an overview of principles governing newborn screening pilots and programs in NSW. The Newborn Screening (NBS) National Policy Framework (NPF), outlines principles and guides policies surrounding newborn screening programs in Australia [14]. This framework reflects the 10 principles developed by Wilson and Jungner in 1968 [15], principles that are widely accepted in public health for guiding population-based screening decisions. Individual states and territories refer to this framework when considering the addition or removal of NBS programs for a particular medical condition within their healthcare jurisdictions. For a state or territory to implement NBS for a condition, the NPF requires evidence that demonstrates need, cost-benefit, and efficacy of the proposed screening tool and program. However, such evidence that the benefits of a screening program outweigh the potential harms may be unavailable without ample data that investigates the outcomes of pre-symptomatic detection and diagnosis for the test condition. As a result, pilot NBS programs aim to bridge this hurdle and support research that builds an evidence base for an implemented NBS program.
In NSW, the Office for Health and Medical Research (OHMR) has recently proposed guidelines to assist researchers design and implement a state-wide pilot NBS program. The guidelines aim to support research into the development of an evidence base, treatments, and/or ongoing screening for a condition. The proposals must also aim to meet one of the following aims outlined in the National Policy Framework:
I. Reducing morbidity and mortality by early detection and early treatment
II. Reduce the incidence of a condition by identifying and treating its precursors
III. Reduce the severity of a condition by identifying people with the condition and offering effective treatment
IV. Increase choice by identifying conditions or risk factors at an early stage in life course when more options are available
To address the wider epidemiological and reproductive benefits of screening and build readiness for individuals to access emerging therapies, the implementation NBS for DMD pilot was underpinned by these new guidelines. Pertinently, a recent epidemiological study revealed an unchanging disease incidence of DMD in NSW across the last two decades, despite dedicated and integrated genetic cascade screening and counselling services and incorporation of modern genetic diagnostic algorithms [16]. These results suggest that to effect epidemiological change, current screening models should be augmented by newborn and reproductive carrier screening. Subsequent presentations throughout the workshop discussed the applicability of implementing a NBS program for DMD based on these guidelines.
THE RATIONALE FOR NEWBORN SCREENING IN DUCHENNE MUSCULAR DYSTROPHY: EXPLORATION OF THE POTENTIAL FACILITATORS AND BARRIERS TO EFFECTIVE AND ETHICAL IMPLEMENTATION OF THE PILOT PROGRAM
Associate Professor of Genetic Medicine, and Director of the NSW NBS programme, Veronica Wiley, reported on the principles governing inclusion of a condition for newborn screening As a condition, currently with no disease modifying interventions commonly initiated in early life (neonatal or infancy), DMD does not fit the traditional screening criteria established by Wilson and Jungner. However, through the modern perspective of NBS for DMD, net health and psychosocial benefits have been ascribed to the early diagnosis of children with the condition, potentially outweighing the risks of receiving a diagnosis without an accepted therapeutic intervention. Secondary benefits which are now just as pertinent to establishing the rationale for NBS in DMD extend outside that of the affected individual and include the capacity to restore reproductive confidence to carriers of DMD, improve the psychological well-being of the family unit and reduce the health economic implication of disease. Klair Bayley described the rationale and need for the development of a NBS program, from the perspective as a patient advocate at Duchenne Australia, a consumer, and as a parent of a child with DMD. Prior studies have ascertained that from a consumer perspective, presymptomatic screening program stems from an urgency to limit the diagnostic odyssey, facilitate family planning, restore reproductive confidence, and promote reproductive options for genetic carriers, and commence clinical intervention early.
Despite advances in diagnostic technologies and an increase in DMD awareness [21], the age of diagnosis has remained stagnant in Australia. According to the McKell Report (2020), the average age of diagnosis for DMD was 4.39 years of age, often coming 1-2 years after familial concerns over symptom onset. For some individuals the diagnostic delay extended up to 10 years [21]. For 173 families surveyed in the McKell report, on average 3 health professionals were seen before reaching a diagnosis for their child. Concerningly, for nearly 30% of participants, 4 or more different clinicians were required before a diagnosis was reached [21].
Prompt detection and diagnosis of DMD first and foremost allows for the commencement of medical intervention and early surveillance for an individual. Emerging evidence supports an increase in favourable outcomes and symptom progression with an early diagnosis and subsequent intervention [17, 18]. Early diagnosis also favours access to early allied health support, including speech therapy and physiotherapy, which can considerably improve mobility, communication difficulties and improve an individual’s quality of life. As discussed in the treatment segment of the workshop, early diagnosis also broadens the timeframe available to participate in clinical trials, and access emerging therapies as they become available.
For the family unit, earlier detection of DMD can also facilitate reproductive planning [19]. As a X-linked disease, confirmed female carriers of mutations to the dystrophin gene have a 50% chance of any biological male child being affected by DMD, and a 50% chance of any biological female child being a carrier. Diagnostic delays placed families at risk of having a second (or third) affected child with DMD. Accordingly, an earlier diagnosis better ensures access to genetic counselling, and information about reproductive options and technologies that can allow families the choice to prevent multiple diagnoses among relatives. According to the McKell report, 23% of respondents had more than one child with DMD, with reports of families with up to 5 children living with the disease [20]. Previous studies have indicated that earlier detection of children with terminal genetic conditions, and thus genetic carriers may prevent future siblings being born with the same condition [21–23].
Finally, early detection may also aid in preventative health practices for DMD relatives of a diagnosed individual. Despite DMD being widely considered a disease that manifests in males, female carriers of the abnormal dystrophin gene may also develop a treatable cardiomyopathy and/or muscle weakness at some point in their lifetime [24, 25]. A recent survey demonstrated that improved identification of female carriers allowed for more effective surveillance of symptoms and informed reproductive options [26].
Taken together these support the broader impacts of DMD NBS valued by families, with restoration of reproductive confidence and the ability to access disability support services, valued by families. Despite advances in genomic technologies, the age of diagnosis for those with DMD has remained relatively constant. These diagnostic delays continue to have negative impacts on individuals and their families, only highlighting the urgency for a routine, newborn/presymptomatic screening program not only across NSW, but Australia wide.
PUBLIC ACCEPTABILITY OF NEWBORN SCREENING IN DUCHENNE MUSCULAR DYSTROPHY
From a consumer’s perspective, Klair Bailey also introduced the discussion surrounding broader community attitudes around NBS for DMD in Australia. There is theoretical support for NBS for DMD from within the DMD and NBS communities as evidenced across jurisdictions that have similar populations and healthcare systems as Australia. In prior pilot programs, addition of DMD to the NBS panel did not coincide with a drop in participation rates [27–30].
An Australian mixed methods study reported the experiences of 62 families undergoing the DMD diagnostic process, to help understand the perceived health, economic and psychosocial impact that a population screening program may have [31]. The families participating in this study described mixed experiences regarding their child’s diagnostic journey. Approximately 50% reported a level of frustration that came with believing their child could have been diagnosed earlier. In these families that yearned for an earlier diagnosis, (preferably prior to symptom onset) perceived benefits included early access to therapies, financial preparedness, and informed reproductive decisions.
Nonetheless, in conjunction with these perceived benefits, was the perceived fear of bonding issues with their child if a positive screening and subsequent diagnoses were to occur so early in life. Whilst the potential disruption of familial bonds was recognised in this study, several studies have not identified negative psychosocial impacts of NBS among families who received a diagnosis through NBS [32, 33].
Another qualitative study of 97 families demonstrated that most families of affected boys were in favour of newborn screening on the grounds of reproductive choice and time to prepare emotionally and practically. There was no evidence of long term disruption to the familial relationships [32]. In a 2014 USA based survey, 95.9% of parents with an affected child were in favour of NBS programs for neuromuscular disease, even in the absence of viable therapeutic advances [34]. Collectively these figures highlight theoretical public acceptability of NBS programs for DMD.
THE COST OF ILLNESS IN DUCHENNE MUSCULAR DYSTROPHY
Substantial evidence worldwide continues to determine that DMD is associated with a significant economic burden, not only on an individual basis but to the wider family, society, and healthcare system [12, 35]. Klair Bailey presented the costs associated with managing the care of an individual which expands throughout their lifetime, and is comprised of healthcare and social costs, and informal liabilities that run concurrently with an escalating requirement for multiple care interventions and increasing disability.
The estimated lifetime total costs for an individual with DMD who lives to their mid-thirties is approximately $2.25 million AUD [12]. In addition, it is estimated that a lifetime total of $630,000 AUD reflects the burden of informal care undertaken by immediate family members for an affected individual. Healthcare costs alone are estimated to fall between $300,000- $600,000 per individual over their lifetime. In NSW, monthly out of pocket medical costs average $430.43 per month, but nationally can be as high as $1800 per month. Furthermore, informal individual, familial and wider societal economic burdens such as lost productivity, reduced civic participation, mental health implications, and relationship burdens, are likely vastly underestimated.
In terms of health system costs, an Australian study on healthcare utilisation reported the mean annual health system cost of individuals with DMD to be substantially higher than the average annual healthcare costs across all age groups [36]. In general, costs increased with age, coinciding with the time interval in the natural history of the disease, which leads to the accrual of comorbidities requiring cardiorespiratory and orthopaedic interventions and reductions in independent ambulation. The study estimated total health system costs to exceed $26 million AUD annually.
BUILDING HEALTH SYSTEM READINESS FOR PROACTIVE SURVEILLANCE AND EXPERIMENTAL THERAPIES
Doctor Michelle Lorentzos, paediatric neurologist, described current management and the therapeutic pipeline for DMD. Novel therapies and clinical trials are embedded in, and not a replacement of, the current management approach. Early diagnosis through a NBS for DMD pilot program has the potential of facilitating management that is personalised, preventative and precise; with the correct diagnosis allowing for correct approach to treatment.
Historically DMD was typically fatal in the later teenage years. With the development of evidenced based surveillance and symptomatic interventions, as well as standardised steroid regimens, boys with DMD are now remaining ambulant for longer and the mean life expectancy has increased to 28 years [37–39]. Lifelong multidisciplinary care remains essential as novel therapies emerge to optimise outcomes. The diagnosis initiates a long-term partnership between the individual with DMD, their family and healthcare team. Aspects of care included in this partnership are education and counselling, targeted physiotherapy and occupational health interventions, targeted learning and behaviour interventions, medical surveillance, and treatment for comorbidities, evidenced based pharmacotherapy and access to peer and community support.
Multidisciplinary medical care incorporates neurological, genetic, cardiac, respiratory, endocrinological, and orthopaedic input, as well as allied health involvement from physiotherapists, occupational therapists, speech therapists, psychologists, social workers, and genetic counsellors. Glucocorticoids remain a key approved pharmacotherapy for slowing disease progression and extending life expectancy, ambulation, upper limb strength and cardiac function. Data is emerging that steroid initiation in the presymptomatic phase of disease i.e. prior to the plateau in motor skills may potentially be beneficial to maintain muscle function and strength over the longer term [5].
The therapeutic landscape of DMD has changed rapidly in the past five years with a robust clinical trial pipeline targeting multiple physiological pathways to safely reduce progression, stabilise or improve function. These include dystrophin restoration or replacement (gene therapy, small molecules targeting nonsense mutations or antisense oligonucleotides for out of frame mutations) and downstream treatments combating fibrosis, reducing inflammation, regulating calcium balance, improving muscle growth and protection, and restoring energy. Australia continues to participate in the clinical development of these, recognising the high unmet need in DMD. Early treatment has the potential to prevent muscle deterioration, fibrosis and other damage and therapeutic interventions may be optimally effective the earlier they can be offered. Early diagnosis will be important in enhancing opportunities of early intervention and access to DMD clinical trials, as inclusion criteria expands to boys with DMD at younger ages. The evaluation of several promising therapies in pivotal (late phase) trials were presented; forthcoming results may rapidly change the clinical landscape and implementation of DMD NBS programs. These include vamorolone, a novel dissociative steroid that has been shown to provide the therapeutic benefits of corticosteroids with less adverse effects in a phase 2 study [40]. Ataluren, remains a pharmacotherapy designed to address stop or non-sense deletions in boys with DMD, with the aim of converting boys with DMD to a less severe phenotype. A meta-analysis of Phase IIb and phase III data showed increased 6-minute walk test for boys in the ambulatory transition phase with a baseline between 300 and 400 metres [41]. Further analyses have suggested that ataluren may prolong ambulation for up to 5 years [42]. Results from two phase 2 trials quantifying dystrophin in muscle biopsies is awaited.
Exon skipping agents include antisense oligonucleotides that are designed to modulate splicing of pre-mRNA in a mutation specific manner and produce truncated but partially functional dystrophin transcripts. Currently exon skipping agents target mutations in exons 45 to 55, collectively accounting for approximately 30% of people with DMD. There are various phase trials occurring for these exon skipping agents and five have conditional U.S Food and Drug Administration (FDA) approval (ataluren, eteplirsen, golodirsen, viltolarsen, and casimersen) [43]. As the therapeutic pipeline progresses, inclusion criteria have been expanded to include younger boys with DMD, with some studies enrolling from age 6 months. Second generation exon skipping agents, allowing greater affinity for muscle cells, and reducing potential toxicity, are also in development.
Lastly, there are three ongoing adeno-associated virus (AAV) mediated micro and mini dystrophin gene therapy programs for DMD evaluating safety and efficacy (PF-0639926, SGT-001, SRP-9001) across a range of mutations, ages, and functional status. Among the challenges for the development of a one-time AAV mediated gene therapy, the optimal age for administration is not yet known. Spinal muscular atrophy (SMA) has clearly demonstrated how a treatment (targeting terminally differentiated motor neurons) with minimal effect in advanced disease can be transformative if administered at an earlier age. AAVs are non-integrating vectors, bringing an extra layer of complexity in DMD as muscle cell divisions with growth may “dilute” the effect and limit durability, while factors such as weight and disease severity may also impact safety and efficacy. The Pfizer program includes a study enrolling boys aged 2-4 years; an age group that is often prior to symptomatic diagnosis, with consideration to extend the lower age range to infants before 1 year of age.
THE FEASIBILITY OF NEWBORN SCREENING FOR DUCHENNE MUSCULAR DYSTROPHY: ESTABLISHING THE SCREENING PROTOCOL FOR THE PILOT PROGRAM
The utility and feasibility of using ck-mm as a first-tier screening test for duchene muscular dystrophy
Associate Professor Veronica Wiley reported the outcomes of two previous studies undertaken within the NSW/ACT NBS laboratory using CK-MM assays, acknowledging the work of the NSW/ACT NBS laboratory staff and Tiffany Wotton.
In the past, screening for DMD has been based on the measurement of total creatine kinase (CK), which exists in three isoenzyme forms (CK-MM, CK-MB and CK-BB; found in skeletal muscle, heart and brain/lungs respectively). CK is raised in concentrations in the blood after muscle damage. For those with DMD, CK is often significantly elevated, however this biomarker alone is vastly inadequate for a diagnosis as other conditions that cause muscle damage may lead to similarly raised levels. Two de-identified pilot studies have been conducted within the NSW Newborn Screening program laboratory and demonstrated the feasibility of including CK-MM as a biomarker of DMD from birth, establishing a population-based range of CK and cut-off thresholds for second tier testing. In 2013-2014, the first pilot study used a manual assay to measure total CK on de-identified DBS samples of various ages: 10,000 DBSs collected from neonates aged 48 to 72 hours, 100 DBSs collected from neonates aged 6 to 7 days, 100 DBSs collected from infants aged 6 to 12 weeks, 19 DBSs collected from boys with a confirmed genetic diagnosis of DMD and 7 DBSs from known female carriers of DMD mutations. A second pilot laboratory validation study within the NSW/ACT NBS Service demonstrated feasibility of including automated CK-MM as a biomarker for DMD from birth as part of the routine NBS program and established cut-off thresholds for second tier analysis. This pilot collaborated with Cardiff and PerkinElmer, screening for muscle specific CK-MM on 51,647 de-identified DBS samples from infants 48-72 hours old. From these, a 3 millimetres (mm) blood disc was punched in triplicate and plates were analysed on the PerkinElmer Genetic Screening Processor (GSP) instrument using the PerkinElmer kit. Any sample that identified a CK-MM higher than 1000 nanograms per millilitre (ng/mL) was retested. A further 2000 samples were collected on infants six days to 12 weeks old to better validate the results at later time points. In addition, 20 males with DMD and 20 known female carriers were included. The pilot discovered 59 males with a CK-MM > 1000 ng/mL and 29 females with a CK > 1000 ng/mL. In addition, the pilot determined that the efficacy of the test reduced with postnatal age and that readings on average were higher in males. CK-MM levels were consistently lower for infants with a birthweight of < 2 kilogram (kg). This pilot established the threshold for CK-MM assay (> 1000 ng/mL) above which second tier mutation testing would be triggered. All DMD affected specimens were detected with the CK-MM assay using the 99th percentile cut-off. Further analysis of a presumed positive screening result at this threshold to detect Becker muscular dystrophy (BMD), female carriers and/or other types of muscular dystrophy patients was not possible as the study design included de-identified samples. As the CK-MM assay is used within a screening model, the objective is to limit harms by balancing risks of false negatives against the number of false positives. The accuracy and performance of the two-tier screening test will be evaluated over the duration of the pilot NBS for DMD study.
The Parent Project Muscular Dystrophy (PPMD) in collaboration the Newborn Screening Translational Research Network (NBSTRN) and New York State have been undertaking a 2-year pilot from October 2019. As of July 2021, more than 36,000 babies born in New York State have had CK-MM analysed and 34 babies have been referred for significantly elevated levels. Four of these babies have been confirmed to have Duchenne/Becker muscular dystrophy, and one baby was identified as a carrier female.
A recent systematic review has evaluated the accuracy of the CK assay in neonatal screening for DMD and the accuracy of this methodology [44]. Across 11 studies and a sample of 1,416,123 newborns, CK showed good accuracy and performance.
THE UTILITY AND FEASIBILITY OF USING MASSIVE PARALLEL SEQUENCING AS A SECOND-TIER TEST SCREENING TEST FOR DUCHENNE MUSCULAR DYSTROPHY
Doctor Anja Ravine, Molecular Geneticist, discussed the molecular genomics of DMD newborn screening. The dystrophin gene is the largest gene in the human genome, comprising 79 exons across 2.2 million base pairs. There is a wide spectrum of pathogenic variants, encompassing 60% deletions, 5-10% duplications and approximately 35% -point mutations.
Recent advances in genomic technologies have brought massive parallel sequencing (MPS) to the point where it can be implemented into the NBS laboratory workflow. Accordingly, MPS has the potential to be incorporated into the screening algorithm for DMD on samples found to have raised CK-MM levels as a second-tier screening test and limit false positives. However, this is yet to be verified in a high-volume routine newborn screening service. Various MPS approaches in the context of current international DMD NBS pilot studies were reviewed. The potential to establish industry partnerships was identified.
A targeted screening approach has been developed using multiplex quantitative polymerase chain reaction (qPCR) to identify deletions amenable to exon skipping therapy and thereby detect male newborns who could benefit from early initiation of DMD therapies [45]. This would detect approximately 70% of cases. In this model, boys without a known dystrophin deletion and persistently elevated CK-MM on repeat DBS at 6 weeks could be referred for clinical evaluation at a neuromuscular centre. Full genomic testing could be undertaken in families providing informed consent. This would reduce the specificity for DMD, however gain an understanding of alternate conditions that could be identified.
PerkinElmer have developed a single next generation sequencing (NGS) based assay covering the entire genomic sequence of the dystrophin gene [46]. Dystrophin molecular testing was undertaken on 772 individuals, including 480 with clinical suspicion of DMD. Definitive molecular diagnosis was established in 86% (n = 413) cases, however only 5 samples were derived from DBSs. Further validation of the assay on DBSs and training of NBS scientists is needed to establish methodologies for DNA extraction, accurate sequencing and variant interpretation and classification. Copy number variant (CNV) analysis as facilitated by the PerkinElmer DMD mutation kit detects the majority of duplications and deletions, three exons or greater in size. Additional smaller (single nucleotide) CNVs can be detected but require diagnostic analysis. The limitations of the kit include inability to detect variants in deep intronic, promoter and enhancer regions and areas with tandem repeats. However, low coverage regions if any are limited to < 1% of nucleotides in the screening test. The assay also does not detect cases of mosaicism. As a screening tool, the consensus was that despite the limitations, this second tier screening tool allowed for specific targeting of the vast majority of primarily pathogenic variants of DMD.
The application of whole genome or exome sequencing in NBS is being applied in several research projects and commercial NBS genomic panels [47]. Analysis could target solely the DMD genomic sequence, while also serving as proof of principle for the potential application of NGS in both first and second tier future NBS practices for a range of conditions.
ESTABLISHING A SCREENING PROTOCOL FOR THE NBS IN DMD PILOT PROGRAM
Consensus of the workshop was that a two-tiered screen approach would be methodologically sound, reducing the incidence of false positives and false negatives. Prior population pilot programs have used two serial CK-MM measurements over an established threshold, two weeks apart to denote screen positivity, as birth trauma can lead to high CK measurements within the first days of life, leading to a high false-positive rate on single DBS CK-MM measurement [48]. Diagnostic confirmation of disease was through DBS whole exome sequencing for those with ongoing hyperCkaemia on serial DBS’ [48]. The consensus of the workshop however was to include a screening protocol with comprehensive sequencing of the dystrophin gene on first DBS, using the PerkinElmer NEXTflex DMD NGS kit, for all infants with a CK-MM cut off≥1000 ng/mL on first DBS. Screen positivity would be deemed as an infant with a CK-MM≥1000 ng/mL and an identified pathogenic variant previously reported in DMD or predicted to result in DMD on first DBS (the latter verified as disease causing by the expertise of molecular pathologist) (Fig. 1). The involvement of a molecular pathologist to aid in interpretation of possible pathogenic variants at this stage, is key to identifying and flagging screen positive children with possible DMD-causing phenotypes, whether that be through in-frame or out of frame mutations in the dystrophin gene.
Fig. 1
Pilot newborn screening pathway for Duchenne muscular dystrophy.
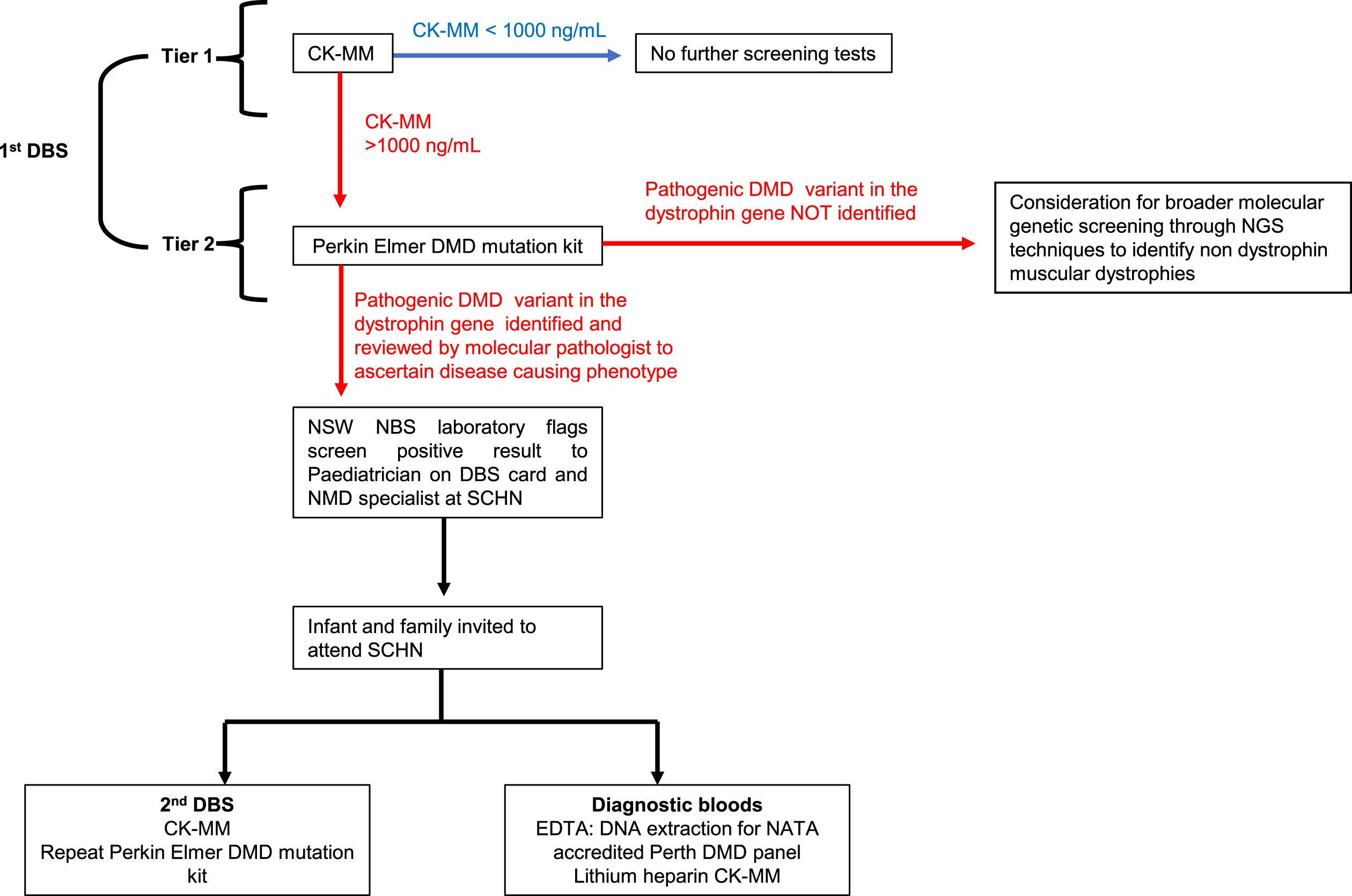
PerkinElmer has a close and long association with NBS internationally. Specific to DMD, the company developed the first commercial CK screening assay, used in the large New York pilot NBS program for DMD as a first-tier assay. This is the only regulatory approved assay for DBS. In terms of using the PerkinElmer test for second tier testing, the NSW NBS laboratory has considerable experience with utilising gene variant kits through the first Australian pilot newborn screening program for SMA which have been incorporated into existing laboratory processes and are seen as feasible and reproducible screening tools. The existence of an established (PerkinElmer) DMD specific kit was seen as advantageous by the consensus group, to optimise screening workflow. Whilst a preliminary study using of 20 known children with DMD determined that pathogenic variants were feasibly identified from DBS, this pilot study will further determine the sensitivity and specificity of incorporating the PerkinElmer DMD mutation kit as standard of care within the screening pathway.
This workflow was chosen to filter out the false positives created by screening for CK-MM early in postnatal age, using targeted DMD specific genetic methodologies. This methodology was considered the most appropriate and efficient, chosen in anticipation of the evolving NBS landscape which has already started to incorporate massive parallel sequencing technologies into its pathways [49]. In addition, second tier genomic screening in infants with CK levels above the pre-specified threshold, was considered as the optimal methodology to circumvent detection of mild muscular dystrophy phenotypes, such as BMD and to aid in variant interpretation. A screening algorithm was developed to depict the screening pathway and reviewed by all workshop participants (Fig. 1) and estimated associated costs over the two years of the proposed pilot established (Fig. 2).
Fig. 2
Estimated cost analysis for the screening and diagnostic pathway for Duchenne muscular dystrophy over the course of the two-year pilot program. Costs are based on prior annual newborn screening numbers across NSW/ACT. DMD = Duchenne muscular dystrophy, DBS = Dried blood spot.

It was considered that this screening pathway has the potential to identify rare variants that may have a heterogenous phenotype (where some individuals have BMD and others DMD), highlighting the importance of variant curation and interpretation by a molecular pathologist within the NBS service. Dependent on the results of the pilot, it was proposed that the pathway could be revised prior to future clinical translation, to include other muscular dystrophies characterised by CK-MM > 1000 ng/ml without a pathogenic DMD variant in the dystrophin gene, using broader molecular genetic analysis such as whole exome or genome sequencing on 2nd tier screening. The pilot NBS for DMD study would concomitantly be used to identify the incidence of these cases, guiding the establishment of infrastructure required for comprehensive diagnostic confirmation and clinical management of non-DMD muscular dystrophies that are associated with markedly elevated CK in the newborn period.
The NSW/ACT NBS program has established several pivotal structures in place for the addition of DMD to population screening, which were established at the time of a previous pilot program for SMA. This includes information on the NBS website for paediatric neuromuscular disorders. Additionally, handouts for families and healthcare professionals (HCPs) already describe NBS for ‘neuromuscular disorders’ enabling the addition of other conditions.
ESTABLISHING REFERRAL PATHWAYS FOR DIAGNOSTIC CONFIRMATION OF DISEASE AND CARE PLANNING
Infants and families of those deemed screen positive would be flagged by the NSW NBS Screening laboratory to the named Paediatrician on the DBS card and the tertiary neuromuscular disease (MND) specialist within Sydney Children’s Hospital Network (SCHN). All screen positive infants would be referred and reviewed within established paediatric tertiary neuromuscular clinics with access to multidisciplinary care including expertise in genetic counselling, diagnosing and treating neuromuscular disorders in line with international standards of care.
At the initial consultation at SCHN, infants would undergo clinical examination and diagnostic blood tests. Diagnostic bloods would be completed (whole blood sampling) for CK-MM and DNA extraction for dystrophin sequencing, the latter completed through established National Association of Testing Authority (NATA) accredited DMD mutation laboratories. To validate the NBS for DMD laboratory processes a second DBS would be taken at this time and analysed by NSW NBS laboratory to confirm CK-MM results and validate the genetic screening results from the first DBS DMD mutation kit (Fig. 1). Families of diagnostically confirmed children with DMD would be integrated into the neuromuscular service at SCHN and would have access to supportive international standards of care as the foundation of management, facilitated by a multidisciplinary team of clinicians and allied health professionals. Embedded in this model of care, continued access to genetic counselling and carrier screening to restore reproductive confidence in parents and provide early options for family planning, alongside psychosocial input through a dedicated social work service, was considered integral to support families through this pathway.
To mitigate the impact of early diagnosis on subpopulations and considering the individual biopsychosocial aspects of the child and their family, the clinical referral pathway would be underpinned by access to interpreters for non-English speakers, social work support for families to help them navigate through health pathways and psychological support for families receiving with an early diagnosis of DMD. For Indigenous and Torres Strait Islander subpopulations, the impact of diagnoses would be managed within the framework of the SCHN Aboriginal Health Strategic Management plan 2018.
Two-tier scerening for DMD would be completed on all DBS collected in the state of New South Wales/Australian Capital Territory for the duration of the two year NBS for DMD pilot program. If CK-MM on first dried blood spot is≥1000 ng/mL, dystrophin sequencing would be completed on the first dried blood spot as a second tier screening test. A screen positive result would be flagged by NSW NBS laboratory to relevant HCPs and the family contacted and invited for review at a specialist neuromuscular disease clinic. Diagnostic bloods would include NATA dystrophin mutation testing and CK-MM on whole blood and 2nd dried blood spot for CK-MM and dystrophin sequencing.
CONSENSUS ON FUTURE PRIORITIES FOR THE NBS FOR DMD PILOT PROGRAM
The pilot would establish the best practice for DMD NBS in Australia. A hybrid implementation-effectiveness study design was considered as the optimal study design to evaluate outcomes from the NBS for DMD cohort (time frames, coverage, sensitivity, specificity, false positive rate, false negative rate, positive predictive value) and processes of genomic NBS for muscular dystrophies.
The NSW/ACT NBS for DMD pilot program aimed to establish the feasibility, acceptability, impact on health and psychosocial outcomes, sensitivity and specificity of NBS for DMD utilizing a biochemical CK-MM assay and second tier NGS method on DBS.
The pilot objectives included:
a. Design and validation of an automated high throughput NGS protocol for DMD and optimisation using DBS.
b. Establishing a NGS based workflow (including analytical and bio-informatic pipeline) in the NSW Newborn Screening laboratory using DMD as a paradigm
c. Assessing the feasibility of screening DBS for DMD using CK-MM and massive parallel sequencing in a NBS laboratory and population pilot.
d. Assessing the accuracy of massive parallel sequencing and variant interpretation for DMD in a NBS setting
e. Establishing the workload demand, bioinformatics and artificial intelligence needs to undertake DMD screening in a highly reproducible manner in line with established standards in the NSW Newborn Screening Service laboratory to meet demands of DMD newborn screening for NSW and ACT
f. Establishing follow up care of positively identified newborns as well as false positive and false negative results. Establish pathways and processes required for children with CK over 1000 ng/ml (tier 1) but who do not have pathogenic variants identified on DNA sequencing on DBS (tier 2) (i.e., those who are not deemed to be screened positive on the tier 2 screening test).
g. Assessing ethical issues of NBS in DMD in an Australian setting
h. Building robust processes for the responsible incorporation of DMD and other emerging technologies (e.g. long read sequencing) into NBS to inform the future trajectory of genomic-based NBS and implementation into health practice and policy.
i. Establishing and evaluating the clinical pathways (temporal course, processes and quality of screening, diagnostic and clinical management frameworks.
j. Establishing the perspectives of stakeholders i.e. consumers and health professionals, including the risks, benefits, facilitators and barriers to the program.
k. Establishing the impact of NBS for DMD as an intervention on the health and wellbeing of affected children and their families.
l. Establishing the cost-effectiveness of NBS for DMD using a cost of illness health economics analysis.
CONCLUSION
As a new era of individualised medicine dawns, the advent of novel genetic technologies and drug discovery confer unprecedented opportunities to fundamentally change the traditional approach to the diagnosis and management of DMD. Challenging the traditional paradigm of supportive care based on the individual, a paradigm shift has emerged in DMD, focussing on proactive models of care that improve health outcomes and mitigate risks not only for the individual, but address the wider ethical, social, epidemiological, and reproductive benefits of screening.
Subsequently, there is impetus and a collective responsibility to support the development of earlier and possibly more effective screening methods for DMD, moving in line with international best practice that has started to use public health (screening) measures to facilitate a personalised approach to diagnosis and management of neuromuscular disease. A timely diagnosis, enabled through NBS for DMD is envisaged to promote health choices for the individual and their family, to build readiness for affected children to access a growing therapeutic repertoire, facilitate early rehabilitation through supportive models of care, and offer timely psychosocial support. Consequently, implementation of a NBS DMD pilot realises multiple core priorities of the 2021 National Strategic action plan for Rare Disease, addressing translational challenges for rare diseases in Australia by enabling timely and equitable diagnosis, care and support, treatment, and research opportunities for individuals.
Beyond DMD, as we move towards a future where genomic capabilities expand, understanding the utility, feasibility, acceptability and implementation of NBS for DMD through a pilot program may be a blueprint for considering other neurodegenerative conditions that have similar genetic complexities and may not fulfil standard principles and practices of newborn screening [50].
ACKNOWLEDGMENTS
This work was supported by the Duchenne Muscular Dystrophy Working Group and workshop attendees including; A/Prof Michelle Farrar, A/Prof Veronica Wiley, A/Prof Kristi Jones, Dr Michelle Lorentzos, Dr Didu Kariyawasam, Prof Craig Munns, Prof Chris Cowell, Dr Anja Ravine, Dr Mark Davis, Klair Bayley, Sarah Grattan, Sandra Holland, Lisa Tang, Lyndal Douglas, Anastasia Loannou, Dr Julia Warning, Sarah Alland, Lisa Quirk, Meg Chard, and Laura Collie. We also thank the NSW Ministry of Health for their time and input to this important workshop, and their ongoing support moving forward. Michele Farrar is the recipient of a National Health and Medical Research Council of Australia Investigator grant (APP1194940).
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest with respect to the research, authorship, or publication of this article.
REFERENCES
[1] | Towbin JA , Hejtmancik JF , Brink P , Gelb B , Zhu XM , Chamberlain JS , et al X-linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993;87. |
[2] | Gao QQ , McNally EM The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr Physiol. (2015) ; 5: (3): 1223–39. |
[3] | Allen DG , Whitehead NP , Froehner SC Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiol Rev. (2016) ; 96: (1): 253–305. |
[4] | Grosse SD , Boyle CA , Kenneson A , Khoury MJ , Wilfond BS From public health emergency to public health service: the implications of evolving criteria for newborn screening panels. Pediatrics. (2006) ;117: (3):923–9. |
[5] | Mendell JR , Shilling C , Leslie ND , Flanigan KM , al-Dahhak R , Gastier-Foster J , et al Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. (2012) ;71: (3):304–13. |
[6] | Timonen A , Lloyd-Puryear M , Hougaard DM , Meriö L , Mäkinen P , Laitala V , et al Duchenne Muscular Dystrophy Newborn Screening: Evaluation of a New GSP(®) Neonatal Creatine Kinase-MM Kit in a US and Danish Population. Int J Neonatal Screen. (2019) ;5: (3):27. |
[7] | PerkinElmer I PerkinElmer Launches First FDA-Approved Assay Kit to Screen for Duchenne Muscular Dystrophy in Newborns 2021 December 15, 2021. Available from: https://ir.perkinelmer.com/news-releases/news-release-details/perkinelmer-launches-first-fda-approved-assay-kit-screen. |
[8] | Ke Q , Zhao ZY , Griggs R , Wiley V , Connolly A , Kwon J , et al Newborn screening for Duchenne muscular dystrophy in China: follow-up diagnosis and subsequent treatment. World J Pediatr. (2017) ;13: (3):197–201. |
[9] | Vita GL , Vita G Is it the right time for an infant screening for Duchenne muscular dystrophy? Neurol Sci (2020) ;41: (7):1677–83. |
[10] | Al-Zaidy SA , Lloyd-Puryear M , Kennedy A , Lopez V , Mendell JR A Roadmap to Newborn Screening for Duchenne Muscular Dystrophy. Int J Neonatal Screen. (2017) ;3: (2):8. |
[11] | Campbell E , Ross LF Parental attitudes regarding newborn screening of PKU and DMD. Am J Med Genet A. (2003) ;120a: (2):209–14. |
[12] | Institute’ TM Living with Duchenne & Becker in Australia: Supporting Families waiting for a Cure. 2020. |
[13] | Wong SH , McClaren BJ , Archibald AD , Weeks A , Langmaid T , Ryan MM , et al A mixed methods study of age at diagnosis and diagnostic odyssey for Duchenne muscular dystrophy. Eur J Hum Genet. (2015) ;23: (10):1294–300. |
[14] | Health ADo Newborn Bloodspot Screening National Policy Framework. 2018. |
[15] | Wilson JMG , Jungner G , World Health O Principles and practice of screening for disease J. M. G. Wilson, G. Jungner. Geneva: World Health Organization; 1968. |
[16] | Kariyawasam DS , Samapio H , Mowat D , Farrar M 023 Genetic carrierscreening for duchenne muscular dystrophy: the outcome of overtwenty years of genetic counselling on disease epidemiology in asingle-centre cohort study in new south wales (NSW), australia. Journal of Neurology, Neurosurgery & Psychiatry. (2019) ;90: (e7):A8–A9. |
[17] | Quinlivan R Early diagnosis of Duchenne muscular dystrophy is essential to improve long term outcomes. Archives of Disease in Childhood. (2014) ;99: (12):1061. |
[18] | Laing NG , Davis MR , Bayley K , Fletcher S , Wilton SD Molecular diagnosis of duchenne muscular dystrophy: past, present and future in relation to implementing therapies. Clin Biochem Rev. (2011) ;32: (3):129–34. |
[19] | McGuire AL , Joffe S , Koenig BA , Biesecker BB , McCullough LB , Blumenthal-Barby JS , et al Point-counterpoint. Ethics and genomic incidental findings. Science. (2013) ;340: (6136):1047–8. |
[20] | Wilfond BS , Fernandez CV , Green RC Disclosing Secondary Findingsfrom Pediatric Sequencing to Families: Considering the “Benefit toFamilies”. J Law Med Ethics. (2015) ;43: (3):552–8. |
[21] | Bombard Y , Miller FA , Hayeems RZ , Avard D , Knoppers BM Reconsidering reproductive benefit through newborn screening: a systematic review of guidelines on preconception, prenatal and newborn screening. European Journal of Human Genetics. (2010) ;18: (7):751–60. |
[22] | Campbell E , Ross LF Parental attitudes regarding newborn screening of PKU and DMD. American Journal of Medical Genetics Part A. (2003) ;120A: (2):209–14. |
[23] | Hutton EM , Thompson MW Carrier detection and genetic counselling in Duchenne muscular dystrophy: a follow-up study. Can Med Assoc J. (1976) ;115: (8):749–52. |
[24] | Finsterer J , Stöllberger C , Freudenthaler B , Simoni DD , Höftberger R , Wagner K Muscular and cardiac manifestations in a Duchenne-carrier harboring a dystrophin deletion of exons 12-29. Intractable Rare Dis Res. (2018) ;7: (2):120–5. |
[25] | Lim KRQ , Sheri N , Nguyen Q , Yokota T Cardiac Involvement in Dystrophin-Deficient Females: Current Understanding and Implications for the Treatment of Dystrophinopathies. Genes (Basel). (2020) ;11: (7):765. |
[26] | Han S , Xu H , Zheng J , Sun J , Feng X , Wang Y , et al Population-Wide Duchenne Muscular Dystrophy Carrier Detection by CK and Molecular Testing. BioMed Research International. (2020) ;2020: 8396429. |
[27] | Drousiotou A , Ioannou P , Georgiou T , Mavrikiou E , Christopoulos G , Kyriakides T , et al Neonatal screening for Duchenne muscular dystrophy: a novel semiquantitative application of the bioluminescence test for creatine kinase in a pilot national program in Cyprus. Genet Test. (1998) ;2: (1):55–60. |
[28] | Eyskens F , Philips E GP 10 10 Newborn screening for Duchenne muscular dystrophy. The experience in the province of AntwerNeuromuscular Disorders. (2006) ;16: (9):721. |
[29] | Greenberg CR , Jacobs HK , Halliday W , Wrogemann K Three years’ experience with neonatal screening for Duchenne/Becker muscular dystrophy: gene analysis, gene expression, and phenotype prediction. Am J Med Genet. (1991) ;39: (1):68–75. |
[30] | Moat SJ , Bradley DM , Salmon R , Clarke A , Hartley L Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK). European journal of human genetics: EJHG. (2013) ;21: (10):1049–53. |
[31] | Wong SH , McClaren BJ , Archibald AD , Weeks A , Langmaid T , Ryan MM , et al A mixed methods study of age at diagnosis and diagnostic odyssey for Duchenne muscular dystrophy. European Journal of Human Genetics. (2015) ;23: (10):1294–300. |
[32] | Parsons EP , Clarke AJ , Hood K , Lycett E , Bradley DM Newborn screening for Duchenne muscular dystrophy: a psychosocial study. Arch Dis Child Fetal Neonatal Ed. (2002) ;86: (2):F91–5. |
[33] | Chung J , Smith AL , Hughes SC , Niizawa G , Abdel-Hamid HZ , Naylor EW , et al Twenty-year follow-up of newborn screening for patients with muscular dystrophy. Muscle Nerve. (2016) ;53: (4):570–8. |
[34] | Wood MF , Hughes SC , Hache LP , Naylor EW , Abdel-Hamid HZ , Barmada MM , et al Parental attitudes toward newborn screening for Duchenne/Becker muscular dystrophy and spinal muscular atrophy. Muscle Nerve. (2014) ;49: (6):822–8. |
[35] | Landfeldt E , Lindgren P , Bell CF , Schmitt C , Guglieri M , Straub V , et al The burden of Duchenne muscular dystrophy: an international, cross-sectional study. Neurology. (2014) ;83: (6):529–36. |
[36] | Teoh LJ , Geelhoed EA , Bayley K , Leonard H , Laing NG Health care utilization and costs for children and adults with duchenne muscular dystrophy. Muscle Nerve. (2016) ;53: (6):877–84. |
[37] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Colvin MK , et al Diagnosis and management of Duchenne muscular dystrophy, part primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. (2018) ;17: (5):445–55. |
[38] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Brumbaugh D , et al Diagnosis and management of Duchenne muscular dystrophy, part diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. (2018) ;17: (3):251–67. |
[39] | Birnkrant DJ , Bushby K , Bann CM , Alman BA , Apkon SD , Blackwell A , et al Diagnosis and management of Duchenne muscular dystrophy, part respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. (2018) ;17: (4):347–61. |
[40] | Conklin LS , Damsker JM , Hoffman EP , Jusko WJ , Mavroudis PD , Schwartz BD , et al Phase IIa trial in Duchenne muscular dystrophy shows vamorolone is a first-in-class dissociative steroidal anti-inflammatory drug. Pharmacol Res. (2018) ;136: , 140–50. |
[41] | Campbell C , Barohn RJ , Bertini E , Chabrol B , Comi GP , Darras BT , et al Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J Comp Eff Res. (2020) ;9: (14):973–84. |
[42] | Mercuri E , Muntoni F , Osorio AN , Tulinius M , Buccella F , Morgenroth LP , et al Safety and effectiveness of ataluren: comparison of results from the STRIDE Registry and CINRG DMD Natural History Study. J Comp Eff Res. (2020) ;9: (5):341–60. |
[43] | Markate T , Oskoui M , Farrar M , Duong T , Goemens N , Servais L Emerging therapies for Duchenne Muscular Dystrophy. TBC. (Under Review). 2022. |
[44] | de Freitas Nakata KC , da Silva Pereira PP , Salgado Riveros B Creatine kinase test diagnostic accuracy in neonatal screening for Duchenne Muscular Dystrophy: A systematic review. Clin Biochem. (2021) ;98: , 1–9. |
[45] | Beckers P , Caberg JH , Dideberg V , Dangouloff T , den Dunnen JT , Bours V , et al Newborn screening of duchenne muscular dystrophy specifically targeting deletions amenable to exon-skipping therapy. Sci Rep. (2021) ;11: (1):3011. |
[46] | Nallamilli BRR , Chaubey A , Valencia CA , Stansberry L , Behlmann AM , Ma Z , et al A single NGS-based assay covering the entire genomic sequence of the DMD gene facilitates diagnostic and newborn screening confirmatory testing. Hum Mutat. (2021) ;42: (5):626–38. |
[47] | Xiao T , Wu B , Cao Y , Liu R , Cheng G , Wang L , et al Genetic identification of pathogenic variations of the DMD gene: a retrospective study from 10,481 neonatal patients based on next-generation sequencing data. Ann Transl Med. (2021) ;9: (9):766. |
[48] | Chien Y-H , Lee N-C , Weng W-C , Chen L-C , Huang Y-H , Wu C-S , et al Duchenne muscular dystrophy newborn screening: the first 50,000 newborns screened in Taiwan. Neurological Sciences. 2022. |
[49] | Roman TS , Crowley SB , Roche MI , Foreman AKM , O’Daniel JM , Seifert BA , et al Genomic Sequencing for Newborn Screening: Results of the NC NEXUS Project. Am J Hum Genet. (2020) ;107: (4):596–611. |
[50] | Health Do National Strategic Action Plan for Rare Disease. 2020. |


