
Eteplirsen Treatment Attenuates Respiratory Decline in Ambulatory and Non-Ambulatory Patients with Duchenne Muscular Dystrophy
Abstract
Background:
Duchenne muscular dystrophy (DMD) patients experience skeletal muscle degeneration, including respiratory muscles. Respiratory decline in glucocorticoid-treated DMD patients, measured by percent predicted forced vital capacity (FVC% p), is typically 5% annually in patients aged 10 to 18 years.
Objective:
Evaluate the effects of eteplirsen on FVC% p annual change in 3 trials versus matched Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS) controls.
Methods:
Eteplirsen studies 201/202 evaluated eligible ambulatory DMD patients for at least 4 years, study 204 evaluated primarily non-ambulatory DMD patients for 2 years, and ongoing study 301 is evaluating ambulatory DMD patients for 2 years (interim analysis is included). Eteplirsen-treated patients (n = 74) were amenable to exon 51 skipping and were receiving glucocorticoids. Three CINRG DNHS cohorts included: glucocorticoid-treated patients amenable to exon 51 skipping (Exon 51 CINRG DNHS; n = 20), all glucocorticoid-treated CINRG patients (All CINRG DNHS; n = 172), and all glucocorticoid-treated genotyped CINRG DNHS patients (Genotyped CINRG DNHS; n = 148). FVC% p assessments between ages 10 and <18 years were included for all patients; mixed-model analyses characterized FVC% p annual change.
Results:
FVC% p annual change was greater for CINRG DNHS Exon 51 controls (– 6.00) versus patients in studies 201/202, study 204, and study 301 (– 2.19, P < 0.001; – 3.66, P 0.004; and – 3.79, P 0.017, respectively). FVC% p annual change in all eteplirsen studies suggested treatment benefit compared with the Genotyped CINRG DNHS (– 5.67) and All CINRG DNHS (– 5.56) cohorts (P < 0.05, all comparisons).
Conclusions:
Significant, clinically meaningful attenuation of FVC%p decline was observed in eteplirsen-treated patients versus CINRG DNHS controls.
INTRODUCTION
Duchenne muscular dystrophy (DMD) is an irreversible, progressive neuromuscular disease caused by out-of-frame mutations in the DMD gene [1–4]. As dystrophin is a critical structural protein in the sarcolemma of myocytes, near absence of dystrophin results in inflammation, necrosis, and eventual replacement of functional muscle tissue with fibrosis and fat [5]. All skeletal muscles are affected, including the respiratory muscles [6]. Consequently, progressive respiratory decline is associated with increased morbidity and mortality in patients with DMD [2, 7–9].
Respiratory decline in DMD patients can be assessed using several measures, including forced vital capacity (FVC), the total amount of air that can be moved through the lungs after a maximal inspiration followed by exhalation. In turn, this can be compared with the predicted FVC for a patient with a similar given age and physical characteristics to generate the percent predicted FVC (FVC% p). In untreated patients with DMD, the FVC% p decline begins early, typically between the ages of 7 and 10 years; glucocorticoid treatment slightly delays the onset of this decline [7, 10]. However, between the ages of 10 and 18 years, the annual FVC% p declines linearly at a clinically significant rate of approximately 5% per year, regardless of glucocorticoid treatment [1, 10–13]. This steady decline requires increased levels of clinical intervention as the FVC% p drops below critical thresholds and as reduced respiratory function notably impacts quality of life [14, 15]. It follows that reducing the annual rate of FVC% p decline to less than 5% may meaningfully extend time to critical milestones of morbidity and mortality.
Eteplirsen is a phosphorodiamidate morpholino oligomer designed to restore out-of-frame DMD gene mutations and produce internally truncated dystrophin protein in patients amenable to exon 51 skipping, estimated to be 13% to 14% of DMD patients [16, 17]. The promise of this approach is evident in the remarkably milder DMD phenotype in patients with mutations amenable to exon 44 skipping [18, 19]. These patients experience natural endogenous exon skipping and produce small levels of internally truncated dystrophin, attenuating the rate of functional decline (in some cases by many years) compared with other DMD patients.
The long-term effect of eteplirsen on a number of pharmacodynamic and functional outcomes has been described previously [20]. In addition, the long-term effect of eteplirsen on respiratory function has been previously described compared with natural history and an external control group [21]. However, more recent literature characterizes the linear respiratory decline with glucocorticoid treatment administration between the ages of 10 and 18 years in patients with DMD, and is the basis for study designs of current clinical trials evaluating effect of therapeutics in DMD on pulmonary decline (eg, clinicaltrials.gov identifier NCT02814019) and the rationale for this new analysis [7, 10]. The impact of eteplirsen treatment on FVC% p decline during the expected linear respiratory decline phase (ages 10 to 18 years) was assessed in 74 patients across 3 studies compared with 3 matched cohorts of patient-level natural history data from the Cooperative International Neuromuscular Research Group Duchenne Natural History Study (CINRG DNHS). Consecutive open-label studies 201 and 202 were previously described [20] and were conducted over a total of 4 years, with an initial 24-week placebo-controlled period. Parameters of respiratory function, including FVC% p, were assessed as exploratory endpoints. All patients (N = 12) in consecutive studies 201/202 received eteplirsen for at least 216 weeks and had FVC% p assessments between the ages of 10 and 18 years. Study 204 was a 2-year, open-label study in minimally ambulatory and non-ambulatory patients with DMD (N = 24), and 20 of these patients aged 10 to 18 years had longitudinal FVC% p data. Study 301 is an ongoing phase 3, open-label, 2-year study, with an interim analysis presented here for patients aged 10 to 18 years with FVC% p assessments (n = 42).
MATERIALS AND METHODS
Eteplirsen-treated Patients
Study 201 (NCT01396239), a single-center, randomized, double-blind, placebo-controlled study, enrolled male patients aged 7 to 13 years with a diagnosis of DMD and DMD gene mutations amenable to exon 51 skipping. Patients had stable respiratory function (FVC% p≥50%), were receiving treatment with oral glucocorticoids (a stable dose for≥24 weeks before study entry), and completed an average distance on the 6-minute walk test (6MWT) between 180 and 440 meters. Patients were randomly assigned to 1 of 3 treatment groups (eteplirsen 30 mg/kg, eteplirsen 50 mg/kg, or placebo). Patients randomized to eteplirsen received a once weekly intravenous (IV) infusion for 28 consecutive weeks. Patients randomized to the placebo group received placebo for the first 24 weeks then were re-randomized to receive either eteplirsen 30 mg/kg or 50 mg/kg for the last 4 weeks of study 201 and continued to receive treatment for the duration of study 202.
Study 202 (NCT01540409) was an open-label, multiple dose extension study designed to assess the ongoing efficacy, safety, and tolerability of weekly IV infusions of eteplirsen. Patients received the same dose of eteplirsen they had received at week 28 in study 201 (eteplirsen 30 mg/kg or 50 mg/kg).
Six patients in studies 201/202 received each dose of eteplirsen and no differences in dystrophin expression were observed across the doses; thus, patients receiving once-weekly eteplirsen 30 mg/kg and 50 mg/kg were grouped together into a 12-patient cohort. Respiratory function tests (including FVC% p) were conducted while patients were on eteplirsen therapy, at least every 24 weeks through 216 weeks, for a total duration of at least 4 years (Fig. 1, Table 1).
Fig.1
Study design for eteplirsen studies 201/202. Study 201 was a 24-week, double-blind, placebo-controlled study that randomized 12 patients to receive placebo or eteplirsen (30 or 50 mg/kg/wk). At week 25, patients originally randomized to eteplirsen treatment continued with the same dosage in study 202, an open-label extension study; patients originally randomized to receive placebo were randomized to eteplirsen 30 or 50 mg/kg. All patients were followed for 4 years. Respiratory function assessments included FVC% p as an exploratory endpoint. FVC% p, percent predicated forced vital capacity; OL, open-label; PTP, primary treatment period; R, randomized.
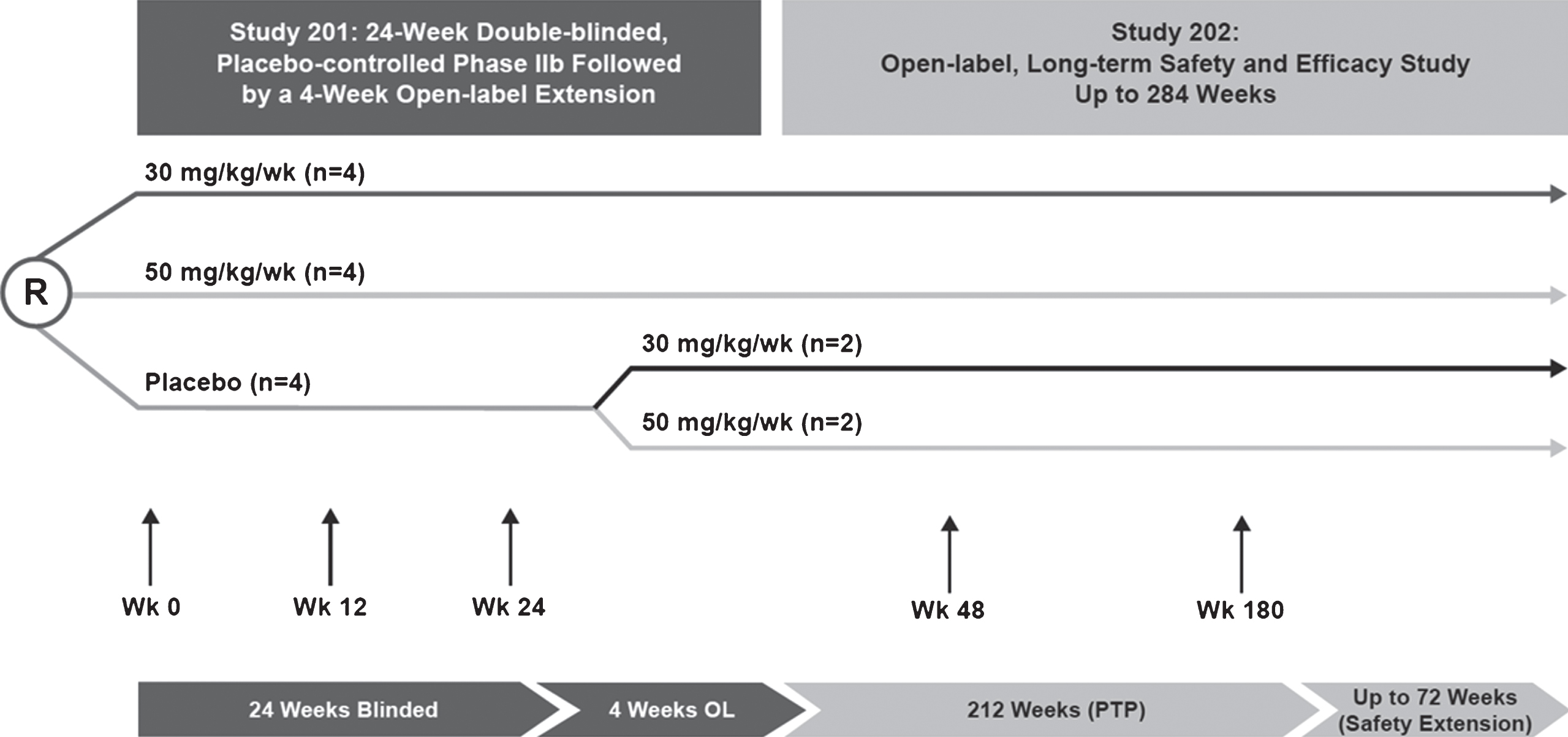
Table 1
Comparison of the key enrollment criteria by study
| Parameter | Studies 201/202 | Study 204 | Study 301 | CINRG DNHS |
| Age | 7–13 years, inclusive | 7–21 years | 7–16 years, inclusive | 2–28 years |
| Genotype | Male with DMD out-of-frame deletion(s) that may be corrected by skipping exon 51 | Male with DMD mutation that may be amenable to exon 51 skipping | Male with DMD out-of-frame deletion that may be treated by exon 51 skipping | 2 to <5 years: + gene deletion or dystrophin gene sequencing expected to preclude production of dystrophin≥5 to <29 years: above criteria or documented clinical DMD symptoms |
| Walking ability | Baseline 6MWT within 180 m and 440 m | Baseline non-ambulatory, or 6MWT < 300 m | Baseline 6MWT≥300 m | None |
| Glucocorticoids | Stable dose of oral glucocorticoids for at least 24 weeks before study entry | Stable dose of oral glucocorticoids or no corticosteroids for at least 24 weeks prior to study drug administration | Stable dose of oral glucocorticoids prior to entry to study | None |
| Respiratory function | Stable respiratory function (FVC≥50% of predicted) | Respiratory function unlikely to decompensate over study period | Stable respiratory function (FVC% p≥50%) | None |
CINRG DNHS, Cooperative International Neuromuscular Research Group Duchenne Natural History Study; 6MWT, 6-Minute Walk Test; DMD, Duchenne muscular dystrophy; FVC% p, percent predicted forced vital capacity.
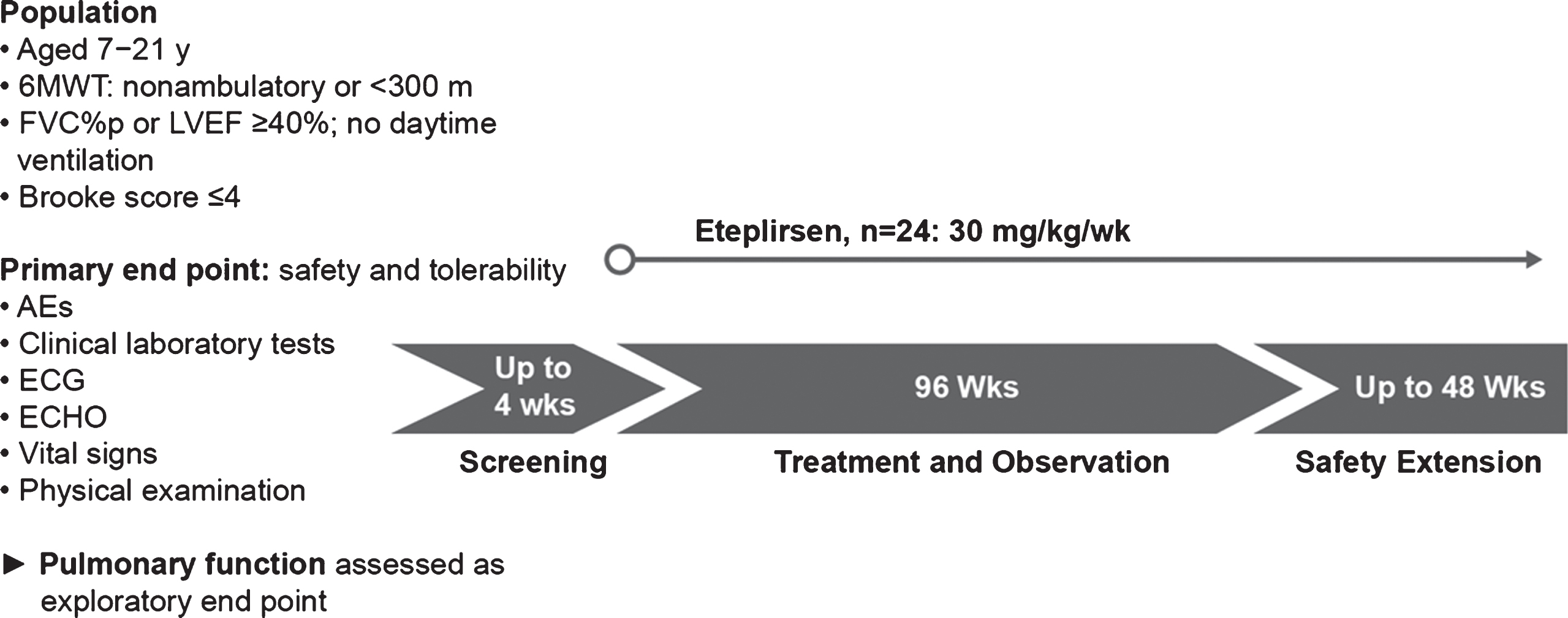
Study 204 (NCT02286947), an open-label, multicenter study, was designed to evaluate the safety and tolerability of eteplirsen treatment in boys aged 7 to 21 years with a diagnosis of DMD and DMD gene mutation(s) amenable to exon 51 skipping. All patients were taking a stable dose of oral glucocorticoids or had not received glucocorticoids for at least 24 weeks prior to study drug administration, were minimally ambulatory or non-ambulatory (defined as taking longer than 30 seconds to independently walk 10 meters or being incapable of walking≥300 meters on the 6MWT), had stable cardiac and respiratory function, and had a score of 1 to 4 on the Brooke Upper Extremity Scale [22] (able to reach fully overhead, reach with compensation to the scalp, or bring their hand to the mouth, with or without a 240-g object). Eligible patients received once-weekly IV infusions of eteplirsen 30 mg/kg for 96 weeks, followed by a safety extension period. Respiratory function tests (including FVC% p) were assessed every 12 weeks for the first year of the study, and every 24 weeks thereafter through 96 weeks, for a total duration of 2 years (Fig. 2, Table 1). Regardless of ambulatory status, patient height was estimated based on ulnar length [23].
Fig.2
Study design for eteplirsen study 204. In this 2-year (96-week), open-label study, patients with advanced-stage DMD (minimally ambulatory to non-ambulatory patients; N = 24) received weekly infusions of eteplirsen 30 mg/kg. Respiratory function assessments included FVC% p as an exploratory endpoint. AEs, adverse events; ECG, electrocardiogram; FVC% p, percent predicated forced vital capacity; ECHO, echocardiogram; LVEF, left ventricular ejection fraction.
Study 301 (NCT02286947) is an ongoing, phase 3, open-label, multicenter study designed to evaluate the efficacy and safety of eteplirsen in boys aged 7 to 16 years with a diagnosis of DMD and DMD gene mutation(s) amenable to exon 51 skipping. Patients were taking a stable dose of oral glucocorticoids for at least 24 weeks prior to study drug administration, had a 6MWT distance of at least 300 meters, and had respiratory function above the recently recommended threshold for assessment of nocturnal ventilation (FVC% p≥50%) [15]. Eligible patients receive once-weekly IV infusions of eteplirsen 30 mg/kg for 96 weeks, followed by a safety extension. FVC% p is assessed every 12 weeks for the first year and every 24 weeks thereafter, through 96 weeks, for a total duration of up to 2 years in the interim analysis (Fig. 3, Table 1).
Fig.3
Study design for eteplirsen study 301. In this open-label study, patients with DMD (N = 71) received weekly IV infusions of eteplirsen 30 mg/kg and had both baseline and post-baseline FVC% p. An interim analysis was conducted at year 2 (96 weeks) for all patients who reached that time point. Respiratory function assessments included FVC% p as an exploratory endpoint. BL, baseline; FVC% p, percent predicted forced vital capacity.

Natural History Patients
The CINRG DNHS, one of the largest prospective natural history studies of DMD conducted to date [8], comprises more than 400 DMD patients with complete characterization of demographic data, along with assessments of clinical parameters affected by DMD [7, 8, 10, 24–26].
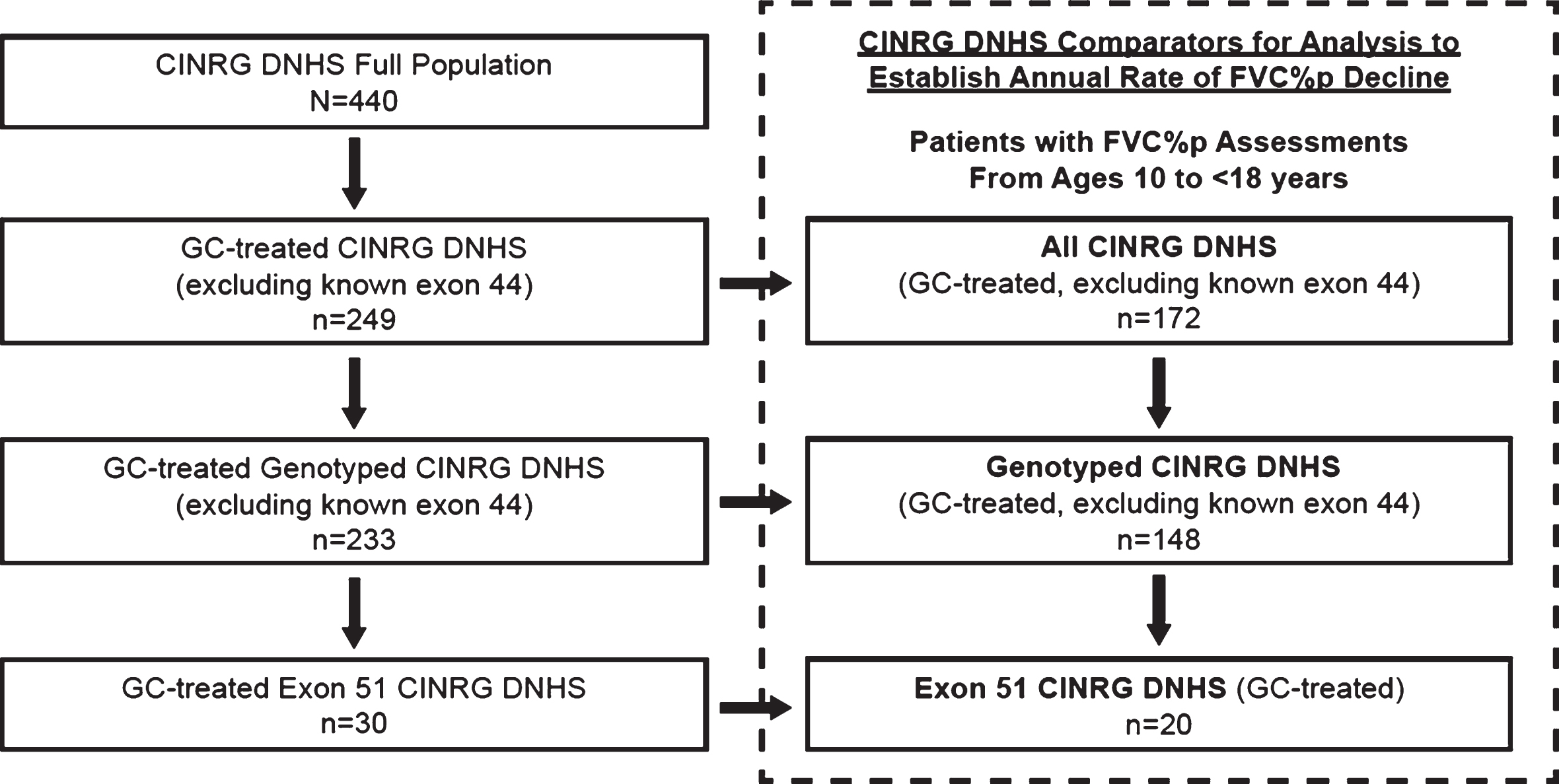
CINRG DNHS patient-level data [24] were obtained for use as the natural history comparators. Upon entry into the natural history study, assessments of respiratory function (including FVC% p) were conducted every 3 months for 1 year, then at 18 and 24 months, and annually thereafter. Subsets of patients from the CINRG DNHS were identified as external controls for comparison with eteplirsen-treated patients. The method for identification and comparison with patients from CINRG DNHS was pre-specified in a statistical analysis plan (SAP; see Supplemental Materials). Although the comparators would ideally be a matched genotype (amenable to exon 51 skipping), at the time of the SAP completion, it was not known whether there would be a sufficient number of exon 51 patients. As such, 3 CINRG DNHS cohorts were predefined based on genetic testing for DMD (Fig. 4). These 3 cohorts included: (1) all glucocorticoid-treated CINRG DNHS patients aged 10 to <18 years, regardless of whether DMD was confirmed by genetic testing (All CINRG DNHS cohort; n = 172); (2) all glucocorticoid-treated, genotyped CINRG DNHS patients aged 10 to <18 years with genetically confirmed DMD (Genotyped CINRG DNHS; n = 148); and (3) glucocorticoid-treated CINRG DNHS patients aged 10 to <18 years with mutations amenable to skipping exon 51 (Exon 51 CINRG DNHS; n = 20). Patients known to be amenable to exon 44 skipping were excluded from all 3 cohorts, as it has been established that they have a phenotypically milder disease [18, 19]. Key inclusion criteria for the eteplirsen studies and for the CINRG DNHS study are summarized in Table 1.
Fig.4
Derivation of CINRG DNHS cohorts for comparison of eteplirsen studies. The CINRG DNHS database contains data from 440 patients with DMD across all ages. The All CINRG DNHS cohort (n = 172) comprised glucocorticoid-treated patients with FVC% p assessments obtained between the ages of 10 and 18 years, and excluded any known exon 44 skip–amenable patients. The Genotyped CINRG DNHS cohort (n = 148) comprised glucocorticoid-treated, genetically confirmed patients with FVC% p assessments between the ages of 10 and 18 years and excluded exon 44 skip-amenable patients. The Exon 51 CINRG DNHS cohort (n = 20) comprised glucocorticoid-treated patients between the ages of 10 and 18 years with mutations amenable to exon 51 skipping. CINRG DNHS, Cooperative International Neuromuscular Research Group Duchenne Natural History Study; FVC% p, percent predicted forced vital capacity; GC, glucocorticoid. Dashed box represents pre-specified comparator.
In all 3 CINRG DNHS cohorts, patients were required to be receiving glucocorticoids. If a patient was using glucocorticoids intermittently, then only those FVC% p assessments collected during the interval in which the patient was receiving glucocorticoid therapy were used.
Conversion of FVC to FVC% p
Forced vital capacity typically increases with normal growth until attainment of adult stature, typically in the mid-to-late teens. As such, age, height, and ethnicity all influence this measure, and normalizing FVC performance for those characteristics yields an FVC% p [27]. Different equations can be used to normalize FVC, resulting in slightly different FVC% p values for the same FVC [27, 28]. As such, the same equation (NHANES III) was used for normalization of FVC to FVC% p for CINRG DNHS and eteplirsen-treated cohorts in studies 204 and 301, with height calculated using ulnar lengths for all patients owing to lack of standing ability in non-ambulatory patients [10, 23, 24]. However, FVC% p was calculated differently for studies 201/202, as these earlier studies evaluated ambulatory patients treated with eteplirsen. As reported on the case report form, the methodology employed was that described by Polgar and Promadhat [28] (height was measured using standing height, and ulnar lengths were not generally collected).
Statistical Analyses
All eteplirsen study patients who received at least 1 dose of eteplirsen of at least 30 mg/kg, and had both a baseline and at least 1 post-baseline FVC% p value were included in the analyses. For patients in studies 201/202 originally randomized to placebo, who began open-label eteplirsen treatment at week 25, FVC% p values at baseline and during eteplirsen treatment were included. For these placebo-to-eteplirsen patients, week 24 FVC% p values were used as baseline values to ensure that each patient had just one untreated baseline value, and all subsequent assessments of FVC% p were taken while patients were on eteplirsen treatment. All CINRG DNHS cohort patients who had at least 2 FVC% p values were included in the analyses.
As the linear decline phase of respiratory function occurs between 10 and 18 years of age [1, 10–13], with a negligible decline seen prior to the age of 10 years in glucocorticoid-treated patients with DMD [7, 10], the analysis presented includes patients aged 10 to <18 years who were in the expected linear decline phase of the disease.
The annual rate of FVC% p change was estimated and compared between eteplirsen-treated and CINRG DNHS cohorts using mixed-model for repeated measures (MMRM) analyses. The MMRM fit FVC% p as the response variable, treatment group (eteplirsen vs CINRG DNHS), age (at visit), and treatment group by age interaction as the fixed effects, and patient as a random effect. To account for potential differences in variability within the eteplirsen and CINRG DNHS groups, the patient variance component and the residual variance were estimated for each treatment group. The coefficient for age (ie, the slope of FVC% p versus age) for each treatment group was interpreted as the annual rate of FVC% p change for each treatment group. This analysis method is an extension to two treatment groups of the statistical method for one treatment group described by McDonald et al. [10] for CINRG DNHS. Each of the 3 eteplirsen studies was compared with each of the 3 CINRG DNHS cohorts.
For each of the MMRM analyses comparing eteplirsen studies with CINRG DNHS cohorts, the numbers of patients and observations, the estimated annual rate of FVC% p change, and the corresponding 95% confidence interval (CI) were computed for each treatment group. Additionally, for each comparison, the estimated difference in annual rates of FVC% p change between the 2 treatment groups, the associated 95% CI, and the nominal P value to test the equality of the annual rates of FVC% p change between the 2 treatment groups were computed. The P values were considered nominal and descriptive; thus, no adjustments were made for multiple testing.
Two sensitivity analyses were performed to assess the robustness of the primary analysis for each eteplirsen study vs CINRG DNHS comparator. The first sensitivity analysis was an MMRM with random slopes for individual patients. The MMRM fit FVC% p as the response variable; fit treatment group, age (at visit), and treatment group by age interaction as the fixed effects; and included a random intercept and random slope for age for each patient. The second sensitivity analysis was the same as the primary analysis except it included baseline height as a covariate. This MMRM fit FVC% p as the response variable; fit treatment group, age (at visit), treatment group by age interaction, and baseline height as the fixed effects; and included patient as a random effect.
In an alternative analysis that included all patients in study 204, regardless of age, the 2-year change from baseline in FVC% p was assessed using descriptive statistics (mean and standard error [SE]) for all (N = 24) study 204 patients, regardless of age. The mean annual change was derived from this value to determine if this was in line with the MMRM analysis.
RESULTS
Demographics and baseline characteristics for the eteplirsen-treated patients across the 4 studies and for the 3 CINRG DNHS natural history cohorts are presented in Table 2 (baseline and last visit characteristics are compared in Supplemental Table S1). The eteplirsen study populations differed between studies 201/202 and 301 compared with study 204 in that the population of study 204 was older, primarily non-ambulatory, and had a greater degree of disease progression (as measured by FVC% p; Table 2). This is consistent with the differences in inclusion criteria for the studies (Table 1). In general, mean values of the baseline demographic and baseline characteristics of all 3 CINRG DNHS cohorts were within the range of values across the eteplirsen studies. Notably, the Exon 51 CINRG DNHS cohort at baseline consisted mainly of ambulatory patients (17/20; 85%) with a median age of 10.4 years, which is most similar to the population of ambulatory patients from eteplirsen studies 201/202 and 301.
Table 2
Eteplirsen-treated patients and CINRG DNHS controls: demographics and other variables at baseline
| Parameter | Patients Aged 10 to < 18 Years | Studies 201/202 (N = 12) | Study 204 (N = 20) | Study 301 (N = 42) | Exon 51 CINRG DNHS (n = 20) | Genotyped CINRG DNHS (n = 148) | All CINRG DNHS (N = 172) |
| Race, n (%) | White | 11 (92) | 19 (95) | 35 (83) | 16 (80) | 116 (78) | 134 (78) |
| Asian | 1 (8) | 1 (5) | 2 (5) | 3 (15) | 19 (13) | 24 (14) | |
| Black | 0 | 0 | 1 (2) | 0 | 2 (1) | 2 (1) | |
| Native Hawaiian or other Pacific Islander | 0 | 0 | 2 (5) | 0 | 2 (1) | 2 (1) | |
| Other | 0 | 0 | 2 (5) | 1 (5) | 9 (6) | 10 (6) | |
| Ethnicity, n (%) | Hispanic or Latino | 0 | 1 (5) | 4 (10) | 1 (5) | 20 (14) | 23 (13) |
| Not Hispanic or Latino | 12 (100) | 18 (90) | 37 (88) | 19 (95) | 128 (86) | 149 (87) | |
| Not reported | 0 | 1 (5) | 0 | 0 | 0 | 0 | |
| Unknown | 0 | 0 | 1 (2) | 0 | 0 | 0 | |
| Age at baseline, years | Mean (SD) | 10.32 (0.301) | 13.04 (2.276) | 11.07 (1.440) | 11.78 (2.235) | 11.73 (1.971) | 11.65 (1.909) |
| Median | 10.24 | 12.97 | 10.37 | 10.40 | 10.86 | 10.83 | |
| Min, max | 10.0, 11.0 | 10.0, 17.3 | 10.0, 16.3 | 10.0, 17.9 | 10.0, 17.9 | 10.0, 17.9 | |
| Standing height at baseline, cm | n | 11 | 8 | 39 | 13 | 87 | 106 |
| Mean (SD) | 126.12 (7.596) | 127.30 (11.286) | 130.46 (7.244) | 131.45 (7.604) | 130.54 (8.381) | 129.88 (8.195) | |
| Median | 124.90 | 126.50 | 130.70 | 131.60 | 130.00 | 129.75 | |
| Min, max | 116.0, 140.5 | 113.6, 150.0 | 116.7, 148.5 | 119.0, 145.0 | 114.0, 157.3 | 114.0, 157.3 | |
| Calculated height at baseline, cm | n | 0 | 20 | 41 | 20 | 148 | 172 |
| Mean (SD) | 141.67 (10.742) | 137.74 (7.891) | 140.76 (12.063) | 139.76 (10.595) | 139.21 (10.411) | ||
| Median | 138.94 | 138.97 | 140.65 | 138.25 | 138.20 | ||
| Min, max | 126.7, 158.5 | 122.4, 155.2 | 124.0, 178.1 | 120.4, 178.1 | 120.4, 178.1 | ||
| Weight at baseline, kg | Mean (SD) | 35.78 (6.510) | 48.71 (12.620) | 39.83 (11.159) | 38.87 (14.783) | 40.03 (12.602) | 39.96 (12.348) |
| Median | 34.75 | 48.50 | 39.30 | 32.25 | 37.35 | 37.50 | |
| Min, max | 27.9, 46.0 | 25.6, 69.6 | 22.2, 67.2 | 23.5, 79.1 | 18.0, 79.5 | 18.0, 79.5 | |
| Ambulatory status at baseline, n (%) | Not ambulatory | 0 | 15 (75) | 0 | 3 (15) | 49 (33) | 52 (30%) |
| Ambulatory | 12 (100) | 5 (25) | 42 (100) | 17 (85) | 99 (67) | 120 (70) | |
| FVC% p at baseline | Mean (SD) | 96.92 (14.042) | 65.94 (16.601) | 78.48 (15.691) | 79.60 (13.300) | 78.81 (19.497) | 78.45 (19.245) |
| Median | 92.00 | 72.25 | 80.31 | 81.00 | 79.00 | 78.00 | |
| Min, max | 84.0, 121.0 | 31.4, 89.7 | 52.6, 127.0 | 50.0, 106.0 | 14.0, 140.0 | 14.0, 140.0 |
CINRG DNHS, Cooperative International Neuromuscular Research Group Duchenne Natural History Study; DMD, Duchenne muscular dystrophy; FVC% p, percent predicted forced vital capacity; max, maximum; min, minimum; SD, standard deviation.
At the completion of studies 201/202, the 12 patients had 132 FVC% p assessments conducted in the age group of 10 to 18 years. Eteplirsen-treated patients had a statistically significant attenuation of the annual rate of FVC% p decline of 2.19% compared with 6.00% for the Exon 51 CINRG DNHS cohort (P < 0.001; Table 3). Comparisons with the broader CINRG DNHS cohorts (Genotyped and All CINRG DNHS) were consistent with this result (P < 0.001 for both comparisons).
Table 3
FVC% p slope model: studies 201/202, 204, and 301 versus the CINRG DNHS cohort
| Exon 51 CINRG DNHS (N = 20) | Genotyped CINRG DNHS (N = 148) | ALL CINRG DNHS (N = 172) | ||||
| Number of observations | 88 | 707 | 803 | |||
| FVC% p annual change | – 6.00 (0.408) | – 5.67 (0.188) | – 5.56 (0.179) | |||
| 95% CI | – 6.80, – 5.19 | – 6.04, – 5.30 | – 5.91, – 5.21 | |||
| Studies 201/202 | Number of observations | 132 | ||||
| FVC% p annual change | – 2.19 (0.710) | Difference in FVC% p annual change (SE) | 3.80 (0.819) | 3.48 (0.735) | 3.37 (0.733) | |
| 95% CI | – 3.60, – 0.79 | 95% CI | 2.19, 5.42 | 2.03, 4.92 | 1.93, 4.80 | |
| P value | <0.001 | <0.001 | <0.001 | |||
| Study 204 | Number of observations | 117 | ||||
| FVC% p annual change | – 3.66 (0.680) | Difference in FVC% p annual change (SE) | 2.34 (0.793) | 2.01 (0.706) | 1.90 (0.703) | |
| 95% CI | – 5.00, – 2.32 | 95% CI | 0.77, 3.90 | 0.62, 3.40 | 0.52, 3.28 | |
| P value | 0.004 | 0.005 | 0.007 | |||
| Study 301 | Number of observations | 184 | ||||
| FVC% p annual change | – 3.79 (0.824) | Difference in FVC% p annual change (SE) | 2.21 (0.919) | 1.89 (0.845) | 1.77 (0.843) | |
| 95% CI | – 5.41, – 2.16 | 95% CI | 0.40, 4.02 | 0.23, 3.54 | 0.12, 3.43 | |
| P value | 0.017 | 0.026 | 0.036 |
CI, confidence interval; CINRG DNHS, Cooperative International Neuromuscular Research Group Duchenne Natural History Study; FVC% p, percent predicted forced vital capacity; SE, standard error.
At the completion of Study 204, 20 patients had 117 FVC% p assessments conducted in the age group of 10 to 18 years. Eteplirsen-treated patients had a statistically significant attenuation of the annual rate of FVC% p decline of 3.66% compared with 6.00% for the Exon 51 CINRG DNHS cohort (P 0.004; Table 3). Again, comparisons with the broader CINRG DNHS cohorts (Genotyped and All CINRG DNHS) were consistent with this result (P 0.005 and P 0.007, respectively). In the alternative analysis that included all patients in study 204, regardless of age, the mean baseline FVC% p (SE) for all study 204 patients (N = 24) was 65.53% (3.365); this declined over the 2-year course of the study to 58.16% (3.847; n = 23, as one patient did not have an assessment at year 2).
At the time of the study 301 interim analysis, 42 patients had 184 FVC% p assessments conducted in the age group of 10 to 18 years. Eteplirsen-treated patients had a statistically significant attenuation of the annual rate of FVC% p decline of 3.79% compared with 6.00% for the Exon 51 CINRG DNHS cohort (P 0.017; Table 3). As seen in studies 201/202 and 204, comparisons of eteplirsen-treated patients in the study 301 interim data with the broader CINRG DNHS comparators (Genotyped and All CINRG DNHS) were consistent with the comparison to Exon 51 CINRG DNHS (P 0.026 and P 0.036).
The sensitivity analyses conducted both for studies 201/202 and for study 204 confirmed that the eteplirsen-treated patients had a significant attenuation in the annual rate of FVC% p decline compared with the Exon 51 CINRG DNHS cohort (studies 201/202: P < 0.001 for both analyses; study 204: P 0.009 and 0.027, respectively), supporting the robustness of the primary analysis (Supplemental Tables S2–S5). Similarly, the baseline height sensitivity analysis for study 301 also confirmed that the eteplirsen-treated patients had an attenuation in the annual rate of FVC% p decline compared with the Exon 51 CINRG DNHS cohort (P 0.018); however, the results of the random slope effect sensitivity analysis for study 301 approached, but did not achieve statistical significance (P 0.058) (Supplemental Tables S6 and S7).
DISCUSSION
Patients with DMD experience predictable, progressive loss of functions, ultimately leading to premature death most commonly due to respiratory or cardiac complications [8, 10, 29, 30]. Decline in respiratory function begins in the first decade of life as respiratory muscles deteriorate along with other skeletal muscles. While glucocorticoid treatment effectively delays the onset of decline initially, it does not slow the rate of decline in respiratory function between the ages of 10 and 18 years compared with patients not taking glucocorticoids [7].
Slowing the rate of respiratory decline in this age range is particularly important, as critical milestones associated with increased morbidity are typically experienced during this time period [15]. For example, declining below FVC% p of 80% is associated with prolonged respiratory illness. As FVC% p drops below 60%, daily cough assistance and deep lung inflation techniques are recommended. At FVC% p of 50%, the recent 2018 Duchenne Care Considerations [15, 31, 32] recommend initiation of noninvasive nocturnal ventilation, and sleep studies are recommended to assess the presence of nocturnal hypoventilation. As the typical annual decline in FVC% p is greater than 5% between the ages of 10 and 18 years, patients can progress rapidly between thresholds (eg, 2 years between 60% and 50% FVC% p) [1, 7, 10–13]. As such, any slowing of the rate of this progression is clinically meaningful, as time to these milestones of decline may be extended. Moreover, as respiratory function continues to decline in the later years, delaying time to established prognostic indicators of mortality, such as FVC% p less than 30% and FVC <1 L, may improve quality of life and extend the lifespan of patients with DMD [8, 10, 29].
The analyses presented here show that eteplirsen treatment in 74 patients with DMD resulted in significant slowing of respiratory decline when compared with glucocorticoid-, age-, and matched exon skipping–amenable CINRG DNHS external controls, across studies 201/202, 204, and 301. The magnitude of attenuation of FVC% p decline due to eteplirsen treatment (– 2.19 to – 3.79) versus untreated controls (– 6.00) is meaningful, and may substantially extend the time between progressive milestones of decline. Such a reduction in the rate of pulmonary decline will likely impact both the quality of life and the duration of life in patients with DMD.
The significant attenuation of FVC% p decline in eteplirsen-treated patients was observed for both ambulatory (n = 54) and primarily non-ambulatory (n = 20) patients, highlighting treatment benefit in all patients between the ages of 10 and 18, including more advanced patients who had lost ambulation. Moreover, higher levels of respiratory function are closely linked to better upper limb function in non-ambulatory patients [8, 10], as both are skeletal muscle outcomes. Thus, slowing the decline in respiratory function in non-ambulatory patients may indicate a general slowing of overall disease progression, including the slowing of upper limb function decline, and further study of the effect of eteplirsen on preservation of upper limb function is needed.
The duration of eteplirsen treatment may be linked to the magnitude of attenuation of FVC% p decline. Patients in studies 201/202 received 4 years of eteplirsen treatment and the annual decline in FVC% p was reduced to 2.19%. In contrast, in the 2-year study 204 and the interim analysis of 2-year study 301, annual declines of 3.66% and 3.79%, respectively, were observed. These findings suggest that the longer duration of eteplirsen treatment may result in progressive attenuation of muscle function decline; however, further analysis of the duration of eteplirsen treatment is warranted.
A potential limitation of this work is that a natural history study is used as a comparator rather than a randomized comparator. The differences between the clinical trial and natural history settings may have contributed to the observed disparities in pulmonary function decline. However, the 3 CINRG DNHS cohorts were selected using pre-specified criteria with respect to age range for pulmonary decline (10 to <18 years) and glucocorticoid treatment to match those of eteplirsen-treated patients, and they were contemporaneous cohorts. Further, the Exon 51 CINRG DNHS cohort and the eteplirsen-treated patients were matched for exon skipping–amenable mutations, making the natural history cohort an appropriate comparator. Comparisons of demographic and clinical characteristics between eteplirsen-treated patients and the CINRG DNHS cohort showed important differences. Eteplirsen-treated patients in studies 201/202 were younger and had higher FVC% p values than the CINRG DNHS cohort patients, while eteplirsen-treated patients in study 204 were older, with more advanced-stage disease. Interestingly, eteplirsen-treated patients from both studies showed slower decline in FVC% p than the CINRG DNHS cohort comparators. The frequency and schedule of assessments were key protocol differences between the eteplirsen studies and the CINRG DNHS natural history study, which were addressed by the slope model analyses. However, the statistical models accounted for differences in variability between the eteplirsen studies and CINRG DNHS cohorts, and statistical significance was achieved in all cases despite these potential sources of variability. Another limitation of this work is that data from an interim analysis from one of the 3 eteplirsen trials (study 301) were used for the current analysis; further analysis at study completion is planned.
In conclusion, significant and clinically meaningful attenuation of FVC% p decline was observed in eteplirsen-treated patients versus matched CINRG DNHS controls over follow-up ranging from 2 to 4 years. This treatment effect of eteplirsen was clinically meaningful and may translate to prolonged time to required mechanical airway clearance, noninvasive ventilation, reduced risk of hospitalization due to respiratory illnesses, improved quality of life, and improved survival.
COLLABORATIVE STUDY GROUP AUTHORS
Eteplirsen Study Group Investigators
Study 201: Jerry R. Mendell (Nationwide Children’s Hospital).
Study 202: Jerry R. Mendell (Nationwide Children’s Hospital); Kevin O’Brien (Miller Children’s Hospital); Ludwig Matthew Frank (Children’s Hospital of the King’s Daughter); Barry Byrne (University of Florida); Erika S. Helgerson (Osceola Medical Center); Amy Harper (Levine Children’s Hospital at the Carolinas Medical Center); Peter Heydemann (Rush University); Hoda Abdel-Hamid (Children’s Hospital of Pittsburgh – UPMC); Robert Dracker (Summerwood Pediatrics/Infusacare); Anne Connolly (Washington University School of Medicine); Linda Specht (Dartmouth – Hitchcock Medical Center); Fawn Leigh (Massachusetts General Hospital).
Study 204: Jerry Mendell (Nationwide Children’s Hospital); Katherine Matthews (University of Iowa); Susan Apkon (Seattle Children’s Hospital); Fawn Leigh (Massachusetts General Hospital); Emma Ciafaloni (University of Rochester Medical Center); Anne Connolly (Washington University School of Medicine); Craig McDonald (University of California, Davis); Perry Shieh (UCLA, Los Angeles); Kathryn Wagner (Johns Hopkins Medical).
Study 301: Jerry Mendell (Nationwide Children’s Hospital); Katherine Matthews (University of Iowa); Barry Byrne (University of Florida Health, Shands Hospital); Susan Iannaccone (The University of Texas Southwestern Medical Center - Children’s Medical Center); Susan Apkon (Seattle Children’s Hospital); Fawn Leigh (Massachusetts General Hospital); Basil Darras (Harvard Medical School, Boston Children’s Hospital); Emma Ciafaloni (University of Rochester Medical Center); Julie Parsons (Children’s Hospital Colorado); Anne Connolly (Washington University School of Medicine); Craig McDonald (University of California, Davis); Ibrahim Binalsheikh (Levine Childrens Hospital, Carolinas Medical Center); Cuixia Tian (Cincinnati Children’s Hospital Medical Center); Hoda Abdel-Hamid (Children’s Hospital of Pittsburgh – UPMC); Erika Finager (Shriners Hospital for Children); Nancy Kuntz (Ann and Robert H. Lurie Children’s Hospital of Chicago); Peter Karachunski (University of Minnesota); J. Ben Renfroe (NW FL Clinical Research Group, LLC); Russell Butterfield (University of Utah, Clinical Neurosciences Center); Richard Haas (Rady Children’s Hospital, U.C. San Diego); Linda Specht (Dartmouth – Hitchcock Medical Center); Richard Barohn (University of Kansas Clinical Research Center); John Day (Stanford University School of Medicine/Medical Center); Richard Finkel (Nemours Children’s Hospital); Michael Hehir (University of Vermont Health Network); Tim Lotze (Texas Children’s Hospital); Darryl De Vivo (Columbia University Medical Center); Perry Shieh (UCLA, Los Angeles); Kumaraswamy Sivakumar (Neuromuscular Research Center); Gihan Tennekoon (Children’s Hospital of Philadelphia); Diana Bharucha-Goebel (Children’s National Health System); Kathryn Wagner (Johns Hopkins Medical); Ashutosh Kumar (Penn State Hershey Medical Center); Mitchel Williams (Wayne State University, School of Medicine); William Bryan Burnette (Vanderbilt University Medical Center, Monroe Carell Jr. Children’s Hospital); David Wolf (Emory University); Gyula Ascadi (Connecticut Children’s Medical Center); Khema Ram Sharma (University of Miami, Miller School of Medicine); Han Phan (Emory University School of Medicine).
Cooperative International Neuromuscular Research Group (CINRG) Investigators
V. Vishwanathan, S. Chidambaranathan (Sundaram Medical Foundation and Apollo Children’s Hospital, Chennai, India); W. Douglas Biggar, Laura C. McAdam (Holland Bloorview Kids Rehab Hospital, Toronto, Ontario, Canada); Jean K. Mah (Alberta Children’s Hospital, Calgary, Alberta, Canada); Mar Tulinius (Queen Silvia Children’s Hospital, Göteborg, Sweden); Avital Cnaan, Lauren P. Morgenroth, Robert Leshner, Carolina Tesi-Rocha, Mathula Thangarajh, Tina Duong (Children’s National Medical Center, Washington DC, USA); Andrew Kornberg, Monique Ryan (Royal Children’s Hospital, Melbourne, Victoria, Australia); Yoram Nevo (Hadassah Hebrew University Hospital, Jerusalem, Israel); Alberto Dubrovsky (Instituto de Neurosciencias Fundacion Favaloro, Buenos Aires, Argentina); Paula R. Clemens, Hoda Abdel-Hamid (University of Pittsburgh and Department of Veterans Affairs, Pittsburgh, Pennsylvania, USA); Anne M. Connolly, Alan Pestronk (Washington University in St Louis, St Louis, Missouri, USA); Jean Teasley (Children’s Hospital of Virginia, Richmond, Virginia, USA); Tulio E. Bertorini (University of Tennessee, Memphis, Tennessee, USA); Richard Webster (Children’s Hospital at Westmead, Sydney, New South Wales, Australia); Hanna Kolski, (University of Alberta, Edmonton, Alberta, Canada); Nancy Kuntz, Sherilyn Driscoll, John B. Bodensteiner (Mayo Clinic, Rochester, Minnesota, USA); Jose Carlo (University of Puerto Rico, San Juan, Puerto Rico), Ksenija Gorni (University of Pavia and Niguarda Ca’ Granda Hospital, Milan, Italy); Timothy Lotze (Texas Children’s Hospital, Houston, Texas, USA); John W. Day, Peter Karachunski (University of Minnesota, Minneapolis, Minnesota, USA); Erik K. Henricson, Richard T. Abresch, Nanette C. Joyce, and Craig M. McDonald (University of California, Davis, Sacramento, California, USA).
CONFLICTS OF INTEREST
This work was supported by Sarepta Therapeutics, Inc. Editorial support was provided by Peloton Advantage, LLC, an OPEN Health Company, and was funded by Sarepta Therapeutics, Inc. N. Khan, H. Eliopolous, and L. Han were employees of Sarepta Therapeutics, Inc. at the time of this study and during manuscript preparation, and may own stock/option in the company. T. B. Kinane, C. McDonald, E. Henricson, L. P. Lowes, and J. R. Mendell serve as remunerated consultants to Sarepta Therapeutics, Inc. H. Gordish-Dressman serves as a remunerated consultant for Agada Biosciences and Solid GT. No author received an honorarium for participation in this project.
Employees of Sarepta Therapeutics, Inc. contributed to the study design, data analysis, and critical review of the manuscript. The decision to submit resided solely with the authors.
DATA DISCLOSURE STATEMENT
Study results and individual de-identified patient data will not be available in a publicly accessible repository to protect the interests of the patients and investigators, in accordance with the policies of Sarepta Therapeutics, Inc.
ACKNOWLEDGMENTS
This study was sponsored by Sarepta Therapeutics, Inc. Editorial support was provided by Peloton Advantage, LLC, an OPEN Health company, and was funded by Sarepta Therapeutics, Inc.
The Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (DNHS) was funded by the U.S. Department of Education/NIDRR (#H133B031118, #H133B090001); U.S. Department of Defense (#W81XWH-12-1-0417); National Institutes of Health/NIAMS (#R01AR061875); Parent Project Muscular Dystrophy. The authors thank the dedicated CINRG DNHS researchers. Additional CINRG DNHS network sites and researchers who contributed to this project include: University of California, Davis – Michelle Cregan, Erica Goude, Alina Nicorici, Merete Glick, Linda Johnson, Jay Han; Holland Bloorview Kids Rehabilitation – Laila Eliasoph, Elizabeth Hosaki, Angela Gonzales, Vivienne Harris; Alberta Children’s Hospital – Angela Chiu, Karla Sanchez, Natalia Rincon, Tiffany Haig; Queen Silvia Children’s Hospital – Anne-Christine Alhander, Lisa Wahlgren, Anne-Berit Ekstrom, Anna-Karin Kroksmark, Ulrika Sterky; Children’s National Medical Center – Marissa Birkmeier, Sarah Kaminski, Allyn Toles; Royal Children’s Hospital – Kate Carroll, Katy DeValle, Rachel Kennedy, Andrew Kornberg, Dani Villano; Hadassah Hebrew University Hospital – Adina Bar Leve, Elana Wisband, Debbie Yaffe; Instituto de Neurosciencias Fundacion Favaloro – Luz Andreone, Jose Corderi, Lilia Mesa, Lorena Levi; Children’s Hospital of Pittsburgh of UPMC and the University of Pittsburgh – Hoda Abdel-Hamid, Christopher Bise, Ann Craig, Casey Nguyen, Andrea Smith, Jason Weimer; Washington University, St. Louis – M. Al-Lozi, Julaine Florence, Betsy Malkus, Renee Renna, Jeanine Schierbacker, Catherine Seiner, Charlie Wulf; Children’s Hospital of Virginia – Susan Blair, Barbara Grillo, Karen Jones, Eugenio Monasterio; University of Tennessee, Memphis – Meegan Barrett-Adair, Judy Clift, Cassandra Feliciano, Rachel Young; Children’s Hospital of Westmead – Kristy Rose, Richard Webster, Stephanie Wicks; University of Alberta – Lucia Chen, Cameron Kennedy; the CINRG DNHS Coordinating Center – Adrienne Arrieta, Tanisha Brown-Caines, Avital Cnaan, Tina Duong, Fengming Hu, Lauren Morgenroth, Wenze Tang.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JND-180351.
REFERENCES
[1] | McDonald CM , Abresch RT , Carter GT , Fowler WM Jr. V Johnson ER , Kilmer DD , Sigford BJ . Profiles of neuromus-cular diseases. Duchenne muscular dystrophy. Am J Phys Med Rehabil. (1995) ;74: (5 Suppl):S70–92. |
[2] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , Kaul A , Kinnett K , McDonald C , Pandya S , Poysky J , Shapiro F , Tomezsko J , Constantin C , DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and pharmacological and psychosocial management. Lancet Neurol. (2010) ;9: (1):77–93. |
[3] | Connolly AM , Florence JM , Cradock MM , Malkus EC , Schierbecker JR , Siener CA , Wulf CO , Anand P , Golumbek PT , Zaidman CM , Philip Miller J , Lowes LP , Alfano LN , Viollet-Callendret L , Flanigan KM , Mendell JR , McDonald CM , Goude E , Johnson L , Nicorici A , Karachunski PI , Day JW , Dalton JC , Farber JM , Buser KK , Darras BT , Kang PB , Riley SO , Shriber E , Parad R , Bushby K , Eagle M , and MDA DMD Clinical Research Network. Motor and cognitive assessment of infants and young boys with Duchenne muscular dystrophy: Results from the Muscular Dystrophy Association DMD Clinical Research Network. Neuromuscul Disord. (2013) ;23: (7):529–39. |
[4] | Hoffman EP , Fischbeck KH , Brown RH , Johnson M , Medori R , Loike JD , Harris JB , Waterston R , Brooke M , Specht L , et al. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N Engl J Med. (1988) ;318: (21):1363–8. |
[5] | Peverelli L , Testolin S , Villa L , D’Amico A , Petrini S , Favero C , Magri F , Morandi L , Mora M , Mongini T , Bertini E , Sciacco M , Comi GP , Moggio M . Histologic muscular history in steroid-treated and untreated patients with Duchenne dystrophy. Neurology. (2015) ;85: (21):1886–93. |
[6] | Goldstein JA , McNally EM . Mechanisms of muscle weakness in muscular dystrophy. J Gen Physiol. (2010) ;136: (1):29–34. |
[7] | Mayer O , Henricson E , McDonald C , Buyse G . Advances in pulmonary care in Duchenne muscular dystrophy. US Neurology. (2017) ;13: (1):7. |
[8] | McDonald CM , Henricson EK , Abresch RT , Duong T , Joyce NC , Hu F , Clemens PR , Hoffman EP , Cnaan A , Gordish-Dressman H , CINRG Investigators. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: A prospective cohort study. Lancet. (2018) ;391: (10119):451–61. |
[9] | Bushby K , Finkel R , Birnkrant DJ , Case LE , Clemens PR , Cripe L , Kaul A , Kinnett K , McDonald C , Pandya S , Poysky J , Shapiro F , Tomezsko J , Constantin C , DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: Implementation of multidisciplinary care. Lancet Neurol. (2010) ;9: (2):177–89. |
[10] | McDonald C , Gordish-Dressman H , Henricson E , Duong T , Joyce NC , jhawar S , Hu F , Connolly AM , Cnaan A , Abresch R . Longitudinal pulmonary function testing outcome measures in Duchenne muscular dystrophy: Long-term natural history with and without glucocorticoids. Neuromuscul Disord. (2018) ;28: (11):897–909. |
[11] | Hahn A , Bach JR , Delaubier A , Renardel-Irani A , Guillou C , Rideau Y . Clinical implications of maximal respiratory pressure determinations for individuals with Duchenne muscular dystrophy. Arch Phys Med Rehabil. (1997) ;78: (1): 1–6. |
[12] | Khirani S , Ramirez A , Aubertin G , Boule M , Chemouny C , Forin V , Fauroux B . Respiratory muscle decline in Duchenne muscular dystrophy. Pediatr Pulmonol. (2014) ;49: (5):473–81. |
[13] | Mayer OH , Finkel RS , Rummey C , Benton MJ , Glanzman AM , Flickinger J , Lindstrom BM , Meier T . Characterization of pulmonary function in Duchenne muscular dystrophy. Pediatr Pulmonol. (2015) ;50: (5):487–94. |
[14] | Miller JR , Colbert AP , Schock NC . Ventilator use in progressive neuromuscular disease: Impact on patients and their families. Dev Med Child Neurol. (1988) ;30: (2):200–7. |
[15] | Birnkrant DJ , Bushby K , Bann CM , Alman BA , Apkon SD , Blackwell A , Case LE , Cripe L , Hadjiyannakis S , Olson AK , Sheehan DW , Bolen J , Weber DR , Ward LM , DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: Respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. (2018) ;17: (4):347–61. |
[16] | Aartsma-Rus A , Fokkema I , Verschuuren J , Ginjaar I , van Deutekom J , van Ommen GJ , den Dunnen JT . Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat. (2009) ;30: (3):293–9. |
[17] | Bladen CL , Salgado D , Monges S , Foncuberta ME , Kekou K , Kosma K , Dawkins H , Lamont L , Roy AJ , Chamova T , Guergueltcheva V , Chan S , Korngut L , Campbell C , Dai Y , Wang J , Barisic N , Brabec P , Lahdetie J , Walter MC , Schreiber-Katz O , Karcagi V , Garami M , Viswanathan V , Bayat F , Buccella F , Kimura E , Koeks Z , van den Bergen JC , Rodrigues M , Roxburgh R , Lusakowska A , Kostera-Pruszczyk A , Zimowski J , Santos R , Neagu E , Artemieva S , Rasic VM , Vojinovic D , Posada M , Bloetzer C , Jeannet PY , Joncourt F , Diaz-Manera J , Gallardo E , Karaduman AA , Topaloglu H , El Sherif R , Stringer A , Shatillo AV , Martin AS , Peay HL , Bellgard MI , Kirschner J , Flanigan KM , Straub V , Bushby K , Verschuuren J , Aartsma-Rus A , Beroud C , Lochmuller H . The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. (2015) ;36: (4):395–402. |
[18] | Bello L , Morgenroth LP , Gordish-Dressman H , Hoffman EP , McDonald CM , Cirak S , CINRG Investigators. DMD genotypes and loss of ambulation in the CINRG Duchenne Natural History Study. Neurology. (2016) ;87: (4):401–9. |
[19] | Ricotti V , Ridout DA , Pane M , Main M , Mayhew A , Mercuri E , Manzur AY , Muntoni F , UK NorthStar Clinical Network. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: Considerations for the design of clinical trials. J Neurol Neurosurg Psychiatry. (2016) ;87: (2):149–55. |
[20] | Mendell JR , Rodino-Klapac LR , Sahenk Z , Roush K , Bird L , Lowes LP , Alfano L , Gomez AM , Lewis S , Kota J , Malik V , Shontz K , Walker CM , Flanigan KM , Corridore M , Kean JR , Allen HD , Shilling C , Melia KR , Sazani P , Saoud JB , Kaye EM , Eteplirsen Study Group. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol. (2013) ;74: (5):637–47. |
[21] | Kinane TB , Mayer OH , Duda PW , Lowes LP , Moody SL , Mendell JR . Long-term pulmonary function in Duchenne muscular dystrophy: Comparison of eteplirsen-treated patients to natural history. J Neuromuscul Dis. (2018) ;5: (1):47–58. |
[22] | Brooke MH , Griggs RC , Mendell JR , Fenichel GM , Shumate JB , and Pellegrino RJ . Clinical trial in Duchenne dystrophy. I. The design of the protocol. Muscle Nerve. (1981) ;4: (3):186–97. |
[23] | Gauld LM , Kappers J , Carlin JB , Robertson CF . Height prediction from ulna length. Dev Med Child Neurol. (2004) ;46: (7):475–80. |
[24] | McDonald CM , Henricson EK , Abresch RT , Han JJ , Escolar DM , Florence JM , Duong T , Arrieta A , Clemens PR , Hoffman EP , Cnaan A , CINRG Investigators. The Cooperative International Neuromuscular Research Group Duchenne Natural History Study-a longitudinal investigation in the era of glucocorticoid therapy: Design of protocol and the methods used. Muscle & Nerve. (2013) ;48: (1):32–54. |
[25] | Henricson EK , Abresch RT , Cnaan A , Hu F , Duong T , Arrieta A , Han J , Escolar DM , Florence JM , Clemens PR , Hoffman EP , McDonald CM , Investigators C. The cooperative international neuromuscular research group Duchenne natural history study: Glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle & Nerve. (2013) ;48: (1):55–67. |
[26] | Duchenne Natural History. The Cooperative International Neuromuscular Research Group, 2017. Available at: http://www.cinrgresearch.org/duchenne-natural-history/. Accessed: November 6, 2018. |
[27] | Hankinson JL , Odencrantz JR , Fedan KB . Spirometric reference values from a sample of the general U.S. population. Am J Respir Crit Care Med. (1999) ;159: (1):179–87. |
[28] | Polgar G , Varuni P Pulmonary Function Testing in Children: Techniques and Standards. Philadelphia, PA: Saunders; (1971) . pp. 273. |
[29] | Phillips MF , Quinlivan RC , Edwards RH , Calverley PM . Changes in spirometry over time as a prognostic marker in patients with Duchenne muscular dystrophy. Am J Respir Crit Care Med. (2001) ;164: (12):2191–4. |
[30] | Roberto R , Fritz A , Hagar Y , Boice B , Skalsky A , Hwang H , Beckett L , McDonald C , Gupta M . The natural history of cardiac and pulmonary function decline in patients with Duchenne muscular dystrophy. Spine. (2011) ;36: (15):E1009–17. |
[31] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Brumbaugh D , Case LE , Clemens PR , Hadjiyannakis S , Pandya S , Street N , Tomezsko J , Wagner KR , Ward LM , Weber DR , Group DCCW . Diagnosis and management of Duchenne muscular dystrophy, part 1: Diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. (2018) ;17: (3):251–67. |
[32] | Birnkrant DJ , Bushby K , Bann CM , Apkon SD , Blackwell A , Colvin MK , Cripe L , Herron AR , Kennedy A , Kinnett K , Naprawa J , Noritz G , Poysky J , Street N , Trout CJ , Weber DR , Ward LM , DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 3: Primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. (2018) ;17: (5):445–55. |


