
Towards Central Nervous System Involvement in Adults with Hereditary Myopathies
Abstract
There is increasing evidence of central nervous system involvement in numerous neuromuscular disorders primarily considered diseases of skeletal muscle. Our knowledge on cerebral affection in myopathies is expanding continuously due to a better understanding of the genetic background and underlying pathophysiological mechanisms. Intriguingly, there is a remarkable overlap of brain pathology in muscular diseases with pathomechanisms involved in neurodegenerative or neurodevelopmental disorders. A rapid progress in advanced neuroimaging techniques results in further detailed insight into structural and functional cerebral abnormalities. The spectrum of clinical manifestations is broad and includes movement disorders, neurovascular complications, paroxysmal neurological symptoms like migraine and epileptic seizures, but also behavioural abnormalities and cognitive dysfunction. Cerebral involvement implies a high socio-economic and personal burden in adult patients sometimes exceeding the everyday challenges associated with muscle weakness. It is especially important to clarify the nature and natural history of brain affection against the background of upcoming specific treatment regimen in hereditary myopathies that should address the brain as a secondary target. This review aims to highlight the character and extent of central nervous system involvement in patients with hereditary myopathies manifesting in adulthood, however also includes some childhood-onset diseases with brain abnormalities that transfer into adult neurological care.
Introduction
Clinical experience of health care professionals around the world and systematic neuropsychological and neuroimaging studies have long suggested the presence of central nervous system (CNS) involvement in neuromuscular disorders traditionally characterized by skeletal muscle weakness and muscle wasting [1]. The knowledge on cerebral affection in neuromuscular diseases is expanding continuously due to a better understanding of the genetic background and underlying pathophysiological mechanisms. Moreover, there is a rapid progress in up to date neuroimaging techniques now often able to demonstrate even marginal abnormalities. Many hereditary myopathies have multisystemic involvement and in some diseases the brain is the most frequently affected organ besides skeletal muscle [2]. Functional and structural CNS manifestations of neuromuscular disorders are highly variable and include e.g. epilepsy, movement disorders, neurovascular complications, headaches, stroke-like episodes, behavioural abnormalities and cognitive impairment ranging from minimal intellectual disability to severe dementia. Neuroimaging in neuromuscular disorders strongly focuses on structural brain magnetic resonance imaging (MRI) techniques that have provided the majority of data from the clinical practice and the literature to date. However, neuroimaging makes use of a plethora of numerous different techniques investigating structural and functional brain abnormalities from cerebral B-Mode ultrasound and standard cerebral computed tomography (CCT) to advanced functional methods like positron emission tomography (PET) or functional MRI. Imaging findings range from grey to white matter changes, disturbances of cerebral metabolism and perfusion to disorders of cerebral connectivity.
This review addresses adult neurologists treating adolescent or adult patients with neuromuscular disorders. We focus on hereditary myopathies with clinical brain involvement excluding optic nerve and retinal CNS manifestations. Our aim is to highlight the character and extent of brain involvement in patients with myopathies manifesting in adulthood, however also to selectively include childhood-onset diseases with CNS abnormalities that transfer into adult neurological care.
CNS manifestations in adult patients with muscular disorders often come with a high socio-economic and personal burden sometimes even exceeding the everyday challenges associated with muscle weakness. This is particularly well examined in myotonic dystrophies that demonstrate in an exemplary manner that specific personality traits and cognitive dysfunction – not muscular weakness- may represent major obstacles for social and occupational integration ultimately resulting in exclusion from social and working life.
Impairment of cognition and intellectual capabilities can be seen in patients not having reached a sufficient level in their development as well as in those that in maturity have lost these abilities. The former is usually referred to as intellectual disability or by the older term, mental retardation. Formal aspects include that the deficit has its origin prior to adulthood and takes adaptive behaviour into account as well as the measured intelligence [3]. Unfortunately, despite standard IQ testing, the tests and criteria used by many researchers differ, as definitions did over time. Assessment of adaptive behaviour in a child with a neurological, neuromuscular disability is a very difficult matter, and this part of the modern definition does only play a marginal role in most reports so far.
Loss of intellectual abilities in maturity defines dementia. While the intellectual decline can be measured globally, some forms of dementia will be hard to detect in early stages unless specific tests are used and behavioural components are included [4]. Again, there is the problem of using a general score threshold for individual performances on a highly variable individual background.
At first glance, the pathophysiological CNS processes discussed here could be divided neatly into (i) the developmental causes, when a molecular defect will lead to a permanently altered structure and function of the immature CNS – corresponding to intellectual disability – and (ii) the ongoing causes of damage acquired in the mature organ due to the disease process, indicating progression – corresponding to dementia. While this may prove correct for the majority of disorders focussing exclusively on the general course of intellectual performance, the cases in which for example episodes of general or focal metabolic crisis (like epilepsy, stroke-like episodes) may further impair the performance of an abnormally developed CNS, will not fit well into these categories. Even more problematic is the question of normal or abnormal ageing in these disorders.
Thus, while it is of great importance to understand the cause and course of the pathologies involved, in particular with regard to therapeutic options, we would caution against a binary, developmental versus degenerative, system to categorise these disorders.
Though causes of death in myopathies with multiorgan affection seem to be dominated by cardiorespiratory failure like aspiration pneumonia and sudden cardiac death, CNS manifestations including neurovascular symptoms and, most frequently, status epilepticus are important causes of early mortality in some disorders [5, 6].
Since multidisciplinary symptomatic treatments including mechanical ventilation and cardioprotective therapies have improved life expectancy in many muscular disorders and advanced medical devices become increasingly available to assist in muscular weakness, CNS disturbances such as intellectual disability or refractory epilepsy may dominate the clinical burden of these chronic diseases in the future.
Thus, current or future treatment strategies like gene therapy should address the CNS as a secondary target for a more curative treatment approach. This however requires a most clear definition of CNS affection as well as natural history data on CNS involvement, a prerequisite for the identification of suitable surrogate marker of brain abnormalities. Finally, understanding CNS disturbances in myopathies and providing specific care is critical for improving prognosis and quality of life in adulthood.
For many of the disorders discussed below, the adult neurologist is faced with two questions. First, will CNS signs be associated with an adult-onset myopathy at all? Second, how will the CNS signs of a patient with a known childhood-onset myopathy affect the course of disease and care in adulthood? Looking for the answer to the former, one may enviously eye the neuropaediatric’s wealth of data, only to be then reduced to slotting disorders as non-progressive versus progressive in answering the latter. In other words, we face the dilemma of having plenty of data that has so far not entered the everyday practice of the adult muscle clinic for most disorders.
Disorders of dystrophin-glycoprotein complex (DGC) - extracellular matrix (ECM) interaction
In skeletal muscle, fully glycosylated α-dystroglycan binds to laminin-211 (i.e. a heterotrimer formed from laminin α2, β1 and γ1 chains, but with α2 as the site of functional interaction), thereby linking the muscle fibre actin cytoskeleton via the dystrophin-glycoprotein complex (DGC) to the fibre’s basement membrane and thereby the extracellular matrix (ECM). This “lateral” adhesion protects the muscle fibre membrane from damage during contraction and the decrease of this stabilisation due to loss or dysfunction of one of the involved proteins is considered the classical pathomechanism of a range of muscular dystrophies, including Duchenne muscular dystrophy (DMD).
Many of the proteins involved are also found in similar arrangements in other tissues, serving – in the absence of the mechanical strains of skeletal muscle – more or less different purposes. In the CNS, the spectrum of interacting proteins for each of these players appears to be both different and wider. In many cases, the “CNS variant” of the protein itself is different or at least shows more variety. As mentioned, the skeletal muscle damage will often only manifest during the use of the developed organ due to the normal wear and tear. In contrast to this, in the CNS, many of these proteins play important roles in the development of the organ leading to structural and functional defects if not fulfilled in a correct and timely manner. Frequently, this damage will be established before the organ works its mature function. Whether this will immediately have clinical consequences or whether these manifest at a later stage is very different for individual genes and mutations, creating a gamut of presentations of CNS involvements in addition to possible muscle signs.
The normal binding partners for a range of differently glycosylated α-dystroglycan variants in the CNS are still laminins. In studies with animal material, laminin-10 (α5β1γ1) and -11 (α5β2γ1) seemed to rather bind brain isoforms of α-dystroglycan, while laminin-1 (α1β1γ1), -2 (α2β1γ1), and -8 (α4β1γ1) bound equally both, brain and skeletal muscle, α-dystroglycan [7]. This binding again plays a role at basement membranes. Animal studies have identified a role of α-dystroglycan on radial glia endfeet for the integrity of the pial basal membrane. If this is compromised, neuronal over-migration will lead to cobblestone malformations as in the type II lissencephaly seen in many of the congenital human diseases caused by perturbed α-dystroglycan function [8]. Isolated pachygyria, polymicrogyria, cerebellar cysts and subcortical heterotopia as described in patients with α-dystroglycanopathies may also be consequences of this [9]. Neuronal α-dystroglycan in contrast appears to have a function in synaptic plasticity [10], which would fit to clinical presentations where brain MRI is normal, but CNS function abnormal. In mice lacking laminin-α2, the development and function of the blood brain barrier was found abnormal and the distribution and polarization of aquaporin 4 channels appeared to be dependent on laminin-α2 α-dystroglycan interaction as well, considered to be responsible for increased water content of the brain [11].
Disorders due to laminin-α2 pathology
The LAMA2 gene encodes for the laminin α2-chain. The autosomal-recessive congenital muscular dystrophy caused by LAMA2 mutations (congenital muscular dystrophy type 1A; MDC1A) is considered the most frequent form in this group of disorders outside of Japan. White matter changes on brain MRI due to increased water content but not myelination abnormalities according to T2- and diffusion-weighted images [12] [Fig. 1], are considered typical (>80%), while epilepsy, intellectual disability or neuronal migration defects are each seen in less than 10% [13]. It should be noted that the white matter changes frequently become apparent during the first six to twelve months of life [14].
Fig.1
Brain MRI in adult LAMA2-MD: axial T2 diffusion (a) and coronal FLAIR (b) images showing confluent, mostly supra-, but also infratentorial white matter signal changes.
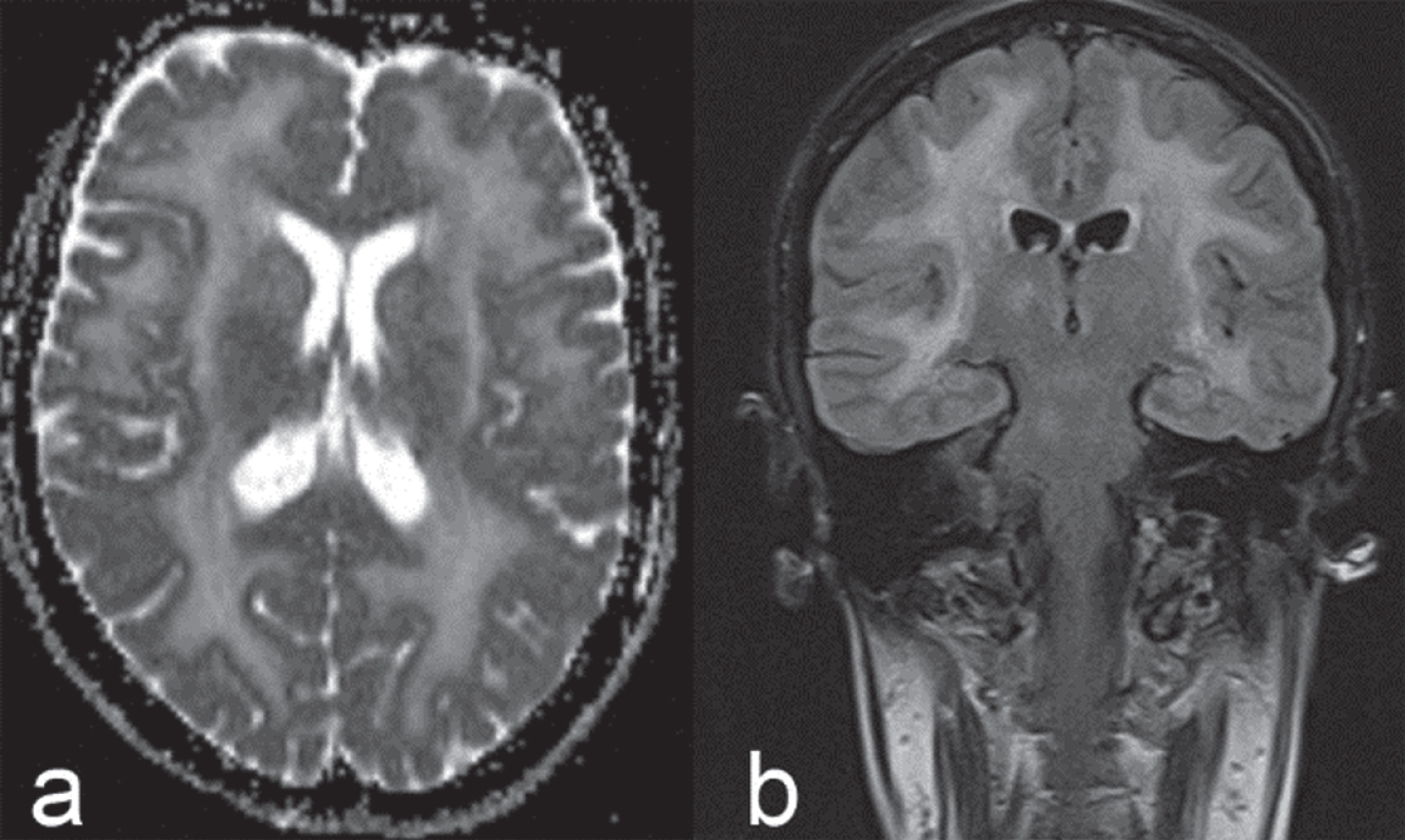
Though usually presenting as a congenital disorder, another approximately 10% of cases manifest after six months of age [13], but manifestation in adulthood is rare. Milder, atypical and therefore also late-onset cases are now often referred to as LAMA2-related muscular dystrophies (LAMA2-MD), probably to avoid the “congenital” label in the clinical setting of the adult neurologist.
Despite a rise of detection of LAMA2 variants due to increased use of next generation sequencing techniques, genotype-phenotype correlation remains unclear. Milder/later onset muscle phenotypes are however more frequently seen in patients with splicing variants or missense substitutions, presumably non-truncating alleles [14]. Some mutations have been found repeatedly in the later-onset cases (e.g. missense variant p.Thr821Pro [14]). The presence and extent of the white matter lesions do not correlate well with the clinical CNS signs, mostly seizures and intellectual disability [13]. Neuronal migration defects, an obvious risk factor for the development of epileptic seizures, have been seen in cases without white matter changes, and tend to be present in those patients with focal cerebral atrophies [13]. Alternative splicing [15] and variants in interacting protein domains may offer clues to this variability, in particular if the attribution of different laminin dystroglycan interactions on different cell types from animal studies – as mentioned above – to these elements of pathology are correct in the human.
In conclusion, the combination of abnormally high T2 signal in the periventricular and subcortical white matter in brain MRI with high CK levels in blood is typical enough that the diagnosis has to be considered in an adult patient. The main obstacle here is that the treating physician fails to grasp the concept that these findings might have a common cause. Misattribution is frequent (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy/CADASIL or other vasculopathies, mitochondrial encephalomyopathy, or “delayed myelination”). A different problem in confirming the diagnosis is the fact that regularly only one mutation in LAMA2 can be found – exactly as in the congenital muscular dystrophy. This will lead rightly to muscle biopsy for confirmation. It is important that both, the 80 kDa and the 300 kDa isoform of the laminin α2 chain, are investigated and occasionally both, immunohistochemisty and western blotting, will be necessary to confirm an abnormality [16].
While the majority of the late-onset patients show the white matter changes and some exhibit clinical signs of CNS affection (mostly epilepsy), a progression of cerebral affection during adulthood does not appear to be typical.
Table 1
Genes associated with α-dystrogylcanopathies and clinical CNS signs in clinical LGMD variants caused by mutations in these genes. Note autosomal-recessive inheritance throughout
| HGNC Approved Gene Symbol | MDDGC phenotype | CNS signs in the LGMD variant described as |
| B3GALNT2 | Not reported | |
| B4GAT1 | Not reported | |
| CRPPA | MDDGC7/LGMDR20 | No |
| DAG1 | MDDGC9/LGMDR16 | “severe cognitive impairment” [21] |
| DOLK | Not reported | |
| DPM1 | Not reported | |
| DPM2 | Not reported | |
| DPM3 | MDDGC15 | “a stroke-like episode” [22] |
| FKRP | MDDGC5/LGMDR9 | “mild impairment in executive functions and visuo-spatial planning” [20] |
| FKTN | MDDGC4/LGMDR13 | No |
| GMPPB | MDDGC14/LGMDR19 | No |
| LARGE1 | Not reported | |
| POMGNT1 | MDDGC3/LGMDR15 | No |
| POMGNT2 | MDDGC8/LGMDR24 | No |
| POMK | MDDGC12 | “Cognitive impairment” given as “IQ was tested at 80, [... ], but she required special schooling”, “requires special schooling and his IQ is 83”, “severely delayed psychomotor development” “tonic seizures” [23] |
| POMT1 | MDDGC1/LGMDR11 | “severe mental retardation and autistic features” [24] |
| POMT2 | MDDGC2/LGMDR14 | “learning difficulties and [... ] presented [... ] with developmental delay” [18] |
| RXYLT1 | Not reported | |
| TRAPPC11 | LGMDR18 | “intellectual disability including cerebral atrophy”, “IQ below 50” [25] |
Disorders due to α-dystroglycan pathology
The α-dystroglycanopathies are due to a wide spectrum of gene defects [Table 1], all leading to a lack of fully glycosylated α-dystroglycan protein, the binding partner of laminin-α2 (s. above). These disorders of α-dystroglycan show prominent clinical involvement of the CNS in their forms presenting with congenital or childhood onset, i.e. Walker-Warburg syndrome (WWS), muscle-eye-brain disease (MEB), and Fukuyama muscular dystrophy, the latter being the most common congenital muscular dystrophy in Japan. While these phenotypes are severe forms of the dystroglycanopathies, the interplay between causative gene, muscle and CNS affection is more complex. E.g. while Fukuyama muscular dystrophy caused by the common Japanese retrotransposal insertion founder mutation in the 3’UTR of FKTN shows a severe, congenital brain and muscle phenotype, the rarer FKTN mutations within the clinical limb-girdle muscular dystrophy (LGMD) spectrum hardly cause any CNS manifestation, and POMGNT1 cases seem to be similar in this. In contrast, POMT1 and POMT2 mutations frequently result in structural or functional CNS involvement even in patients at the milder end of the myopathy spectrum [18]. The most frequent dystroglycanopathy gene in the adult neurologist routine, FKRP, can cause WWS, MEB, but also milder CNS phenotypes, in particular cerebellar cysts and structural abnormalities of brainstem and pons [9] in the congenital manifestations. The vast majority of brain MRI data, however, refers to childhood-onset cases with the latest disease manifestation or highest age at MRI scan in early teenage. In a report collecting MRI data from a natural history study [19] abnormal brain MRI (mutations in FKTN, POMT1, POMT2, POMGNT1, and GMPPB) correlated to 100% with cognitive impairment (mutations in FKTN, FKRP, POMT1, POMT2, POMGNT1, and GMPPB) and 15/15 patients with reported normal development had normal MRI findings (mutations in FKRP, FKTN, and ISPD/CRPPA). Three out of 17 patients with abnormal development had normal MRI (mutations in FKRP, POMT2, and POMGNT1), but none vice versa. While some of these MRI scans were acquired from adult patients, the age average was six years. Data on the brains of those manifesting in adulthood remain hard to find. A systematic study on ten adult FKRP patients ranging from childhood-onset LGMD to asymptomatic adults with high CK reported mildly impaired executive functions and visuospatial planning with normal IQ as well as mild abnormalities in 6/9 cMRI [20]. Some neuropsychological performances were worse in those with early disease onset, but without overall correlation of performance with age at investigation or the individual mutations. Our clinical experience argues against a progression of CNS symptoms in adult life in these disorders.
In Online Mendelian Inheritance in Man (OMIM), this group of disorders has now been termed Muscular Dystrophy-DystroGlycanopathies (MDDG), separated into three subgroups: (1) MDDGA: Congenital muscular dystrophy with brain & eye abnormalities, (2) MDDGB: Congenital muscular dystrophy, less severe±intellectual disability, (3) MDDGC: Limb-girdle muscular dystrophy (LGMD) phenotype with dystroglycanopathy. To provide an overview, in Table 1 the currently known genes of the α-dystrogylcanopathy spectrum are listed with respect to whether clinical CNS signs have been reported in a LGMD variant caused by mutations in this gene.
Disorders due to dystrophin pathology
Duchenne (DMD) and Becker (BMD) muscular dystrophy are X-linked recessive disorders caused by mutations in the DMD gene in males. In a rough, dichotomic model, DMD patients have no functional dystrophin protein while BMD patients have some or partially functional dystrophin, leading to a milder phenotype.
The brain manifestations of dystrophinopathies have long been known due to the intellectual impairment in Duchenne boys, of whom about a third have an IQ below 70 [26]. Further neurocognitive and behavioural disabilities are less frequent but well described. Interest in this part of the dystrophinopathy conundrum became more urgent with the discovery of a group of patients with DMD mutations from the autism disorder spectrum, X-linked intellectual disability without muscular dystrophy [27] and of course the advent of gene therapy options.
Briefly, the DMD gene has seven independent promoter regions and two polyA-addition sites. Thus, even without further splicing etc., these promoters with their unique first exons will produce seven dystrophin isoforms [28]. From 5’ to 3’ and named after their proteins’ molecular weights, these are: 1. The brain full length (Dp427c), found in cortical neurons, skeletal and heart muscle. 2. The muscle full length (Dp427m), found in skeletal and heart muscle, but also glial cells. 3. The Purkinje cell full length (Dp427p), reported in Purkinje cells and skeletal muscle. 4. The retinal isoform (Dp260), highly expressed in retina, but also in brain and cardiac muscle. 5. Dp140, “the CNS isoform”, also found in kidney. 6. Dp116, the peripheral nerve isoform, found in Schwann cells. 7. Dp71 or G-dystrophin, ubiquitous but for the exception of skeletal muscle.
The affection of the distal isoforms Dp140 and Dp71 have long been considered to be linked to the clinical signs of brain affection in DMD. While some researchers reported just a trend for lower IQ with involvement of the distal isoforms [29, 30], others found significant association between distal macrodeletions and cognitive impairment, with relevance of Dp140 [31]. In particular, within a group of DMD and BMD patients, lack of Dp140 was linked to worse performance in neuropsychological tests [32]. DMD patients with impaired Dp140 expression showed lower total brain and grey matter volumes and worse neuropsychological scores than those with preserved Dp140 in comparison to healthy controls [33]. In a large, multinational group of DMD patients, those with mutations affecting the 3’ isoforms were more likely to show neurodevelopmental, behavioural, and emotional symptoms - and “clusters” of these findings - than those with mutations affecting only the Dp427 isoforms [34]. In DMD and BMD, loss of Dp71 as the most distal isoform in question has been found to contribute to signs of intellectual disability [35]. Intellectual disability when all isoforms are affected is considered to be typically severe [29, 35, 36]. Thus, as a guide in dystrophinopathies, the severity of the CNS symptoms – if present – correlates to the number of dystrophin isoforms affected by the mutation.
A recent study on the dystrophin isoforms [37] in normal human development and maturity has now provided a detailed picture. However, as recognised early in studies of cognitive impairment [38], the theoretical correlation of genotype and phenotype is challenged by patients with apparently identical mutations showing different clinical manifestations [28].
For the concerns of the adult neurologist, the CNS manifestations of DMD mutations are generally considered non-progressive, despite an early report of DMD cranial CT abnormalities correlating with age [39], in contrast to the muscle pathology. This probably highlights again the difference between the developmental CNS disorder and the ongoing, primarily mechanical muscle pathology. However, the call for longitudinal data [37] appears justified, in particular with regard to the aspect of prolonged survival in DMD due to new treatments.
Still, the majority of dystrophinopathy patients in the adult clinics will be BMD cases and in contrast to the wealth of studies old and new in DMD, even as some of these included BMD patients [32, 35, 38, 40], detailed neuropsychological data on BMD patients is harder to find. One specific study hints to a shift away from cognitive impairment with low IQ in comparison to DMD, but still showing learning difficulties (appr. 30%) and behavioural problems (>60% with appr. 8% autism) [41]. Data on schooling and employment seems to support this [42]. The applicability of these studies to the full spectrum of BMD has been called into question for their small and on average rather young study populations [37]. Yet, another recent study on 76 adult BMD patients from Japan [43] confirmed behavioural, psychological and social issues together with normal mean IQ. Of the 40 patients of this group with full psychological testing, half were found to show anxiety and a third to have depressive symptoms, while of the 76 cases 16 had been diagnosed with a psychiatric disorder. This study also related their findings to whether or not the mutation in a given case affected Dp140 and found no statistically significant correlation. Yet, developmental disorders, epilepsy, and problematic behaviour were only found in those with Dp140 affection, and only problematic behaviour was significantly correlated with affection of Dp71. Intriguingly, of 14 cases in this study with previous brain MRI, however performed due to neurocognitive and psychiatric symptoms, 11 showed brain atrophy [43].
It seems safe to conclude that the quality of life of adult BMD patients is affected regularly by CNS symptoms related to their dystrophinopathy, whether directly or indirectly. Further practical aspects are that no clinical – but clear electroretinographic – correlates are known for the retinal manifestations of dystrophinopathies [28] and that epilepsy occurs in about 8% of BMD [44].
One study reported minor learning disabilities or behavioural problems in a subset (29%) of a small group of pediatric manifesting female DMD carriers [45], but we were unable to find data on CNS signs in adult manifesting carriers.
The sarcoglycans
Interestingly, there are no reported CNS manifestations of the “muscle sarcoglycans” (alpha, beta, gamma, and delta sarcoglycan), important parts of the DGC. However, mutations in epsilon sarcoglycan, present in muscle but not associated with a myopathy, lead to myoclonus– dystonia syndrome (DYT11) [46], which also includes psychiatric abnormalities. An explanation for the limitation of the disorder to the CNS despite the protein being widely expressed in the body, including skeletal muscle, has been found in a specific splice variant with regard to exon 11b in the brain [47].
DNA repeat disorders
Oculopharyngeal muscular dystrophy (OPMD)
OPMD is caused by expansions of a GCG repeat in exon 1 of the PABPN1 gene on chromosome 14q. Up to 6 repeats are normally found, and the presence of 8 to 13 repeats on one allele will cause autosomal dominant OPMD, while 7 repeats on both alleles are sufficient to cause the more severe and rare recessive form. The ultrastructural hallmarks of the disease are nuclear inclusions consisting of tubulofilaments with 8.5 nm outer diameter forming palisades and tangles. As one would expect from a disease with autosomal dominant as well as recessive presentation, there is loss of function of PABPN1 as well as pathological aggregation leading to toxic gain of function. There are few reports about brain pathology or neuropsychological abnormalities in OPMD. While multisystem involvement in DNA repeat disorders is far from unusual, only a few patients have been investigated in detail. In the more severely affected homozygous patients, Blumen et al. [48] described progressive cognitive impairment and raised an interesting point about the deleterious effect this might have on treatments decisions of the patient, possible contributing to lower life span. Later, cognitive impairment and psychological disorders attributed to alterations of prefrontal-subcortical circuits were found in neuropsychological and neuropsychiatric examination of heterozygous OPMD patients with 13 to 16 repeats [49].
There appeared to be no overall association of neuropsychological abnormalities with brain MRI findings and so far, a relation of brain dysfunction to brain pathology has not been established, despite one report pointing to deposits in the CNS predating the aforementioned studies [50]. Homozygous OPMD patients are mostly found in French Canadian and Jewish Uzbek populations, therefore the average neurologist will not expect their OPMD patients to decline quickly into dementia, but taking a closer look at neuropsychological aspects in heterozygous OPMD patients may be warranted in the future.
Myotonic dystrophies
Myotonic dystrophies type 1 and 2 (DM1, DM2) are autosomal dominantly inherited multisystemic repeat expansion disorders and represent the most common muscular dystrophies in adulthood. DM1 is caused by an expanded CTG repeat within the noncoding 3′ untranslated region of the myotonic dystrophy protein kinase (DMPK) gene, whereas DM2 is caused by an expanded CCTG repeat in the first intron of the zinc finger protein 9 (CNBP, ZNF9). Both the molecular genetic background and pathophysiological mechanisms have largely been investigated in both disorders [51]. Multiorgan affection is characteristic for these prototypic repeat expansion disorders. Brain involvement is one of the most frequent non-muscular features in DM1 and DM2 and is primarily associated with cognitive and behavioural abnormalities. In contrast to other myopathies and mitochondrial disorders, focal neurological deficits, epilepsy, ataxia, and/or headache are not characteristic, and clinical involvement of central motor and extrapyramidal pathways is not reported. With new therapeutic developments targeting multiple organ systems it is mandatory to increase the understanding of CNS affection and its natural history to appropriately monitor potential future treatment effects on the brain. Indeed, CNS dysfunction is one of the most relevant issues affecting quality of life in DM1 and DM2 patients and also implies major socioeconomic drawbacks [52, 53]. The neuropathological correlate of CNS symptoms in myotonic dystrophies is not entirely clear. Neuropathological findings in DM1 brains include intracytoplasmic inclusions in thalamus, striatum, cerebral cortex, and brainstem [54, 55]. Most changes are located in neurons, but white matter alterations have also been described including disordered arrangement of myelin sheaths and axons [56– 58]. Importantly, neurofibrillary degeneration with intraneuronal accumulation of abnormally modified microtubuli-associated tau protein has been demonstrated in the brains of DM1 and DM2 patients. Experimentally, aberrant tau expression by dysregulated alternative splicing has been shown allocating myotonic dystrophies to tauopathies and other neurodegenerative diseases [59].
DM1 can present with a severe congenital form, a childhood-onset form, and the classic adult-onset form which however may show first symptoms in adolescence. In DM1, the classic disease range of CTG repeat numbers is 50 to 4,000, in which repeat sizes of 50 to 80 are usually associated with mild clinical phenotypes, and large repeat expansions up to 4,000 or more are often found in severe, mostly congenital forms of DM1. According to the age at onset, disease severity and/or CTG repeat size, functional and structural brain involvement differs highly between the various forms of the disease. In congenital and childhood-onset forms of DM1, intellectual disability, psychomotor delay, and neuropsychiatric symptoms like autism-spectrum disorders or attention-deficit syndromes are characteristic, whereas brain affection in the adult-onset form is different and usually less severe. Mildly affected DM1 patients may present without any functional CNS affection. In contrast to DM1, there is no congenital or childhood-onset form in DM2 which typically manifests in adulthood, sometimes at advanced ages.
Many methods have been applied to assess the neuropsychological functioning in DM1 and DM2 patients. The instruments most used in neuropsychological studies on DM1 to date have been listed in a recent review with respect to the examined cognitive domains [60]. Many different neuropsychological test batteries have been used ranging from standardized assessments of frontal and executive functioning to psychomotor speed and intelligence tests. Behavioural impairments, personality profiles, social cognition and Quality-of-Life aspects have been measured with distinct neuropsychological and psychological instruments. Various personality inventories, social cognition test batteries and Quality-of-Life questionnaires have been applied from the Minnesota Multiphasic Personality Inventory (MMPI), Facial Emotion Recognition Tests, Individualized Neuromuscular Quality of Life questionnaire (INQoL) to the Myotonic Dystrophy Health Index (MDHI) as self-reported multi-domain rating scale [61]. As in neuroimaging techniques, an international consensus on obligatory neuropsychological test batteries as outcome measures in myotonic dystrophies does not exist which may be due to the complex nature of the diseases and variable findings across different studies.
Various neuroimaging techniques have been applied in myotonic dystrophies over the years to examine the structure and function of the brain [62, 63]. Methods range from CCT, voxel-based morphometry (VBM), diffusion-tensor-imaging (DTI) to most advanced functional MRI techniques (fMRI). Fluorodeoxyglucose positron emission tomography (FDG-PET) has been used to investigate the cerebral glucose metabolism. 99mTc-emission computed tomography (ECD) and 99mTc hexamethyl propylenamine oxime (HMPAO) single-photon emission computed tomography (SPECT) as well as H215O-PET have been applied to analyse cerebral perfusion. Analyses of cellular markers have been performed by Proton-MR-spectroscopy (1H-MRS). fMRI using e.g. motor tasks and resting-state analyses were introduced to study cerebral networks. Further, quantitative mapping of the magnetic susceptibility and the effective transverse relaxation rate were recently applied to assess the iron content in distinct brain regions in DM1 and DM2 [64]. As bedside technique, transcranial B-mode sonography has been used in adult-onset DM1 and DM2 patients to evaluate ventricle diameters and the echogenicity of brainstem and basal ganglia [65, 66].
Fatigue, excessive daytime sleepiness, depression and anxiety in myotonic dystrophies
Behaviourally, fatigue, apathy and depression are defined by a significant reduction in self-initiated voluntary action. Fatigue is one of the most frequently reported symptoms in almost all neuromuscular diseases. The phenomenon of fatigue is not fully understood yet. However, a unifying definition of chronic pathological fatigue as a disease-independent mechanism has recently been proposed based on a more physiological standpoint [67]. Whereas signs of depression and apathy can be identified and measured by external observers, fatigue is always self-reported and supposed to be primarily a perceptual phenomenon arising from dysfunctional activational systems and aberrant sensory attenuation.
Sleepiness and fatigue constitute major complaints in DM1, but also in DM2 [68, 69]. According to our findings and data from the literature, daytime sleepiness is widely restricted to DM1, whereas fatigue is a major complaint in DM1 and DM2 patients. Previous data suggested the absence of excessive daytime sleepiness in DM2 to be a discriminative feature between both disorders [69]. Symptoms of fatigue are rather stable over time in DM1 and DM2. These findings underline former data postulating that fatigue and daytime sleepiness but also apathy in myotonic dystrophies more likely result from CNS dysfunction than from respiratory muscle weakness and/or sleep-related breathing disorders [70]. A recent randomized controlled treatment trial targeting the CNS in fatigued DM1 patients indicated that implementation of cognitive behavioural therapy is able to lead to a higher capacity for activity and social participation [71].
Anxiety has been reported in DM1 and DM2, but does not seem to be a prominent feature according to data in the literature and our personal experience.
Depression is present in DM1 and DM2, however seems to be rather stable over time [72]. Depressed mood in DM1 might be more pronounced in earlier disease stages, whereas depression is more likely to be found in later disease stages of DM2 [73, 74]. Thus, DM1 patients might adapt to their symptoms earlier or show a lack of self-awareness at later stages of the disorder.
Myotonic dystrophy type 1 (DM1)
In DM1, there is longstanding evidence of functional cerebral involvement [52, 75]. The cognitive profile of DM1 has been characterised extensively in various neuropsychological studies over recent decades. Cognitive deficits, especially executive dysfunction, mental slowing, social cognition impairment, impairments in theory-of-mind variables, behavioural abnormalities and specific personality traits have been described and may deteriorate over time [76– 80]. Reported behavioural abnormalities and personality patterns are consistent in DM1 and include reduced initiative and inactivity, apathy, avoidant and passive personality features, less frequently obsessive-compulsive and sometimes passive-aggressive behaviour. Signs of a personality disorder are present in only a minority of patients [81]. Avoidant and passive behavioural patterns may be due to a frontal dysexecutive syndrome that is associated with various morphological and functional neuroimaging findings in DM1 [52]. DM1 patients may show low social engagement and difficulties in social-cognitive functions. Recently, social cognition impairment due to basic deficits in emotion recognition measured by facial emotion recognition, e.g. of anger and disgust, have shown to be core deficits in the adult-onset disease [82, 83]. Neural substrates associated with social cognition deficits in DM1 have recently been described by use of resting-state fMRI [84].
In DM1 patients, cognitive deficits include a variable combination of cognitive impairment across different domains [60]. Cognitive deficits in the classic adult-onset form may affect memory, attention, visuospatial construction and verbal ability. According to most neuropsychological studies, executive dysfunction, attentional and/or visuospatial/visuoconstructional impairments constitute the predominant deficits [52, 74, 85]. Both earlier onset and longer duration of the disease seem to be indicative of more severe cognitive deficits [80]. Due to small sample sizes and different neuropsychological methods applied, neuropsychological testing profiles in DM1 show variable results. In neuropsychological longitudinal studies, cognitive impairment worsened over time in the majority of patients affecting different domains and suggesting an accelerated normal aging process [78, 80, 86]. However, some studies found only minor and rather stable neuropsychological deficits in adult-onset DM1 that predominantly affected executive functioning [74, 77]. In the clinical practice, DM1 patients may wrongly give the impression of having reduced overall intelligence because of “mental and psychomotor slowing”, and the facial expression combined with a lack of initiative [52].
There is also ample evidence for structural brain involvement in DM1 and DM2. Transcranial B-mode sonography showed that brainstem raphe hypoechogenicity was more common in DM1 patients than in controls, and both hypoechogenicity and hyperechogenicity of the substantia nigra were more frequent. Third ventricle diameters were increased in DM1 patients [65, 66]. Cross-sectional neuroimaging studies demonstrated brain atrophy, focal subcortical and cortical grey matter reduction, ubiquitous white matter reduction and white matter lesions in DM1 more than DM2 patients. White matter lesions have frequently been reported in DM1 [Fig. 2a], do not seem to be associated with vascular risk factors and are predominantly located in frontal and temporal lobes [74, 87, 88]. Particularly, anterior temporal white matter lesions (ATWML) are a rather specific presentation in DM1 especially when compared to DM2 [Fig. 2b], where this characteristic brain affection is not present [87, 89– 91]. Thinning or atrophy of the callosal body has been reported most frequently in congenital DM1 but also in adult-onset forms of the disease [91]. Global brain atrophy has been described in DM1 very early. Grey matter abnormalities include ventricular enlargement, diffuse cortical atrophy, global grey matter reduction, focal brain atrophy in various cerebral lobes, the hippocampus and basal ganglia [74, 79, 92– 94]. VBM studies confirmed a widespread cortical involvement of grey matter affecting all lobes. Volume reductions of subcortical grey matter were reported in striatum, thalamus, and cerebellum [74, 79, 90, 93– 97]. DTI showed a widespread degradation of white matter fibre tracts in DM1, and recently, DTI and network measures gave evidence of brain involvement in DM1 as a complex network disorder characterised by white matter network alterations [72, 74, 91, 98, 99]. White matter exceeded grey matter changes by far in adult-onset DM1 and DM2 suggesting a predominant white matter disease [72, 74, 94, 95].
Fig.2
1.5 T brain MRI in DM1, FLAIR sequences. a) 41-year-old adult-onset male DM1 patient with moderate confluent periventricular and mild subcortical white matter lesions. b) 47-year-old adult-onset female DM1 patient with bilateral anterior temporal white matter lesions (ATWML) and marked thickening of the skull [87].
![1.5 T brain MRI in DM1, FLAIR sequences. a) 41-year-old adult-onset male DM1 patient with moderate confluent periventricular and mild subcortical white matter lesions. b) 47-year-old adult-onset female DM1 patient with bilateral anterior temporal white matter lesions (ATWML) and marked thickening of the skull [87].](https://content.iospress.com:443/media/jnd/2020/7-4/jnd-7-4-jnd200507/jnd-7-jnd200507-g002.jpg)
PET studies in myotonic dystrophies gave evidence of glucose hypometabolism in prefrontal, frontal, temporal, and pericentral regions in DM1 more than DM2 [79, 100, 101]. ”Perfusion“-PET and SPECT studies showed regional cerebral blood flow deficits in frontal, temporo-parietal, and less pronounced in parieto-occipital regions in DM1 [76, 88, 100, 102]. Brain cellular and neuronal markers were mainly reduced in occipital and temporo-parietal cortical regions and frontal white matter in DM1 more than DM2 [103]. Resting-state fMRI studies in DM1 showed a reduced connectivity in specific cerebral networks associated with behavioural abnormalities [84]. In classic adult-onset DM1 patients, widespread iron accumulation in deep grey matter was detected and significantly associated with clinical symptoms including muscular weakness, daytime sleepiness, and specific cognitive deficits [64].
However, the functional relevance of structural brain abnormalities is still under discussion, and results of correlation analyses between brain morphology, neuropsychological, clinical, and genetic data are highly controversial to date [74, 79, 90, 91, 93, 95, 96, 98, 99, 104]. While some studies showed correlations of brain morphological changes with neuropsychological and clinical parameters in DM1, others failed to do so.
The natural history of structural brain changes in myotonic dystrophies is not clear yet [62, 63]. A more neurodegenerative than neurodevelopmental origin was hypothesized, since MRI changes correlated with disease duration in cross-sectional analyses [74]. A cross-sectional study on juvenile and adult DM1 postulated a degenerative, premature aging origin of grey matter changes, however a more neurodevelopmental origin of white matter alterations [95]. This is partly in line with first longitudinal brain imaging data that demonstrated a lack of significant disease-related progression of grey and white matter involvement over a period of five years. This finding suggests a slowly progressive or even stable course of cerebral changes in middle-aged adult-onset DM1 patients [72] [Fig. 4].
Myotonic dystrophy type 2 (DM2)
There is clear evidence that neuropsychological and cognitive deficits may be present in DM2 patients, though usually to a minor degree when compared to DM1 [52, 72, 74, 77, 88]. The earliest evidence of cognitive impairment in DM2 came from studies from Meola et al. demonstrating deficits in visual-spatial performance and attention [76, 102]. Subsequent neuropsychological examinations detected no general cognitive decline, but focal cognitive deficits predominantly affecting executive functions like focussed attention and interference, but also verbal memory [74, 77, 85, 101]. These findings correspond well with predominant frontal and parietal lobe dysfunction. Some degree of behavioural abnormalities has been reported in DM2 primarily associated with a dysexecutive syndrome [76, 102]. An avoidant personality profile has early been demonstrated, whereas recent examinations did not show statistically significant personality impairments in DM2. However, compulsive and paranoid personality features were present in some patients. In contrast to former studies, the most common neuropsychological symptoms in DM2 were anxiety and somatization [76, 105].
As in DM1, the discrepancy regarding the presence and extent of cognitive and behavioural deficits between different studies might partially be explained by methodological differences. Though DM2 might be as prevalent as DM1 in some regions, there are much less neuroimaging studies on DM2. The first brain imaging study on DM2 was performed in 1997 [106]. Since then, less than 20 cross-sectional and one longitudinal brain imaging studies have been reported [63, 66, 72, 107, 108].
Transcranial B-mode sonography in DM2 patients demonstrated higher frequencies of brainstem raphe hypoechogenicity and substantia nigra hyperechogenicity and increased diameters of the third ventricle [66, 107]. Cross-sectional neuroimaging studies showed patchy, sometimes confluent periventricular white matter lesions not clearly attributed to a vascular origin and no ATWML [Fig. 3a]. The amount of white matter lesions in DM2 was less compared to DM1 [74, 87]. Further brain MRI studies revealed focal and/or general brain atrophy in DM2 [Fig. 3b], including cortical grey matter reduction, but also subcortical grey matter reduction in hypothalamus, thalamus, brainstem and adjacent midline brain regions [79, 108]. Some authors reported a more pronounced grey matter loss in DM2 compared to DM1, affecting cuneus, temporal regions and amygdala [96]. In contrast, subsequent VBM analyses in a larger group of DM2 patients detected no grey matter decrease compared to controls [74]. However, VBM studies revealed predominant white matter alterations along corpus callosum and in all lobes and in the cerebellum [74, 108]. DTI studies demonstrated microstructural dysintegrity predominantly of the corpus callosum, but also of other association and projection fibres, including the limbic system [72, 74]. Most imaging studies suggested a predominant white matter disease similar to DM1, however, less pronounced. Functional brain imaging studies showed glucose hypometabolism and reduced perfusion mainly in frontal regions, but also in temporal and parieto-occipital areas, in general less pronounced compared to DM1 [76, 79, 102]. As in DM1, brain cellular and neuronal markers were reduced mainly in occipital and temporo-parietal cortical regions and frontal white matter, again much less pronounced [103]. Iron accumulation in deep grey matter, e.g. in the putamen and accumbens, could be identified, however to a minor extent compared to DM1 [64].
Fig.3
1.5 T brain MRI in DM2, FLAIR sequences. a) 68-year-old female DM2 patient with severe confluent periventricular white matter lesions and general moderate brain atrophy. Cerebrovascular risk factors like hypertension or diabetes were not present. b) 65-year-old female DM2 patient with general brain atrophy [87].
![1.5 T brain MRI in DM2, FLAIR sequences. a) 68-year-old female DM2 patient with severe confluent periventricular white matter lesions and general moderate brain atrophy. Cerebrovascular risk factors like hypertension or diabetes were not present. b) 65-year-old female DM2 patient with general brain atrophy [87].](https://content.iospress.com:443/media/jnd/2020/7-4/jnd-7-4-jnd200507/jnd-7-jnd200507-g003.jpg)
In contrast to DM1, repeat expansion sizes are not routinely analysed in DM2 and do not correlate well with clinical symptoms. Thus, studies on the association of neuroimaging and neuropsychological findings with genetic results are missing. As in DM1, the functional relevance of structural brain abnormalities is still unclear, and results of correlation analyses between brain morphology, neuropsychological, and clinical data in DM2 are controversial or even contradictory [64, 74, 76, 79, 88, 96, 101]. A recent functional imaging study however demonstrated that FDG-PET findings corresponded well with the results of neuropsychological testing in DM2. This suggests that functional neuroimaging techniques might be more suitable to identify potential associations of brain alterations with neuropsychological performing, therefore serving as more suitable biomarkers [53, 72, 101].
Fig.4
3.0 T brain MRI, voxel-based morphometry (VBM) in DM1 and DM2. Longitudinal MRI data at baseline and follow-up after 5.5±0.4 years. Group comparisons at baseline and follow-up. Displayed results of VBM analyses are based on a threshold of pfalse discovery rate < 0.05 at voxel-level with an extended cluster threshold of 10 voxels. The coordinates refer to the MNI reference space. VBM analyses were performed in 13 DM1, 15 DM2 patients, and 13 controls. (a) Grey matter decrease in DM1 patients compared with controls at baseline (light red) and at follow-up (light and dark red). Dark red areas had not been affected at baseline. (b) Grey matter decrease in DM2 patients compared with controls at baseline and at follow-up (no clusters detected). (c) White matter decrease in DM1 patients compared with controls at baseline (light blue) and at follow-up (light and dark blue). (d) White matter decrease in DM2 patients compared with controls at baseline (light blue) and at follow-up (light and dark blue). Dark blue areas had not been affected at baseline [72].
![3.0 T brain MRI, voxel-based morphometry (VBM) in DM1 and DM2. Longitudinal MRI data at baseline and follow-up after 5.5±0.4 years. Group comparisons at baseline and follow-up. Displayed results of VBM analyses are based on a threshold of pfalse discovery rate < 0.05 at voxel-level with an extended cluster threshold of 10 voxels. The coordinates refer to the MNI reference space. VBM analyses were performed in 13 DM1, 15 DM2 patients, and 13 controls. (a) Grey matter decrease in DM1 patients compared with controls at baseline (light red) and at follow-up (light and dark red). Dark red areas had not been affected at baseline. (b) Grey matter decrease in DM2 patients compared with controls at baseline and at follow-up (no clusters detected). (c) White matter decrease in DM1 patients compared with controls at baseline (light blue) and at follow-up (light and dark blue). (d) White matter decrease in DM2 patients compared with controls at baseline (light blue) and at follow-up (light and dark blue). Dark blue areas had not been affected at baseline [72].](https://content.iospress.com:443/media/jnd/2020/7-4/jnd-7-4-jnd200507/jnd-7-jnd200507-g004.jpg)
The first longitudinal brain imaging data on DM2 demonstrated that white matter affection was prominent, but less pronounced compared to DM1 with few additional white matter changes after a period of five years [Fig. 4]. As in DM1, there was a lack of significant disease-related progression of morphological brain involvement over time which suggests a slow progress or even stable disease course [72]. Nevertheless, longitudinal data on brain involvement in DM2 is highly limited and further studies are needed.
Facioscapulohumeral muscular dystrophy (FSHD)
FSHD is a dominant disease caused by re-expression of the DUX4 gene on chromosome 4q. The most frequent cause (FSHD1) of this is contraction of D4Z4 macrosatellite repeats by deletion in a “permissive” 4A haplotype (mostly 4qA161, alternatively 4qA159, 4qA166H or 4qA168 [109]). Normally, 11 to 100 repeats are found. Repeat numbers below 11 on one allele lead to FSHD1 with the repeat size often correlating with age at onset and inversely with disease severity. The diagnostic standard therefore is the determination of the repeat length by southern blotting with the p13E-11 probe after DNA digestion with EcoRI/BlnI (and possibly XapI to double-check against repeats from chromosome 10) [110]. Thus, in most of the literature the extent of the D4Z4 repeats is given as “fragment size”, with one D4Z4 repeat corresponding roughly to 3.3 kB. When in doubt, however, additional haplotype analysis will be required.
In early onset FSHD patients, high-frequency hearing loss is considered to be present as part of the disorder in more than half of the patients [111]. It is usually apparent before age seven years and progressive in some patients [112]. Fitting well with an occurrence in early onset cases, a threshold fragment size of 20 kb (five D4Z4 repeats) has been suggested [112]. Patients with larger fragments do not seem to suffer from hearing loss in childhood. Further investigations argue for this to be a disorder of the cochlea and thereby not a CNS manifestation [113, 114]. However, early onset cases with short fragments may also show intellectual disability and/ or epilepsy in particular with fragments of 10 or 11 kB (two repeats) [114]. Due to the relation of short fragments, i.e. large deletions, to early onset, it seems unlikely to encounter these problems in an adult onset case, and in adults at least the occurrence of hearing loss is reported to be no higher than in the age control [115].
This is different for brain MRI findings, where white matter changes have been described repeatedly, but without correlation to epilepsy, intellectual disability or indeed disease severity. It should be noted that some frequently quoted early brain MRI and neurophysiology data were gathered without genetic confirmation of the diagnosis [e.g. 116]. Even more puzzling, one study [117] found significantly lower grey matter volumes in FSHD in comparison to controls, localised to the left precentral cortex, the anterior cingulate, and the right frontopolar region by VBM. These changes did not correlate with disease duration, deletion size, age at onset or presence of white matter hyperintensities. Nor did this group contain cases with epilepsy or intellectual disability. There was however correlation to disease severity (Ricci scale [118]). The demographics of this group were given as: mean age at onset: 15.8 years (range 10– 57) and D4Z4 fragment size 25.9±6.9 kb (range 11– 35), i.e. two to nine repeats. Unfortunately, data on hearing loss was not given.
We are not aware of CNS data on – much rarer – FSHD2 patients, in the vast majority of whom a SMCHD1 mutation of chromosome 18 leads to hypomethylation of more than 10 D4Z4 repeats, which in a 4A haplotype will again cause DUX4 expression [119]. FSHD2 patients have been judged “clinically indistinguishable” [120] from FSHD1 patients. This report found 18% patients reporting symptomatic hearing loss (the average age at disease onset was given with 26 years, range 0– 60, but no correlation of age with hearing loss), higher than in FSHD1 adult populations [115], but the clinical CNS signs appear so far to be a feature of the FSHD1 cases with large D4Z4 deletions.
In conclusion, an adult-onset FSHD1 patient may have asymptomatic brain MRI abnormalities associated with the disease. In childhood-onset cases, cochlear hearing loss is frequently present and has been found to be progressive in some cases [112].
Metabolic disorders and enzymatic dysfunctions
Obviously, many of the genes causing α-dystroglycanopathies are thought to code for glycosyltransferases and could be listed here, but in the interest of gathering disorders of common pathophysiologies these genes appear above in the DGC-ECM interaction-associated disease section.
Mitochondrial encephalomyopathies
In its most narrow definition, mitochondrial disorders are clinical syndromes that are associated with a primary dysfunction of the respiratory chain. The oxidative phosphorylation (OXPHOS) system is under dual genetic control of the mitochondrial (mt) and nuclear DNA. Mitochondrial disorders result either from primary mtDNA mutations or from pathogenic mutations of nuclear genes, the latter displaying an emerging group of adult mitochondrial diseases. Nuclear DNA mutations may directly or indirectly affect respiratory chain enzyme activities [121]. Mitochondrial respiratory chain defects are an important cause of genetically determined neurological disorders in adulthood, and the prevalence of adult mitochondrial disease is comparable with the most common forms of inherited neurological disorders in adulthood [122]. Though almost all organs can be affected, OXPHOS defects most frequently and severely strike organs with high energy demands like skeletal muscles and the CNS [123–125]. Neurons are heavily dependent on mitochondria for the energy production, but mitochondria also play an important role in apoptosis, iron-sulphur cluster biogenesis and calcium buffering in neuronal cells.
Clinical symptoms of CNS involvement include epilepsy, migraine, stroke-like episodes, generalised dystonia, cerebellar ataxia, afferent ataxia due to peripheral neuropathies and/or posterior column affection of the spinal cord, spastic paraplegia, psychiatric disorders, behavioural abnormalities, cognitive dysfunction, dementia, and brainstem symptoms. In this review, ophthalmological symptoms originating from optic nerve and/or retinal affection are excluded for the reason of limited space. The neuropathological correlates of brain affection in mitochondrial disorders are heterogeneous. Neuronal loss, cortical laminar necrosis, deep grey matter vasculo-necrotic changes, spongy degeneration, gliosis, demyelination, and vasogenic edema have been described affecting grey and white matter of the brain and spinal cord [126, 127].
Epilepsy
Mitochondria are intimately involved in pathways leading to neuronal cell death seen in experimental and human epilepsy. Mitochondrial dysfunction plays a direct pathogenic role in the process of epileptogenesis in certain types of epilepsy [128]. On the other hand, epileptic seizures can be the presenting feature of mitochondrial disorders in adulthood usually related to nuclear DNA or pathogenic mtDNA point mutations. Mitochondrial disorders in adulthood most frequently related with epilepsy are myoclonus epilepsy with ragged red fibres (MERRF), mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS), mitochondrial DNA polymerase gamma (POLG)-associated diseases as well as non-classic encephalomyopathic overlap phenotypes.
Focal seizures characterise the epileptic presentation in the majority of adult patients and often show an occipital lobe predilection. They present as motor seizures, simple or complex focal seizures, or epilepsia partialis continua, and may evolve to generalised tonic-clonic seizures, or segmental and generalized myoclonic seizures [129, 130]. In contrast, generalised tonic-clonic seizures and progressive myoclonus epilepsy are an early and consistent symptom in specific mitochondrial encephalopathies in adults including patients harbouring the m.8344A>G MT-TK point mutation associated with MERRF. Generalised progressive myoclonus epilepsy can also be the presenting feature in patients harbouring mutations in the POLG gene or carrying particular mtDNA tRNA mutations, e.g. in MT-TL1 or MT-TF genes [128, 131, 132]. Mutations in POLG are also attributed to a phenotype called Mitochondrial Spinocerebellar Ataxia and Epilepy (MSCAE) [133]. POLG-associated diseases often present with a focal occipital lobe semiology that may coincidence with headache and/or vomiting clinically resembling migraine with aura. Disease courses in POLG-associated epilepsy are frequently complicated by epilepsia partialis continua, refractory epileptic seizures and status epilepticus with high morbidity and mortality [129, 134, 135]. Mitochondrial disorders attributed to mutations in COQ8A (ADCK3, CABC1) can also present with epilepsy, stroke-like episodes and ataxia mimicking POLG-associated diseases [136]. COQ8A is one of several genes associated with primary coenzyme Q10 (CoQ10) deficiency. Defects of the CoQ10 metabolism and biosynthesis pathways cause a variety of disorders ranging from isolated myopathy to multisystem involvement with CNS symptoms [137]. Onset is typically during infancy or childhood, but adult-onset cases are increasingly reported with epilepsy, tremor, ataxia, or coordination problems as first symptoms.
Classic MELAS is frequently associated with focal epileptic seizures predominantly affecting the occipital and temporal lobes. According to a recent European consensus statement, stroke-like episodes should be newly defined in terms of an epileptic encephalopathy and usually origin in focal epileptic activity of the brain [138].
Cerebellar ataxia
CoQ10 deficiency due to recessive COQ8A mutations has also been described as a potentially treatable cause of adult-onset ataxia [139]. Cerebellar ataxia is a common CNS symptom in various mitochondrial disorders in adulthood. Ataxia of cerebellar origin may be present in combination with afferent ataxia due to spinal cord affection and/or peripheral neuropathies usually axonal in nature. Adult-onset ataxia is usually progressive over time and a characteristic finding in the clinical syndromes of MERRF, POLG-associated diseases (“POLG ataxia“ [140]) like MSCAE or Sensory Ataxic Neuropathy, Dysarthria, and Ophthalmoparesis (SANDO), and adult-onset mitochondrial syndromes associated with mutations in the mtDNA encoded ATP6 gene (MT-ATP6) like Neuropathy, Ataxia, and Retinitis Pigmentosa (NARP). According to data from the German mitoNET registry and other local databases from Europe, USA, Japan, and China, ataxia is present in more than half of MERRF patients harbouring the m.8344A>G MT-TK point mutation and in more than 80% of patients with MT-ATP6-associated mitochondrial disorders [141, 142]. The latter finding is underlined by a recent epidemiological study in the U.K. showing that ataxia presented the most common symptom in MT-ATP6-associated mitochondrial disorders [143]. Ataxia is also frequently reported in patients harbouring many different mtDNA mutations including mutations in MT-TL1 and even mtDNA single deletions. Kearns-Sayre Syndrome (KSS) usually originates in mtDNA single deletions, and the ataxia is one of the prominent clinical features [144]. Ataxia is also a common “plus” symptom in chronic progressive external ophthalmoplegia (CPEO) that is frequently associated with mtDNA single or multiple deletions [145].
Stroke-like episodes
Stroke-like episodes are often present in mitochondrial disorders and one of the main syndromic features of patients with MELAS due to the m.3243A>G MT-TL1 (“common MELAS“) point mutation [146]. However, stroke-like episodes are not restricted to classic MELAS but may equally occur in patients with other mtDNA point mutations like MT-TK and MT-ND5 mutations or in POLG-associated disorders. Stroke-like episodes are typically characterised by a subacute onset, headache, nausea and vomiting, confusion, visual disturbances and focal-onset seizures with neurological deficits originating from the occipital lobes. Brain MRI shows transient, sometimes persistent, cortical and subcortical signal abnormalities not confined to vascular territories. Characteristic stroke-like lesions appear as T2-weighted signal hyperintensities in brain MRI typically located in posterior brain regions, however often migrating to other regions of the brain in the course of the episode. Data on MR apparent diffusion coefficients (ADC) in the acute phase of a stroke-like episode are conflicting, however increased instead of reduced ADC values reflecting vasogenic instead of ischemia-induced cytotoxic oedema have frequently been reported [147] [Fig. 5]. The underlying mechanism of stroke-like episodes is still not fully understood. The vascular theory emphasises the potential role of mitochondrial angiopathy and impaired autoregulation of small cerebral vessels resulting in ischemia additionally linking nitric oxide deficiency with the pathogenesis of stroke-like episodes [148]. However, the current understanding of stroke-like episodes focusses on an underlying focal cerebral metabolic crises usually triggered by epileptic seizures [138].
Fig.5
1.5 T brain MRI. A 53-year-old female patient with MELAS and multiple stroke-like lesions showing an encephalopathic episode with epileptic seizures, mild left-sided hemiparesis, and dysarthria since 1 week: Diffusion-weighted (a, e) images with corresponding ADC maps (b, f) show restricted diffusion in the right temporal cortex (arrows). ADC maps are indicative of mixed cytotoxic and vasogenic edema in these areas (b, f; arrows). Additional non-acute lesions are demonstrated in FLAIR (c, g; arrowheads) and ADC maps (b, f; arrowheads) in the left temporal lobe. Unenhanced CT (d) shows no calcification and no hemorrhage. There is no enhancement on T1-weighted post-contrast image (h; arrows) [161].
![1.5 T brain MRI. A 53-year-old female patient with MELAS and multiple stroke-like lesions showing an encephalopathic episode with epileptic seizures, mild left-sided hemiparesis, and dysarthria since 1 week: Diffusion-weighted (a, e) images with corresponding ADC maps (b, f) show restricted diffusion in the right temporal cortex (arrows). ADC maps are indicative of mixed cytotoxic and vasogenic edema in these areas (b, f; arrows). Additional non-acute lesions are demonstrated in FLAIR (c, g; arrowheads) and ADC maps (b, f; arrowheads) in the left temporal lobe. Unenhanced CT (d) shows no calcification and no hemorrhage. There is no enhancement on T1-weighted post-contrast image (h; arrows) [161].](https://content.iospress.com:443/media/jnd/2020/7-4/jnd-7-4-jnd200507/jnd-7-jnd200507-g005.jpg)
Extrapyramidal symptoms
Extrapyramidal symptoms including focal or generalised dystonia, choreoathetosis, dyskinesia, akathisia and Parkinson features have been described in adult-onset mitochondrial disorders, in particular in the context of POLG-associated diseases [149, 150]. Genetically determined diseases of mtDNA maintenance most frequently manifest with parkinsonism, and idiopathic Parkinson symptoms have likewise been related to a loss of mtDNA integrity in nigrostriatal neurons [151]. However, Parkinson symptoms in classic adult-onset mitochondrial syndromes are very rare in the clinical practice and seem to respond well to levo-DOPA treatment. In patients with childhood-onset Leigh syndrome who survive into young adulthood, marked generalised dystonia may be a common challenging symptom.
Psychiatric symptoms
Psychiatric symptoms are common in mitochondrial disorders in adulthood. To date, this issue has received little attention in the literature, and primary mitochondrial disorders are likely underdiagnosed in psychiatric patients [152]. Psychiatric disorders in mitochondrial diseases in adults include behavioural abnormalities, personality disorders, anxiety, mood disorders like depression, and psychosis. According to our clinical experience, serious psychiatric symptoms like psychotic episodes are frequently related to pathogenic mtDNA point mutations in MT-TL1 including MELAS and MT-TK including MERRF. Further, POLG-associated diseases commonly present with psychiatric symptoms. Psychiatrists should be aware of typical non-psychiatric clinical features of mitochondrial dysfunction (“red flags“) that may be indicative of an underlying primary mitochondrial disease. These clinical signs and symptoms can be minimal like multiple lipoma in MT-TK-related diseases or deafness, diabetes and/or short stature in MT-TL1 mutations, nevertheless can be highly indicative in individual cases. Importantly, non-convulsive seizures may cause subacute-onset neuropsychiatric symptoms in mitochondrial diseases, especially in stroke-like episodes. Some of these patients present with anxiety, aggressiveness, agitation or psychosis with auditory or visual hallucinations and are in acute need of anticonvulsive treatment [138].
Cognitive decline
Cognitive decline and/or early-onset dementia are widely reported in mitochondrial disorders in adults. Nevertheless, data is very limited and there are only few studies on cognitive function in adult-onset mitochondrial diseases. Recently, a first review and systematic search of the literature has been published. Results were variable, but demonstrated deficits in visuospatial tasks, memory, attention, processing speed, and executive functions. Conclusions from various studies have been compromised by small sample sizes, diverse genotypes and variability of neuropsychologcial test batteries applied [153]. In our practical experience, cognitive impairment is -similar to psychiatric symptoms quite prevalent in patients with MT-TL1 and MT-TK mutations presenting with MELAS or MERRF. POLG-associated diseases are often complicated by cognitive dysfunction, too. However, this might be the consequence of refractory epilepsy in many cases. According to data from national registries like the German mitoNET and databases from Europe, USA, Japan, and China, cognitive decline is present in almost 50% of patients harbouring mitochondrial MT-ATP6 mutations [142]. In CPEO and KSS patients due to mtDNA single deletions or the m.3243A>G MT-TL1 mutation, neuropsychological testing did not reveal general intellectual deterioration, but specific cognitive deficits, particularly in visual construction, attention and abstraction/flexibility [154]. In contrast, patients with MELAS due to MT-TL1 mutations often present with a more global cognitive deterioration [155]. The cognitive decline in MELAS may rapidly progress to early-onset dementia.
Headache
Headache is a common complaint in adulthood. A German cross-sectional questionnaire-based study on patients with various mitochondrial diseases and different genotypes showed that headache was present in 70% of patients. Tension-type headache showed the highest prevalence, followed by migraine and probable migraine [156]. However, migraine with or without aura is most frequently reported in mitochondrial diseases often affecting patients in early adulthood or adolescence. Migraine or migraine-like headache is frequently associated with the onset of a stroke-like episode or a focal epileptic seizure, but may also occur independently of other neurological features [157].
Neuroimaging studies in mitochondrial disorders show heterogeneous results, including stroke-like lesions, laminar cortical necrosis esp. of occipital lobes usually following prolonged epileptic seizure activity, cerebellar atrophy, calcification of deep cerebellar nuclei, diffuse cerebellar white matter changes, global brain atrophy, cerebral deep grey matter changes, cerebral white matter lesions, leukoencephalopathy, and rarely brainstem or spinal cord leukodystrophy and atrophy [126, 158]. Brainstem and spinal cord leukodystrophy is pathognomonic for “leukoencephalopathy with brainstem and spinal cord involvement with lactate elevation (LBSL)” due to mutations in the DARS2 gene encoding the mitochondrial aspartyl-tRNA synthetase, a childhood-onset, often fatal mitochondrial disorder that may be diagnosed in more mildly affected adolescent and adult patients, too [159]. Beyond LBSL, there are other mitochondrial diseases related to mutated mitochondrial aminoacyl tRNA synthetases (mt-aaRSs) associated with specific syndromes that affect the CNS and show rather characteristic MRI patterns, including EARS2 and AARS2 leukodystrophies caused by mutations in mitochondrial glutamyl-tRNA synthetase, and mitochondrial alanyl-tRNA synthetase, respectively [160]. Stroke-like lesions are usually present in MELAS patients suffering from stroke-like episodes. However, in MELAS and in MELAS overlap phenotypes without stroke-like episodes, deep grey matter changes are a distinctive finding in many patients. Global brain atrophy is also a prominent feature and often accentuated in the cerebellum in m.3243A>G MT-TL1 mutation carriers with encephalomyopathic phenotypes [161].
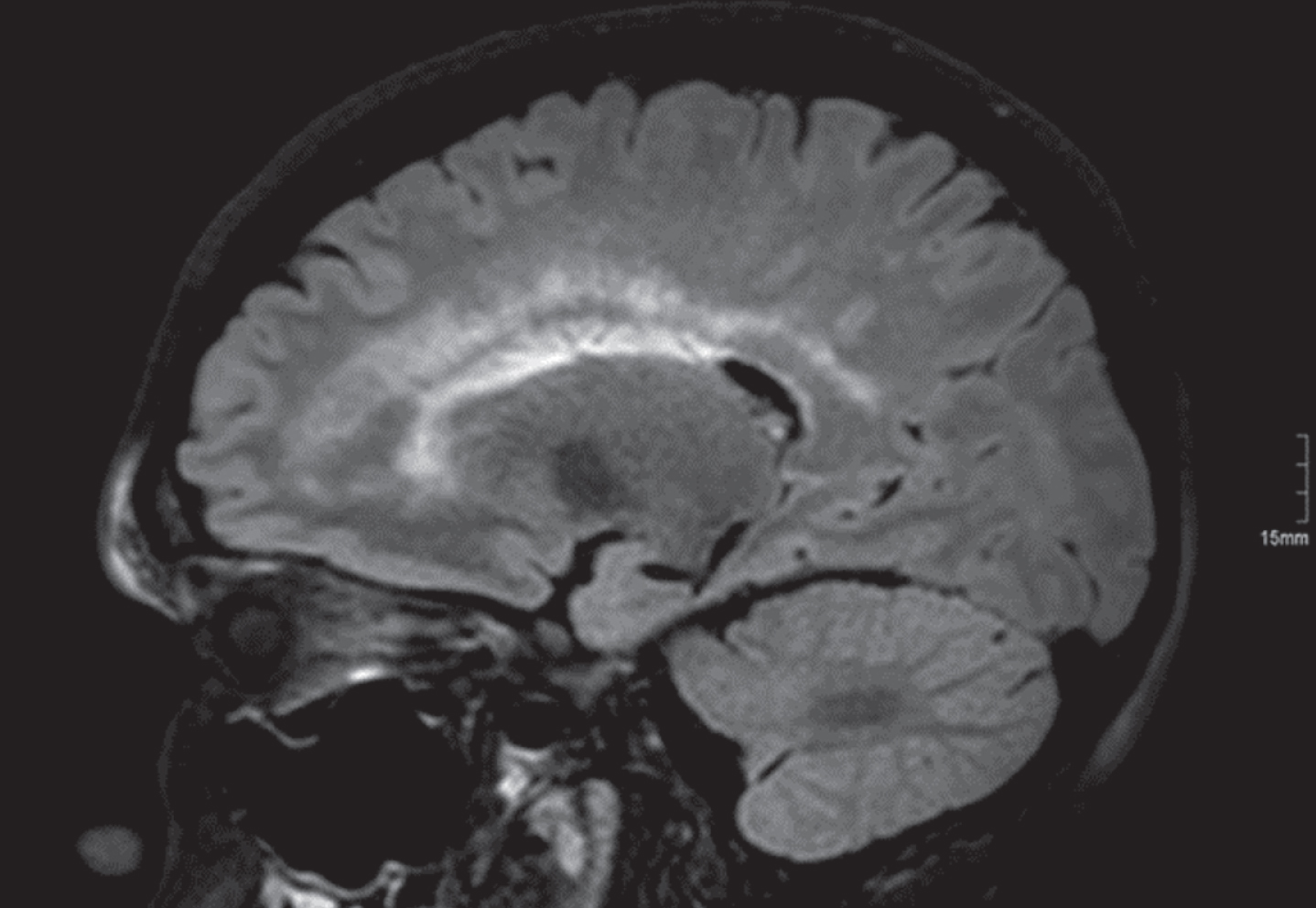
CNS symptoms are not necessarily associated with characteristic neuroimaging findings, and neuroimaging findings are not implicitly related to characteristic clinical phenotypes vice versa. Thus, a leukoencephalopathy is present in mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) per definition [Fig. 6], however usually not associated with any clinical symptom [162].
Fig.6
1.5 T brain MRI, FLAIR sequences. A 25-year-old male patient with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) harbouring two pathogenic heterozygeous TYMP mutations.
Late-onset Pompe Disease (LOPD)
Pompe disease, or glycogen storage disease type II, is a rare, autosomal recessive, multisystemic disorder caused by a deficiency of lysosomal acid α-glucosidase due to pathogenic GAA gene mutations. The disorder presents either as severe infantile onset Pompe disease (IOPD) with generalized muscle weakness, hypotonia, respiratory distress, and hypertrophic cardiomyopathy. Further disease forms, childhood-onset and late onset Pompe disease (LOPD), are characterized by later onset from childhood to adulthood, mild to moderate CK elevation, limb-girdle and axial muscle weakness, and respiratory muscle affection. Enzyme replacement therapy with human recombinant acid α-glucosidase is available for all disease forms. In adults, this therapy improves or stabilizes skeletal muscle strength, muscle function, respiratory function, and survival [163].
Fig.7
3.0 T brain MRI. 61-year-old male late-onset Pompe disease patient harbouring two pathogenic heterozygous GAA mutations, enzyme replacement therapy with glucosidase alfa since 9 years. a) FLAIR sequences. Subacute paramedian pontine hemorrhage, chronic ischemic stroke lesion affecting the left cerebellar hemisphere. b) Susceptibility-weighted images (SWI). Subacute paramedian pontine hemorrhage, mild ectasia of the basilar artery and dilative angiopathy of vertebral arteries, chronic ischemic stroke lesion affecting the left cerebellar hemisphere. c) Susceptibility-weighted images (SWI). Multiple microbleeds presenting predominantly as vertebrobasilar circulation disorder. d) MR angiography shows mild ectasia of the basilar artery and dilative angiopathy of vertebral arteries.
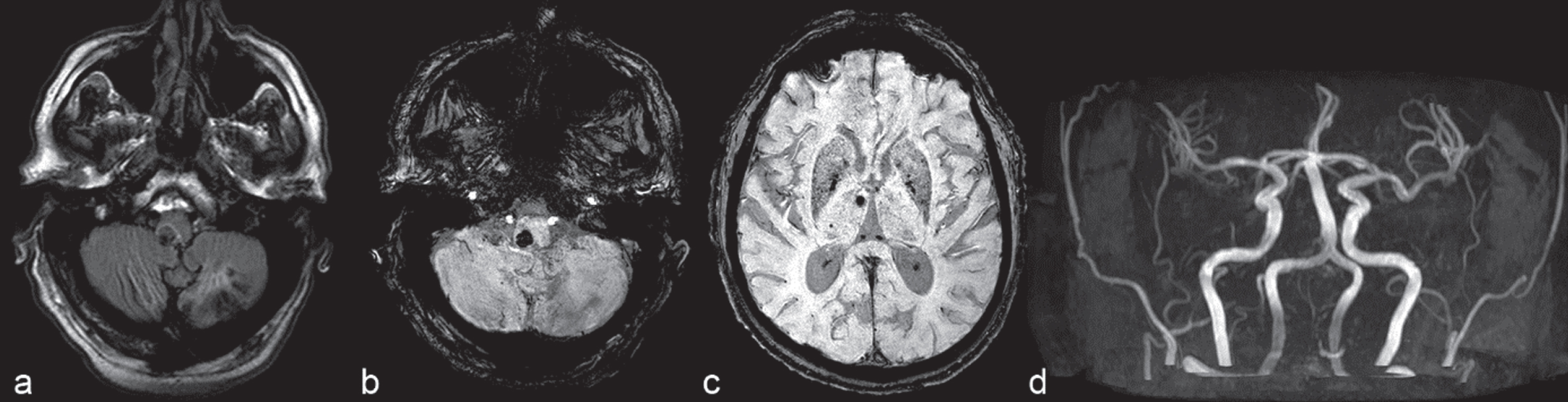
However, LOPD may present with various ages-at-onset and a broad clinical spectrum [164]. Although Pompe disease has been considered primarily a neuromuscular disorder, it is now evident that it is a multisystem disease. Involvement of several other organs including smooth muscles, motor neurons and also the CNS is frequently present. In fact, the central, peripheral, and autonomous nervous systems are often affected, and vascular abnormalities like aneurysms and cerebral small vessel disease resulting in minor strokes or cerebral haemorrhage are frequently observed [Fig. 7a,b]. Recent studies on CNS affection in LOPD patients applied different brain MRI techniques and neuropsychological testing [165, 166]. In the examined LOPD cohorts, morphological and functional brain alterations were present including mild neuropsychological dysfunction and cerebrovascular alterations not related to common vascular risk factors, suggesting a major role of enzyme deficiency in the pathogenesis of brain abnormalities. In detail, signs of cerebral vasculopathy (cerebral small vessel disease), a dolichoectasia of the vertebrobasilar system (esp. the basilar artery), intracranial aneurysms and dilative angiopathy could be identified in LOPD patients [Fig. 7c,d]. Grey matter atrophy was also present correlating with age and disease duration, and resting-state fMRI demonstrated decreased brain connectivity. Neuropsycholocical functional testing showed that a mild impairment in executive functions may be present [166].
Moreover, CNS affection including white matter lesions, a more severe leucoencephalopathy and/or cognitive dysfunction has recently been shown in IOPD patients reaching adulthood under long-term enzyme replacement therapy that in general is not able to cross the blood-brain barrier [167]. Therefore, it is mandatory to include the brain as an additional target in the development of next-generation therapeutic strategies for classic IOPD, but also for LOPD [168].
Phosphoglycerate Kinase Deficiency
The X-chromosomal disorder that leads to deficiency of this enzyme of the glycolytic pathway presents with a spectrum of manifestations ranging from hereditary nonspherocytic haemolytic anaemia, myopathy (frequently with exertional rhabdomyolysis [169]), intellectual disability to seizures and possibly juvenile-onset Parkinsonism [170]. Initially, a haemolytic, a myopathic and a mixed type were defined. In a review of 33 cases, haemolytic anaemia with CNS involvement was the most prevalent combination (one third of patients), with the isolated myopathic presentation somewhat rarer, while isolated haemolysis and myopathy with CNS dysfunction were rare, and anaemia with myopathy or a combination of haemolytic anaemia, myopathy and CNS symptoms very rare presentations [171]. Genotype-phenotype correlation in that collection pointed to a clustering of mutations affecting the C-terminal domain of the protein in the isolated myopathic cases. Age at onset in the myopathic cases was frequently in the patients’ teenage, but occasionally in adulthood. Affected females have been reported, but no abnormal brain imaging.
It should be noted that some myopathic cases show very little glycogen accumulation only detectable in electron microscopy, e.g. [172], or may even considered entirely normal [171]. Baseline CK levels in blood may be normal or high [169]. Biochemical or genetic analysis triggered by the clinical presentation appears to be the way to establish this diagnosis.
Others
Inclusion body myopathy associated with Paget disease of bone (PDB) and/or frontotemporal dementia (IBMPFD)
This is an adult-onset, autosomal-dominant disease caused by mutations in the VCP, the HNRNPA1 or the HNRNPA2B1 gene. Only a minority of patients will develop all three of the eponymous manifestations. Studies on VCP mutation caused cases – the most frequent cause – have shown myopathy as the most frequent (>80%) and earliest sign. This is followed by Paget disease, with frontotemporal dementia being the rarest and having the latest clinical onset, typically about a decade after the muscle and/or bone manifestations [173, 174]. There is some overlap with amyotrophic lateral sclerosis, which for classic VCP IBMPFD is an allelic disease for a small portion of familial [175] and an even smaller portion of sporadic cases [176]. There has been some debate whether the cases of Parkinson’s disease within the collectives of VCP IBMPFD should not be considered coincidental, due to classical clinical presentation and response to medication. However, at 4% patients with Parkinson’s disease in one North American study [174] with 36 families, the numbers are clearly above the general prevalence in that age group.
From a neuropathological point of view, the disease has been categorised as TDP-43 associated frontotemporal lobar degeneration FTLD-TDP/neurodegeneration [177], ubiquitin-positive intranuclear inclusions can be found in both, affected neurons and skeletal muscle. Obviously, brain imaging has been applied to these cases [178] and dedicated imaging and neuropsychological examination will discover early signs of FTD [173, 179], if present, prior to detection of brain atrophy.
This is a disease that will face the adult neurologist with the possibility of diagnosing not only a myopathy with a frequently unfavourable course due to respiratory and cardiac involvement, but an autosomal-dominant neurodegenerative disorder along with it when attending a case of muscle weakness with onset beyond the third decade of life. In our experience, the necessary extensive and repeated counselling about the meaning of molecular diagnosis and its impact on the patient’s plans and family can be challenging, especially with regard to the wide variability of the clinical manifestations even within families and what may have been early signs of FTD in the consulter.
Neuroacanthocytosis disorders
Neuroacanthocytosis syndromes show basal ganglia degeneration and red blood cell acanthocytosis. In this heterogeneous group of disorders, the X-linked McLeod Syndrome (MLS) and the autosomal recessive Chorea-Acanthocytosis (ChAc) are also considered myopathic diseases. This designation is based on frequently elevated CK in a range of 300 – 3,000 U/l [180], thereby often in excess of what is accepted as a result of denervation, the rare case of rhabdomyolysis [181], EMG results, and the distribution of damage in muscle imaging [182]. Despite this, there is plenty of evidence pointing towards a major component of nerve damage in the peripheral pathophysiology of these disorders, particularly in MLS [183].
While genetic testing of XK (MLS) and VPS13A gene (ChAc), respectively, will secure the diagnosis, both disorders can be tested for with high sensitivity and specificity with blood- and antibody-based protein testing. In MLS, the Kx antigen will be absent and the Kell antigens expressed weakly (referred to as “McLeod phenotype”, regardless of the presence of symptoms) [184], while in ChAc western blotting for the chorein protein will show loss or weak signal of its 360 kDa band [185]. Thus, blood typing will lead to the detection of neurologically asymptomatic males with the McLeod phenotype that are certainly at risk of transfusion complications and will most likely later develop symptoms making them MLS patients, as penetrance is complete by age 60 but for rare missense mutation cases [186]. Affected female MLS carriers are rare.
As first symptoms range from movement disorder via psychiatric illness to muscle weakness, the individual patient’s path to the neuromuscular clinic will be variable. As far as a typical neurological disease course for MLS can be described, this would be the development of movement disorder (predominantly limb chorea) in the third to sixth decade of life, followed by progressive, predominantly lower limb weakness involving first distal and later proximal muscles in 60 to 80% of the cases [182, 183]. Generalized seizures occur in about 50% [187]. Sensorimotor axonal neuropathy is a common finding.
In comparison, ChAc can manifest in childhood or late in life, but usually at around 30 years with the movement disorder (limb chorea, oral/ tongue dystonia, but also parkinsonism) [188] or occasionally with seizures (found in almost 50%) of temporal lobe origin [189]. Predominantly distal muscle atrophy and weakness are seen in more than half of the cases, while some signs of neuropathy like loss of ankle reflexes are almost always found [188].
In both disorders, psychiatric symptoms are frequently apparent before onset of the movement disorder. While the range of symptoms reported is wide, obsessive-compulsive disorder and schizophrenia-like psychosis seem to be typical, particularly in ChAc [190]. These are thought to be caused by disruption of frontal-subcortical loops due to the degeneration in the caudate nucleus and putamen, together with executive impairment and impulsive behaviour and – later in the disease course – a cognitive decline similar to frontotemporal dementia [190].
Brain imaging will show progressive atrophy of caudate nuclei and putamen in both diseases [180]. In ChAc further atrophy of frontal lobes as well as hippocampi and increased T2 signal of caudate nuclei and putamen have been described [188, 189].
The two disorders can be difficult to differentiate without genetic or protein diagnostics which is crucial for genetic counselling and cardiac surveillance since cardiac manifestations like arrhythmia, sudden cardiac death or dilated cardiomyopathy are found in more than half of the cases of MLS, but rarely in ChAc [187].
Note that more widespread changes to the X chromosome can lead to a contiguous gene syndrome, combining MLS with chronic granulomatous disease, retinitis pigmentosa and even DMD [191].
Conclusion
CNS affection is frequent in adult patients with chronic childhood- or late-onset diseases traditionally classified as muscular disorders with pure muscle phenotypes. Progress in the treatment of some conditions with previous early fatality like infantile Pompe disease or congenital myotonic dystrophy now lead to “new”, emerging phenotypes of CNS manifestations of these disorders. While this implies that these patients will already have an established diagnosis, it might lead to other challenges in the care for this group. This will not only result in the need to better define these CNS manifestations to differentiate them from independent disorders, but to a potential burden on the care providers. More detailed knowledge on brain affection in adult patients with muscle diseases will have to create a feed-back on the paediatric treatment decisions and of course the treatment methods in neuromuscular disorders.
With the increasing evidence of structural or functional CNS involvement in so many disorders considered primarily diseases of skeletal muscle, it is time to apply this understanding to our clinical practice. This involves systematically assessing CNS symptoms, attributing them to the disorder and integrating them in the care for our patients. It also implies that many “muscle diseases” for which we consider tissue for research – skeletal muscle biopsies – readily available are in fact lacking the collection of brain tissue even though in many cases there is a direct overlap with mechanisms involved in neurodegenerative disorders.
It is of high importance to clarify the nature and natural history of brain affection against the background of currently upcoming specific treatment studies in hereditary myopathies that should include the CNS as a secondary target. A most clear definition of cerebral changes and its surrogate markers is of urgent need not only to design optimal therapy compounds but also for the identification of best fitting study protocols, biomarkers, and appropriate clinical trial outcome parameters.
Conflicts of interest
The author declare no conflict of interest.
ACKNOWLEDGMENTS
The authors are members of the European Reference Network for Rare Neuromuscular Diseases ERN EURO-NMD.
Cornelia Kornblum is co-coordinator and member of mitoNET, the German Network for mitochondrial disorders, funded by the Federal Ministry of Education and Research (BMBF), project number 01GM1906E.
The authors declare no financial or material support for this review article.
REFERENCES
[1] | D’Angelo MG , Bresolin N . Cognitive impairment in neuromuscular disorders. Muscle Nerve. (2006) ;34: (1):16–33. |
[2] | Keshavan N , Rahman S . Natural history of mitochondrial disorders: A systematic review. Essays Biochem. (2018) ;62: (3):423–42. doi: 10.1042/EBC20170108. |
[3] | Patel DR , Cabral MD , Ho A , Merrick J . A Clinical Primer on Intellectual Disability. Transl Pediatr. (2020) ;9: (Suppl 1):S23–S35. doi: 10.21037/t2020.02.02. |
[4] | Rossor MN , Fox NC , Mummery CJ , Schott JM , Warren JD . The Diagnosis of Young-Onset Dementia. Lancet Neurol. (2010) ;9: (8):793–806. doi: 10.1016/S1474-4422(10)70159-9. |
[5] | Klopstock T , Jaksch M , Gasser T . Age and cause of death in mitochondrial diseases. Neurology. (1999) ;53: (4):855–85. |
[6] | Turner C , Hilton-Jones D . Myotonic dystrophy: Diagnosis, management and new therapies. Curr Opin Neurol. (2014) ;27: (5):599–606. doi: 10.1097/WCO.0000000000000128. |
[7] | McDearmon EL , Combs AC , Sekiguchi K , Fujiwara H , Ervasti JM . Brain alpha-dystroglycan displays unique glycoepitopes and preferential binding to laminin-10/11. FEBS Lett. (2006) ;580: (14):3381–5. |
[8] | Myshrall TD , Moore SA , Ostendorf AP , Satz JS , Kowalczyk T , Nguyen H , et al. Dystroglycan on Radial Glia Endfeet Is Required For Pial Basement Membrane Integrity and Columnar Organization of the Developing Cerebral Cortex J Neuropathol Exp Neurol. J Neuropathol Exp Neurol. (2012) ;71: (12):1047–63. doi: 10.1097/NEN.0b013e318274a128. |
[9] | Clement E , Mercuri E , Godfrey C , Smith J , Robb S , Kinali M , et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann Neurol. (2008) ;64: (5):573–82. doi: 10.1002/ana.21482. |
[10] | Satz JS , Ostendorf AP , Hou S , Turner A , Kusano H , Lee JC , et al. Distinct functions of glial and neuronal dystroglycan in the developing and adult mouse brain. J Neurosci. (2010) ;30: (43):14560–72. doi: 10.1523/JNEUROSCI.3247-10.2010. |
[11] | Menezes MJ , McClenahan FK , Leiton CV , Aranmolate A , Shan X , Colognato H . The extracellular matrix protein laminin α2 regulates the maturation and function of the blood-brain barrier. J Neurosci. (2014) ;34: (46):15260–80. doi: 10.1523/JNEUROSCI.3678-13.2014. |
[12] | Alkan A , Sigirci A , Kutlu R , Aslan M , Doganay S , Yakinci C . Merosin-negative congenital muscular dystrophy: Diffusion-weighted imaging findings of brain. J Child Neurol. (2007) ;22: :655–9. |
[13] | Jones KJ , Morgan G , Johnston H , Tobias V , Ouvrier RA , Wilkinson I , et al. The expanding phenotype of laminin alpha2 chain (merosin) abnormalities: Case series and review. J Med Genet. (2001) ;38: (10):649–57. |
[14] | Oliveira J , Gruber A , Cardoso M , Taipa R , Fineza I , Gonçalves A , et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum Mutat. (2018) ;39: (10):1314–37. doi: 10.1002/humu.23599. |
[15] | Pegoraro E , Marks H , Garcia CA , Crawford T , Mancias P , Connolly AM , et al. Laminin alpha2 muscular dystrophy: Genotype/phenotype studies of 22 patients. Neurology. (1998) ;51: (1):101–10. |
[16] | Bushby K , Anderson LV , Pollitt C , Naom I , Muntoni F , Bindoff L . Abnormal merosin in adults. A new form of late onset muscular dystrophy not linked to chromosome 6q2. Brain. (1998) ;121: (4):581–8. |
[17] | Kobayashi K , Nakahori Y , Miyake M , Matsumura K , Kondo-Iida E , Nomura Y , et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. (1998) ;394(6691):388–92. |
[18] | Godfrey C , Clement E , Mein R , Brockington M , Smith J , Talim B , et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. (2007) ;130: (10):2725–35. |
[19] | Brun BN , Mockler SR , Laubscher KM , Stephan CM , Wallace AM , et al. Comparison of brain MRI findings with language and motor function in the dystroglycanopathies. Neurology. (2017) ;88: (7):623–9. doi: 10.1212/WNL.0000000000003609. |
[20] | Palmieri A , Manara R , Bello L , Mento G , Lazzarini L , Borsato C , et al. Cognitive profile and MRI findings in limb-girdle muscular dystrophy 2I. J Neurol.. (2011) ;258: (7):1312–20. doi: 10.1007/s00415-011-5930-3. |
[21] | Hara Y , Balci-Hayta B , Yoshida-Moriguchi T , Kanagawa M , Beltrán-Valero de Bernabé D , Gündeşli H , et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N Engl J Med. (2011) ;364: (10):939–46. doi: 10.1056/NEJMoa1006939. |
[22] | Lefeber DJ , Schönberger J , Morava E , Guillard M , Huyben KM , Verrijp K , et al. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am J Hum Genet. (2009) ;85: (1):76–86. doi: 10.1016/j.ajhg.2009.06.006. |
[23] | Di Costanzo S , Balasubramanian A , Pond HL , Rozkalne A , Pantaleoni C , Saredi S , et al. POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum Mol Genet. (2014) ;23: (21):5781–92. doi: 10.1093/hmg/ddu296. |
[24] | Bello L , Melacini P , Pezzani R , D’Amico A , Piva L , Leonardi E , et al. Cardiomyopathy in patients with POMT1-related congenital and limb-girdle muscular dystrophy. Eur J Hum Genet. (2012) ;20: (12):1234–9. doi: 10.1038/ejhg.2012.71. |
[25] | Koehler K , Milev MP , Prematilake K , Reschke F , Kutzner S , Jühlen R , et al. A novel TRAPPC11 mutation in two Turkish families associated with cerebral atrophy, global retardation, scoliosis, achalasia and alacrima. J Med Genet. (2017) ;54: (3):176–85. doi: 10.1136/jmedgenet-2016-104108. |
[26] | Emery AE , Muntoni F , Duchenne Muscular Dystrophy, 3rd ed, Oxford: Oxford University Press, (2003) . |
[27] | de Brouwer AP , Nabuurs SB , Verhaart IE , Oudakker AR , Hordijk R , Yntema HG , et al. A 3-base pair deletion, c9711_9713del, in DMD results in intellectual disability without muscular dystrophy. Eur J Hum Genet. (2014) ;22: (4):480–5. doi: 10.1038/ejhg.2013.169. |
[28] | Muntoni F , Torelli S , Ferlini A . Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol. (2003) ;2: (12):731–40. |
[29] | Moizard MP , Billard C , Toutain A , Berret F , Marmin N , Moraine C . Are Dp71 and Dp140 brain dystrophin isoforms related to cognitive impairment in Duchenne muscular dystrophy? Am J Med Genet ((1998) ;80: (1):32–41. |
[30] | Banihani R , Smile S , Yoon G , Dupuis A , Mosleh M , Snider A , et al. Cognitive and Neurobehavioral Profile in Boys With Duchenne Muscular Dystrophy. J Child Neurol. (2015) ;30: (11):1472–82. doi: 10.1177/0883073815570154. |
[31] | Felisari G , Martinelli Boneschi F , Bardoni A , Sironi M , Comi GP , Robotti M , et al. Loss of Dp140 dystrophin isoform and intellectual impairment in Duchenne dystrophy. Neurology. (2000) ;55: (4):559–64. |
[32] | Chamova T , Guergueltcheva V , Raycheva M , Todorov T , Genova J , Bichev S , et al. Association between loss of dp140 and cognitive impairment in duchenne and becker dystrophies. Balkan J Med Genet. (2013) ;16: (1):21–30. doi: 10.2478/bjmg-2013-0014. |
[33] | Doorenweerd N , Straathof CS , Dumas EM , Spitali P , Ginjaar IB , Wokke BH , et al. Reduced cerebral gray matter and altered white matter in boys with Duchenne muscular dystrophy. Ann Neurol. (2014) ;76: (3):403–11. doi: 10.1002/ana.24222. |
[34] | Ricotti V , Mandy WP , Scoto M , Pane M , Deconinck N , Messina S , et al. Neurodevelopmental, emotional, and behavioural problems in Duchenne muscular dystrophy in relation to underlying dystrophin gene mutations. Dev Med Child Neurol. (2016) ;58: (1):77–84. doi: 10.1111/dmcn.12922. |
[35] | Daoud F , Angeard N , Demerre B , Martie I , Benyaou R , Leturcq F , et al. Analysis of Dp71 contribution in the severity of mental retardation through comparison of Duchenne and Becker patients differing by mutation consequences on Dp71 expression. Hum Mol Genet. (2009) ;18: (20):3779–94. doi: 10.1093/hmg/ddp320. |
[36] | Moizard MP , Toutain A , Fournier D , Berret F , Raynaud M , Billard C , et al. Severe cognitive impairment in DMD: Obvious clinical indication for Dp71 isoform point mutation screening. Eur J Hum Genet. (2000) ;8: (7):552–6. |
[37] | Doorenweerd N , Mahfouz A , van Putten M , Kaliyaperumal R , T’ Hoen PAC , Hendriksen JGM , et al. Timing and localization of human dystrophin isoform expression provide insights into the cognitive phenotype of Duchenne muscular dystrophy. Sci Rep. (2017) ;7: (1):1257–5. doi: 10.1038/s41598-017-12981-5. |
[38] | Hodgson SV , Abbs S , Clark S , Manzur A , Heckmatt JZ , Dubowitz V , et al. Correlation of clinical and deletion data in Duchenne and Becker muscular dystrophy, with special reference to mental ability. Neuromuscul Disord. (1992) ;2: (4):269–76. |
[39] | Yoshioka M , Okuno T , Honda Y , Nakano Y . Central nervous system involvement in progressive muscular dystrophy. Arch Dis Child. (1980) ;55: (8):589–94. |
[40] | Bardoni A , Felisari G , Sironi M , Comi G , Lai M , Robotti M , et al. Loss of Dp140 regulatory sequences is associated with cognitive impairment in dystrophinopathies. Neuromuscul Disord. (2000) ;10: (3):194–9. |
[41] | Young HK , Barton BA , Waisbren S , Portales Dale L , Ryan MM , Webster RI , North KN . Cognitive and psychological profile of males with Becker muscular dystrophy. J Child Neurol. (2008) ;23: (2):155–62. |
[42] | Bushby KM , Gardner-Medwin D . The clinical, genetic and dystrophin characteristics of Becker muscular dystrophy. I. Natural history. J Neurol. (1993) ;240: (2):98–104. |
[43] | Mori-Yoshimura M , Mizuno Y , Yoshida S , Ishihara N , Minami N , Morimoto E , et al. Psychiatric and neurodevelopmental aspects of Becker muscular dystrophy. Neuromuscul Disord. (2019) ;29: (12):930–9. doi: 10.1016/j.nmd.2019.09.006. |
[44] | Goodwin F , Muntoni F , Dubowitz V . Epilepsy in Duchenne and Becker muscular dystrophies. Eur J Paediatr Neurol. (1997) ;1: (4):115–9. |
[45] | Papa R , Madia F , Bartolomeo D , Trucco F , Pedemonte M , Traverso M , et al. Genetic and Early Clinical Manifestations of Females Heterozygous for Duchenne/Becker Muscular Dystrophy. Pediatr Neurol. (2016) ;55: :58–63. doi: 10.1016/j.pediatrneurol.2015.11.004. |
[46] | Zimprich A , Grabowski M , Asmus F , Naumann M , Berg D , Bertram M , et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet. (2001) ;29: (1):66–9. |
[47] | Ritz K , van Schaik BD , Jakobs ME , van Kampen AH , Aronica E , Tijssen MA , et al. SGCE isoform characterization and expression in human brain: Implications for myoclonus-dystonia pathogenesis? Eur J Hum Genet. (2011) ;19: (4):438–44.doi: 10.1038/ejhg.2010.206 |
[48] | Blumen SC , Bouchard JP , Brais B , Carasso RL , Paleacu D , Drory VE , et al. Cognitive impairment and reduced life span of oculopharyngeal muscular dystrophy homozygotes. Neurology. (2009) ;73: (8):596–601. doi: 10.1212/WNL.0b013e3181b388a3. |
[49] | Dubbioso R , Moretta P , Manganelli F , Fiorillo C , Iodice R , Trojano L , et al. Executive functions are impaired in heterozygote patients with oculopharyngeal muscular dystrophy. J Neurol. (2012) ;259: (5):833–7. doi: 10.1007/s00415-011-6255-y. |
[50] | Dion P , Shanmugam V , Gaspar C , Messaed C , Meijer I , Toulouse A , et al. Transgenic expression of an expanded (GCG)13 repeat PABPN1 leads to weakness and coordination defects in mice. Neurobiol Dis. (2005) ;18: (3):528–36. |
[51] | Day JW , Ranum LP . RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord. (2005) ;15: :5–16. |
[52] | Meola G , Sansone V . Cerebral involvement in myotonic dystrophies. Muscle Nerve. (2007) ;36: (3):294–306. |
[53] | Bugiardini E , Meola G . Consensus on cerebral involvement in myotonic dystrophy: Workshop report: May 24– 27, Ferrere (AT), Italy. Neuromuscul Disord. (2014) ;24: (5):445–52. doi: 10.1016/j.nmd.2014.01.013. |
[54] | Wiśniewski HM , Berry K , Spiro AJ . Ultrastructure of thalamic neuronal inclusions in myotonic dystrophy. J Neurol Sci. (1975) ;24: :321–9. |
[55] | Ono S , Inoue K , Mannen T , Kanda F , Jinnai K , Takahashi K . Neuropathological changes of the brain in myotonic dystrophy–some new observations. J Neurol Sci. (1987) ;81: :301–20. |
[56] | Abe K , Fujimura H , Toyooka K , Yorifuji S , Nishikawa Y , Hazama T , et al. Involvement of the central nervous system in myotonic dystrophy. J Neurol Sci. (1994) ;127: :179–85. |
[57] | Ogata A , Terae S , Fujita M , Tashiro K . Anterior temporal white matter lesions in myotonic dystrophy with intellectual impairment: An MRI and neuropathological study. Neuroradiology. (1998) ;40: :411–5. |
[58] | Itoh K , Mitani M , Kawamoto K , Futamura N , Funakawa I , Jinnai K , et al. Neuropathology does not Correlate with Regional Differences in the Extent of Expansion of CTG Repeats in the Brain with Myotonic Dystrophy Type 1. Acta Histochem Cytochem. (2010) ;43: :149–56. |
[59] | Fernandez-Gomez F , Tran H , Dhaenens CM , Caillet-Boudin ML , Schraen-Maschke S , Blum D , et al. Myotonic Dystrophy: An RNA Toxic Gain of Function Tauopathy? Adv Exp Med Biol. (1184) ;207–16. doi: 10.1007/978-981-32-9358-8_17. |
[60] | Okkersen K , Buskes M , Groenewoud J , Kessels RPC , Knoop H , van Engelen B , et al. The cognitive profile of myotonic dystrophy type A systematic review and meta-analysis. Cortex. (2017) ;95: :143–55. doi: 10.1016/j.cortex.2017.08.008. |
[61] | Heatwole C , Bode R , Johnson N , Dekdebrun J , Dilek N , Heatwole M , et al. 3rd. Myotonic Dystrophy Health Index: Initial evaluation of a disease-specific outcome measure. Muscle Nerve. (2014) ;49: (6):906–14. doi: 10.1002/mus.24097. |
[62] | Okkersen K , Monckton DG , Le N , Tuladhar AM , Raaphorst J , van Engelen BGM . Brain imaging in myotonic dystrophy type A systematic review. Neurology. (2017) ;89: (9):960–9. doi: 10.1212/WNL.0000000000004300. |
[63] | Minnerop M , Gliem C , Kornblum C . Current Progress in CNS Imaging of Myotonic Dystrophy. Front Neurol. (2018) ;9: :646. doi: 10.3389/fneur.2018.00646. |
[64] | Ates S , Deistung A , Schneider R , Prehn C , Lukas C , Reichenbach JR , et al. Characterization of Iron Accumulation in Deep Gray Matter in Myotonic Dystrophy Type 1 and 2 Using Quantitative Susceptibility Mapping and R2* Relaxometry: A Magnetic Resonance Imaging Study at 3 Tesla. Front Neurol. (2019) ;10: :1320. doi: 10.3389/fneur.2019.01320. |
[65] | Peric S , Pavlovic A , Ralic V , Dobricic V , Basta I , Lavrnic D , et al. Transcranial sonography in patients with myotonic dystrophy type 1. Muscle Nerve. (2014) ;50: (2):278–82. doi: 10.1002/mus.24162. |
[66] | Krogias C , Bellenberg B , Prehn C , Schneider R , Meves SH , Gold R , et al. Evaluation of CNS involvement in myotonic dystrophy type 1 and type 2 by transcranial sonography. J Neurol. (2015) ;262: (2):365–74. doi: 10.1007/s00415-014-7566-6. |
[67] | Kuppuswamy A . The fatigue conundrum. Brain.. (2017) ;140: (8):2240–5. doi: 10.1093/brain/awx153. |
[68] | Giubilei F , Antonini G , Bastianello S , Morino S , Paolillo A , Fiorelli M , et al. Excessive daytime sleepiness in myotonic dystrophy. J Neurol Sci. (1999) ;164: :60–3. |
[69] | Tieleman AA , Knoop H , van de Logt AE , Bleijenberg G , van Engelen BG , Overeem S . Poor sleep quality and fatigue but no excessive daytime sleepiness in myotonic dystrophy type 2. J Neurol Neurosurg Psychiatry. (2010) ;81: :963–7. doi: 10.1136/jnn2009.192591. |
[70] | Laberge L , Begin P , Dauvilliers Y , Beaudry M , Laforte M , Jean S , et al. A polysomnographic study of daytime sleepiness in myotonic dystrophy type 1. J Neurol Neurosurg Psychiatry. (2009) ;80: (6):642–6. doi: 10.1136/jnn2008.165035. |
[71] | Okkersen K , Jimenez-Moreno C , Wenninger S , Daidj F , Glennon J , Cumming S , et al. Cognitive behavioural therapy with optional graded exercise therapy in patients with severe fatigue with myotonic dystrophy type A multicentre, single-blind, randomised trial. The Lancet Neurology. (2018) ;17: (8):671–80. doi: 10.1016/S1474-4422(18)30203-5. |
[72] | Gliem C , Minnerop M , Roeske S , Gärtner H , Schoene-Bake JC , Adler S , et al. Tracking the brain in myotonic dystrophies: A 5-year longitudinal follow-up study. PLoS One. (2019) ;14: (3):e3810213. doi: 10.1371/journal.pone.0213381. |
[73] | Winblad S , Jensen C , Månsson JE , Samuelsson L , Lindberg C . Depression in Myotonic Dystrophy type Clinical and neuronal correlates. Behav Brain Funct. (2010) ;6: :25. doi: 10.1186/1744-9081-6-25. |
[74] | Minnerop M , Weber B , Schoene-Bake JC , Roeske S , Mirbach S , Anspach C , et al. The brain in myotonic dystrophy 1 and Evidence for a predominant white matter disease. Brain. (2011) ;134: (12):3530–46. doi: 10.1093/brain/awr299. |
[75] | Machuca-Tzili L , Brook D , Hilton-Jones D . Clinical and molecular aspects of the myotonic dystrophies: A review. Muscle Nerve. (2005) ;32: :1–18. |
[76] | Meola G , Sansone V , Perani D , Scarone S , Cappa S , Dragoni C , et al. Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM-1) and in proximal myotonic myopathy (PROMM/DM-2). Neuromuscul Disord. (2003) ;13: (10):813–21. |
[77] | Gaul C , Schmidt T , Windisch G , Wieser T , Müller T , Vielhaber S , et al. Subtle cognitive dysfunction in adult onset myotonic dystrophy type 1 (DM1) and type 2 (DM2). Neurology. (2006) ;67: :350–2. |
[78] | Sansone V , Gandossini S , Cotelli M , Calabria M , Zanetti O , Meola G . Cognitive impairment in adult myotonic dystrophies: A longitudinal study. Neurol Sci. (2007) ;28: (1):9–15. |
[79] | Weber YG , Roebling R , Kassubek J , Hoffmann S , Rosenbohm A , Wolf M , et al. Comparative analysis of brain structure, metabolism, and cognition in myotonic dystrophy 1 and 2. Neurology. (2010) ;74: :1108–17. doi: 10.1212/WNL.0b013e3181d8c35f. |
[80] | Winblad S , Samuelsson L , Lindberg C , Meola G . Cognition in myotonic dystrophy type A 5-year follow-up study. Eur J Neurol. (2016) ;23: (9):1471–6. doi: 10.1111/ene.13062. |
[81] | Winblad S , Lindberg C , Hansen S . Temperament and character in patients with classical myotonic dystrophy type 1 (DM-1). Neuromuscul Disord. (2005) ;15: (4):287–92. |
[82] | Winblad S , Hellström P , Lindberg C , Hansen S . Facial emotion recognition in myotonic dystrophy type 1 correlates with CTG repeat expansion. J Neurol Neurosurg Psychiatry. (2006) ;77: (2):219–23. |
[83] | Labayru G , Arenzana I , Aliri J , Zulaica M , López de Munain A , Sistiaga AA . Social cognition in myotonic dystrophy type Specific or secondary impairment? PLoS One. (2018) ;13: (9):e2270204. doi: 10.1371/journal.pone.0204227. |
[84] | Serra L , Cercignani M , Bruschini M , Cipolotti L , Mancini M , Silvestri G , et al. “I Know that You Know that I Know”: Neural Substrates Associated with Social Cognition Deficits in DM1 Patients. PLoS One. (2016) ;11: (6):e9010156. doi: 10.1371/journal.pone.0156901. |
[85] | Peric S , Mandic-Stojmenovic G , Stefanova E , Savic-Pavicevic D , Pesovic J , Ilic V , et al. Frontostriatal dysexecutive syndrome: A core cognitive feature of myotonic dystrophy type 2. J Neurol. (2015) ;262: (1):142–8. doi: 10.1007/s00415-014-7545-y. |
[86] | Gallais B , Gagnon C , Mathieu J , Richer L . Cognitive decline over time in adults with myotonic dystrophy type A 9-year longitudinal study. Neuromuscul Disord. (2017) ;27: (1):61–72. doi: 10.1016/j.nmd.2016.10.003. |
[87] | Kornblum C , Reul J , Kress W , Grothe C , Amanatidis N , Klockgether T , et al. Cranial magnetic resonance imaging in genetically proven myotonic dystrophy type 1 and 2. J Neurol. (2004) ;251: (6):710–4. |
[88] | Romeo V , Pegoraro E , Ferrati C , Squarzanti F , Sorarù G , Palmieri A , et al. Brain involvement in myotonic dystrophies: Neuroimaging and neuropsychological comparative study in DM1 and DM2. J Neurol. (2010) ;257: (8):1246–55. |
[89] | Abe K , Fujimura H , Soga F . The fluid-attenuated inversion-recovery pulse sequence in assessment of central nervous system involvement in myotonic dystrophy. Neuroradiology. (1998) ;40: (1):32–5. |
[90] | Baldanzi S , Cecchi P , Fabbri S , Pesaresi I , Simoncini C , Angelini C , et al. Relationship between neuropsychological impairment and grey and white matter changes in adult-onset myotonic dystrophy type 1. NeuroImage Clinical. (2016) ;12: :190–7. doi: 10.1016/j.nicl.2016.06.011. |
[91] | Cabada T , Iridoy M , Jerico I , Lecumberri P , Seijas R , Gargallo A , et al. Brain Involvement in Myotonic Dystrophy Type A Morphometric and Diffusion Tensor Imaging Study with Neuropsychological Correlation. Archives of clinical neuropsychology: The official journal of the National Academy of Neuropsychologists. (2017) ;32: (4):401–12. doi: 10.1093/arclin/acx008. |
[92] | Kassubek J , Juengling FD , Hoffmann S , Rosenbohm A , Kurt A , Jurkat-Rott K , et al. Quantification of brain atrophy in patients with myotonic dystrophy and proximal myotonic myopathy: A controlled 3-dimensional magnetic resonance imaging study. Neurosci Lett. (2003) ;348: (2):73–6. |
[93] | Serra L , Petrucci A , Spano B , Torso M , Olivito G , Lispi L , et al. How genetics affects the brain to produce higher-level dysfunctions in myotonic dystrophy type 1. Functional neurology. (2015) ;30: (1):21–31. |
[94] | Zanigni S , Evangelisti S , Giannoccaro MP , Oppi F , Poda R , Giorgio A , et al. Relationship of white and gray matter abnormalities to clinical and genetic features in myotonic dystrophy type 1. NeuroImage Clinical. (2016) ;11: :678–85. doi: 10.1016/j.nicl.2016.04.012. |
[95] | Caso F , Agosta F , Peric S , Rakocevic-Stojanovic V , Copetti M , Kostic VS , et al. Cognitive impairment in myotonic dystrophy type 1 is associated with white matter damage. PloS one. (2015) ;9: (8):e971046. doi: 10.1371/journal.pone.0104697. |
[96] | Schneider-Gold C , Bellenberg B , Prehn C , Krogias C , Schneider R , Klein J , et al. Cortical and Subcortical Grey and White Matter Atrophy in Myotonic Dystrophies Type 1 and 2 Is Associated with Cognitive Impairment, Depression and Daytime Sleepiness. PloS one. (2015) ;10: (6):e3520130. doi: 10.1371/journal.pone.0130352. |
[97] | Sugiyama A , Sone D , Sato N , Kimura Y , Ota M , Maikusa N , et al. Brain gray matter structural network in myotonic dystrophy type 1. PLoS One. (2017) ;12: (11):e3430187. doi: 10.1371/journal.pone.0187343. |
[98] | Wozniak JR , Mueller BA , Lim KO , Hemmy LS , Day JW . Tractography reveals diffuse white matter abnormalities in Myotonic Dystrophy Type 1. Journal of the Neurological Sciences. (2014) ;341: (1-2):73–8. doi: 10.1016/j.jns.2014.04.005. |
[99] | van Dorst M , Okkersen K , Kessels RPC , Meijer FJA , Monckton DG , van Engelen BGM , et al. Structural white matter networks in myotonic dystrophy type 1. Neuroimage Clin. (2019) ;21: :1016–15. doi: 10.1016/j.nicl.2018.101615. |
[100] | Romeo V , Pegoraro E , Squarzanti F , Sorarù G , Ferrati C , Ermani M , et al. Retrospective study on PET-SPECT imaging in a large cohort of myotonic dystrophy type 1 patients. Neurol Sci. (2010) ;31: (6):757–63. doi: 10.1007/s10072-010-0406-2. |
[101] | Peric S , Brajkovic L , Belanovic B , Ilic V , Salak-Djokic B , Basta I , et al. Brain positron emission tomography in patients with myotonic dystrophy type 1 and type 2. J Neurol Sci. (2017) ;378: :187–92. doi: 10.1016/j.jns.2017.05.013. |
[102] | Meola G , Sansone V , Perani D , Colleluori A , Cappa S , Cotelli M , et al. Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology. (1999) ;53: (5):1042–50. |
[103] | Vielhaber S , Jakubiczka S , Gaul C , Schoenfeld MA , Debska-Vielhaber G , Zierz S , et al. Brain 1H magnetic resonance spectroscopic differences in myotonic dystrophy type 2 and type 1. Muscle Nerve. (2006) ;34: (2):145–52. |
[104] | Serra L , Silvestri G , Petrucci A , Basile B , Masciullo M , Makovac E , et al. Abnormal functional brain connectivity and personality traits in myotonic dystrophy type 1. JAMA Neurology. (2014) ;71: (5):603–11. doi: 10.1001/jamaneurol.2014.130. |
[105] | Paunic T , Peric S , Parojcic A , Savic-Pavicevic D , Vujnic M , Pesovic J , et al. Personality traits in patients with myotonic dystrophy type 2. Acta Myol. (2017) ;36: (1):14–8. |
[106] | Hund E , Jansen O , Koch MC , Ricker K , Fogel W , Niedermaier N , et al. Proximal myotonic myopathy with MRI white matter abnormalities of the brain. Neurology. (1997) ;48: (1):33–7. |
[107] | Rakocevic-Stojanovic V , Peric S , Savic-Pavicevic D , Pesovic J , Mesaros S , Lavrnic D , et al. Brain sonography insight into the midbrain in myotonic dystrophy type 2. Muscle Nerve. (2016) ;53: (5):700–4. doi: 10.1002/mus.24927. |
[108] | Minnerop M , Luders E , Specht K , Ruhlmann J , Schneider-Gold C , Schroder R , et al. Grey and white matter loss along cerebral midline structures in myotonic dystrophy type 2. J Neurol. (2008) ;255: (12):1904–9. doi: 10.1007/s00415-008-0997-1. |
[109] | Larsen M , Rost S , El Hajj N , Ferbert A , Deschauer M , Walter MC , et al. Diagnostic approach for FSHD revisited: SMCHD1 mutations cause FSHD2 and act as modifiers of disease severity in FSHD1. Eur J Hum Genet. (2015) ;23: (6):808–16. doi: 10.1038/ejhg.2014.191. doi: 10.1038/ejhg.2014.191. |
[110] | Lemmers RJL , de Kievit P , van Geel M , van der Wielen MJ , Bakker E , Padberg GW , et al. Complete allele information in the diagnosis of facioscapulohumeral muscular dystrophy by triple DNA analysis. Ann Neurol. (2001) ;50: (6):816–9. |
[111] | Brouwer OF , Padberg GW , Bakker E , Wijmenga C , Frants RR . Early onset facioscapulohumeral muscular dystrophy. Muscle Nerve Suppl. (1995) )2:S67–72. |
[112] | Lutz KL , Holte L , Kliethermes SA , Stephan C , Mathews KD . Clinical and genetic features of hearing loss in facioscapulohumeral muscular dystrophy. Neurology. (2013) ;81: (16):1374–7. doi: 10.1212/WNL.0b013e3182a84140. |
[113] | Voit T , Lamprecht A , Lenard HG , Goebel HH . Hearing loss in facioscapulohumeral dystrophy. Eur J Pediatr. (1986) ;145: (4):280–5. |
[114] | Trevisan CP , Pastorello E , Tomelleri G , Vercelli L , Bruno C , Scapolan S et al . Facioscapulohumeral muscular dystrophy: Hearing loss and other atypical features of patients with large 4q35 deletions. Eur J Neurol. (2008) ;15: (12):1353–8. doi: 10.1111/j.1468-1331.2008.02314.x. |
[115] | Trevisan CP , Pastorello E , Ermani M , Angelini C , Tomelleri G , Tonin P , et al. Facioscapulohumeral muscular dystrophy: A multicenter study on hearing function. Audiol Neurootol. (2008) ;13: (1):1–6. |
[116] | Fierro B , Daniele O , Aloisio A , Buffa D , La Bua V , Oliveri M , et al. Evoked potential study in facio-scapulo-humeral muscular dystrophy. Acta Neurol Scand. (1997) ;95: (6):346–50. |
[117] | Quarantelli M , Lanzillo R , Del Vecchio W , Mollica C , Prinster A , Iadicicco L , et al. Modifications of brain tissue volumes in facioscapulohumeral dystrophy. Neuroimage. (2006) ;32: (3):1237–42. |
[118] | Ricci E , Galluzzi G , Deidda G , Cacurri S , Colantoni L , Merico B , et al. Progress in the molecular diagnosis of facioscapulohumeral muscular dystrophy and correlation between the number of KpnI repeats at the 4q35 locus and clinical phenotype. Ann Neurol. (1999) ;45: (6):751–7. |
[119] | Lemmers RJ , Tawil R , Petek LM , Balog J , Block GJ , Santen GW , et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat Genet. (2012) ;44: (12):1370–4. doi: 10.1038/ng.2454. |
[120] | de Greef JC , Lemmers RJ , Camaño P , Day JW , Sacconi S , Dunand M , et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. (2010) ;75: (17):1548–54. doi: 10.1212/WNL.0b013e3181f96175. |
[121] | DiMauro S , Schon EA , Carelli V , Hirano M . The clinical maze of mitochondrial neurology. Nat Rev Neurol. (2013) ;9: (8):429–44. doi: 10.1038/nrneurol.2013. |
[122] | Gorman GS , Schaefer AM , Ng Y , Gomez N et al . Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann Neurol. (2015) ;77: (5):753–9. doi: 10.1002/ana.24362. |
[123] | Munnich A , Rötig A , Chretien D , Saudubray JM , Cormier V , Rustin P . Clinical presentations and laboratory investigations in respiratory chain deficiency. Eur J Pediatr. 155. (1996) )262–74. |
[124] | Baertling F , Klee D , Haack TB , Prokisch H , Meitinger T , Mayatepek E , et al. The many faces of paediatric mitochondrial disease on neuroimaging. Childs Nerv Syst. (2016) ;32: (11):2077–83. |
[125] | Lax NZ , Gorman GS , Turnbull DM . Review: Central nervous system involvement in mitochondrial disease. Neuropathol Appl Neurobiol. (2017) ;43: (2):102. doi: 10.1111/nan.12333. |
[126] | Finsterer J , Zarrouk Mahjoub S . Leukoencephalopathies in mitochondrial disorders: Clinical and MRI findings. J Neuroimaging. (2012) ;22: (3):e1–11. doi: 10.1111/j.1552-6569.2011.00693.x. |
[127] | Filosto M , Tomelleri G , Tonin P , Scarpelli M , Vattemi G , Rizzuto N , Padovani A , Simonati A . Neuropathology of mitochondrial diseases. Biosci Rep. (2007) ;27: (1-3):23–30. |
[128] | Zsurka G , Kunz WS . Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. (2015) ;14: (9):956–66. doi: 10.1016/S1474-4422(15)00148-9. |
[129] | Bindoff LA and Engelsen BA . Mitochondrial disease and epilepsy. Epilepsia. (2012) ;53: (Suppl. 4):92–7. doi: 10.1111/j.1528-1167.2012.03618.x. |
[130] | Canafoglia L , Franceschetti S , Antozzi C , Carrara F , Farina L , Granata T , et al. Epileptic phenotypes associated with mitochondrial disorders. Neurology. (2001) ;56: (10):1340–6. |
[131] | Nguyen KV , Ostergaard E , Ravn SH , Balslev T , Danielsen ER , Vardag A , et al. POLG mutations in Alpers syndrome. Neurology. (2005) ;65: :1493–5. |
[132] | Zsurka G , Hampel KG , Nelson I , Jardel D , Mirandola S , Sassen R , et al. Severe epilepsy as the major symptom of new mutations in the mitochondrial tRNAPhe gene. Neurology. (2010) ;74: (6):9507–12. doi: 10.1212/WNL.0b013e3181cef7ab. |
[133] | Engelsen BA , Tzoulis C , Karlsen B , Lillebo A , Laegreid LM , Aasly J , et al. POLG1 mutations cause a syndromic epilepsy with occipital lobe predilection. Brain. (2008) ;131: :818–28. doi: 10.1093/brain/awn007. |
[134] | Tzoulis C , Engelsen BA , Telstad W , Aasly J , Zeviani M , Winterthun S , et al. The spectrum of clinical disease caused by the A467T and W748S POLG mutations: A study of 26 cases. Brain. (2006) ;129: :1685–92. |
[135] | Rahman S , Copeland WC . POLG-related disorders and their neurological manifestations. Nat Rev Neurol. (2019) ;15: (1):40–52. doi: 10.1038/s41582-018-0101-0. |
[136] | Hikmat O , Tzoulis C , Knappskog PM , Johansson S , Boman H , Sztromwasser P , et al. ADCK3 mutations with epilepsy, stroke-like episodes and ataxia: A POLG mimic? Eur J Neurol. (2016) ;23: (7):1188–94. doi: 10.1111/ene.13003. |
[137] | Desbats MA , Lunardi G , Doimo M , Trevisson E , Salviati L . Genetic bases and clinical manifestations of coenzyme Q10 (CoQ 10) deficiency. J Inherit Metab Dis. (2015) ;38: :145–56. doi: 10.1007/s10545-014-9749-9. |
[138] | Ng YS , Bindoff LA , Gorman GS , Horvath R , Klopstock T , Mancuso M , et al. Consensus-based statements for the management of mitochondrial stroke-like episodes [version 1; peer review: 1 approved] Wellcome Open Research. 201. (2019) ;4: :201. doi: 10.12688/wellcomeopenres.15599.1. |
[139] | Horvath R , Czermin B , Gulati S , Demuth S , Houge G , Pyle A , et al. Adult-onset cerebellar ataxia due to mutations in CABC1/ADCK3. J Neurol Neurosurg Psychiatry. (2012) ;83: (2):174–8. doi: 10.1136/jnnp-2011-301258. |
[140] | Winterthun S , Ferrari G , He L , Taylor RW , Zeviani M , Turnbull DM , et al. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology. (2005) ;64: :1204–8. |
[141] | Altmann J , Büchner B , Nadaj-Pakleza A , Schäfer J , Jackson S , Lehmann D , et al. Expanded phenotypic spectrum of the m. A>G “MERRF” mutation: Data from the German mitoNET registry. J Neurol. (2016) ;263: (5):961–72. doi: 10.1007/s00415-016-8086-3. |
[142] | Stendel C , Neuhofer C , Floride E , Yuqing S , Ganetzky RD , Park J , et al. Delineating MT-ATP6-associated disease: From isolated neuropathy to early onset neurodegeneration. Neurol Genet. (2020) ;6: (1):e393. doi: 10.1212/NXG.0000000000000393. |
[143] | Ng YS , Martikainen MH , Gorman GS , Blain A , Bugiardini E , Bunting A , et al. Pathogenic variants in MT-ATP A United Kingdom-based mitochondrial disease cohort study. Ann Neurol. (2019) ;86: (2):310–5. doi: 10.1002/ana.25525. |
[144] | Lestienne P , Ponsot G . Kearns-Sayre syndrome with muscle mitochondrial DNA deletion. Lancet. (1988) ;1: :885–. |
[145] | Moraes CT1 , DiMauro S , Zeviani M , Lombes A , Shanske S , Miranda AF , et al. Mitochondrial DNA deletions in progressive external ophthalmoplegia and Kearns-Sayre syndrome. N Engl J Med. (1989) ;320: :1293–9. |
[146] | Goto Y , Nonaka I , Horai A . A mutation in tRNALeu(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. (1990) ;348: :651–3. |
[147] | Yoneda M , Maeda M , Kimura H , Fujii A , Katayama K , Kuriyama M . Vasogenic edema on MELAS: A serial study with diffusion-weighted MR imaging. Neurology. (1999) ;53: (9):2182–4. |
[148] | El-Hattab AW , Adesina AM , Jones J , Scaglia F . MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. (2015) ;116: (1-2):4–12. doi: 10.1016/j.ymgme.2015.06.004. |
[149] | Luoma P , Melberg A , Rinne JO , Kaukonen JA , Nupponen NN , Chalmers RM , et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet. (2004) ;364: (9437)875–82. |
[150] | Paus S , Zsurka G , Baron M , Deschauer M , Bamberg C , Klockgether T , et al. Apraxia of lid opening mimicking ptosis in compound heterozygosity for A467T and W748S POLG1 mutations. Mov Disord. (2008) ;23: (9):1286–8. doi: 10.1002/mds.22135. |
[151] | Giannoccaro MP , La Morgia C , Rizzo G , Carelli V . Mitochondrial DNA and primary mitochondrial dysfunction in Parkinson’s disease. Mov Disord. (2017) ;32: (3):346–63. doi: 10.1002/mds.26966. |
[152] | Anglin RE , Garside SL , Tarnopolsky MA , Mazurek MF , Rosebush PI . The psychiatric manifestations of mitochondrial disorders: A case and review of the literature. J Clin Psychiatry. (2012) ;73: (4):506–12. doi: 10.4088/JCP.11r07237. |
[153] | Moore HL , Blain AP , Turnbull DM , Gorman GS . Systematic review of cognitive deficits in adult mitochondrial disease. Eur J Neurol. (2020) ;27: (1):3–17. doi: 10.1111/ene.14068. |
[154] | Bosbach S , Kornblum C , Schröder R , Wagner M . Executive and visuospatial deficits in patients with chronic progressive external ophthalmoplegia and Kearns-Sayre syndrome. Brain. (2003) ;126: (5):1231–40. |
[155] | Neargarder SA , Murtagh MP , Wong B , Hill EK . The neuropsychologic deficits of MELAS: Evidence of global impairment. Cogn Behav Neurol. (2007) ;20: (2):83–92. |
[156] | Kraya T , Deschauer M , Joshi PR , Zierz S , Gaul C . Prevalence of Headache in Patients With Mitochondrial Disease: A Cross-Sectional Study. Headache. (2018) ;58: (1):45–52. doi: 10.1111/head.13219. |
[157] | Finsterer J , Zarrouk-Mahjoub S . Headache in mitochondrial disorders. Clin Neurol Neurosurg. (2018) ;166: :44–9. doi: 10.1016/j.clineuro.2018.01.020. |
[158] | Mascalchi M , Montomoli M , Guerrini R . Neuroimaging in mitochondrial disorders. Essays Biochem. (2018) ;62: (3):409–421. doi: 10.1042/EBC20170109. |
[159] | van Berge L , Hamilton EM , Linnankivi T , Uziel G , Steenweg ME , Isohanni P , et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: Clinical and genetic characterization and target for therapy. Brain. (2014) ;137: (4):1019–29. doi: 10.1093/brain/awu026. |
[160] | Fine AS , Nemeth CL , Kaufman ML , Fatemi A . Mitochondrial aminoacyl-tRNA synthetase disorders: An emerging group of developmental disorders of myelination. J Neurodev Disord. (2019) ;11: (1):29. doi: 10.1186/s11689-019-9292-y. |
[161] | Tschampa HJ , Urbach H , Greschus S , Kunz WS , Kornblum C . Neuroimaging characteristics in mitochondrial encephalopathies associated with the m.A>G MTTL1 mutation. J Neurol. (2013) ;260: (4):1071–80. doi: 10.1007/s00415-012-6763-4. |
[162] | Nishino I , Spinazolla A , Hirano M . Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science. (1999) ;283: :689–92. |
[163] | van der Ploeg AT , Kruijshaar ME , Toscano A , Laforêt P , Angelini C , Lachmann RH , et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: A 10-year experience. Eur J Neurol. (2017) ;24: (6):768–e31. doi: 10.1111/ene.13285. |
[164] | Toscano A , Rodolico C , Musumeci O . Multisystem late onset Pompe disease (LOPD): An update on clinical aspects. Ann Transl Med. (2019) ;7: (13):284. doi: 10.21037/atm.2019.07.24. |
[165] | Hensel O , Schneider I , Wieprecht M , Kraya T , Zierz S . Decreased outlet angle of the superior cerebellar artery as indicator for dolichoectasia in late onset Pompe disease. Orphanet J Rare Dis. (2018) ;13: (1):57. doi: 10.1186/s13023-018-0794-6. |
[166] | Musumeci O , Marino S , Granata F , Morabito R , Bonanno L , Brizzi T , et al. Central nervous system involvement in late-onset Pompe disease: Clues from neuroimaging and neuropsychological analysis. Eur J Neurol. (2019) ;26: (3):442–e35. doi: 10.1111/ene.13835. |
[167] | Ebbink BJ , Poelman E , Aarsen FK , Plug I , Régal L , Muentjes C , et al. Classic infantile Pompe patients approaching adulthood: A cohort study on consequences for the brain. Dev Med Child Neurol. (2018) ;60: (6):579–86. doi: 10.1111/dmcn.13740. |
[168] | Byrne BJ , Fuller DD , Smith BK , Clement N , Coleman K , Cleaver B , et al. Pompe disease gene therapy: Neural manifestations require consideration of CNS directed therapy. Ann Transl Med. (2019) ;7: (13):290. doi: 10.21037/atm.2019.05.56. |
[169] | Scalco RS , Gardiner AR , Pitceathly RD , Zanoteli E , Becker J , Holton JL , et al. Rhabdomyolysis: A genetic perspective. Orphanet J Rare Dis. (2015) ;10: :51. doi: 10.1186/s13023-015-0264-3. |
[170] | Sotiriou E , Greene P , Krishna S , Hirano M , DiMauro S . Myopathy and parkinsonism in phosphoglycerate kinase deficiency. Muscle Nerve. (2010) ;41: (5):707–10. doi: 10.1002/mus.21612. |
[171] | Spiegel R , Gomez EA , Akman HO , Krishna S , Horovitz Y , DiMauro S . Myopathic form of phosphoglycerate kinase (PGK) deficiency: A new case and pathogenic considerations. Neuromuscul Disord. (2009) ;19: (3):207–11. doi: 10.1016/j.nmd.2008.12.004. |
[172] | Sugie H , Sugie Y , Nishida M , Ito M , Tsurui S , Suzuki M , et al. Recurrent myoglobinuria in a child with mental retardation: Phosphoglycerate kinase deficiency. J Child Neurol. (1989) ;4: (2):95–9. |
[173] | Stojkovic T , Hammoudael H , Richard P , López de Munain A , Ruiz-Martinez J , Camaño P , Laforêt P , et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord. (2009) ;19: (5):316–23. doi: 10.1016/j.nmd.2009.02.012. |
[174] | Al-Obeidi E , Al-Tahan S , Surampalli A , Goyal N , Wang AK , et al. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin Genet. (2018) ;93: (1):119–25. doi: 10.1111/cge.13095. |
[175] | Johnson JO , Mandrioli J , Benatar M , Abramzon Y , Van Deerlin VM , Trojanowski JQ , et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. (2010) ;68: (5):857–64. doi: 10.1016/j.neuron.2010.11.036. |
[176] | Abramzon Y , Johnson JO , Scholz SW , Taylor JP , Brunetti M , Calvo A , et al. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. (2231) ;33: (9):e1–e6. doi: 10.1016/j.neurobiolaging.2012.04.005. |
[177] | Neumann M , Tolnay M , Mackenzie IR . The molecular basis of frontotemporal dementia. Expert Rev Mol Med. (2009) ;11: :e23. doi: 10.1017/S1462399409001136. |
[178] | Krause S , Göhringer T , Walter MC , Schoser BG , Reilich P , Linn J , et al. Brain imaging and neuropsychology in late-onset dementia due to a novel mutation (R93C) of valosin-containing protein. Clin Neuropathol. (2007) ;26: (5):232–40. |
[179] | Kalbe E , Onur OA , Minnerop M , Reimann J , Althaus A , Ahmadzadehfar H , et al. Early signs of VCP-related frontotemporal dementia: A neuropsychological, FDG-PET and fMRI study. J Neurol. (2011) ;258: (3):515–8. doi: 10.1007/s00415-010-5774-2. |
[180] | Jung HH , Danek A , Walker RH . Neuroacanthocytosis syndromes. Orphanet J Rare Dis. (2011) ;6: :68–76. doi: 10.1186/1750-1172-6-68. |
[181] | Jung HH , Brandner S . Malignant McLeod myopathy. Muscle Nerve. (2002) ;26: (3):424–7. doi: 10.1002/mus.10199. |
[182] | Ishikawa S , Tachibana N , Tabata KI , Fujimori N , Hayashi RI , Takahashi J , et al. Muscle CT scan findings in McLeod syndrome and chorea-acanthocytosis. Muscle Nerve. (2000) ;23: (7):1113–1116. doi: 10.1002/1097-4598(200007)23:7<1113::aid-mus15>3.0.co;2-6. |
[183] | Hewer E , Danek A , Schoser BG , Miranda M , Reichard R , Castiglioni C , et al. McLeod myopathy revisited: More neurogenic and less benign. Brain. (2007) ;130: (12):3285–96. doi: 10.1093/brain/awm269. |
[184] | Allen FH Jr , Krabbe SM , Corcoran PA . A new phenotype (McLeod) in the Kell blood-group system. Vox Sang.. (1961) ;6: :555–60. doi: 10.1111/j.1423-0410.1961.tb03203.x. |
[185] | Dobson-Stone C , Velayos-Baeza A , Filippone LA , Westbury S , Storch A , Erdmann T , et al. Chorein detection for the diagnosis of chorea-acanthocytosis. Ann Neurol. (2004) ;56: (2):299–302. doi: 10.1002/ana.20200. |
[186] | Walker RH , Danek A , Uttner I , Offner R , Reid M , Lee S . McLeod phenotype without the McLeod syndrome. Transfusion. (2007) ;47: (2):299–305. doi: 10.1111/j.1537-2995.2007.01106.x. |
[187] | Danek A , Rubio JP , Rampoldi L , Ho M , Dobson-Stone C , Tison F , et al. McLeod neuroacanthocytosis: Genotype and phenotype. Ann Neurol. (2001) ;50: (6):755–64. doi: 10.1002/ana.10035. |
[188] | Velayos Baeza A , Dobson-Stone C , Rampoldi L , Bader B , Walker RH , Danek A , et al. Chorea-Acanthocytosis. In: Adam MP , Ardinger HH , Pagon RA , Wallace SE , Bean LJH , Stephens K , Amemiya A , editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2020. (2002) [updated 2019]. Access from: www.ncbi.nlm.nih.gov/books/NBK1387/. |
[189] | Bader B , Vollmar C , Ackl N , Ebert A , la Fougère C , Noachtar S , et al. Bilateral temporal lobe epilepsy confirmed with intracranial EEG in chorea-acanthocytosis. Seizure. (2011) ;20: (4):340–2. doi: 10.1016/j.seizure.2010.12.007. |
[190] | Walterfang M , Evans A , Looi JC , Jung HH , Danek A , Walker RH , et al. The neuropsychiatry of neuroacanthocytosis syndromes. Neurosci Biobehav Rev. (2011) ;35: (5):1275–83. doi: 10.1016/j.neubiorev.2011.01.001. |
[191] | Francke U , Ochs HD , de Martinville B , Giacalone J , Lindgren V , Distèche C , et al. Minor Xp21 chromosome deletion in a male associated with expression of Duchenne muscular dystrophy, chronic granulomatous disease, retinitis pigmentosa, and McLeod syndrome. Am J Hum Genet. (1985) ;37: (2):250–67. |


