
A Multisystem Mitochondrial Disease Caused by a Novel MT-TL1 mtDNA Variant: A Case Report
Abstract
Background:
Mitochondrial tRNA (MTT) genes are hotspot for mitochondrial DNA mutation and are responsible of half mitochondrial disease. MTT mutations are associated with a broad spectrum of phenotype often with complex multisystem involvement and complex genotype-phenotype correlations. MT-TL1 mutations, among which the m.3243A>G mutation is the most frequent, are associated with myopathy, maternal inherited diabetes and deafness, MELAS, cardiomyopathy, and focal segmental glomerulosclerosis.
Case study:
Here we report the case of an Italian 49-years old female presenting with encephalomyopathy, chronic proteinuric kidney disease and a new heteroplasmic m.3274_3275delAC MT-TL1 gene mutation.
Conclusions:
Our case demonstrates a systemic mitochondrial disease caused by the heteroplasmic m.3274_3275delAC MT-TL1 gene mutation, not yet described in the literature. A mitochondrial disease should be suspected in case of complex multisystem phenotypes, including steroid-resistant nephrotic syndrome with multisystemic involvement.
INTRODUCTION
Primary mitochondrial disorders (PMDs) are complex diseases due to a defective oxidative phosphorylation [1]. In adulthood, PMDs are often caused by mitochondrial DNA (mtDNA) mutations, with mitochondrial tRNA (MTT) genes hotspot for mtDNA mutations, and cause frequently multisystemic involvement of tissues with greater aerobic metabolism [1]. MT-TL1 mutations, among which the m.3243A>G is the most frequent, are associated with a broad spectrum of clinical manifestations, ranging from primary mitochondrial myopathy, mitochondrial inherited diabetes and deafness to the full blown MELAS phenotype [1]; kidney involvement is frequently reported, with proteinuria and chronic kidney disease up to end-stage renal failure [2]. Here we describe a 49-year-old woman presenting with a complex multisystemic phenotype characterized by encephalomyopathy, chronic proteinuric kidney disease and an undescribed heteroplasmic m.3274_3275delAC MT-TL1mutation.
CASE PRESENTATION
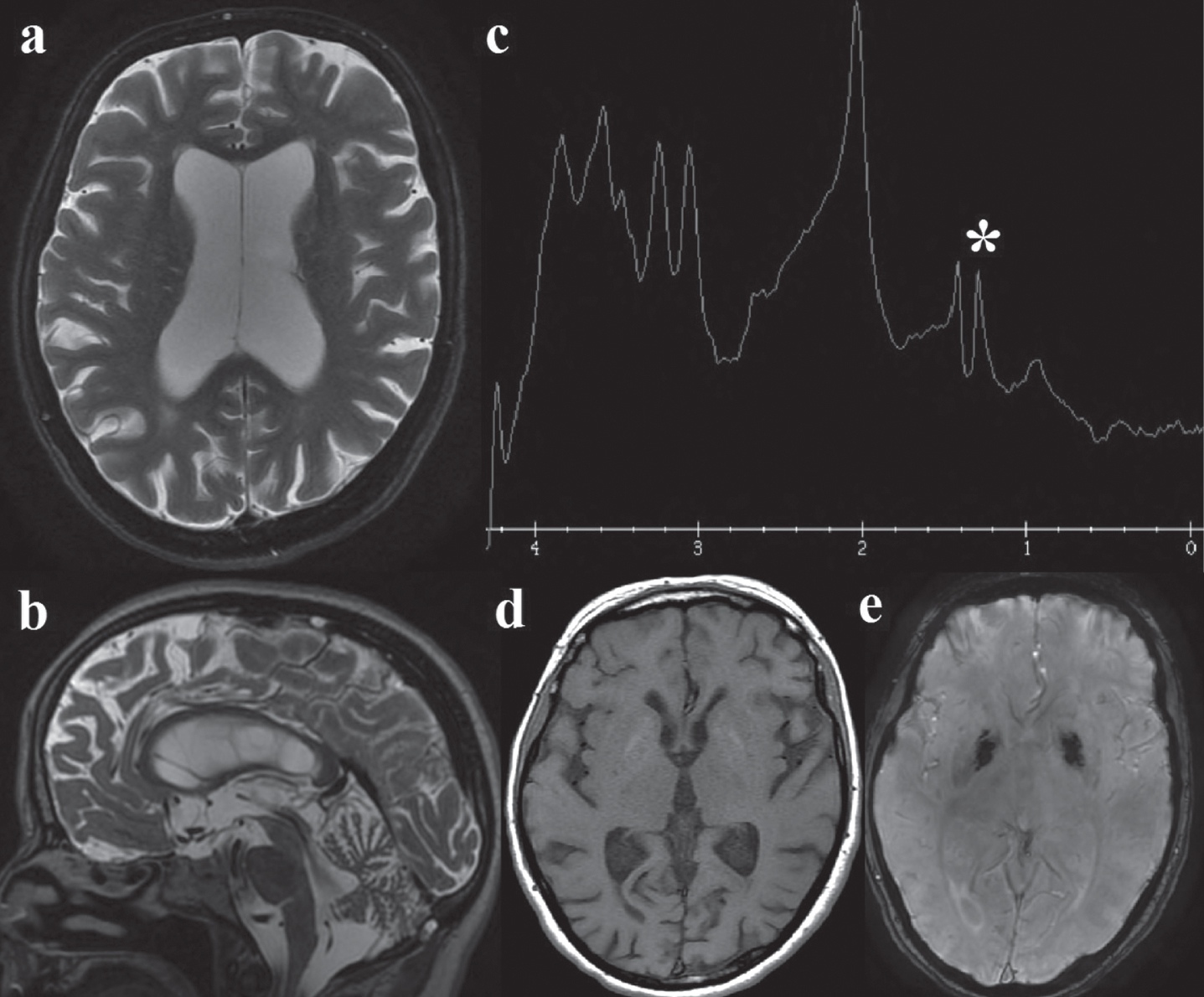
A 49 years-old Italian woman, married with no children, was referred to our center for a complex multisystemic disease. She presented migraine from adolescence and from 38-years of age a progressive sensorineural hearing loss, fatigue, bilateral cataracts and pigmented retinopathy. Her mother died affected by diabetes, migraine, deafness and not investigated chronic kidney disease. Neurological examination in our patient showed mild cerebellar signs (widened base of support, mild dysmetria). Electromyography showed a myopathic pattern. A short PR interval (100 msec) with normal echocardiogram were detected. Spirometry showed no abnormalities. The brain magnetic resonance imaging showed a global cerebral atrophy, cerebellar atrophy, white matter hyperintensities, bipallidal calcifications with a lactate peak on spectroscopy (Fig. 1).
Fig. 1
Brain MRI: a. Axial T2, showing global cerebral atrophy with ex-vacuo lateral ventricles dilatation; b. Sagittal T2, cerebellar atrophy; c. MR spectroscopy: the white asterisk (*) marks the lactate peak; d. axial T1 and e. susceptibility-weighted angiography (SWAN): mild bipallidal calcifications.
Muscle biopsy revealed myopathic changes with scattered ragged red and blue fibers, as well as COX negative fibers (Fig. 2). Sequencing of the whole muscular mtDNA, performed using Nextera XT technology (Illumina, San Diego, CA), showed the heteroplasmic (57%) m.3274_3275delAC MT-TL1 gene variant (Supplementary Figure 1). This variant has a high score of pathogenicity (94.2%, according to MitoTip [3]). No other family members were tested for this mtDNA variant.
Fig. 2
Muscle biopsy reveals mild proliferation of mitochondria (a), COX negative fibers (b) and (c) one ragged blue fiber.

The patient also presented proteinuria up to 2100 mg/24 hours and albuminuria 1600 mg/24 hours, creatinine 0,79 mg/l; urinalysis showed proteinuria 100 mg/dl and absence of white and red cell. Kidney biopsy (Supplementary Figure 2) demonstrated secondary focal segmental glomerulosclerosis associated to proximal tubulopathy: microscopy showed 7 glomeruli with global mesangial expansion and 2 of these with synecheal capsular adhesions with consensual parietal epithelial proliferation, which were not classifiable as tip lesion or vascular variants; mild focal tubular atrophy (5%) with associated mild fibrosis was also present. Immunofluorescence microscopy revealed IgM mesangial deposition in the glomeruli. Moreover, electron microscopy showed proximal tubulopathy with dysmorphic, abnormal mitochondria; tubulocytes had an increased number of mitochondria with polymorphous size and irregular shapes; at higher magnification, despite the suboptimal morphology due to the retrieval procedure, the mitochondria showed a reduction in cristae and the presence of clear vesicles related to the degeneration of the cristae. Finally, glomeruli showed 80% of fusion of podocyte foot processes.
The mutation was detected also in the kidney tissue with a 40% heteroplasmy and blood (10%) heteroplasmy. Therefore, the patient was treated with renin angiotensin system blocking therapy and low-sodium diet.
DISCUSSION AND CONCLUSIONS
Our case demonstrates a systemic mitochondrial disease caused by the heteroplasmic m.3274_3275delAC MT-TL1 gene mutation, not yet reported in the literature. The dysfunction affects central nervous system and muscle (encephalomyopathy), kidney (both tubular and glomerular site), hearth (short PR interval), sense organs (sensory neural hearing loss and pigmentous retinopathy) and cause chronic kidney disease with no-nephrotic proteinuria. The kidney dysfunction observed in our patient is in our opinion a primary consequence of the PMD as it shows a no-complete podocyte effacement, for the presence of capsular adhesions and for the association to the typical ultrastructural features of PMD with proximal tubular involvement.
Kidney involvement in PMDs due to mutations in the mtDNA more frequent occurs in childhood, usually in association with neurological and muscular involvement [4]. The proximal tubules, due to their intense metabolic activity to modify urine according to systemic requirement, contain more mitochondria than the other part of the kidney [5]. In fact, in case of mitochondrial dysfunction, proximal tubular injury is the most frequent manifestation, characterized by Fanconi syndrome and in few cases by partial tubular dysfunction (tubular acidosis, aminoaciduria, glycosuria, Bartter-like syndrome) [4, 6]. Other manifestations of the tubular involvement are tubulointerstitial damage, characterized by tubular atrophy and interstitial fibrosis, and cystic renal disease [6, 7].
Glomerular injury is also observed in PMDs. It can be secondary to tubular damage and atrophy (global glomerulosclerosis), but it can be linked with direct podocyte dysfunction (focal and segmental glomerulosclerosis) [7, 8]. The tubular and glomerular involvement may be observed either individually or simultaneously. Absence of response to steroid therapy and multisystemic involvement are criteria to suspect a kidney involvement caused by a primary mitochondrial dysfunction [9].
MT-TL1 mutations are frequently associated with kidney involvement; the m.3243A>G leads both to tubulopathy and to glomerulosclerosis characterized by not-nephrotic steroid-resistant proteinuria. Up to 30% of mitochondrial inherited diabetes and deafness (MIDD) have chronic kidney disease with a variable age at onset and disease progression [2, 10]. Not only podocyte or tubular damage can be found in patients with PMDs. In fact, vascular nephropathy (arterioles nephrosclerosis) with renal ischemic changes detected in a MELAS patients suggested the hypothesis that vascular kidney alterations could be early histologic markers of kidney PMDs before glomerular or tubular patterns [11]. This is an intriguing observation since also a cerebral mitochondrial microangiopathy may play a role into the pathogenesis of stroke like episode and migraine in MELAS [12]. In a variable proportion of cases, kidney involvement progresses to chronic kidney disease and kidney failure requiring renal replacement therapy [13].
Therapy of kidney involvement in PMDs involves mainly supportive measures. Coenzyme Q10 treatment decreased proteinuria in primary coenzyme Q10 deficiency [14], while cyclosporine therapy showed some efficacy on nephrotic syndrome in COQ6 mutated patients [15]. The response to steroid therapy is generally poor and a positive effect of angiotensin-converting-enzyme inhibitors (ACEi) or Ang-II receptor blockers (ARB) on mitochondrial function and oxidative stress has been hypothesized only in vitro or in experimental models of hypertension [16], but not in vivo.
This variant should be considered pathogenic according to Di Mauro and Schon criteria [17]: it is not a known neutral polymorphism, it is heteroplasmic on multiple and affected tissue (blood, muscle and kidney); moreover, even if the mutation does not affect a highly conserved residue, the micro-deletion of two bases modifies the sequence of the anticodons with important deleterious consequences on the tRNA function. Finally, this variant has a high score of pathogenicity (94.2%, according to MitoTip [3]). Since we cannot provide single fiber, steady-state level, or trans-mitochondrial cybrid studies, according to the scoring system of Yarham [18], our mutation should be considered probably pathogenetic.
A nucleotide change at the same 3274 nucleotide was described in 2001 [19] in a patient with progressive myopathy, deafness, ataxia, psychomotor retardation, retinopathy, and psychosis; that variant affected a non-conserved Watson-Crick base pair in the anticodon stem of tRNA and was associated with Complex I deficiency and an high score of pathogenicity (77,1% according to MitoTip).
In conclusions, we report a novel mtDNA leading to a complex multisystem phenotype; a PMD should be suspected in case of complex phenotypes, including steroid-resistant nephrotic syndrome with multisystemic involvement.
ACKNOWLEDGMENTS
This work was partially supported by Telethon Grants (GUP09004 and GSP16001) and by EJP RD Joint Transnational Call (JTC2019) GENOMIT.
We are grateful to the European Reference Network ERN NMD and RND (MM, VM, PL, GS) as representatives for the Italian HCP partners.
The authors declare that they did not receive any funding for this paper.
ETHICS APPROVAL
We hereby confirm that the present study conforms to the ethical standards and guidelines of the journal.
CONFLICT OF INTEREST
The authors have no conflict of interest.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-221526.
REFERENCES
[1] | Ng YS , Bindoff LA , Gorman GS , Klopstock T , Kornblum C , Mancuso M , et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. (2021) ;20: :573–584. https://doi.org/10.1016/S1474-4422(21)00098-3. |
[2] | Guillausseau PJ , Massin P , Dubois-LaForgue D , Timsit J , Virally M , Gin H , et al. Maternally inherited diabetes and deafness: A multicenter study. Ann Intern Med. (2001) ;134: :721–728. https://doi.org/10.7326/0003-4819-134-9_part_1-200105010-00008. |
[3] | Sonney S , Leipzig J , Lott MT , Zhang S , Procaccio V , Wallace DC , et al. Predicting the pathogenicity of novel variants in mitochondrial tRNA with MitoTIP. PLoS Comput Biol. (2017) ;13. https://doi.org/10.1371/journal.pcbi.1005867. |
[4] | Emma F , Pizzini C , Tessa A , Di Giandomenico S , Onetti-Muda A , Santorelli FM , et al. ⪡Bartter-like⪢ phenotype in Kearns-Sayre syndrome. Pediatr Nephrol. (2006) ;21: :355–360. https://doi.org/10.1007/s00467-005-2092-5. |
[5] | Bhargava P , Schnellmann RG . Mitochondrial energetics in the kidney. Nat Rev Nephrol. (2017) ;13: :629–646. https://doi.org/10.1038/nrneph.2017.107. |
[6] | Schijvens AM , van de Kar NC , Bootsma-Robroeks CM , Cornelissen EA , van den Heuvel LP , Schreuder MF . Mitochondrial disease and the kidney with a special focus on CoQ10 deficiency. Kidney Int Reports. (2020) ;5: :2146–2159. https://doi.org/10.1016/j.ekir.2020.09.044. |
[7] | Govers LP , Toka HR , Hariri A , Walsh SB , Bockenhauer D . Mitochondrial DNA mutations in renal disease: An overview. Pediatr Nephrol. (2021) ;36: :9–17. https://doi.org/10.1007/s00467-019-04404-6. |
[8] | Emma F , Montini G , Parikh SM , Salviati L . Mitochondrial dysfunction in inherited renal disease and acute kidney injury. Nat Rev Nephrol. (2016) ;12: :267–280. https://doi.org/10.1038/nrneph.2015.214. |
[9] | Preston R , Stuart HM , Lennon R . Genetic testing in steroid-resistant nephrotic syndrome: Why, who, when and how? Pediatr Nephrol. (2019) ;34: :195–210. https://doi.org/10.1007/s00467-017-3838-6. |
[10] | Iwasaki N , Babazono T , Tsuchiya K , Tomonaga O , Suzuki A , Togashi M , et al. Prevalence of A-to-G mutation at nucleotide 3243 of the mitochondrial tRNALeu(UUR) gene in Japanese patients with diabetes mellitus and end stage renal disease. J Hum Genet. (2001) ;46: :330–334. https://doi.org/10.1007/s100380170068. |
[11] | Piccoli G , Bonino L , Campisi P , Vigotti F , Ferraresi M , Fassio F , et al. Chronic kidney disease, severe arterial and arteriolar sclerosis and kidney neoplasia: On the spectrum of kidney involvement in MELAS syndrome. BMC Nephrol. (2012) ;13. https://doi.org/10.1186/1471-2369-13-9. |
[12] | Smeitink J , Koene S , Beyrath J , Saris C , Turnbull D , Janssen M . Mitochondrial Migraine: Disentangling the angiopathy paradigm in m.3243A>G patients. JIMD Rep. (2019) ;46: :52–62. https://doi.org/10.1002/jmd2.12017. |
[13] | De Laat P , Van Engelen N , Wetzels JF , Smeitink JAM , Janssen MCH . Five non-mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes phenotype adult patients with m.3243A>G mutation after kidney transplantation: Follow-up and review of the literature. Clin Kidney J. (2019) ;12: :840–846. https://doi.org/10.1093/ckj/sfz020. |
[14] | Atmaca M , Gülhan B , Atayar E , Bayazıt AK , Candan C , Arıcı M , et al. Long-term follow-up results of patients with adck4 mutations who have been diagnosed in the asymptomatic period: Effects of early initiation of coq10 supplementation. Turk J Pediatr. (2019) ;61: :657–663. https://doi.org/10.24953/turkjped.2019.05.003. |
[15] | Sadowski CE , Lovric S , Ashraf S , Pabst WL , Gee HY , Kohl S , et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. (2015) ;26: :1279–1289. https://doi.org/10.1681/ASN.2014050489. |
[16] | Eirin A , Lerman A , Lerman LO . Mitochondria: A pathogenic paradigm in hypertensive renal disease. Hypertension. (2015) ;65: :264–270. https://doi.org/10.1161/HYPERTENSIONAHA.114.04598. |
[17] | DiMauro S , Schon EA . Mitochondrial DNA mutations in human disease. Am J Med Genet - Semin Med Genet. (2001) ;106: :18–26. https://doi.org/10.1002/ajmg.1392. |
[18] | Yarham JW , Al-Dosary M , Blakely EL , Alston CL , Taylor RW , Elson JL , et al. A comparative analysis approach to determining the pathogenicity of mitochondrial tRNA mutations. Hum Mutat. (2011) ;32: :1319–1325. https://doi.org/10.1002/humu.21575. |
[19] | Jaksch M , Lochmuller H , Schmitt F , Volpel B , Obermaier-Kusser B , Horvath R . A mutation in mt tRNALeu(UUR) causing a neuropsychiatric syndrome with depression and cataract. Neurology. (2001) 57: : 1930–1931. https://doi.org/10.1212/wnl.57.10.1930. |


