
Revertant Phenomenon in DMD and LGMD2I and Its Therapeutic Implications: A Review of Study Under Mentorship of Terrence Partridge
Abstract
This review recollects my initial research focus on revertant fibers (expressing dystrophin in the background of frame-shifting mutation) in Duchenne muscular dystrophy (DMD) muscles in Professor Terrence Partridge’s Muscle Cell Biology Laboratory in MRC Clinical Research Science Center, Harmmersmith Hospital, London, UK. Our data indicated that revertant fibers are most likely resulted from epigenetic random events which skip exon(s) flanking the mutated exon, leading to the restoration of the reading frame. Some of these events establish themselves as relatively permanent skipping patterns, a mechanism similar to multiple transcript species established in various cell types. With this hypothesis, antisense oligonucleotide-mediated exon skipping is likely to have a great chance to achieve restoration of therapeutic levels of dystrophin in DMD muscles. This leads to our first reports of local and systemic efficacy of antisense oligonucleotide-mediated exon skipping for DMD treatment. The experience under Terry’s mentorship shaped my thinking and led me to explore another revertant feature in the dystroglycanopathy caused by mutations in the Fukutin Related Protein (FKRP) gene which functions as a glycosyltransferase. Mutant FKRPs retain partial function and produce a fraction of normal to no detectable levels of laminin-binding α-dystroglycan (matriglycan) in most of the muscle fibers. Reversion to near normal levels of matriglycan expression in muscles with FKRP mutations depends on muscle regeneration and in muscles of neonate mice, suggesting that changes in metabolism and gene expression could be sufficient to compensate for the reduced function of mutant FKRP genes even those associated with severe congenital muscular dystrophy (CMD). This is now supported by our successful demonstration that supply of FKRP mutant mice with ribitol, a precursor for substrate of FKRP, is sufficient to restore the levels of matriglycan with therapeutic significance. Our data overall suggest that rare events of reversion in muscular dystrophy, and likely other diseases could provide unique insight for mechanisms and therapeutic exploitation.
INTRODUCTION
Mutations in the genes of the dystrophin glycoprotein complex (DGC) are the most common causes of muscular dystrophy [1, 2]. The nature of these mutations creates a wide variation in the levels of gene expression and function of gene products, the two principal factors determining disease severity. The best example is mutations in the dystrophin gene causing Duchenne muscular dystrophy (DMD) with out-of-frame and nonsense mutations and Becker muscular dystrophy (BMD) with largely in-frame deletions. Biochemically, DMD muscles lack dystrophin expression whereas BMD muscles presents truncated dystrophin with reduced function of varying degree, yet insufficient to maintain muscle integrity and prevent muscle from degeneration. Mutations in genes modifying DGC proteins cause secondary muscular dystrophy, which is exemplified by dystroglycanopathies resulting from reduction to loss of glycosylation of alpha dystroglycan (α-DG) [3, 4]. This laminin binding glycan (matriglycan) is the key component linking DGC proteins to extracellular matrix, and the muscles lacking matriglycan suffer similar consequence of muscle degeneration as those muscles lacking DGC proteins [5]. The structure of matriglycan chains on α-DG has recently been elucidated and individual genes identified for their role in the synthesis of the glycan. One of them is Fukutin-Related Protein (FKRP) gene which functions as ribitol-5-phosphate (ribitol-5P) transferase, adding the pentose phosphate to the glycan structure [6–8]. Almost all FKRP mutations are missense point mutations, but mutations at different sites cause huge variation in disease phenotype from mild Limb Girdle Muscular Dystrophy (LGMD) 2I to severe congenital muscular dystrophy (CMD) with brain and eye involvement. As secondary myopathies, biochemical feature of all dystroglycanopathies including FKRP muscular dystrophy is represented by reduced or lack of matriglycan rather than the mutated proteins [9–11]. Nevertheless, both dystrophin and matriglycan serve as their respective biomarker of the diseases, and restoration of their expression is the fundamental goal of experimental therapies.
One shared and interesting feature of DMD and FKRP muscular dystrophy is that muscles from both diseases can spontaneously correct their own biochemical defects with the restoration of either dystrophin in DMD, or matriglycan in FKRP muscular dystrophy. Muscle fibers with restored expression of the missing protein in diseased muscles are therefore named as revertant fibers (RF) and the protein as revertant protein [12, 13]. My interest in revertant fibers started when I joined Terry’s Muscle Cell Biology (MCB) Laboratory more than 20 years ago, at that time I knew little about muscular dystrophy. The MCB laboratory routinely used mdx mice to study satellite cells and disease mechanisms of muscular dystrophy. Trained as a pathologist and loving immunohistochemistry for disease characterization, I was immediately fascinated by the presence of RFs in the muscles of the mice.
Revertant fibers in DMD muscles, mechanism(s) and oligonucleotide-mediated exon skipping
The presence of RFs was recognized soon after the identification of dystrophin gene mutations as the cause of DMD. Early studies reported RFs in the majority of DMD muscle biopsies. The incidence of RF in DMD muscles is generally low, less than 7% [13–15]. However, relatively large numbers of RFs, up to 70%, were also reported from individual biopsy samples. RFs were also present in DMD mouse models, but generally comprised less than 1% of fibers in the mdx mouse [16, 17]. Immunostaining with antibodies against dystrophin showed signal intensity of RFs was equal or weaker than that in control normal muscles. Interestingly, RFs frequently appeared as small clusters although singular RFs were also common (Fig. 1) Revertant dystrophins are likely functional as the same fibers also restore membrane-localized proteins of the DGC. A few previous reports have investigated mechanisms and clinical significance of RFs. Hoffman et al showed that revertant fibers in mdx muscles increases after irradiation [16]. Further, Morris’s group mapped individual revertant fibers from patient muscles by exon specific monoclonal antibodies and RTPCR and reported deletion of exons flanking the frame-shift mutation sites [18]. Wilton et al used nested RTPCR to examine dystrophin gene transcripts around the mutated exon 23 in mdx mice and revealed several alternatively processed transcript species with skipping of additional exons neighbouring the mutated exon 23 [19]. Interestingly, these shortened transcripts were also present in normal adult and embryonic mouse muscle tissues. These studies overall, based mainly on cluster and clonal assumption, concluded that somatic mutations, specifically genomic deletion, are the most likely mechanism for the restoration of reading frames in DMD. Variation in signal intensity also led Nicholson et al. [17] to propose a modified hypothesis that strong positive revertant dystrophin signals might be a result of somatic mutations whereas weak dystrophin signals could be a result of exon splicing.
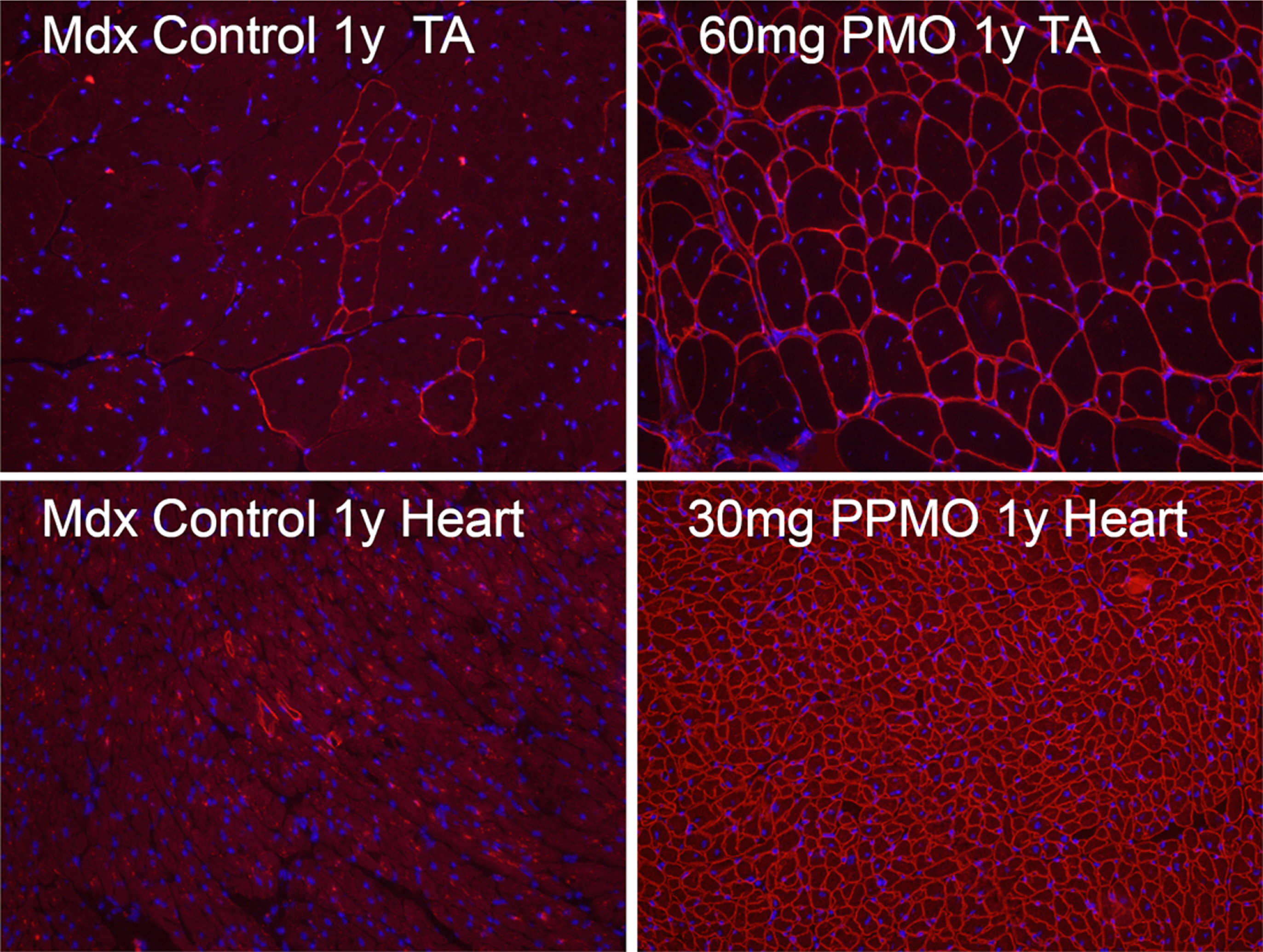
Fig. 1
Revertant fibers in skeletal and cardiac muscles (Control, left panel) of mdx mice and mdx mice treated for 1 year (1y) with 60 mg/kg morpholino (PMO, upper right) and 30 mg/kg peptide-conjugated morpholino (PPMO, lower right) antisense oligonucleotide targeting exon 23 of dystrophin gene. Dystrophin is recognized with primary rabbit anti-dystrophin antibody P7 and detected by donkey anti-rabbit Alexa Fluor 594. Nuclei (blue) are stained with DAPI.
With this background data in mind, Terry and I discussed the problems with these hypotheses and difficulties we faced to further explore mechanisms involved in the production of revertant dystrophins. The high frequency, even 1% of RFs disfavours a somatic deletion hypothesis. As mutations are most likely to be random events, only a fraction of random deletional mutations could possibly correct existing reading frames shift in DMD. Thus, many folds more of the observed mutational events would be required to create one genome sequence with the dystrophin gene back in-frame. Further, mRNA transcripts detected by RTPCR from whole muscle tissue are largely irrelevant to the production of dystrophin in such small number of RFs [16, 17], nor be considered as an evidence of secondary somatic mutations in the absence of direct evidence of corresponding genomic rearrangement. Truncated new transcripts could simply be the result of low-level random splicing events. We reasoned that the high frequency of RF and existence of truncated transcripts in normal muscles favours splicing events.
However, further probing the mechanism(s) involved in the generation of revertant dystrophins faced several technical hurdles. First, revertant fibers are mainly present as small clusters with only one or a few fibers spanning different lengths and can only be visualized by immunohistochemistry with specific antibodies. The total amount of revertant dystrophin is very limited and cannot be reliably measured by western blots. This plus the syncytial nature of muscle fibers makes it almost impossible to isolate the correct fibers or nucleus and keep the material at a condition suitable for further analysis once the fibers are already immunohistochemically stained. Second, there were technical limits for a single cell to be analysed by genomics, transcriptomics and proteomics in combination, essential for study of revertant fibers in DMD. Additionally, the number of such revertant fibers varies greatly from muscle to muscle across patients’ biopsy samples. Therefore, the functional significance of RFs has been uncertain despite the fact that revertant dystrophin undoubtedly possesses functional characteristics as evidenced with co-restoration of membrane DGC proteins. This leads us to believe that production of revertant dystrophin might not be of significant value, especially for development of experimental therapies. This view is now considered too simplistic as the presence and pattern of truncated revertant dystrophin could provide guidance for gene therapy vector design, such as mini-dystrophin for AAV-mediated delivery, for maximal efficacy with minimal size of the dystrophin coding sequence. Perhaps more importantly as reported by Nagaraju’s group in a study of morpholino antisense oligonucleotide therapy in DMD mouse model, de novo dystrophin could elicit humoral responses with serum-circulating antibodies and more antigen-specific T-cell cytokine responses (e.g., IFN-g, IL-2), leading to significant and persistent muscle inflammation in treated mouse muscles [20]. Also important, mouse anti-dystrophin antibodies recognize dystrophin protein from canine and human muscles as well as mouse muscle, suggesting that immunodominant epitopes are likely conserved in all three species. However, no dystrophin-specific antibodies were observed against expanded RFs expressing revertant dystrophin. It is therefore desirable to select truncated forms of dystrophin with least prospect to elicit immune response as coding sequence for gene therapy by examining revertant dystrophins.
With the aim to validate either somatic mutation or exon skipping mechanism, we initially attempted to culture individual revertant myoblasts from muscle of mdx mouse, but failed to establish any clones producing revertant dystrophin. We therefore applied 2 unique methods in combination to examine the nature of revertant fibers in situ, mapping the entire dystrophin protein with exon specific monoclonal antibodies, and DNA in situ hybridization for the detection of genomic sequence corresponding to the missing exons on revertant dystrophin proteins on serial sections [21]. The results showed that many RF clusters expressed revertant dystrophin proteins missing multiple exons around the mutated exon 23, but genomic sequence coding for the protein sequences was readily detected. This data would rule out a genomic deletion mechanism and imply a splicing-based exon skipping for the production of revertant dystrophin. This conclusion was further supported by the detection of RFs with missing exons in two non-contiguous regions of the revertant proteins. Furthermore, almost all revertant dystrophins in muscles of mdx mice lack exon 23, yet retain intact NH2-domain and C-terminal domain, ruling out somatic suppression and alternative use of downstream promoters as causes of the revertant phenotype [21].
Understanding the complexity of revertant dystrophin and with the hypothesis that spontaneous exon skipping could rescue the frame-shift and nonsense mutations prompted us to explore the possibility of inducing specific exon skipping in the dystrophin gene by antisense oligonucleotides. Consequently, we were able to demonstrate targeted skipping of the mutated exon 23 by antisense oligonucleotide and achieve restoration of therapeutic levels of truncated dystrophin by either local or systemic delivery. Importantly, almost normal levels of dystrophin with the target exon 23 skipped can be realized in cardiac and skeletal muscles with significant improvement in muscle function by modified antisense oligonucleotides [22–24].
Several questions to RFs remain to be answered. First, direct evidence confirming revertant dystrophin expression from individual nucleus without further dystrophin gene mutation remains to be collected. It may now be possible to further explore the phenomenon with single cell genomics, proteomics and metabolomics. Further, mechanism(s) permitting individual RF to expand into a cluster is not clearly understood although a positive selection environment appears to be required. This is supported by the fact that the number of RFs accumulates in muscles of old mice expressing non-functional or no dystrophin. However, such accumulation is not observed in muscles of old mice expressing normal dystrophin [25]. Understanding the process could lead to new experimental therapy. Interestingly, RFs can also be detected in cardiac muscle (Fig. 1) No study has been conducted to explore its mechanisms.
Revertant fibers in FKRP muscular dystrophy, mechanism(s) and ribitol-mediated restoration of matriglycan expression
Revertant fibers expressing matriglycan in FKRP muscular dystrophies is associated with missense mutations in the FKRP gene, rather than out-frame and nonsense mutations in DMD. The wide spectrum of disease phenotypes of the FKRP muscular dystrophy is closely related to mutation sites, which affect FKRP function differentially, and consequently levels of matriglycan in muscles. In general, the common L276I mutation is associated with mild LGMD2I, whereas mutations such as P448L is associated with severe CMD type disease [10–11, 26]. Consistently, muscles with mild disease phenotype express wide-spread, but reduced levels of matriglycan in the majority of muscle fibers. In contrast, muscles from severe disease phenotypes often lack clearly detectable matriglycan in almost all muscle fibers. Under a background of weak to no matriglycan expression in the majority of muscle fibers, RFs can be easily identified as they stand out with signal intensity similar to that of normal muscle as we reported in the P448L mutant muscles. RFs are present in diseased muscles with FKRP missense mutations, but not present in muscles with a nonsense mutation, suggesting some minimum levels of FKRP function is required [27, 28]. Early investigation from Campbell’s group suggests that some FRs in dystroglycanopathy are associated with the expression of embryonic myosin heavy chains, thus possibly with muscle regeneration [29].
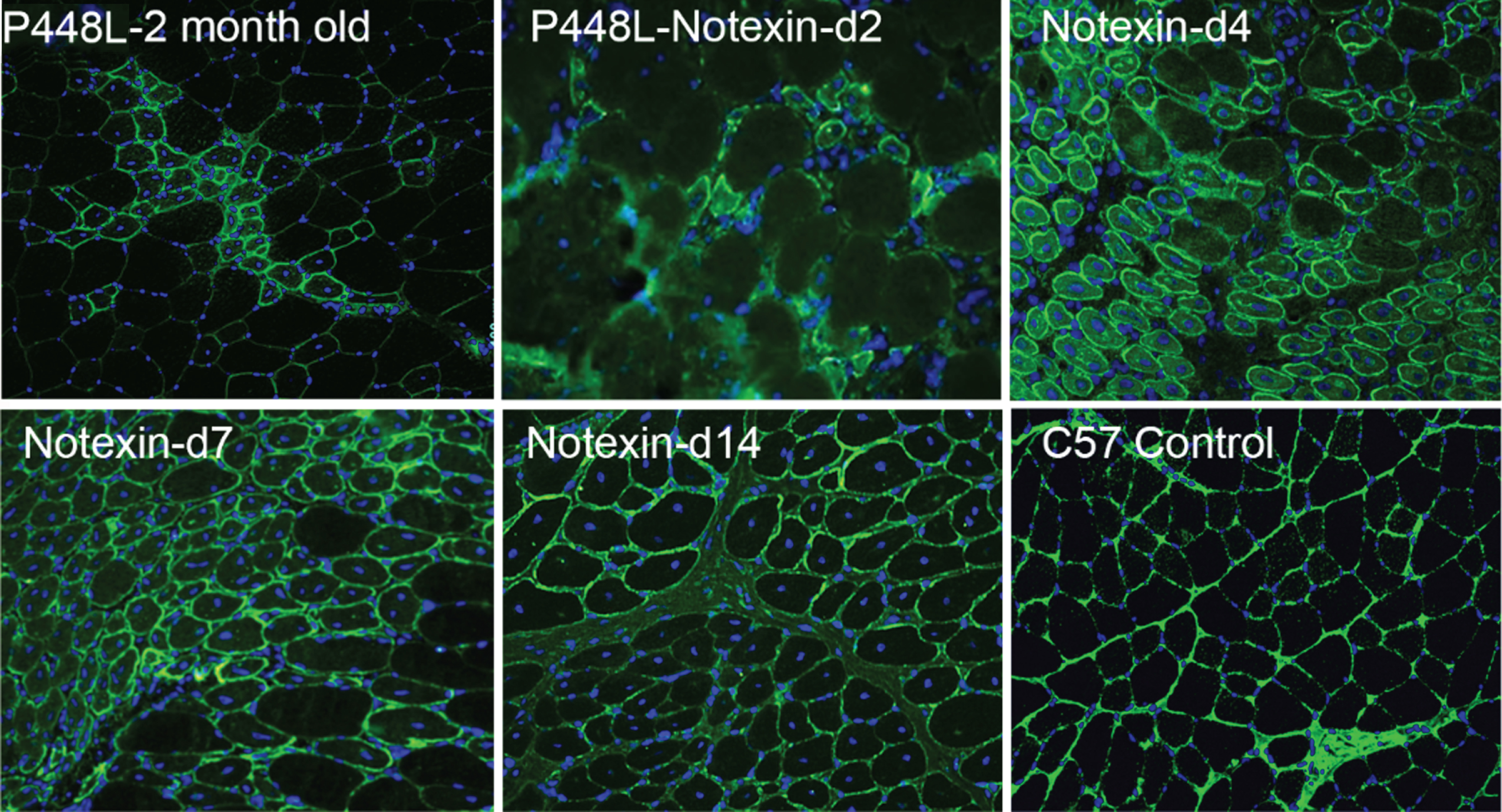
RFs were easily identified in our FKRP mutant mice models. Interestingly, RFs were more readily identified in the adult muscles of P448L homozygotes clinically associated with severe CMD phenotypes. RFs were not detected before 3 weeks of age when muscle degeneration has not started in this mutant strain, suggesting again that the events are degeneration and regeneration related. We then conducted a Notexin-induced muscle damage test in the P448L mutant mice with a timed follow-up from day 2, 4, 7, 14 to 28. The results clearly showed that restoration of matriglycan is associated with muscle regeneration and matriglycan levels in the regenerating fibers reaching normal levels within 1 week. The expression then starts to decline and became hardly detectable after 4 weeks (Fig. 2) Restoration of matriglycan depends on the expression of both LARGE and partially functional FKRP. However, up-regulation of the LARGE and FKRP was not observed during the period of matriglycan expression [30]. The results indicate that FKRP mutations even clinically associated with severe CMD retain at least partial functions which can be sufficient for normal levels of matriglycan expression. This led us to examine the potential of using a mutated FKRP gene for AAV-mediated gene therapy. Indeed, even the P448L mutant FKRP can effectively restore up to normal levels of matriglycan in body-wide muscles and alleviate disease phenotype. Applying a mutant FKRP as therapeutic transgene could avoid any potential immune response against novel antigenic epitope on normal FKRP transprotein which is not present in endogenous mutant FKRP proteins in patients [31].
Fig. 2
Revertant fibers in muscles of P448L FKRP mutant mice (Upper left) and restoration of matriglycan expression in the regenerating muscle fibers after Notexin induced damage from day 2 to day 14 (d2-d14). Matriglycan (green) is detected with primary monoclonal antibody IIH6 (EMD Millipore) (1:500) and detected with secondary Alexa Fluor 488 goat anti-mouse (Invitrogen) and nuclei (blue) are stained with DAPI.
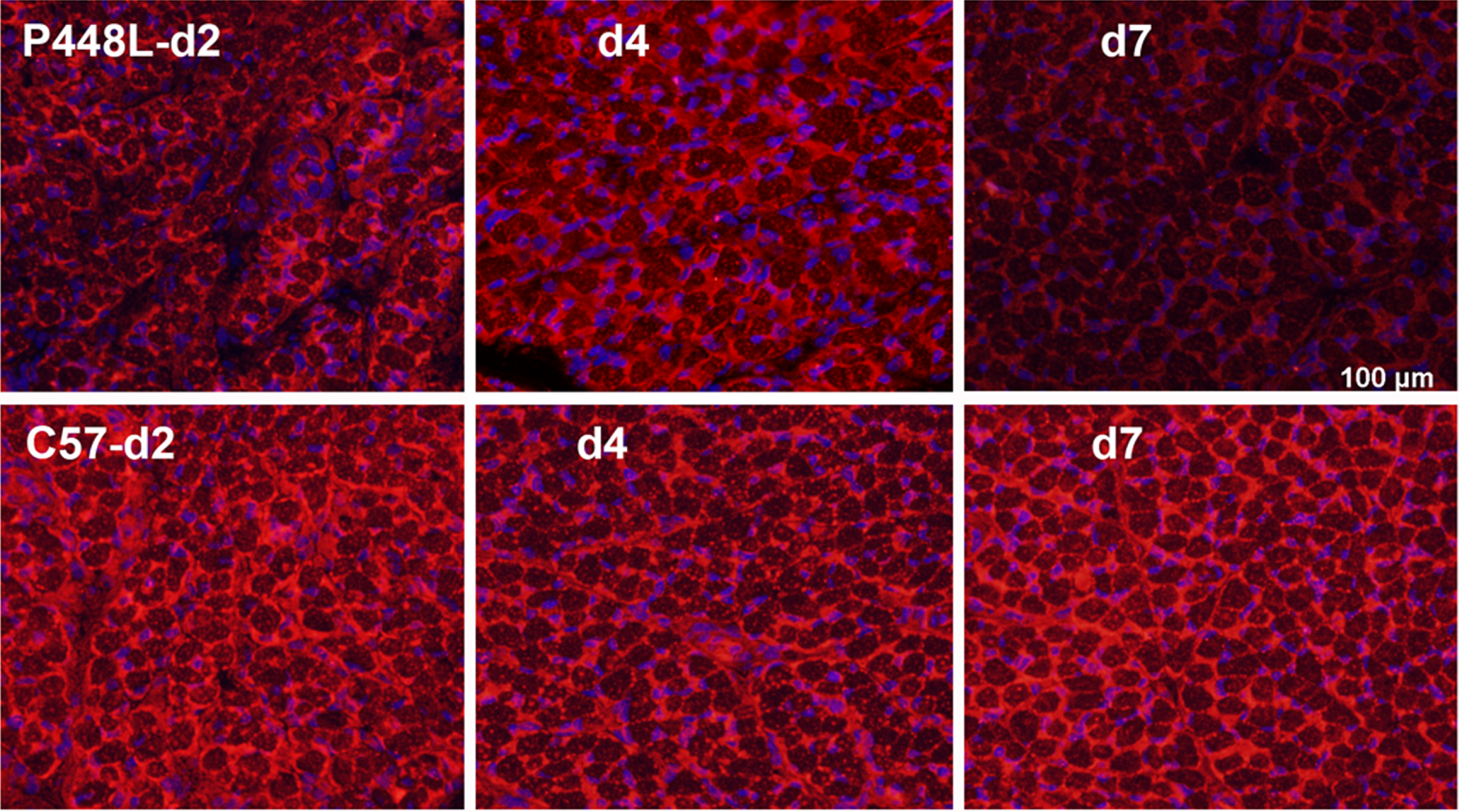
Having established that restoration of matriglycan starts from early stage of regenerating muscle fibers and diminishes with maturation prompted us to consider that muscles at certain stage of development might also be conducive to matriglycan restoration in FKRP mutant subjects. We therefore examine the leg, diaphragm, and cardiac muscles of neonatal P448L mice. Indeed, all three muscles express clearly detectable although slightly lower levels of matriglycan when compared to the age-matched correspondent tissue of normal C57 mice. However, the expression diminishes rather quickly at about 6-7 days after birth (Fig. 3) [32].
Fig. 3
Matriglycan expression in new-born P448L FKRP mutant mice at postnatal day 2 (d2) to day 7 (d7) in comparison with C57 normal mice. Matriglycan (red) on α-DG is recognized by primary monoclonal antibody IIH6((EMD Millipore) (1:500) and detected with secondary Alexa Fluor 594 goat anti-mouse (Invitrogen). Except membrane localized signal, there is also diffuse cytoplasmic staining of the early myofibers as observed in the newly regenerated myofibers. Nuclei (blue) are stained with DAPI.
Normal muscles from day 7 onwards continue to express matriglycan at even higher levels than that of the muscle within the first 7 days after birth. It is therefore unlikely that changes in expression levels of those matriglycan-related glycosyltransferases including FKRP are responsible for the restoration of matriglycan during that short perinatal period of the P448L mutant mice. We therefore hypothesized that conditions other than levels of FKRP expression, including difference in metabolic composition could enhance the function of FKRP and compensate for the reduced functionality of mutant FKRP, thus achieving near normal levels of matriglycan in the neonatal mutant muscles. We then decided to conduct a cell-based large-scale drug screening to identify potential candidates for enhancing matriglycan expression. Unfortunately, this effort was not successful, probably due to lack of cell models representing cellular condition in muscles and lack of drug candidates from metabolites and their analogues.
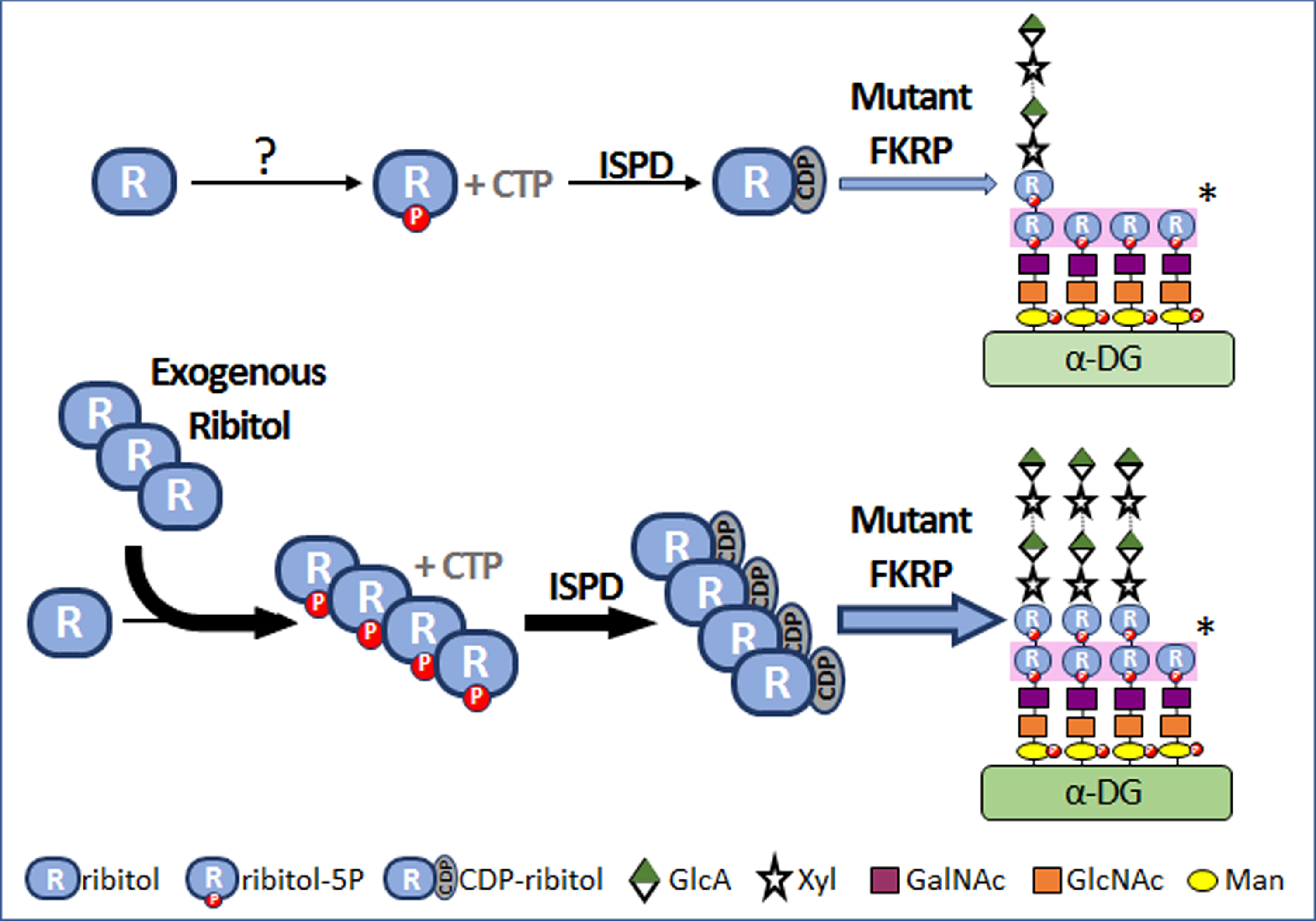
Excitingly, recent advance finally revealed that FKRP functions as a glycosyltransferase and uses CDP-ribitol as a substrate to add ribitol–5-phosphate (ribitol-5P) to the matriglycan structure [6, 7]. Further, ribitol can be used as precursor for the synthesis of CDP-ribitol [8]. This prompted us to examine the hypothesis that, if conversion of ribitol to CDP-ribitol is not a rate-limiting process in muscles as suggested in the normal mice, then an increase in intracellular levels of ribitol could increase the levels of CDP-ribitol. Since most mutant FKRPs retain at least partial function, an increase in the levels of CDP-ribitol substrate might enhance the efficiency of remaining function of mutant FKRPs, thus compensating for reduced function of mutant FKRPs and enhancing matriglycan (Fig. 4). Indeed, our results from the P448L mutant mice showed that ribitol treatment systemically in drinking water increases levels of ribitol-5P and CDP-ribitol in muscle tissues and effectively restores therapeutic levels of matriglycan. This is associated with significant improvement in muscle pathology and functions. The results also support the hypothesis that metabolic alteration could be a mechanism for the reversion of matriglycan expression in neonatal and regenerating FKRP mutant muscle fibers [33–35].
Fig. 4
Mechanism of ribitol-induced matriglycan expression in FKRP mutant cells. “?”, mechanism(s) is not understood. “*”, Each monosaccharide is transferred to the glycan of α-DG by specific glycosyltransferase and the first and second ribitol-5P on the glycan chains are transferred by Fukutin and FKRP respectively. P, phosphate. ISPD, isoprenoid synthase domain containing gene. CTP, cytidine triphosphate. CDP, cytidine diphosphate.
SUMMARY
Absence or reduced expression of proteins or their functions are the results of gene mutations and direct causes of recessive muscular dystrophies. Spontaneous correction or compensation (reversion) of mutated genes or their functions is a common phenomenon, but usually not sufficient to prevent disease progression. There are several mechanisms responsible for such reversion. Reversion in DMD with frame-shift mutations is likely caused by spontaneous splicing events as well as rare secondary genomic mutations. Reversion in FKRP-related dystroglycanopathy with reduction in gene functions represents compensation for the diseased gene. This can be achieved through alteration in gene expression, post translational modifications and metabolism related to cells at different stage of differentiation, metabolic status and other unknown conditions. Current experimental therapies, including gene therapy, gene editing, exon skipping and ribitol treatment can all be considered the utilization of advanced technologies to fulfil the promise of those naturally occurring phenomenon.
Finally, my experience in Terry’s MCB laboratory and later collaboration have benefited my research career immensely. Terry’s working ethics, paying great attention to details of interesting phenomenon even it appears trivial and well-known without losing sight of their broad significance, has helped me to continue the search for possible answers and applications related to revertant fibers in muscular dystrophies.
ACKNOWLEDGMENTS
Qi Long Lu is supported by the Carolinas Muscular Dystrophy Research Endowment through the Atrium Health Foundation at Charlotte, NC. The authors would like to thank the reviewers and Dr Jason Tucker, McColl Lockwood Laboratory, Atrium Health, Charlotte, NC, for their comments and editing.
REFERENCES
[1] | Koenig M , Hoffman EP , Bertelson CJ , Monaco AP , Feener C , Kunkel LM . Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. (1987) ;50: :509–17. |
[2] | Amalfitano A , Rafael JA , Chamberlain JS . Structure and mutation of the dystrophin gene. In: Brown S.C., Lucy-JackA., editors. Dystrophin Gene, Protein and Cell Biology. Cambridge University Press; Cambridge, UK: (1997) ; pp. 1–26. |
[3] | Ervasti JM , Campbell KP . Membrane organization of the dystrophin-glycoprotein complex. Cell.. (1991) ;66: :1121–31. |
[4] | Ervasti JM , Campbell KP . A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. (1993) ;122: :809–23. |
[5] | Kanagawa M , Toda T . The genetic and molecular basis of muscular dystrophy: Roles of cell-matrix linkage in the pathogenesis. J Hum Genet. (2006) ;51: :915–26. |
[6] | Kanagawa M , Kobayashi K , Tajiri M , Manya H , Kuga A , Yamaguchi Y , et al. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep. (2016) ;14: :2209–23. |
[7] | Praissman JL , Campbell L . The functional mannose glycan on alpha-dystroglycan contains a phosphoribitol primed for matriglycan addition. 2016;eLife 5. |
[8] | Gerin I , Ury B , Breloy I , Bouchet-Seraphin C , Bolsée J , Halbout M , et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat Commun. (2016) ;7: :11534. |
[9] | Brockington M , Blake DJ , Prandini p , Brown SC , Torelli S , Benson MA , et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystoglycan. Am J Hum Genet. (2001) ;69: :1198–209. |
[10] | Beltran-Valero de Bernabé D , Voit T , Longman C , Steinbrecher A , Straub V , Yuva Y , et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet. (2004) ;41: :e61. |
[11] | Brockington M , Yuva Y , Prandini P , Brown SC , Torelli S , Benson MA , et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet. (2001) ;10: :2851–9. |
[12] | Burrow KL , Coovert DD , Klein CJ , Bulman DE , Kissel JT , Rammohan KW , Burghes AHM , Mendell JR . Dystrophin expression and somatic reversion in prednisone-treated and untreated Duchenne dystrophy. Neurology. (1991) ;41: :661–6. |
[13] | Fanin M , Danieli GA , Cadaldini M , Miorin M , Vitiello L , Angelini C . Dystrophin positive fibers in Duchenne dystrophy: Origin and correlation to clinical course. Muscle Nerve. (1995) ;18: :1115–20. |
[14] | Klein CJ , Coovert DD , Bulman DE , Ray PN , Mendell JR , Burghes AHM . Somatic reversion/suppression in Duchenne muscular dystrophy (DMD)evidence supporting a frame-restoring mechanism in rare dystrophin-positive fibers. Am J Hum Genet. (1992) ;50: :950–9. |
[15] | Uchino M , Tokunaga M , Mita S , Uyama E , Ando Y , Teramoto H , et al. PCR and immunocytochemical analyses of dystrophin-positive fibers in Duchenne muscular dystrophy. J Neurol Sci. (1995) ;129: :44–50. |
[16] | Hoffman EP , Morgan JE , Watkins SC , Partridge TA . Somatic reversion/suppression of the mouse mdx phenotype in vivo. J Neurol Sci. (1990) ;99: :9–25. |
[17] | Nicholson LVB , Johnson MA , Bushby KMD , Gardner-Medwin D . Functional significance of dystrophin positive fibres in Duchenne muscular dystrophy. Arch Dis Child. (1993) ;68: :632–6. |
[18] | Thanh LT , Nguyen TM , Helliwell TR , Morris GE . Characterization of revertant muscle fibers in Duchenne muscular dystrophy, using exon-specific monoclonal antibodies against dystrophin. Am J Hum Genet. (1995) ;56: (3):725–31. |
[19] | Wilton SD , Dye DE , Blechynden LM , Laing NG . Revertant fibres: A possible genetic therapy for Duchenne muscular dystrophy? Neuromuscul Disord. (1997) ;7: :329–35. |
[20] | Vila MC , Novak JS , Klimek MB , Li N , Morales M , Fritz AG , et al. Morpholino-induced exon skipping stimulates cell-mediated and humoral response to dystrophin in mdx mice. J Pathol. (2019) ;248: (3):339–51. |
[21] | Lu QL , Morris GE , Wilton SD , Ly T , Artem’yeva OV , Strong P , et al. Massive idiosyncratic exon skipping corrects the nonsense mutation in dystrophic mouse muscle and produces functional revertant fibers by clonal expansion. J Cell Biol. (2000) ;148: :985–96. |
[22] | Lu QL , Mann CJ , Lou F , Bou-Gharios G , Morris GE , Xue SA , et al. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat Med. (2003) ;9: :1009–15. |
[23] | Lu QL , Rabinowitz A , Chen YC , Toshifumi Y , Yin HF , Alter J , et al. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc Natl Acad Sci USA. (2005) ;102: :198–203. |
[24] | Alter L , Lou F , Rabinowitz A , Yin HF , Rosenfeld J , Wilton SD , et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med. (2006) ;12: (2):175–7. |
[25] | Crawford GE , Lu QL , Partridge TA , Chamberlain JS . Suppression of revertant fibers in mdx mice by expression of a functional dystrophin. Hum Mol Genet.. (2001) ;10: (24):2745–50. |
[26] | Brown SC , Torelli S , Brockington M , Yuva Y , Jimenez C , Feng L , et al. Abnormalities in α-Dystroglycan Expression in MDC1C and LGMD2I Muscular Dystrophies. Am J Pathol. (2004) ;164: (2):727–37. |
[27] | Chan YM , Keramaris-Vrantsis E , Lidov H , Norton JH , Zinchenko N , Gruber HE , et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum Mol Genet. (2010) ;19: :3995–4006. |
[28] | Blaeser A , Keramaris E , Chan YM , Sparks S , Cowley D , Xiao X , et al. Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum Genet. (2013) ;132: :923–34. |
[29] | Cohn RD , Henry MD , Michele DE , Barresi R , Saito F , Moore SA , et al. Disruption of DAG1 in differentiated skeletal muscle reveals a role for dystroglycan in muscle regeneration. Cell. (2002) ;11: (5):639–48. |
[30] | Awano N , Blaeser A , Keramaris E , Xu L , Tucker J , Wu B , et al. Restoration of Functional Glycosylation of alpha-Dystroglycan in FKRP Mutant Mice Is Associated with Muscle Regeneration. Am J Pathol. (2015) ;185: :2025–37. |
[31] | Tucker JD , Lu PJ , Xiao X , Lu QL . Overexpression of Mutant FKRP Restores Functional Glycosylation and Improves Dystrophic Phenotype in FKRP Mutant Mice. Mol Ther Nucleic Acids. (2018) ;11: :216–27. |
[32] | Keramaris E , Lu PJ , Tucker J , Lu QL . Expression of glycosylated alpha-dystroglycan in newborn skeletal and cardiac muscles of fukutin related protein (FKRP) mutant mice. Muscle & nerve. (2017) ;55: :582–90. |
[33] | Cataldi MP , Lu P , Blaeser A , Lu QL . Ribitol restores functionally glycosylated α-dystroglycan and improves muscle function in dystrophic FKRP-mutant mice. Nat Commun. (2018) ;9: (1):3448. |
[34] | Cataldi MP , Blaeser A , Lu P , Leroy V , Lu QL . ISPD Overexpression Enhances Ribitol-Induced Glycosylation of α-Dystroglycan in Dystrophic FKRP Mutant Mice. Mol Ther Methods Clin Dev. (2019) ;17: :271–80. |
[35] | Lu PJ , Tucker JD , Branch EK , Guo F , Blaeser AR , Lu QL . Ribitol enhances matriglycan of α-dystroglycan in breast cancer cells without affecting cell growth. Sci Rep. (2020) ;10: (1):4935. |


