
An Extended Targeted Copy Number Variation Detection Array Including 187 Genes for the Diagnostics of Neuromuscular Disorders
Abstract
Background:
Our previous array, the Comparative Genomic Hybridisation design (CGH-array) for nemaline myopathy (NM), named the NM-CGH array, revealed pathogenic copy number variation (CNV) in the genes for nebulin (NEB) and tropomyosin 3 (TPM3), as well as recurrent CNVs in the segmental duplication (SD), i.e. triplicate, region of NEB (TRI, exons 82–89, 90–97, 98–105). In the light of this knowledge, we have designed and validated an extended CGH array, which includes a selection of 187 genes known to cause neuromuscular disorders (NMDs).
Objective:
Our aim was to develop a reliable method for CNV detection in genes related to neuromuscular disorders for routine mutation detection and analysis, as a much-needed complement to sequencing methods.
Methods:
We have developed a novel custom-made 4×180 k CGH array for the diagnostics of NMDs. It includes the same tiled ultra-high density coverage of the 12 known or putative NM genes as our 8×60 k NM-CGH-array but also comprises a selection of 175 additional genes associated with NMDs, including titin (TTN), at a high to very high coverage. The genes were divided into three coverage groups according to known and potential pathogenicity in neuromuscular disorders.
Results:
The array detected known and putative CNVs in all three gene coverage groups, including the repetitive regions of NEB and TTN.
Conclusions:
The targeted neuromuscular disorder 4×180 k array-CGH (NMD-CGH-array v1.0) design allows CNV detection for a broader spectrum of neuromuscular disorders at a high resolution.
INTRODUCTION
Congenital myopathies are hereditary disorders within the heterogeneous group of neuromuscular disorders (NMDs). They may be caused by mutations, including copy number variations (CNVs), in genes coding for structural proteins of the sarcomere, genes for membrane or matrix proteins, or genes affecting muscular development by playing a role in e.g. protein turnover or muscle differentiation [1]. The wide array of causative genes and mutations leads to a widely varying group of disorders; they may be passed on according to all modes of inheritance and they display a multitude of clinical phenotypes, with variation in progression, severity and distribution of muscle weakness [2, 3].
It is important to note that the phenotypes of congenital myopathies may overlap with each other and with other neuromuscular disorders, and that different mutations in the same gene may cause various phenotypes, thus challenging diagnostics. Specifically designed follow-up and specific putative future therapies can be administered only when the correct molecular diagnosis and the underlying causative mechanisms of the disorder have been determined [3, 4].
CNVs are structural changes of the genome, in which a certain genomic sequence is present in the studied genome at an aberrant copy number compared with the reference genome. CNVs were initially described as variations of at least 1 kb in size [5], but modern technology has allowed CNV detection at a higher resolution, and the minimum size for a deleted or duplicated sequence has been suggested to be as small as 50 bp [6]. Estimations are that de novo locus-specific CNVs have a higher mutation rate compared with nucleotide substitutions [5].
While thousands of regions are prone to benign CNVs [7], variants that interfere with normal gene expression or function can be potentially pathogenic [8]. For example, Duchenne muscular dystrophy (DMD; MIM ID: DMD #310200) is commonly caused by CNVs in the DMD gene [9, 10]. Furthermore, pathogenic CNVs have been shown to contribute to roughly 6.4% of nemaline myopathy cases (NM; MIM IDs: NEM1 #609284, NEM2 #256030, NEM3 #161800, NEM4#609285, NEM5 #605355, NEM6 #609273, NEM7 #610687, NEM8 #615348, NEM9 #615731, NEM10 #616165, NEM11 #617336) [11].
Array Comparative Genomic Hybridisation (aCGH) constitutes the standard method for research and diagnostics into disorders with pathogenetic mechanisms involving CNVs. Copy number variation analysis methods based on Next-Generation Sequencing (NGS) are evolving [12], but the precision and reliability of aCGH remains to be matched by computer algorithms.
Especially regions of segmental duplication (SD) pose a challenge to NGS based CNV analysis methods [13], and are in fact often excluded from commercial CGH arrays and sequencing panels. Such regions are found both in the nebulin gene (NEB) [14] and the titin gene (TTN) [15], the products of which are enormous sarcomeric proteins. In NEB, this region holds a threefold repetition of an 8-exon block (exons 82–89, 90–97 98–105), referred to as the triplicate region (TRI). The NEB TRI region is known to harbour CNVs – gains and losses of one 8-exon block are considered benign, while gains of more than one 8-exon block are considered to be pathogenic [16]. In TTN, the SD constitutes a threefold repetition of 9 exons (exons 166–174, 175–183, and 184–192) within the PEVK segment of the gene, followed by fourth copies of the first two exons of the repeated block [15]. CNVs in this type of region are all but rare, and may potentially be pathogenic [16].
Here we describe our custom targeted neuromuscular disorder CGH array (NMD-CGH array v1.0), targeting 187 different genes related to NMDs. As NM can be caused by CNVs [11, 17, 18], especially common in NEB, we hypothesised that CNVs may be disease-causing also in other genes related to NMDs – consequently, that there probably are undiscovered pathogenic CNVs to be identified in these genes.
The array targets a wide variety of genes related to NM and other NMDs. The design includes our previously published, targeted 8×60 k nemaline myopathy CGH-array (NM-CGH array) [11] in an updated version. In addition to the 11 published and one unpublished NM-causing gene; ACTA1, NEB, TPM3, TPM2, TNNT1, CFL2, KBTBD13, KLHL40, KLHL41, LMOD3, MYPN and YBX3, the array includes 175 carefully chosen genes related to other NMDs – amongst these, the TTN, dystrophin (DMD), the ryanodine receptor (RYR1), and obscurin (OBSCN) genes.
METHODS
Samples
Forty-six DNA samples from 19 different clinicians in 11 countries were acquired for the validation of the array. The 46 DNA samples included 3 positive controls with known CNVs, 9 blind cross-validation samples, and 30 patient samples. In addition, we acquired 4 healthy control samples, of which 3 from the Finnish Red Cross Blood Service and one commercial reference sample (Male reference DNA, Promega Corporation, Madison, WI, USA). (Table 1).
Table 1
The representative numbers of samples and families included in the validation process
| Samples | Families | |
|---|---|---|
| Healthy controls | 4 | 4 |
| Positive controls | 3 | 3 |
| Cross-validation samples | 9 | 9 |
| Patient samples, of which | 30 | 20 |
| First mutation previously identified | 10 | 8 |
| Candidate mutation previously identified | 7 | 3 |
| Both mutations unidentified | 13 | 9 |
| Altogether | 46 | 36 |
The study was approved by the local Ethics Committees and samples were obtained according to the Declaration of Helsinki of 1975.
Microarray design, protocol, and data analysis
The CGH array was designed with the support of Oxford Gene Technology IP Limited Ltd (Oxford, UK) (Human reference sequence, GRCh37/Hg19). The array covers 187 genes associated with muscular disorders, divided into 3 different categories of highly targeted levels.
The array includes the known NM genes (ACTA1, CFL2, KBTBD13, KLHL40, KLHL41, LMOD3, NEB, TNNT1, TNNT3, TPM2, TPM3, MYPN) and 176 known or candidate neuromuscular disorder genes, including those of the 1st version of the targeted NGS MyoCap panel [19]. The current number of targeted probes chosen for the design is 150,300/180,000, leaving space for future additions and updates.
The genes have been divided into three gene coverage groups of varying coverage according to the known and potential NMD-causing pathogenicity harboured by the genes. Coverage group 1 includes the NM genes at the same ultra-highly tiled coverage as the previously validated and published 8×60 k NM-CGH array [11]. Coverage group 2 consists of 29 other characterised muscle disorder genes (ANO5, BAG3, BIN1, CRYAB, DES, DNM2, DYSF, FLNC, GNE, HSPB8, KLHL9, LDB3, MTM1, MYBPC1, MYBPC2, MYH1, MYH13, MYH2, MYH3, MYH4, MYH7, MYH8, MYOT, OBSCN, OBSL1, RYR1, SEPN1, TIA1, TTN) at a very high coverage. The remaining 147 genes (Supplementary Table 1) of coverage group 3 were also targeted at a high level of coverage, with approximately 4 probes covering each exon. The coverage group 2 and 3 genes are either known neuromuscular disorder genes or strong candidates for such. Of these, we selected genes coding for structural proteins of the sarcomere to be targeted at group 2 level as their putative impact on muscular structure is clearer than that of regulatory genes such as transcription factors, which were targeted at group 3 level. Exceptions to this rule are genes that are known to harbour pathogenic or putative pathogenic CNVs in group 2.
In all the groups, a minimum of one probe per exon and per intron was targeted in each gene. Flanking regions were included 25 kb upstream and downstream of the targeted genes. The coverage level of the three gene groups is presented in Table 2.
Table 2
The coverage of exon, intron and flanking regions in the three subgroups
| Coverage by gene coverage group | |||
|---|---|---|---|
| Group 1 | Group 2 | Group 3 | |
| Exon | 10 bp tiling, +/– | Adjacent probes, +/– | ∼4 probes/exon |
| Intron | 20 bp tiling, +/– | Adjacent probes, +/– | ∼1–3 probes/intron |
| Flanking | ±25 kb | ±25 kb | ±25 kb |
+/– signifies probe targeting on both the forward and the reverse strand. ±signifies the region covered both upstream and downstream of the targeted genes.
In addition, the backbone coverage was increased from approximately 1 probe per Mb to 1 probe per 300 kb, in order to allow for the detection of large genomic rearrangements, as well as better alignment throughout the genome. The 12,300 backbone probes are spaced evenly across the non-targeted regions.
The USCS Genome Browser (https://genome.ucsc.edu/) online application was used to visualise the probe distribution in the designing process. All genes were targeted at least at the exon level. In addition to the Oxford Gene Technology in-house probes, new custom probes were designed for the regions of interest, especially in the regions of repetitive sequences.
Labelling, hybridisation, scanning and analysis of the array was performed according to the manufacturer’s protocol (Oxford Gene Technology Ltd, ISCA array, Cytosure Genomic DNA labelling kit protocol, 4×180 k format, code 020020). We used a commercial male DNA reference for all the studied samples (Promega Corporation). The array slides were scanned using the Agilent DNA Microarray Scanner G2505C with 2μm resolution, Agilent Scan Control version A.8.5.1 (Agilent Technologies Inc, Santa Clara, CA, USA) and normalised and further analysed in Feature Extraction software v.12.0.0.7 (Agilent Technologies). The Cytosure Software v.4.6.85 (Hg19) (Oxford Gene Technology Ltd) was used for graphic analysis of the data. A minimum of 5 probes was used as a threshold for making a positive call for an aberration. The array profiles and aberration calls were also manually checked.
Validation
The initial validation of the NMD-CGH array was made using 4 healthy controls and 3 positive controls (identified with the NM-CGH array). In addition, we performed a blind cross-validation process with 9 samples. In these, CNVs had been detected with 4 different NGS-based CNV detection algorithms (CoNIFER v0.2.2, XHMM v1.1, ExomeDepth v1.1.10 and CODEX v1.4.0). The CNV detection by algorithms was performed as described in Välipakka et al., 2017 [20].
Subsequently, we ran 30 patient samples from 20 families, which had not previously been analysed for CNVs by any method.
RESULTS
The validation runs on the 7 control samples confirmed the coverage, specificity, and resolution of the array and the quality of the probes chosen. In the 4 healthy controls (samples 9–12), we identified a gain in the TTN SD region (coverage group 2) in 1 sample. In the 3 positive controls (samples 1-2 and 8), we identified 1 one-copy loss in the NEB TRI region (coverage group 1) and 3 losses in the TTN SD region, as well as the known pathogenic changes in NEB (coverage group 1) and DMD (coverage group 3).
In the blind cross-validation samples (16–23, 46), we were able to detect the expected CNVs. The changes include a partial duplication of the adjacent coverage group 2 genes MYH6 and MYH7 (Fig. 1A) and duplications in coverage group 3 genes MYOM and PYGM (Fig. 1B-C). Moreover, deletions in the genes TTN (Fig. 1D), DMD, LDHB, and the upstream region of COL6A3 were detected (all but TTN belong to coverage group 3). A supposedly benign gain was detected in the TTN SD region of one sample (Fig. 1F). The NGS-based analysis programmes had flagged one change that was not detected on the array and was concluded to be a false positive. The cross-validation process confirmed the ability of the array to detect CNVs in all gene coverage groups.
Fig.1
A demonstration of the array results. NEB is targeted at Coverage group 1 level, TTN and MYH7 are targeted at Coverage group 2 level and MYOM1 and PYGM are targeted at Coverage group 3 level.
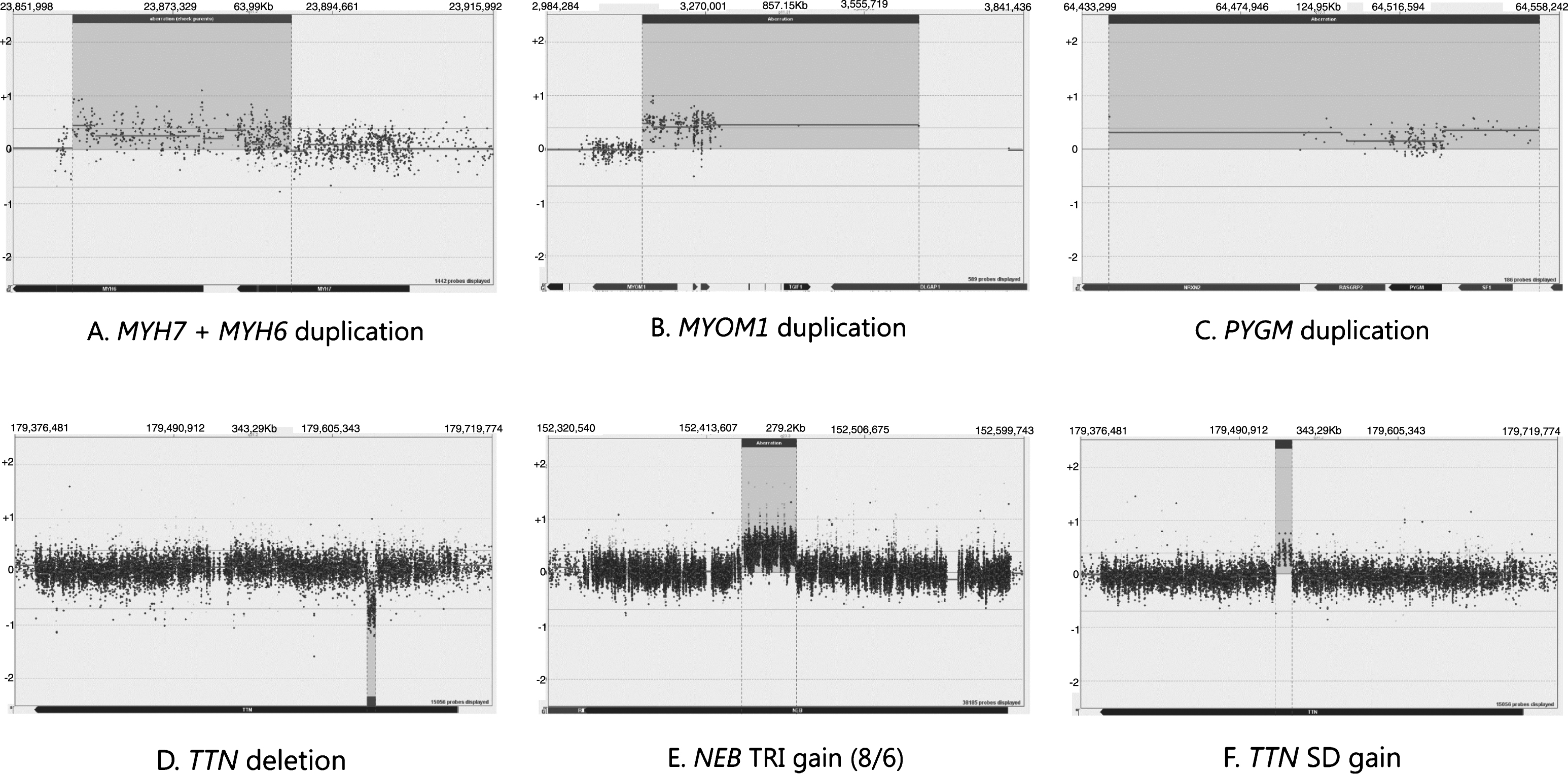
The diagnostic runs performed on patient samples (samples 3–7, 13–15, 24–45) revealed two aberrations of unknown significance in coverage group 2 genes, one upstream deletion of ANO5 and one upstream duplication of OBSL1 partially covering the gene DNPEP. Benign variation of the NEB TRI region was detected in 2 samples altogether, an example of which is presented in Fig. 1E. Copy number variation in the TTN SD region was seen in 8 diagnostic samples, of which 3 were losses and 5 were gains.
In addition to the known and putative pathogenic aberrations and known benign variations detected, the novel NMD-CGH array revealed previously unrecorded variation in the TTN SD region (Table 3). Variation in the TTN SD region was seen in 13 samples altogether. These findings warrant further studies and will be the subject of another publication.
Table 3
The prevalence of gains and losses in the NEB triplicate region (TRI) and TTN segmental duplication (SD) region in the different sample categories.
| Positive | Healthy | Patients and | Total | |
| controls | controls | blind validation | ||
| samples | ||||
| NEB TRI gain | 0 | 0 | 1 | 1 |
| NEB TRI loss | 0 | 0 | 1 | 1 |
| TTN SD gain | 0 | 1 | 6 | 7 |
| TTN SD loss | 3 | 0 | 3 | 6 |
All changes identified, including the controls and cross-validation samples, are presented in Table 4.
Table 4
Copy number variations identified on the NMD-CGH-array during the validation process and the subsequent diagnostic runs
| Sample ID | Gene | Variation | Size | Coverage group |
|---|---|---|---|---|
| 1 | DMD partial deletion | ChrX.hg19:g.(32,551,900_32,563,471)_(32,953,942_32,978,285)del | 390,471–466,385 bp | 3 |
| 1, 2, 8, 13 | TTN SD loss | Chr2.hg19:g.(179,516,753_179,517,001)_(179,528,302_179,528,842)del | 11,301–12,089 bp | 2 |
| 5, 9, 19, 25 | TTN SD gain | Chr2.hg19:g.(179,516,799_179,517,346)_(179,528,170_179,529,256)dup | 10,824–12,457 bp | 2 |
| 29 | NEB TRI loss | Chr2.hg19:g.(152,433,608_152,434,494)_(152,464,242_152,464,694)del | 29,748–31,086 bp | 1 |
| 25 | NEB TRI gain | Chr2.hg19:g.(152,433,608_152,434,494)_(152,466,075_152,466,085)dup | 31,581–32,477 bp | 1 |
| 3, 38 | OBSL1 upstream duplication | Chr2.hg19:g.(220,438,234_220,439,293)_(220,460,509_220,569,618)dup | 21,216–131,384 bp | 2 |
| 8 | NEB partial deletion | Chr2.hg19:g.(152,434,836_152,435,622)_(152,554,712_152,561,404)del | 109,090–126,568 bp | 1 |
| 15 [20] | TTN partial deletion | Chr2.hg19:g.(179,630,403_179,630,386)_(179,636,145_179,636,177)del | 5,754–5,774 bp | 2 |
| 16 | LDBH partial deletion | Chr12.hg9:g.(21,803,624_ 21,806,090)_(21,811,051_ 21,812,528)del | 4,961–8,904 bp | 3 |
| 17 | DMD partial deletion | ChrX.hg19:g.(30,843,289_31,086,990)_(31,313,100_31,327,408)del | 226,110–484,119 bp | 3 |
| 18 | MYOM1 partial duplication | Chr18.hg19:g.(3,155,151_3,155,985)_(3,653,512_3,815,684)dup | 497,527–660,533 bp | 3 |
| 20 | PYGM duplication | Chr11.hg19:g.(64,155,908_64,440,343)_(64,553,037_64,758,928)dup | 112,694–603,020 bp | 3 |
| 21 | MYH6+MYH7 partial duplication | Chr14.hg19:g.(23,859,986_23,859,988)_(23,889,063_23,889,443)dup | 29,075–29,457 bp | 2 |
| 36 | ANO5 upstream deletion | Chr11.hg19:g.(21,579,750_21,931,190)_(22,207,427_22,208,122)del | 276,237–628,372 bp | 2 |
| 46 | COL6A3 upstream deletion | Chr2.hg19:g.(238,318,105_238,322,413)_(238,346,871_238,440,079)dup | 23,458–121,974 bp | 3 |
To date, we have run around 410 samples from approximately 300 families with NM or related disorders using the NM-CGH or NMD-CGH-array. The yield of pathogenic CNVs in these samples has been altogether around 13%, of which 4.5% are pathogenic CNVs within the NEB TRI region.
DISCUSSION
The design of the novel NMD-CGH array allows us to reliably identify CNVs in genes associated with muscle disorders, on a resolution varying from circa 70 bp (unpublished data) to the exon level, depending on the gene coverage group. The design can and will be updated based on new findings: we can move genes to a higher-tiled coverage group, or add altogether new genes to the array. Any recurrent, unclear or putatively pathogenic variation identified on our array or by a different method can be basis enough to move a gene to the higher coverage group. It is thus a dynamic method for the research and diagnostics of CNVs in genes causing neuromuscular disorders.
The human genome harbours more than 1000 regions prone to benign variation in terms of copy number, and CNVs have already been proven to constitute part of the mutation spectra behind muscle disorders, e.g. NM [1, 17, 18]. In patients lacking a molecular diagnosis for their disorders or in whom only one recessive mutation has been identified, the NMD-CGH array allows for precise and reliable detection of deletions and duplications in a wide range of NMD genes and NMD candidate genes. The genes were carefully picked out and divided into the coverage groups based on the current knowledge of proven and putative NMD genes, and the resolution at which these genes and their CNVs can be detected on the array.
Despite advancements in NGS data analysis of CNVs, aCGH still remains the gold standard method for CNV detection and analysis, especially in diagnostics, due to the method being well-established and reliable, and its quick sample turnover time. The repetitive nature of the SD regions in certain genes, e.g. NEB and TTN, present challenges for analysis by conventional NGS methods. Algorithms can predict CNVs, but whole-exome, targeted exome, or whole-genome data are not as clear, precise and sensitive as a well-designed CGH array. The number of false positives and false negatives is higher, and the findings still require validation by other methods, such as aCGH. Repetitive regions may harbour CNVs in unconventional numbers [16], with the baseline deviating from the normal diploid copy number of two, and these can be difficult to determine using NGS-based methods and read counts only. In addition, the CNV breakpoints may be pinpointed with higher precision using a densely tiled aCGH design compared with sequence-based methods. However, for determining exact breakpoints at the base-pair level, Sanger sequencing is still the only reliable method.
Our array allows for the detection of CNVs, not only in NMD genes in general, but also in the repetitive NEB TRI region [16], and the SD region of TTN. These regions are typically excluded from commercial CGH-array designs because of the difficulty their repetitive nature imposes on the analysis. In terms of the NEB TRI region, our array has been validated [16], and we are currently validating the analysis of the SD region of TTN.
Based on our results so far, we expect the yield of pathogenic CNVs to be similar in range (∼13%) as more samples will be run.
CONCLUSIONS AND FUTURE PROSPECTS
The NMD-CGH-array constitutes a dependable CNV analysis method for the diagnostics of NMDs. The use of the NMD-CGH array will continue in our laboratory as a first tier diagnostic tool for patients and families whose disease-causing variants remain unidentified.
The recurrent CNV in the SD region of TTN will be the topic of a further project. Preliminary data suggest that the region is highly prone to variation in a similar fashion as the NEB TRI region. It is possible that this region, too, will be found in some patients to contain modifying or pathogenic CNVs.
CONFLICT OF INTEREST
The authors declare no conflict of interest to report.
AUTHOR CONTRIBUTIONS
LS ran the array hybridisations, carried out analysis and interpretation of the data and prepared the manuscript. LS and KK designed the extended 4×180 k NMD-CGH-array. SV performed the NGS-based CNV-analysis for the blind cross-validation. BU provided samples for the blind cross-validation. KK set up the array method in the laboratory and prepared the manuscript. V-LL, LS, KK and SV selected samples for the array. CW-P was responsible for the project as PI. KP was responsible for the project as Co-PI. All authors have participated in the planning of the study, interpretation of the results, and in the writing of the manuscript.
ACKNOWLEDGMENTS
We thank Dr. Nina Horelli-Kuitunen for sharing samples for the validation process, Marco Savarese, Mridul Johari, and Merja Soininen for their contributions to the validation process, and Marilotta Turunen and Sampo Koivunen for outstanding technical assistance. This study was supported by grants from Muscular Dystrophy UK, the Sigrid Jusélius Foundation, the Folkhälsan Research Foundation, the Finska Läkaresällskapet, the Medicinska understödsföreningen Liv och Hälsa, l’Association Française contre les Myopathies, the Jane and Aatos Erkko Foundation, the Academy of Finland (no. 138491, B.U.), and the Orion Research Foundation sr.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: 10.3233/JND-170298.
REFERENCES
[1] | Kiiski K , Lehtokari VL , Manzur AY , Sewry C , Zaharieva I , Muntoni F , . A Large deletion affecting tpm3, causing severe nemaline myopathy. J Neuromuscul Dis. ((2015) ); 2: (4): 433–8. doi: 10.3233/jnd-150107 |
[2] | North K What’s new in congenital myopathies? Neuromuscul Disord. ((2008) ); 18: (6) 433–42. doi: 10.1016/j.nmd.2008.04.002 . |
[3] | Jungbluth H , Ochala J , Treves S , Gautel M Current and future therapeutic approaches to the congenital myopathies. Semin Cell Dev Biol. ((2017) ); 64: : 191–200. doi: 10.1016/j.semcdb.2016.08.004 |
[4] | North KN , Wang CH , Clarke N , Jungbluth H , Vainzof M , Dowling JJ , . Approach to the diagnosis of congenital myopathies. Neuromuscul Disord. ((2014) ); 24: (2) 97–116. doi: 10.1016/j.nmd.2013.11.003 |
[5] | Redon R , Ishikawa S , Fitch KR , Feuk L , Perry GH , Andrews TD , . Global variation in copy number in the human genome. Nature. (2006) ;444: (7118): 444–54. doi: 10.1038/nature05329 . |
[6] | Alkan C , Coe BP , Eichler EE Genome structural variation discovery and genotyping. Nat Rev Genet.363-76. ((2011) ); 12: (5): doi: 10.1038/nrg2958 |
[7] | McCarroll SA , Kuruvilla FG , Korn JM , Cawley S , Nemesh J , Wysoker A , . Integrated detection and population-genetic analysis of SNPs and copy number variation. Nat Genet.. ((1166) ); 40: (10): 1166–74. doi: 10.1038/ng.238 . |
[8] | Freeman JL , Perry GH , Feuk L , Redon R , McCarroll SA , Altshuler DM , . Copy number variation: New insights in genome diversity. Genome Res.. ((2006) ); 16: (8)949–61. doi: 10.1101/gr.3677206 |
[9] | Koenig M , Beggs AH , Moyer M , Scherpf S , Heindrich K , Bettecken T , . The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am J Hum Genet. ((1989) ); 45: (4), 498–506. |
[10] | Den Dunnen JT , Grootscholten PM , Bakker E , Blonden LA , Ginjaar HB , Wapenaar MC , . Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications. Am J Hum Genet. ((1989) ); 45: (6), 835–47. |
[11] | Kiiski K , Laari L , Lehtokari V-L , Lunkka-Hytönen M , Angelini C , Petty RK , . Targeted array comparative genomic hybridization - A new tool for the detection of large copy number variations in nemaline myopathy-causing genes. Neuromuscular Disorders. ((2013) ); 23: (1):10. doi: 10.1016/j.nmd.2012.07.007 |
[12] | Chen Y , Zhao L , Wang Y , Cao M , Gelowani V , Xu M , . SeqCNV: A novel method for identification of copy number variations in targeted next-generation sequencing data. BMC Bioinformatics. ((2017) ); 18: (1): 147. doi: 10.1186/s12859-017-1566-3 |
[13] | Ravenscroft G , Davis MR , Lamont P , Forrest A , Laing NG New era in genetics of early-onset muscle disease: Breakthroughs and challenges. Semin Cell Dev Biol.. ((2017) ); 64: 160–70. doi: 10.1016/j.semcdb.2016.08.002 |
[14] | Donner K , Sandbacka M , Lehtokari VL , Wallgren-Pettersson C , Pelin K Complete genomic structure of the human nebulin gene and identification of alternatively spliced transcripts. Eur J Hum Genet.. ((2004) ); 12: (9)744–51. doi: 10.1038/sj.ejhg.5201242 |
[15] | Bang ML , Centner T , Fornoff F , Geach AJ , Gotthardt M , McNabb M , et al The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res.. ((2001) ); 89: (11)1065–72. doi: 10.1161/hh2301.100981 |
[16] | Kiiski K , Lehtokari V-L , Löytynoja A , Ahlstén L , Laitila J , Wallgren-Pettersson C , . A recurrent copy number variation of the NEB triplicate region: Only revealed by the targeted nemaline myopathy CGH array. European Journal of Human Genetics. ((2016) ); 24: (4)7. doi: 10.1038/ejhg.2015.166 |
[17] | Anderson SL , Ekstein J , Donnelly MC , Keefe EM , Toto NR , LeVoci LA , . Nemaline myopathy in the Ashkenazi Jewish population is caused by a deletion in the nebulin gene. Hum Genet.. ((2004) ); 115: (3)185–90. doi: 10.1007/s00439-004-1140-8 |
[18] | Lehtokari VL , Greenleaf RS , DeChene ET , Kellinsalmi M , Pelin K , Laing NG , . The exon 55 deletion in the nebulin gene–one single founder mutation with world-wide occurrence. Neuromuscul Disord. ((2009) ); 19: (3)179–81. doi: 10.1016/j.nmd.2008.12.001 . |
[19] | Evilä A , Arumilli M , Udd B , Hackman P Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscular Disorders. ((2016) ); 26: (1)9.. doi: 10.1016/j.nmd.2015.10.003 |
[20] | Välipakka S , Savarese M , Johari M , Sagath L , Arumilli M , Kiiski K , . Copy number variation analysis increases the diagnostic yield in muscle diseases. Neurology Genetics. ((2017) );3: (6): doi: 10.1212/nxg.0000000000000204 |


