
A Multidisciplinary Perspective Addressing the Diagnostic Challenges of Late-Onset Pompe Disease in the Arabian Peninsula Region Developed From an Expert Group Meeting
Abstract
Pompe disease is a rare, metabolic, autosomal recessive disorder. Early diagnosis is critical for progressive Pompe disease as delays can significantly alter the clinical course of the disease. Diagnostic modalities, including dried blood spot testing and genetic testing, are available and are effective for diagnosing patients with late-onset Pompe disease (LOPD). However, clinicians face numerous clinical challenges related to the diagnosis of the disease. Two expert group committee meetings, involving 11 experts from the United Arab Emirates, Kuwait, the Kingdom of Saudi Arabia, and Oman, were convened in October 2019 and November 2020 respectively to develop a uniform diagnostic algorithm for the diagnosis of pediatric and adult LOPD in the Arabian Peninsula region. During the first meeting, the specialty-specific clinical presentation of LOPD was defined. During the second meeting, a diagnostic algorithm was developed after a thorough validation of clinical presentation or symptoms, which was performed with the aid of existing literature and expert judgement. A consensus was reached on the diagnostic algorithm for field specialists, such as neurologists, rheumatologists, general practitioners/internal medicine specialists, orthopedic specialists, and pulmonologists. This specialty-specific diagnostic referral algorithm for pediatric and adult LOPD will guide clinicians in the differential diagnosis of LOPD.
HIGHLIGHTS
• In the Arabian Peninsula region, clinicians face numerous clinical challenges related to the diagnosis of late-onset Pompe disease (LOPD).
• Suggestive symptoms of LOPD include limb– girdle muscles weakness, unexplained hyperCKemia, and respiratory insufficiency.
• Considering the high consanguinity rate in the region, physicians are encouraged to set a low threshold for LOPD screening in the appropriate clinical setting.
• Dried blood spot testing for GAA activity should be performed first in the appropriate clinical setting, followed by confirmatory genetic testing.
ABBREVIATIONS
ALT | Alanine aminotransferase |
AST | Aspartate aminotransferase |
BA | Basilar artery |
CK | Creatine kinase |
DBS | Dried blood spot testing |
DMD | Duchenne muscular dystrophy |
EMG | Electromyography |
FVC | Forced vital capacity |
GAA | Acid alpha-glucosidase |
GPs/IMs | General practitioners/internal medicine specialists |
KSA | Kingdom of Saudi Arabia |
LDH | Lactate dehydrogenase |
LOPD | Late-onset Pompe disease |
LGMW | Limb–girdle muscle weakness |
MG | Myasthenia gravis |
NBS | Newborn screening |
NMDs | Neuromuscular disorders |
RCTs | Randomized-controlled trials |
SDB | Sleep-disordered breathing |
SMA | Spinal muscular atrophy |
PAS | Positive vacuoles |
INTRODUCTION
Pompe disease is a lysosomal storage disorder characterized by a deficiency in the lysosomal enzyme acid alpha-glucosidase (GAA) due to mutations in the GAA gene [1]. This deficiency leads to the accumulation of glycogen in multiple organs, specifically in the skeletal and respiratory muscles [1]. The estimated incidence of Pompe disease is reported to be 1 in 40,000 live births [2, 3]. Late-onset Pompe disease (LOPD) is a progressive muscle disorder affecting individuals of varying ages, particularly from early childhood (above 12 months of age) to late adulthood [4, 5]. The estimated global incidence of LOPD is 1 in 57,000 people [6]. Two prospective trials conducted in Taiwan (N = 344,056) and Austria (N = 34,736) in the context of newborn screening (NBS) depicted the high incidence of LOPD in Taiwan (1 in 26,466 live births) [7] and Austria (1 in 8684 live births) [8]. These results indicated that LOPD is underestimated or underdiagnosed in countries without national screening programs. Underdiagnosis in countries which lack national screening program constitutes the most significant barrier to the management of LOPD. Therefore, an effective and early screening strategy is essential to overcome underdiagnosis in countries without national screening programs. It is noteworthy that early diagnosis can lead to early initiation of treatment, which may improve outcomes in LOPD patients [9].
In the Middle East and North Africa (MENA) region, consanguineous marriages are culturally favored and are considered traditional in most of the communities [10]. A study conducted on 3212 Saudi families showed that the overall rate of consanguinity was 57.7% with a high frequency of first-cousin marriages (28.4%) [11]. Even a cross-sectional study that was carried out in a Tehrani population (n = 93) demonstrated the high degree of consanguinity (43%) in this region and that the incidence of LOPD (n = 3) was reported in families with consanguinity [12]. Therefore, consanguinity could be one of the determinants of the prevalence of LOPD in the MENA region [10]. However, there is a lack of awareness of LOPD among healthcare professionals (HCPs) in the MENA region, which is a significant challenge that may lead to a delay in diagnosis, missed diagnosis, and delay in treatment [9].
The important signs and symptoms that occur in 80% of patients with LOPD include musculoskeletal and respiratory symptoms [5, 13]. The gastrointestinal symptoms that are commonly observed in LOPD patients include constipation, diarrhea, poor weight gain, and hepatomegaly [5, 13]. The other clinical manifestations are ptosis, brain aneurysms, arrhythmias, facial muscle weakness, dysphagia, dysarthria, a rigid spine, and macroglossia [13]. The symptoms of LOPD may mimic that of other diseases, including unspecified myopathy, limb–girdle muscular dystrophy, and inflammatory myopathy [9, 14–24]. This broad clinical spectrum of manifestations that mimic symptoms of other diseases is a significant barrier that hinders initial diagnosis and thereby causes diagnostic delays. Therefore, differential diagnoses play a crucial role in the accurate diagnosis of LOPD.
Due to the broad clinical spectrum of presenting symptoms, which are of a wide range of complexity, there is a necessity for a high degree of suspicion for the accurate diagnosis of LOPD [9]. The diagnostic tests that are performed as a part of clinical evaluation in suspected individuals include: (1) measurement of creatine kinase, which alters from normal (60–305 IU/L) to 15 times the upper limit of the normal value; (2) measurement of lactate dehydrogenase, alanine transaminase, and aspartate transaminase, which are frequently increased; (3) assessment of forced vital capacity (FVC), which is reduced in the majority of LOPD patients; (4) electromyography (EMG), in which suggestive signs of LOPD include myopathic EMG, specifically in proximal muscles, myotonic discharges without clinical myotonia, particularly in paraspinal muscles, fibrillation potentials, positive sharp waves, and complex repetitive discharges; and (5) dried blood spot testing (DBS), in which GAA activity is reduced [9].
A positive DBS testing should be followed by a confirmatory test for the diagnosis of LOPD [9]. The array of confirmatory tests encompasses (1) lymphocyte GAA activity, in which GAA is assayed in purified lymphocyte preparations; (2) measurement of GAA activity in skin fibroblast cultures, in which GAA is assayed in cultured fibroblasts from skin biopsy; (3) molecular genetic testing (DBS-extracted DNA) or gene sequencing for detecting variations that include targeted mutational analysis, deletion/duplication analysis, and full sequence analysis; and (4) muscle tissue biopsy with or without GAA enzyme analysis [9]. Pathogenic mutations can be detected through genetic testing, and in muscle biopsy, vacuoles that stain positively for glycogen with glycogen accumulation in the lysosomes and cytosol are observed during the advanced stages of the disease [9]. Although several novel diagnostic strategies are available for diagnosing LOPD, an appropriate diagnostic criterion, including DBS testing of lymphocyte GAA activity and skin fibroblast GAA activity, for clinicians is still lacking in the Arabian Peninsula region.
Overall, under-recognition, missed diagnosis, delayed diagnosis, lack of appropriate diagnostic facilities, and lack of awareness about LOPD among HCPs and families of LOPD patients are the unmet clinical challenges that are widespread across this region. Additionally, LOPD diagnosis is often challenging due to the complex and broad spectrum of clinical symptoms, which overlap with other diseases [9, 14].
With this background, the unmet clinical challenges of diagnostic clues for LOPD patients were examined, particularly from the perspective of the Arabian Peninsula region. The present manuscript aims to establish diagnostic clues for clinicians across the Arabian Peninsula region to better diagnose LOPD patients, that is pediatric LOPD patients and adult LOPD patients, by developing a diagnostic algorithm for different specialists, including neurologists, pulmonologists, rheumatologists, orthopedic specialists, and general practitioners/internal medicine specialists (GPs/IMs).
METHODS
Two expert group committee meetings involving 11 experts with expertise in the department of neurology from the United Arab Emirates, Kuwait, the Kingdom of Saudi Arabia (KSA), and Oman, were convened in October 2019 and November 2020. The committee aimed to develop a consensus-based diagnostic algorithm for pediatric and adult LOPD that would aid clinicians in the early diagnosis of LOPD in the Arabian Peninsula region.
During the first meeting, the clinical presentation and recommendations for the diagnosis of pediatric LOPD were stratified according to specialty, and a clear demarcation of symptoms for different specialists, including neurologists, pulmonologists, GPs/IMs, and rheumatologists, was developed. A similar stratification was developed for the diagnosis of adult LOPD, in addition to a clear-cut symptom demarcation for different specialists, including neurologists, pulmonologists, GPs/IMs, and orthopedics.
After the development of diagnostic criteria for pediatric and adult LOPD, a literature search was performed using PubMed for randomized control trials to identify the prevalence of clinical symptoms in patients with LOPD. The search terms included late-onset Pompe disease and clinical symptoms associated with LOPD.
During the second meeting, all the experts reviewed and validated the evidence and critically discussed the applicability of the literature for LOPD in the Arabian Peninsula region.
A diagnostic algorithm was developed after validation of clinical presentation or symptoms, which was performed with the aid of existing literature and expert judgement.
RESULTS
Development of a consensus-based diagnostic algorithm
The proposed pediatric and adult LOPD clinical presentation and recommendations for different specialties were developed during the expert group committee meetings and are presented in Fig. 1 and Fig. 2, respectively. The literature supporting clinical presentation and recommendations for pediatric and adult LOPD, which was reviewed and validated by experts, is depicted in Table 1 and Table 2, respectively. Finally, a consensus-based diagnostic algorithm was developed for field specialists based on the clinical experiences of experts and a summary of evidence (Fig. 3 and Fig. 4).
Fig. 1
Proposed pediatric LOPD* clinical presentation and recommendations by experts during the first Expert Group Committee Meeting.
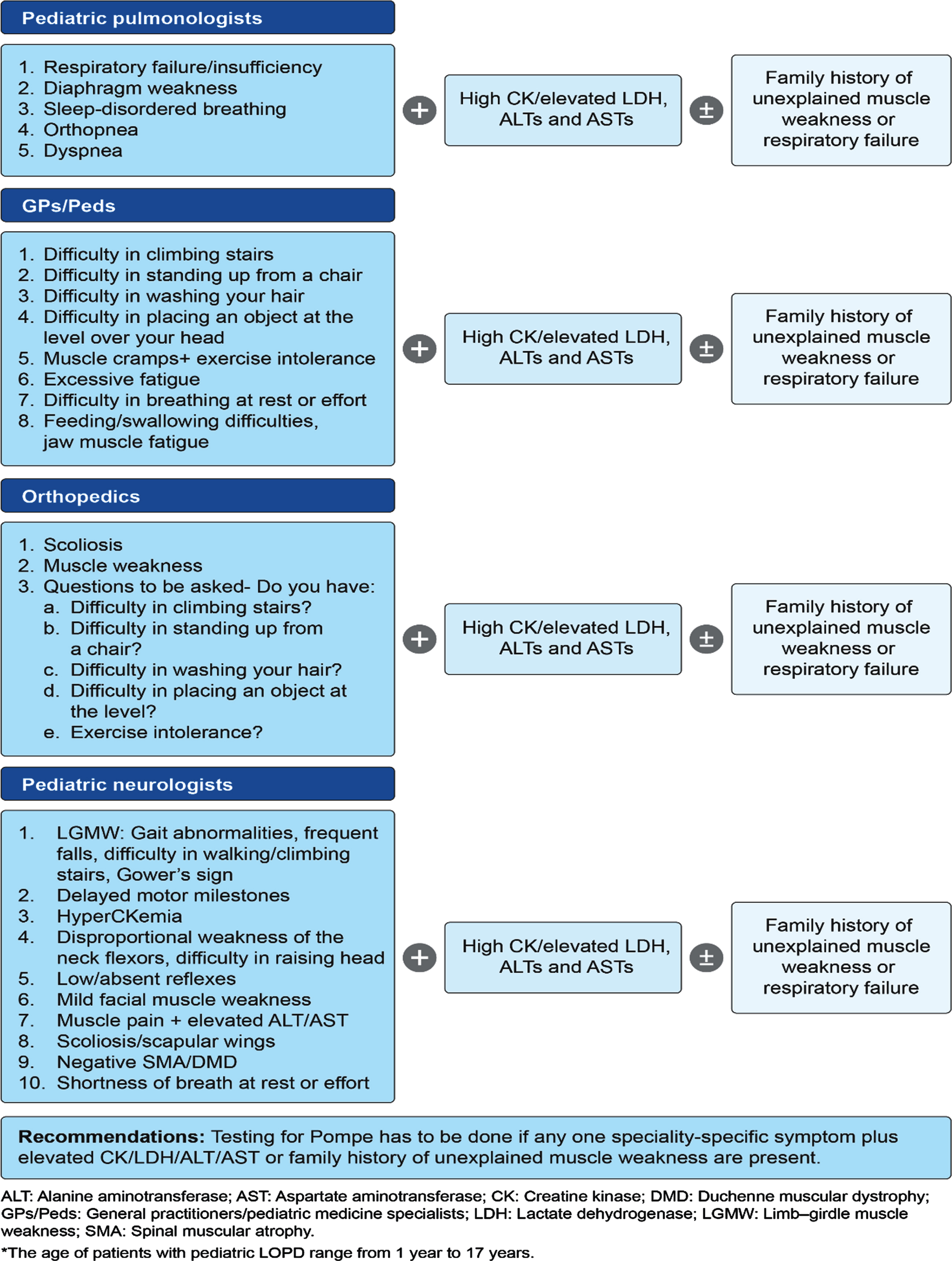
Fig. 2
Proposed adult LOPD* clinical presentation and recommendations by experts during the first expert group committee meeting.
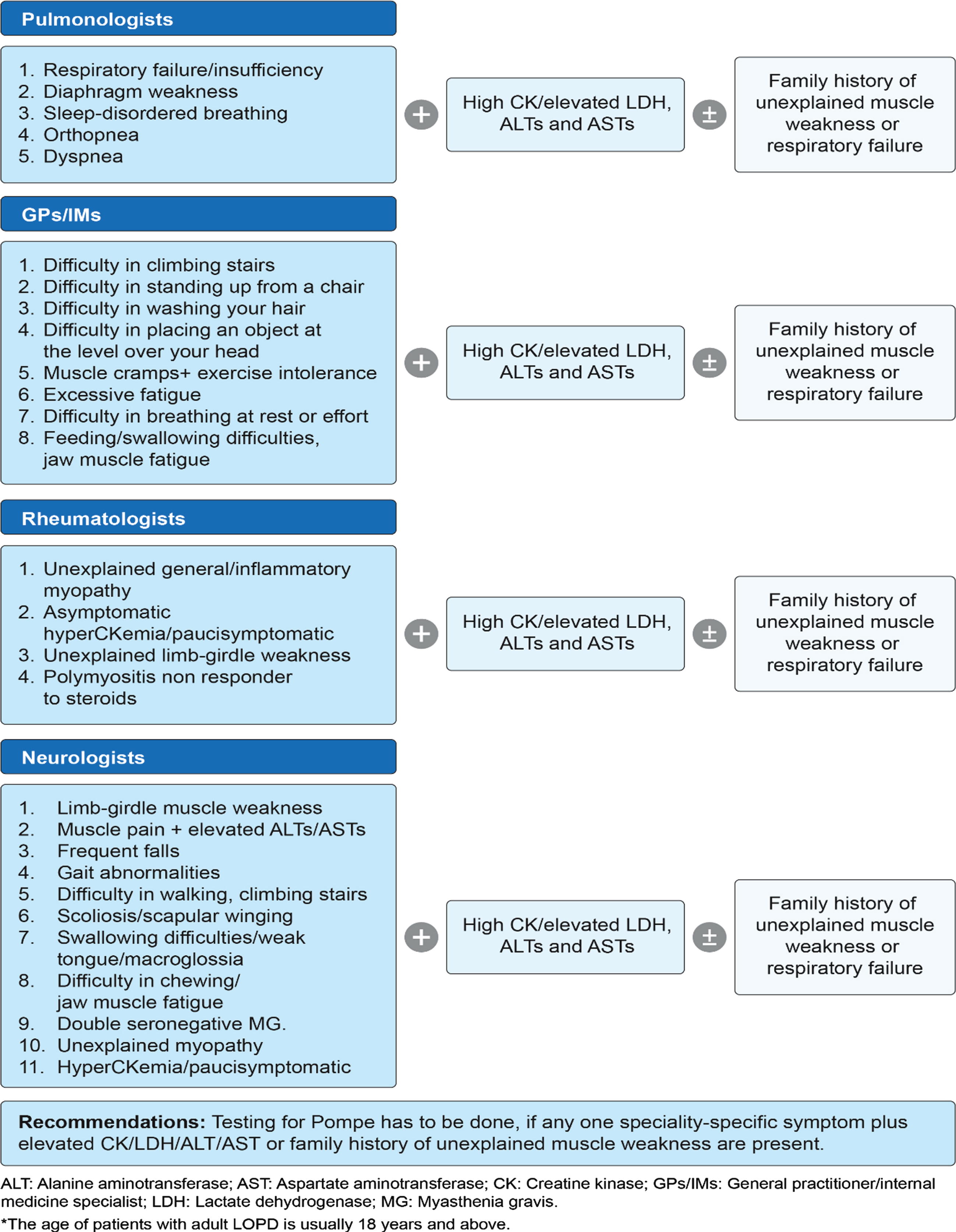
Table 1
Clinical evidence rationalizing the importance of clinical presentation and recommendations for each specialty for the diagnosis of pediatric LOPD
| Specialty * | Number of studies reflecting the importance of clinical presentation and recommendations | Symptoms observed | References |
| Neurology | 11 (1 RCT; 10 observational studies [cohort studies/screening studies]) | 1. LGMW: Gait abnormalities, frequent falls, difficulty in walking/climbing stairs, Gower’s sign | [25–35] |
| 2. Delayed motor milestones | |||
| 3. HyperCKemia | |||
| 4. Disproportional weakness of the neck flexors, difficulty in raising the head | |||
| 5. Low/absent reflexes | |||
| 6. Mild facial muscle weakness | |||
| 7. Muscle pain+elevated ALT/AST | |||
| 8. Scoliosis/scapular wings | |||
| 9. Negative SMA/DMD | |||
| 10. Shortness of breath at rest or effort | |||
| Pulmonology | 7 observational studies (cohort studies/screening studies) | 1. Respiratory failure/insufficiency | [29–31, 33, 34, 36, 37] |
| 2. Diaphragm weakness | |||
| 3. Sleep-disordered breathing | |||
| 4. Orthopnea | |||
| 5. Dyspnea | |||
| 6. Elevated biochemical markers | |||
| a. Serum creatine kinase | |||
| b. ALT | |||
| c. AST | |||
| Orthopedics | 11 observational studies (cohort studies/screening studies) | 1. Scoliosis | [25–35] |
| 2. Elevated CK/ALT/AST | |||
| 3. Muscle weakness | |||
| 4. Questions to be asked: | |||
| Do you have: | |||
| a. Difficulty in climbing stairs? | |||
| b. Difficulty in standing up from a chair? | |||
| c. Difficulty in washing your hair? | |||
| d. Difficulty in placing an object at a level? | |||
| e. Exercise intolerance? |
*The clinical presentation and recommendations for the internal medicine are the same for the diagnosis of pediatric and adult LOPD and thus combined clinical validation by existing literature was performed for pediatric and adult LOPD symptoms. ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; CK: Creatine kinase; DMD: Duchenne muscular dystrophy; LGMW: Limb–girdle muscle weakness; LOPD: Late-onset Pompe disease; SMA: Spinal muscular atrophy.
Table 2
Clinical evidence rationalizing the importance of clinical presentation and recommendations for each specialty for the diagnosis of adult LOPD
| Specialty | Number of studies reflecting the importance of clinical presentation and recommendations | Symptoms observed | References |
| Neurology | 25 (5 RCTs; 20 observational studies [Cohort studies/screening studies]) | 1. Limb–girdle muscle weakness | [12, 25–36, 38–49] |
| 2. Muscle pain + elevated ALT/AST | |||
| 3. Frequent falls | |||
| 4. Gait abnormalities | |||
| 5. Difficulty in waIking and climbing stairs | |||
| 6. Scoliosis/scapular winging | |||
| 7. Swallowing difficulties/weak tongue/macroglossia | |||
| 8. Difficulty in chewing/jaw muscle fatigue | |||
| 9. Double seronegative MG | |||
| 10. Unexplained myopathy | |||
| 11. HyperCKemia/paucisymptomatic | |||
| Pulmonology | 15 (4 RCTs; 11 observational studies [Cohort studies/screening studies]) | 1. Respiratory failure/insufficiency | [29–31, 36, 38–41, 50–56] |
| 2. Diaphragm weakness | |||
| 3. Sleep-disordered breathing | |||
| 4. Orthopnea | |||
| 5. Dyspnea | |||
| 6. Elevated biochemical markers | |||
| a. Serum creatine kinase | |||
| b. ALT | |||
| c. AST | |||
| Rheumatology | 12 observational studies (cohort studies/screening studies) | 1. Unexplained general/inflammatory myopathy | [12, 25–27, 36, 38, 39, 42, 49, 57–59] |
| 2. Asymptomatic hyperCKemia/Paucisymptomatic | |||
| 3. Unexplained limb–girdle weakness | |||
| 4. Polymyositis nonresponder to steroids | |||
| Internal medicine | 27 (5 RCTs; 23 observational studies [cohort studies/ screening studies]) | 1. Difficulty in climbing stairs | [12, 25–27, 29–31, 36, 38–44, 50–56, 60–64] |
| 2. Difficulty in standing up from a chair | |||
| 3. Difficulty in washing your hair | |||
| 4. Difficulty in placing an object at the level over the head | |||
| 5. Muscle cramps+exercise intolerance | |||
| 6. Excessive fatigue | |||
| 7. Difficulty in breathing at rest or effort | |||
| 8. Feeding/swallowing difficulties, jaw muscle fatigue | |||
| 9. High CK/elevated ALT and AST |
ALT: Alanine aminotransferase; AST: Aspartate aminotransferase; CK: Creatine kinase; MG: Myasthenia gravis; RCTs: Randomized-controlled trials.
Fig. 3
Proposed diagnostic algorithm for all specialists* except GPs/IMs/Peds /orthopedics.
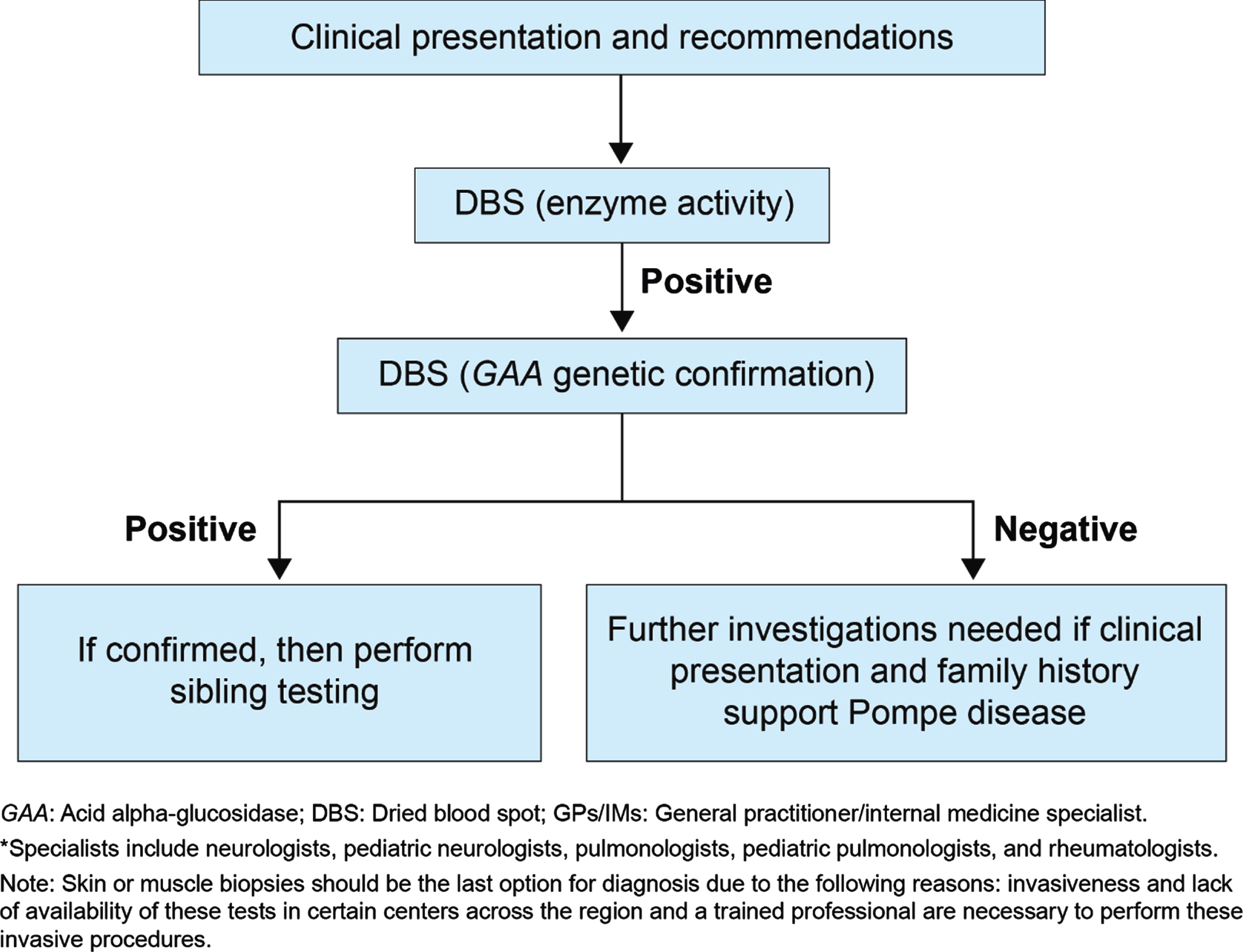
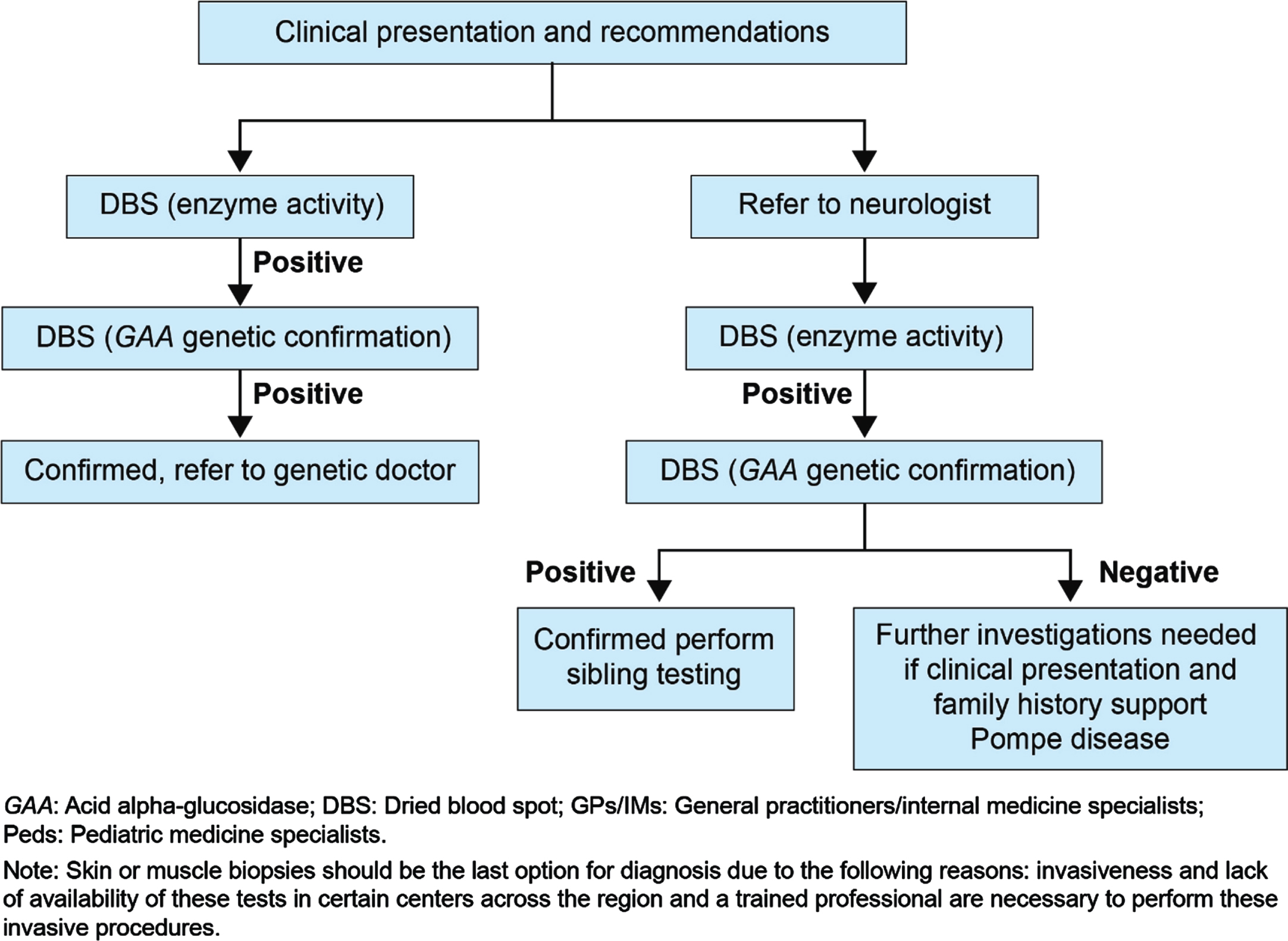
Fig. 4
Proposed diagnostic algorithm for GPs/IMs/ Peds/ orthopedics.
DISCUSSION
Late-onset Pompe disease is a progressive and debilitating disorder. Delays in diagnosis and treatment have negative consequences on a patient’s morbidity and mortality. Therefore, an appropriate diagnostic approach is essential for the early and accurate diagnosis of LOPD. Although international [14] and regional guidelines [9] are available for the diagnosis of LOPD, clinicians in the Arabian Peninsula region face numerous diagnostic challenges. The goal of this manuscript is to provide solutions to the diagnostic challenges of LOPD, thereby facilitating early and accurate diagnosis.
Late-onset Pompe disease is characterized by an insidious onset, a slowly progressive course, and clinical variability between patients, wherein symptoms may start at any stage of life, ranging from early childhood to adulthood [4, 5]. The majority of the symptoms of LOPD can mimic other medical conditions, thus creating the biggest challenge for clinicians from various specialties for accurate diagnosis of the disease. In addition, the clinical diagnostic clues of LOPD for various specialties play a crucial role in diagnosing LOPD. The current manuscript emphasizes that no symptom should be overlooked by clinicians as underdiagnosis and a delay in diagnosis may lead to fatal consequences and early referral to neurologists may enhance accurate diagnosis of LOPD. These specialty-specific diagnostic algorithms of pediatric and adult LOPD for field specialists, such as neurologists, rheumatologists, GPs/IMs, orthopedic specialists, and pulmonologists, based on clinical judgement of experts and summary of the evidence may facilitate the early and accurate diagnosis of the disease.
Consensus-based diagnostic approach for pediatric neurologists and neurologists
• Neurologists and pediatric neurologists should suspect LOPD in patients whose clinical manifestation is an ‘unexplained’ limb–girdle muscle weakness [31] as well as asymptomatic hyperCKemia [27].
• Scoliosis is prevalent in LOPD patients and should not be left unnoticed while suspecting pediatric and adult LOPD [35].
• Respiratory problems such as dyspnea and respiratory distress should not be overlooked as these are the most common symptoms of pediatric LOPD and 50% of pediatric LOPD patients require respiratory assistance [27].
• Patients with pediatric LOPD present with frequent falls [34], have difficulty in walking, and require wheelchair assistance [32]. Therefore, these symptoms should not be overlooked by pediatric neurologists while suspecting LOPD.
• Tongue abnormalities on brain MRI are common in adult LOPD patients compared to other neuromuscular disorders (NMDs). Particular attention to the tongue while reviewing brain MRIs may be a crucial clue for the diagnosis of muscle weakness in adult LOPD patients [45].
• Dolichoectasia of the basilar artery (BA) is the most striking cerebrovascular finding in adult LOPD patients as Pompe disease is associated with BA dilation, elongation, and elevated bifurcation height of the BA, resulting in cerebrovascular complications [46]. Therefore, neurologists should recommend a computer tomography angiography or a magnetic resonance angiography in adult LOPD patients for early detection of cerebrovascular malformations as these could lead to life-threatening events such as subarachnoid hemorrhage or brainstem compression [33].
Consensus-based diagnostic approach for pulmonologists and pediatric pulmonologists
Pulmonologists play a key role in diagnosing and managing Pompe patients as Pompe disease can heavily impact respiratory function.
• Pompe disease weakens the diaphragm muscles, including expiratory muscles, inspiratory muscles, and upper airway muscles. Diaphragm weakness initially manifests during sleep, leading to sleep-disordered breathing (SDB). Weakening of the diaphragm muscles leads to respiratory insufficiency, hypercapnia, shallow breathing, orthopnea, impaired cough, impaired speech, impaired swallowing, increased risk of atelectasis, and chest infections. Respiratory muscle weakness causes respiratory dysfunction [65].
• Approximately, 60% of LOPD patients have a mild reduction (< 80% predicted) and 30% –40% have a moderate reduction in vital capacity (< 60% predicted) [14], indicating the progressive loss of pulmonary function. While assessing the pulmonary function in infants, certain clinical factors, such as daytime energy level and the degree of fatigability (i.e. the ability to feed without developing increased work of breathing) should be additionally assessed as the evaluation of pulmonary function in infants is technically difficult [14].
• Weakness of the diaphragm can be an early and major finding in Pompe disease, and a reduction in vital capacity, sleep hypoventilation, and respiratory failure often develops following respiratory dysfunction. Pulmonologists should look for the aforementioned symptoms for the accurate diagnosis of LOPD.
Consensus-based diagnostic approach for rheumatologists
Untreated LOPD results in significant disability, including impairment in muscle structure and function; deterioration in mobility, respiration, and activities; and handicap in social participation (e.g. work, study, and recreation) [66]. The clinical clues that can help rheumatologists distinguish LOPD from autoimmune myopathy include [67]:
• Myotonic discharges on EMG.
• The diaphragm tends to be more severely involved than other skeletal muscles in Pompe; therefore, a low FVC should raise suspicion for Pompe disease.
• Muscle biopsies from patients with Pompe disease usually reveal periodic acid-Schiff (PAS)-positive vacuoles and do not have inflammatory infiltrates as seen in patients with autoimmune myopathy.
• Pompe patients do not respond to steroid treatment, which is the treatment strategy for inflammatory myopathy.
Consensus-based diagnostic approach for general practitioners/internal medicine specialists
• General practitioners/internal medicine specialists should not overlook respiratory symptoms because SDB is highly prevalent in pediatric and adult LOPD and may comprise alveolar hypoventilation or obstructive sleep apnea or both [53].
• Fatigue is a common and disabling problem in patients with early and advanced stages of Pompe disease [60].
• The follow-up of patients with LOPD should focus on respiratory and limb–girdle muscle function, the capacity to perform daily activities, and the presentation of fatigue and pain [62]. Overall, GPs/IMs should look for LOPD in patients whose clinical manifestation is an “unexplained” limb–girdle muscular weakness even without vacuolar myopathy in muscle biopsy, respiratory symptoms, hyperCKemia, and fatigue [53, 60, 62].
Consensus-based diagnostic approach for orthopedics
• Untreated pediatric LOPD results in significant disability, including impairment in muscle structure and function; deterioration in mobility, respiration, and activities; and handicap in social participation (e.g. work, study, and recreation) [66]. Therefore, muscle weakness and difficulty in walking should not be overlooked by orthopedics while suspecting pediatric LOPD.
• Scoliosis poses a significant burden in pediatric LOPD patients and thus the presence of this symptom in suspected pediatric LOPD patients should be considered an important sign of LOPD and require a confirmatory test for pediatric LOPD [35].
CONCLUSION
The heterogeneous clinical presentation, slow progression, and similarity of LOPD to other NMDs can create a diagnostic challenge for clinicians. Therefore, there is a need for increased awareness of LOPD among clinicians. In clinical practice, there appears to be inconsistency in the patterns of patient evaluation and a lack of standardized diagnostic testing, specifically in the Arabian Peninsula region. Although guidelines are available that aid in the diagnosis of LOPD, several challenges are faced by clinicians for the early and accurate diagnosis of LOPD. Moreover, the phenotype of LOPD is often difficult to distinguish from that of other genetic and acquired NMDs. The goal of this manuscript was to provide solutions to the diagnostic challenges of LOPD and to develop a consensus-based simplified screening algorithm for different specialties to facilitate timely and accurate diagnosis in all patients who may have LOPD. This consensus-based simplified screening algorithm, which is supported by evidence, will guide clinicians in the differential diagnosis of LOPD. It will also facilitate direct referral, which may assist in the early and accurate diagnosis of LOPD.
ACKNOWLEDGMENTS
BioQuest Solutions has provided the editorial support for the preparation of this review manuscript.
FUNDING
The funding for logistics and editorial support for the development of this consensus manuscript was provided by Sanofi Genzyme.
AUTHOR CONTRIBUTIONS
All authors reviewed, provided critical revisions, and approved the final manuscript.
POTENTIAL CONFLICTS OF INTEREST
The authors declare no conflict of interest.
DECLARATION OF INTERESTS
All the authors have received personal fees in the form of honoraria for attending two Sanofi Genzyme advisory board meetings, however, they were not paid for writing the manuscript.
REFERENCES
[1] | McCall AL , Salemi J , Bhanap P , Strickland LM , Elmallah MK , The impact of Pompe disease on smooth muscle: A review. J Smooth Muscle Res. (2018) ;54: :100–18. doi: https://doi.org/10.1540/jsmr.54.100. |
[2] | Taverna S , Cammarata G , Colomba P , Sciarrino S , Zizzo C , Francofonte D , et al., Pompe disease: Pathogenesis, molecular genetics and diagnosis. Aging (Albany NY). (2020) ;12: :15856–74. doi: https://doi.org/10.18632/aging.103794. |
[3] | Meena NK , Raben N , Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules. (2020) ;10: :1339. doi: https://doi.org/10.3390/biom10091339. |
[4] | Feeney EJ , Austin S , Chien YH , The value of muscle biopsies in Pompe disease: Identifying lipofuscin inclusions in juvenile- and adult-onset patients. Acta Neuropathol Commun. (2014) ;2: :2. doi: https://doi.org/10.1186/2051-5960-2-2. |
[5] | Toscano A , Rodolico C , Musumeci O , Multisystem late onset Pompe disease (LOPD): An update on clinical aspects. Ann Transl Med. (2019) ;7: :284. doi: https://doi.org/10.21037/atm.2019.07.24. |
[6] | Ausems MG , Verbiest J , Hermans MP , Kroos MA , Beemer FA , Wokke JH , et al., Frequency of glycogen storage disease type II in The Netherlands: Implications for diagnosis and genetic counselling. Eur J Hum Genet. (1999) ;7: :713–6. doi: https://doi.org/10.1038/sj.ejhg.5200367. |
[7] | Chien YH , Lee NC , Huang HJ , Thurberg BL , Tsai FJ , Hwu WL , Later-onset Pompe disease: Early detection and early treatment initiation enabled by newborn screening. J Pediatr.. (2011) ;158: :1023–7. e1021. doi: https://doi.org/10.1016/j.jpeds.2010.11.053. |
[8] | Mechtler TP , Stary S , Metz TF , Neonatal screening for lysosomal storage disorders: Feasibility and incidence from a nationwide study in Austria. Lancet. (2012) ;379: :335–41. doi: https://doi.org/10.1016/S0140-6736(11)61266-X. |
[9] | MENA Pompe Working Group Al Jasmi F , Al Jumah M , Diagnosis and treatment of late-onset Pompe disease in the Middle East and North Africa region: Consensus recommendations from an expert group. BMC Neurol. (2015) ;15: :205. doi: https://doi.org/10.1186/s12883-015-0412-3. |
[10] | Hamamy H , Antonarakis SE , Cavalli-Sforza LL . Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop Report. Genet Med. (2011) ;13: :841–7. doi: https://doi.org/10.1097/GIM.0b013e318217477f. |
[11] | el-Hazmi MA , al-Swailem AR , Warsy AS , al-Swailem AM , Sulaimani R , al-Meshari AA , Consanguinity among the Saudi Arabian population. J Med Genet. (1995) ;32: :623–6. doi: https://doi.org/10.1136/jmg.32.8.623. |
[12] | Tehrani KHN , Sakhaeyan E , Sakhaeyan E , Evaluation prevalence of Pompe disease in Iranian patients with myopathies of unknown etiology. Electron Physician.. (2017) ;9: :4886–9. doi: https://doi.org/10.19082/4886. |
[13] | Signs and Symptoms Checklist–Late-onset Pompe Disease. Available at: https://www.pompe.com/-/media/ems/conditions/rarediseases/brands/pompe-us/hcp/pdf/Signs%20&%20Symptoms LOPD%203.11.19.pdf. Accessed on:06 October 2021. |
[14] | Kishnani PS , Steiner RD , Bali D , Pompe disease diagnosis and management guideline [published correction appears in Genet Med Jun382. ACMG Work Group on Management of Pompe Disease [removed]; Case, Laura [corrected to Case, Laura E]]. . Genet Med. (2006) ;8: (5):267–88. doi:https://doi.org/10.1097/01.gim.0000218152.87434.f3. |
[15] | Chaudhuri A , Behan PO , Fatigue in neurological disorders. Lancet. (2004) ;363: :978–88. doi: https://doi.org/10.1016/S0140-6736(04)15794-2. |
[16] | Jaradeh S Muscle disorders affecting oral andpharyngeal swallowing. Goyal & Shaker GI Motility, 2006 Available at: http://www.nature.com/gimo/contents/pt1/full/gimo35.html. [Published May 16, 2006]. Accessed on: March 3, 2020. |
[17] | Féasson L , Camdessanche J-P , El Mandhi L , Calmels P , Millet GY , Fatigue et affections neuromusculaires. Ann Readopt Med Phys. (2006) ;49: :289–300. doi: https://doi.org/10.1016/j.annrm2006.04.015. |
[18] | Mah JK , Korngut L , Dykeman J , Day L , Pringsheim T , Jette N , A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscu Disord. (2014) ;24: :482–91. doi: https://doi.org/10.1016/j.nmd.2014.03.008. |
[19] | Barnabei MS , Martindale JM , Townsend D , Metzger JM , Exercise and Muscular Dystrophy: Implications and Analysis of Effects on Musculoskeletal and Cardiovascular Systems. Compr Physiol. (2011) ;1: :1353–63. doi: https://doi.org/10.1002/cphy.c100062. |
[20] | Gilchrist JM , Overview of Neuromuscular Disorders Affecting Respiratory Function. Semin Respir Grit Care Med. (2002) ;23: :191–200. doi: https://doi.org/10.1055/s-2002-33027https://doi.org/10.1055/s-2002-33027. |
[21] | Furst DE , Amato AA , Iorgo ŞR , Gajria K , Fernandes AW , Epidemology of adult idiopathic inflammatory myopathies in a US.. managed care plan. Muscle Nerve. (2012) ;45: :676–83. doi: https://doi.org/10.1002/mus.23302. |
[22] | Smoyer-Tomic KE , Amato AA , Fernandes AW , Incidence and prevalence of idiopathic inflammatory myopathies among commercially insured, Medicare supplemental insured, and Medicaid enrolled populations: An administrative claims analysis. BMC Musculoskelet Disord. (2012) ;13: :103. doi: https://doi.org/10.1186/1471-2474-13-103. |
[23] | Mastaglia FL , Phillips BA , Idiopathic inflammatory myopathies: Epidemiology, classification, and diagnostic criteria. Rheum Dis Clin North Am . (2002) ;28: :723–41. doi: https://doi.org/10.1016/s0889-857x(02)00021-2. |
[24] | Oh TH , Brumfield KA , Hoskin TL , Stolp KA , Murray JA , Basford JR , Clinical Characteristics, Treatment Strategies and Outcome in 62 Patients. Mayo Clin Proc. (2007) ;82: :441–7. doi: https://doi.org/10.4065/82.4.441. |
[25] | Musumeci O , Marino S , Granata F , Central nervous system involvement in late-onset Pompe disease: Clues from neuroimaging and neuropsychological analysis. Eur J Neurol. (2019) ;26: :442–e35. doi: https://doi.org/10.1111/ene.13835. |
[26] | Ünver O , Hacıfazlıoglu NE , Karatoprak E , The frequency of late-onset Pompe disease in pediatric patients with limb-girdle muscle weakness and nonspecific hyperCKemia: A multicenter study. Neuromuscul Disord. (2016) ;26: :796–800. doi: https://doi.org/10.1016/j.nmd.2016.09.001. |
[27] | Löscher WN , Huemer M , Stulnig TM , Pompe disease in Austria: Clinical, genetic and epidemiological aspects. J Neurol. (2018) ;265: :159–64. doi: https://doi.org/10.1007/s00415-017-8686-6. |
[28] | Almeida V , Conceicão I , Fineza I , Screening for Pompe disease in a Portuguese high risk population. Neuromuscul Disord. (2017) ;27: :777–81. doi: https://doi.org/10.1016/j.nmd.2017.03.010. |
[29] | Jastrzebska A , Potulska-Chromik A , Łusakowska A , Screening for late-onset Pompe disease in Poland. Acta Neurol Scand. (2019) ;140: :239–43. doi: https://doi.org/10.1111/ane.13133. |
[30] | Lee JH , Shin JH , Park HJ , Targeted population screening of late onset Pompe disease in unspecified myopathy patients for Korean population. Neuromuscul Disord. (2017) ;27: :550–6. doi: https://doi.org/10.1016/j.nmd.2017.03.005. |
[31] | Fukuhara Y , Fuji N , Yamazaki N , A molecular analysis of the GAA gene and clinical spectrum in 38 patients with Pompe disease in Japan. Mol Genet Metab Rep. (2017) ;14: :3–9. doi: https://doi.org/10.1016/j.ymgmr.2017.10.009. |
[32] | Jones HN , Crisp KD , Asrani P , Sloane R , Kishnani PS , Quantitative assessment of lingual strength in late-onset Pompe disease. Muscle Nerve. (2015) ;51: :731–5. doi: https://doi.org/10.1002/mus.24523. |
[33] | Montagnese F , Granata F , Musumeci O , Intracranial arterial abnormalities in patients with late onset Pompe disease (LOPD). J Inherit Metab Dis. (2016) ;39: :391–8. doi: https://doi.org/10.1007/s10545-015-9913-x. |
[34] | Bertoldo F , Zappini F , Brigo M Prevalence of asymptomatic vertebral fractures in late-onset Pompe disease. J Clin Endocrinol Metab. (2015) ;100: :401–6. doi: https://doi.org/10.1210/jc.2014-2763. |
[35] | Roberts M , Kishnani PS , van der Ploeg AT , The prevalence and impact of scoliosis in Pompe disease: Lessons learned from the Pompe Registry. Mol Genet Metab. (2011) ;104: :574–82. doi: https://doi.org/10.1016/j.ymgme.2011.08.011. |
[36] | Gaeta M , Barca E , Ruggeri P , Late-onset Pompe disease (LOPD): Correlations between respiratory muscles CT and MRI features and pulmonary function. Mol Genet Metab. (2013) ;110: :290–6. doi: https://doi.org/10.1016/j.ymgme.2013.06.023. |
[37] | Reyes-Leiva D , Alonso-Pérez J , Mayos M , Correlation Between Respiratory Accessory Muscles and Diaphragm Pillars MRI and Pulmonary Function Test in Late-Onset Pompe Disease Patients. Front Neurol. (2021) ;12: :6212–57. doi: https://doi.org/10.3389/fneur.2021.621257. |
[38] | Confalonieri M , Vitacca M , Scala R , Is early detection of late-onset Pompe disease a pneumologist’s affair? A lesson from an Italian screening study. Orphanet J Rare Dis. (2019) ;14: :62. doi: https://doi.org/10.1186/s13023-019-1037-1. |
[39] | Montagnese F , Barca E , Musumeci O , Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): Unusual features and response to treatment. J Neurol. (2015) ;262: :968–78. doi: https://doi.org/10.1007/s00415-015-7664-0. |
[40] | Ruggeri P , Lo Monaco L , Musumeci O , Ultrasound assessment of diaphragm function in patients with late-onset Pompe disease. Neurol Sci. (2020) ;41: (8):2175–84. doi: https://doi.org/10.1007/s10072-020-04316-6. |
[41] | Gaeta M , Musumeci O , Mondello S , Clinical and pathophysiological clues of respiratory dysfunction in late-onset Pompe disease: New insights from a comparative study by MRI and respiratory function assessment. Neuromuscul Disord. (2015) ;25: :852–8. doi: https://doi.org/10.1016/j.nmd.2015.09.003. |
[42] | Lukacs Z , Nieves Cobos P , Wenninger S , Prevalence of Pompe disease in 3,076 patients with hyperCKemia and limb-girdle muscular weakness. Neurology. (2016) ;87: :295–8. doi: https://doi.org/10.1212/WNL.0000000000002758. |
[43] | Lorenzoni PJ , Kay CSK , Higashi NS , D’Almeida V , Werneck LC , Scola RH , Late-onset Pompe disease: What is the prevalence of limb-girdle muscular weakness presentation? Arq Neuropsiquiatr. (2018) ;76: :247–51. doi: https://doi.org/10.1590/0004-282x20180018. |
[44] | Teodoro J , Silva M , Zimerman L Neuromuscular disorders. 2019:29 (Suppl 1) [Abstract only]. Available at: https://www.nmd-journal.com/article/S0960-8966(19)30483-3/fulltext. Accessed on: 5 July 2021. |
[45] | Karam C , Dimitrova D , Yutan E , Chahin N , Bright tongue sign in patients with late-onset Pompe disease. J Neurol. (2019) ;266: :2518–23. doi: https://doi.org/10.1007/s00415-019-09455-1. |
[46] | Hensel O , Schneider I , Wieprecht M , Kraya T , Zierz S , Decreased outlet angle of the superior cerebellar artery as indicator for dolichoectasia in late onset Pompe disease. Orphanet J Rare Dis. (2018) ;13: :57. doi: https://doi.org/10.1186/s13023-018-0794-6. |
[47] | Pichiecchio A , Sacco S , De Filippi P , Caverzasi E , Ravaglia S , Bastianello S , et al., Late-onset Pompe disease: A genetic-radiological correlation on cerebral vascular anomalies. J Neurol. (2017) ;264: :2110–8. doi: https://doi.org/10.1007/s00415-017-8601-1. |
[48] | Hensel O , Hanisch F , Stock K , Stoevesandt D , Deschauer M , Müller T , Morphology and function of cerebral arteries in adults with pompe disease. JIMD Rep. (2015) ;20: :27–33. doi: https://doi.org/10.1007/8904_2014_385. |
[49] | Pérez-López J , Selva-O’Callaghan A , Grau-Junyent JM , Delayed diagnosis of late-onset Pompe disease in patients with myopathies of unknown origin and/or hyperCKemia. Mol Genet Metab. (2015) ;114: :580-–3. doi: https://doi.org/10.1016/j.ymgme.2015.02.004. |
[50] | Wenninger S , Greckl E , Babacić H , Stahl K , Schoser B , Safety and efficacy of short- and long-term inspiratory muscle training in late-onset Pompe disease (LOPD): A pilot study. J Neurol. (2019) ;266: :133–47. doi: https://doi.org/10.1007/s00415-018-9112-4. |
[51] | Smith BK , Allen S , Mays S , Martin AD , Byrne BJ , Dynamic respiratory muscle function in late-onset Pompe disease. Sci Rep. (2019) ;9: :19006. doi: https://doi.org/10.1038/s41598-019-54314-8. |
[52] | Berger KI , Chan Y , Rom WN , Oppenheimer BW , Goldring RM , Progression from respiratory dysfunction to failure in late-onset Pompe disease. Neuromuscul Disord. (2016) ;26: :481–89. doi: https://doi.org/10.1016/j.nmd.2016.05.018. |
[53] | Boentert M , Dräger B , Glatz C , Young P , Sleep-Disordered Breathing and Effects of Noninvasive Ventilation in Patients with Late-Onset Pompe Disease. J Clin Sleep Med. (2016) ;12: :1623–32. doi: https://doi.org/10.5664/jcsm.6346. |
[54] | Wens SC , Ciet P , Perez-Rovira A , Lung MRI and impairment of diaphragmatic function in Pompe disease. BMC Pulm Med. (2015) ;15: :54. doi: https://doi.org/10.1186/s12890-015-0058-3. |
[55] | Guimarães MJ , Winck JC , Conde B , Prevalence of late-onset pompe disease in Portuguese patients with diaphragmatic paralysis - DIPPER study. Rev Port Pneumol. (2017) ;23: :208–15. doi: https://doi.org/10.1016/j.rppnen.2017.02.004. |
[56] | De Vito EL , Monteiro SG , Aruj PK , Blunted Hypercapnic Respiratory Drive Response in Subjects With Late-Onset Pompe Disease. Respir Care. (2016) ;61: :930–5. doi: https://doi.org/10.4187/respcare.03940. |
[57] | Figueroa-Bonaparte S , Segovia S , Llauger J , Muscle MRI Findings in Childhood/Adult Onset Pompe Disease Correlate with Muscle Function. PLoS One. (2016) ;11: :e4930163. doi: https://doi.org/10.1371/journal.pone.0163493. |
[58] | Gutiérrez-Rivas E , Bautista J , Vílchez JJ , Targeted screening for the detection of Pompe disease in patients with unclassified limb-girdle muscular dystrophy or asymptomatic hyperCKemia using dried blood: A Spanish cohort. Neuromuscul Disord. (2015) ;25: :548–53. doi: https://doi.org/10.1016/j.nmd.2015.04.008. |
[59] | Rairikar MV , Case LE , Bailey LA , Insight into the phenotype of infants with Pompe disease identified by newborn screening with the common c-32-13T > G “late-onset” GAA variant. Mol Genet Metab. (2017) ;122: :99–107. doi: https://doi.org/10.1016/j.ymgme.2017.09.008. |
[60] | Güngör D , de Vries JM , Brusse E , Enzyme replacement therapy and fatigue in adults with Pompe disease. Mol Genet Metab. (2013) ;109: :174–8. doi: https://doi.org/10.1016/j.ymgme.2013.03.016. |
[61] | Scheidegger O , Leupold D , Sauter R , Findling O , Rösler KM , Hundsberger T , 36-Months follow-up assessment after cessation and resuming of enzyme replacement therapy in late onset Pompe disease: Data from the Swiss Pompe Registry. J Neurol. (2018) ;265: :2783–8. doi: https://doi.org/10.1007/s00415-018-9065-7. |
[62] | Hagemans ML , Winkel LP , Van Doorn PA , Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain. (2005) ;128: :671–7. doi: https://doi.org/10.1093/brain/awh384. |
[63] | Hamed A , Curran C , Gwaltney C , DasMahapatra P , Mobility assessment using wearable technology in patients with late-onset Pompe disease. NPJ Digit Med. (2019) ;2: :70. doi: https://doi.org/10.1038/s41746-019-0143-8. |
[64] | Boentert M , Karabul N , Wenninger S , Sleep-related symptoms and sleep-disordered breathing in adult Pompe disease. Eur J Neurol. (2015) ;22: :369–e27. doi: https://doi.org/10.1111/ene.12582. |
[65] | Perrin C , Unterborn JN , Ambrosio CD , Hill NS , Pulmonary complications of chronic neuromuscular diseases and their management. Muscle Nerve. (2004) ;29: :5–27. doi: https://doi.org/10.1002/mus.10487. |
[66] | Hagemans ML , Laforêt P , Hop WJ , Impact of late-onset Pompe disease on participation in daily life activities: Evaluation of the Rotterdam Handicap Scale. Neuromuscul Disord. (2007) ;17: :537–43. doi: https://doi.org/10.1016/j.nmd.2007.03.006. |
[67] | Mammen AL . Which nonautoimmune myopathies are most frequently misdiagnosed as myositis? Curr Opin Rheumatol. (2017) ;29: (6):618–22. doi: https://doi.org/10.1097/BOR.0000000000000441. |


