
Treatment Algorithm for Infants Diagnosed with Spinal Muscular Atrophy through Newborn Screening
Abstract
Background:
Spinal muscular atrophy (SMA) is an autosomal recessive disease characterized by the degeneration of alpha motor neurons in the spinal cord, leading to muscular atrophy. SMA is caused by deletions or mutations in the survival motor neuron 1 gene (SMN1). In humans, a nearly identical copy gene, SMN2, is present. Because SMN2 has been shown to decrease disease severity in a dose-dependent manner, SMN2 copy number is predictive of disease severity.
Objective:
To develop a treatment algorithm for SMA-positive infants identified through newborn screening based upon SMN2 copy number.
Methods:
A working group comprised of 15 SMA experts participated in a modified Delphi process, moderated by a neutral third-party expert, to develop treatment guidelines.
Results:
The overarching recommendation is that all infants with two or three copies of SMN2 should receive immediate treatment (n = 13). For those infants in which immediate treatment is not recommended, guidelines were developed that outline the timing and appropriate screens and tests to be used to determine the timing of treatment initiation.
Conclusions:
The identification SMA affected infants via newborn screening presents an unprecedented opportunity for achievement of maximal therapeutic benefit through the administration of treatment pre-symptomatically. The recommendations provided here are intended to help formulate treatment guidelines for infants who test positive during the newborn screening process.
INTRODUCTION
SMA clinical features and spectrum of severity
Spinal muscular atrophy (SMA) is an autosomal recessive disorder predominately caused by bi-allelic deletion of the survival motor neuron 1 (SMN1) gene. It is characterized by dysfunction and then loss of the alpha motor neurons in the spinal cord that causes progressive muscle atrophy and weakness [1, 2]. A large study in a broad cohort from the United States identified an overall carrier frequency of 1 in 54, with a calculated incidence of 1 in 11,000 [3]. Historically, SMA has been the leading monogenic cause of death in infancy, but there is reason for hope that this will greatly change with widespread early administration of newly approved disease modifying therapies.
SMA manifests across a continuous gradient of phenotype severity, separated by functional “type” based on age of onset and maximum motor milestones achieved [4]. Individuals with onset of weakness in the first six months of infancy who never achieve an ability to sit independently, once known as “Werdnig-Hoffmann disease” but now classified as having “SMA type 1”, constitute approximately 60% of all individuals with SMA [2, 5, 6–10]. Approximately 30% of patients are diagnosed with “SMA type 2”. These patients present with weakness recognized in later infancy and achieve the ability to sit, but not walk, independently. Those able to walk are grouped under the “SMA type 3” or “Kugelberg-Welander” label and constitute approximately 10% of the patient population. Some clinicians include outlier groups labeled as “SMA type 0”, referring to fetal onset with severe weakness, joint contractures, and respiratory compromise presenting at birth; and “SMA type 4” denoting a small group who first show weakness in adult years. Across the range of SMA, at all levels of severity, there is a common path of relatively greater progression of weakness, or departure from a normal pattern of developmental gain of milestones, followed thereafter by a slower plateau of relative stability with very slow worsening.
Approximately 95% of all individuals with SMA, at all levels of phenotypic severity, have a homozygous SMN1 gene deletion, detection of which serves as the primary diagnostic assay for the disorder. All individuals with SMA have a variable copy number SMN2, a paralog of SMN1, that produces low, but essential levels of SMN protein. Copy number of SMN2 correlates inversely with SMA phenotype severity, as greater SMN2 copy number is associated with milder phenotypic presentation [11]. At the most severe end, a single copy of SMN2 is associated with very severe weakness and a limited duration of survival after birth with SMA type 0 [12, 13]. In one large German study, 80% of those with SMA type 1 have two or fewer copies of SMN2, 82% of those with SMA type 2 have three copies of SMN2, and 96% of those with SMA type 3 have three or four copies of SMN2. This relationship of phenotype to genotype thus enables prediction of SMA type from the SMN2 copy number before the onset of symptoms. Infants found to have two or three copies of SMN2 are highly likely to manifest SMA type 1 or 2, which are associated with high early mortality and substantial morbidity. Conversely, those with four or more copies of SMN2 are extremely unlikely (<0.5%, or 1:298 in combined cohorts; [12–15]) to present with the most severe SMA type 1. In addition, all of these studies predict that less than 10% of SMA cases would first present in children older than three years, and these would typically be classified as having SMA type 3. Thus, identification of homozygous deletion of SMN1 combined with determination of SMN2 copy number is a powerful predictor of disease and identifies a group who would benefit substantially from new and emerging therapies.
SMA treatments and therapies
SMA directly causes muscle weakness, leading to numerous downstream complications, many of which are amenable to supportive care that has been outlined in standard-of-care consortia [5, 16, 17]. Respiratory treatment with bi-level positive airway pressure support when appropriate, orthopedic management of scoliosis and other deformities, and nutritional support have made meaningful differences in clinical outcome [11, 16–18]. SMA has also been advantaged by a robust drug development pipeline involving several different lines of specific therapeutic strategies evolving over the last decade. One therapy called nusinersen, directed at improving functional SMN protein expression by altering SMN2 transcript splicing using an antisense oligonucleotide approach, was recently approved by the Food and Drug Administration (FDA) and is now commercially available. Other approaches to therapy at the level of increasing SMN protein levels, including a gene transfer approach using an AAV9 vector [19] or small molecule modification of SMN2 splicing [20], are in promising clinical trials. Additional approaches, including putative neuroprotective agents and therapies intending to increase muscle function directly, are also in development [20].
Rationale for early intervention
Thus far, both clinical and preclinical data indicate that early treatment will be critical to modulate the rapid and progressive degeneration seen in SMA, especially in type 1. There is strong evidence that the irreversible loss of motor neurons in humans with SMA type 1 begins early in the perinatal period, with severe denervation in the first three months and loss of more than 90% of motor units within six months of age [11]. Furthermore, preclinical studies looking at the timing of drug delivery in severe mouse models of SMA consistently show that the best results occur when drugs are given as early as possible, before significant motor weakness or loss is present [21, 22]. Despite such evidence, diagnostic delay is very common in SMA. A recent systematic literature search covering 21 reports for studies published between 2000 and 2014 showed that the mean ages of symptom onset were 2.5, 8.3, and 39.0 months for SMA types 1, 2, and 3 respectively, whereas the weighted mean ages of confirmed SMA diagnosis were 6.3, 20.7, and 50.3 months for types 1, 2, and 3, respectively [23]. Because of these diagnostic delays and the lack of newborn screening (NBS) for SMA, most patients have progressed past the point where maximal benefit is achievable before therapeutic interventions occur. Given that most affected individuals seek treatment when diagnosed, NBS has the potential to increase the benefit of these therapies without increasing the cost of therapy. Furthermore, NBS may substantially decrease the cost of support needed to help those with functional impairment.
The role of timing in successful drug intervention is also apparent from two Biogen-sponsored clinical trials testing nusinersen in symptomatic and pre-symptomatic infants. The ENDEAR trial was a randomized, placebo-controlled clinical trial that included 121 subjects with infantile-onset SMA and two copies of SMN2 who were diagnosed before 6 months of age and who were less than 7 months old at the time of their first dose. In the final analysis, 51% of subjects treated with nusinersen achieved improvement in motor milestones, whereas none of the control subjects did (p < 0.001). Additionally, only 32% of infants with a disease duration of more than 12 weeks responded positively on the Hammersmith Infant Neurological Examination (HINE) [24] motor milestone, compared to 75% of infants with disease duration of 12 weeks or less. Moreover, nusinersen demonstrated a statistically significant 47% reduction in the risk of death or permanent ventilation (p = 0.005) and had a favorable safety profile [25].
In contrast, the NURTURE trial is an ongoing open-label clinical trial that enrolled pre-symptomatic, genetically diagnosed infants with SMA having two or three copies of SMN2 who were less than 6 weeks of age at first dose. Interim analysis showed that 100% of the infants were still alive and did not require invasive ventilation at all or non-invasive ventilation for greater than 6 hours per day continuously for more than 7 days [26]. In comparison, the total mean HINE score improvement was substantially higher in the pre-symptomatic subjects of NURTURE compared to the symptomatic subjects of ENDEAR. Overall, greater attainment of specific motor milestones was achieved with pre-symptomatic treatment compared to symptomatic treatment, and many of these subjects have achieved motor skills at a developmentally appropriate age and have remained free of the need for ventilation or feeding support (Table 1). The NURTURE study results are a strong indicator of the importance of NBS in achieving maximal efficacy with SMN enhancers in treating SMA.
Table 1
Summary of motor milestone achievements of infants receiving nusinersen in ENDEAR versus NURTURE clinical trials
| Milestone | Total number of infants achieving milestone, n/N (%) | |
| ENDEARa (Symptomatic patients; N = 73) | NURTUREb (Pre-symptomatic patients; N = 13) | |
| Head control (full) | 16/73 (22) | 5/9 (55) |
| Sitting (independent: stable, pivot) | 6/73 (8) | 6/13 (46) |
| Standing (stands with support, unaided) | 1/73 (1) | 4/13 (31) |
| Walking (cruising, walking) | 0/73 (0) | 2/13 (15) |
n = number of patients with milestone; N = number of patients analyzed for that milestone. a[25]. b[26]. Note: Only infants with two copies of SMN2 were included in this table (no three-copy SMN2 patients were included from the NURTURE trial). All infants who enrolled in ENDEAR had two copies of SMN2. Note: The ENDEAR interim was performed when 51 subjects who received nusinersen had the opportunity to be treated and observed for at least 183 days and up to 394 days. The NURTURE interim analysis data cutoff date was October 31, 2016.
Newborn screening assays
As described above, to achieve maximal therapeutic benefit, early identification of affected infants in the pre-symptomatic period is critical. Therefore, the need for reliable and well validated newborn screening assays is paramount.
An SMN1 assay has been developed that utilizes a modified, multiplexed real-time polymerase chain reaction (PCR) to detect the presence of SMN1 from a dried blood spot. Initial results show that the assay identified SMN1 exon 7 deletions in all SMA-affected patients, while all unaffected individuals showed the presence of exon 7. This molecular assay is inexpensive, as it can be multiplexed at minimal additional cost to a broadly accepted assay for severe combined immunodeficiency (SCID) [27]. There have been two pilot studies using this or similar assays in Taiwan and New York State. The Taiwan pilot study detected homozygous deletions in SMN1 intron 7, while the New York state pilot study is detecting homozygous and heterozygous (carrier) deletions in SMN1 exon 7. From November 2014 to September 2016, a total of 120,267 infants had been tested in the Taiwan pilot study. From January to December 2016, a total of 3,264 infants were tested at three hospitals in New York State [28]. In both studies, the assays have shown 100% positive predictive value using DNA extracted from dried blood spot punches.
A second, commercial real-time PCR assay has been developed by PerkinElmer that detects homozygous and heterozygous deletions in SMN1 exon 7 and SMN2 copy number from a dried blood spot punch. This assay was also designed to be multiplexed with the SCID assay. An ongoing study is currently being conducted to screen over 3,000 samples, with analyses thus far showing 100% positive predictive value [29]. Research and development studies are currently ongoing for this assay, and pilot studies are being planned.
SMA Recently suggested as a condition on the recommended uniform screening panel
In order for new conditions to be added to the RUSP for newborns, several elements are required. These include a body of evidence on the condition itself and treatment options supported by evidence-based information, including validation of a laboratory test, widely available confirmatory testing with a sensitive and specific diagnostic test, and a prospective population-based pilot study. For SMA, there is now strong natural history data that indicate typical progression as well as an approved treatment that demonstrates higher therapeutic value when administered in pre-symptomatic infants. Adequate pilot screening data from state public labs is available utilizing an inexpensive and reliable diagnostic test for broad-scale use. The SMN1 deletion testing has been shown to be highly sensitive and specific, and the SMN2 copy determination will provide important prognostic information. In February 2018, the Advisory Committee on Heritable Disorders in Newborns and Children (ACHDNC) voted to recommend SMA for addition to the RUSP. The recommendation now goes to the Health and Human Services secretary for final approval. In further support of the addition of SMA to the RUSP, we report here guidelines of the SMA NBS Multidisciplinary Working Group describing a recommended treatment algorithm for those infants identified as having a positive SMA newborn screen.
MATERIALS AND METHODS
SMA NBS multidisciplinary working group and mission
The SMA NBS Multidisciplinary Working Group, supported by a major patient advocacy group, CureSMA, brought together clinicians and geneticists with SMA expertise and patient advocacy representatives to formulate a treatment algorithm for individuals who have a positive SMA NBS test. The working group consisted of a total of 15 members, five of whom were organizing members, and 13 of whom participated in voting. All working group members participated voluntarily without compensation.
Achieving consensus through the delphi technique
The working group employed a modified version of the Delphi technique [30, 31] to reach consensus on treatment guidelines. Data was collected using multiple iterative rounds [32] of an online survey. The sequential surveys started with a few broader questions that then motivated more specific questions related to SMA treatment. Voting members answered survey questions anonymously by choosing from a selection of responses. Following each survey, the group response was reported back to the voting members. During this discussion, voting members considered whether to keep their original answers or change their opinions after seeing the overall group’s response. An expert in the Delphi process served as a neutral non-voting, third party member who moderated discussions and had no stake in the decisions made.
This technique’s advantages include allowing voting members to provide their opinions anonymously, without undue influence from more outspoken individuals within the group. This method allowed for easy identification of topics for which the working group did not initially reach consensus for further examination. Another advantage of using this technique is the ease in using online tools that allow global communication among physically distant voting members. Consequently, the Delphi method has been increasingly used to reach consensus in many fields, including education, medicine, and research [33]. In order to achieve consensus on the treatment algorithm and SMN-up-regulating therapy guidelines, four conference calls were held following administration of the online surveys, in addition to a final live polling to address outstanding questions.
RESULTS
Treatment algorithm for SMN-up-regulating therapies
SMA has multiple genotypes and associated phenotypes, resulting in a spectrum of severity that may include some asymptomatic individuals at its mildest, albeit rarest, end. The first question to be addressed was thus directed to which screen-identified SMA individuals should be treated immediately with FDA-approved therapies in the context of NBS, and which should be monitored carefully with treatment initiated later in life. This is of critical importance, as some genotypes of SMA are imminently life threatening without treatment.
The working group first developed a treatment algorithm for the administration of an SMN-up-regulating treatment based upon genotype following a positive NBS result (Fig. 1). The initial decisions for this algorithm were based on the correlation of SMN genotype to phenotype across multiple studies. SMA types 1 and 2 represent a large majority of SMA cases and account for the bulk of those who screen positive for SMA and have three or fewer copies of SMN2. The working group unanimously recommends immediate treatment for these individuals to achieve a maximal response to treatment, as supported by the strong positive results arising from pre-symptomatic infants in the NURTURE trial for individuals predicted to manifest SMA by qualifying genotype who have either two and three copies of SMN2 [26].
Fig.1
SMA Newborn Screening Treatment Schematic for SMN-Up-Regulating Therapy. SMA=spinal muscular atrophy; SMN=survival motor neuron.
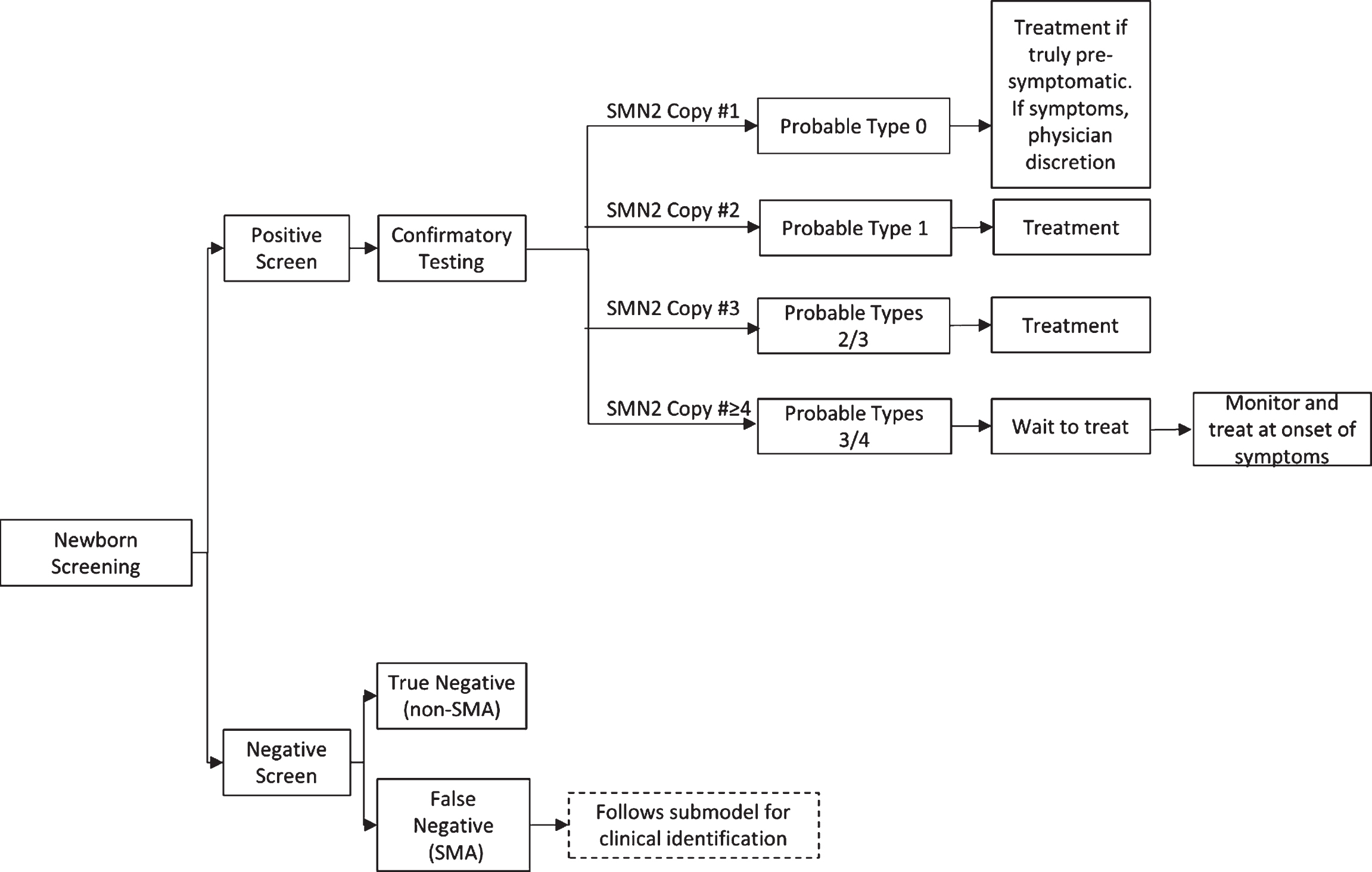
Recommendation for treatment of individuals who screen positive for SMA and have low (one copy) or high (four or more copies) SMN2 copy number is more complicated, and summarized in Table 2 and Table 3. The working group recognizes that the majority of infants with SMA who have one copy of SMN2 will be symptomatic at birth [34]. In this case, the consensus is to defer to the attending physician to determine if the infant and family would benefit from treatment given his or her current disease state. In the rare event that an SMA infant with one copy of SMN2 is truly pre-symptomatic, the strong consensus is that the infant should be treated immediately. The working group was evenly divided as to whether infants with four SMN2, as identified by NBS, should be treated immediately or instead screened carefully for the first signs of mild symptoms to initiate treatment (n =13). The committee did reach consensus that patients with more than 4 copies should not be treated immediately but screened carefully for the first presentation of symptoms. Recommendations were then developed for what screens and tests should be carefully monitored (Table 3) and how often they should occur to determine when treatment should be initiated in infants and children undergoing watchful waiting.
Table 2
Summary of working group’s voting responses concerning tests used to monitor patients with ≥4 SMN2 copies for whom treatment is not initiated immediately
| Survey Question Possible Responses | Group Voting (%) |
| When thinking about patients with four SMN2 copies, what is the greater risk? (N = 9 voting members) | |
| Not treating the child early enough who then develops symptoms, which may be more refractory to treatment at that point | 56 |
| Treating the child who is subjected to the risks and burden of treatment and yet might not have exhibited signs of SMA for many years | 44 |
| As a group developing a treatment algorithm for infants with SMA identified through NBS, do you feel we should provide: (N = 9 voting members) | |
| A prescriptive recommended battery of tests/evaluations with changes or early plateau values that would trigger a recommendation of treatment | 11 |
| A list of possible appropriate tests where a change on any would allow informed clinical judgment to trigger a recommendation of treatment | 89 |
| Which of the following would you recommend as follow-up testing/assessment in patients not initially treated, assuming the patient was the correct age for the test? (N = 9 voting members) | |
| Motor Function Scales | 0 |
| Myometry | 0 |
| EMG | 0 |
| CMAP | 0 |
| Physical Assessment, including reflexes | 0 |
| Any of the following | 100 |
| What level of change/results on an EMG should cause initiation of treatment? (N = 9 voting members) | |
| Any active or chronic neurogenic change | 100 |
| Abnormal spontaneous activity in one proximal and one distal muscle | 0 |
| Abnormal spontaneous activity in two different muscle groups | 0 |
| Abnormal spontaneous activity in two limbs and an axial region | 0 |
| Any abnormal spontaneous activity/fibrillations | 0 |
| What level of change/results on a CMAP test should cause initiation of treatment? (N = 9 voting members) | |
| Other (please specify) | 0 |
| 20% decrease in amplitude from a prior test of that child | 0 |
| 10% decrease in amplitude from a prior test of that child | 0 |
| 20% below normative values for an age-matched child | 0 |
| Below normative values for an age-matched child | 100 |
| What level of change/results on a physical exam should cause initiation of treatment? Please select all applicable choices. (N = 9 voting members) | |
| Loss of reflexes | 0 |
| Weakness in trunk right/derotation | 0 |
| Proximal weakness defined as developmentally appropriate | 0 |
| Regression in ability to perform motor milestones | 0 |
| Failure to meet developmental motor milestones | 0 |
| All of the above | 100 |
| What level of otherwise unexplained decline or early plateau in age appropriate motor function assessments (e.g., Bayley Scales, Hammersmith scales, CHOP INTEND, 6MWT, and WHO motor milestones) should cause a recommendation of treatment? (N = 9 voting members) | |
| A. A failure to gain motor functions with age in keeping with normal development | 0 |
| B. A drop in total score | 0 |
| Either A or B | 100 |
| Physicians should instructs parents/caregivers to contact them immediately if they see any of the following: (N = 9 voting members) | |
| Significant change in child’s movement, feeding, or breathing pattern during time of illness | 100 |
| Observed abdominal breathing | 89 |
| Failure to gain weight appropriately | 89 |
| Change in voice/weak cry | 100 |
| Increased fatigue without increased activity | 100 |
| Trouble feeding in young children or infants | 100 |
| Decline or loss of function in previously attained motor ability or failure to show progress in expected motor ability | 100 |
CHOP INTEND = Children’s Hospital of Philadelphia Infants Test of Neuromuscular Disorders; CMAP = compound muscle action potential; EMG = electromyograph; N = number of voting members; NBS = newborn screening; SMA = spinal muscular atrophy; SMN = survival motor neuron; 6MWT = six-minute walk test; WHO = World Health Organization.
Table 3
Summary of tests used to monitor patients with ≥4 SMN2 copies for whom treatment is not initiated immediately
| Test or Outcome Measure | Level of Change/Results Which Would Prompt Initiation of Treatment | Appropriate Age of Patient for Test |
| EMG/nerve conduction | Any active or chronic neurogenic change | All |
| CMAP | Below normative values for an age-matched child | All |
| Myometry | Decrease in extent of muscle contraction | ≥4 years |
| Physical Exam/Reflexes | Any of the following: loss of reflexes, failure to meet or regression in ability to perform motor milestones, proximal weakness, and weakness in trunk righting/de-rotation | All |
| CHOP INTEND | A failure to gain motor functions with age in keeping with normal development or a drop in total score from previous assessment | Infants |
| HINE | A failure to gain motor functions with age in keeping with normal development or a drop in total score from previous assessment | Infants |
| Hammersmith Functional Motor Scale – Expanded | A failure to gain motor functions with age in keeping with normal development or a drop in total score from previous assessment | ≥2 years |
| 6MWT | A failure to gain motor functions with age in keeping with normal development or a drop in total score from previous assessment | ≥5 years |
| Bayley Scales of Infant and Toddler Development | A failure to gain motor functions with age in keeping with normal development or a drop in total score from previous assessment | Infants/Toddlers (Recommended 1 to 42 months) |
CHOP INTEND = Children’s Hospital of Philadelphia Infants Test of Neuromuscular Disorders; CMAP = compound muscle action potential; EMG = electromyograph; HINE = Hammersmith Infant Neurological Exam; SMN2 = spinal motor neuron 2; 6MWT = six-minute walk test.
Guidelines for follow up of follow up of infants identified as having SMA with four copies of SMN2
The following treatment recommendations are intended to advise the follow-up and initiation of an SMN-up-regulating therapy in infants and children with four or more copies of SMN2, who are not immediately treated with a FDA-approved disease modifying therapy for SMA. These recommendations are intended as guidelines only. The working group reached full consensus that the attending physician’s clinical judgment, as well as the patient’s and/or the patient’s family’s wishes, should be the deciding factor on when to initiate treatment (n = 13). In patients with fewer than four copies of SMN2, it should be noted that disease onset would be expected to be more rapid and/or severe: deferring treatment is not recommended for these patients, and thus the following guidelines would not apply to such cases.
GUIDELINE NO. 1: FOR THOSE PATIENTS IN WHOM TREATMENT IS NOT INITIATED IMMEDIATELY, ROUTINE FOLLOW-UP CARE SHOULD IDEALLY BE PROVIDED BY A NEUROMUSCULAR SPECIALIST
Given the options of a pediatrician, a neuromuscular specialist, and a general child neurologist, the working group reached full consensus that a neuromuscular specialist would be able to provide the best routine follow-up care in individuals with four or more SMN2 copies (n = 9). A pediatrician’s expertise in child healthcare may be broad and not cover the unique features of a rare neuromuscular disorder, while a general child neurologist may not specialize in the role of the neuromuscular system in the patient’s symptomatology and diagnosis or have the knowledge to administer the specific tests being recommended here. The working group also acknowledges that it is typically considered standard practice through NBS programs to refer patients to a geneticist, and this would also be advised in all cases of SMA detected through NBS. A neuromuscular specialist would have the deepest knowledge of the clinical manifestations of SMA in order to detect the earliest symptomatology, in addition to experience with administering the highly sensitive assessments of motor neuron function and SMA specific motor function. This recommendation is offered knowing that not all patients live in communities with easy access to a neuromuscular specialist.
GUIDELINE NO. 2: INFANTS IDENTIFIED AS HAVING FOUR OR MORE COPIES OF SMN2 SHOULD BE REFERRED TO SOMEONE WHO CAN IDENTIFY THEIR EXACT COPY NUMBER
Currently, not all commercial laboratories distinguish SMN2 copy number precisely at four or greater copies, reporting those cases as “≥4 SMN2 copies”. Given this imprecision, about 89% of the working group (n = 9 [voting members]) felt that determining the patient’s exact copy number would allow for a more informed prediction of when disease symptoms may appear. In addition, the working group discussed the importance of checking for certain known disease modifying mutations, such as the SMN2 c.859G>C mutation in exon 7. This rare variant regulates the splicing of SMN2 pre-mRNAs such that a greater proportion of SMN2 transcripts contain exon 7, resulting in a milder phenotype [35–37]. Generally, because SMN2 copy number is a strong predictor of disease severity, the group felt that identifying the exact copy number, if that was not originally provided, would greatly help inform an attending physician in the best approach to monitor the patient’s disease progression, as well as provide the best testing strategies and follow-up care possible. In cases identified as having ≥4 SMN2 copies, there is no specific urgency to confirm exact copy number in a reference laboratory, as these patients present with less rapid disease progression than those with three or fewer SMN2 copies.
GUIDELINE NO. 3: FOR THOSE PATIENTS IN WHOM TREATMENT IS NOT INITIATED IMMEDIATELY, ROUTINE FOLLOW-UP CARE SHOULD IDEALLY OCCUR EVERY THREE TO SIX MONTHS UNTIL THE PATIENT REACHES TWO YEARS OF AGE AND EVERY SIX TO 12 MONTHS THEREAFTER
The working group reached full consensus (n = 9 [voting members]) that a higher frequency of visits early on is essential. This was recommended to ensure the detection of any rare cases of children with a severe SMA type 1 or 2, who have four or more copies of SMN2. While a type 1 or 2 phenotype arising from a genotype of four or more SMN2 copies is very rare, the group agreed that due to the rapid progression and severe morbidity of these types of SMA, earlier more frequent monitoring to ensure that the child does not have a more severe form of disease. Once the child reaches two years of age having achieved motor milestones, an early severe form of SMA can be considered excluded and the follow-up frequency can be reduced, as less severe forms of disease are known to have later onset and slower functional decline. Therefore, a diminishing frequency algorithm for follow-up visits is recommended, as frequency of visits should correlate with the predicted severity of the disease that drops as a function of age of onset. Such an algorithm also provides parents and caregivers flexibility in terms of the burden of follow-up visits, while also attempting to minimize treatment-related risks in a child with less severe SMA. The schedule balancing these concerns can be modified by caring physicians in the context of individual circumstances.
GUIDELINE NO. 4: ASSUMING THE PATIENT IS THE APPROPRIATE AGE FOR A SPECIFIC TEST, THE FOLLOWING ARE RECOMMENDED AS FOLLOW-UP ASSESSMENTS IN PATIENTS NOT INITIALLY TREATED: ELECTROMYOGRAPHY (EMG), COMPOUND MUSCLE ACTION POTENTIAL (CMAP) MONITORING, MYOMETRY, PHYSICAL EXAMINATIONS, AND MOTOR FUNCTION SCALES
The working group reached full consensus (n = 9 [voting members]) that EMG, CMAP monitoring, myometry, physical examinations, and motor function scales are each of potential value in follow-up visits for children not initially treated (Table 2). The group recommended a variety of tools to use for assessment, knowing these tests will vary in availability, physician expertise and preference, and the patient’s ability, based on age, to participate. This allows for the inclusion of assessments that show the highest sensitivity toward early changes in pre-symptomatic children, like CMAP and EMG, in addition to the utilization of physical assessments and motor function scales that are less specialized, require less skill, and are more broadly utilized across clinical practices. It should also be noted that not all of these assessments are appropriate for all age ranges, and that tests should be carefully selected based on the age of the patient. The working group’s recommendation was that a change on any of these assessments could be used as the basis to initiate treatment.
The following sections outline guidelines for the amount of change on each of the assessments that is recommended to trigger initiation of treatment in pre-symptomatic children with four or more SMN2 copies.
Any active or chronic neurogenic change in EMG recordings
The working group reached full consensus (n = 9 [voting members]) that any active or chronic neurogenic change in EMG recordings should prompt initiation of treatment. EMG can detect neurogenic changes in muscles and hence can be used to monitor motor neuron or motor unit health in SMA patients [38]. Due to the high sensitivity of this assessment, any neurogenic change can indicate denervation of muscle fibers and warrants further examination and treatment in a patient with a known SMA genotype. However, the working group also acknowledges variability in individual sites’ experience and comfort with EMG in children. For example, the burden of sedating a patient, based on the age of the child, in regards to the frequency and use of EMG as an assessment for monitoring for onset of disease must be balanced.
Below normative values on a CMAP test for an age-matched child
Similar to EMG, the working group reached full consensus (n = 9 [voting members]) that CMAP amplitudes that are below normative values for an age-matched child should initiate treatment. As a reliable electrophysiological measure of muscle function in SMA patients [39], this assessment is also highly sensitive and an early indicator of disease onset in a pre-symptomatic child. Many voting members also felt that any drop in CMAP amplitudes from a prior test of that individual should trigger initiation. Due to the challenging technical nature of these electrophysiological assessments, the guidance for these two assessments (EMG and CMAP) is not to look for a particular percentage change but rather for any results below normative values in an age-matched child. The working group is aware that a threshold determination of the difference needed to be meaningful is not available, and likely differs between institutions. This determination is thus left to the discretion of the consulting neuromuscular specialist.
Decrease in extent of muscle contraction as seen in myometric measures
The working group acknowledges that there is not a large body of evidence on normative values for myometric measures in SMA patients. Thus, the working group reached full consensus (n = 9 [voting members]) that any clinically meaningful myometric changes [40] that indicate reduced muscle force should initiate treatment. When considering age appropriateness of this test, it should be noted that this test is not recommended for children under the age of 5 years. Additionally, the group acknowledges that myometric testing can be technically challenging in children given limited cooperation and need for specialized equipment. Given these difficulties, the working group noted the benefit of coupling myometry evaluation with other physical and electrophysiological examinations as part of a comprehensive, watchful waiting regimen.
Changes in physical examinations, including loss of reflexes, failure to meet or regression in ability to perform motor milestones, proximal weakness, and weakness in trunk righting/de-rotation
The working group reached full consensus (n = 9 [voting members]) that loss of previously identified tendon reflexes, failure to meet, or regression in the ability to perform motor milestones, proximal weakness, and weakness in trunk righting or de-rotation were all important physical examination findings that should initiate treatment. Such changes are relatively straightforward and easily identified by both neuromuscular specialists and general physicians alike. These measures are generally accessible without specialized equipment, and the assessments are non-invasive and can easily be done in an office setting, thus allowing for a broader application across all communities. These assessments may, however, be less sensitive to the early changes that develop in early symptomatic SMA. The working group recommends that these assessments accompany the more sensitive electrophysiological tests as part of a comprehensive follow-up.
A failure to gain motor function with age in keeping with normal development or a drop in total score in motor function assessments
The working group recommends that either a failure to gain age-appropriate motor function or a drop in total score of in any motor function assessment should trigger the initiation of treatment. When thinking about the appropriate assessment for an individual patient, the working group advises that the age of the patient (Table 3), along with physician’s own familiarity with the administration of the assessment be considered to allow for the most accurate and sensitive monitoring. The following briefly describes the motor function assessments considered by the working group for use in SMA patients.
The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND) was initially developed to assess motor skills in infants with SMA type 1 with a mean age of 11.5 months. The assessment includes 16 items selected by an expert panel and scored from a range of 0 (no response) to 4 (complete response) comprising a 64-point scale. The items include both active, spontaneous movements as well as elicited, reflex movements. As it does not include respiratory or feeding behaviors, the CHOP INTEND is both easy and quick to administer and is well tolerated by infants [41]. Because this assessment was designed for SMA type 1 infants with severe muscle weakness, it is only recommended for patients ages birth to two years of age. In addition, asymptomatic children within this age range may reach a ceiling prior to age two.
The HINE consists of 37 items, divided into three sections. The first section contains 26 items assessing cranial nerve function, movements, muscle tone, posture, and reflexes. The second section includes eight items assessing motor function development, and lastly, the third section contains three items for the assessment of behavioral state. All items may be scored individually or added together to generate a global score, providing a useful quantitative measure of motor development [24]. This assessment is relatively user-friendly and can be utilized in both clinical and research settings with less technical skill required and is recommended for infants under the age of two years.
The Hammersmith Functional Motor Scale – Expanded (HFMSE) is an SMA-specific scale of motor function, including a 13-item expansion module added to the original HFMS. The HFMS assesses motor skills using 20 items scored on a 3-point Likert scale. Though it is easy to administer, requires minimal equipment and technical skills, and is reproducible, the range of skills that can be examined in a patient (independent sitting to taking four steps) is limited and can cause a ceiling effect in higher functioning SMA patients. Thus, the HFMSE was created to assess more detailed aspects of certain motor skills, including lying supine; high and half kneeling; going from standing to squatting; and standing while holding one rail and walking up steps. Such items were validated as showing clinically meaningful changes in patients with later-onset SMA types 2 and 3 [42]. Consequently, this assessment is recommended for children two years of age and older who are able to perform such activities.
The 6-minute walk test (6MWT) is easy and safe to administer and is well-tolerated by most patients. Originally used to assess functional capabilities in adult heart and lung disease patients, it has since been modified to assess walking abnormalities in neuromuscular disorders. Further modifications have allowed for its adaptation to children for the assessment of gait abnormalities [43]. This assessment is recommended for children five years of age and older, for which normative values are available.
The Bayley Scales of Infant and Toddler Development are individually administered instruments used primarily to assess the development of infants and toddlers between the ages of 1 and 42 months. These measures consist of a series of developmental play tasks that take between 45–60 minutes to administer and derive a developmental quotient (DQ) rather than an intelligence quotient (IQ). Raw scores of successfully completed items are converted both to scale scores and to composite scores, which are then used to determine the child’s performance compared with norms taken from typically developing children of their age (in months). The most recent edition, the Bayley-III has three main subtests: the Cognitive Scale, the Language Scale, and the Motor Scale [44]. These scores are largely used for screening and identifying the need for further observation and intervention in infants who score outside the normative values. The Bayley has been validated in many studies of children with motor delay in the first years of life [45] and was used in a recent SMA clinical trial assessing an SMN replacement therapy [19].
GUIDELINE NO. 5: PHYSICIANS SHOULD INSTRUCT PARENTS/CAREGIVERS TO CONTACT THEM IMMEDIATELY IF THEY SEE ANY OF THE FOLLOWING
• Significant change in child’s movement, feeding, or breathing pattern
• Change in voice/weak cry
• Increased fatigue without increased activity
• Trouble feeding in young children or infants
• Decline or loss of function in previously attained motor ability or failure to show progress in expected motor ability
• Abdominal breathing
• Failure to thrive
The working group recommended development of materials or a written checklist that parents or caregivers would utilize at home to aid in the surveillance of potential SMA signs triggering immediate re-evaluation. The working group reached full consensus (n = 9 [voting members]) that the observation between clinical visits of any of the first five manifestations of SMA listed above should trigger patients and caregivers to immediately contact their neuromuscular specialist or attending physician. Consensus was not reached on whether the last two manifestations should be included in such materials, not because they are not important symptoms of SMA, but rather that several voting members felt that patients should be identified well before their manifestation. Overall, these seven items are overt signs of SMA that traditionally have been used to diagnose the most severe form of the disease during infancy. SMA is often first detected in patients during the course of an acute illness in the hospital or clinic setting after presenting with these symptoms or those of an associated respiratory illness. The working group anticipates that in the context of NBS, pharmacological drug intervention should be initiated well before the onset of these signs and symptoms if clinicians are successful in their surveillance. Thus, the voting members recommended that parental observation of any of these should prompt immediate re-evaluation of the patient.
This final recommendation underlies the critical need for educational materials to be provided to all parents whose children are diagnosed with SMA via NBS, as the disease course will be different than children diagnosed with SMA after symptom presentation.
DISCUSSION
In summary, the SMA NBS Multidisciplinary Working Group reached consensus on a variety of topics related to the follow-up care and treatment of SMA patients. The working group acknowledges that the vast majority of SMA patients will have three or fewer copies of SMN2. Lower SMN2 copy number correlates with increased disease severity, rapid disease onset, and poor prognosis [11, 13, 15], causing the working group to recommend these patients be treated immediately. In addition, there is clinical trial data supporting the enhanced efficacy of pre-symptomatic treatment in patients with two and three copies of SMN2.
The working group did not reach consensus on the immediate treatment of patients with four or more SMN2 copies, as these patients do not typically present as early or with as severe forms of SMA as patients with a lower copy number. Given that the voting members were split on their responses to this question (Table 2), they developed an algorithm for an effective means of surveillance in patients with four or more copies of SMN2 who may not be treated immediately. In doing so, they had to balance the risks of treatment and the demands of follow-up visits, when symptoms may not present for years, versus not treating the child early enough to achieve maximal benefit from a pharmacological drug treatment. Thus, the group recommended that follow-up visits should ideally happen every three to six months until the patient reaches two years of age and every six to 12 months thereafter.
Given that very few patients with four or more copies of SMN2 have been followed pre-symptomatically, the protocol for following such patients follows recommendation of clinical acumen rather than a data-driven path. This group of experts recommended that in ideal situations, a neuromuscular specialist would monitor patients for follow-up care, including EMG, CMAP, myometry, physical examinations, and age-appropriate motor function scales. The use of a variety of assessments allows the physician more latitude and reliance on clinical judgment. In addition, in regions where neuromuscular experts are not readily available, not all assessments may be easily or reliably administered. It is hoped that application of NBS will generate data about the full range of phenotypes associated with individuals identified to have SMA with four or more copies of SMN2, and thereby help refine these recommendations made largely from arguments of plausibility even further.
Strong evidence warrants the need for early identification and treatment of SMA children. The co-identification of SMN1 deletion and SMN2 copy number often allows clinicians to predict the SMA type and corresponding disease severity in order to select the best timing for FDA-approved drug treatments for SMA. The guidelines noted here were created by the SMA NBS Multidisciplinary Working Group to serve as recommendations for which infants identified through NBS should be immediately treated with SMN-up-regulating therapy. Additionally, the guidelines provide recommendations for a surveillance regimen for infants not immediately treated. The working group acknowledges that the advent of new FDA-approved therapies in the future will prompt the need for additional consideration by both physicians and patients alike, as each drug will present unique risks, burdens, and benefits to the patient, including comparative level of efficacy, route of administration, frequency of administration, known side effect profile, and time to clear the drug in cases of an adverse event. Finally, given universal NBS has already begun in several states in the US, both the collection of natural history data and the assessment of long-term term cost effectiveness of NBS for SMA will be critical to further inform treatment and care of SMA patients.
CONFLICT OF INTEREST
JJ and JG: Employees of Cure SMA and have no financial stake in any SMA drug or company involved in developing SMA drugs.
JS: Co-investigator on SMA clinical trials sponsored by Biogen, Roche, AveXis, and Cytokinetics. No grants or advisory boards, no stock held.
AP: Employee of the Muscular Dystrophy Association and has no financial stake in any SMA drug or company involved in developing SMA drugs.
AC: Reports advisor/consultant for AveXis, Bristol Meyer Squibb, Cytokinetics, Genzyme, Roche, and Sarepta; principal investigator for clinical studies for PTC Therapeutics, Pfizer, Fibrogen, NS Pharma; Biogen, AveXis, Cytokinetics, Sarepta, Cytokinetics, and Italefarmeco and member of data management and safety monitoring board for Catabasis.
BD: Serves on advisory boards for AveXis, Biogen, Bristol Myers Squibb, Cytokinetics, Marathon, PTC, Roche, and Sarepta; grants from Cytokinetics, FibroGen, Ionis/Biogen Pharmaceuticals, Inc. (during ENDEAR, CHERISH, CS12, CS11), National Institutes of Health (National Institute of Neurological Disorders and Stroke), PTC, Sarepta, Slaney Fund for SMA, SMA Foundation, and Summit.
JD: Has received grants for research trials from: Biogen; Ionis Pharmaceuticals; Cytokinetics; Roche Pharmaceuticals; aTyr; AveXis; Bristol Meyers Squibb; Sanofi-Genzyme; AMO Pharmaceuticals and Sarepta Therapeutics in addition to NIH, MDA, PPMD, MDF, CureSMA and SMA. He has served as a consultant for: Biogen; Ionis Pharmaceuticals; Roche Pharmaceuticals; Cytokinetics; Pfizer; AveXis; AMO Pharmaceuticals; Sarepta Therapeutics; Santhera Pharmaceuticals. He has patents licensed to Athena Diagnositcs for genetic testing of myotonic dystrophy type 2 (US patent 7442782) and spinocerebellar ataxia type 5 (US patent 7527931).
RF: Grants/advisor fees from Ionis Pharmaceuticals. Inc. during ENDEAR and CHERISH; grants/advisor fees from Biogen; grants from Cytokinetics; advisor to AveXis, Roche and Novartis outside the submitted work; advisor to nonprofit organizations: Muscular Dystrophy Association, Cure SMA, SMA Europe, Spinal Muscular Atrophy Foundation, and SMA Reach (UK); data safety monitoring board for the AveXis gene transfer study and Roche Moonfish study.
RRH: Serves on the advisory boards of Baebies, Inc., Sarepta Therapeutics, Amicus Therapeutics, and Biomarin. He served as Founding Chair of the HHS Secretary’s Advisory Committee on Heritable Disorders in Newborns and Children (2004-2012). He is a Board Member of the following non-Profit Organizations: Muscular Dystrophy Association (Board Chair), International Society of Neonatal Screening (President), National PKU Alliance, Save Babies Through Screening Foundation, Newborn Foundation, Dr. John T. MacDonald Foundation, and the American College of Medical Genetics Foundation.
KK: Employee of Sanofi, a company which has in the past had therapeutic programs in SMA, and may have such programs in the future; and has served on advisory boards/committees for CureSMA and the SMA Foundation.
NK: Serves on advisory boards for Biogen; advisory boards and consulting fees for AveXis, Catalyst, Cytokinetics, Marathon, PTC, and Sarepta outside the submitted work; advisory capacity to CureSMA and the Myasthenia Gravis Foundation of America.
TP: No conflicts to report.
PS: Serves on ad hoc advisory boards for Biogen and AveXis; outside the submitted work: advisory boards for PTC and Sarepta; speaker for Alexion and Grifols; research funding from NIH/NINDS, Biogen, AveXis, Audentes, Bristol-Myers Squibb, Cytokinetics, Catalyst, Fibrogen, Ionis Pharmaceuticals, Inc., Marathon, Pfizer, PTC, Sarepta, Santhera, Summit, Sanofi/Genzyme, and Ultragenyx.
TC: Advisor/consultant to AveXis, Biogen, Catalyst, Cytokinetics, Marathon, Novartis, Pfizer, Roche, Sarepta, CureSMA, and the SMA Foundation.
DK: Employee and holds equity in Generation Bio, a company that does not have an SMA program in development; and has no financial stake in any SMA drug or company involved in developing SMA drugs.
ACKNOWLEDGMENTS
We would like to thank the SMA NBS Coalition members, Biogen and AveXis, for funding this project. These funders did not play any role, however, in designing the consensus process, participating in debates, or in crafting or reviewing the guidelines. We also thank Sudipta Chakraborty, PhD, at Synchrogenix for writing the early drafts of this manuscript. We also acknowledge the key role of Susan Martin at RTI for serving as the expert in the Delphi process as a neutral non-voting, third party member who moderated discussions and had no stake in the decisions made.
REFERENCES
[1] | Lefebvre S , Burglen L , Reboullet S , Clermont O , Burlet P , Viollet L , et al. Identification and characterization of a spinal muscular atrophy - determining gene. Cell. (1995) ;80: (1):155–65. |
[2] | Arnold WD , Kassar D , Kissel JT . Spinal muscular atrophy: Diagnosis and management in a new therapeutic era. Muscle Nerve. (2015) ;51: (2):157–67. |
[3] | Sugarman EA , Nagan N , Zhu H , Akmaev VR , Zhou Z , Rohlfs EM , et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. Eur J Hum Genet. (2012) ;20: (1):27–32. |
[4] | Munsat TL , Davies KE . International SMA consortium meeting (26-28 June Bonn, Germany). Neuromuscul Disord. (1992) ;2: (5-6):423–28. |
[5] | Wang CH , Finkel RS , Bertini ES , Schroth M , Simonds A , Wong B , et al. Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. (2007) ;22: :1027–49. |
[6] | Ogino S , Leonard DG , Rennert H , Ewens WJ , Wilson RB . Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet. (2002) ;110: (4):301–7. |
[7] | Verhaart IE , Robertson A , Wilson IJ , Aartsma-Rus A , Cameron S , Jones CC , et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. (2017) ;12: (1):124. |
[8] | Finkel RS , McDermott MP , Kaufmann P , Darras BT , Chung WK , Sproule DM , et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. (2014) ;83: (9):810–7. |
[9] | Thomas NH , Dubowitz V . The natural history of type I (severe) spinal muscular atrophy. Neuromuscul Disord. (1994) ;4: (5-6):497–502. |
[10] | Zerres K , Rudnik-Schoneborn S . Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. (1995) ;52: (5):518–23. |
[11] | Swoboda KJ , Prior TW , Scott CB , McNaught TP , Wride MC , Reyna SP , et al. Natural history of denervation in SMA: Relation to age, SMN2 copy number, and function. Ann Neurol. (2005) ;57: (5):704–12. |
[12] | Mailman MD , Heinz JW , Papp AC , Snyder PJ , Sedra MS , Wirth B , et al. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet Med. (2002) ;4: :20–6. |
[13] | Wadman RI , Stam M , Gijzen M , Lemmink HH , Snoeck IN , Wijngaarde CA , et al. Association of motor milestones, SMN2 copy and outcome in spinal muscular atropy types 0-4. J Neurol Neurosurg Psychiatry. (2017) ;88: :365–7. |
[14] | Cuscó I , Barceló MJ , Rojas-García R , Illa I , Gamez J , Cervera C , et al. SMN2 copy number predicts acute or chronic spinal muscular atrophy but does not account for intrafamilial variability in siblings. J Neurol. (2006) ;253: :21. |
[15] | Feldkotter M , Schwarzer V , Wirth R , Wienker TF , Wirth B . Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. (2002) ;70: (2):358–68. |
[16] | Mercuri E , Finkel RS , Muntoni F , Wirth B , Montes J , Main M , et al. Diagnosis and management of spinal muscular atrophy: Part 1: Recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. (2017) . doi: 10.1016/j.nmd.2017.11.005 |
[17] | Finkel RS , Mercuri E , Meyer OH , Simonds AK , Schroth MK , Graham RJ , et al. Diagnosis and management of spinal muscular atrophy. Part 2: Pulmonary and acute care; medications, supplements and immunizations; other organ systems; and ethics. Neuromuscul Disord. (2017) . doi: 10.1016/j.nmd.2017.11.004 |
[18] | Oskoui M , Levy G , Garland CJ , Gray JM , O’Hagen J , De Vivo DC , et al. The changing natural history of spinal muscular atrophy type 1. Neurology. (2007) ;69: (20):1931–6. |
[19] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , et al. Single-dose gene-replacement therapy for spinal muscular atrophy. New Engl J Med. (2017) ;377: (18):1713–22. |
[20] | Scoto M , Finkel RS , Mercuri E , Muntoni F . Therapeutic approaches for spinal muscular atrophy (SMA). Gene Ther. (2017) ;24: :514–9. |
[21] | Phan HC , Taylor JL , Hannon H , Howell R . NBS for spinal muscular atrophy: Anticipating an imminent need. Semin Perinatol. (2015) ;39: (3):217–29. |
[22] | Farrar MA , Park SB , Vucic S , Carey KA , Turner BJ , Gillingwater TH , et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. (2016) . doi: 10.1002/ana.24864 |
[23] | Lin CW , Kalb SJ , Yeh WS . Delay in diagnosis of spinal muscular atrophy: A systematic literature review. Pediatr Neurol. (2015) ;53: (4):293–300. |
[24] | Haataja L , Mercuri E , Regev R , Cowan F , Rutherford M , Dubowitz V , et al. Optimality score for the neurologic examination of the infant at 12 and 18 months of age. J Pediatr. (1999) ;135: :153–61. |
[25] | Finkel RS , Mercuri E , Darras BT , Connolly AM , Kuntz NL , Kirschner J , et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N Engl J Med. (2017) ;377: :1723–32. |
[26] | De Vivo DC , Hwu W-L , Reyna SP , Farwell W , Gheuens S , Sun P , et al. on behalf of the NURTURE Study Group. Interim efficacy and safety results from the Phase 2 NURTURE study evaluating nusinersen in presymptomatic infants with spinal muscular atrophy In: American Academy of Neurology; 2017 April 22-28; Boston, MA. |
[27] | Taylor JL , Lee FK , Yazdanpanah GK , Staropoli JF , Liu M , Carulli JP , et al. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin Chem. (2015) ;61: (2):412–9. |
[28] | Chien YH , Chiang SC , Weng WC , Lee NC , Lin CJ , Hsieh WS , et al. Presymptomatic diagnosis of spinal muscular atrophy through NBS. J Pediatr. (2017) ;190: :124–9. |
[29] | Gutierrez-Mateo C , Baker M , Mochal S , Moore K , Filippov G , Dallaire S . A five-plex qPCR assay that measures copy numbers of SMN1, SMN2, TREC, KREC and RPP30. In: American College of Medical Genetics and Genomics; 2017 March 21-25; Phoenix, AZ. |
[30] | Delbecq A , Van de Ven A , Gustafson D . The Delphi technique. In: Delbecq A, Van de Ven A, Gustafson D, editors. Group techniques for program planning: A guide to nominal group and Delphi processes. Madison, WI: Scott Foresman; (1975) . pp. 83–107. |
[31] | Rowe G , Wright G . Expert opinions in forecasting. Role of the Delphi technique. In: Armstrong JS, editor. Principles of forecasting: A handbook of researchers and practitioners. Boston: Kluwer Academic Publishers; (2001) . |
[32] | Hsu CC , Sandford BA . The Delphi technique: Making sense of consensus practical assessment. Res Eval. (2007) ;12: :1–10. |
[33] | Sinha IP , Smyth RL , William PR . Using the Delphi technique to determine which outcomes to measure in clinical trials: Recommendations for the future based on an systematic review of existing studies. PLoS Med. (2011) ;8: (1). doi: 10.1371/journal.pmed.1000393 |
[34] | MacLeod MJ , Taylor JE , Lunt PW , Mathew CG , Robb SA . Prenatal onset spinal muscular atrophy. Eur J Paediatr Neurol. (1999) ;3: :65–72. |
[35] | Prior TW , Krainer AR , Hua Y , Swoboda KJ , Snyder PC , Bridgeman SJ , et al. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. (2009) ;85: :408–13. doi: 10.1016/j.ajhg.2009.08.002 |
[36] | Bernal S , Alías L , Barceló MJ , Also-Rallo E , Martínez-Hernández R , Gámez J , et al. The c.859G>C variant in the SMN2 gene is associated with types II and III SMA and originates from a common ancestor. J Med Genet. (2010) ;47: :640–2. |
[37] | Vezain M , Saugier-Veber P , Goina E , Touraine R , Manel V , Toutain A , et al. A rare SMN2 variant in a previously unrecognized splicing element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat. (2010) ;31: (1):E1110–25. doi: 10.1002/humu.21173 |
[38] | Kolb SJ , Coffey CS , Yankey JW , Krosschell K , Arnold WD , Rutkove SB , et al. Baseline results of the NeuroNEXT spinal muscular atrophy infant biomarker study. Ann Clin Transl Neurol. (2016) ;3: (2):132–145. doi: 10.1002/acn3.283 |
[39] | Lewelt AJ , Krosschell KJ , Scott C , Sakonju A , Kissel JT , Crawford TO , et al. Compound muscle action potential and motor function in children with spinal muscular atrophy. Muscle Nerve. (2010) ;42: (5):703–8. |
[40] | Sloan C . Review of the reliability and validity of myometry with children. Phys Occup Ther Pediatr. (2002) ;22: (2):79–93. |
[41] | Glanzman AM , Mazzone E , Main M , Pelliccioni M , Wood J , Swoboda KJ , et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscul Disord. (2010) ;20: (3):155–61. |
[42] | O’Hagen JM , Glanzman AM , McDermott MP , Ryan PA , Flickinger J , Quigley J , et al. An expanded version of the Hammersmith Functional Motor Scale for SMA II and III patients. Neuromuscul Disord. (2007) ;17: (9-10):693–7. |
[43] | McDonald CM , Henricson EK , Han JJ , Abresch RT , Nicorici A , Elfring GL , et al. The 6-minute walk test as a new outcome measure in Duchenne muscular dystrophy. Muscle Nerve. (2010) ;41: (4):500–10. |
[44] | Bayley N . Bayley-III: Bayley Scales of infant and toddler development. 3rd ed. San Antonio, TX: Pearson; (2006) . |
[45] | Connolly AM , Florence JM , Cradock MM , Malkus EC , Schierbecker JR , Siener CA , et al. Motor and cognitive assessment of infants and young boys with duchenne muscular dystrophy; results from the Muscular Dystrophy Association DMD Clinical Research Network. Neuromuscul Disord. (2013) ;23: (7):529–39. |


