
One Year of Newborn Screening for SMA – Results of a German Pilot Project
Abstract
Objective:
Spinal muscular atrophy (SMA) is the most common neurodegenerative disease in childhood. The study was conducted to assess the impact of early detection of SMA by newborn screening (NBS) on the clinical course of the disease.
Methods:
Screening was performed in two federal states of Germany, Bavaria and North Rhine Westphalia, between January 2018 and February 2019. The incidence in the screening population was calculated as number of detected patients with a homozygous deletion in the SMN1-gene per number of screened patients. To get an idea about the incidence of newly diagnosed SMA in the year prior to screening a survey covering all neuropediatric centers in the state of Bavaria was conducted, identifying all SMA-cases in 2017 and 2018. Following positive NBS and confirmatory diagnostic test, treatment was advised according to the recommendations of the “American SMA NBS Multidisciplinary Working Group”. Immediate treatment with Nusinersen was recommended in children with 2 and 3 SMN2 copies and a conservative strict follow-up strategy in children with ≥4 copies. All children underwent regular standardized neuropediatric examination, CHOP INTEND and HINE-2 testing as well as electrophysiological exams every 2-3 months.
Results:
165,525 children were screened. 22 cases of SMA were identified, meaning an incidence rate of 1:7524. SMN2 copy number analysis showed 2 SMN2 copies in 45% of patients, 3 SMN2 copies in 19 % and 4 SMN2 copies in 36%. These findings are confirmed in the most recent statistical data-cut from 31st August 2019 (incidence 1:7089, 2 SMN2 copies in 44%, 3 in 15% and 4 in 38%). Comparison with up-to-date German data on SMA incidence and the Bavarian survey give evidence that NBS did not lead to a relevant increase in incidence. 10 patients with 2 or 3 SMN2 copies were treated with Nusinersen, starting between 15– 39 days after birth, in 7/10 patients before onset of symptoms. Presymptomatically treated patients (age at last examination: 1– 12 months, median 8 months) showed no muscle weakness by the age of one month to one year. One child with 4 SMN2 copies became symptomatic at the age of 8 months.
Conclusions:
Newborn screening, resulting in presymptomatic treatment, improves outcome in children with genetically proven SMA. Newborn screening for SMA should be introduced in all countries where therapy is available. An immediate therapy in cases with 4 SMN2 copies should be considered.
INTRODUCTION
Spinal muscular atrophy (SMA) is the most common neurodegenerative disease in childhood (1). SMA is classified by age of onset and the motor milestones achieved (2).
SMA type 1 (Werdnig-Hoffmann disease) manifests in the first six months of life, and children are unable to sit. The vast majority of children die before the age of 2 years due to respiratory failure if mechanical ventilation is not initiated. SMA type 2 (intermediate form) shows first signs between 6 and 18 months, patients achieve the ability to sit but cannot walk. SMA type 3 (Kugelberg-Welander disease) manifests after ambulation has been acquired. Ambulation may be lost over time (3) (4). The classification originates from the time when pharmacological treatment was not available and refers to the natural history of the disease.
A homozygous deletion in the SMN1 gene, encoding the SMN (“survival motor neuron”)-protein, is responsible for the autosomal recessive disorder in more than 95% of cases (5).
Reduced levels of SMN protein result in motor neuron death in the spinal cord. SMN is a ubiquitously expressed, 32-kDa protein. It has an important housekeeping role in all cells regulating the biogenesis of ribonucleoprotein (RNP) complexes. SMN depletion affects a number of other cellular pathways that are of particular interest for the maintenance of neuronal homeostasis (6).
In contrast to animals, humans have a nearly identical homologous gene copy of the SMN1 gene, called survival motor neuron 2 (SMN2) gene. Whereas the transcription of SMN1 leads to full-length mRNA, transcription of SMN2 primarily generates a shortened mRNA lacking exon 7 (due to a c.840C>T silent mutation) and only 5– 10% amount of full-length transcript (7) (8). The severity of symptoms is influenced by a number of genetic modifiers (9). It largely depends on the copy number of the SMN2 gene; its existence rescues an otherwise lethal phenotype as shown in mice where only one SMN gene exists (10).
In recent years it was shown in the mouse model that successful treatment is possible with gene therapy, replacing the SMN1 gene, and with antisense oligonucleotides, which modify SMN2 splicing to include exon 7 (11) (12) (13). Clinical studies have demonstrated that both strategies are able to significantly alter the course of SMA in humans (14) (15) (16).
Nusinersen (Spinraza®) was approved as the first drug for SMA in the USA in December 2016 and in the European Union in July 2017.
It is an antisense oligonucleotide drug that blocks an intronic silencer in SMN2, thereby facilitating exon 7 inclusion (17). In May 2019, Zolgensma, a systemically delivered AAV9-mediated gene therapy to replace SMN1 in infants with SMA type 1 (16) has been approved by the FDA in the US. In addition, trials using orally available splicing modifiers are on the way (18).
Since the lack of SMN leads to an irreversible loss of motor neurons, the timing of treatment prior to the onset of symptoms is crucial for a good outcome (19). In patients with SMA type 1, about 95% of motor neurons are lost within the first 6 months of life (20). Consequently, there is a need for newborn screening, similar to other treatable inborn diseases (21).
Patients with 2 and 3 SMN2 copies are at high risk of developing SMA type 1 or type 2. Preliminary data of the NURTURE study, in which pre-symptomatic children with 2 and 3 SMN2 copies were treated with Nusinersen, showed a significantly better motor development compared to children who were treated after the onset of symptoms (22) (23). 25 SMA patients with 2 or 3 SMN2 copies had a significantly better outcome with regard to motor and respiratory function as compared to the natural history and to the results of the controlled studies in symptomatic children. All pre-symptomatically treated patients with 3 SMN2 copies are symptom-free, and also the patients with 2 SMN2 copies show either normal development or profoundly attenuated symptoms of SMA (15).
In this study, we present the clinical outcome of children, detected through a pilot screening project for SMA in Germany, between January 2018 and February 2019. The method for quantitative PCR from DNA extracted from dried blood spots to screen for a homozygous deletion of exon 7, has already been published (24).
METHODS
The screening was carried out in two German states, Bavaria and North Rhine Westphalia, from January 2018. The clinical and electrophysiological data cut-off for this paper was February 2019. Statistical data is available until August 2019. The DBS samples mainly came from maternity clinics, with approximately 42% of the samples coming from North Rhine-Westphalia and 58% from Bavaria. The pilot NBS program covered about 80% of the newborns in Bavaria and 40% in North Rhine-Westphalia.
Patients in whom a homozygous deletion of exon 7 of the SMN1-gene was detected in the NBS program were immediately referred to one of the participating neuropediatric departments (Dr. v. Hauner Children’s Hospital, University of Munich; Childrens Hospital, University of Essen; Children’s Hospital, University of Münster) to inform the parents and to draw a second blood sample for the confirmation of the NBS result. The method for the NBS is described in detail in an independent manuscript (24). In short, DNA was extracted from punches acquired from dried blood spot cards which were part of the standard NBS screening program for inborn metabolic and endocrinological diseases. A primer specifically targeting the SMN1 c.840C position was used to detect a homozygous deletion of exon 7 of the SMN1-gene. QPCR experiments were evaluated based on cycle quantification. The homozygous SMN1 deletion was verified in all newborns via Multiplex Ligation-dependent Probe Amplification (MLPA) test for SMN1 and SMN2 copy number based on a second blood sample as previously described (25). SMN2 copy numbers were double-checked in a second laboratory.
After positive screening from dried blood spots and confirmation diagnostics, a second appointment with the family was set within a few days to discuss the results of the gene test, to inform in detail about SMA and to determine the treatment plan together with the parents.
The treatment proposal was based on the recommendations of the American SMA NBS Multidisciplinary Working Group (26). Early treatment with Nusinersen was recommended to all children with 2 and 3 SMN2 copies. Parents were free to decide if, and when they wanted to start with the treatment. Children with 4 or more SMN2 copies were monitored with a conservative strict follow-up strategy.
Clinical assessment
All children underwent careful neuropediatric examination by experienced pediatric neurologists with expertise in SMA. CHOP INTEND (27) and HINE-2 (28) were performed by trained physiotherapists at every visit. In the treatment group, examinations of the children were performed at intervals of 3 months. In the conservative strict follow-up group, children were examined at monthly intervals during the first six months of life and every two or three months thereafter.
Electrophysiology
CMAPs were obtained from the abductor digiti V, and abductor hallucis brevis muscles. Care was taken to do all measurements at standard skin temperature. In selected cases, needle electromyography was performed on the quadriceps femoris and/or the anterior tibial muscle.
Children were considered being asymptomatic/presymtomatic at first investigation when clinical assessment revealed normal movements of extremities and ulnar CMAPs were >1 mV. Patients were considered being affected, when ulnar CMAP was <1 mV and/or CHOP Intend was <30 points.
SMA survey
In the State of Bavaria (South Germany) we conducted a survey to determine the incidence of newly diagnosed SMA cases in the year prior to NBS (2017) and in the first year of NBS (2018). A total of 24 questionnaires was sent to all pediatric neurological hospitals and outpatient centers. The response rate was 100%. The survey covered all treatment centers for SMA in Bavaria (all neuropediatric departments in hospitals and all social pediatric centers), which usually are involved in the diagnostic and therapeutic workup of patients with SMA. Duplications can be excluded since anonymized but patient-specific code was exchanged.
The clinical follow-up study of the patients was approved by the local ethics committee of the participating universities (ethics committee project no. 18– 269, LMU).
DATA AVAILABILITY STATEMENT
All data from CHOP INTEND, HINE-2, electrophysiology and from the survey can be shared and is available at the author’s institution in Munich, Dr. v. Haunersches Kinderspital, Lindwurmstr. 4, 80337 München, Germany.
RESULTS
Incidence and SMN2-copy number
Among 165,525 newborn children screened for SMA, a homozygous SMN1-deletion was found in 22 children from 21 families (there was one dizygotic twin pair). MLPA confirmed the diagnosis of homozygous deletion of exon 7 in all patients (19 with a deletion of exon 7 and 8; 3 with a deletion of exon 7, only). The incidence rate of a homozygous deletion of exon 7 of the SMN1 gene in our cohort is 1:7524 (22 in 165,525) after 13 months of screening.
SMN2-copy number analysis showed 2 SMN2 copies in 10 patients (45%), 3 SMN2 copies in 4 patients (18%) and 4 SMN2 copies in 8 patients (36%). Preliminary statistical data from the 31st of August 2019 revealed 34 children, detected in a screened cohort of 241.270. The incidence of 1:7096, as well as the distribution of the SMN2 copy numbers with 2 SMN2 copies in 15 patients (44%), 3 SMN2 copies in 5 patients (15%) and 4 SMN2 copies in 13 patients (38%) remained in the same range.
Results from the comprehensive survey involving all Bavarian neuropediatric treatment centers revealed 15 newly diagnosed patients all over Bavaria in 2017, which was the year before the screening started. Out of these 15 children, 10 patients met the clinical criteria of SMA type 1, two of SMA type 2 and three of SMA type 3. The number of childbirths in Bavaria in 2017 was 126,191, thus the incidence rate of SMA in the pre-screening year was 1:8413.
In 2018, when the screening started, 7 symptomatic SMA patients were found in Bavaria in addition to 11 patients diagnosed in the NBS project. 3/7 were patients with SMA type 1, born in 2018 but not screened in the screening lab that runs the SMA project. 4/7 patients were born before 2018 (one with SMA type 2 and 3 with SMA type 3). The number of childbirths in Bavaria in 2018 was 127.616. Thus, the estimated incidence rate of SMA in Bavaria was 1:7089 in 2018.
Clinical findings and treatment decisions
Patient’s characteristics and follow-up results are presented in Table 1.
Table 1
Clinical and electrophysiological findings, follow-up, treatment and compliance of 22 SMA patients, screened between January 2018 and February 2019. Mo = months, d = days
| Pat Nr. | SMN2 copy Nr. | Status at first exam | CHOP Intend first exam | Ulnar CMAP at first investigation | Motor detoriation | Deceasedage | Treatment with Nusinersen from age | Age at last exam | CHOP Intend at last exam | Ulnar CMAP at last exam (mV) | Respiratory involvement | Compliance for visits |
| 1 | 2 | symptomatic | 33 | 0,6 | – | – | 39 d | 13 mo | 60 | n.a. | no | good |
| 2 | 3 | unaffected | 52 | 1,6 | – | – | 24 d | 13 mo | 64 | 9,0 | no | good |
| 3 | 4 | unaffected | 54 | 7,6 | – | – | – | 8 mo | 64 | 6,1 | no | poor, lost to follow-up |
| 4 | 2 | ? (CMAP n.a.) | 39 | n.a. | 2 weeks | 5,5 mo | Lack of reimbursement | – | – | – | yes | – |
| 5 | 2 | unaffected | 40 | 1,7 | – | – | 15 d | 10 mo | 64 | 6,8 | no | good |
| 6 | 3 | unaffected | 50 | 1,2 | – | – | 24 d | 10 mo | 64 | 4,2 | no | good |
| 7 | 4 | unaffected | 47 | 4,9 | – | – | – | 9 mo | 64 | 7,1 | no | good |
| 8 | 4 | unaffected | 60 | 5,8 | – | – | – | 9 mo | 64 | 7,4 | no | good |
| 9 | 4 | unaffected | 44 | 5,2 | – | – | – | 9 mo | 64 | 8,3 | no | good |
| 10 | 2 | unaffected | 54 | n.a. | – | – | 35 d | 7 mo | 64 | 5,2 | no | good |
| 11 | 4 | unaffected | 55 | 3,4 | 8 mo | – | 9 mo | 9 mo | 62 | 5,8 | no | good |
| 12 | 4 | unaffected | 62 | 4,1 | 7 mo | 64 | 5,8 | no | good | |||
| 13 | 4 | unaffected | 38 | 4,2 | – | – | – | 5 mo | 58 (age 3 months) | 4,7 | no | good |
| 14 | 4 | unaffected | 57 | 3,2 | – | – | – | 5 mo | 64 | 7,1 | no | good |
| 15 Twin | 3 | unaffected | 61 | 6,4 | – | – | Refused by parents | 4 mo | 64 | 6,7 | no | good |
| 16 Twin | 3 | unaffected | 60 | 6,3 | – | – | Refused by parents | 4 mo | 64 | 5,9 | no | good |
| 17 | 2 | unaffected | 53 | 1,9 | 12 weeks | 5 mo | Refused by parents | 3 mo | 21 | n.a. | yes | – |
| 18 | 2 | unaffected | 59 | 3,2 | – | – | 25 d | 4 mo | 64 | 6,5 | no | good |
| 19 | 2 | symptomatic | 35 | 0,6 | – | – | 22 d | 3 mo | 48 | n.a. | no | good |
| 20 | 2 | symptomatic | 9 | 0,4 | – | – | 17 d | 2 mo | 22 | 0,7 | no | good |
| 21 | 2 | symptomatic | 48 | 0,8 | 2 weeks | – | 15 d | 6 weeks | 21 | 0,1 | no | good |
| 22 | 2 | unaffected | n.a. | 6,4 | – | – | 17 d | 4 weeks | 58 | n.a. | No | good |
According to the current recommendations, immediate treatment has been proposed to patients with two or three SMN2 copies (14/22).
Two children with two SMN2 copies could not be treated, one due to lack of reimbursement and one due to the parent’s decision. The patients died at the age of 5 and 5,5 months, respectively.
A second family with twins with 3 SMN2 copies decided against immediate treatment and wished to follow a “conservative strict follow-up” strategy due to fears of a painful lumbar puncture.
The twins were still without symptoms at their last follow-up at the age of 4 months.
Ten patients were treated with Nusinersen according to the standard protocol with 4 dosages á 12 mg Nusinersen within the first 63 days of life and then 4 months intervals of medication. Treatment was started at the age of 15– 39 days (median 24 days) after birth.
The use of sedation has been adapted to the difficulty of lumbar puncture. If possible, sedation was avoided. Nevertheless, sedation was necessary for 3 patients from the first intrathecal injection of Nusinersen, in one starting at the second injection, in one at the fourth and in another one starting at the fifth intrathecal injection. The anesthesia or sedation did not follow a prescribed protocol, but was at the discretion of the anesthetist. No relevant complications related to the drug delivery or to the drug itself have occurred in our cohort so far.
Four of the treated patients (all with 2 SMN2 copies), were already symptomatic at the first examination with low ulnar CMAPs <1 mV (Patient 1, 19, 20 and 21 (Table 1)). Two of them presented with moderate hypotonia an initial score of 35 / 33 points (patient 1 and 19), one with a severe proximal weakness and a CHOP INTEND score of only 9 points (patient 20), and the fourth with the score falling rapidly from initially 47 to 21 points within 3 weeks. Treatment was started at the age of 39, 17, 22 and 15 days, respectively. Currently the observation period is still too short to judge the final outcome, but by the last investigation, all three (age 2, 3 and 3 months, respectively) showed an increase in CHOP INTEND score under treatment (Fig. 1). Patient 1 learned to sit independently but developed a tremor at the age of 10 months. Presymptomatically treated patients (age at last examination: 1– 12 months, median 8 months) have showed no muscular weakness so far. Data from CHOP-INTEND, HINE-2 and ulnar CMAPs are presented in Figs. 1–3.
Fig. 1
CHOP-INTEND a) patients with 2 SMN2 copies b) patients with 3 SMN2 copies c) patients with 4 SMN2 copies.
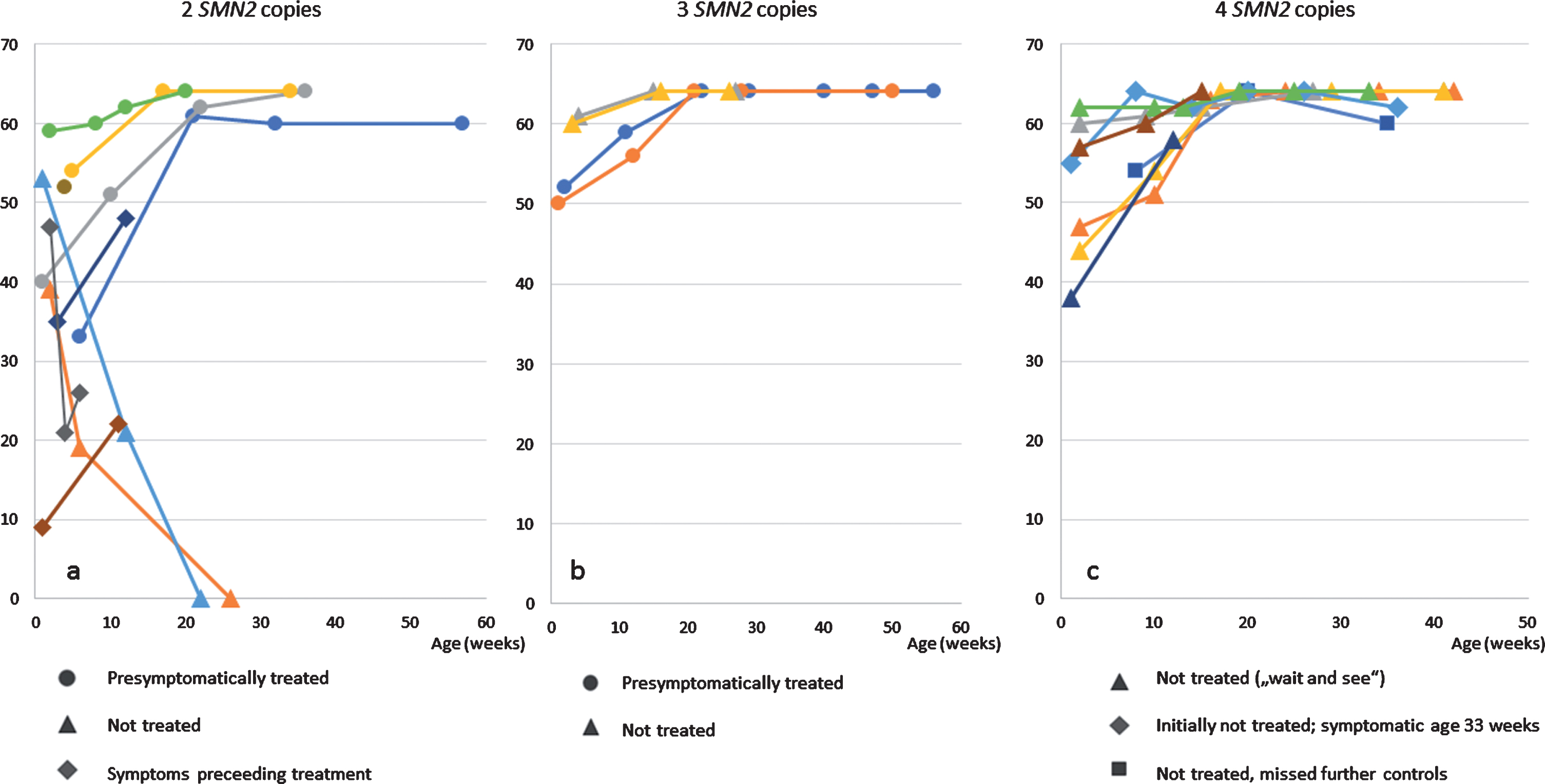
Fig. 2
HINE-2 a) patients with 2 SMN2 copies b) patients with 3 SMN2 copies c) patients with 4 SMN2 copies.
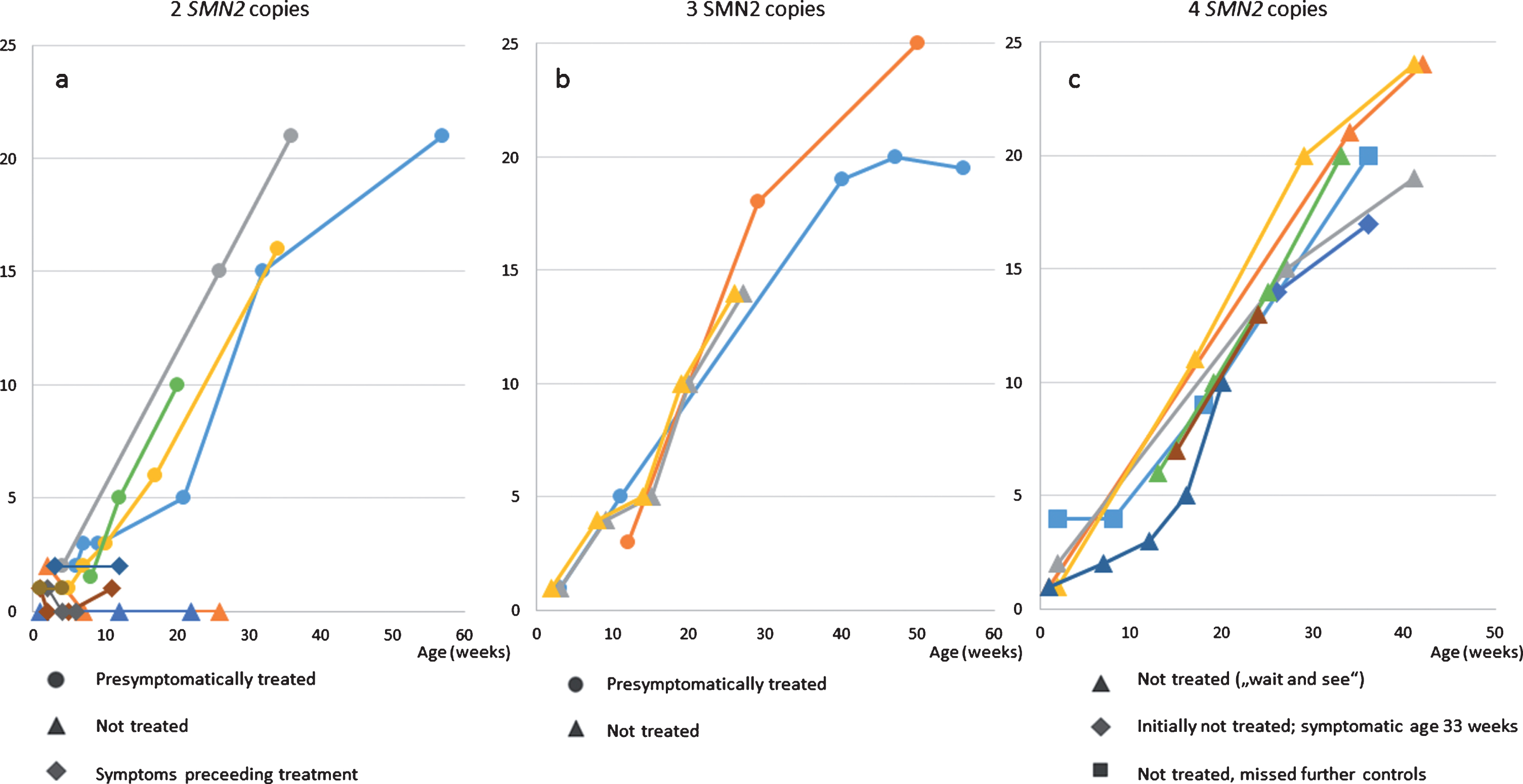
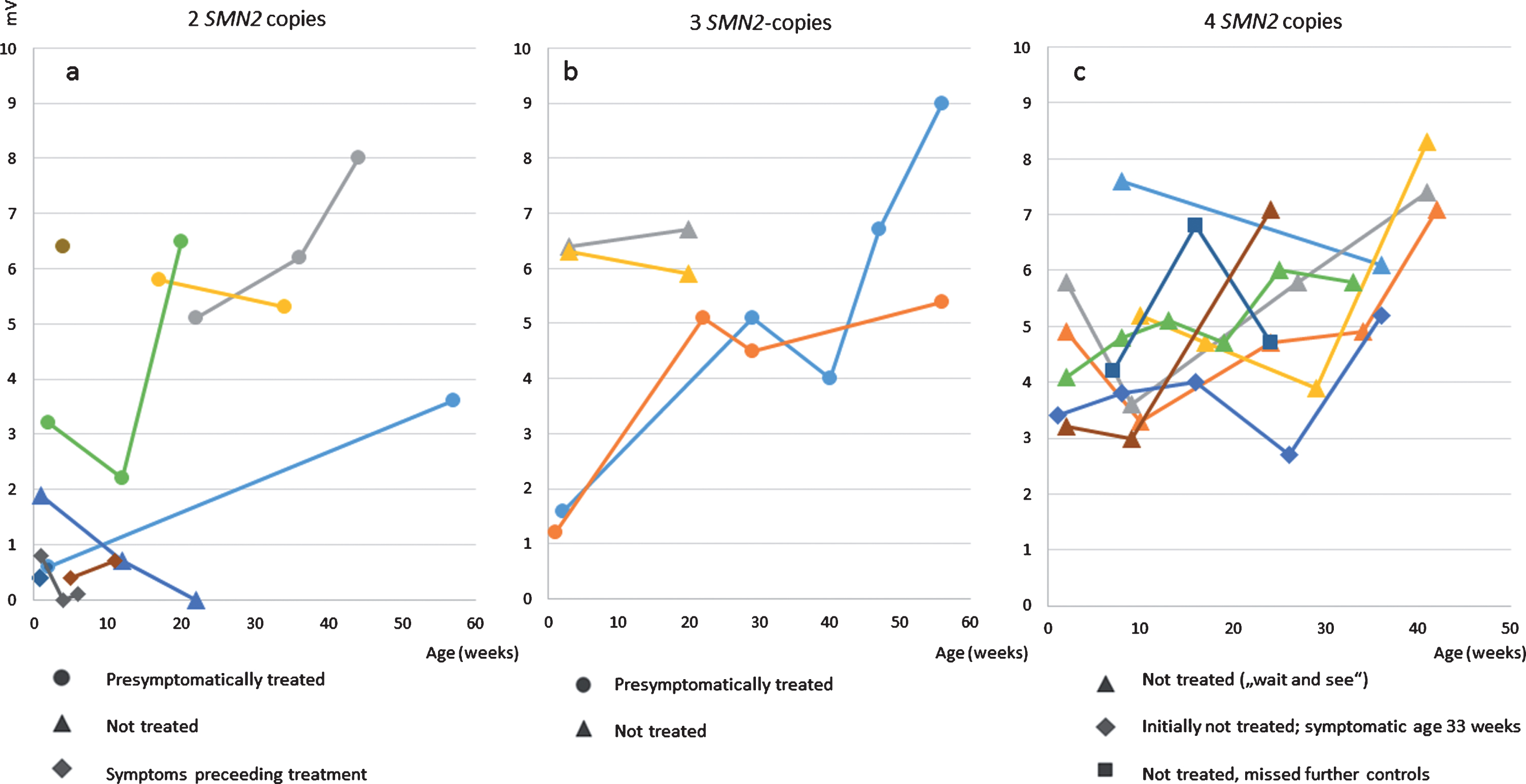
Fig. 3
ulnar nerve CMAPs a) patients with 2 SMN2 copies b) patients with 3 SMN2 copies c) patients with 4 SMN2 copies.
Eight patients with 4 SMN2 copies were treated with a conservative strict follow-up strategy and with regular clinical, electrophysiological and sonographic controls. The oldest patient with 4 SMN2 copies, born in February 2018, missed the recommended follow-up investigations from age 8 months on. The second oldest patient with 4 SMN2 copies (patient 11) showed first clinical symptoms at the age of 8 months with a new proximal weakness of the legs. EMG confirmed a neurogenic lesion and treatment was initiated. Time of follow-up after start of treatment is still too short to judge the outcome. After a baby with 4 SMN2 copies was detected due to NGS, the 5 year old brother was also diagnosed as SMA type 3 with hypotonia and tremor, he retrospectively developed first symptoms at the age of 3 years. The other 6 patients with 4 SMN2 copies were between 3 and 9 months of age at last examination and have not developed symptoms hitherto (Figs. 1, 2).
Neurographic investigation
Figure 2 shows CMAPS of the ulnar nerve in patients after birth and the course under treatment or observation, respectively. In summary, CMAPs in patients with 2 and 3 SMN2 copies were lower than in those with 4 copies. One untreated patient with 2 SMN2 copies, symptomatic 10 weeks of age showed a decrease of the distal CMAP (measured at 12 weeks of age).
In the patient with 4 SMN2 copies who showed symptoms at the age of 8 months chronic neurogenic changes in the EMG of the quadriceps preceded the decrease of tibial and ulnar CMAPS, which were still in the normal range 14 days after the family had noted first symptoms. Deep tendon reflexes were absent at that time.
DISCUSSION
This paper presents clinical data of patients discovered in a newborn screening pilot project for SMA in Germany. After more than one year of screening, out of 165,525 investigated newborns, 22 children with a homozygous deletion of exon 7 or exon 7-8 in the SMN1 gene were identified. Ten of these children had 2, four children 3, and eight children 4 copies of the SMN2 gene. These findings could be confirmed in an update statistical result from August 2019.
Our preliminary data indicate that the incidence for SMN-related SMA in Germany was 1:7524 after 13 months of screening and 1:7096 after 19 months of screening. This figure is consistent with estimations, which were published in previous studies. Most recently, König et al published de-duplicated incidence rates from independent data sources for SMA in Germany, covering the last decade. They found, e.g. in 2014, a minimal incidence of 1.36/10000, (1:7353). This is the most current and probably most accurate calculation and confirms our incidence quite precisely. In 2017, Verhaart et al. reanalyzed prevalence, incidence and carrier frequency of 5q-linked SMA in Europe. They found 4653 patients who were genetically diagnosed between 2011 and 2015. The data did not exclude duplications and is more vague, providing an estimated incidence of SMA in Europe of 1 in 3900– 16,000 live births. They included two older studies from Germany that provided evidence that in Germany the incidence of SMA might be higher than in other countries in Western Europe (29). The fact that the incidence in the NBS population is similar to the figures mentioned above is a strong argument against the view that NBS detects children with a homozygous deletion of SMN1, which otherwise would not have come to clinical attention. To get further insight into this problem we conducted additionally a survey involving all Bavarian neuropediatric centers to compare the number of patients with newly diagnosed SMA prior and after the start of the NBS program. All centers dealing with neuromuscular or neuropediatric patients in Bavaria contributed information about new cases of SMA, which guarantees full coverage. Duplications were excluded. The data revealed occurrence of SMA in 2017 in Bavaria of 1:8413, which is in the same range (similar fluctuations between individual years were observed in the paper by König et al), which gives further evidence that NBS does not lead to the detection of a high number of probably healthy individuals with genetic changes of an SMA. Thus, we assume a high probability that children which are identified by NBS will develop symptoms during infancy and childhood, irrespective of the number of SMN2 copies. This is in line with the fact that despite extensive genetic work in the past decades only few asymptomatic cases with a homozygous deletion of the SMN1 gene a have been published (30) (31) (32) (33) (34). In part of them two protective genetic modifiers, PLS3 and NCALD, have been identified (35) (36).
However, single publications measured carrier frequencies, which was determined to be 1:35 based on direct genetic testing of control individuals (37) in Germany, in Poland, a similar discrepancy was described (38). If this figure is correct, an even higher incidence for SMA up to 1:4,900 or a certain number of late-onset, mild or unaffected patients would be expected. This incidence rate would be much higher then the indidence which we have found in our population. Long term data will be needed to verify these numbers.
The survey demonstrates that, to date, in Bavaria so far no SMA patient was diagnosed who had been overlooked by the NBS. Patients who were newly diagnosed in 2018 apart from those detected by NBS were either born before 2018 or not screened by the participating laboratory. Nevertheless, since the detection of SMA in our project is based on the detection of a homozygous deletion in the SMN1 gene, about 2– 5% of SMA patients with a heterozygous deletion combined with an additional point mutation will be missed (39). As well, rarer milder forms of adult onset SMA may not be detected clinically in the pediatric age range.
A central question facing the field is whether it is or shall be possible to forecast the severity of the disease and the onset of symptoms of pre-symptomatic individual genotyped as SMN1 – /–, that is with a genetically proven SMA. Current knowledge is mainly based on existing meta-analyses of multiple studies describing clinical and genetic findings in symptomatic patients. Prospective population-based data do not exist apart from a former screening project in Taiwan (40) with a total number of 120,267 children screened. They found an incidence of SMA in Taiwan being 1 in 17,181 and identified a total of 7 SMA patients, among them 3 with 2 SMN2 copies, 2 with 3 SMN2 copies, and 2 with 4 SMN2 copies. Similar to our results, one of their patients with 2 SMN2 copies already had clinical signs at birth. A much smaller cohort was screened in New York (41) with 3,826 children, in which the authors identified a patient with 2 SMN2 copies.
According to a big meta-analysis of Verhaart et al. in 2017, 50– 60% of SMA patients are known to suffer from type 1 (29).
The study pointed out that many of the published prevalence or incidence rates predated genetic testing and that classification schemes have changed over the years, highlighting the need for contemporary data (29). Another recent study (Calucho et al. 2018), who reviewed a worldwide case series of more than 3,400 SMA patients, reported 36% to suffer from type 1, 33% from type 2, and 31% from type 3 (42). The study measured prevalence, not incidence, which leads to a decrease of type 1 cases due to early death.
The number of SMN2 copies still holds for the major disease modifier in SMA (43); a higher SMN2 copy number correlates with a larger amount of full-length SMN protein and a milder clinical phenotype (42) (37) (44). However, the SMN2 copy number does not allow to precisely predict the course of the disease in a given individual (42) (45) (46). In the study of Calucho et al., 2 SMN2 copies were present in 33% of individuals, 3 SMN2 copies in 48% and 4 SMN2 copies in 15% of cases. The existence of only one SMN2 copy was rarely found (3%) and 5 or 6 copies in SMA could be detected in only 0.7% (percentages recalculated from their graphing data (42)).
In our cohort, (statistical data is available until August 2019), 44% of patients have 2 SMN2 copies, 15% have 3 SMN2 copies and 38% have 4 SMN2 copies. One explanation for this difference to previous data is, that SMA type 3 might be underdiagnosed in the population. However, in our study we found a higher number of 4 SMN2 copies and a lower number of patients with 3 SMN2 copies than known in the literature. Only recently, Schorling et al were able to show that the reliability of SMN2 copy number determination was low especially in previous times (47). Thus a possible explanation for the difference between the studies can be that in former studies a considerable number of patients with 4 copies have been wrongly classified as patients with 3 copies of SMN2. If this is true, the current guideline could underestimate the risk for patients with 4 SMN2 copies. Keeping in mind that the copy number is highly dependent on methodological aspects we confirmed all numbers by a second independent laboratory. At the moment our number of cases is still not high enough to determine the final distribution of SMN2 copy numbers in the population. However, the figures remained quite stable during various time points of the study.
The study of Calucho et al., mentioned above, analyzing the distribution of SMA types according to the number of SMN2 copies (42), showed that 95% of the patients with 2 SMN2 copies suffer either from SMA type 1 or type 2. In 5% of patients with 2 SMN2 copies, SMA type 3 occurred. About the half of these mild cases with a type 3 SMA phenotype and 2 SMN2 copies carry the SMN2-c.859G>C variant (42), which increases full-length SMN2 transcripts and act protective in SMA (43). 87% of the patients with 4 SMN2 copies suffer from the milder SMA type 3-phenotype. Nevertheless, a severe SMA type 2-phenotype is seen in at least 3% of patients with 4 SMN2 copies.
For the time being, our decision to treat or to monitor patients in a conservative strict follow-up strategy was according to the published treatment algorithm of the North American SMA NBS Multidisciplinary Working Group (26). This algorithm is based exclusively on expert opinion but not on data from prospective studies. All families with children with 2 and 3 SMN2 copies were advised to treat their children with Nusinersen.
In this study, we can confirm the favourable results of the Nurture study. In our NBS cohort, so far, all pre-symptomatically treated patients remained without motor symptoms; of note, the oldest children with 2 SMN2 copies were 13 month-old at the last examination. As expected, the 2 untreated children (one due to lack of medical insurance, one due to the decision of the parents) with 2 SMN2 copies showed onset of disease before 3 months of age and clinically deteriorated rapidly (Figs. 1 and 2), which underlines the strong argument for immediate post-natal treatment. The untreated monozygotic twins with 3 SMN2 copies (according to the decision of the parents) remained asymptomatic so far. However, the future occurrence of a severe type 2 SMA is still not excluded. Three children with clinical symptoms within the first 2 weeks of life show an increase in CHOP INTEND under treatment, which reflects the known effect of the therapy in symptomatic SMA type 1 (48, 49), but are still too young to judge final motor outcome.
In this, even to the specialist, very new cohort of newborn SMA patients, the role of the different evaluation methods to detect the onset of disease in time has to be determined. Our results give first evidence, that in the very beginning of the severe types, the CMAP is more sensitive than the clinical evaluation: Table 1 shows that in all children with early symptom onset, the CMAP was <1 mV at first investigation, while motor detoriation did sometimes follow shortly after or could even have been prevented. However, regarding the follow-up of in the group with 4 copies of SMN2, the assessment of the motor status seems to be predicting. EMG is a very sensitive parameter, but is invasive and not highly available. In contrast, in our patient with onset of proximal weakness at age 8 months CMAPs remained unchanged (patient 11) even a few weeks after disease onset.
In patients with 4 SMN2 copies, a conservative strict follow-up approach has been proposed. The reason for this is that the number of patients with a mild disease is still unknown and that intrathecal therapy with Nusinersen is not free from side effects. Whereas the overall frequency of drug-related side effects seems to be low, some cases of hydrocephalus necessitating a shunt procedure are known.
On the other hand, as mentioned above, the probability of all children with a homozygous deletion of SMN1 to develop clinical symptoms is high. In our series, a child with 4 SMN2 copies developed first symptoms at the age of 8 months. A sibling of another child with 4 SMN2 copies was identified retrospectively as developing signs of SMA type 3 age 3 years. In the course of our study it will have to be seen whether the recommendations have to be adapted. The use of biomarkers like neurofilaments in addition to EMG, which seems to be the most sensitive parameter for early detection at the moment, might be helpful for decision making in this group of patients (50).
Even for those patients who do not qualify for an immediate treatment, early diagnosis is useful. It is known that treatment with Nusinersen can be effective in symptomatic late onset patients (15). An earlier onset of treatment results in a better outcome than a late onset. Especially in SMA type 3, there is a considerable delay of the diagnosis up to some years (51). Thus, early detection of the mutation allows an adequate monitoring of the children and an early start of treatment if necessary.
Pathophysiological considerations are still in favor of an early treatment for patients with more SMN2 copies (11). It might be reasonable to treat them early and to stop treatment at a time when there is less need for SMN protein for the developing nervous system (52).
Compliance in SMA is new challenge with NBS. In our study, compliance was generally good; however, in a relevant number of patients (10% of families), medical advice concerning treatment was not accepted in 2/22 families, and one family did not comply with the strict medical appointments and was lost to follow up while still asymptomatic. The psychosocial impact of early detection of a genetically confirmed diagnosis has yet to be evaluated, as will have the different factors that lead to incompliance. Since it is known that compliance after NBS is better diseases which can easily be treated like connatal hypothyreosis or biotinidase-deficiency (53) whereas it is much less (especially with increasing age) in disorders like phenylketonuria (54), which require a very restrictive diet, the further development of less invasive treatments in SMA would probably improve compliance.
CONCLUSION
With the advent of effective pharmacological treatment for SMA (55) (14), there is a worldwide discussion about strategies to identify patients as early as possible. This is especially so for children with an expected severe form of SMA who in our view should be treated immediately (26) (19).
Our preliminary results show that newborn screening for SMA, resulting in pre-symptomatic treatment, can prevent the disease and partially rescue motor neuron function. Up to now, there has been normal motor development in all pre-symptomatically treated children despite tremor in one. The incidence rate of SMA did not increase following initiation of NBS. Newborn screening for SMA should be therefore introduced in countries with available medical treatment. Long-term data are needed to establish the most appropriate treatment for patients with 4 or more SMN2 copies.
CONFLICT OF INTEREST/FINANCIAL DISCLOSURES
1. Serving on a scientific advisory board or data safety monitoring board.
2. Gifts (other than travel or compensation for consulting or for educational efforts) worth more than USD $1000.
3. Funding for travel or speaker honoraria to the individual from a commercial or non-profit entity not included in the study funding [Exclude CME activities and Grand Rounds].
4. Serving as a journal editor, an associate editor, or editorial advisory board member. This may include a journal published by your national medical/scientific organization. Please include regardless of whether you receive compensation.
5. Patents issued or pending.
6. Publishing Royalties (do not include honoraria for occasional writing).
7. Employment. If you are currently employed by a commercial entity, please disclose below. In addition, if your past employment at a commercial entity is directly related to this manuscript, please disclose below.
8. Consultancies.
9. Speakers’ bureau.
10. Other activities not covered in designations above (if in doubt, provide full disclosure).
11. Some published work has potential for financial gain for the study investigators or the sponsor. The following question seeks to provide transparency regarding any financial benefits to investigators or sponsors.
Katharina Vill, Heike Kölbel, Oliver Schwartz, Astrid Blaschek, Bernhard Olgemöller, Erik Harms, Uta Nennstiel, and Beate Jensen have nothing to declare.
Siegfried Burggraf, Wulf Röschinger, Jürgen Durner and Marc Becker are employed by/owner of a commercial entity (Laboratory Becker and colleagues MVZ GbR, Führichstraße 70, 81871 München, Germany).
Dieter Gläser: employed by/owner of a commercial entity (Genetikum®, Wegenerstr. 15, 89231 Neu-Ulm, Germany).
Ulrika Schara is serving on a scientific advisory board or data safety monitoring board for Biogen, Avexis and Novartis.
Brunhilde Wirth is serving on a scientific advisory board or data safety monitoring board for SMA Europe and received travel and speaker honoraria from Biogen.
Katharina Hohenfellner received commercial travel support and speaker honoraria from Ortphan Europe, Chiesi, and non-profit travel support from Cystinosis Foundation and Nephie.
Wolfgang Müller-Felber is serving on a scientific advisory board or data safety monitoring board for Biogen, Avexis, PTC, Sanofi-Aventis and Cytokinetics and received travel and speaker honoraria from Biogen, Avexis, PTC and Sanofi-Aventis.
ACKNOWLEDGMENTS
The study was funded by the German Cystinosis Foundation (Cystinose Stiftung, DSZ-Regional Office Munich, Widenmayerstr. 10, 80538 Munich, Germany). The funders had no influence in study design, interpretation and publication of these data.
REFERENCES
[1] | Sugarman EA , Nagan N , Zhu H , Akmaev VR , Zhou Z , Rohlfs EM , et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. European Journal of Human Genetics: EJHG. (2012) ;20: (1):27–32. |
[2] | Wang CH , Finkel RS , Bertini ES , Schroth M , Simonds A , Wong B , et al. Consensus statement for standard of care in spinal muscular atrophy. Journal of Child Neurology. (2007) ;22: (8):1027–49. |
[3] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , et al. Diagnosis and management of spinal muscular atrophy: Part Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscular Disorders: NMD. (2018) ;28: (2):103–15. |
[4] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , et al. Diagnosis and management of spinal muscular atrophy: Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscular disorders: NMD. (2018) ;28: (3):197–207. |
[5] | Lefebvre S , Burglen L , Reboullet S , Clermont O , Burlet P , Viollet L , et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. (1995) ;80: (1):155–65. |
[6] | Shorrock HK , Gillingwater TH , Groen EJN . Overview of current drugs and molecules in development for spinal muscular atrophy therapy. Drugs. (2018) ;78: (3):293–305. |
[7] | Lorson CL , Hahnen E , Androphy EJ , Wirth B . A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. (1999) ;96: (11):6307–11. |
[8] | Helmken C , Hofmann Y , Schoenen F , Oprea G , Raschke H , Rudnik-Schoneborn S , et al. Evidence for a modifying pathway in SMA discordant families: Reduced SMN level decreases the amount of its interacting partners and Htra2-beta1. Hum Genet. (2003) ;114: (1):11–21. |
[9] | Wirth B , Garbes L , Riessland M . How genetic modifiers influence the phenotype of spinal muscular atrophy and suggest future therapeutic approaches. Current Opinion in Genetics & Development. (2013) ;23: (3):330–8. |
[10] | Schrank B , Gotz R , Gunnersen JM , Ure JM , Toyka KV , Smith AG , et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. (1997) ;94: (18):9920–5. |
[11] | Foust KD , Wang X , McGovern VL , Braun L , Bevan AK , Haidet AM , et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nature Biotechnology. (2010) ;28: (3):271–4. |
[12] | Passini MA , Bu J , Richards AM , Kinnecom C , Sardi SP , Stanek LM , et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. (2011) ;3: (72):72ra18. |
[13] | Hua Y , Sahashi K , Rigo F , Hung G , Horev G , Bennett CF , et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. (2011) ;478: (7367):123–6. |
[14] | Finkel RS , Chiriboga CA , Vajsar J , Day JW , Montes J , De Vivo DC , et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet (London, England). (2017) ;388: (10063):3017–26. |
[15] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , et al. Nusinersen versus sham control in later-onset spinal muscular atrophy. The New England Journal of Medicine. (2018) ;378: (7):625–35. |
[16] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. The New England Journal of Medicine. (2017) ;377: (18):1713–22. |
[17] | Hua Y , Sahashi K , Hung G , Rigo F , Passini MA , Bennett CF , et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. (2010) ;24: (15):1634–44. |
[18] | Talbot K , Tizzano EF . The clinical landscape for SMA in a new therapeutic era. Gene Therapy. (2017) ;24: (9):529–33. |
[19] | Tizzano EF , Finkel RS . Spinal muscular atrophy: A changing phenotype beyond the clinical trials. Neuromuscular Disorders: NMD: (2017) . |
[20] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Natural history of infantile-onset spinal muscular atrophy. Annals of Neurology. (2017) ;82: (6):883–91. |
[21] | Fingerhut R , Olgemoller B . Newborn screening for inborn errors of metabolism and endocrinopathies: An update. Anal Bioanal Chem. (2009) ;393: (5):1481–97. |
[22] | De Vivo DC . Data from the Nurture Study (Biogen), presented at the MDA meeting in Arlington. (2018) . |
[23] | Parsons J De Vivo DC BE, Hwu W-L , Crawford TO , Swoboda KJ , Finkel RS , Kirschner J , Kuntz N , Ryan MM , Butterfield RJ , Topaloglu H , Ben Omran T , 14 Sansone VA , Jong Y-J , Shu F , Reyna SP , Johnson K , Foster R , Bhan I , Fradette S , Farwell W , on behalf of the NURTURE Study Investigators. Nusinersen in Infants Who Initiate Treatment in a Presymptomatic Stage of Spinal Muscular Atrophy (SMA): Interim Efficacy and Safety Results From the Phase 2 NURTURE Study. 23rd Annual Spinal Muscular Atrophy Researcher Meeting, Anaheim, CA. (2019) . |
[24] | Czibere L , Burggraf S , Fleige T , Gluck B , Keitel LM , Landt O , et al. High-throughput genetic newborn screening for spinal muscular atrophy by rapid nucleic acid extraction from dried blood spots and 384-well qPCR. European Journal of Human Genetics: EJHG. (2019) . |
[25] | Arkblad EL , Darin N , Berg K , Kimber E , Brandberg G , Lindberg C , et al. Multiplex ligation-dependent probe amplification improves diagnostics in spinal muscular atrophy. Neuromuscular Disorders: NMD. (2006) ;16: (12):830–8. |
[26] | Glascock J , Sampson J , Haidet-Phillips A , Connolly A , Darras B , Day J , et al. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J Neuromuscul Dis. (2018) . |
[27] | Glanzman AM , Mazzone E , Main M , Pelliccioni M , Wood J , Swoboda KJ , et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscular Disorders: NMD. (2010) ;20: (3):155–61. |
[28] | Bishop KM , Montes J , Finkel RS . Motor milestone assessment of infants with spinal muscular atrophy using the hammersmith infant neurological Exam-Part Experience from a nusinersen clinical study. Muscle & Nerve. (2018) ;57: (1):142–6. |
[29] | Verhaart IEC , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet Journal of Rare Diseases. (2017) ;12: (1):124. |
[30] | Jedrzejowska M , Szczaluba K , Sielska D . Homozygous deletion in the SMN1 gene in asymptomatic individual - genetic counselling issues in SMA-risk families. Medycyna Wieku Rozwojowego. (2011) ;15: (2):126–31. |
[31] | Hahnen E , Forkert R , Marke C , Rudnik-Schoneborn S , Schonling J , Zerres K , et al. Molecular analysis of candidate genes on chromosome 5q13 in autosomal recessive spinal muscular atrophy: Evidence of homozygous deletions of the SMN gene in unaffected individuals. Human Molecular Genetics. (1995) ;4: (10):1927–33. |
[32] | Riessland M , Kaczmarek A , Schneider S , Swoboda KJ , Lohr H , Bradler C , et al. Neurocalcin delta suppression protects against spinal muscular atrophy in humans and across species by restoring impaired endocytosis. American Journal of Human Genetics. (2017) ;100: (2):297–315. |
[33] | Wang CH , Xu J , Carter TA , Ross BM , Dominski MK , Bellcross CA , et al. Characterization of survival motor neuron (SMNT) gene deletions in asymptomatic carriers of spinal muscular atrophy. Human Molecular Genetics. (1996) ;5: (3):359–65. |
[34] | Cobben JM , van der Steege G , Grootscholten P , de Visser M , Scheffer H , Buys CH . Deletions of the survival motor neuron gene in unaffected siblings of patients with spinal muscular atrophy. American Journal of Human Genetics. (1995) ;57: (4):805–8. |
[35] | Oprea GE , Krober S , McWhorter ML , Rossoll W , Muller S , Krawczak M , et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. (2008) ;320: (5875):524–7. |
[36] | Heesen L , Peitz M , Torres-Benito L , Holker I , Hupperich K , Dobrindt K , et al. Plastin 3 is upregulated in iPSC-derived motoneurons from asymptomatic SMN1-deleted individuals. Cell Mol Life Sci. (2016) ;73: (10):2089–104. |
[37] | Feldkotter M , Schwarzer V , Wirth R , Wienker TF , Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. American Journal of Human Genetics. (2002) ;70: (2):358–68. |
[38] | Jedrzejowska M , Milewski M , Zimowski J , Zagozdzon P , Kostera-Pruszczyk A , Borkowska J , et al. Incidence of spinal muscular atrophy in Poland–more frequent than predicted? Neuroepidemiology. (2010) ;34: (3):152–7. |
[39] | Wirth B . An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Human Mutation. (2000) ;15: (3):228–37. |
[40] | Chien YH , Chiang SC , Weng WC , Lee NC , Lin CJ , Hsieh WS , et al. Presymptomatic diagnosis of spinal muscular atrophy through newborn screening. The Journal of Pediatrics. (2017) . |
[41] | Kraszewski JN , Kay DM , Stevens CF , Koval C , Haser B , Ortiz V , et al. Pilot study of population-based newborn screening for spinal muscular atrophy in New York state. Genet Med. (2018) ;20: (6):608–13. |
[42] | Calucho M , Bernal S , Alias L , March F , Vencesla A , Rodriguez-Alvarez FJ , et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of reported cases. Neuromuscular Disorders: NMD. (2018) ;28: (3):208–15. |
[43] | Prior TW , Krainer AR , Hua Y , Swoboda KJ , Snyder PC , Bridgeman SJ , et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. American Journal of Human Genetics. (2009) ;85: (3):408–13. |
[44] | Wirth B , Brichta L , Schrank B , Lochmuller H , Blick S , Baasner A , et al. Mildly affected patients with spinal muscular atrophy are partially protected by an increased SMN2 copy number. Hum Genet. (2006) ;119: (4):422–8. |
[45] | Cusco I , Barcelo MJ , Rojas-Garcia R , Illa I , Gamez J , Cervera C , et al. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. Journal of Neurology. (2006) ;253: (1):21–5. |
[46] | Ogino S , Gao S , Leonard DG , Paessler M , Wilson RB . Inverse correlation between SMN1 and SMN2 copy numbers: Evidence for gene conversion from SMN2 to SMN1. European Journal of Human Genetics: EJHG. (2003) ;11: (3):275–7. |
[47] | Schorling DC , Becker J , Pechmann A , Langer T , Wirth B , Kirschner J . Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology. (2019) ;93: (6):267–9. |
[48] | Finkel RS , Chiriboga CA , Vajsar J , Day JW , Montes J , De Vivo DC , et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet (London, England). (2016) ;388: (10063):3017–26. |
[49] | Pane M , Palermo C , Messina S , Sansone VA , Bruno C , Catteruccia M , et al. Nusinersen in type 1 SMA infants, children and young adults: Preliminary results on motor function. Neuromuscular Disorders: NMD. (2018) ;28: (7):582–5. |
[50] | Verde F , Steinacker P , Weishaupt JH , Kassubek J , Oeckl P , Halbgebauer S , et al. Neurofilament light chain in serum for the diagnosis of amyotrophic lateral sclerosis. Journal of Neurology, Neurosurgery, and Psychiatry. (2019) ;90: (2):157–64. |
[51] | Lin CW , Kalb SJ , Yeh WS . Delay in diagnosis of spinal muscular atrophy: A systematic literature review. Pediatric Neurology. (2015) . |
[52] | Kariya S , Obis T , Garone C , Akay T , Sera F , Iwata S , et al. Requirement of enhanced Survival Motoneuron protein imposed during neuromuscular junction maturation. The Journal of Clinical Investigation. (2014) ;124: (2):785–800. |
[53] | Wolf B . Successful outcomes of older adolescents and adults with profound biotinidase deficiency identified by newborn screening. Genet Med. (2017) ;19: (4):396–402. |
[54] | Mlcoch T , Puda R , Jesina P , Lhotakova M , Sterbova S , Dolezal T . Dietary patterns, cost and compliance with low-protein diet of phenylketonuria and other inherited metabolic diseases. Eur J Clin Nutr. (2018) ;72: (1):87–92. |
[55] | Scoto M , Finkel RS , Mercuri E , Muntoni F . Therapeutic approaches for spinal muscular atrophy (SMA). Gene Therapy. (2017) ;24: (9):514–9. |


