
Anoctamin 5 Knockout Mouse Model Recapitulates LGMD2L Muscle Pathology and Offers Insight Into in vivo Functional Deficits
Abstract
Mutations in the Anoctamin 5 (Ano5) gene that result in the lack of expression or function of ANO5 protein, cause Limb Girdle Muscular Dystrophy (LGMD) 2L/R12, and Miyoshi Muscular Dystrophy (MMD3). However, the dystrophic phenotype observed in patient muscles is not uniformly recapitulated by ANO5 knockout in animal models of LGMD2L. Here we describe the generation of a mouse model of LGMD2L generated by targeted out-of-frame deletion of the Ano5 gene. This model shows progressive muscle loss, increased muscle weakness, and persistent bouts of myofiber regeneration without chronic muscle inflammation, which recapitulates the mild to moderate skeletal muscle dystrophy reported in the LGMD2L patients. We show that these features of ANO5 deficient muscle are not associated with a change in the calcium-activated sarcolemmal chloride channel activity or compromised in vivo regenerative myogenesis. Use of this mouse model allows conducting in vivo investigations into the functional role of ANO5 in muscle health and for preclinical therapeutic development for LGMD2L.
INTRODUCTION
Muscular dystrophies are a diverse group of inherited diseases that result in progressive loss of muscle structure and function, that leads to weakness and wasting of skeletal muscle. Among these, the Limb-girdle muscular dystrophies (LGMD) represent a group of myopathies where severely affected muscles include the hip and shoulder girdles, with subsequent involvement of other limb muscles. LGMD results in progressive muscle weakness from early childhood to late adulthood. Over two dozen genes responsible for LGMD have been identified, which lead to either recessive or dominant inheritance [1, 2]. LGMD2L/ LGMDR12 is a recessive disorder with a prevalence of 0.2–2 patients / 100,000 that is amongst the 5 most common LGMDs [3–5]. It is caused by mutations in the gene that encodes the Anoctamin 5 (ANO5) or the Transmembrane16E (TMEM16E) protein [6–9]. ANO5/TMEM16E protein belongs to a family of 10 related transmembrane proteins that function either as calcium-activated ion channels, lipid scramblases, or both [10, 11]. Among these, ANO1 (TMEM16A) and ANO2 (TMEM16B) encode calcium-activated chloride channels, while ANO6 (TEME16F) and ANO10 (TMEM16K) are phospholipid scramblases (PLS) [10–22]. ANO5 is the only member of this family that is associated with muscular dystrophy. This gene is expressed in bones, skeletal muscles, testes, and cardiac muscles [23–25]. Unlike the recessive Ano5 mutations, dominant mutations in Ano5 lead to the bone disorder, gnathodiaphyseal dysplasia 1 (GDD1) [25, 26]. While GDD1 is characterized by bone fragility and jawbone lesions, LGMD2L/R12 is characterized by increased serum level of muscle enzyme Creatine Kinase, myofiber damage, sporadic rhabdomyolysis, exercise-induced myalgia, proximal limb muscle pain and weakness, and difficulty walking and standing on toes [6, 8]. Many of these clinical features are shared with other muscular dystrophies such as LGMD2B/R2, where mutations reduce or prevent expression of the membrane protein dysferlin, leading to increased myofiber death and muscle degeneration [27–29].
Endogenous ANO5 protein localizes to the Sarco/Endoplasmic Reticulum (SER) membrane, but exogenously expressed ANO5 is detected at the plasma membrane where it can exhibit calcium-activated scramblase as well as ion channel activity [21, 25, 30–34]. We recently identified the requirement of endogenously expressed ANO5 for calcium-activated calcium uptake by the SER during cellular calcium overload [24, 35]. The ion channel and lipid scramblase activities of ANO5 have been implicated in sarcolemmal repair, myoblast fusion during muscle regeneration, and mouse sperm motility [24, 34–39]. Further, biochemical studies of ANO5 and targeted GDD1 and LGMD2L patient mutations suggest that while the GDD1 associated mutations result in gain of ANO5 function, LGMD2L/R12 mutations are associated with the loss of ANO5 activity [30]. This view is supported by the observation that patient cells lacking detectable ANO5 protein exhibit poor membrane repair [24, 35], indicating that Ano5 knockout would be a suitable animal model for LGMD2L/R12.
Knockout animal models targeting different regions of Ano5 gene have been generated previously. While deletion of the first two exons of Ano5 results in no detectable muscle deficits [23, 39], Ano5 disruption in mouse by insertional deletion of exons 8–9 results in notable muscle pathology [37], and deletion of exons 11–12 leads to bone weakness [40]. Deletion of exons 12–13, with consequent disruption of the Ano5 reading frame in rabbits faithfully recapitulates the dystrophic muscle features [41]. With these diverse outcomes identified from Ano5 knockout animal models, here we describe a ANO5 knockout mouse model to investigate the ANO5 function in muscular dystrophy. Our work builds on two previous findings – symptomatic animal models involve disruption of Ano5 gene in the region spanning exons 8–12, and cells lacking ANO5 protein exhibit ion homeostasis and sarcolemmal repair deficit [30, 35]. With ANO5 function linked to muscle cell membrane repair, in vitro myoblast fusion, and plasma membrane ion channel activity [24, 30–39], we have examined these activities in vivo and assessed their impact on muscle pathology in our model. Our findings establish a new mouse model of LGMD2L and the characterization we present here offers insights into the in vivo relevance of ANO5 function for muscle pathology in LGMD2L.
METHODS
Animals and knockout mouse generation
All animal procedures were conducted in accordance with guidelines for the care and use of laboratory animals and were approved by the Children’s National Research Institute Animal Care and Use Committee (#00030709), the local animal ethics committee of University Lyon 1 and Ethical Committee for Animal Experimentation C2EA-51 of Evry (#APAFIS#01304.01). C57BL/6J (WT) mice were obtained from the Jackson Laboratory (Bar Harbor, ME) and maintained in our animal facility for the purpose of this study. All animals were maintained in an individually vented cage system under a controlled 12 h light/dark cycle with free access to food and water and animals of both genders were used for experiments.
Construction of the targeting vector and generation of the ANO5 knockout mouse was performed by Genoway (Lyon). A bacterial artificial chromosome (BAC) library was screened using Ano5 primers allowing the identification of 3 clones covering the genomic region around exons 11 to 13 of the Ano5 gene. These BAC were used to construct the targeting vector, which was electroporated into ES cells. After selection and analysis of the homologous recombination events, two positive ES clones were selected and then injected into C57BL/6J blastocysts that were reimplanted into foster mothers to generate chimeric mice. Five highly chimeric males were obtained and bred first with the deleter mice, constitutively expressing the Flp recombinase for deletion of the neomycin selection cassette. Resulting animals were mated with mice transgenic for CMV-CRE, which permits the excision of the floxed Ano5 segment. The Cre transgene was segregated by a first cross on C57BL/6 background and the resulting heterozygous mice were backcrossed for 10 generations on the C57Bl/6 and then interbred. For genotyping, genomic DNA from mouse tail was extracted and amplified using KAPA2G Fast HotStart Genotyping Mix, (Sigma, St. Louis, MO, USA) with the following: 49683cre-IRII.F: attcctgagaatatgtgtaattgtggcagc 49698flp-IRII.R: 5′-ccctagaactacataatcttggtgtggtggtag -3′. A PCR fragment of 2,68 kb is generated for the WT allele and of 890 bp for the mutant allele.
In vivo muscle injury, bromodeoxyuridine (BrdU) labelling, and immunostaining
Muscles were injured by local injection of notexin in 10-month-old animals under isofluorane anaesthesia (42). Following removal of fur from the anterior hindlimb, 40 μl notexin (5 μg/ml, Latoxan, #L8104) was delivered by intramuscular injection into the tibialis anterior (TA) using a 0.3 ml ultrafine insulin syringe (BD Biosciences, #324906). Immediately prior to injection, the needle was dipped in green tattoo dye (Harvard Apparatus, #72-9384) to mark the needle track. For the first 7 days post injury, BrdU (Sigma-Aldrich, B9285) was administered ad libitum in sterile drinking water at a concentration of 0.8 mg/ml. Animals were euthanized either 7- or 14-days post-injury, and tissues were harvested for analysis [43, 44].
Skeletal muscles were dissected out and frozen in isopentane cooled in liquid nitrogen. Transverse cryosections (8-μm thickness) were prepared from frozen muscles and were processed for hematoxylin and eosin (H&E) and Laminin staining. Frozen sections were cut and fixed in ice-cold acetone for 10 min, followed by incubation in 2 N HCl at 37°C for 30 min, and then briefly neutralized with 0.15 M sodium tetraborate (Sigma-Aldrich, MO). Following this, sections were blocked for 1 h in phosphate-buffered saline (PBS) supplemented with 20% goat serum (GeneTex, CA), 0.1% tween-20 (Sigma-Aldrich, MO), and 10 mg/ml BSA (Sigma-Aldrich, MO). Primary antibodies against BrdU (B35138, 1 : 100, Life Technologies, CA) and laminin (L9393, 1 : 400, Sigma-Aldrich, MO) were incubated overnight at 4°C. Sections were then washed and probed with the appropriate Alexa Fluor secondary antibody (Life Technologies, MA) at a dilution of 1 : 500 for 1 h at room temperature. Prior to mounting, nuclei were counterstained with propidium iodide (P4170, 2.5 μg/ml, Sigma-Aldrich, MO). Digital images were captured with a VS120 virtual slide microscope, and images were processed and quantified using CellSens and ImageJ software.
Muscle force measurements
Forelimb and hindlimb grip-strength measurement (GSM) were carried out using a grip strength meter (Columbus Instruments, Columbus, OH, USA) as previously described [45]. The animals were acclimatized for 3 days before actual data collection. The forelimb and hindlimb grip-strength data were then collected over 5 consecutive days. Data were represented as averaged grip strength/kg body weight over 5 days.
To measure in vivo torque production of the anterior crural muscles (TA, extensor digitorum longus (EDL), peroneus tertius, and extensor hallucis longus), mice were anesthetized with 1.5% isoflurane-mixed O2 and hair was removed from the lower hind limbs, while the foot was attached to the dual-mode lever and maintained at a 90° angle for isometric torque assessment (Aurora Scientific, Aurora, Canada). Isometric muscle contractions were stimulated at 1.0–2.0 mA using Pt-Ir needle electrodes inserted percutaneously adjacent to the peroneal nerve. Peak isometric torque was measured in response to tetanic stimulations at 20, 40, 60, 80, 100, 120, 140, 160, 180, and 200 Hz, providing a 60s rest period between stimuli. The rate of rise in torque was modeled using the exponential equation T = C(1-e-Df), where T = torque produced at the given frequency (f), C = maximal torque, and D = the rate of rise in torque [46]. Here, we tested 10-month-old, male WT and ANO5-/- mice (n = 5).
Myofiber isolation and electrophysiology
Mice were euthanized by cervical dislocation followed by removal of flexor digitorum brevis (FDB) muscles. Single fibers were isolated by a 50-minute enzymatic treatment at 37° C using a Tyrode solution containing 2 mg/mL collagenase type I (Sigma). Fibers were voltage-clamped using the silicone clamp technique as previously described [47]. Briefly, a major part of a single fiber was electrically insulated with silicone grease and a micropipette was inserted into the fiber through the silicone layer to voltage clamp the portion of the fiber free of grease (50 to 150 μm length) using a patch-clamp amplifier (Bio-Logic RK-400, Claix, France) in whole-cell configuration. Analog compensation was systematically used to decrease the effective series resistance. The tip of the micropipette was then crushed into the dish bottom to allow intracellular dialysis of the fiber with the intra-pipette solution. Cell capacitance was determined by integration of a current trace obtained with a 10-mV hyperpolarizing pulse from the holding potential and was used to calculate the density of currents (A/F). Currents were acquired at a sampling frequency of 10 kHz. Data are given as means±S.E.M.
The external solution contained (in mM) 140 TEA-MeSO3 (9 mM Cl- containing solution) or 140 TEA-Cl (149 mM Cl- containing solution), 2.5 CaCl2, 2 MgCl2, 0.002 tetrodotoxin, 1 4-aminopyridine and 10 HEPES adjusted to pH 7.2 with TEA-OH. The internal dialyzed solution contained (in mM) 140 K-glutamate, 2 EGTA, 5 Na2-ATP, 5 Na2-phosphocreatine, 5 MgCl2, 5 glucose and 10 HEPES adjusted to pH 7.2 with K-OH. The 2 mM internal [EGTA] prevented deterioration of the muscle fiber in response to large depolarizing pulses but preserved fiber contraction upon suprathreshold depolarizations. Fibers were dialyzed with the intracellular solution through the micropipette during 10 min prior starting the experiments.
RNA isolation and quantitative RT-PCR
RNA was extracted by the Trizol method from muscles previously sampled and frozen in liquid nitrogen. Residual DNA was removed from the samples using Free DNA kit or Turbo DNA-free Kit (Ambion). One μg of RNA was reverse transcribed using the SuperScript II first strand synthesis kit (Invitrogen) or revertAid H Minus First Strand cDNA Synthesis kit (ThermoFisher) and random hexamers. Real-time PCR was performed using LightCycler480 29437 (Roche) Taqman Gene Expression or miR Assays (ThermoFisher) or 0.2 μM of each primer and 0.1 μM of the probe according to the protocol Absolute QPCR Rox Mix (ThermoFisher).
Endogenous gene expression was quantified using Taqman Gene Expression Assay: ANO5: Mm00624629_m1; ANO6: Mm00614693_m1; ANO8: Mm01343244_m1, MYMK Mm00481256_m1, CD11b Mm00434455_m1, MYH3 Mm01332463_m1, MYH8 Mm01329494_m1, CD3G Mm00438095_m1, TIMP-1 Mm0131 Mm01341361_m1, IL1β Mm00434228_m1, IL6 Mm00446190_m1, PLIN5 Mm00508852_m1, and COL6A3 Mm00711678_m1. The ubiquitous acidic ribosomal phosphoprotein (P0) was used to normalize the data across samples. The primer pairs and Taqman probe used for P0 amplification were: m181PO.F (5′-CTCCAAGCAGATGCAGCAGA-3′), m267PO.R (5′-ACCATGATGCGCAAGGCCAT-3′), m225PO.P (5′-CCGTGGTGCTGATGGGCAAGAA-3′) and each experiment was separately replicated.
Expression of miRNA were performed using TaqMan Assays miRNA: miR-21 (hsa-miR-21-5p) ref: 000397, miR-142 (hsa-miR142-3p) ref: 000464, miR-31 (mmu-miR-31-5p) ref: 000185, miR 1(hsa-miR1-3p) ref: 000385, miR-29a (hsa-miR29a-3p) ref: 002112, and normalized using the expression of U6 (U6 snRNA) ref: 001973. Fold change in RNA expression (Fc) in tissues from ANO5-KO mice was calculated using the traditional 2∧(–ΔΔCt) method: Fc = 2∧(–(ΔCt – Avg ΔCt WT)), allowing comparison of Ct value with that obtained from tissue of WT animals. Evaluation of consequences at RNA level of the mutation in the model was performed by RT-PCR on muscle extracts with the following primers (Ex6.F: GAAGACGAGAGTTTGAACAAAATCTCAGAAAAACAG, Ex14.R: CAAAGTACCATGGGATGCGATGGC). The PCR generated fragments of 1080 bp in WT and 778 bp in ANO5–/–.
Statistical analysis
The statistical analysis was carried out using the GraphPad 8.0 Prism Software, where the data were examined by pairwise testing by Mann–Whitney U test or by Analysis of Variance (ANOVA). Outcome of the statistical test is represented in the figures by way of p values as indicated in figure legends. Each plot shows the individual data point with the average representing median value, unless noted otherwise.
RESULTS
Generation of ANO5 knockout mouse model for LGMD2L
To generate the mouse model of LGMD2L/R12, we synthesized a mouse Ano5 targeting vector composed of a long homology arm of 5.7 kb and a short arm of 1.9 kb on each side of a region encompassing exons 11–13 flanked by LoxP sites. Positive selection by neomycin gene flanking by FRT sequences was also added in the vector. Through homologous recombination using this vector we disrupted the predicted transmembrane domain of the mouse ANO5 protein, by out of frame deletion of exons 10 to 12 (Fig. 1A). In the resulting chimeric animals, the neo gene and rest of the insertional cassette was excised by crossing with Flp and CRE recombinase under the control of the ubiquitous CMV promoter. Ano5 deficient mice were generated by a targeted 1793 bp deletion in the Ano5 genomic loci, which was confirmed by PCR genotyping (Fig. 1B). Absence of the 301 bp spanning exons 10–12 in the resulting mRNA transcript was confirmed by RT-PCR analysis and RNA sequencing (Fig. 1C–E). Congenic ANO5 deficient mice were backcrossed onto the C57Bl/6 genetic background for 10 generations with subsequent interbreeding. Homozygous Ano5-/- mice are viable and fertile with no gross abnormalities or increased mortality up to 1 year of age.
Fig. 1
Generation and genetic characterization of ANO5–/– mouse model. (A) Schematic showing the genetic modifications used to disrupt the mouse Ano5 gene in the ANO5–/– mouse model. WT gene and the homologous recombination of the genome that led to the ANO5–/– mice. (B) PCR analysis of the genomic region containing the deleted exonic regions shown in panel A. (C) Schematic of mRNAs resulting from WT and Ano5-/- allele. Arrows indicate the region around which primers are designed for PCR amplification and sequencing. (D) Gel image showing PCR amplified product of the marked region of Ano5 gene in panel C from mRNAs isolated from WT and ANO5–/– mice. (E) Chromatogram showing the sequence of disrupted Ano5 allele in the ANO5–/– mouse. Plots showing qRT-PCR quantification of (F) Ano5, (G) Ano6, and (H) Ano8, in 9-months-old male mouse muscles (quadriceps, LA (EDL+TA), gastrocnemius). Each dot on the plot represents an individual muscle and the black bar indicates median of these values. p values are measured by unpaired Mann-Whitney t test and indicated by ****p < 0.0001.
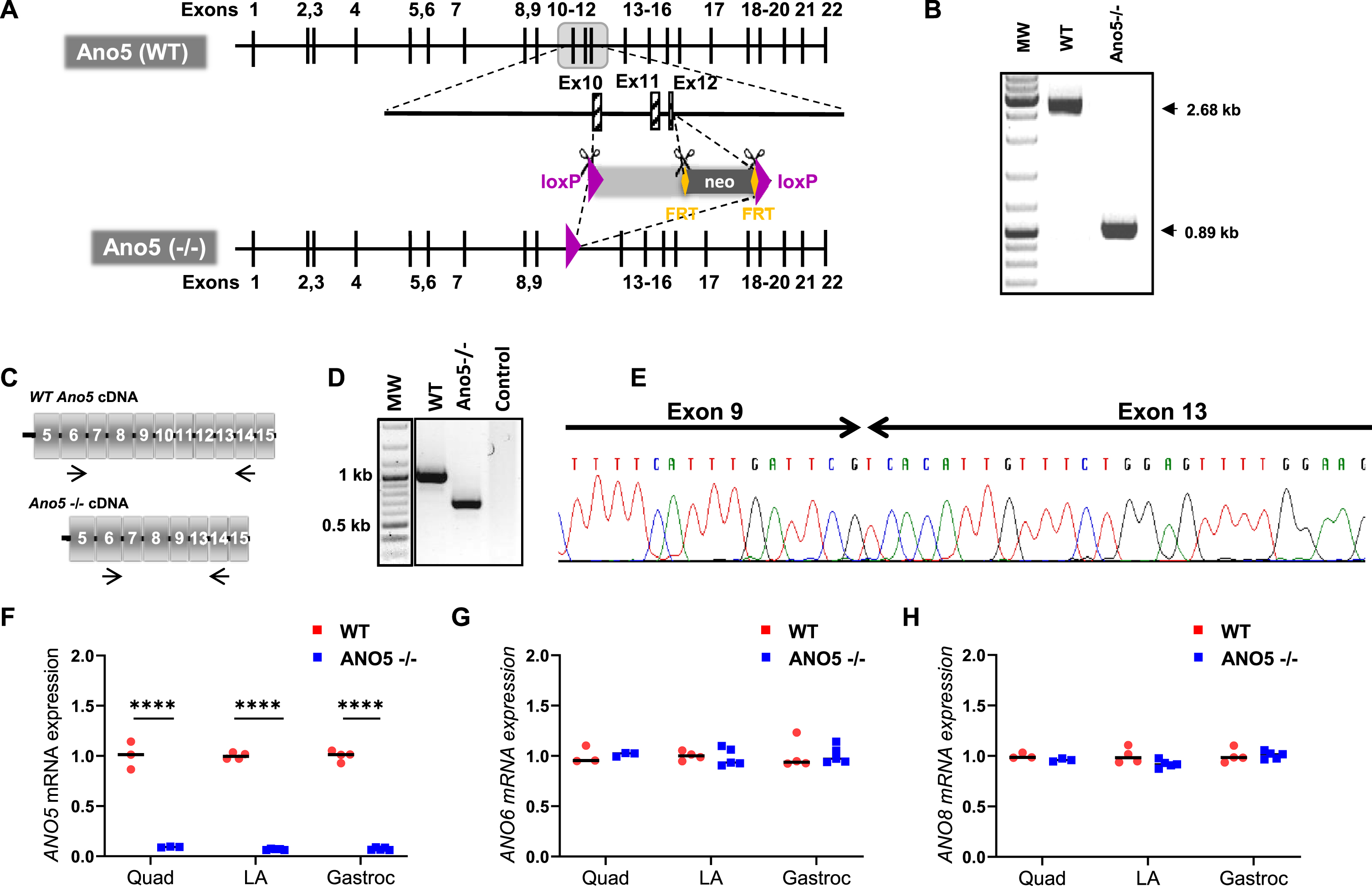
To assess the consequences of the targeted knockout of Ano5, we quantified Ano5 transcript levels in skeletal muscle by quantitative RT-PCR (qRT-PCR) analysis. This revealed low-levels of Ano5 transcript (< 10 % of WT level) in various muscles (Fig. 1F). In view of the role of ANO5 protein in regulating plasma membrane (PM) and Sarco/Endoplasmic Reticulum (SER) function [24, 48, 49], we used qRT-PCR analysis to assess the expression of an anoctamin localized to the PM (ANO6), and one localized to the SR/PM (ANO8). Neither of these transcripts were found to be altered in their expression in the ANO5–/– muscles, suggesting no compensatory change in the levels of these anoctamins in ANO5 deficient skeletal muscles (Fig. 1G, H).
Characterization of ANO5 deficient muscle
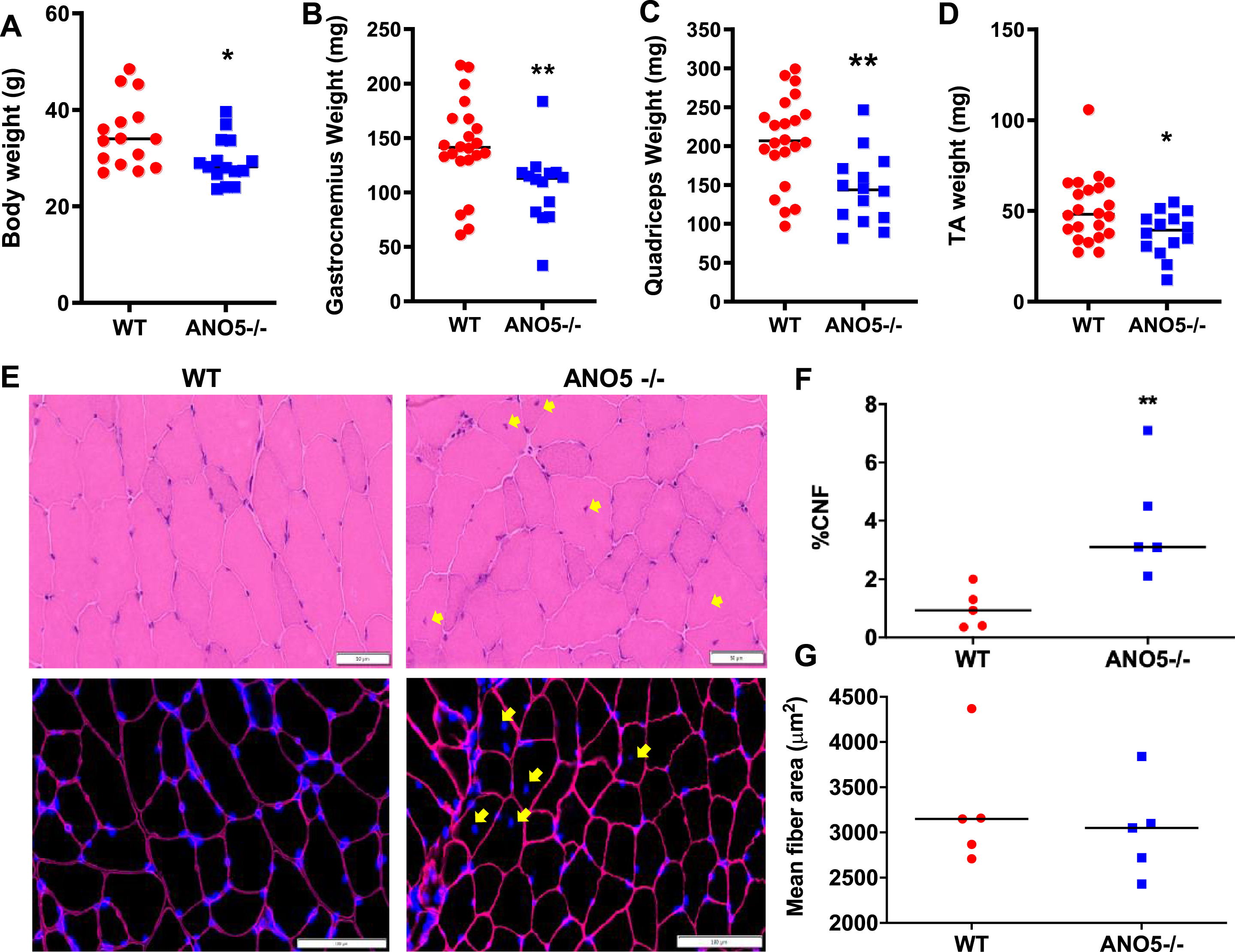
Mutations leading to loss of ANO5 protein in patients result in damage, weakness, and wasting of muscle starting from late adulthood to middle age [8, 9, 50]. Given the relative age match of middle-aged human with 9–10 month old mice, we assessed mice at this age [51]. Analysis of the body and muscle weight in ANO5–/– mice compared to WT mice showed a significant drop in both body weight and weights of multiple muscles including quadriceps, gastrocnemius, and TA (Fig. 2A–D). In view of the muscle loss induced by ANO5 deficit, we next examined if this is associated with changes in muscle histology. Cross sections of quadriceps were stained with H&E and independently immunostained to mark the basement membrane (laminin) and nuclei (DAPI) (Fig. 2E). These analyses identified the presence of a significant increase in the number of centrally nucleated myofibers in the ANO5–/– muscles when compared to WT, but there were no signs of overt muscle inflammation (Fig. 2E, F). Further, the increase in regenerated (centrally nucleated) fibers occurred without any corresponding decrease in myofiber cross-sectional area of ANO5–/– muscles (Fig. 2E, G). Taken together, the data suggest a lack of myofiber atrophy and/or a high rate of myofiber turnover, which would result in accumulation of small caliber regenerated myofibers. As an independent assessment of inflammation and extracellular matrix (ECM) remodeling, we performed qRT-PCR analysis to assess the expression of different regulators of inflammation, including CD3G, interleukin 1β, interleukin 6, CD11b, and miR-142. We observed no indication for altered inflammation in the Psoas (Fig. S1A) and other ANO5–/– muscles examined (Fig. S1B). Similar analysis of the expression of ECM modulating genes – TIMP-1, Perilipin, collagen, and micro RNAs – miR-21, miR-29a, showed lack of ANO5 did not detectably alter the genes responsible for ECM remodeling in the Psoas (Figure S1C) and other muscles we examined (Figure S1D) in ANO5–/– mice.
Fig. 2
Effect of ANO5 deficit on muscle size and histopathology. Plots showing (A) body weight and weights of (B) Gastrocnemius, (C) Quadriceps, and (D) TA muscles. Each dot represents an individual mouse/muscle. Images showing cross sections of quadriceps muscle (E) stained with H&E (top) and for nuclei (DAPI) and basement membrane (Laminin immunostain) (bottom). Yellow arrows mark the centrally nucleated fiber (CNF) and these were quantified to measure (F) proportion of CNFs and (G) myofiber cross-sectional areas. Each dot represents value averaged from multiple cross sections per muscle, black line represents the median value of the distribution. Scale bars are 50 μm (top) and 100 μm (bottom). p values are measured by unpaired Mann-Whitney t test and indicated by *p < 0.05; **p < 0.01; A–D (n > 15), F, G (n = 5).
Next, we examined the expression of multiple myogenic regulators to assess the extent of ongoing regenerative myogenesis. The expression of the myogenic indicators – embryonic myosin heavy chain (MYH8), myomaker (MYMK), embryonic myosin (MYH3), miR-01, and miR-31 were unaltered in the ANO5-deficient muscles (Figure S2A-C). Independently, to examine if the muscle of the 10-months old ANO5–/– mice undergo spontaneous myofiber damage and regeneration in vivo, we labeled spontaneously regenerating myonuclei over a 1-week period to mark all nascent myonuclei produced during this period with the nucleotide analogue BrdU delivered through the drinking water [43, 44]. As can be expected, WT mice showed no spontaneous BrdU-labeled myonuclei over this period, and we found the same is true in case of the ANO5–/– mice (Figure S2D). Thus, quantification of myogenic gene expression and spontaneous in vivo regenerative myogenesis in ANO5–/– mice showed a low-level spontaneous myofiber regeneration, without chronic inflammation. This is unlike the severe muscular dystrophies that are associated with extensive muscle regeneration, chronic inflammation, and excessive ECM remodeling [43, 44].
Effect of ANO5 deficit on muscle strength and sarcolemmal ion channel activity
With some of the previous ANO5 null models having reported underwhelming muscle histopathology and weakness [23, 39], we next examined muscle functional deficits in our ANO5–/– mouse model. For this we measured force production by grip strength analysis of the forelimb and hindlimb muscles of 10-month-old ANO5–/– mice. Similar to the reduced muscle strength noted in LGMD2L patients, we found ANO5–/– mice demonstrated reduced grip strength of both the forelimb (by 4.5 KgF/Kg) and hindlimb (by 8.5 KgF/Kg), in comparison to WT controls (Fig. 3A, B). To further characterize the muscle force deficits in our ANO5–/– model, we evaluated in vivo muscle torque generated in response to increasing tetanic stimulations of the anterior crural muscles. Here we elicited isometric contractions by subcutaneous stimulation of the peroneal nerve across a range of frequencies from 20–200 Hz to generate a force-frequency plot. The muscles of ANO5–/– mice generated contractile force similar to the WT mice at stimulation frequencies below 80 Hz, but at tetanic stimulation frequencies (> 100 Hz) contractile force of the ANO5–/– muscle was reduced (1.2 mN-m) as compared to the WT muscle (1.6 mN-m) (Fig. 3C). These results independently demonstrate greater weakness of ANO5–/– limb muscle and reduced contractile force of these muscle during tetanic stimulation.
Fig. 3
Muscle strength and chloride currents in ANO5 deficient muscle. Plots for (A) hindlimb and (B) forelimb grip strength of the mice (each dot representing individual animal, black line represents the median value of the distribution.). (C) Plot showing the force-frequency relationship for the TA of mice (n = 5; mean±SD). Difference between genotypes along the frequencies was significant beyond 80Hz (Two-way ANOVA). (D) Cl- currents were recorded in the same wild type (upper traces) and ANO5–/– myofibers (middle traces) in the presence of either 149 mM or 9 mM external Cl-, in response to the voltage protocol shown in the lower traces. Voltage pulses were delivered every 5 s. (E) Relationships between the voltage and the mean end-pulse amplitude of the current measured in the presence of 149 mM Cl- and of the current difference (current in 149 mM Cl- minus current in 9 mM Cl-) in 12 fibers from wild type and in 13 fibers from two ANO5-KO mice. p values are measured by unpaired Mann-Whitney t test (A, B) or 2-way ANOVA (C) and indicated by **p < 0.01, n≥5.
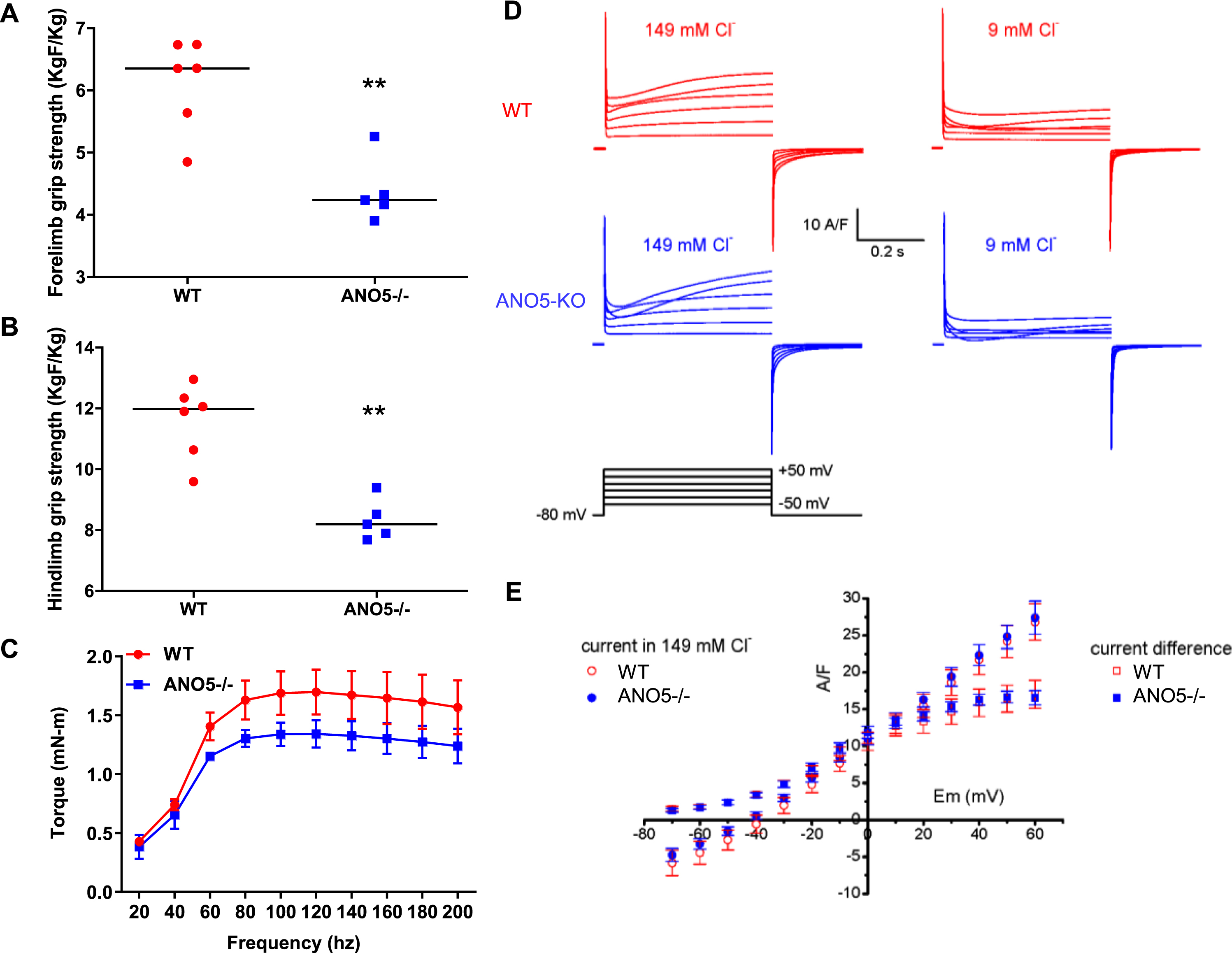
ANO5 protein has been suggested to operate as a plasma membrane ion channel that can be activated by a rise in intracellular Ca2+[31, 34, 52]. Thus, we examined if weakness of ANO5–/– muscle is related to altered anion channel activity in the myofiber sarcolemma. For this we recorded plasma membrane currents elicited by 500 ms-duration depolarizing voltage pulses in isolated muscle fibers from WT and ANO5–/– mice in the presence of an external solution containing 149 mM or 9 mM Cl- and blockers of voltage-gated Na+ and K+ channels (Fig. 3D). Depolarizations of increasing amplitudes in the presence of 149 mM Cl- elicited currents displaying an early phase during which L-type voltage-gated Ca2+ currents activated, followed by a late phase during which voltage-gated Ca2+ currents inactivated and positive currents developed. These late phase positive currents were strongly reduced in the presence of 9 mM Cl- in wild type and in ANO5-KO fibers indicating that the positive current recorded in the presence of the 149 mM Cl- solution was mostly carried by Cl- ions. In each fiber, the remaining current recorded in the presence of 9 mM Cl- was subtracted from the current recorded in the presence of 149 mM Cl- to extract the Cl- current. The amplitude of these Cl- current differences, and of the currents recorded in the presence of 149 mM Cl- were measured at the end of voltage pulses in each fiber and plotted as a function of voltage. The relationships between mean current amplitudes and voltages obtained in ANO5–/– myofibers were indistinguishable from the WT myofibers (Fig. 3E). It is also noteworthy that all fibers we tested contracted in response to voltage pulses given above –30 mV, allowing us to exclude the possibility that ANO5 did not activate due to absence of intracellular Ca2+ rise. Lack of detectable difference in depolarization evoked Cl- currents on myofiber sarcolemma between WT and ANO5–/– myofibers indicates that ANO5 does not function as a sarcolemmal Cl- channel in muscle fibers and that weakness of ANO5–/– myofibers cannot be attributed to altered sarcolemmal Cl- channel activity.
In vivo role of ANO5 on muscle regeneration
The ability to regulate myoblast fusion is another role attributed to ANO5 [34, 37]. In previous analysis of ANO5 deficient patient myoblasts we did not observe a myogenic deficit in vitro [35]. With the availability of the ANO5–/– model, we next examined the role of ANO5 in regenerative myogenesis in vivo. For this we used BrdU-labeling of activated myogenic cells to monitor spontaneous regenerative myogenesis [43, 44]. We used this approach in combination with notexin-based, sterile injury to investigate myogenic cell fusion after synchronized muscle damage [42, 53]. Here, the quantification of BrdU-labeled central nuclei in recently regenerated myofibers provides a readout of satellite cell activation and myogenic cell fusion in response to in vivo muscle injury. Following injury, BrdU was administered to the WT and the ANO5–/– mice for 7- or 14-days and the muscle cross-sections were scored for presence of BrdU stained central-myonuclei to identify the newly regenerated myofibers, while all nuclei were stained with propidium iodide and myofiber boundary was marked with laminin staining (Fig. 4A). Both WT and ANO5–/– muscle showed abundant BrdU labeled myofibers at 7-days and at 14-days post injury (Fig. 4A). Quantification of the number of BrdU-labeled myofibers identified no difference between the WT and ANO5–/– muscles at either 7-days or 14-days post injury (Fig. 4B). This indicated no detectable deficit in regenerative myogenesis on account of in vivo myoblast fusion deficit in ANO5–/– mouse muscle. Previous studies identified that the size of the newly regenerated myofibers was reduced at 30 days or 90 days post myotoxin
Fig. 4
Analysis of regenerative myogenesis in vivo. (A) Images of NTX-injured TA muscle cross-sections stained for regenerated myonuclei (Brdu), nuclei (propidium iodide – PI) and basement membrane (Laminin) from WT and ANO5–/– mice at 7 days (upper panel) or 14 days (lower panel) post single bout of injury. (B) Plot showing number of myofibers in individual muscle cross section that contained BrdU-labeled myonuclei (C, D) Plot showing mean fiber cross-sectional area for (C) myofibers containing BrdU-labeled nuclei. (D) all myofibers in the muscle cross-section. Scale bar - 100 μm. Data represents mean±SD with each dot representing value from whole muscle cross-section from individual mouse muscle. p values are measured by two-way ANOVA with Tukey’s multiple comparisons test and indicated by **p < 0.01, ***p < 0.001, n = 4.
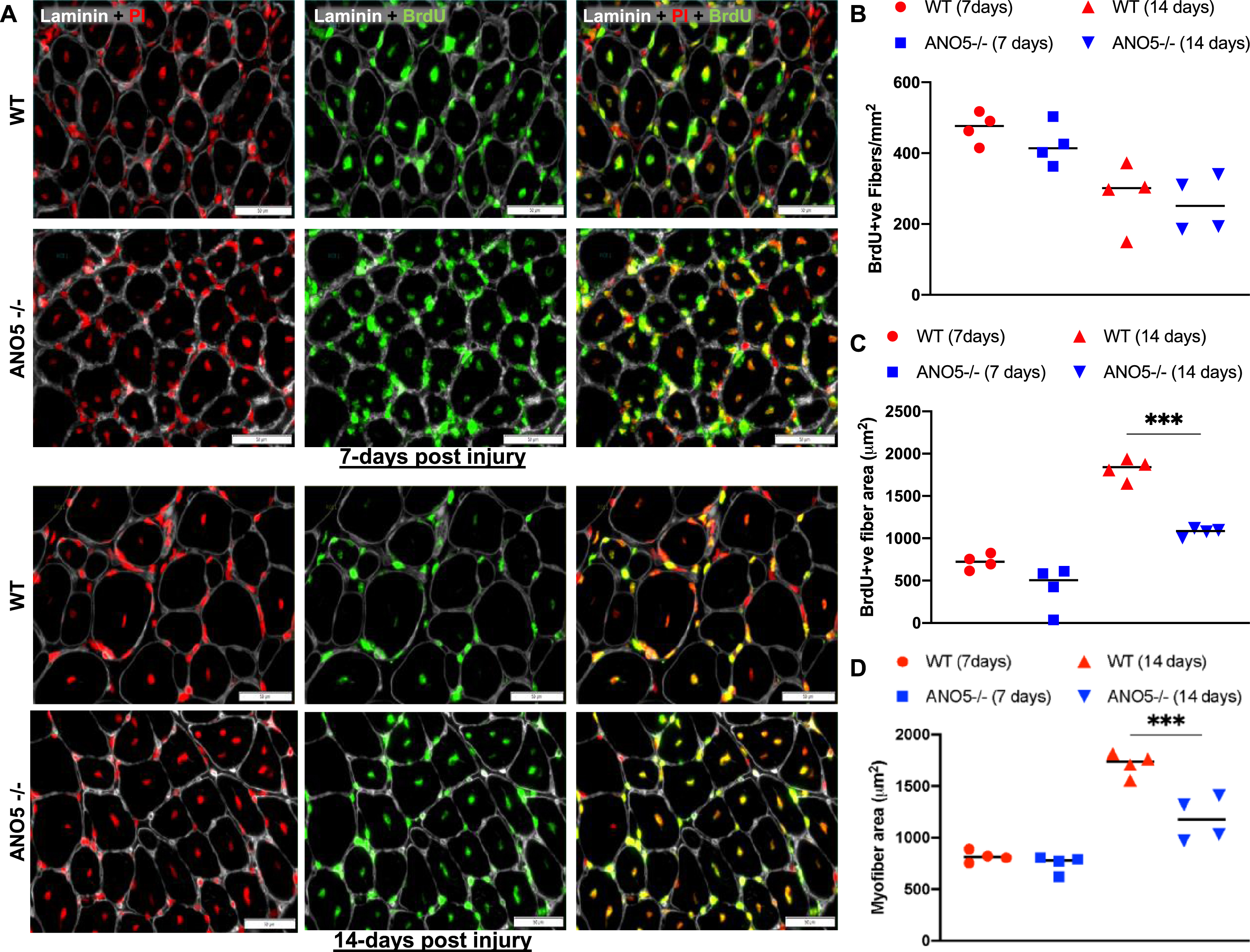
injury [37]. We thus measured growth of newly regenerated myofibers at 7- and 14-days post injury. While average myofiber size at 7-days post injury was not different between the WT and ANO5–/– mouse muscles, the average size of freshly regenerated (BrdU-labeled) ANO5–/– fibers was lower at 14-days post injury relative to control (Fig. 4C). This difference was significant even when fibers that did not contain a BrdU labeled nuclei were also included in the quantification of the myofiber cross-sectional area (Fig. 4D). These findings indicate that while the lack of ANO5 does not compromise myoblast fusion in vivo, it slows subsequent myofiber growth.
DISCUSSION
With the increasing identification of LGMD2L/R12 muscular dystrophy patients [54–56], there is a growing need to develop suitable animal models to help understand the in vivo role of ANO5 protein and test therapies which target this deficit. Our study has generated such a mouse model that mimics several clinical features of ANO5 deficit in LGMD2L/R12 ranging from muscle weakness, myofiber damage, and progressive muscle loss. We created this model by the deletion of exons 10–12 of mouse Ano5 gene, which selectively prevented the expression of this gene without affecting the expression of the other anoctamin family members tested. This is different from a previous ANO5 mouse model with a reading frameshift caused by exon 11–12 deletion that results in the loss of Ano5 transcript in bone and 71% reduction in muscle, leading to GDD-like bone defects [40]. Our observation of muscle pathology caused by the deletion of exons 10–12 aligns with muscle pathology caused by the deletion of exons 8–9 in the mouse, and of exons 12–13 in the rabbit, but not in mice where exons 1, 2 are deleted [23, 37, 39, 41].
The mouse model we describe shows reduced total body mass and reduced muscle mass, recapitulating the muscle wasting and loss reported in the human patients [6, 8, 57]. These ANO5-deficient mouse muscles also showed reduced muscle strength reported in the patients [6, 8]. In addition to the reduced forelimb and hindlimb grip strength in ANO5–/– mice, the TA muscle of these mice also fatigue faster and show reduced isometric force at tetanic stimulation as compared to the WT mice. This latter response of ANO5–/– muscle is in addition to our recent observation that lengthening contraction (LC) of the EDL muscle leads to greater muscle force drop in the ANO5–/– muscle as compared to WT muscle [24]. Poor recovery of ANO5–/– muscle from LC injury occurs due to impaired ability of these myofibers to undergo sarcolemmal repair – a deficit documented in patient muscle cells and another ANO5-deficient mouse model [24, 35, 37, 38]. Poor sarcolemmal repair caused by ANO5 deficit could also contribute to muscle loss and to muscle weakness observed here in the ANO5–/– mice.
Impaired myofiber sarcolemmal repair in the LGMD2L/R12 mouse model is shared with the LGMD2B/R2 mouse model, where mutations affect the dysferlin protein [27, 58]. Similar to dysferlin, ANO5 protein also translocate to the injured plasma membrane in myoblasts and in mature myofibers [27, 35, 38, 59]. However, unlike the LGMD2B patients and mice, which show adipogenic muscle loss [42, 57], we did not observe this as a feature of ANO5–/– muscle. This suggests that dysferlin and ANO5 protein have different functions leading to different manifestation of disease symptoms. In support of this, our previous work has shown that AAV-based expression of ANO5 in dysferlin-deficient mouse muscle fails to rescue the sarcolemmal repair and other symptoms of the dysferlin-deficient mouse [60]. Indeed, ANO5 and dysferlin have distinct roles in sarcolemmal repair. While dysferlin regulates membrane repair through regulation of lysosome fusion, loss of ANO5 compromises handling of cytosolic Ca2+ and impairs membrane repair mediated by annexin, mitochondrial signaling, and phosphatidylserine lipids [24, 35–38]. Dysferlin deficit alters the homeostasis of another membrane lipid – sphingomyelin, and use of the sphignomyelinase enzyme as well as improving the stability of the dysferlinopathic myofiber membrane improves repair and reduces muscle loss [45, 58]. Aside from membrane lipid alteration, dysferlin- and ANO5-deficient muscles also show cellular Ca2+ dysregulation upon myofiber stress/damage [24, 61, 62].
The above role of ANO5 in SR Ca2+ homeostasis is due to its ability to function as an anion channel at the ER membrane [24]. Cellular models with exogenous ANO5 overexpression leading to the presence of ANO5 at the plasma membrane enables Ca2+-activated ion channel activity [30, 31, 63]. However, our analysis of the chloride channel activity at the plasma membrane of ANO5-deficient mouse myofibers showed no difference in this activity between ANO5–/– and WT myofibers. This could be either due to the lack of anion channel activity of the plasma membrane-localized ANO5, or that endogenous ANO5 protein shows little (or no) expression at the plasma membrane, resulting in no detectable channel activity at the sarcolemma. Indeed, ANO5 localizes at the ER membrane and alters ER ion homeostasis when absent [24, 26, 35]. Aside from ion channel activity, ANO5 also possesses lipid scramblase activity, which has been implicated in regulation of myoblast fusion in vitro [34, 37]. Our in vivo analysis shows no significant spontaneous regenerative myogenesis in the adult ANO5–/– muscle and no difference in the ability of the satellite cells in the injured muscle to undergo fusion to regenerate the lost myofibers, which is in agreement with the in vitro studies using patient-derived myoblasts [35]. Interestingly, we observed that growth of the freshly regenerated ANO5–/– myofibers is slower as compared to the matched WT myofibers, recapitulating a similar observation in another ANO5-deficient mouse model [37]. Thus, while ANO5 deficit in mouse myoblast was found to impair their myogenic fusion in vitro, this role of ANO5 does not extend in vivo in mouse muscle nor to in vitro patient cell fusion. Additional studies will be needed to extend this analysis to other patient mutations and to determine the basis of such in vivo versus in vitro differences. Lack of myogenic fusion deficit in human myoblasts in vitro, and in mouse myofibers in vivo indicate that poor myogenesis may not be the basis for muscle loss in the LGMD2L/R12 patients, but the slower growth of nascent regenerated myofibers could contribute to the muscle weakness.
In summary, the findings we report in this study establishes a new mouse model for LGMD2L/R12 that manifests multiple muscle pathologies reported in ANO5 deficient muscular dystrophy patients. Description of these muscle pathologies and physiological deficits reported here and our earlier studies identifying a therapeutic approach to improve repair of ANO5–/– myofibers demonstrate the utility of this model to improve our understanding of the mechanisms of ANO5 function in skeletal muscle and testing therapies to treat muscular dystrophy caused by its deficit.
ACKNOWLEDGMENTS
This study was supported by funds from Association Française contre les Myopathies (AFM) to IR. JKJ acknowledges financial support from the National Institute of Health grants (R01AR055686, R21HD103993, P50HD105328), and thanks Dr. Terry Partridge for his insights into myogenesis, studying its role using mouse models of diseases, and the use of BrdU-labeling approach employed in this study. IR and JKJ conceived this study, obtained funds for this study and supervised the work. GT and SSC conducted all the functional studies with help from GC, JN, and MH for muscle exercise and myogenesis analysis. IR, KC, LS-P generated the knockout mouse and conducted its molecular characterization. BA conducted and described the electrophysiological analysis. GT and JKJ wrote the manuscript with help from IR and all the other authors.
CONFLICT OF INTEREST
Authors declare no conflict of interests.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-210720.
REFERENCES
[1] | Bushby K . Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC consortium on limbgirdle dystrophies. Neuromuscular Disorders. (1995) ;5: (1):71–4. |
[2] | Angelini C . LGMD. Identification, description and classification. Acta Myol. (2020) ;39: (4):207–17. |
[3] | Georganopoulou DG , Moisiadis VG , Malik FA , Mohajer A , Dashevsky TM , Wuu ST , et al. A Journey with LGMD: From Protein Abnormalities to Patient Impact. Protein J. (2021) ;40: (4):466–88. |
[4] | Ten Dam L , Frankhuizen WS , Linssen W , Straathof CS , Niks EH , Faber K , et al. Autosomal recessive limb-girdle and Miyoshi muscular dystrophies in the Netherlands: The clinical and molecular spectrum of 244 patients. Clin Genet. (2019) ;96: (2):126–33. |
[5] | Nallamilli BRR , Chakravorty S , Kesari A , Tanner A , Ankala A , Schneider T , et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol. (2018) ;5: (12):1574–87. |
[6] | Bolduc V , Marlow G , Boycott KM , Saleki K , Inoue H , Kroon J , et al. Recessive mutations in the putative calcium-activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. American journal of human genetics. (2010) ;86: (2):213–21. |
[7] | Sarkozy A , Hicks D , Hudson J , Laval SH , Barresi R , Hilton-Jones D , et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Human mutation. (2013) ;34: (8):1111–8. |
[8] | Hicks D , Sarkozy A , Muelas N , Koehler K , Huebner A , Hudson G , et al. A founder mutation in Anoctamin 5 is a major cause of limb-girdle muscular dystrophy. Brain: a journal of neurology. (2011) ;134: (Pt 1):171–82. |
[9] | Mahjneh I , Jaiswal J , Lamminen A , Somer M , Marlow G , Kiuru-Enari S , et al. A new distal myopathy with mutation in anoctamin 5. Neuromuscular disorders: NMD. (2010) ;20: (12):791–5. |
[10] | Whitlock JM , Hartzell HC . Anoctamins/TMEM16 Proteins: Chloride Channels Flirting with Lipids and Extracellular Vesicles. Annu Rev Physiol. (2017) ;79: :119–43. |
[11] | Boccaccio A , Di Zanni E , Gradogna A , Scholz-Starke J . Lifting the veils on TMEM16E function. Channels. (2019) ;13: (1):33–5. |
[12] | Tian Y , Schreiber R , Kunzelmann K . Anoctamins are a family of Ca2+-activated Cl- channels. Journal of cell science. (2012) ;125: (Pt 21):4991–8. |
[13] | Berg J , Yang H , Jan LY . Ca2+-activated Cl–channels at a glance. Journal of cell science. (2012) ;125: (6):1367–71. |
[14] | Yang YD , Cho H , Koo JY , Tak MH , Cho Y , Shim W-S , et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. (2008) ;455: (7217):1210–5. |
[15] | Schroeder BC , Cheng T , Jan YN , Jan LY . Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. (2008) ;134: (6):1019–29. |
[16] | Caputo A , Caci E , Ferrera L , Pedemonte N , Barsanti C , Sondo E , et al. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. (2008) ;322: (5901):590–4. |
[17] | Pifferi S , Dibattista M , Menini A . TMEM16B induces chloride currents activated by calcium in mammalian cells. Pflugers Archiv: European journal of physiology. (2009) ;458: (6):1023–38. |
[18] | Stohr H , Heisig JB , Benz PM , Schoberl S , Milenkovic VM , Strauss O , et al. TMEM16B, a novel protein with calcium-dependent chloride channel activity, associates with a presynaptic protein complex in photoreceptor terminals. J Neurosci. (2009) ;29: (21):6809–18. |
[19] | Tsuji T , Cheng J , Tatematsu T , Ebata A , Kamikawa H , Fujita A , et al. Predominant localization of phosphatidylserine at the cytoplasmic leaflet of the ER, and its TMEM16K-dependent redistribution. Proceedings of the National Academy of Sciences. (2019) ;116: (27):13368–73. |
[20] | Petkovic M , Oses-Prieto J , Burlingame A , Jan LY , Jan YN . TMEM16K is an interorganelle regulator of endosomal sorting. Nature communications. (2020) ;11: (1):1–16. |
[21] | Suzuki J , Fujii T , Imao T , Ishihara K , Kuba H , Nagata S . Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. The Journal of biological chemistry. (2013) ;288: (19):13305–16. |
[22] | Alvadia C , Lim NK , Mosina VC , Oostergetel GT , Dutzler R , Paulino C . Cryo-EM structures and functional characterization of the murine lipid scramblase TMEM16F. Elife. (2019) ;8: :e44365. |
[23] | Xu J , El Refaey M , Xu L , Zhao L , Gao Y , Floyd K , et al. Genetic disruption of Ano5 in mice does not recapitulate human ANO5-deficient muscular dystrophy. Skelet Muscle. (2015) ;5: :43. |
[24] | Chandra G , Sreetama SC , Mázala DA , Charton K , VanderMeulen JH , Richard I , et al. Endoplasmic reticulum maintains ion homeostasis required for plasma membrane repair. Journal of Cell Biology. (2021) ;220: (5). |
[25] | Mizuta K , Tsutsumi S , Inoue H , Sakamoto Y , Miyatake K , Miyawaki K , et al. Molecular characterization of GDD1/TMEM16E, the gene product responsible for autosomal dominant gnathodiaphyseal dysplasia. Biochemical and biophysical research communications. (2007) ;357: (1):126–32. |
[26] | Tsutsumi S , Kamata N , Vokes TJ , Maruoka Y , Nakakuki K , Enomoto S , et al. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD). American journal of human genetics. (2004) ;74: (6):1255–61. |
[27] | Bansal D , Miyake K , Vogel SS , Groh S , Chen CC , Williamson R , et al. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. (2003) ;423: (6936):168–72. |
[28] | Ho M , Post CM , Donahue LR , Lidov HG , Bronson RT , Goolsby H , et al. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet. (2004) ;13: (18):1999–2010. |
[29] | Liu J , Aoki M , Illa I , Wu C , Fardeau M , Angelini C , et al. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. (1998) ;20: (1):31–6. |
[30] | Di Zanni E , Gradogna A , Picco C , Scholz-Starke J , Boccaccio A . TMEM16E/ANO5 mutations related to bone dysplasia or muscular dystrophy cause opposite effects on lipid scrambling. Human mutation. (2020) ;41: (6):1157–70. |
[31] | Di Zanni E , Gradogna A , Scholz-Starke J , Boccaccio A . Gain of function of TMEM16E/ANO5 scrambling activity caused by a mutation associated with gnathodiaphyseal dysplasia. Cellular and molecular life sciences: CMLS. (2018) ;75: (9):1657–70. |
[32] | Gyobu S , Ishihara K , Suzuki J , Segawa K , Nagata S . Characterization of the scrambling domain of the TMEM16 family. Proc Natl Acad Sci U S A. (2017) ;114: (24):6274–9. |
[33] | Duran C , Qu Z , Osunkoya AO , Cui Y , Hartzell HC . ANOs 3-7 in the anoctamin/Tmem16 Cl- channel family are intracellular proteins. American journal of physiology Cell physiology. (2012) ;302: (3):C482–93. |
[34] | Whitlock JM , Yu K , Cui YY , Hartzell HC . Anoctamin 5/TMEM16E facilitates muscle precursor cell fusion. J Gen Physiol. (2018) ;150: (11):1498–509. |
[35] | Chandra G , Defour A , Mamchoui K , Pandey K , Mishra S , Mouly V , et al. Dysregulated calcium homeostasis prevents plasma membrane repair in Anoctamin 5/TMEM16E-deficient patient muscle cells. Cell death discovery. (2019) ;5: (1):1–15. |
[36] | Jaiswal JK , Marlow G , Summerill G , Mahjneh I , Mueller S , Hill M , et al. Patients with a non-dysferlin Miyoshi myopathy have a novel membrane repair defect. Traffic. (2007) ;8: (1):77–88. |
[37] | Griffin DA , Johnson RW , Whitlock JM , Pozsgai ER , Heller KN , Grose WE , et al. Defective membrane fusion and repair in Anoctamin5-deficient muscular dystrophy. Human molecular genetics. (2016) ;25: (10):1900–11. |
[38] | Foltz SJ , Cui YY , Choo HJ , Hartzell H . ANO5 ensures trafficking of annexins in wounded myofibers. Journal of Cell Biology. (2021) ;220: (3). |
[39] | Gyobu S , Miyata H , Ikawa M , Yamazaki D , Takeshima H , Suzuki J , et al. A Role of TMEM16E Carrying a Scrambling Domain in Sperm Motility. Molecular and cellular biology. (2016) ;36: (4):645–59. |
[40] | Wang X , Liu X , Dong R , Liang C , Reichenberger EJ , Hu Y . Genetic disruption of anoctamin 5 in mice replicates human gnathodiaphyseal dysplasia (GDD). Calcified tissue international. (2019) ;104: (6):679–89. |
[41] | Sui T , Xu L , Lau YS , Liu D , Liu T , Gao Y , et al. Development of muscular dystrophy in a CRISPR-engineered mutant rabbit model with frame-disrupting ANO5 mutations. Cell Death Dis. (2018) ;9: (6):609. |
[42] | Hogarth MW , Defour A , Lazarski C , Gallardo E , Diaz Manera J , Partridge TA , et al. Fibroadipogenic progenitors are responsible for muscle loss in limb girdle muscular dystrophy 2B. Nat Commun. (2019) ;10: (1):2430. |
[43] | Mazala DA , Novak JS , Hogarth MW , Nearing M , Adusumalli P , Tully CB , et al. TGF-beta-driven muscle degeneration and failed regeneration underlie disease onset in a DMD mouse model. JCI Insight. (2020) ;5: (6). |
[44] | Novak JS , Hogarth MW , Boehler JF , Nearing M , Vila MC , Heredia R , et al. Myoblasts and macrophages are required for therapeutic morpholino antisense oligonucleotide delivery to dystrophic muscle. Nat Commun. (2017) ;8: (1):941. |
[45] | Sreetama SC , Chandra G , Van der Meulen JH , Ahmad MM , Suzuki P , Bhuvanendran S , et al. Membrane Stabilization by Modified Steroid Offers a Potential Therapy for Muscular Dystrophy Due to Dysferlin Deficit. Mol Ther. (2018) ;26: (9):2231–42. |
[46] | Bittel AJ , Sreetama SC , Bittel DC , Horn A , Novak JS , Yokota T , et al. Membrane Repair Deficit in Facioscapulohumeral Muscular Dystrophy. Int J Mol Sci. (2020) ;21: (15). |
[47] | Idoux R , Fuster C , Jacquemond V , Dayal A , Grabner M , Charnet P , et al. Divalent cations permeation in a Ca(2+) non-conducting skeletal muscle dihydropyridine receptor mouse model. Cell Calcium. (2020) ;91: :102256. |
[48] | Jha A , Chung WY , Vachel L , Maleth J , Lake S , Zhang G , et al. Anoctamin 8 tethers endoplasmic reticulum and plasma membrane for assembly of Ca2+signaling complexes at the ER/PM compartment. The EMBO journal. (2019) ;38: (12):e101452. |
[49] | Wu N , Cernysiov V , Davidson D , Song H , Tang J , Luo S , et al. Critical role of lipid scramblase TMEM16F in phosphatidylserine exposure and repair of plasma membrane after pore formation. Cell reports. (2020) ;30: (4):1129–40. e5. |
[50] | Witting N , Duno M , Petri H , Krag T , Bundgaard H , Kober L , et al. Anoctamin 5 muscular dystrophy in Denmark: prevalence, genotypes, phenotypes, cardiac findings, and muscle protein expression. Journal of neurology. (2013) ;260: (8):2084–93. |
[51] | Rae EA , Brown RE . The problem of genotype and sex differences in life expectancy in transgenic AD mice. Neurosci Biobehav Rev. (2015) ;57: :238–51. |
[52] | Tran TT , Tobiume K , Hirono C , Fujimoto S , Mizuta K , Kubozono K , et al. TMEM16E (GDD1) exhibits protein instability and distinct characteristics in chloride channel/pore forming ability. Journal of cellular physiology. (2014) ;229: (2):181–90. |
[53] | Leikina E , Defour A , Melikov K , Van der Meulen JH , Nagaraju K , Bhuvanendran S , et al. Annexin A1 deficiency does not affect myofiber repair but delays regeneration of injured muscles. Scientific reports. (2015) ;5: (1):1–12. |
[54] | Savarese M , Di Fruscio G , Tasca G , Ruggiero L , Janssens S , De Bleecker J , et al. Next generation sequencing on patients with LGMD and nonspecific myopathies: Findings associated with ANO5 mutations. Neuromuscular disorders: NMD. (2015) ;25: (7):533–41. |
[55] | Kuhn M , Glaser D , Joshi PR , Zierz S , Wenninger S , Schoser B , et al. Utility of a next-generation sequencing-based gene panel investigation in German patients with genetically unclassified limb-girdle muscular dystrophy. Journal of neurology. (2016) ;263: (4):743–50. |
[56] | Bohlega S , Monies DM , Abulaban AA , Murad HN , Alhindi HN , Meyer BF . Clinical and genetic features of anoctaminopathy in Saudi Arabia. Neurosciences (Riyadh). (2015) ;20: (2):173–7. |
[57] | Ten Dam L , van der Kooi AJ , Rovekamp F , Linssen WH , de Visser M . Comparing clinical data and muscle imaging of DYSF and ANO5 related muscular dystrophies. Neuromuscular disorders: NMD. (2014) ;24: (12):1097–102. |
[58] | Defour A , Van der Meulen JH , Bhat R , Bigot A , Bashir R , Nagaraju K , et al. Dysferlin regulates cell membrane repair by facilitating injury-triggered acid sphingomyelinase secretion. Cell Death Dis. (2014) ;5: :e1306. |
[59] | Bittel DC , Chandra G , Tirunagri LMS , Deora AB , Medikayala S , Scheffer L , et al. Annexin A2 Mediates Dysferlin Accumulation and Muscle Cell Membrane Repair. Cells. (2020) ;9: (9). |
[60] | Monjaret F , Suel-Petat L , Bourg-Alibert N , Vihola A , Marchand S , Roudaut C , et al. The phenotype of dysferlin-deficient mice is not rescued by adeno-associated virus-mediated transfer of anoctamin 5. Human gene therapy Clinical development. (2013) ;24: (2):65–76. |
[61] | Kerr JP , Ziman AP , Mueller AL , Muriel JM , Kleinhans-Welte E , Gumerson JD , et al. Dysferlin stabilizes stress-induced Ca2+signaling in the transverse tubule membrane. Proc Natl Acad Sci U S A. (2013) ;110: (51):20831–6. |
[62] | Chandra G , Mazala DAG , Jaiswal JK . Coping with the calcium overload caused by cell injury: ER to the rescue. Cell Stress. (2021) ;5: (5):73–5. |
[63] | Schreiber R , Ousingsawat J , Kunzelmann K . Targeting of Intracellular TMEM16 Proteins to the Plasma Membrane and Activation by Purinergic Signaling. International journal of molecular sciences. (2020) ;21: (11):4065. |


