
Orphan Peripheral Neuropathies
Abstract
Objectives:
Generally, neuropathies of peripheral nerves are a frequent condition (prevalence 2–3%) and most frequently due to alcoholism, diabetes, renal insufficiency, malignancy, toxins, or drugs. However, the vast majority of neuropathies has orphan status. This review focuses on the etiology, frequency, diagnosis, and treatment of orphan neuropathies.
Methods:
Literature review
Results:
Rareness of diseases is not uniformly defined but in the US an orphan disease is diagnosed if the prevalence is <1:200000, in Europe if <5:10000. Most acquired and hereditary neuropathies are orphan diseases. Often the causative variant has been reported only in a single patient or family, particularly the ones that are newly detected (e.g. SEPT9, SORD). Among the complex neuropathies (hereditary multisystem disorders with concomitant neuropathies) orphan forms have been reported among mitochondrial disorders (e.g. NARP, MNGIE, SANDO), spinocerebellar ataxias (e.g. TMEM240), hereditary spastic paraplegias (e.g UBAP1), lysosomal storage disease (e.g. Schindler disease), peroxisomal disorders, porphyrias, and other types (e.g. giant axonal neuropathy, Tangier disease). Orphan acquired neuropathies include the metabolic neuropathies (e.g. vitamin-B1, folic acid), toxic neuropathies (e.g. copper, lithium, lead, arsenic, thallium, mercury), infectious neuropathies, immune-mediated (e.g. Bruns-Garland syndrome), and neoplastic/paraneoplastic neuropathies.
Conclusions:
Though orphan neuropathies are rare per definition they constitute the majority of neuropathies and should be considered as some of them are easy to identify and potentially treatable, as clarification of the underlying cause may contribute to the knowledge about etiology and pathophysiology of these conditions, and as the true prevalence may become obvious only if all ever diagnosed cases are reported.
ABBREVIATIONS
AD | Autosomal dominant |
AHSCT | Allogenic hematopoetic stem cell transplantation |
AIDP | Acute inflammatory demyelinating polyneuropathy |
AR | Autosomal recessive |
ASCT | Autologous stem cell transplantation |
BGS | Bruns Garland syndrome |
CANOMAD | Chronic ataxic neuropathy with ophthalmoplegia, M-proteins, cold agglutinins and disialosyl antibodies |
CANVAS | Cerebellar ataxia, neuropathy, vestibular areflexia syndrome |
CASPR-1 | Contactin-associated protein-1 |
CIDP | Chronic, inflammatory demyelinating polyneuropathy |
CIPN | Chemotherapy-induced polyneuropathy |
CMT | Charcot-Marie-Tooth |
CNS | Central nervous system |
CNTN-1 | Contactin-1 |
DADS | Distal, acquired, demyelinating, symmetric neuropathy |
EGPA | Eosinophilic granulomatosis with polyangitis |
EHE | Epithelioid hemangio-endothelioma |
FRDA | Friedreich ataxia |
GAN | Giant axonal neuropathy |
GBS | Guillain-Barre syndrome |
GFAP | Glial fibrillary acidic protein |
HDL | High density lipoprotein |
HMN | Hereditary motor neuropathy |
HMSN | Hereditary motor and sensory neuropathy |
HNA | Hereditary neuralgic amyotrophy |
HNPP | Hereditary neuropathy with liability to pressure palsies |
HSAN | Hereditary sensory and autonomic neuropathy |
HSP | Hereditary spastic paraplegia |
IF | Intermediate filaments |
LFN | Large-fiber neuropathy |
LSDs | Lysosomal storage disorders |
MADSAM | Multifocal acquired demyelinating sensory and motor |
MAG | Myelin associated glycoprotein |
MID | Mitochondrial disorder |
MFS | Miller-Fisher syndrome |
MEMSA | Myoclonus epilepsy, myopathy, and sensory ataxia |
MGUS | Monoclonal gammopathy of unknown significance |
MMN | Multifocal motor neuropathy |
MNGIE | Mitochondrial neuropathy, gastrointestinal encephalopathy |
MT | Maternal transmission |
NARP | Neuropathy, ataxia, retinitis pigmentosa |
NCS | Nerve conduction study |
NF-155 | Neurofascin-155 |
NGS | Next generation sequencing |
NSVN | Non-systemic, vasculitic neuropathy |
PME | Progressive myoclonus epilepsy |
PNP | Polyneuropathy |
PNS | Peripheral nervous system |
POEMS | Polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes |
SANDO | Sensory ataxic neuropathy with dysarthria and ophthalmoparesis |
SCA | Spinocerebellar ataxia |
SCAE | Spinocerebellar ataxia with epilepsy |
SCLC | Small cell lung carcinoma |
SFN | Small fiber neuropathy |
SSN | Subacute sensory neuropathy |
SSR | Sympathetic skin response |
TTR | Transthyretin-related |
VEGF | Vascular endothelial growth factor |
WES | Whole exome sequencing |
XL | X-linked |
INTRODUCTION
Generally, neuropathies of peripheral nerves are a highly prevalent neurological problem, due to the high prevalence of diabetes, alcoholism, and renal insufficiency [1]. The remaining neuropathies are rare and there are a number of neuropathies, which have been only reported in a few patients or a single patient (orphan neuropathies). Concerning these rare neuropathies, prevalence figures are hardly available and often questionable due to limited coverage of a region or insufficient work-up or cooperation between centres. Generally, rare neuropathies occur among all subtypes, hereditary and acquired (metabolic, immune, neoplastic, infectious, toxic). This review focuses on recent advances concerning the etiology, clinical presentation, diagnosis, treatment (if available), and outcome of rare neuropathies of peripheral nerves.
DEFINITION OF RARENESS
The term “rare” is poorly defined in Medicine. It may implicate that only a “few” cases have been reported worldwide so far or it may implicate that the condition can be hardly found in a particular region but may occur with a much higher frequency in another region. Subjectively, a rare disease may be one that an individual physician has not encountered during his practice yet but this depends strongly on the duration of his experience and the setting he is working in. Quantitatively, rareness of a disease can be defined as a disorder with <10 hits on a broad PubMed search. With regard to prevalence rareness is defined as neuropathy occurring in less than 1:10000 subjects. The definition of an orphan disease is not uniform and varies from country to country [2]. Figures vary from 1:1000 to 1:200000 (e.g. USA) people nationwide [3]. In Europe, orphan diseases are those with a prevalence of <5:10000. Ultra-rare diseases have a prevalence of <1:50000. It is estimated that there are about 6000 (5000–7000) orphan diseases [3]. For the majority of rare neuropathies, figures about the true prevalence and incidence are unavailable. Thus, frequency figures largely rely only on estimations [4].
CLASSIFICATION OF NEUROPATHIES
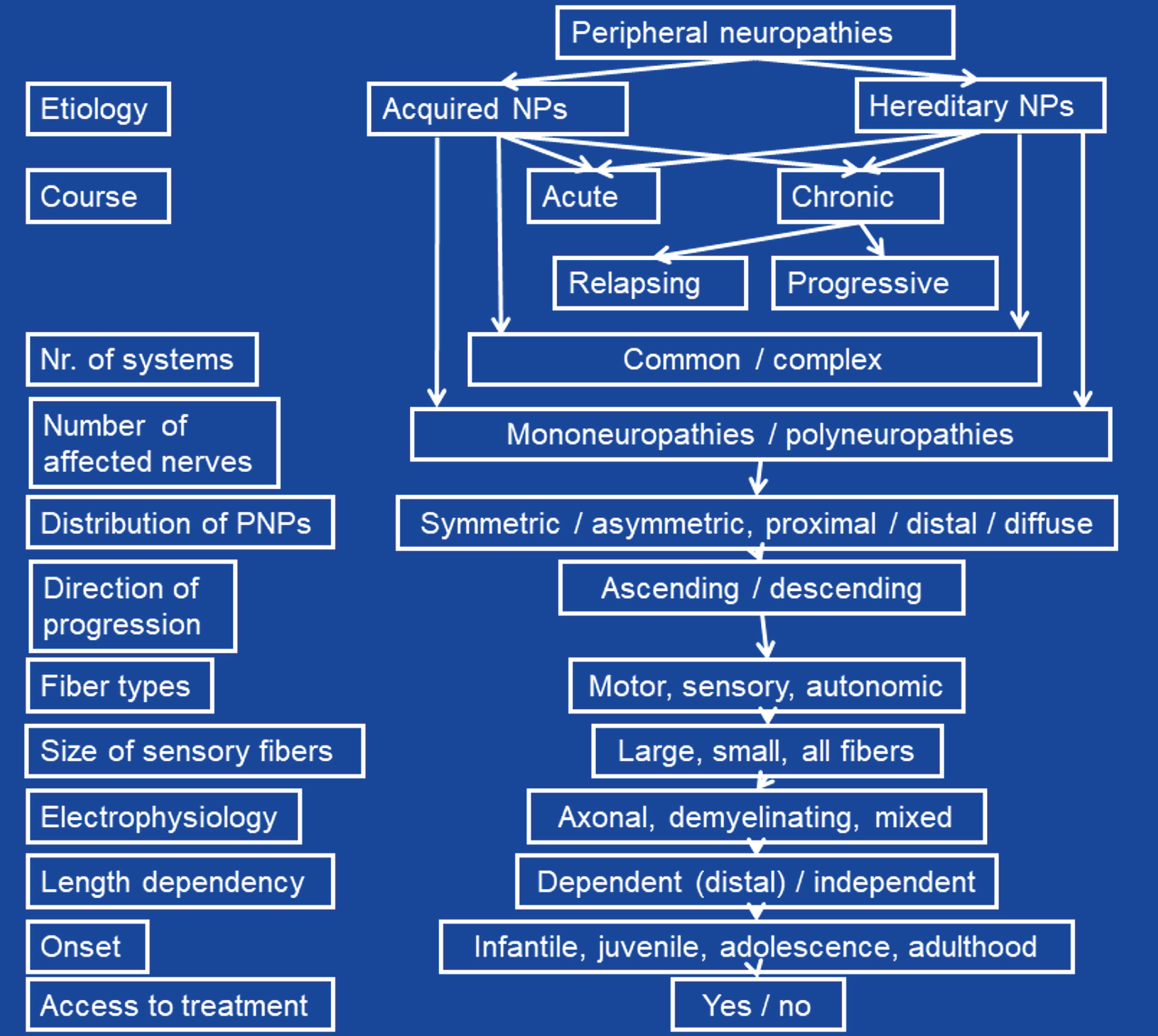
Neuropathies of peripheral nerves may be classified according to various criteria (Fig. 1). The most important classification is the one according to the etiology, which differentiates between acquired or inherited forms. Among acquired and hereditary neuropathies further subdivision have been proposed (see below). Acquired and hereditary neuropathies are classified according to the course as acute or chronic. Acquired and hereditary neuropathies may be accompanied by involvement of other systems/organs/tissues or not. The latter are further classified according to the degree of co-affection of other organs or tissues as dominant or non-dominant. In non-dominant cases, neuropathy is overshadowed by manifestations of other organs. The course may be chronic progressive or relapsing remitting (Tangier’s disease, porphyrias). Progressive neuropathies may be slowly or rapidly progressive. According to the number of affected nerves, mono-neuropathies and polyneuropathies (PNPs) are distinguished. Among PNPs those with uniform affection of all nerves and those with predominant affection of several single nerves (multiplex neuropathy) are differentiated. PNPs are further classified according to the distribution as symmetric or asymmetric and as proximal or distal. Furthermore, neuropathies of the cranial nerves and the peripheral nerves are delineated. According to the direction of progression neuropathies can be ascending or descending. According to the types of nerve fibers affected they may be classified as motor, sensory, autonomic, or mixed neuropathies. According to the size of affected fibers, large- and small-fiber neuropathies (LFNs, SFNs) are delineated. Upon electrophysiological findings, axonal, demyelinating (with or without conduction block), or mixed neuropathies are differentiated. According to the prevalence, they may be classified as frequent or infrequent (rare). Neuropathies can be painful or painless. Neuropathies can be length-dependent (i.e. distal, the longer the more severe, dying-back), which is the case for most of the neuropathies, or length-independent. Lastly, neuropathies may have an infantile/childhood onset or may have an onset in adolescence/adulthood. Some neuropathies may be accessible to treatment, others may not.
Fig. 1
Classification of orphan peripheral neuropathies according to various criteria.
FREQUENCY OF PNPS
It is estimated that the prevalence of peripheral neuropathies is 2–3% in the general population [5]. However, in patients >55 y of age the prevalence increases to 8% [5]. Most of the neuropathies of undetermined cause are rare neuropathies but many of the acquired and hereditary neuropathies are rare either. Exact prevalence/incidence data about rare neuropathies are hardly available. In the rare case that prevalence/incidence data about neuropathies are available, they are representative only for a certain region but usually not for a nation, a continent, or the globe. Low prevalence data may result from inadequate effort to detect the cause or insufficient tools to search for the etiology. For example, transthyretin-related (TTR)-amyloid neuropathy is infrequent (estimated world-wide prevalence: >10000) because it may remain undetected for some time as long as TTR is considered as a differential diagnosis of neuropathy of undetermined cause. The prevalence of TTR-amyloid neuropathy is 22.93/100000 in Portugal [6], 8.7–11/100000 in Japan [7], and 91–104/100000 in Sweden [8]. The prevalence of chronic, inflammatory demyelinating polyneuropathy (CIDP) is calculated as 2.8–3.0/100000.
DIAGNOSIS OF PERIPHERAL NEUROPATHIES
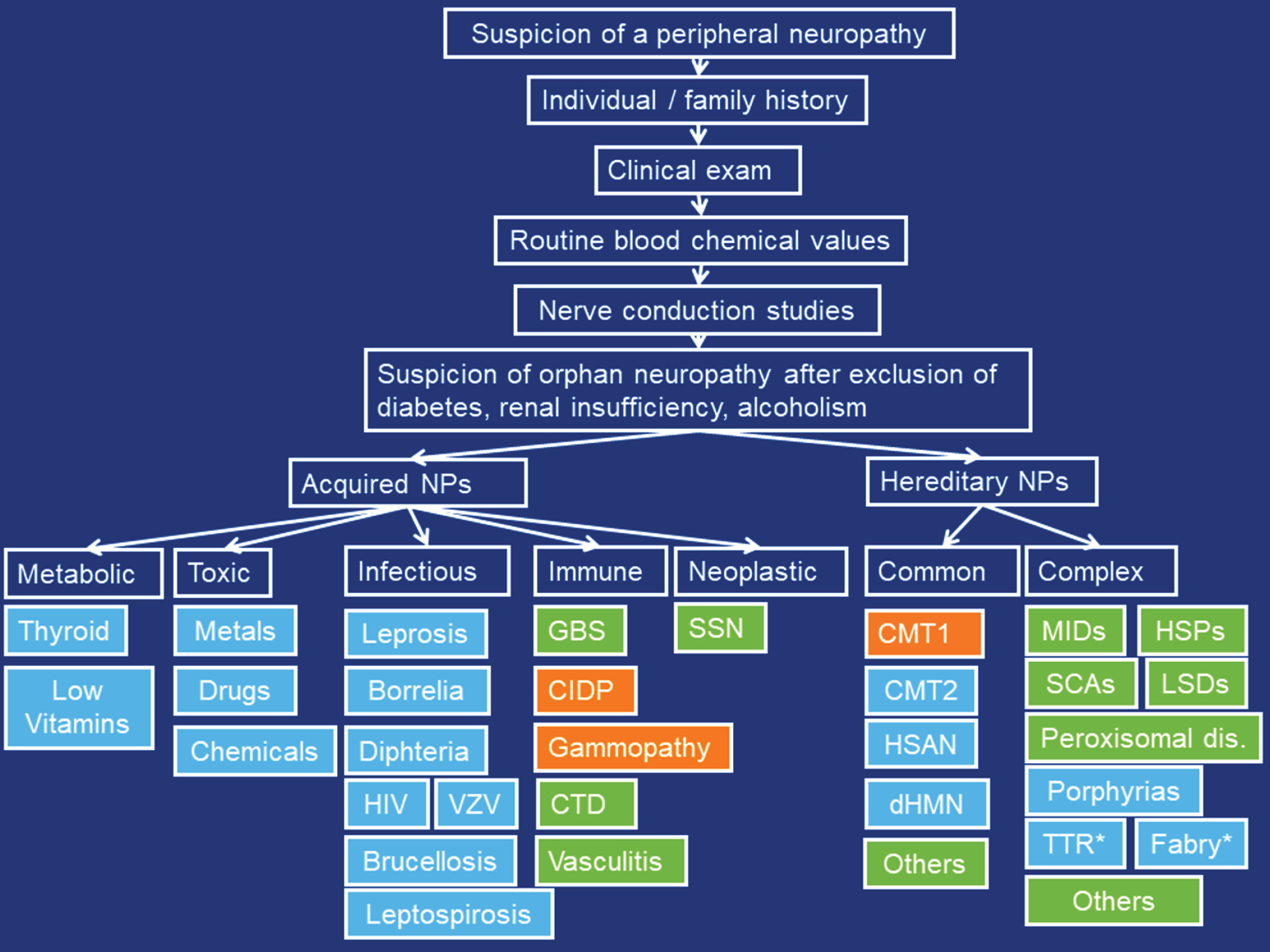
Multiple algorithms for diagnostic work-up of neuropathies are available [9]. They all rely on the history, clinical exam, blood tests, NCSs, CSF investigations, imaging, biopsy and genetic investigations. Selection of laboratory investigations is variable, depending on the clinical presentation, availability, costs, and expertise of the referring neurologist. Generally, answers to the questions “what” (which nerve fiber modalities are involved), “where” (which are the complaints), “when” (which is the temporal evolution), and “which setting” (unique clinical circumstances of a patient) should be obtained to classify the type of neuropathy [5]. For work-up of hereditary neuropathies, it is crucial that a thorough individual and family history is taken and that apparently asymptomatic family members are seen by the managing physician for subclinical weakness, wasting, or foot deformities, since the affected subject may not recognise subtle deficiencies himself. As soon as hereditary neuropathy is suspected, genetic work-up (e.g. multigene panel, next generation sequencing (NGS)) should be initiated. NGS has the disadvantage that it does not reliably detect duplications or deletions, splice-site variants, or intronic variants and is difficult to interpret in the context of non-specific point mutations (variants of unknown significance) with oligogenic inheritance [10]. Additionally, NGS has a cost-depth trade-off [10], that is, it can screen a small number of genes with good read depth or a large number with less depth [10]. A list of Charcot-Marie-Tooth (CMT) genes can be accessed via the inherited neuropathy variant browser (http://hihg.med.miami.edu/neuropathybrowser). Work-up for SFNs relies on skin biopsy as golden standard but can be supported by quantitative sensory testing, electrophysiology, and the skin-responsive Sudoscan®. After diagnosing SFN, work-up for the underlying cause must follow. In patients with prominent muscle atrophy but little muscle weakness or sensory deficits and preserved tendon reflexes, a vascular neuropathy should be considered. In asymmetric PNPs an infectious, immune-mediated, or neoplastic etiology should be considered. Nerve biopsy is indicated for vasculitis, amyloidosis, sarcoidosis, or leprosy, but may be helpful also for CIDP. A proposal for the diagnostic work-up of orphan neuropathies is provided in Fig. 2.
Fig. 2
Proposed diagnostic work-up for orphan peripheral neuropathies. Orange: demyelinating, blue: axonal, green: mixed. CTD: connective tissue diseases, SSN: subacute sensory neuropathy, MIDs: mitochondrial disorders, HSPs: hereditary spastic paraplegias, SCAs: spinocerebellar ataxias, LSDs: lysosomal storage diseases, TTR: transthyretin-related neuropathy, *: Treatable.
ACQUIRED NEUROPATHIES
Acquired neuropathies include the metabolic, toxic, infectious, immune-mediated, and neoplastic/paraneoplastic neuropathies [5]. As with hereditary neuropathies, acquired neuropathies can be the exclusive manifestation of a disease or may occur together with affection of other organs. The majority of acquired neuropathies is frequently diagnosed but some have to be assessed as rare. Generally, acquired neuropathies are more frequent than hereditary neuropathies.
Metabolic neuropathies
Generally, metabolic neuropathies include diabetic, uremic, endocrine, and nutritive neuropathies. Particularly, among the nutritive neuropathies rare forms are known. Nutritive neuropathies are most frequently length-dependent, sensory neuropathies with the exception of vitamin-B12 deficiency neuropathy [11]. While diabetic and uremic neuropathies are highly prevalent, most of the other metabolic neuropathies are rare.
Endocrine
Hypothyroidism: Though hypothyroidism is a frequent abnormality worldwide, neuropathy attributable to hypothyroidism is extremely rare. The reason is that hypothyroidism is well-controlled in the majority of the cases, why the prevalence of neuropathy has declined. Onset of hypothyroid neuropathy is usually in early adulthood. Neuropathy due to hypothyroidism usually starts as SFN and may affect large fibers with progression of the disease [12]. Patients initially complain about paresthesias and numbness of the feet. Hypothyroid neuropathy may manifest as mononeuropathy in the form of a carpal tunnel syndrome. NCSs reveal a length-dependent, sensory-motor axonopathy [13]. Hypothyroid neuropathy can be complicated by myopathy, which is more frequent than neuropathy.
Hyperthyroidism: Hyperthyroidism as a cause of neuropathy is extremely rare and debated. The clinical presentation can be similar to that of hypothyroidism but may also show up as a Guillain-Barre syndrome (GBS)-like presentation [14]. NCSs reveal a symmetric, sensori-motor, mixed, neuropathy with active denervation in lower extremity muscles. More frequent than neuropathy is myopathy due to hypothyroidism.
Vitamin deficiencies
Though vitamin deficiencies are frequently detected in the general population, neuropathy due to vitamin deficiency, with/without involvement of other systems, has been only rarely reported. Low vitamin levels may be due to dietary causes (malnutrition, malabsorption, diarrhea), autoimmune conditions (e.g. perniciosa), certain drugs, alcoholism, chronic colitis, bariatric surgery, or due to other gastrointestinal compromise. Low vitamins that can go along with neuropathy include vitamin-B12, folic acid, thiamine (vitamin-B1), vitamin-B6, and vitamin-E. Why only a small portion of patients with vitamin deficiency develops neuropathy, remains speculative but one hypothesis is that pre-existing nerve pathology may be necessary for the development of a vitamin-deficiency-related neuropathy.
Vitamin-B12: Vitamin-B12-deficiency may be associated with distal, symmetric PNP and mainly, as in funicular myelosis, proprioceptive dysfunction. About 4% of the distal symmetric PNPs are caused by vitamin-B12-deficiency [15]. Normal blood levels of vitamin-B12 do not exclude a clinically relevant, relative vitamin-B12-deficiency [16]. In such cases, increased methyl-malonate and homocysteine levels can support the diagnosis of relative vitamin-B12-deficiency [16]. However, methyl-malonate and homocysteine can be also increased in hypothyroidism, renal insufficiency, hypovolemia, vitamin-B6 deficiency, or in patients with heterozygous homocysteinemia [16, 17]. Contrary to other nutrition neuropathies, vitamin-B12-deficiency manifests with a length-independent, sensory neuropathy [11]. Patients with vitamin-B12-deficiency usually present with concomitant myelopathy [11].
Folic acid: PNP due to folate deficiency is rare and manifests with slowly progressive distal symmetric, sensory neuropathy [18]. Folate-deficient neuropathy predominantly affects the lower limbs. Cognitive and affective symptoms of folate-deficiency are far more common than PNP and the laboratory test for folate-levels is only relevant in this clinical context [19]. Since folate deficiency causes secondary thiamine deficiency, neuropathy in folate deficiency can be blended by thiamine deficiency neuropathy [20].
Thiamine: Not only central nervous system (CNS) symptoms occur with thiamin-deficiency (Wernicke encephalopathy, Marchiafava-Bignami, dry beriberi), but also distal axonal, sensory-motor, length-dependent PNP has been described. Thiamine deficiency-related PNP is often painful and has been described in the context of bariatric surgery or severe alcohol-abuse [21].
Vitamin-B6: Vitamin-B6 deficiency almost exclusively develops in the context of drugs that reduce vitamin-B6 resorption (e.g. isoniazid) [22] and cause axonal distal sensory-motor PNP. A further cause of vitamin-B6 deficiency can be chronic dialysis [23].
Vitamin-E: Vitamin-E deficiency may cause a distal symmetric PNP with severe, sensory ataxia. Since vitamin-E deficiency is frequently associated with a spinocerebellar syndrome, concomitant cerebellar ataxia may occur [11, 24]. However, vitamin-E deficiency is a rare cause of PNP and serum levels of vitamin-E should be determined only in patients with expected vitamin-E deficiency.
Toxic neuropathies
Metals
Metal intoxications most frequently not only cause isolated neuropathy but also systemic manifestations. Thus, systemic features should be considered in the diagnostic work-up of suspected metal intoxications.
Copper: Generally, copper deficiency (hypocupremia) manifests with bone marrow dysplasia, myelopathy, and neuropathy [25]. Neuropathy due to hypocupremia is rare and predominantly occurs after bariatric surgery, gastrectomy, or due to alcoholism [26]. Neuropathy is progressive, ascending and associated with gait ataxia, cerebellar ataxia, fatigue, dyspnea, macrocytic anemia, neutropenia, and unintentional weight loss. Neuropathy due to copper deficiency is usually sensory and symmetric. The clinical presentation is similar to that of funicular myelosis. Diagnostic is hypocupremia and low copper in the bone marrow biopsy. Serum copper should be particularly determined in patients with a funicular myelosis presentation but normal folic acid and cobalamin levels [13].
Lithium: Lithium has long been used in psychiatry as an adjuvant therapy for bipolar disorders. Occasionally, long-term treatment with lithium results in chronic intoxication, clinically manifesting as nystagmus, ataxia, tremor, memory impairment, myoclonus with generalised triphasic epileptiform discharges, dysarthria, fasciculations, clonus, and PNP [27]. PNP is usually a length-dependent, sensorimotor axonopathy. Frequently, lithium neuropathy shows a rapidly progressive course [28]. Patients under lithium therapy with symptoms and signs of PNP require quantification of the lithium serum level, NCSs, and eventually replacement of the causative agent. Discontinuation of lithium is usually followed by improvement or resolution of the PNP [13].
Lead: Lead intoxication most frequently results from industrial exposure and can cause a progressive, mostly asymmetric motor PNP that affects the upper extremities more than the lower extremities. Classical lead neuropathy manifests with weakness of the wrist and finger extensors [29]. Lead intoxication often also causes anemia, gingival, dental, and cerebral changes (lead encephalopathy). The most well-known gingival abnormality is Burton’s line [30]. The prognosis of recovery is favorable as long as exposure is terminated promptly [29].
Arsenic: The causes of arsenic intoxication are poisoning with contaminated water, ingestion of traditional medicine for obesity, or attempted homicide. Neuropathy is one among other manifestations of arsenic intoxication and presents as subacute, progressive, painful, sensory-motor neuropathy with autonomic dysfunction [31]. In some cases, neuropathy can be the only manifestation of the intoxication [31].
Thallium: Thallium intoxication has been reported in cases with accidental ingestion, suicide attempt, or criminal adulteration. Thallium intoxication may cause a progressive painful, sensory-motor PNP with autonomic dysfunction. In addition to neuropathy patients may develop visual loss, myalgia, leg weakness, acute gastrointestinal symptoms, or alopecia [13].
Mercury: Mercury intoxication causes an axonal, sensory PNP with small-fiber involvement and autonomic dysfunction [19, 32]. In addition to neuropathy, patients with mercury intoxication may present with diffuse full-body rash, fever, myalgias, headache, oral paresthesias, and tender cervical posterior lymphadenopathy.
Cobalt: Cobalt toxicity usually results from failed total hip replacement. Cobalt metallosis is rare but can be devastating [33]. Clinical manifestations of cobalt toxicity include cardiomyopathy, hypothyroidism, skin rash, visual and hearing impairment, polycythemia, muscle weakness, fatigue, cognitive impairment, and neuropathy [33]. Neuropathy is a progressive, length-dependent, sensory axonopathy with normal motor function [13].
Drugs
Neuropathy can be a side effect of several drug intoxications. Most well-known for their neuropathic toxicity are chemotherapeutics and nucleoside analogs. However, neuropathy may be also a side effect of non-chemotherapeutic drugs.
Non-chemotherapeutic drugs: Among the non-chemotherapeutic drugs neuropathy is most frequently caused by phenytoin, disulfiram, fluoroquinolones, linezolide, metronidazole, chloroquine, dapson, isoniazide, ethambutol, nitrofurantoin, nucleoside-analogs (dideoxycytidine, zalcitabine, dideoxiinosine, stavudine, lamivudine, thalidomide, tacrolimus, leflunomide, amiodarone, or colchicine. Rarely, toxic neuropathy occurs in association with vitamin-B6, L-DOPA, or statin overdose [34].
Vitamin-B6: Vitamin-B6 is the only vitamin that may cause PNP in the context of increased blood levels, which is mostly due to excess vitamin supplementation. Vitamin-B6 levels need to be elevated 50–100 times the upper level of normal to induce neuropathy. Vitamin-B6 toxicity causes a non-length dependent, sensory PNP with marked ataxia or a painful SFN [11, 35].
L-DOPA: Neuropathy due to L-DOPA is a length-dependent, sensory axonopathy, possibly related to elevated homocysteine or neuronal deposition of α-synuclein. In a study of 73 Parkinson patients without a history of previous PNP, 67.3% of those taking L-DOPA developed a PNP, whereas in the non-L-DOPA group only one patient developed PNP [36]. The results imply that long-term L-DOPA intake can secondarily cause vitamin-B12 and folate deficiency and consecutively PNP [36, 37].
Chemotherapeutics: Chemotherapy-induced peripheral neuropathy (CIPN) is a frequent complication of various chemotherapeutics. Most frequently neuropathy has been described in association with taxanes, vinca-alkaloids, platanes, and bortezomib. However, there are chemotherapeutics, such as the immune checkpoint inhibitors or eribulin, which have been only rarely described to be complicated by neuropathy. Immune checkpoint inhibitors (e.g. pembrolizumap, nivolumab, ipilimumab) are a new class of chemotherapeutic drugs being effective with refractory cancers [13]. Peripheral nervous system (PNS) side effects are rare and occur in <1% of the patients. PNS adverse events include symmetric, distal, axonal PNP, GBS, CIDP, and radiculitis [13]. Neuropathy occurs within 2–3 weeks after initiation of treatment. Neuropathy can be severe but resolves upon discontinuation of the drugs, and treatment with steroids, IVIG, or plasmapheresis [13]. Eribulin is a chemotherapeutic agent used to treat acute lymphatic leukaemia, myeloma, and metastatic breast cancer [38]. The most frequent non-hematological side effect of eribulin is an axonal, length-dependent, sensory PNP of mild to moderate severity [38]. Neuropathy resolves after discontinuation of the compound. Vinca-alkaloids, platins, taxanes, and thalidomide are more neurotoxic than eribulin. The initial manifestation of a CIPN may be SFN [39].
Chemicals and plants
A number of chemicals (e.g. nitric oxide, vinyl-benzene, di-ethylene-glycol, hexa-carbon, acryl-amide, carbon-disulfide, and many others), is toxic to peripheral nerves [13]. Neuropathy due to intoxication with any of these molecules has been only rarely described. For example, acryl-amide causes, in addition to encephalopathy, dermatitis, and hyperhidrosis, a sensori-motor axonopathy [13]. Acryl-amide is neurotoxic as it directly attacks DNA or is transformed into glycidamid by liver enzymes [40]. Acrylamid and glycidamide bind to aminoacids and nucleotides and thus directly impair DNA functions. Hexa-carbon causes a subacute, length-dependent, sensori-motor axonopathy with secondary demyelination [13]. A length-dependent, sensori-motor axonopathy is also caused by carbon-disulfide. Diethylene-glycol causes an acute, rapidly progressive, sensori-motor axonopathy with demyelination [13]. Toxicity of vinyl-benzene manifests as painful, sensory, length-dependent SFN [13]. An example of a plant causing neuropathy is the buckthorn fruit, which occurs in south-west US and Mexico. Buckthorn causes a rapidly progressive, length-dependent, sensori-motor axonopathy with secondary demyelination [13]. The neuro-toxic agent in Buckthorn intoxication has been identified as T-544 [13].
Infectious neuropathies
The prevalence of infectious neuropathies is quite variable and strongly depends on the region of investigation [41]. What may be rare in one region, country, or continent, may be highly prevalent in another country. The prevalence of infectious neuropathies may also increase in association with epidemic outbreaks. Infectious agents most frequently causing neuropathy include M. Leprae (South America, Asia, Africa), Borrelia burgdorferi (Europe, America), C. diphteriae (Asia), HIV (worldwide), VZV (worldwide), hepatitis-C (worldwide), HTLV1 (worldwide), Zika (GBS) (South America), influenza-A (GBS) (Europe), CMV (Europe, America), dengue (GBS) (Asia, Africa), Toscana-virus (Europe, Africa), and other arboviruses.
Immune-mediated neuropathies
Guillain Barre syndrome
The term GBS includes a group of autoimmune disorders that share a common presentation of acute/subacute, progressive poly-radiculo-neuropathy [42]. The incidence of GBS varies between regions and countries from 1.1–1.8/100000/y to 2.66/100000/y [42]. There are several subtypes of GBS classified according to the underlying pathology, clinical presentation, and NCS features [42]. The most common subtype in Europe is acute, inflammatory, demyelinating poly-radiculo-neuropathy (AIDP) with primarily demyelinating features and a favourable prognosis [42]. Less common is acute, axonal, motor neuropathy (AMAN) with primary axonal injury, pure motor involvement, and a worse prognosis [42]. AMAN is the most prevalent subtype in East Asia. A rare cause of dysimmune and acquired neuropathy is acute painful autoimmune neuropathy, an acute small fiber neuropathy resembling GBS [43]. Acute, motor, sensory, axonal neuropathy (AMSAN) shares a similar pathogenesis with AMAN with additional sensory involvement. Miller-Fisher syndrome (MFS), is characterised by opthalmoparesis, areflexia, and ataxia. Bickkerstaff encephalitis presents similarly to MFS but additionally with impaired consciousness due to brainstem involvement. Less common variants are the pharyngeal-cervico-brachial variant, associated with GQ1b and GD1a antibodies and pandysautonomia, associated with GT1a antibodies [42, 44]. GBS is often preceded by an infection with Campylobacter jejunii, mycoplasma pneumoniae, EBV, CMV, influenza-A, or hepatitis-E [42, 45]. Micro-organisms more rarely triggering GBS include scrub typhus (Tsutsugamishi fever) [46], Chikungunya [47], rubella virus [45], VZV, Toscana virus, hepatitis-C, hepatitis-A, hepatitis-B, West Nile, COVID-19, SARS-CoV-2, or Zika [48]. Rare, non-infectious triggers of GBS include hyponatriemia [49], bariatric surgery [50], lupus, pregnancy [42], or bacterial meningitis [51]. Antibodies associated with GBS include GM1/GD1a (AIDP), GM1/GT1a/GD1a (AMAN), or GD1a/GQ1b (AMSAN) [42].
CIDP
CIDP is a chronic, demyelinating, poly-radiculo-neuropathy, which manifests clinically with symmetric weakness and sensory disturbances. CIDP is diagnosed according to the EFNS/AAN/INCAT criteria if there is progressive or recurrent symmetric weakness of proximal and distal upper and lower extremities for at least 2 months. Frequently, there is dissociation cytoalbuminique and there is demyelination on NCSs (reduced or normal motor conduction velocity, conduction block, temporal dispersion, increased F-wave latency, prolongation of distal latency and of the CMAP duration) [52]. According to these findings definite, probable, and possible CIDP are delineated. Several rare variants have been identified since the original description, including multifocal acquired demyelinating sensory and motor (Lewis-Sumner syndrome, MADSAM) neuropathy, a pure sensory form, distal, acquired, demyelinating, symmetric CIDP (DADS) neuropathy, and syndromes associated with monoclonal gammopathy [44]. Rarely, nerves innervating respiratory muscles or involvement of the autonomic nerves (bowel or bladder dysfunction, dry mouth/eyes, orthostatic intolerance, sexual dysfunction, flushing, hyperhidrosis) have been described [52]. In 2–6% of the cases CIDP is associated with elevated titers of auto-antibodies against the paranodal protein contactin-1 (CNTN-1), neurofascin-155 (NF-155), or contactin-associated protein-1 (Caspr1) [52]. These patients more frequently present with an acute onset, tremor and ataxia than CIDP patients without these antibodies and often poorly respond to standard immunotherapy [52]. Anti-CNTN-1 antibodies have been shown to interfere with the structural integrity of the paranodal region and are known to induce conduction block [53]. Occasionally, antibodies against gangliosides or MAG, are elevated in CIDP [44]. Most frequently these antibodies are found in patients with multifocal motor neuropathy (MMN) (GM1), DADS (anti-MAG), or chronic ataxic neuropathy with ophthalmoplegia, M-proteins, cold agglutinins and disialosyl antibodies (CANOMAD). The other parameters lack sufficient sensitivity and specificity to be used as reliable biomarkers. First-line treatment of CIDP includes steroids, intravenous immunoglobulins, and plasmapheresis.
Neuropathies related to monoclonal gammopathies (paraproteinemias)
Monoclonal gammopathies are a heterogeneous group of lymphoproliferative disorders characterised by overproduction and deposition of paraproteins (light chains, immunoglobulins). Monoclonal gammopathies include monoclonal gammopathy of unknown significance (MGUS), multiple myeloma, amyloidosis, Waldenström macroglobulinemia, and CANOMAD, polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, and skin changes (POEMS) syndrome [13]. Neuropathy is a common manifestation in monoclonal gammopathies and attributed to a cross-reaction of the paraprotein with neural antigens, deposition of paraproteins in nerve or myelin, or due to a neurotoxic effect of the paraprotein [13]. Other pathomechanisms include vasculitis due to cryoglobulinemia or infiltration of the neve roots by a lymphoma. Neuropathy is particularly common in IgM-MGUS, multiple myeloma, and amyloidosis. More rarely, neuropathy develops in the remaining four types.
Waldenström’s macroglobulinemia: Waldenström’s macroglobulinemia is a rare subtype of B-cell lymphoma with elevated monoclonal IgM and clinically characterised by hyperviscosity syndrome, swelling of lymph nodes, cold agglutinine disease, anemia, thrombocytopenia, elevated IgM, amyloidosis, splenomegaly, Bing-Neel syndrome (infiltration of the CNS with malignant B-lymphocytes), and neuropathy [54]. Neuropathy in Waldenström’s macroglobulinemia occurs in one fifth to one third of the patients [54]. Neuropathy is usually a chronic, progressive, symmetric, predominantly distal PNP [55]. NCSs show demyelinating features. Anti-MAG antibodies are elevated in about 50% of cases. Treatment relies on rituximab in combination with cyclophosphamide, hydroxy-doxorubicin, vincristine, and prednisone (R-, therapy) [54]. Treatment with drugs that induce toxic neuropathy such as vincristine, should be avoided. R-CHOP therapy is recommended but the use of vincristine may worsen neuropathy. Treatment should be discussed with hematologists in terms of risk benefit.
POEMS syndrome: POEMS syndrome is a rare paraneoplastic (lymphoma, Castleman syndrome, myeloma, plasmocytoma) condition, characterised by PNP, organomegaly due to elevation of serum vascular endothelial growth factor (VEGF), endocrinopathy, M-gradient λ-light chains derived from the germlines IGLV1–40 and IGLV1–44, extravascular volume overload, and skin changes [56]. Two thirds experience clonal plasma cell bone marrow infiltration and one third develops a plasmocytoma. Neuropathy in POEMS syndrome is severe, distal, sensory-motor, and predominantly of the demyelinating type [57]. Treatment is directed towards targeting monoclonal plasma cells with thalidomide or autologous stem cell transplantation (ASCT) [56]. Additionally, melphalan and dexamethason or lenalidomide and dexamethasone can be beneficial. If myeloma is of the osteosclerotic type radiation therapy can be beneficial. Whether these types of treatment are also effective for neuropathy is unknown but conceivable.
CANOMAD: CANOMAD is an ultra-rare paraproteinemia, clinically manifesting with chronic ataxic neuropathy and ophthalmoparesis leading to gait disturbance. In some patients cranial nerves may be additionally affected [58]. NCSs may show a mixed axonal/demyelinating pattern, pure axonal lesions, or rarely pure demyelinating lesions [59]. Ultrasound may show nerve enlargement patterns as in acquired demyelination [59]. Antibodies against GD1b and other disialosyl anti-ganglioside antibodies can be elevated [52]. Cold agglutinins may be positive. Patients with CANOMAD may respond favourably to intravenous immunoglobulins [60] or rituximab [61]. Autopsy in these patients may reveal dorsal column atrophy and dorsal root ganglionopathy, and infiltration of clonal B-lymphocytes within the endoneurium, perineurium, and leptomeninges [62].
Connective tissue disorders
Connective tissue disorders are multisystem, autoimmune disorders that show common features of organ inflammation (most commonly joints and skin) [13]. In many of the connective tissue disorders the CNS or PNS are involved. Neuropathy has been most frequently reported in lupus erythematosus and rheumatoid arthritis (75% of patients). Rarely, neuropathy occurs in sarcoidosis, Sjögren syndrome, or systemic sclerosis (scleroderma). Neuropathy in sarcoidosis frequently starts as SFN with prominent pain, areas of tenderness, numbness, and dysesthesias, responding only to immunoglobulins or TNF-alpha blockers [13]. LFN is much less common and manifests as length-independent PNP or polyradiculopathy, or rarely as mononeuritis multiplex, GBS (AIDP), symmetric sensori-motor neuropathy, or as pure sensory neuropathy [13]. Neuropathy in Sjögren’s syndrome is usually a sensory, length-dependent PNP. Occasionally, sensory symptoms are patchy and asymmetric, suggesting ganglionopathy [13]. More rarely, neuropathy is sensori-motor taking a subacute or chronic course. Rarely, neuropathy presents as polyradiculopathy, mononeuritis multiplex, or autonomic neuropathy [13]. Neuropathy in systemic sclerosis (scleroderma) manifests as length-independent numbness and paresthesias of hands and feet [13].
Vasculitis
Vasculitic neuropathies result from non-infectious inflammation of the vasa nervorum [63]. Vasculitic neuropathy may occur in the context of systemic inflammation or from vasculitis confined to the peripheral nerves (non-systemic vasculitic neuropathy (NSVN)) [63]. Vasculitis may be primary (arteritis originates from the arteries) or secondary (due to conditions such as connective tissue diseases, infections, paraneoplastic, immune-mediated conditions, or drug-induced) [63]. Vasculitic neuropathy typically presents with an acute or subacute onset of multiple, painful, sensory, or sensori-motor mononeuropathies [63]. All sensory modalities are affected as pure SFNs are rarely vasculitic [63].
Primary vasculitis
Systemic vasculitis: Systemic vasculitis manifesting with neuropathy includes panarteriitis nodosa, microscopic polyangiitis, eosinophilic granulomatosis with polyangitis (EGPA, formerly Churg-Strauss), granulomatosis with polyangitis (formerly Wegener granulomatosis), Hennoch-Schönlein purpura, giant cell arteritis, and essential mixed cryoglobulinemia [63]. The prevalence of vasculitic neuropathy is 65% in EGPA (Churg-Strauss), 23% in microscopic polyangiitis, and 19% in granulomatosis with polyangiitis (Wegener) [64]. c-ANCA and p-ANCA may be positive in Wegener granulomatosis and Churg-Strauss syndrome.
Non-systemic vasculitic neuropathy (NSVN): NSVN is a single-organ vasculitis of the peripheral nerves that can be diagnosed only by nerve biopsy [65]. Subtypes of NSVN include Wartenberg migratory sensory neuropathy (Wartenberg syndrome), non-diabetic, lumbosacral, radiculo-plexus neuropathy, postsurgical inflammatory neuropathy, and Parsonage-Turner syndrome (neuralgic amyotrophy) [65]. Cutaneous polyarteritis nodosa and other skin-nerve vasculitides overlap with NSVN. Clinically, three patterns of involvement are delineated, multifocal neuropathy, distal symmetric PNP, and asymmetric PNP [65]. NSVN and neuropathy-predominant systemic vasculitis respond more favourably to steroids plus an immunosuppressant than to steroids alone [65]. After treatment, NSVN rarely spreads to other organs but one third of the patients experience relapses [65]. Long-term outcome is favourable but chronic pain is common [65].
Secondary vasculitis: Secondary vasculitis may be due to conditions such as metabolic disease, connective tissue diseases, infections, paraneoplastic conditions, immune-mediated conditions, or drug-induced. An example of non-systemic, secondary vasculitis of the PNS is Bruns-Garland syndrome (BGS), also known as diabetic amyotrophy. BGS is a rare proximal neuropathy clinically characterised by sudden onset asymmetric proximal pain and weakness [66, 67]. BGS presents with stepwise or rapid progression within 2 to 18 months [67]. The contralateral extremity becomes involved within 3d to 8 months with affection of proximal and distal muscles [67]. Neuropathy in BGS is of the axonal type. Sural nerve biopsy shows multifocal variability of nerve fiber density with non-random fiber loss [67].
Neoplastic
Neuropathies due to neoplasms are rare but have been occasionally reported as acute, demyelinating neuropathy in neurolymphomatosis due to direct infiltration of the PNS [68], as upper limb neuropathies in epithelioid hemangio-endothelioma (EHE) [69], as infiltration of the plexus by lung cancer (Pancoast) [70], as plexopathy in breast cancer, or as peripheral nerve metastasis [71].
Paraneoplastic
Paraneoplastic neuropathies are regarded as a secondary complication of a primary neoplasm due to reaction of the immune system against the neoplasm. Paraneoplastic neuropathies develop prior or after detection of a malignoma and are attributed to a remote effect of the malignoma, irrespective of neoplastic infiltration, chemotherapy, infection, or metabolic complications [72–74]. Paraneoplastic neuropathies occur in <1% of patients with malignoma [72]. Definite and possible paraneoplastic neuropathies are delineated [72]. Definite paraneoplastic neuropathies include those with a direct link between malignoma and neuropathy in the presence of an antibody, those representing a well-established paraneoplastic syndrome but without an identified antibody, and those which improve upon successful treatment of malignancy [72]. With regard to the antigen localisation, two types of antigens are delineated, intracellular and membrane-bound antigens [72]. The most frequent intracellular antigens are HuD and CRMP5. The most frequent membrane bound antigen is Caspr2 [72].
Paraneoplastic neuropathies are classified as neuronopathies (subacute sensory neuropathy (SSN (Denny Brown syndrome), lower motor neuron disease, sensory-motor neuropathy, autonomic neuropathy), as sensorimotor neuropathies without gammopathy (axonal, mixed axonal/demyelinating, demyelinating, vasculitic), as sensorimotor neuropathies with gammopathy (axonal, sensory and painful (AL-amyloidosis, myeloma), demyelinating (Waldenström, non-Hodgkin lymphoma), POEMS, vasculitic), and as neuromyotonia. An estimation of the number of these subtypes so far reported is provided by Antoine et al. 2017 [72].
The most common of the paraneoplastic neuropathies is SSN [72]. It is probably T-cell mediated and targets neurons in dorsal root ganglia [72]. Sensory disturbances are multifocal, asymmetric, involve the upper limbs in a length-independent manner, and progress rapidly over weeks [72]. NCSs show reduced or absent sensory nerve action potentials. SSN is associated with small cell lung cancer (SCLC) in 70–80% of cases [75]. Most patients have anti-Hu antibodies, some CRMP5 antibodies, and in 10% no antibodies are found. Among the sensory-motor neuropathies without gammopathy the mixed axonal/demyelinating subtype is the most frequent and associated with CRMP5 antibodies. Among those with CRMP5 antibodies about 50% develop neuropathy. Only anecdotally anti-Yo, or anti-Ma2 antibodies have been reported.
Acquired neuro-myotonia (peripheral nerve hyper-excitability), clinically characterised by cramps, stiffness, twitching, spasms, weakness, paresthesias, hyperhidrosis, and abnormal relaxation, occurs most frequently in association with thymoma, SCLC, or lymphoma. Frequently, Caspr2 antibodies and rarely netrin-1 antibodies can be found.
Pure sensory neuropathies
Pure sensory neuropathies (PSNs) are usually acquired but can be also hereditary. A subtype of acquired PSN is sensory neuropathy with anti-FGFR3-autoantibodies. PSN can be also associated with vitamin-B12 deficiency. Chronic immune sensory polyradiculopathy (CISP) is a pure sensory form of CIDP. There are pure sensory forms of LFNs associated with connective tissue disorders. PSN may be a manifestation of NSVN. Among the mononeuritis multiplex neuropathies, a pure sensory form with IgG1 deficiency has been reported. As a counterpart to MMN, multifocal sensory demyelinating neuropathy has been described. Statins are known to cause a pure sensory and autonomic ganglionopathy. Pure sensory GBS is a rare entity.
HEREDITARY NEUROPATHIES
Hereditary neuropathies can be common or complex (Tables 1 and 2). Common hereditary neuropathies are characterised by isolated or predominant affection of the peripheral nerves, whereas complex hereditary neuropathies are characterised by being part of a hereditary multisystem disorder, which overshadows neuropathy [10]. Generally, hereditary neuropathies are classified according to the 1) clinical manifestations (hereditary sensory-motor neuropathies (HMSN), hereditary sensory-autonomic neuropathies (HSAN), distal hereditary motor neuropathies (dHMN)), 2) nerve conduction studies (NCSs, axonal, demyelinating, mixed), 3) mode of inheritance (autosomal dominant (AD), autosomal recessive (AR), X-linked (XL), maternally transmitted), and 4) mutated gene. HMSN types are also known as Charcot-Marie-Tooth (CMT) disease. The most widely used classification system is the one proposed by Dyck et al. [1]. This classification system has been repeatedly revised and the current version also includes the mutated genes.
Table 1
Overview about hereditary neuropathies (updated until 31st August 2020)
| Gene | Disease | Phenotype | Frequency | Therapy |
| CMT/HMSN | ||||
| CMT1 (AD, demyelinating) | ||||
| PMP22 | CMT1A (dupl/del) | Classical CMT1, DSD, CHN | ∼40% of CMTs | symptomatic |
| MPZ | CMT1B | CMT1, DSD, CHN, CMT2 | ∼3% of CMT | symptomatic |
| LITAF | CMT1C | CMT1 | <1% of CMT | symptomatic |
| EGR2 | CMT1D | CMT1, DSD, CHN | <1% of CMT | symptomatic |
| PMP22 (exon 4) | CMT1E | CMT1 | <1% of CMT | symptomatic |
| NEFL | CMT1F | CMT1, CMT2 | <1% of CMT | symptomatic |
| FBLN5 | CMT1H | HMN, macula degeneration | <1% of CMT | symptomatic |
| ARHGEF10 | CMT1 | asymptomatic | <1% of CMT | none required |
| CMT2 (AD, axonal) | ||||
| MFN2 | CMT2A | CMT2, optic atrophy | ∼3% of CMT | symptomatic |
| RAB7 | CMT2B | CMT2, mutilating ulcers | <1% of CMT | symptomatic |
| TRPV4 | CMT2C | CMT2, vocal cord | <1% of CMT | symptomatic |
| GARS | CMT2D | CMT2 | <1% of CMT | symptomatic |
| NEFL | CMT2E | CMT2 | <1% of CMT | symptomatic |
| HSPB1 | CMT2F | CMT2 | <1% of CMT | symptomatic |
| MPZ | CMT2I | CMT2, late onset | <1% of CMT | symptomatic |
| MPZ | CMT2J | CMT2, hypoacusis, | <1% of CMT | symptomatic |
| GDAP1 | CMT2K | CMT2, late onset | <1% of CMT | symptomatic |
| HSPB8 | CMT2L | CMT2 | <1% of CMT | symptomatic |
| DNM2 | CMT2M | CMT2, im CMT, cataract, ptosis | <1% of CMT | symptomatic |
| AARS | CMT2N | CMT2 | <1% of CMT | symptomatic |
| LRSAM1 | CMT2P | CMT2 | <1% of CMT | symptomatic |
| DHTKD1 | CMT2Q | CMT2 | <1% of CMT | symptomatic |
| MME | CMT2T | CMT2 | <1% of CMT | symptomatic |
| MARS | CMT2U | CMT2, congenital | <1% of CMT | symptomatic |
| TFG | CMT2 | CMT2, proximal involvement | <1% of CMT | symptomatic |
| HARS | CMT2 | CMT2 | <1% of CMT | symptomatic |
| VCP | CMT2 | CMT2 | <1% of CMT | symptomatic |
| KIF5A | CMT2 | CMT | <1% of CMT | symptomatic |
| JAG1 | CMT2 | CMT2 | 2 families | symptomatic |
| CMT2 (AR, axonal) | ||||
| LMNA | CMT2B | CMT2 | <1% of CMT | symptomatic |
| TRIM2 | CMT2R | CMT2 | <1% of CMT | symptomatic |
| IGHMBP2 | CMT2S | CMT2 | <1% of CMT | symptomatic |
| HSJ1 | CMT2 | CMT2 | <1% of CMT | symptomatic |
| CMT4 (AR, demyelinating) | ||||
| GDAP1 | CMT4A | CMT2, vocal cord, diaphragm | <1% of CMT | symptomatic |
| MTMR2 | CMT4B1 | CMT1, facial, bulbar | <1% of CMT | symptomatic |
| SBF3 | CMT4B2 | CMT1, glaucoma | <1% of CMT | symptomatic |
| MTMR5 | CMT4B3 | CMT1 | <1% of CMT | symptomatic |
| SH3TC2 | CMT4C | CMT1, scoliosis | <1% of CMT | symptomatic |
| NDGR1 | CMT4D | CMT1, hypoacusis, gypsy | <1% of CMT | symptomatic |
| EGR2 | CMT4E | CMT1, DSD, CHN | <1% of CMT | symptomatic |
| PRX | CMT4F | CMT1, sensory dominant | <1% of CMT | symptomatic |
| HK1 | CMT4G | CMT1, gypsy | <1% of CMT | symptomatic |
| FGD4 | CMT4H | CMT1 | <1% of CMT | symptomatic |
| FIG4 | CMT4J | CMT1 | <1% of CMT | symptomatic |
| CTDP1 | CMT4 | CMT1, cataract, gypsy | <1% of CMT | symptomatic |
| SURF1 | CMT4 | CMT1 | <1% of CMT | symptomatic |
| CMT4 (X-linked) | ||||
| GJB1 | CMTX1 | CMT1 (males), CMT2 (females) | ∼3% of CMT | symptomatic |
| AIFM1 | CMTX4 | CMT2, hypoacusis, development ↓ | <1% of CMT | symptomatic |
| PRPS1 | CMTX5 | CMT2, hypoacusis, optic atrophy | <1% of CMT | symptomatic |
| PDK3 | CMTX6 | CMT2 | <1% of CMT | symptomatic |
| CMT intermediate (AD) | ||||
| DNM2 | CMTDIB | intermediate CMT or CMT2 | <1% of CMT | symptomatic |
| YARS | CMTDIC | intermediate CMT | <1% of CMT | symptomatic |
| MPZ | CMTDID | intermediate CMT | <1% of CMT | symptomatic |
| IFN2 | CMTDIE | intermediate CMT, renal failure | <1% of CMT | symptomatic |
| GNB4 | CMTDIF | intermediate CMT | <1% of CMT | symptomatic |
| CMT intermediate (AR) | ||||
| GDAP1 | CMTRIA | intermediate CMT | <1% of CMT | symptomatic |
| PLEKHG5 | CMTRIC | intermediate CMT | <1% of CMT | symptomatic |
| COX6A1 | CMTAID | intermediate CMT | <1% of CMT | symptomatic |
| SORD | CMTRI | intermediate CMT | <1% of CMT | symptomatic |
| HSN/HSAN | ||||
| HSN/HSAN (AD) | ||||
| SPTLC1 | HSAN1A | HSN, mutilating ulcers | rare | symptomatic |
| SPTLC2 | HSAN1C | HSN, mutilating ulcers | rare | symptomatic |
| RAB7 | CMT2B | HSN, mutilating ulcers | rare | symptomatic |
| ATL1 | HSN1D | HSN, mutilating ulcers | rare | symptomatic |
| DNMT1 | HSN1E | HSN, hypoacusis, dementia | rare | symptomatic |
| ATL3 | HSN1F | HSN, bone destruction | rare | symptomatic |
| SCN11A | HSAN7 | insensitivity to pain | rare | symptomatic |
| PRNP | HSAN | HSN, dementia | rare | symptomatic |
| HSN/HSAN (AR) | ||||
| WNK1 | HSAN2A | HSN, mutilating ulcers | rare | symptomatic |
| FAM134B | HSAN2B | HSN, mutilating ulcers | rare | symptomatic |
| KIF1A | HSAN2C | HSN, mutilating ulcers | rare | symptomatic |
| IKBKAP | HSAN3 | HSN, absent papillae, Jewish | rare | symptomatic |
| SCN9A | HSAN | insensitivity to pain, SFN | rare | symptomatic |
| NTRK1 | HSAN4 | insensitivity t6o pain | rare | symptomatic |
| NGF-B | HSAN5 | insensitivity to pain | rare | symptomatic |
| DST | HSAN6 | HSN, absent papillae, Jewish | rare | symptomatic |
| CCT5 | HSN | HSN, spastic paraplegia | rare | symptomatic |
| HMN | ||||
| HMN (AD) | ||||
| HSPB8 | HMN2A | HMN | rare | symptomatic |
| HSPB1 | HMN2B | HMN | rare | symptomatic |
| HSPB3 | HMN2C | HMN | rare | symptomatic |
| FBXO38 | HMN2D | HMN | rare | symptomatic |
| SETX | HMN | HMN, pyramidal signs | rare | symptomatic |
| GARS | HMN5A | hand wasting | rare | symptomatic |
| REEP1 | HMN5B | hand wasting, pyramidal signs | rare | symptomatic |
| IGHMBP2 | HMN6 | respiratory muscles | rare | symptomatic |
| SLC5A7 | HMN7A | HMN, vocal cord | rare | symptomatic |
| DCTN1 | HMN7B | HMN, facial, bulbar | rare | symptomatic |
| DYNC1H1 | SMALED | contractures, pyramidal signs | rare | symptomatic |
| BICD2 | SMALED2 | contractures, pyramidal signs | rare | symptomatic |
| TRPV4 | PNMHH | MHN, scapular winging | rare | symptomatic |
| AAR5 | HMN | HMN | rare | symptomatic |
| WARS | HMN | distal motor neuropathy | rare | symptomatic |
| HMN (AR) | ||||
| HINT1 | HMN | HMN | rare | symptomatic |
| VRK1 | HMN | distal motor neuropathy | rare | symptomatic |
| SORD | HMN | distal motor neuropathy, diabetes | rare | symptomatic |
| SIGMAR1 | HMN | motor neuropathy, pyramidal signs | rare | symptomatic |
| HMN (XL) | ||||
| LAS1L | SMAX | respiratory muscles | rare | symptomatic |
| ATP7A | SMAX3 | HMN | rare | symptomatic |
| Other | ||||
| GAN | GAN | NP, progressive, multisystem dis | rare | symptomatic |
| PMP22 | HNPP | liability to pressure palsies | frequent | symptomatic |
| PMP22 | plexopathy | painless plexopathy | rare | symptomatic |
| SEPT9 | plexopathy | painless plexopathy | rare | symptomatic |
| TTR | SFN | sensory, not progressing to LFN | rare | tafamidis, patisiran |
| SPTLC2 | SFN | sensory, not progressing to LFN | rare | symptomatic |
| POLG1 | SFN | sensory, not progressing to LFN | rare | symptomatic |
| SCN9A | SFN | sensory, progressing to LFN | rare | symptomatic |
| SCN10A | SFN | sensory, progressing to LFN | rare | symptomatic |
| SCN11A | SFN | sensory, progressing to LFN | rare | symptomatic |
| COL6A5 | SFN | sensory, progressing to LFN | rare | Symptomatic |
| SPTBN4 | axonal neuropathy | motor, myopathy, hypoacusis | rare | Symptomatic |
CHN: congenital hypomyelinating neuropathy, DSD: Dejerine-Sottas disease, HMN: hereditary motor neuropathy, im: intermediate, SFN: small fiber neuropathy, SMA: spinal muscular atrophy.
Table 2
Overview of orphan, complex hereditary neuropathies
| Group | Disorder | Gene | Phenotype | Treatment |
| MIDs | NARP | MT-ATP6 | multisystem | symptomatic |
| MNGIE | TYMP2 | multisystem | ERT | |
| SCAE | POLG1 | multisystem | symptomatic | |
| SANDO | POLG1 | multisystem | symptomatic | |
| Non-syndromic | mtDNA. nDNA | multisystem | symptomatic | |
| HSPs | SPG1 | L1CAM | paraspasticity, MASA syndrome | symptomatic |
| SPG2 | PLP1 | paraspasticity, dementia, optic atrophy | symptomatic | |
| SPG3 | ATL1 | paraspasticity, Silver syndrome | symptomatic | |
| SPG4 | SPAST | paraspasticity, dementia, epilepsy | symptomatic | |
| SPG5 | CYP7B1 | paraspasticity, leucoencephalopathy | symptomatic | |
| SPG6 | NIPA1 | paraspasticity, epilepsy, dystonia | symptomatic | |
| SPG7 | SPG7 | paraspasticity, optic/cerebellar atrophy | symptomatic | |
| SPG9B | ALDH18A1 | paraspasticity, cataract | symptomatic | |
| SPG10 | KIF5A | paraspasticity, Parkinsonism | symptomatic | |
| SPG11 | KIAA1840 | paraspasticity, Parkinsonism, ataxia | symptomatic | |
| SPG14 | SPG14 | paraspasticity, retardation | symptomatic | |
| SPG15 | ZFYVE26 | paraspasticity, retardation, retinopathy | symptomatic | |
| SPG25 | SPG25 | paraspasticity, disc herniation | symptomatic | |
| SPG27 | SPG27 | paraspasticity, retardation | symptomatic | |
| SPG30 | KIF1A | paraspasticity, cerebellar signs | symptomatic | |
| SPG31 | REEP1 | paraspasticity, Silver syndrome, ataxia | symptomatic | |
| SPG36 | SPG36 | paraspasticity | symptomatic | |
| SPG38 | SPG38 | paraspasticity, Silver syndrome | symptomatic | |
| SPG39 | NTE | paraspasticity, spinal cord atrophy | symptomatic | |
| SPG43 | C19orf12 | paraspasticity, Silver syndrome | symptomatic | |
| SPG47 | AP4B1 | paraspasticity, retardation | symptomatic | |
| SPG49 | TECPR2 | paraspasticity | symptomatic | |
| SPG55 | C12orf65 | paraspasticity | symptomatic | |
| SPG56 | CYP3U1 | paraspasticity | symptomatic | |
| SPG57 | TFG | paraspasticity | symptomatic | |
| SPG76 | CAPN1 | paraspasticity | symptomatic | |
| SCAs | SCA1–37 | nDNA genes | ataxia | symptomatic |
| LSDs | Schindler’s disease | NAGA | regression, seizures, cardiomyopathy | symptomatic |
| Ohtahara syndrome | DMXL2 | epilepsy, encephalopathy | symptomatic | |
| Metachromatic leukodystrophy | ARSA | ataxia, spasticity, optic atrophy, dementia | symptomatic | |
| Fabry disease | GLA | corneal, gastrointestinal, cerebral | agalsidase α, β | |
| Beta-mannosidase | MANBA | behavioral change, hypoacusis, keratoma | symptomatic | |
| Krabbe disease | GALC | seizures, cognitive decline | symptomatic | |
| Sandhoff disease | HEXB | motor neuron disease | symptomatic | |
| Gaucher disease | GBA | anemia, thrombopenia, hepatosplenomegaly | ERT | |
| Nieman Pick type-C | NPC1, NPC2 | hepatosplenomegaly, ophthalmoparesis | cyclodextrine | |
| Galactosialidosis | CTSA | hepatosplenomegaly, edema, dysmorphism | symptomatic | |
| Mucolipidosis | MCOLN1 | Sialidosis | symptomatic | |
| Ceroid lipofuscinosis | nDNA | epilepsy, dementia | symptomatic | |
| Pompe disease | GAA | cardiomyopathy, myopathy, aneurysm | ERT | |
| PSDs | Zellweger syndrome | PEX1 | encephalopathy, dysmorphism | symptomatic |
| Adrenoleucodystrophy | ABCD1 | encephalopathy, dementia, hypocorticism | symptomatic | |
| Refsum disease | PDXK | PNP | pyridoxal | |
| Porphyrias | DOSS porphyria | ALAD1 | neuro-visceral attacks | givosiran |
| Acute intermittent porphyria | HMBS | abdominal pain, weakness, hallucinations | givosiran | |
| Hereditary coproporphyria | CPOX | abdominal pain, diarrhea, seizures | hemin | |
| Variegate porphyria | PPOX | gastrointestinal, seizures, hallucinations | liver transplantation | |
| Others | Transthyretin amyloidosis | TTR | cardiomyopathy, PNP | tafamidis, patisiran |
| Sensory neuropathy | RFC1 | cerebellar ataxia, vestibular dysfunction | symptomatic | |
| Tangier disease | ABCA1 | yellow tonsils, hepatosplenomegaly | symptomatic | |
| Braun-Vialetto-Van Laere syndrome | SLC52A2 | riboflavin deficiency, ataxia, hypoacusis | riboflavin | |
| Progressive myoclonus epilepsy | SCARB2 | seizures | symptomatic | |
| Hereditary neuropathy+diabetes | SORD | diabetes | symptomatic | |
| Methyl-malonic aciduria | CB1C | hyperhomocysteinemia | hydroxy-cobalamin | |
| MTHFR deficiency | MTHFR | diabetic neuropathy | folic acid | |
| Neurodevelopmental disease | NARS1 | microcephaly, seizures, ataxia, delay | symptomatic | |
| Waardenburg syndrome | SOX10 | visual impairment, leukodystrophy | symptomatic |
ERT: enzyme replacement therapy, PNP: polyneuropathy, PSD: peroxisomal disorders.
Common hereditary neuropathies
Among common hereditary neuropathies the frequency of rare forms is particularly high. A large number of common hereditary neuropathies has been reported only in a single family or a few families. [76]. Often the pathogenic variant has been detected only in one or a few family members. Rare hereditary neuropathies have been most frequently identified by panel investigations or whole exome sequencing (WES). Common hereditary neuropathies are classified as CMT/HMSN (∼75% of common hereditary neuropathies), with predominant motor and sensory involvement, as HSAN (∼3% of common hereditary neuropathies) with predominant sensory or autonomic involvement, or as distal hereditary motor neuropathy (HMN) (∼3% of common hereditary neuropathies) [77]. There is a significant overlap between HMN, HMSN, and HSAN [10]. Other common hereditary neuropathies include hereditary neuropathy with liability to pressure palsies (HNPP) (∼3% of hereditary neuropathies), hereditary plexopathy, giant axonal neuropathy (GAN), and the hereditary SFNs [77]. Several subtypes derive from inclusion of the underlying mutated gene in the classification. Currently, mutations in >170 genes have been identified as causes of hereditary neuropathies [76].
CMT (HMSN)
More than 120 genes have been identified, which are associated with the CMT/HMSN phenotype (Table 1) [78]. Genes most frequently mutated in CMT1 phenotypes (AD, demyelinating) are PMP22 (duplication/deletion), GJB1, MFN2, and MPZ genes with high variability between regions of investigation. According to a study from the UK, >90% of the CMTs are due to mutations in these four genes [10]. CMT2 (AD or AR, axonal), CMT4 (AR, demyelinating) phenotypes due to mutations in the remaining genes have to be regarded as orphan diseases. In a study of 245 CMT patients from Norway the prevalence of CMT/HMSN in the general population was 1 in 1214 [79]. Contrary to the UK study, the prevalence of the PMP22 duplication, GJB1 mutations, MPZ, and of MFN2 mutations in the cohort was only 13.6% [79]. In a study of 612 patients with hereditary neuropathies the genetic defect was identified in 121 patients (19.8%) [80]. Of these, 54.4% showed an AD, 33.9% an AR, and 11.6% XL inheritance. The most frequently affected genes were PMP22 (16.4%), GJB1 (10.7%), MPZ, and SH3TC2 (both 9.9%), and MFN2 (8.3%) [80]. Overall, a genetic diagnosis can be reached in ∼80% of the demyelinating CMT1s but only in ∼25% of axonal CMTs. CMT2 due to mutations in HINT1 may go along with neuromyotonia [81].
An example of a rare CMT is CMT4C which is due to mutations in SH3TC2. CMT4C is the commonest of the AR transmitted, common hereditary neuropathies. Onset of CMT4C is in infancy. Usually, CMT4C is restricted to the peripheral nerves. In single cases, however, cerebellar involvement may occur. Scoliosis is a major feature. CMT4C patients do not manifest with cardiomyopathy, endocrine abnormalities, or increased iron accumulation in the dentate nuclei [82]. Neuropathy manifests clinically with generalised wasting, muscle hypotonia, pes cavus, and scoliosis. Neuropathy in these patients is of the demyelinating type and slowly progressive. Rarely, cerebellar signs and hypoacusis may be present [82].
HSAN
HSAN represent ∼3% of hereditary neuropathies [77]. Currently, mutations in >25 genes are made responsible for the respective HSAN subtypes (Table 1) [76]. All HSANs have to be regarded as rare or ultra-rare diseases (Table 1).
Distal HMN
Distal HMNs are rare. Like the HSANs, HMNs constitute ∼3% of the hereditary neuropathies [77]. Currently, mutations in >35 genes are made responsible for distal HMNs (Table 1) [76]. All of them have orphan status.
Other common hereditary neuropathies
These include HNPP, GAN, hereditary plexopathy, and the hereditary SFNs. With the exception of HNPP, which is due to duplications/deletions in PMP22 these neuropathies are very rare.
Giant axonal neuropathy (GAN): GAN is a severe, pediatric, AR disorder caused by mutations in GAN, which encodes gigaxonin [83]. Gigaxonin is involved in protein degradation via the ubiquitin-proteasome system [10]. Onset of GAN is usually in childhood. Phenotypically, patients manifest with a progressive multisystem disorder including severe sensorimotor neuropathy, cerebellar ataxia, cranial nerve involvement, bulbar symptoms, intellectual disability, pes cavus, scoliosis, distinctive curly (kinky) hair, and ophthalmologic abnormalities [10, 83]. Usually, patients become wheelchair-bound in the second decade of life [83]. Neuropathologically, accumulation of intermediate filaments (IFs) can be found in neurons [84]. IFs show strong immune-histochemical positivity for glial fibrillary acidic protein (GFAP) [84]. IFs also occur in the lens [84].
Hereditary plexopathy: Hereditary plexopathy is rare although it may occur in the context of HNPP [85]. The most common cause of hereditary plexopathy is a deletion in PMP22 [85]. Recurrent painless plexopathy may be the exclusive manifestation of HNPP [86]. Recently, mutations in SEPT9 have been made responsible for hereditary neuralgic amyotrophy (HNA) [87]. Hereditary plexopathy due to mutations in SEPT9 was first described in 2005 [88] and represents <10% of the hereditary plexopathies.
Hereditary small fiber neuropathies: Small fiber neuropathies (SFNs) are common [89]. The gold standard for diagnosing the condition is skin biopsy showing reduced intra-epidermal nerve fiber density [89]. SFN may affect only small fibers without progression to large fibers during the entire course, or may progress to large-fiber neuropathy (LFN) over time. The most well-known SFN progressing to LFN and the most frequent is the one associated with mutations in the TTR gene. SFN has been additionally reported in the early stages of HSAN1C due to mutations in SPTLC2 [90]. SFNs may not only occur in association with common but also with complex hereditary neuropathies. SFN has been also reported in mitochondrial disorders (MIDs), particularly in MIDs due to POLG1 mutations [91]. In a study of 27 genetically proven MID patients, 33% had a SFN as assessed by the sympathetic skin response (SSR) or the Sudoscan®, [91]. Hereditary SFNs, which do not progress to LFN include SFNs due to mutations in SCN11A, SCN9A, SCN10A, and COL6A5 [92].
Complex hereditary neuropathies
Complex hereditary neuropathies can be more easily diagnosed than common hereditary neuropathies as they go along with concomitant pathology in systems other than the peripheral nerves. Accordingly, not only the neurologist may be involved or the first being confronted with such a phenotype but many other specialities depending on the time point at which other organs/tissues/structure become affected. Since some of the manifestations may favourably respond to symptomatic treatment, it is crucial that the managing physician refers these patients to other responsible specialities. Complex hereditary neuropathies are most prevalent among MIDs, hereditary spastic paraplegias (HSPs), spinocerebellar ataxias (SCAs), lysosomal storage disorders (LSDs), porphyrias, and other rare genetic conditions (Table 2).
Mitochondrial neuropathies
Many of the syndromic and non-syndromic MIDs manifest with neuropathy. Neuropathy may dominate the phenotype or may be only a collateral feature. Particularly, among the non-syndromic MIDs, neuropathy can be the only manifestation, usually at onset of the MID. Generally, the prevalence of neuropathies in MIDs is high but compared with the general population mitochondrial neuropathies are rare. Mitochondrial neuropathies more frequently occur with nDNA than mDNA variants, while neuropathies are rare with single mtDNA deletions.
NARP: Neuropathy, ataxia, and retinitis pigmentosa (NARP) syndrome is a MID due to the variant m.8993T>G in MT-ATP6 with heteroplasmy rates of 70–90% [93]. Onset of NARP is usually in early infancy. NARP is clinically characterised by neuropathy, ataxia in the absence of cerebellar atrophy, and retinitis pigmentosa [93]. Neuropathy in NARP is usually a dominant feature, of the axonal type, sensory-motor, and slowly progressive [94]. Nerve biopsy shows peculiar, nuclear accumulations in Schwann cells, clusters of concentrically arranged Schwann cells in myelinated axons, and degenerated mitochondria [93]. In addition to the classical features, some patients develop leukoencephalopathy, seizures, or psychomotor retardation.
MNGIE: Mitochondrial neuro-gastro-intestinal encephalopathy (MNGIE) is a rare (∼100 cases worldwide) AR, multisystem MID due to mutations in TYMP1 [95]. TYMP1 deficiency results in accumulation of the nucleosides thymidine (dThd) and deoxyuridine (dUrd) [95]. Onset of MNGIE is usually in adolescence. Phenotypic manifestations of MNGIE include ptosis, ophthalmoparesis, severe gastrointestinal dysmotility with consecutive cachexia, leukoencephalopathy, and progressive sensorimotor neuropathy [95]. NCSs usually demonstrate demyelinating PNP [95]. Whether gastrointestinal dysmotility is due to involvement of the autonomic fibers or due to primary affection of the smooth muscle cells is under debate. Whether hemodialysis, liver transplantation, allogenic hematopoetic stem cell transplantation (AHSCT), or treatment with erythrocyte encapsulated thymidine phosphorylase is effective also for neuropathy in MNGIE patients, is unknown but conceivable.
SANDO: Sensory ataxic neuropathy, dysarthria, and ophthalmoparesis (SANDO) syndrome is a rare multisystem MID due to mutations in POLG1 [96]. Both, AR and AD transmission have been reported. Onset of SANDO is in adulthood [97]. Phenotypically, SANDO manifests with dysarthria, dysphagia, ophthalmoplegia, and progressive, sensory, ataxic neuropathy. Additionally, involvement of the heart (e.g. cardiac arrest), liver, brain (depression, anxiety), or the intestines develops [96]. Some patients may present with features of MNGIE [96]. Neuropathy may not only manifest with sensory disturbances and weakness but also with twitches and cramps, and progressive gait disturbance ultimately leading to impaired mobility and requirement of a wheel chair [96].
MEMSA (SCAE): Myoclonus epilepsy, myopathy, sensory ataxia (MEMSA) syndrome previously termed spinocerebellar ataxia with epilepsy (SCAE), is a rare MID due to mutations in POLG1. Onset of MEMSA is in early adulthood. MEMSA includes an overlapping spectrum of abnormalities, including myopathy, epilepsy, and ataxia, in the absence of ophthalmoplegia. It may occur with or without ragged-red fibers [98]. Neuropathy in MEMSA spectrum disorders is rare, sensory, of the axonal type, and progressive. Though POLG1 mutations are frequent, MEMSA has been only rarely reported. MEMSA should be considered in patients with myoclonic epilepsy, mitochondrial myopathy sparing the extra-ocular muscles, and sensory neuropathy.
Other mitochondrial neuropathies: In addition to the syndromic MIDs, a high number of non-syndromic MIDs manifests with neuropathy as a collateral feature [99]. In a study of 108 MID patients, mitochondrial neuropathy was diagnosed in 35% [100]. In a study of 109 MID patients, 45% had neuropathy [99]. In a study of 116 genetically proven MIDs neuropathy predicted the presence of a nuclear defect [101]. Axonal or sensory neuropathy may occur in MELAS and MERRF.
Hereditary spastic paraplegias (HSPs)
HSPs are rare genetic disorders, clinically manifesting with spasticity predominantly of the lower limbs and urinary dysfunction (pure HSPs) [102]. Some of the HSPs, however, may manifest with additional phenotypic features, including neuropathy (HSP-plus) [102]. Currently, 80 different types of HSPs are differentiated, which are classified according to their mode of inheritance as AD, AR, XL, or maternal transmission (MT). HSPs, which not only manifest with paraspasticity and bowel or bladder dysfunction, but also with neuropathy alone or with other manifestations (HSP-plus), include SPG1–7, SPG9–11, SPG14-15, SPG25, SPG27, SPG30-31, SPG36, SPG38-39, SPG43. SPG47. SPG49. SPG55–57, and SPG76 [102]. Except for SPG4, SPG3, SPG31 (most frequent of the AD forms) and SGG11 (most frequent of the AR forms), all other SPGs are regarded as rare.
Spinocerebellar ataxias (SCAs)
Currently (8/2020), 37 types of SCAs (SCA1–SCA37) are known. Some of the SCAs may occasionally go along with neuropathy. These include SCA1 (sensory neuropathy) [103], SCA2 (axonal neuropathy) [104], SCA3 (Machado-Joseph disease) [105], SCA4 [106], SCA6 [78], SCA7 [107], SCA10 [108], SCA11 [109], and SCA18 [110]. Neuropathy may be also a feature in non-classified SCAs, such as those due to mutations in SETX [111]. SCAs with neuropathy are generally rare.
Porphyrias
Porphyrias are a group of ultrarare, mostly hereditary metabolic disorders due to a defective hem biosynthesis pathway [112]. Primary (hereditary) and secondary (acquired) porphyrias are differentiated. Primary porphyrias are subdivided into hepatic and erythropoetic forms. Among the hepatic porphyrias, acute porphyrias (acute intermittent porphyria, variegate porphyria, coproporphyria, DOSS porphyria (ALA-dehydratase (porphobilinogen synthase) deficiency), the most common of the porphyrias termed after Dr. Doss) and chronic porphyrias (porphyria cutanea tarda, hepato-erythropoetic porphyria) are delineated. The phenotype depends on the defective enzyme within the pathway and is due to accumulation of intermediate metabolites. Neuro-visceral porphyrias are characterized by recurrent, acute attacks, triggered by excessive hem synthesis, clinically manifesting as severe abdominal pain, vomiting, tachycardia, hypertension, hyponatremia, mild cognitive impairment, and peripheral neuropathy [112]. Severe attacks may manifest with seizures, psychosis, quadruparesis, respiratory failure, coma, or death [112]. Neuropathy occurs particularly in aminolevulinic acid dehydratase (ALAD1 gene) porphyria (DOSS porphyria), acute intermittent porphyria (HMBS gene), hereditary coproporphyria (CPOX gene), and variegate porphyria (PPOX gene) [112]. Not only peripheral nerves but also cranial nerves can be affected. Neuropathy is usually axonal and develops rapidly over 2–4 weeks with predominant proximal weakness [113]. Progression of neuropathy with affection of the nerves innervating respiratory muscles may lead to respiratory failure [112]. Porphyrias may respond to application of siRNA in the form of givosiran.
Lysosomal disorders
Lysosomal storage disorders (LSDs) predominantly affect the brain but in some of the about 80 subtypes, neuropathy has been occasionally described. Since LSDs are rare conditions and neuropathy does not occur in all of them, neuropathy in LSDs are generally rare. LSDs in which neuropathy has been described include Schindler’s disease [114], Ohtahara syndrome [115], metachromatic leucodystrophy [116], Fabry disease [117], hereditary β-mannosidosis [118], Krabbe disease [119], Sandhoff disease [120], Gaucher disease [121], Niemann-Pick disease type C [122], galactosialidosis [123], mucolipidosis [124], ceroid lipofuscinosis [125], and rarely Pompe disease [126]. In Tay-Sachs disease axonal neuropathy can be found in a quarter of patients. The most frequent of the LSDs with neuropathy is Fabry’s disease. Though Fabry disease is rare, all other LSDs are even rarer and thus neuropathy in these other LSDs has orphan status.
Peroxisomal disorders
Peroxisomal disorders form a group of rare, multisystem metabolic disorders, which are generally classified as either due to impaired peroxisomal biogenesis (PBDs) or due to deficiency of a single perioxismal enzyme (Zellweger spectrum disorders). The prototype of PBDs is Zellweger syndrome, clinically characterised by hypotonia, developmental delay, neuropathy, facial dysmorphism, epilepsy, rhizomelia, abnormal calcification, hepatomegaly, and abnormal liver function. Neuropathy in Zellweger syndrome is not well characterised but in the majority of the cases it is of the axonal type. The most well-known of the Zellweger spectrum disorders are neonatal, X-linked adrenoleukodystrophy and Refsum’s disease. Adenoleukodystrophy is due to mutations in the ABCD1 gene. Neuropathy in adrenoleukodystrophy is a slowly progressive, dying back axonopathy. Refsum disease is either due to mutations in PHYH (90%) or PEX7 (10%). Neuropathy in Refsum disease is sensori-motor and of the axonal type [127]. Phytanic acid levels can be reduced by plasmapheresis or diet.
Other complex hereditary neuropathies
Complex, hereditary neuropathies occur in a number of other genetic conditions. These include Tangier’s disease, TTR-amyloid neuropathy, progressive myoclonus epilepsy due to SCARB2 gene mutations, cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS). CANVAS is clinically characterised by cerebellar ataxia (CA), vestibular areflexia (VA), and neuropathy (N). Vestibular loss is usually bilateral. The prevalence of CANVAS is estimated as 26/100000. Recently, a homozygous, intronic AAGGG-repeat expansion in RFC1 was discovered as the cause of CANVAS [128]. TTR-amyloid neuropathy is a progressive, debilitating, systemic disease due to deposition of misfolded transthyretin protein in the endoneurium. Misfolding results from mutations in the TTR-gene. Early diagnosis is desirable as treatments (e.g. tafamidis, patisiran) are available that slow the progression of the disease. Early diagnosis, however, is hampered by mistaking clinical manifestations for CIDP, idiopathic axonal PNP, diabetic neuropathy, or AL amyloidosis. In endemic countries (Portugal, Japan, Sweden, Brazil) TTR amyloid neuropathy should be suspected in any patient with length-dependent SFN with autonomic dysfunction [129]. In non-endemic countries the disease may manifest with rapidly progressive, sensorimotor, axonal neuropathy or atypical CIDP [129]. Tangier disease is due mutations in ABCA1, encoding the ATP binding cassette transporter. The disease is characterised by deficiency of high-density lipoproteins (HDLs) and accumulation of cholesterol esters in various tissues [130]. Major clinical findings include hypertrophic yellow-orange tonsils [131], hepatosplenomegaly, and neuropathy [132]. The disease course is either relapsing-remitting or chronic progressive. Neuropathy is motor and sensory. Distribution of neuropathy can be multifocal, focal, or distal symmetric [130]. Upon NCSs neuropathy is demyelinating with conduction blocks in 11% of the patients [130]. Another rare, complex neuropathy is Brown-Vialetto-Van Laere syndrome which is due to loss-of-function mutations in the riboflavin transporter genes SLC52A2 or SLC52A3 [133]. In addition to sensorimotor neuropathy, patients present with cranial nerve neuropathies, ataxia, optic atrophy, deafness, and respiratory insufficiency [133]. Patients respond favourably to supplementation with riboflavin [133]. For patients carrying CB1C or MTHFR mutations, hydroxy-cobalamin respectively folic acid may be beneficial. Patients carrying mutations in PDXK profit from supplementation with pyridoxal-phosphate [134].
CONCLUSIONS
Rare (orphan) neuropathies of peripheral nerves need to be studied not only to assess the true prevalence of these conditions but also to delineate similarities and differences concerning clinical presentation and genetic heterogeneity from other more frequent types of neuropathies and to discover etiology and pathophysiology. Diagnosis of orphan neuropathies can be improved by refering neuropathies of unknown etiology to a reference center. Diagnosis of hereditary neuropathies can be improved by application of advanced genetic testing methods as early as possible. Treatment of acquired neuropathies can be improved by accurate, early diagnosis. Among the hereditary neuropathies TTR-PNP, Fabry disease, and neuropathies due to mutations in the riboflavin transporter need to be detected as early as possibly to provide available treatment as early as possible. Though rare neuropathies are challenging with regard to diagnosis, epidemiology, and treatment, we should nonetheless meet this challenge as it may help elucidating etiology, pathophysiology, range of clinical presentation, and potential treatment options for these conditions.
CONFLICTS OF INTEREST
There are no conflicts of interest.
FUNDING
No funding was received.
AUTHOR CONTRIBUTION
JF: design, literature search, discussion, first draft, critical comments.
All authors have read the journal’s position on issues involved in ethical publication.
The authors have read the Journal’s position on issues involved in ethical publication.
REFERENCES
[1] | Dyck P , Thomas PK . Peripheral Neuropathy. Elsevier, Saunders 4th Edition, eBook ISBN: 9781437713473, Hardcover ISBN: 9780721694917, 2005 |
[2] | Blog. Van Swieten . Orphan diseases. https://ub.meduniwien.ac.at/blog/?p=18295 [last accessed: Jannuary 2020] |
[3] | https://www.fda.gov/drugs/drug-information-consumers/orphan-products-hope-people-rare-diseases |
[4] | Kuwabara S . Peripheral nerve disorders as common diseases. Brain Nerve. (2013) ;65: :1071–5. |
[5] | Burns TM , Mauermann ML . The evaluation of polyneuropathies. Neurology. (2011) ;76: (7 suppl 2):S6–13. |
[6] | Inês M , Coelho T , Conceição I , Duarte-Ramos F , de Carvalho M , Costa J . Epidemiology of Transthyretin Familial Amyloid Polyneuropathy in Portugal: A Nationwide Study. Neuroepidemiology. (2018) ;51: :177–82. |
[7] | Kato-Motozaki Y , Ono K , Shima K , Morinaga A , Machiya T , Nozaki I , Shibata-Hamaguchi A , Furukawa Y , Yanase D , Ishida C , Sakajiri K , Yamada M . Epidemiology of familial amyloid polyneuropathy in Japan: Identification of a novel endemic focus. J Neurol Sci. (2008) ;270: :133–40. |
[8] | Sousa A , Andersson R , Drugge U , Holmgren G , Sandgren O . Familial amyloidotic polyneuropathy in Sweden: Geographical distribution, age of onset, and prevalence. Hum Hered. (1993) ;43: :288–94. |
[9] | Siao P , Kaku M . A Clinician’s Approach to Peripheral Neuropathy. Semin Neurol. (2019) ;39: :519–30. |
[10] | Carroll AS , Burns J , Nicholson G , Kiernan MC , Vucic S . Inherited Neuropathies. Semin Neurol. (2019) ;39: :620–39. |
[11] | Gwathmey KG , Grogan J . Nutritional neuropathies. Muscle Nerve. 2019. doi: 10.1002/mus.26783 |
[12] | Sharma S , Tobin V , Vas PRJ , Rayman G . The LDIFLARE and CCM Methods Demonstrate Early Nerve Fiber Abnormalities in Untreated Hypothyroidism: A Prospective Study. J Clin Endocrinol Metab. (2018) ;103: :3094–102. |
[13] | Jin PH , Shin SC . Neuropathy of Connective Tissue Diseases and Other Systemic Diseases. Semin Neurol. (2019) ;39: :651–68. |
[14] | Al-Wahaibi AK , Kumar S , Al-Risi A , Wali F . Thyrotoxic Neuropathy: A rare cause of acute flaccid paraplegia. Sultan Qaboos Univ Med J. (2017) ;17: :e460–e463. doi: 10.18295/squmj.2017.17.04.014 |
[15] | England JD , Gronseth GS , Franklin G , Carter GT , Kinsella LJ , Cohen JA , Asbury AK , Szigeti K , Lupski JR , Latov N , Lewis RA , Low PA , Fisher MA , Herrmann DN , Howard JF Jr , Lauria G , Miller RG , Polydefkis M , Sumner AJ ; American Academy of Neurology. Practice Parameter: Evaluation of distal symmetric polyneuropathy: Role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. (2009) ;72: :185–92. |
[16] | Saperstein DS , Wolfe GI , Gronseth GS , Nations SP , Herbelin LL , Bryan WW , Barohn RJ . Challenges in the identification of cobalamin-deficiency polyneuropathy. Arch Neurol. (2003) ;60: :1296–301. |
[17] | Levine TD , Saperstein DS . Laboratory evaluation of peripheral neuropathy. Neurol Clin. (2013) ;31: :363–76. |
[18] | Koike H TD , Takahashi M TD , Ohyama K TD , Hashimoto R TD , Kawagashira Y , Iijima M , Katsuno M , Doi H , Tanaka F , Sobue G , Clinicopathologic features of folate-deficiency neuropathy. Neurology. (2015) ;84: :1026–33 |
[19] | Huan MC . Laboratory evaluation of peripheral neuropathy. Semin Neurol. (2010) ;30: :337–49. |
[20] | Sechi G , Fois C , Addis A , Sechi E . Clinicopathologic features of folate-deficiency neuropathy. Neurology. (2015) ;85: :1090–1. |
[21] | Shible AA , Ramadurai D , Gergen D , Reynolds PM . Dry Beriberi Due to Thiamine Deficiency Associated with Peripheral Neuropathy and Wernicke’s Encephalopathy Mimicking Guillain-Barré syndrome: A Case Report and Review of the Literature. Am J Case Rep. (2019) ;20: :330–4. |
[22] | Kulkantrakorn K . Pyridoxine-induced sensory ataxic neuronopathy and neuropathy: Revisited. Neurol Sci. (2014) ;35: :1827–30. |
[23] | Moriwaki K , Kanno Y , Nakamoto H , Okada H , Suzuki H . Vitamin B6 deficiency in elderly patients on chronic peritoneal dialysis. Adv Perit Dial. (2000) ;16: :308–12. |
[24] | Wysota B , Michael S , Hiew FL , Dawson C , Rajabally YA . Severe but reversible neuropathy and encephalopathy due to vitamin E deficiency. Clin Neurol Neurosurg. (2017) ;160: :19–20. |
[25] | Rohm CL , Acree S , Lovett L . Progressive myeloneuropathy with symptomatic anaemia. BMJ Case Rep. (2019) ;12: . pii: e230025. doi: 10.1136/bcr-2019-230025 |
[26] | Rapoport Y , Lavin PJ . Nutritional Optic Neuropathy Caused by Copper Deficiency After Bariatric Surgery. J Neuroophthalmol. (2016) ;36: :178–81. |
[27] | Stetkarova I , Bocek V , Gismatullina A , Svobodova Z , Peisker T . Severe chronic lithium intoxication in patient treated for bipolar disorder. Neuro Endocrinol Lett. (2017) ;38: :397–400. |
[28] | Chan CH , Leung AK , Cheung YF , Chan PY , Yeung KW , Lai KY . A rare neurological complication due to lithium poisoning. Hong Kong Med J. (2012) ;18: :343–5. |
[29] | Thomson RM , Parry GJ . Neuropathies associated with excessive exposure to lead. Muscle Nerve. (2006) ;33: :732–41. |
[30] | Helmich F , Lock G . Burton’s Line from Chronic Lead Intoxication. N Engl J Med. (2018) ;379: :e35. |
[31] | Valappil AV , Mammen A . Subacute Arsenic Neuropathy: Clinical and Electrophysiological Observations. J Neurosci Rural Pract. (2019) ;10: :529–32. |
[32] | Hoitsma E , Marziniak M , Faber CG , Reulen JP , Sommer C , De Baets M , Drent M . Small fibre neuropathy in sarcoidosis. Lancet. (2002) ;359: :2085–6. |
[33] | Pelayo-de Tomás JM , Novoa-Parra C , Gómez-Barbero P . Cobalt toxicity after revision total hip replacement due to fracture of a ceramic head. Rev Esp Cir Ortop Traumatol. (2017) ;61: :203–7. |
[34] | Al-Kuraishy HM , Al-Gareeb AI , Hussien NR , Al-Naimi MS , Rasheed HA . Statins an oft-prescribed drug is implicated in peripheral neuropathy: The time to know more. J Pak Med Assoc. (2019) ;69: (suppl 3):S108–S112. |
[35] | Ghavanini AA , Kimpinski K . Revisiting the evidence for neuropathy caused by pyridoxine deficiency and excess. J Clin Neuromuscul Dis. (2014) ;16: :25–31. |
[36] | Vanta OM , Tohanean N , Pintea S , Perju-Dumbrava L . Large-Fiber Neuropathy in Parkinson’s Disease: Clinical, Biological, and Electroneurographic Assessment of a Romanian Cohort. J Clin Med. (2019) ;24;8: (10). |
[37] | Romagnolo A , Merola A , Artusi CA , Rizzone MG , Zibetti M , Lopiano L . Levodopa-Induced Neuropathy: A Systematic Review. Mov Disord Clin Pract. (2018) ;6: :96–103. |
[38] | Islam B , Lustberg M , Staff NP , Kolb N , Alberti P , Argyriou AA . Vinca alkaloids, thalidomide and eribulin-induced peripheral neurotoxicity: From pathogenesis to treatment. J Peripher Nerv Syst. (2019) ;24: (suppl 2):S63–S73. |
[39] | de Greef BTA , Hoeijmakers JGJ , Gorissen-Brouwers CML , Geerts M , Faber CG , Merkies ISJ . Associated conditions in small fiber neuropathy - a large cohort study and review of the literature. Eur J Neurol. (2018) ;25: :348–55. |
[40] | Hersch MI , McLeod JG , Satchell PM , Early RG , Sullivan CE . Breathing pattern, lung inflation reflex and airway tone in acrylamide neuropathy. Respir Physiol. (1989) ;76: :257–76. |
[41] | Anand P , Kharal GA , Reda H , Venna N . Peripheral Neuropathies in Infectious Diseases. Semin Neurol. (2019) ;39: :640–50. |
[42] | Malek E , Salameh J . Guillain-Barre Syndrome. Semin Neurol. (2019) ;39: :589–95. |
[43] | Yuki N , Chan AC , Wong AHY , Inoue T , Yokai M , Kurihara T , Devaux JJ , Wilder-Smith E . Acute painful autoimmune neuropathy: A variant of Guillain-Barré syndrome. Muscle Nerve. (2018) ;57: :320–4. |
[44] | Bourque PR , Chardon JW , Massie R . Autoimmune peripheral neuropathies. Clin Chim Acta. (2015) ;449: :37–42. |
[45] | Hao Y , Wang W , Jacobs BC , Qiao B , Chen M , Liu D , Feng X , Wang Y . Antecedent infections in Guillain-Barré syndrome: A single-center, prospective study. Ann Clin Transl Neurol. (2019) ;6: (12):2510–7. |
[46] | Dev N , Kumar R , Kumar D . Guillain-Barre syndrome: A rare complication of leptospirosis and scrub typhus co-infection. Trop Doct. (2019) ;49: :248–9. |
[47] | Hameed S , Khan S . Rare variant of Guillain-Barré syndrome after chikungunya viral fever. BMJ Case Rep. 2019;12. pii: e228845. doi: 10.1136/bcr-2018-228845 |
[48] | Rivera-Correa J , de Siqueira IC , Mota S , do Rosário MS , Pereira de Jesus PA , Alcantara LCJ , Ernst JD , Rodriguez A . Anti-ganglioside antibodies in patients with Zika virus infection-associated Guillain-Barré Syndrome in Brazil. PLoS Negl Trop Dis. (2019) ;13: (9):e0007695. doi: 10.1371/journal.pntd.0007695 |
[49] | Shah PM , Dhakre VW , Veerasuri R , Bhabhor A . Dysautonomia and hyponatraemia as harbingers of Guillain-Barre syndrome. BMJ Case Rep. (2019) ;12: (4). pii: e226925. doi: 10.1136/bcr-2018-226925 |
[50] | Chang HW , Yang PY , Han TI , Meng NH . Wernicke encephalopathy concurrent with polyradiculoneuropathy in a young man after bariatric surgery: A case report. Medicine (Baltimore). (2019) ;98: (10):e14808. doi: 10.1097/MD.0000000000014808 |
[51] | Ding L , Chen Z , Sun Y , Bao H , Wu X , Zhong L , Zhang P , Lin Y , Liu Y . Guillain-Barré syndrome following bacterial meningitis: A case report and literature review. BMC Neurol. (2018) ;18: :208. doi: 10.1186/s12883-018-1211-4 |
[52] | Shije J , Brannagan TH 3rd . Chronic Inflammatory Demyelinating Polyradiculoneuropathy. Semin Neurol. (2019) ;39: :596–607. |
[53] | Doppler K , Schuster Y , Appeltshauser L , Biko L , Villmann C , Weishaupt A , Werner C , Sommer C . Anti-CNTN1 IgG3 induces acute conduction block and motor deficits in a passive transfer rat model. J Neuroinflammation. (2019) ;16: :73. doi: 10.1186/s12974-019-1462-z |
[54] | Sekiguchi N , Hamano A , Kitagawa T , Kurihara Y , Ito K , Kurimoto M , Watanabe K , Hirano K , Noto S , Yamada K , Takezako N . Impact of rituximab and half-dose CHOP as primary therapy for untreated symptomatic Waldenström Macroglobulinemia: Review of a combined regimen of rituximab with an alkylating agent. Blood Res. (2018) ;53: :117–22. |
[55] | Torrealba-Acosta G , Gadhia R , Leslie-Mazwi T . A rare neurological complication of Waldenstrom’s Macroglobulinemia. J Clin Neurosci. (2018) ;48: :143–6. |
[56] | Nakaseko C . POEMS syndrome: Advances in molecular pathophysiology and treatment. Rinsho Ketsueki. (2019) ;60: :979–87. |
[57] | Byun JM , Kwon YN , Koh Y , Yoon SS , Sung JJ , Kim I . Distinctive patterns of peripheral neuropathy across the spectrum of plasma cell disorders. Sci Rep. (2019) ;9: :16769. doi: 10.1038/s41598-019-53289-w |
[58] | Hamedani AG , Bird SJ , Tamhankar MA . CANOMAD Presenting as Bilateral SixthNerve Palsies. J Neuroophthalmol. (2019) ;39: :397–8. |
[59] | Garcia-Santibanez R , Zaidman CM , Sommerville RB , Lopate G , Weihl CC , Pestronk A , Bucelli RC . CANOMAD and other chronic ataxic neuropathies with disialosyl antibodies (CANDA). J Neurol. (2018) ;265: :1402–9. |
[60] | Boussaïd I , Bouhour F , Vial C , Caudie C . Identification and characterization of a monoclonal IgM reacting with disialylated gangliosides recognizing the CANOMAD syndrome. Ann Biol Clin (Paris). (2011) ;69: :476–80. |
[61] | Delmont E , Jeandel PY , Hubert AM , Marcq L , Boucraut J , Desnuelle C . Successful treatment with rituximab of one patient with CANOMAD neuropathy. J Neurol. (2010) ;257: :655–7. |
[62] | McKelvie PA , Gates PC , Day T . Canomad: Report of a case with a 40-year history and autopsy. Is this a sensory ganglionopathy with neuromuscular junction blockade? Muscle Nerve. (2013) ;48: :599–603. |
[63] | Beachy N , Satkowiak K , Gwathmey KG . Vasculitic Neuropathies. Semin Neurol. (2019) ;39: :608–19. |
[64] | Bischof A , Jaeger VK , Hadden RDM , Luqmani RA , Pröbstel AK , Merkel PA , Suppiah R , Craven A , Collins MP , Daikeler T . Peripheral neuropathy in antineutrophil cytoplasmic antibody-associated vasculitides: Insights from the DCVAS study. Neurol Neuroimmunol Neuroinflamm. (2019) ;6: (6). pii: e615. doi: 10.1212/NXI.0000000000000615 |
[65] | Collins MP , Hadden RD . The nonsystemic vasculitic neuropathies. Nat Rev Neurol. (2017) ;13: :302–16. |
[66] | Jiménez-Ávila JM , Castañeda-Huerta JE , González-Cisneros AC . Bruns Garland syndrome. Report of a case and differential diagnosis with cauda equine syndrome. Acta Ortop Mex. (2019) ;33: :42–5. |
[67] | Barohn RJ , Sahenk Z , Warmolts JR , Mendell JR . The Bruns-Garland syndrome (diabetic amyotrophy). Revisited 100 years later. Arch Neurol. (1991) ;48: :1130–5. |
[68] | Mahmoud RA , Abrams CK . Acute demyelinating neuropathy in a patient with neurolymphomatosis. BMJ Case Rep. 2018;2018. pii: bcr-2017-222814. doi: 10.1136/bcr-2017-222814 |
[69] | Triplett JD , Khor TS , Kermode AG . Recurrent upper limb neuropathies secondary to an epithelioid haemangioendothelioma - A rare mimic of nerve tumours. J Clin Neurosci. 2019. pii: S0967-5868(18)32226-4. |
[70] | Kesikburun S , Omaç ÖK , Taşkaynatan MA , Özgül A , Tan AK . Neoplastic brachial plexopathy detected by ultrasonography in a patient with chronic cervicobrachialgia. J Rehabil Med. (2012) ;44: :181–3. |
[71] | Gwathmey KG . Plexus and peripheral nerve metastasis. Handb Clin Neurol. (2018) ;149: :257–79. |
[72] | Antoine JC , Camdessanché JP . Paraneoplastic neuropathies. Curr Opin Neurol. (2017) ;30: :513–20. |
[73] | Koike H , Sobue G . Paraneoplastic neuropathy. Handb Clin Neurol. (2013) ;115: :713–26. |
[74] | Graus F , Dalmau J . Paraneoplastic neuropathies. Curr Opin Neurol. (2013) ;26: :489–95. |
[75] | Khan U , Warriach SA . Rare case: Paraneoplastic syndrome affecting peripheral nerves, associated with anti-collapsin-response mediator protein-5 (anti-CRMP5) antibodies, as early manifestation of small cell lung cancer confined to a solitary lymph node without evidence of lung mass on routine CT thorax. BMJ Case Rep. (2020) ;13: (2). pii: e232656. doi: 10.1136/bcr-2019-232656 |
[76] | Eggermann K , Gess B , Häusler M , Weis J , Hahn A , Kurth I . Hereditary Neuropathies. Dtsch Arztebl Int. (2018) ;115: :91–7. |
[77] | Fridman V , Bundy B , Reilly MM , Pareyson D , Bacon C , Burns J , Day J , Feely S , Finkel RS , Grider T , Kirk CA , Herrmann DN , Laurá M , Li J , Lloyd T , Sumner CJ , Muntoni F , Piscosquito G , Ramchandren S , Shy R , Siskind CE , Yum SW , Moroni I , Pagliano E , Zuchner S , Scherer SS , Shy ME ; Inherited Neuropathies Consortium. CMT subtypes and disease burden in patients enrolled in the Inherited Neuropathies Consortium natural history study: A cross-sectional analysis. J Neurol Neurosurg Psychiatry. (2015) ;86: :873–8. |
[78] | Linnemann C , Tezenas du Montcel S , Rakowicz M , Schmitz-Hübsch T , Szymanski S , Berciano J , van de Warrenburg BP , Pedersen K , Depondt C , Rola R , Klockgether T , García A , Mutlu G , Schöls L . Peripheral Neuropathy in Spinocerebellar Ataxia Type 1, 2, 3, and 6. Cerebellum. (2016) ;15: :165–73. |
[79] | Braathen GJ . Genetic epidemiology of Charcot-Marie-Tooth disease. Acta Neurol Scand Suppl. (2012) ;193: :iv–22. |
[80] | Dohrn MF , Glöckle N , Mulahasanovic L , Heller C , Mohr J , Bauer C , Riesch E , Becker A , Battke F , Hörtnagel K , Hornemann T , Suriyanarayanan S , Blankenburg M , Schulz JB , Claeys KG , Gess B , Katona I , Ferbert A , Vittore D , Grimm A , Wolking S , Schöls L , Lerche H , Korenke GC , Fischer D , Schrank B , Kotzaeridou U , Kurlemann G , Dräger B , Schirmacher A , Young P , Schlotter-Weigel B , Biskup S . Frequent genes in rare diseases: Panel-based next generation sequencing to disclose causal mutations in hereditary neuropathies. J Neurochem. (2017) ;143: :507–22. |
[81] | Wang Z , Lin J , Qiao K , et al. Novel mutations in HINT1 gene cause the autosomal recessive axonal neuropathy with neuromyotonia. Eur J Med Genet. (2019) ;62: (3):190–4. doi: 10.1016/j.ejmg.2018.07.009 |
[82] | Skott H , Muntean-Firanescu C , Samuelsson K , Verrecchia L , Svenningsson P , Malmgren H , Cananau C , Espay AJ , Press R , Solders G , Paucar M . The cerebellar phenotype of Charcot-Marie-Tooth neuropathy type 4C. Cerebellum Ataxias. (2019) ;6: :9. doi: 10.1186/s40673-019-0103-8 |
[83] | Echaniz-Laguna A , Cuisset JM , Guyant-Marechal L , Aubourg P , Kremer L , Baaloul N , Verloes A , Beladgham K , Perrot J , Francou B , Latour P . Giant axonal neuropathy: Amulticenter retrospective studywith genotypic spectrum expansion. Neurogenetics. 2019. doi: 10.1007/s10048-019-00596-z |
[84] | Armao D , Bouldin TW , Bailey RM , Hooper JE , Bharucha DX , Gray SJ . Advancing the pathologic phenotype of giant axonal neuropathy: Early involvement of the ocular lens. Orphanet J Rare Dis. (2019) ;14: :27. doi: 10.1186/s13023-018-0957-5 |
[85] | Pareyson D , Solari A , Taroni F , Botti S , Fallica E , Scaioli V , Ciano C , Sghirlanzoni A . Detection of hereditary neuropathy with liability to pressure palsies among patients with acute painless mononeuropathy or plexopathy. Muscle Nerve. (1998) ;21: :1686–91. |
[86] | Stögbauer F , Young P , Kerschensteiner M , Ringelstein EB , Assmann G , Funke H . Recurrent brachial plexus palsies as the only clinical expression of hereditary neuropathy with liability to pressure palsies associated with a de novo deletion of the peripheral myelin protein-22 gene. Muscle Nerve. (1998) ;21: :1199–201. |
[87] | Serin HM , Yılmaz S , Kanmaz S , Şimşek E , Aktan G , Tekgül H , Gökben S . A rare cause of brachial plexopathy: Hereditary neuralgic amyotrophy. Turk Pediatri Ars. (2019) ;54: :189–91. |
[88] | Kuhlenbäumer G , Hannibal MC , Nelis E , Schirmacher A , Verpoorten N , Meuleman J , Watts GD , De Vriendt E , Young P , Stögbauer F , Halfter H , Irobi J , Goossens D , Del-Favero J , Betz BG , Hor H , Kurlemann G , Bird TD , Airaksinen E , Mononen T , Serradell AP , Prats JM , Van Broeckhoven C , De Jonghe P , Timmerman V , Ringelstein EB , Chance PF . Mutations in SEPT9 cause hereditary neuralgic amyotrophy. Nat Genet. (2005) ;37: :1044–6. |
[89] | Zhou L . Small Fiber Neuropathy. Semin Neurol. (2019) ;39: :570–7. |
[90] | Suriyanarayanan S , Auranen M , Toppila J , Paetau A , Shcherbii M , Palin E , Wei Y , Lohioja T , Schlotter-Weigel B , Schön U , Abicht A , Rautenstrauss B , Tyynismaa H , Walter MC , Hornemann T , Ylikallio E . The Variant (Arg183Trp) in SPTLC2 Causes Late-Onset Hereditary Sensory Neuropathy. Neuromolecular Med. (2016) ;18: :81–90. |
[91] | Luigetti M , Primiano G , Cuccagna C , Bernardo D , Sauchelli D , Vollono C , Servidei S . Small fibre neuropathy in mitochondrial diseases explored with sudoscan. Clin Neurophysiol. (2018) ;129: :1618–23. |
[92] | Cazzato D , Lauria G . Small fibre neuropathy. Curr Opin Neurol. (2017) ;30: :490–9. |
[93] | Claeys KG , Abicht A , Häusler M , Kleinle S , Wiesmann M , Schulz JB , Horvath R , Weis J . Novel genetic and neuropathological insights in neurogenic muscle weakness, ataxia, and retinitis pigmentosa (NARP). Muscle Nerve. (2016) ;54: :328–33. |
[94] | Rawle MJ , Larner AJ . NARP Syndrome: A 20-Year Follow-Up. Case Rep Neurol. (2013) ;5: :204–7. |
[95] | Falcão de Campos C , Oliveira Santos M , Roque R , Conceição I , de Carvalho M . Mitochondrial Neurogastrointestinal Encephalomyopathy: Novel Pathogenic Mutation in Thymidine Phosphorylase Gene in a Patient from Cape Verde Islands. Case Rep Neurol Med. (2019) ;2019: :5976410. doi: 10.1155/2019/5976410 |
[96] | Gáti I , Danielsson O , Jonasson J , Landtblom AM . Sensory ataxic neuropathy with dysarthria/dysphagia and ophthalmoplegia (SANDO). Two case reports. Acta Myol. (2011) ;30: :188–90. |
[97] | Hanisch F , Kornhuber M , Alston CL , Taylor RW , Deschauer M , Zierz S . SANDO syndrome in a cohort of 107 patients with CPEO and mitochondrial DNA deletions. J Neurol Neurosurg Psychiatry. (2015) ;86: :630–4. |
[98] | Wong LJ , Naviaux RK , Brunetti-Pierri N , Zhang Q , Schmitt ES , Truong C , Milone M , Cohen BH , Wical B , Ganesh J , Basinger AA , Burton BK , Swoboda K , Gilbert DL , Vanderver A , Saneto RP , Maranda B , Arnold G , Abdenur JE , Waters PJ , Copeland WC . Molecular and clinical genetics of mitochondrial diseases due to POLG mutations. Hum Mutat. (2008) ;29: :E150–72. |
[99] | Luigetti M , Sauchelli D , Primiano G , Cuccagna C , Bernardo D , Lo Monaco M , Servidei S . Peripheral neuropathy is a common manifestation of mitochondrial diseases: A single-centre experience. Eur J Neurol. (2016) ;23: :1020–7. |
[100] | Finsterer J . Mitochondrial neuropathy. Clin Neurol Neurosurg. (2005) ;107: :181–6. |
[101] | Horga A , Pitceathly RD , Blake JC , Woodward CE , Zapater P , Fratter C , Mudanohwo EE , Plant GT , Houlden H , Sweeney MG , Hanna MG , Reilly MM . Peripheral neuropathy predicts nuclear gene defect in patients with mitochondrial ophthalmoplegia. Brain. (2014) ;137: :3200–12. |
[102] | Finsterer J , Löscher W , Quasthoff S , Wanschitz J , Auer-Grumbach M , Stevanin G . Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J Neurol Sci. (2012) ;318: :1–18. |
[103] | Scott P , Al Kindi A , Al Fahdi A , Al Yarubi N , Bruwer Z , Al Adawi S , Nandhagopal R . Spinocerebellar ataxia with axonal neuropathy type 1 revisited. J Clin Neurosci. (2019) ;67: :139–44. |
[104] | Pelosi L , Iodice R , Antenora A , Kilfoyle D , Mulroy E , Rodrigues M , Roxburgh R , Iovino A , Filla A , Manganelli F , Santoro L . Spinocerebellar ataxia type 2-neuronopathy or neuropathy? Muscle Nerve ((2019) ;60: :271–8. |
[105] | Paulson H . Spinocerebellar Ataxia Type 3. 1998 Oct 10 [updated 2015 Sep 24]. In:AdamMP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from http://www.ncbi.nlm.nih.gov/books/NBK1196/ |
[106] | Hellenbroich Y , Bubel S , Pawlack H , Opitz S , Vieregge P , Schwinger E , Zühlke C . Refinement of the spinocerebellar ataxia type 4 locus in a large German family and exclusion of CAG repeat expansions in this region. J Neurol. (2003) ;250: :668–71. |
[107] | Salas-Vargas J , Mancera-Gervacio J , Velázquez-Pérez L , Rodrígez-Labrada R , Martínez-Cruz E , Magaña JJ , Durand-Rivera A , Hernández-Hernández O , Cisneros B , Gonzalez-Piña R . Spinocerebellar ataxia type A neurodegenerative disorder with peripheral neuropathy. Eur Neurol. (2015) ;73: :173–8. |
[108] | Matsuura T , Ashizawa T . Spinocerebellar Ataxia Type 10. 2002 Apr 23 [updated 2019 Sep 19]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from http://www.ncbi.nlm.nih.gov/books/NBK1175/ |
[109] | Chen Z , Puzriakova A , Houlden H . Spinocerebellar Ataxia Type 11. 2008 Jul 22 [updated 2019 Oct 31]. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020. Available from http://www.ncbi.nlm.nih.gov/books/NBK1757/ |
[110] | Lin P , Zhang D , Xu G , Yan C . Identification of IFRD1 variant in a Han Chinese family with autosomal dominant hereditary spastic paraplegia associated with peripheral neuropathy and ataxia. J Hum Genet. (2018) ;63: :521–4. |
[111] | Nanetti L , Cavalieri S , Pensato V , Erbetta A , Pareyson D , Panzeri M , Zorzi G , Antozzi C , Moroni I , Gellera C , Brusco A , Mariotti C . SETX mutations are a frequent genetic cause of juvenile and adult onset cerebellar ataxia with neuropathy and elevated serum alpha-fetoprotein. Orphanet J Rare Dis. (2013) ;8: :123. doi: 10.1186/1750-1172-8-123 |
[112] | Edel Y , Mamet R . Porphyria: What Is It and Who Should Be Evaluated? Rambam Maimonides Med J. (2018) ;9: (2). doi: 10.5041/RMMJ.10333 |
[113] | O’Malley R , Rao G , Stein P , Bandmann O . Porphyria: Often discussed but too often missed. Pract Neurol. (2018) ;18: :352–8. |
[114] | Castro RG , Pérez AMG , Curto MCR , Álvarez JC , Ferreirós AC , Cuadros AV , Bueno DM , Fernández AJC . A New Case of Schindler Disease. Eur J Case Rep Intern Med. (2019) ;6: (11):001269. doi: 10.12890/2019_001269 |
[115] | Esposito A , Falace A , Wagner M , Gal M , Mei D , Conti V , Pisano T , Aprile D , Cerullo MS , De Fusco A , Giovedì S , Seibt A , Magen D , Polster T , Eran A , Stenton SL , Fiorillo C , Ravid S , Mayatepek E , Hafner H , Wortmann S , Levanon EY , Marini C , Mandel H , Benfenati F , Distelmaier F , Fassio A , Guerrini R . Biallelic DMXL2 mutations impair autophagy and cause Ohtahara syndrome with progressive course. Brain. (2019) ;142: :3876–91. |
[116] | Beerepoot S , Nierkens S , Boelens JJ , Lindemans C , Bugiani M , Wolf NI . Peripheral neuropathy in metachromatic leukodystrophy: Current status and future perspective. Orphanet J Rare Dis. (2019) ;14: :240. doi: 10.1186/s13023-019-1220-4 |
[117] | Varela P , Mastroianni Kirsztajn G , Ferrer H , Aranda C , Wallbach K , Ferreira da Mata G , Moura LA , Moreira SR , Mendes C , Curiati MA , Martins AM , Bosco Pesquero J . Functional Characterization and Pharmacological Evaluation of a Novel GLA Missense Mutation Found in a Severely Affected Fabry Disease Family. Nephron. 2019:1-9. doi: 10.1159/000503998 |
[118] | Bolfa P , Wang P , Nair R , Rajeev S , Armien AG , Henthorn PS , Wood T , Thrall MA , Giger U . Hereditary β-mannosidosis in a dog: Clinicopathological and molecular genetic characterization. Mol Genet Metab. (2019) ;128: :137–43. |
[119] | Nashabat M , Al-Khenaizan S , Alfadhel M . Report of a Case that Expands the Phenotype of Infantile Krabbe Disease. Am J Case Rep. (2019) ;20: :643–6. |
[120] | Grunseich C , Schindler AB , Chen KL , Bakar D , Mankodi A , Traslavina R , Ray-Chaudhury A , Lehky TJ , Baker EH , Maragakis NJ , Tifft CJ , Fischbeck KH . Peripheral neuropathy in a family with Sandhoff disease and SH3TC2 deficiency. J Neurol. (2015) ;262: :1066–8. |
[121] | Andréasson M , Solders G , Björkvall CK , Machaczka M , Svenningsson P . Polyneuropathy in Gaucher disease type 1 and 3 - a descriptive case series. Sci Rep. (2019) ;9: :15358. doi: 10.1038/s41598-019-51976-2 |
[122] | Zafeiriou DI , Triantafyllou P , Gombakis NP , Vargiami E , Tsantali C , Michelakaki E . Niemann-Pick type C disease associated with peripheral neuropathy. Pediatr Neurol. (2003) ;29: :242–4. |
[123] | Tan EK , Wong MC , Ng I , Teo SH , Lo YL , Cho MM . Unusual pure motor axonal neuropathy in a Burmese family with galactosialidosis. J Inherit Metab Dis. (1998) ;21: :869–70. doi: 10.1023/a:1005431021025 |
[124] | Steet RA , Hullin R , Kudo M , et al. A splicing mutation in the alpha/beta GlcNAc-1-phosphotransferase gene results in an adult onset form of mucolipidosis III associated with sensory neuropathy and cardiomyopathy. Am J Med Genet A. (2005) ;132A: :369–75. doi:10.1002/ajmg.a.30498 |
[125] | Wisniewski KE , Madrid RE , Dambska M , Rapin I , Pullarkat R , Sklower S . Spino-cerebellar degeneration with polyneuropathy associated with ceroid lipofuscinosis in one family. J Child Neurol. (1988) ;3: :33–41. doi: 10.1177/088307388800300108 |
[126] | Lamartine S Monteiro M , Remiche G . Late-onset Pompe disease associated with polyneuropathy. Neuromuscul Disord. (2019) ;29: :968–72. doi: 10.1016/j.nmd.2019.08.016 |
[127] | Dasouki M . Peroxisomal disorders. In: Biomarkers in Inborn Errors of Metabolism, 2017 |
[128] | Cortese A , Tozza S , Yau WY , Rossi S , Beecroft SJ , Jaunmuktane Z , Dyer Z , Ravenscroft G , Lamont PJ , Mossman S , Chancellor A , Maisonobe T , Pereon Y , Cauquil C , Colnaghi S , Mallucci G , Curro R , Tomaselli PJ , Thomas-Black G , Sullivan R , Efthymiou S , Rossor AM , Laurá M , Pipis M , Horga A , Polke J , Kaski D , Horvath R , Chinnery PF , Marques W , Tassorelli C , Devigili G , Leonardis L , Wood NW , Bronstein A , Giunti P , Züchner S , Stojkovic T , Laing N , Roxburgh RH , Houlden H , Reilly MM . Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain. (2020) ;143: :480–90. |
[129] | Adams D , Ando Y , Beirão JM , Coelho T , Gertz MA , Gillmore JD , Hawkins PN , Lousada I , Suhr OB , Merlini G . Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2020. doi: 10.1007/s00415-019-09688-0 |
[130] | Mercan M , Yayla V , Altinay S , Seyhan S . Peripheral neuropathy in Tangier disease: A literature review and assessment. J Peripher Nerv Syst. (2018) ;23: :88–98. doi: 10.1111/jns.12265 |
[131] | Muratsu J , Koseki M , Masuda D , Yasuga Y , Tomoyama S , Ataka K , Yagi Y , Nakagawa A , Hamada H , Fujita S , Hattori H , Ohama T , Nishida M , Hiraoka H , Matsuzawa Y , Yamashita S . Accelerated Atherogenicity in Tangier Disease. J Atheroscler Thromb. (2018) ;25: :1076–85. |
[132] | Burnett JR , Hooper AJ , McCormick SPA , Hegele RA . Tangier Disease. 2019 Nov 21. In: Adam 1853 MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from http://www.ncbi.nlm.nih.gov/books/NBK549920/ |
[133] | Manole A , Jaunmuktane Z , Hargreaves I , Ludtmann MHR , Salpietro V , Bello OD , Pope S , Pandraud A , Horga A , Scalco RS , Li A , Ashokkumar B , Lourenço CM , Heales S , Horvath R , Chinnery PF , Toro C , Singleton AB , Jacques TS , Abramov AY , Muntoni F , Hanna MG , Reilly MM , Revesz T , Kullmann DM , Jepson JEC , Houlden H . Clinical, pathological and functional characterization of riboflavin-responsive neuropathy. Brain. (2017) ;140: :2820–37. |
[134] | Chelban V , Wilson MP , Warman Chardon J , et al. PDXK mutations cause polyneuropathy responsive to pyridoxal 5’-phosphate supplementation. Ann Neurol. (2019) ;86: :225–40. |

