
Beta-2 Adrenergic Receptor Agonists Enhance AChR Clustering in C2C12 Myotubes: Implications for Therapy of Myasthenic Disorders
Abstract
Background:
Congenital myasthenic syndromes (CMS) are a group of inherited neuromuscular transmission disorders causing fatiguable muscle weakness. ADRB2 agonists have been observed to provide therapeutic benefit where destabilisation of NMJ structures is part of the underlying pathology, such as in DOK7, COLQ and MuSK CMS as well as in slow channel syndrome. However, very little is known about the molecular mechanisms underlying the effects of ADRB2 agonists in CMS.
Objective:
In vitro investigation into whether an ADRB2 agonist affects the AChR clustering pathway and has the potential to increase the number and stability of AChR clusters.
Methods:
Cultured C2C12 mouse myotubes overexpressing the common DOK7 frameshift mutation c.1124_1127dupTGCC were incubated with salbutamol sulphate and the effect on AChR cluster numbers were investigated. Moreover, agrin-induced AChR clusters in C2C12 WT cells were left to disperse after agrin-wash-off, and the effects of incubation with salbutamol sulphate on AChR cluster numbers were explored.
Results:
Salbutamol sulphate induced a significant increase in the number of AChR clusters formed on C2C12 cells overexpressing c.1124_1127dupTGCC. Furthermore, significantly more clusters remained in C2C12 WT myotubes incubated with salbutamol sulphate following agrin wash-off.
Conclusions:
The results suggest that ADRB2 agonists directly affect proteins located at the neuromuscular junction and exert a stabilising effect on AChR clusters.
INTRODUCTION
Congenital myasthenic syndrome (CMS) is a group of inherited neuromuscular transmission disorders leading to symptoms of muscle weakness [1]. CMS is caused by mutations in genes that affect proteins located at the neuromuscular junction, and are rare conditions with, in the UK, an overall incidence of approximately 1:100,000 with a recent study giving a prevalence of 9.2 cases per one million children below the age of 18 [2]. As with many genetic disorders, worldwide this will vary depending upon ethnic groupings.
CMS results in a reduced safety factor of neurotransmission, but clinical features, age of onset and treatment strategies differ depending on the mutation [1, 3]. CMS is a very heterogeneous group of disorders, with at least 29 affected genes identified [4, 5]. In about 20% of UK CMS cases, patients have mutations in the gene DOK7 and develop a characteristic limb-girdle pattern of weakness [3, 6]. DOK7, expressed in skeletal muscle, was discovered in 2006 as a key molecule involved in the development and maintenance of the neuromuscular junction [7]. Dimeric DOK7 binds to the juxtamembrane region of muscle specific kinase (MuSK), promotes trans-autophosphorylation and, together with signalling of the nerve-derived protein agrin, activation of MuSK [8]. Subsequent downstream kinase activity of MuSK promotes the formation of a dense array of clustered acetylcholine receptors (AChR) on the postsynaptic membrane beneath the nerve terminal which is required for efficient neurotransmission [9]. Muscle biopsies from DOK7 patients revealed small simplified neuromuscular junctions (NMJ) [6, 10–12]. Postsynaptic junctional folds were often degenerated, and the length of the postsynaptic membrane was significantly reduced. Furthermore, absence or degeneration of nerve terminals was common. Overexpression of mutant DOK7 in cultured C2C12 mouse muscle cells, such as the commonly identified frameshift mutation c.1124_1127dupTGCC, caused a significant reduction in the number of AChR clusters [6].
The choice of treatment for CMS depends on the pathology underlying the disease, and drugs which reduce fatigable weakness in some CMS subtypes may be harmful in others. Neurotransmission enhancing acetylcholinesterase inhibitors, in some cases given in combination with 3,4-diaminopyridine, were, before the advent of modern molecular genetics, first line treatment for CMS other than acetylcholinesterase deficiency caused by mutations in COLQ [13]. While cholinesterase inhibitors are beneficial in many types of CMS such as fast channel syndrome, rapsyn CMS and AChR deficiency mutations [4], they had no effect or caused worsening of symptoms in DOK7 CMS [6].
Instead, a marked response to ephedrine and salbutamol which stimulate the beta-2 adrenergic receptor (ADRB2) system was noticed [6, 10, 14, 15]. This observation sparked a revival of ADRB2 agonists as treatment for neuromuscular disorders, and for many patients a combinatorial treatment approach is now chosen.
Despite the well-documented effects of ADRB2 agonists in CMS, very little is known about how ADRB2 activation affects NMJ functioning. The aim of this paper is to shed light on the molecular mechanisms underlying the dramatic effects of ADRB2 agonists in DOK7 CMS. It was hypothesised that the ADRB2 cAMP/PKA-signalling cascades directly affect proteins located at the NMJ.
ADRB2 agonists appear to provide benefit where destabilisation of NMJ structures is part of the underlying pathology, such as in DOK7, COLQ, MuSK, LRP4 CMS as well as in one reported case of slow channel syndrome [4]. Here, the effects of an ADRB2 agonist on the number and stability of AChR clusters were investigated in cultured C2C12 mouse muscle cells.
MATERIALS AND METHODS
Cell culture
C2C12 myoblasts and Phoenix-ECO cells were purchased from ATCC, HEK293T cells were purchased from ECACC. Cells were passaged using 0.5% Trypsin-EDTA (Invitrogen), diluted 1:10 in PBS, and cultured in growth medium at 37°C and 5.5% CO2.
Production of agrin
Neural agrin was produced by transfection of HEK293T cells with plasmid DNA containing soluble short rat agrin (plasmid kindly donated by the late Dr. Werner Hoch). This truncated form of agrin contains the truncated C-terminus of agrin with a 12 amino acid insert at the x-splice site, a four amino acid insert at the y-splice site and an eight amino acid insert at the z-splice site, and has been shown to be sufficient to induce AChR clustering in C2C12 cells [16]. HEK293T cells were seeded at 2×106 cells in a T75 flask in HEK cell growth medium (DMEM (Sigma-Aldrich), supplemented with 10% FCS (Gibco) and 1% PSA (100x Antibiotic-Antimycotic, Gibco)). The following day, cells were transfected with 18 μg plasmid DNA, using polyethyleneimine and 20% glucose. The following morning, the transfection solution was replaced with 7 ml HEK cell growth medium, and cells were left for incubation for further two days. The conditioned medium containing agrin was centrifuged at 1200 g for 10 min at room temperature, aliquoted and frozen at –20°C. A fresh aliquot was used for each experiment and thawed shortly before use. To determine a suitable agrin concentration for AChR cluster induction, a titration experiment was conducted for each batch of agrin.
Production of retrovirus and retroviral infection
DOK7-encoding cDNA was cloned into the retroviral expression vector pBabe puro EGFP and transfected into Phoenix-ECO cells. Phoenix-ECO cells were seeded at 2×106 in T25 tissue culture flasks in DMEM, supplemented with 10% FCS and 1% PSA. The following day, cells were transfected with 12 μg DNA using polyethyleneimine. Cells were transferred to a T75 flask the following day and incubated in 7 ml of growth medium for another two days. The conditioned medium containing retrovirus was harvested and centrifuged for 10 min at 1200 g at room temperature to remove cell debris. 1 ml aliquots were stored at –80°C and thawed shortly before each infection. Equipment used for handling retrovirus or for cells recently infected with retrovirus was decontaminated with Distel Laboratory Disinfectant (Tristel) for at least 15 min before disposal. C2C12 myoblasts were seeded at 3×105 in T25 tissue culture flasks and infected with 1 ml retrovirus and 1 μl of polybrene (8 μg/ml, Sigma-Aldrich) the following night. The next day, cells were reseeded in a T75 or T175 flask depending on the density and left to grow in growth medium for one day. To select for infected cells, the medium was then supplemented with 1 μg/ml puromycin dihydrochloride (Sigma-Aldrich), and cells were kept under selection during further passaging.
AChR clustering assay
C2C12 myoblasts were seeded at a concentration of 6 × 104 - 105 in 24-well plates. Cells were left to grow for 48 hours in DMEM, supplemented with 15% FCS and with 1% PSA to reach 100% confluency. Differentiation of myoblasts into myotubes was induced by growth factor depletion. Cells were cultured in differentiation medium (DMEM supplemented with 2% FCS and with 1% PSA) for 5–10 days until myotubes formed.
C2C12 cells overexpressing DOK7 form AChR clusters in the absence of agrin [7]. To induce clustering of AChRs in C2C12 WT cells, myotubes were incubated with soluble short rat agrin for 16 h. Cells were washed three times for 5 min in differentiation medium to remove agrin.
AChR clusters were visualised by incubation of myotubes with Alexa Fluor-594 conjugated alpha-bungarotoxin (Invitrogen), diluted 1:500 in differentiation medium, for 1 h at 37°C. Cells were washed three times for 5 min in the dark with differentiation medium and fixed in differentiation medium, supplemented with 3% paraformaldehyde (TAAB) for 20 min at room temperature in the dark. The fixing solution was removed and the cells were stored in PBS (1 ml/24 well) at 4°C.
Images of myotubes were acquired using an Olympus IX71 fluorescence microscope at 20x magnification, using Simple PCI software (purchased from Digital Pixel). At least 20 microscopic fields per experimental condition were selected in the bright field to ensure the presence of AChR myotubes without selecting for areas with AChR clusters. Images in the red fluorescence channel were acquired and analysed manually using the software Volocity (Improvision). Only clusters longer than 3 μm (or 5 μm for AChR dispersion experiments) were included in the statistical analysis.
Application of treatment solutions, microscopy and image analysis were carried out blinded.
Western blot analysis
C2C12 myoblasts were seeded onto 6-well plates at a density of 4×105 cells/well. Cells were differentiated into myotubes which were lysed with 200 μl lysis buffer/well, supplemented with 1:100 protease inhibitor cocktail, for 15 mins under agitation at 4°C. Cells were scraped off the plates and centrifuged for 12 min at 12000 g at 4°C. Protein concentration was determined using a Pierce BCA Protein Assay Kit. 8 μg protein was mixed with 4x NuPAGE LDS sample buffer and 10x NuPAGE sample reducing buffer. Samples were incubated at 95°C for 5 min prior to SDS-PAGE. Proteins were transferred to nitrocellulose and probed with anti-DOK7 antibody H-284 (Santa Cruz) 1:500 and secondary antibody HRP-conjugated anti-rabbit (Dako) 1:1000, followed by detection using ECL reagent (GE Healthcare).
Statistical analysis of the number of AChR clusters
A significance level of 0.05 was selected for all statistical analyses.
Differences in the number of AChR clusters were statistically analysed, using the Software RStudio for Windows (RStudio, Inc., version 0.98.1074).
In the analysis of cluster numbers, the response data are counts with no fixed maximum which can be modelled using a Poisson distribution [17]. A widely used method for the analysis of count data is fitting a generalised mixed effects model (GLMM), which combines the properties of two regression models [18]. As a type of generalised linear model, the GLMM “generalises” ordinary linear regression by allowing the response variable to be other than normally distributed. Secondly, as a type of mixed effect model, the GLMM incorporates both fixed and random effects. The application of an ADRB2 agonist at different concentrations or the expression of a particular DOK7 mutation is the explanatory variable of interest and can be interpreted as a fixed effect. Since experimental results are pooled from at least three experiments and variation between cultured cell samples is common, this variation based on performing separate experiments is incorporated in the model as a random effect. Thus, a Poisson generalised mixed effects model was fitted, with cluster counts as the response variable, drug concentration as a fixed effect coded as a categorical variable, and with experiment as a random intercept (R package MASS, function glmmPQL). Differences between mean cluster counts were tested for significance by performing Wald t-tests (Bonferroni-corrected). The number of clusters in 20 microscopic fields was counted, and the mean number per microscopic field was calculated. This was repeated N number of times as stated in the respective figure legend. The difference in the mean AChR cluster count between untreated and treated cells was determined for each drug concentration and tested for significance. Of note, when Poisson regression models are fitted for the analysis of count data, the Poisson mean representing an expected count has to be positive in value. Therefore, the logarithm of the mean count is modelled, and the results of the significance test are on a log scale accordingly (logarithm to the base e). For straight forward interpretation of the results, the data were unlogged and the “raw” cluster numbers were visualised in a separate bar chart.
RESULTS
The common DOK7 frameshift duplication mutation c.1124_27dupTGCC causes a significant decrease in the number of AChR clusters when overexpressed in C2C12 cells compared to cells overexpressing WT DOK7 [6]. A cell line overexpressing c.1124_27dupTGCC was therefore considered to provide a suitable in vitro model of DOK7 CMS, which could then be utilised to investigate the effects of ADRB2 agonists on the AChR clustering pathway.
To establish this model for DOK7 CMS the pBabe/puro retroviral expression system was used which ensures nearly 100% infection efficiency and allows for the generation of stable cell lines [19]. Retrovirus was produced by transfection of Phoenix-ECO cells with plasmid DNA containing c.1124_27dupTGCC. The plasmids were generated by cloning of human DOK7-encoding cDNA into pBabe puro EGFP, which has an internal ribosomal entry site upstream of enhanced green fluorescent protein (EGFP) such that DOK7 and EGFP are expressed as two separate proteins [20]. C2C12 myoblasts were infected with retrovirus, and the puromycin-resistance gene in the pBabe-vector allowed for selection of cells. The resulting stable cell line is henceforth referred to as C2C12DOK7 c. 1124 _ 27dupTGCC. A stable C2C12 line expressing wild type DOK7, referred to as C2C12DOK7 WT was also generated. To demonstrate over-expression of wild type and mutant DOK7 in both of these cell lines, the myoblasts were differentiated into myotubes and cell lysates were analysed by western blot. DOK7 was robustly expressed in both cell lines compared with C2C12 myotubes which had been infected with control retrovirus expressing EGFP (Fig. 1A)
Fig.1
Effects of salbutamol sulphate on AChR clustering in C2C12DOK7c. 1124 _ 27dupTGCC cells. (A) C2C12DOK7c. 1124 _ 27dupTGCC and C2C12DOKWT myotubes robustly express DOK7 compared with control myotubes infected with pBABE-PURO-EGFP. Myotube lysates were analysed by western blot using anti-DOK7 antibody H-284 (Santa Cruz) 1:500.. (B-D) C2C12DOK7c. 1124 _ 27dupTGCC myotubes were incubated with 0.01, 0.1, 1, 5, 10 or 50 μM salbutamol sulphate for 22 h. (B) Salbutamol sulphate treatment induced a significant increase in the number of clusters at all concentrations tested (difference = 0.31, SE = 0.08, p = 0.001, difference = 0.32, SE = 0.07, p = 0.00002, difference = 0.47, SE = 0.05, p < 0.00005, difference = 0.52, SE = 0.07, p < 0.00005, difference = 0.37, SE = 0.06, p < 0.00005, and difference = 0.68, SE = 0.05, p < 0.00005, respectively). (C) Data back-transformed into cluster numbers. (D) Representative microscopic images of C2C12DOK7c. 1124 _ 27dupTGCC myotubes treated with salbutamol sulphate as indicated. N = 12. Error bars indicate the standard error of the mean. n.s.p > 0.05 ***p≤0.001, ****p≤0.0001.
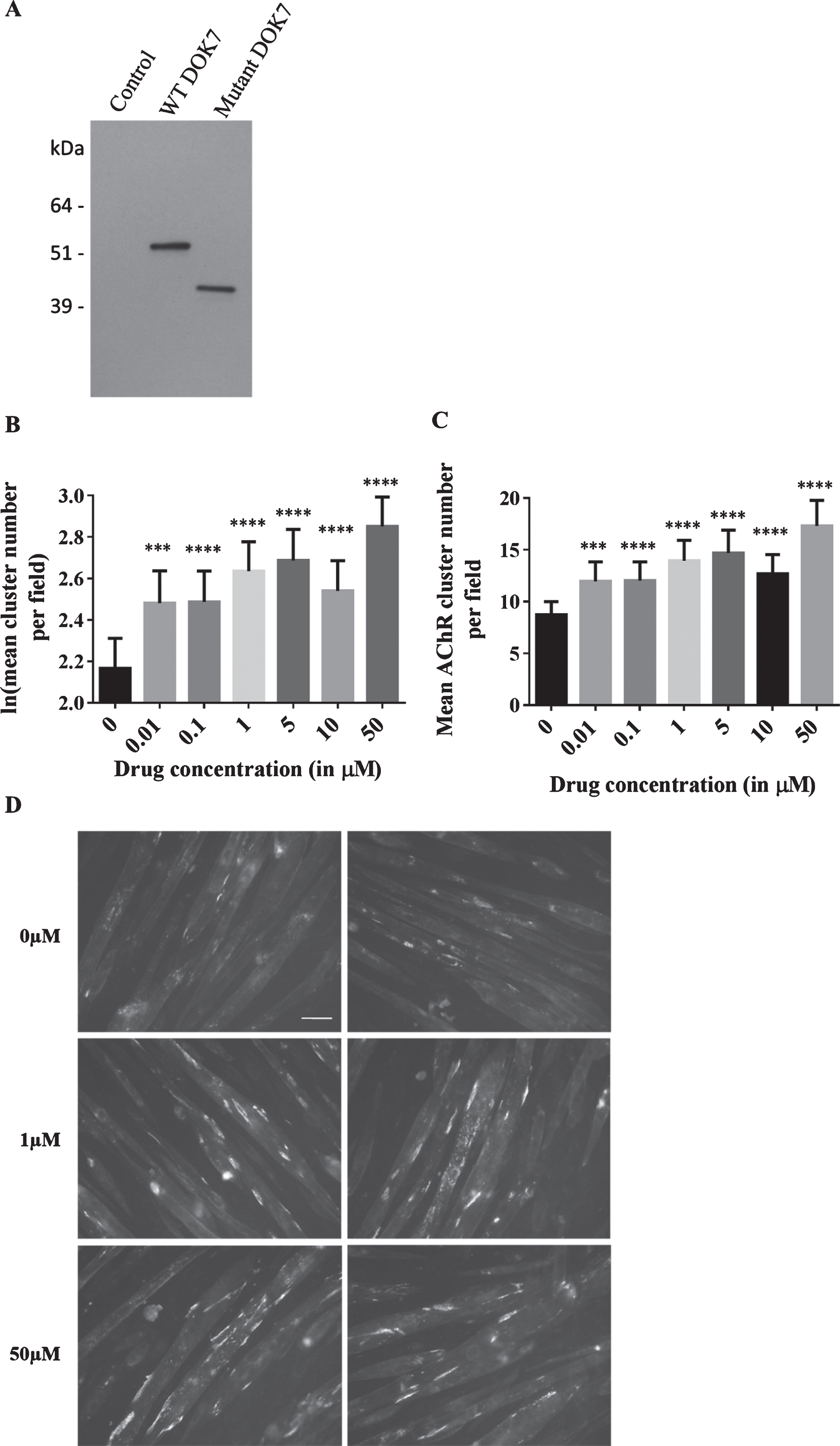
C2C12DOK7 c. 1124 _ 27dupTGCC myoblasts were differentiated into myotubes by serum starvation and treated for 22 hours with 0, 0.01, 0.1, 1, 5, 10 or 50 μM of salbutamol sulphate. Salbutamol sulphate, either added at the initiation of differentiation or when myotubes had formed did not affect either myotube number or diameter. AChR clusters formed on the myotubes were visualised with Alexa Fluor-594 conjugated bungarotoxin. The difference in the mean AChR cluster count between untreated (0 μM) and treated cells was determined for each drug concentration and tested for significance. A significant dose-dependent increase in the number of clusters was observed (Fig. 1). 0.01 and 0.1 μM salbutamol sulphate caused a similar increase in AChR clustering (p = 0.001 and p = 0.00002, respectively). 1 and 5 μM caused a further increase in clustering (p < 0.00005 and p < 0.00005, respectively), whereas a less marked increase in the treatment response was found at 10 μM (p < 0.00005). The largest increase in AChR clustering was obtained after incubation with 50 μM salbutamol sulphate (increase of 99%, p < 0.00005). Baseline levels of AChR cluster numbers in the absence of treatment can vary considerably between stable cell lines, i.e. between retroviral infections (between 5 and 15 clusters per field). Variation also often occurs between experiments on cells from the same batch, and this is likely to be the cause of the less marked increase in cluster number for the 10 μM salbutamol sulphate incubation. Therefore, the experiment was repeated, using both different batches of cells and virus. We also carried out experiments to determine levels of cell-surface AChR expression following incubation with salbutamol and saw no difference AChR cell-surface expression levels between wild type C2C12 cells, C2C12 cells expressing wild type or mutant DOK7 or in addition in the human muscle cell line TE671.
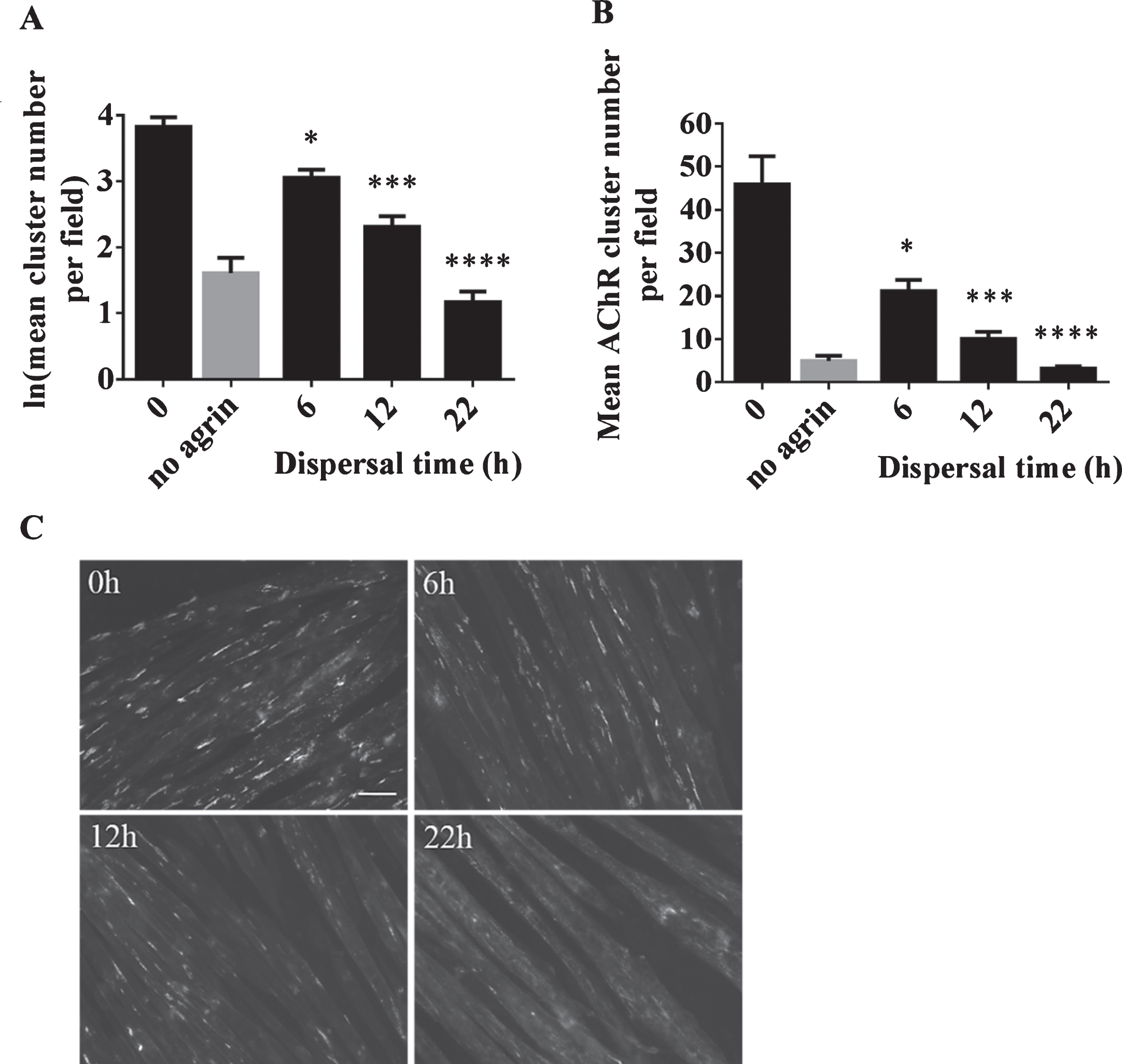
These experiments indicate that ADRB2 agonists have an effect on DOK7-mediated AChR clusters. The observed increase in the number of AChR clusters might be due to a stabilising effect of ADRB2 agonists on forming clusters. To explore this hypothesis in more detail, experiments on the stability of AChR clusters were conducted by studying the effects of salbutamol on dispersing clusters. C2C12 WT myotubes, where cluster formation can be induced by agrin, provides a suitable model for such experiments. Clusters induced by incubation with the soluble short C-terminal region of agrin are known to disperse following the removal of agrin. Accordingly, a reduction in cluster numbers over time was expected. A time course experiment was performed in which clusters were induced with 1:500 dilution of soluble short rat agrin for 16 h. Agrin was removed, and the cells were washed three times in differentiation medium. The cells were then left in differentiation medium for 6, 12 or 22 h, and the number of clusters were compared to cells fixed and stained directly after 16 h of agrin treatment (‘0h’). The results confirmed significant dispersal of AChR clusters in a time-dependent manner (6h: p = 0.02, 12h: p = 0.001, and 22h: p < 0.00005) (Fig. 2). After 22 h, only very few clusters remained and images of myotubes resembled those from the ‘no agrin’ control condition.
Fig.2
Dispersal of AChR clusters in C2C12 WT cells as a result of agrin wash-off. After incubation with soluble short rat agrin, cells were washed to remove agrin and left for 0, 6, 12 or 22 h in differentiation medium for dispersal of AChR clusters. (A) The number of AChR clusters was significantly reduced in a time-dependent manner (6h: difference = 0.77, SE = 0.19, p = 0.02, 12h: difference = 1.51, SE = 0.21, p = 0.001, and 22h: difference = 2.66, SE = 0.21, p < 0.00005) (B) Data shown in A back-transformed into cluster numbers. (C) Representative microscopic images of cells acquired 0, 6, 12 and 22 h after agrin wash-off. Scale-bar=50 μm. Error bars indicate the standard error of the mean. N = 3 (6 and 22 h), N = 2 (0 and 12 h). *≤0.05, ***p≤0.001, ****p≤0.0001.
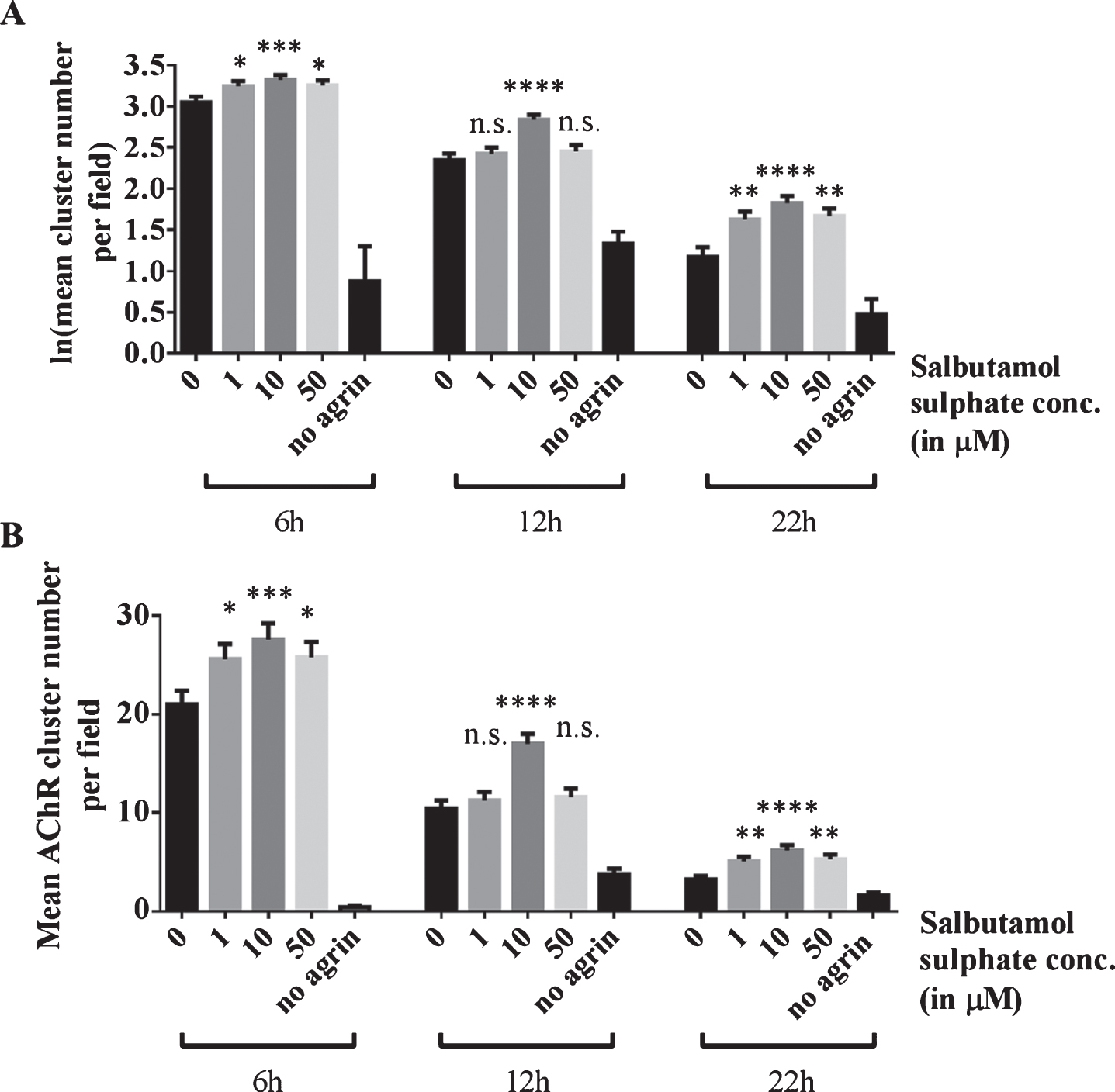
To investigate whether salbutamol has an effect on dispersing clusters, cells were incubated with 0, 1, 10 or 50 μM salbutamol sulphate after agrin wash-off and left in the respective treatment solution for 6, 12 or 22 h. For each treatment duration, the cluster number for salbutamol treated cells was compared to the number for untreated cells (‘0 μM’). The results show that 6 h after agrin-wash off, significantly more clusters remained on myotubes incubated with salbutamol (p = 0.03, p = 0.0006, and p = 0.02, respectively; no agrin control: p < 0.00005) (Fig. 3). Salbutamol had a similar effect on clusters 12 h after agrin wash-off (p > 0.99, p < 0.00005, and p = 0.98, respectively; no agrin control: p < 0.00005) and 22 h after agrin wash-off (p > 0.99, p < 0.00005, and p = 0.98, respectively; no agrin control: p < 0.00005). The most likely explanation for these results is that ADRB2 agonists increase the stability of AChR clusters.
Fig.3
Effects of salbutamol on AChR clusters after agrin wash-off. (A) Clusters were induced by incubation of myotubes with 1:500 soluble short rat agrin. After agrin was removed, AChR clusters were left to disperse for 6, 12 or 22 h in the absence or presence of 1, 10 or 50 μM salbutamol sulphate. Significantly more clusters remained in salbutamol treated cells after a dispersal time of 6 h (difference = 0.2, SE = 0.07, p = 0.027, difference = 0.27, SE = 0.07, p = 0.0006, and difference = 0.2, SE = 0.07, p = 0.02, respectively; no agrin control: difference = 3.92, SE = 0.43, p < 0.00005), 12 h (difference = 0.08, SE = 0.11, p > 0.99, difference = 0.49, SE = 0.1, p < 0.00005, and difference = 0.11, SE = 0.11, p = 0.98, respectively; no agrin control: difference = 1.01, SE = 0.16, p < 0.00005) and 22 h (difference = 0.08, SE = 0.11, p > 0.99, difference = 0.49, SE = 0.1, p < 0.00005, and difference = 0.11, SE = 0.11, p = 0.98, respectively; no agrin control: difference = 1.01, SE = 0.16, p < 0.00005). (B) Data shown in A back-transformed into cluster numbers. N = 3 (6 and 22 h), N = 2 (12 h). Error bars indicate the standard error of the mean. n.s.p > 0.05, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001.
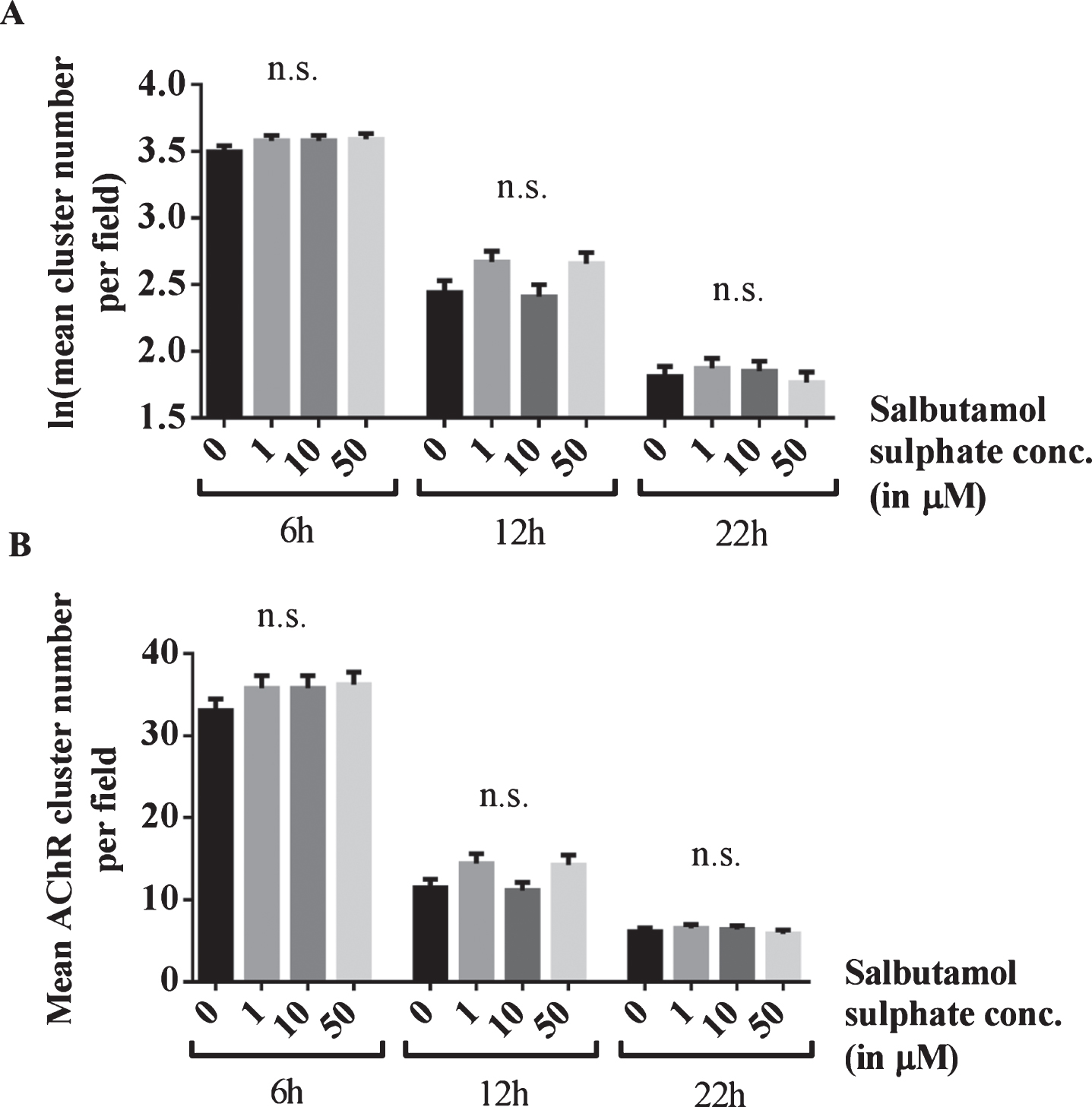
To confirm that this effect is mediated through ADRB2 signalling, the experiment was repeated in the presence of the ADRB2 inhibitor ICI-118,551 (Enzo Life Sciences, Inc.). AChR clusters were induced by incubation of C2C12 WT myotubes with soluble agrin overnight and left to disperse for 6, 12 or 22 h. For each treatment duration, the number of AChR clusters for salbutamol-treated cells was compared to the cluster number for untreated cells (‘0 μM’). In the presence of 1 μM ICI-118,551, salbutamol sulphate (1, 10 or 50 μM) did not have an effect on the number of AChR clusters after agrin-wash off (6h: p = 0.47, p = 0.47, and p = 0.3, 12h: p = 0.06, p > 0.99, and p = 0.08, and 22h: p > 0.99, p > 0.99, and p > 0.99) (Fig. 4).
Fig.4
Blocking of the effects of salbutamol on AChR clusters after agrin wash-off by inhibition of ADRB2 s. (A) C2C12 WT myotubes were incubated with short rat agrin overnight. After agrin wash-off, AChR clusters were left for dispersal for 6, 12 or 22 h, during which myotubes were incubated with a mix of 0, 1, 10 or 50 μM salbutamol sulphate and 1 μM ICI-118,551. The ADRB2 inhibitor abolished the stabilising effect of salbutamol on AChR clusters and no difference occurred between salbutamol-treated and untreated cells (6h: difference = 0.08, SE = 0.06, p = 0.47, difference = 0.08, SE = 0.06, p = 0.47, and difference = 0.09, SE = 0.06, p = 0.3, 12h: difference = 0.23, SE = 0.1, p = 0.06, difference = 0.03, SE = 0.1, p > 0.99, and difference = 0.22, SE = 0.1, p = 0.08, and 22h: difference = 0.06, SE = 0.11, p > 0.99, difference = 0.04, SE = 0.11, p > 0.99, and difference = 0.04, SE = 0.11, p > 0.99) (B) Data shown in A back-transformed into cluster numbers. N = 3 (6 h), N = 2 (12 h and 22 h). Error bars indicate the standard error of the mean. n.s.p > 0.05.
DISCUSSION
As opposed to gene therapy approaches which aim at curing genetic diseases and may find application in the more distant future, drug delivery systems can provide life-changing symptomatic treatment for patients. In order to be able to tailor effective drug treatment for an inherited condition it is of great importance to understand the molecular mechanisms that underlie the disease and the effects a compound has on the disease pathology. Despite some striking recent advances, CMS remains one of few genetic diseases of muscle for which effective drug therapy is available. This is largely due to the detailed understanding of the functioning of the NMJ and the effects of mutations that cause its dysfunction.
The current study aimed at investigating ADRB2 agonists as therapy for disorders of neurotransmission. The effects of the ADRB2 agonist salbutamol on AChR clustering were explored in C2C12 cultures. By studying AChR clusters that form ‘postsynaptically’ on C2C12 cultures, one can mimic some of the key events that take place at the neuromuscular synapse. Under physiological conditions, AChR clusters form on the muscle as a result of a complex signalling cascade, in which agrin, MuSK and DOK7 are key molecules. In vitro, the presynaptic side and extracellular matrix components of the synaptic cleft are missing, and the complex cross-talk beween the pre- and post synaptic sides of the synapse, which is crucial for synaptic integrity cannot take place. Moreover, the structures formed around the AChR clusters on the myotubes lack the complexity of a fully matured synapse, and the C2C12 model system for study does not allow for the very long term application of salbutamol. Nevertheless, AChR cluster formation in C2C12 myotubes has proved an effective model over many years for the study of postsynaptic signalling events.
Our results provide tangible evidence that ADRB2 agonists directly affect proteins located at the postsynaptic side of the NMJ. It was shown that salbutamol significantly increases the number of clusters that form on C2C12 myotubes overexpressing mutant DOK7. Salbutamol appears to be beneficial to neuromuscular disorders characterised by disruption of the endplate structure, such as MUSK/DOK7/COLQ CMS or even MuSK antibody positive myasthenia gravis. Salbutamol sulphate taken in oral tablets at 4–8 mg once or twice daily may reach serum concentrations around 100 ng/ml (∼0.5 μM) with a relatively short half-life of a few hours [21] although this will vary between individuals. Therefore, experiments were performed to explore whether salbutamol has a stabilising effect on the postsynaptic structure. The results are in line with the model proposed by Joshua Sanes and colleagues, suggesting that synaptic maturation and maintenance is regulated by positive and negative nerve-derived signals [22] (Fig. 5). Numerous studies have demonstrated a destabilising effect of ACh, which appears to be counteracted by agrin signalling. In line with the results presented by Lin et al. (2005) [23], it could be shown that agrin-induced AChR clusters disperse in a time-dependent manner if agrin is removed. However, in the presence of salbutamol significantly more clusters remained. In additional experiments (data not shown) salbutamol did not induce the formation of clusters in the absence of agrin. Therefore, induction of as yet unidentified subsynaptic pathways by the ADRB2 agonists act as a positive signal to maintain stability of AChR clusters on the membrane. A stabilising effect of salbutamol on motor endplate structures could also underlie the beneficial effects of ADRB2-treatment observed in patients with severe AChR deficiency due to mutations in CHRNE [24]. In these patients, the initial beneficial response to cholinesterase inhibitors is not always maintained with its longer-term use, which may be caused by a destabilising effect of enhanced ACh signalling on the postsynaptic muscle membrane. It was demonstrated that the addition of salbutamol or ephedrine to chronic treatment with pyridostygmine caused a robust improvement in the treatment response [24].
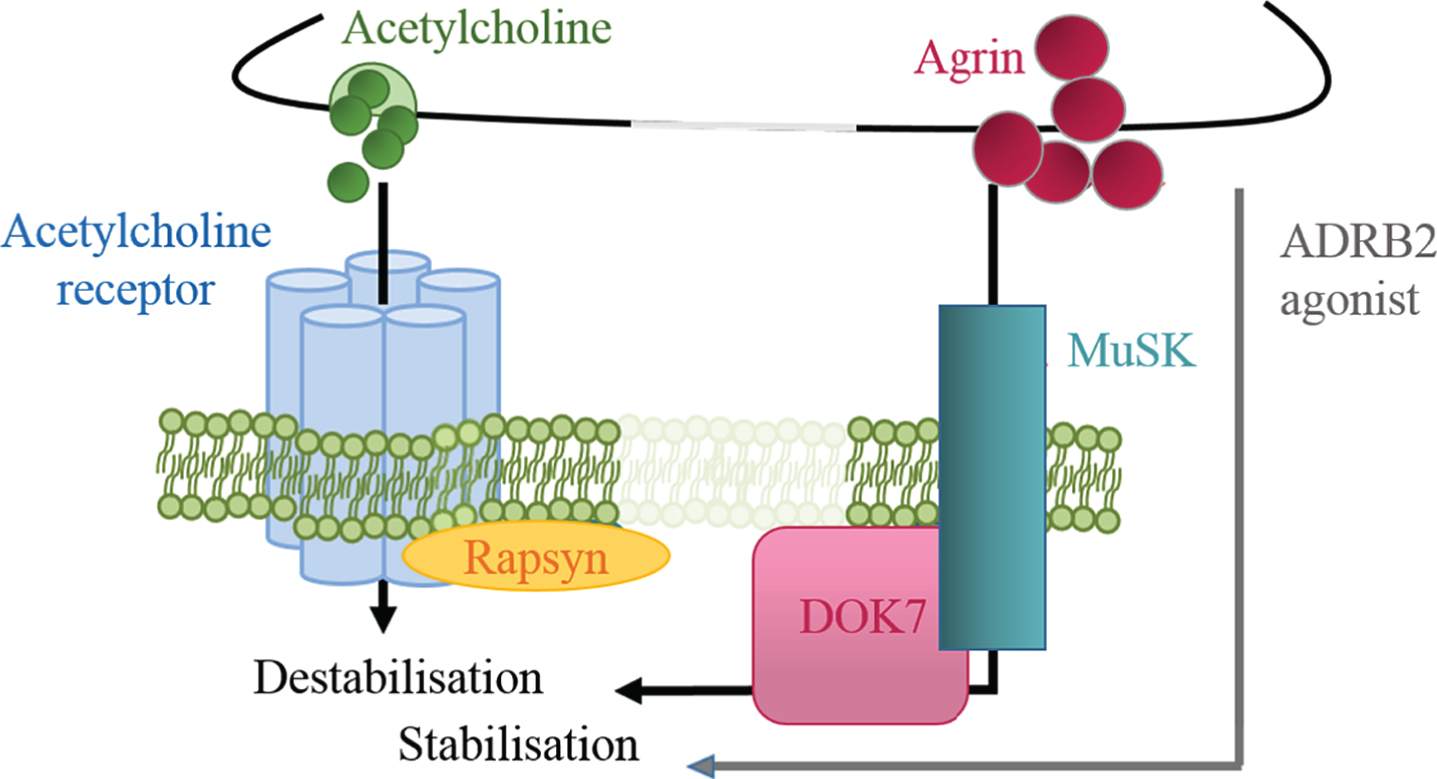
Fig.5
Regulation of postsynaptic stability by positive and negative signals. Agrin is a positive signal for AChR cluster stabilisation, while ACh induces dispersal. ADRB2 agonists might compensate for the partial loss of DOK7 signalling and exert a stabilising effect on the endplate structure.
The results on the effects of ADRB2 agonists on AChR clustering are in line with recent finding on the effects of salbutamol in a mouse model of MuSK MG [25]. Daily injections of mice with IgG from anti-MuSK positive patients for a duration of two weeks induced whole-body muscle weakness. Sections of the diaphragm muscles showed a reduction in the number of AChR-stained endplates per microscopic field, and endplates were fragmented into small AChR clusters. Mice treated with salbutamol during the injection period were significantly stronger and showed less fragmentation of AChR clusters. Alternative studies suggest that there is sympathetic innovation in the vicinity of the neuromuscular junction that contributes towards maintenance of synaptic structures and that ADRB2 agonists could be working through this system [26].
We saw no effect of salbutamol on MuSK tyrosine phosphorylation in cells overexpressing WT or mutant DOK7, or on phosphorylation of the AChR β subunit (data not shown). This suggests that salbutamol is exerting its effects downstream from the AGRN-LRP4-MUSK-DOK7 complex and is unlikely to be acting directly on this pathway. A potential underlying mechanism could be that ADRB2 agonists are affecting the synaptic structure via the downstream cAMP-dependent protein kinase (PKA) type I signalling pathway. In response to adrenergic signalling, PKA activates the proto-oncogene tyrosine kinase SRC [27]. Src-family kinases play a role in the stabilisation of AChR clusters and transfection of mice soleus muscle with kinase-inactive Src constructs resulted in endplate fragmentation, characterised by the disassembly of AChR pretzels and disturbed topology of nerve terminals and synaptic nuclei [28]. Agrin-induced AChR clusters formed on myotubes from Scr and Fyn double mutant mice dispersed more quickly than clusters from control cells when agrin was remove [29].
Another potential mechanism could be a direct interaction of ADRB2-mediated PKA signalling with rapsyn. Recent studies suggested that AChR localisation and recycling is regulated by a complex of AChR, myosin Va and PKA type I [30]. It has been suggested that myosin-Va captures the recycling carriers for AChRs near the membrane and regulates the activation of PKA by capturing PKA and AChRs in a microdomain of subsynaptic cAMP [31]. Rapsyn may be responsible for accumulating PKA in proximity to the NMJ [32]. Alternatively, although we see no effect on AChR expression, cAMP signalling in response to ADRB2 activation could have an effect on transcription of other synaptic components at the NMJ. Some genes coding for proteins expressed at the NMJ, such as MuSK, possess a cAMP response element (CRE)-like element in the promotor, and their expression is regulated by cAMP [33]. Thus there are many different sub-synaptic pathways that could be affected by ADRB2 signaling. In CMS patients with mutations in DOK7 restoring impaired synaptic function through treatment with salbutamol or ephedrine is a gradual process that can take many months, and this would be in accordance with the lack of an immediate direct input into signaling from AGRN-LRP4-MUSK-DOK7 but rather on other factors that contribute to synaptic stability. In turn, the relatively modest immediate affect we see in increasing numbers of clusters for mutant DOK7 as opposed to the increase in cluster numbers that would be required to restore function to the levels obtained for wild type DOK7 suggest a gradual but sustained effect on the postsynaptic environment.
Our findings here show that ADRB2 agonists do affect the postsynaptic AChR clustering in vitro. Further research is required to decipher the precise mechanisms underlying the effects on AChR cluster formation and stability which is likely to involve the complex interaction with kinase pathways involved in AChR aggregation. The AChR clustering experiments conducted here should provide a useful framework for future studies
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGEMENTS INCLUDING SOURCES OF SUPPORT
Supported by Muscular Dystrophy Campaign Studentship RA3/839 and MRC Programme Grant MR/M006824.
REFERENCES
[1] | Engel AG , Shen XM , Selcen D , Sine SM Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. The Lancet Neurology. (2015) ;14: (4):420–34. doi: 10.1016/S1474-4422(14)70201-7 |
[2] | Parr JR , Andrew MJ , Finnis M , Beeson D , Vincent A , Jayawant S How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Archives of Disease in Childhood. (2014) ;99: (6):539–42. doi: 10.1136/archdischild-2013-304788 |
[3] | Finlayson S , Beeson D , Palace J Congenital myasthenic syndromes: An update. Practical Neurology. (2013) ;13: (2):80–91. doi: 10.1136/practneurol-2012-000404 |
[4] | Rodriguez Cruz PM , Palace J , Beeson D Inherited disorders of the neuromuscular junction: An update. Journal of Neurology. (2234) ;261: (11):2234–43. doi: 10.1007/s00415-014-7520-7 |
[5] | Beeson DCongenital myasthenic syndromes: Recent advances. Current Opinion in Neurology. (2016) ;29: (5):565–71. doi: 10.1097/WCO.0000000000000370 |
[6] | Beeson D , Higuchi O , Palace J , Cossins J , Spearman H , Maxwell S , et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. (1975) ) 313–8. doi: 10.1126/science.1130837 |
[7] | Okada K , Inoue A , Okada M , Murata Y , Kakuta S , Jigami T , et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. (1802) )312–5. doi: 10.1126/science.1127142 |
[8] | Bergamin E , Hallock PT , Burden SJ , HubbardSR. The cytoplasmic adaptor protein Dok7 activates the receptor tyrosine kinase MuSK via dimerization. Molecular Cell. (2010) ;39: (1):100–9. doi: 10.1016/j.molcel.2010.06.007 |
[9] | Glass DJ , Bowen DC , Stitt TN , Radziejewski C , Bruno J , Ryan TE , et al. Agrin acts via a MuSK receptor complex. Cell. (1996) ;85: (4):513–23. |
[10] | Palace J , Lashley D , Newsom-Davis J , Cossins J , Maxwell S , Kennett R , et al. Clinical features of the DOK7 neuromuscular junction synaptopathy. Brain: A Journal of Neurology. (2007) ;130: (Pt 6):1507–15. doi: 10.1093/brain/awm072 |
[11] | Selcen D , Milone M , Shen XM , Harper CM , Stans AA , Wieben ED , et al. Dok-7 myasthenia: Phenotypic and molecular genetic studies in 16 patients. Annals of Neurology. (2008) ;64: (1):71–87. doi: 10.1002/ana.21408 |
[12] | Slater CR , Fawcett PR , Walls TJ , Lyons PR , Bailey SJ , Beeson D , et al. Pre- and post-synaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain: A Journal of Neurology. (2006) ;129: (Pt 8):2061–76. doi: 10.1093/brain/awl200 |
[13] | Engel AG , Ohno K , Sine SM Congenital myasthenic syndromes: Progress over the past decade. Muscle & Nerve. (2003) ;27: (1):4–25. doi: 10.1002/mus.10269 |
[14] | Lashley D , Palace J , Jayawant S , Robb S , Beeson D Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology. (2010) ;74: (19):1517–23. doi: 10.1212/WNL.0b013e3181dd43bf |
[15] | Liewluck T , Selcen D , Engel AG Beneficial effects of albuterol in congenital endplate acetylcholinesterase deficiency and Dok-7 myasthenia. Muscle & Nerve. (2011) ;44: (5):789–94. doi: 10.1002/mus.22176 |
[16] | Ferns MJ , Campanelli JT , Hoch W , Scheller RH , Hall Z The ability of agrin to cluster AChRs depends on alternative splicing and on cell surface proteoglycans. Neuron. (1993) ;11: (3):491–502. |
[17] | Mize C , Koehler K , Compton M Statistical considerations for in vitro research: II - Data to presentation. In Vitro Cellular & Developmental Biology - Plant. (1999) ;35: (2):5. |
[18] | Bolker BM , Brooks ME , Clark CJ , Geange SW , Poulsen JR , Stevens MH , et al. Generalized linear mixed models: A practical guide forecology and evolution. Trends in Ecology & Evolution. (2009) ;24: (3):127–35. doi: 10.1016/j.tree.2008.10.008 |
[19] | Morgenstern JP , Land H Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Research. (1990) ;18: (12):3587–96. |
[20] | Cossins J , Liu WW , Belaya K , Maxwell S , Oldridge M , Lester T , et al. The spectrum of mutations that underlie theneuromuscular junction synaptopathy in DOK7 congenital myasthenic syndrome. Human Molecular Genetics. (2012) ;21: (17):3765–75. doi: 10.1093/hmg/dds198 |
[21] | Morgan DJ , Paull J , Richmond B , Wilson-Evered E , Ziccone SP Pharmacokinetics of intravenous and oral salbutamoland its sulphate conjugate. Br J Clin Pharmac. (1986) ;22: :587–93. |
[22] | Kummer TT , Misgeld T , SanesJR. Assembly of the postsynaptic membrane at the neuromuscular junction: Paradigm lost. Current Opinion in Neurobiology. (2006) ;16: (1):74–82. doi: 10.1016/j.conb.2005.12.003 |
[23] | Lin W , Dominguez B , Yang J , Aryal P , Brandon EP , Gage FH , et al. Neurotransmitter acetylcholine negatively regulates neuromuscular synapse formation by a Cdk5-dependent mechanism. Neuron. (2005) ;46: (4):569–79. doi: 10.1016/j.neuron.2005.04.002 |
[24] | Rodriguez Cruz PM , Palace J , Ramjattan H , Jayawant S , Robb SA , Beeson D Salbutamol and ephedrine in the treatment of severe AChRdeficiency syndromes. Neurology. (2015) ;85: (12):1043–7. doi: 10.1212/WNL.0000000000001952 |
[25] | Ghazanfari N , Morsch M , Tse N , Reddel SW , Phillips WD Effects of the ss2-adrenoceptor agonist, albuterol, in a mouse model of anti-MuSK myasthenia gravis. PloS one. (2014) ;9: (2):e08784. 10.1371/journal.pone.0087840 |
[26] | Khan MM , Lustrino D , Silveira WA , Wild F , Straka T , Issop Y , et al. Sympathetic innervation controls homeostasis of neuromuscular junctions in health and disease. Proc Natl Acad Sci U S A. (2016) ;113: (3):746–50. doi: 10.1073/pnas.1524272113 |
[27] | Armaiz-Pena GN , Allen JK , Cruz A , Stone RL , Nick AM , Lin YG , et al. Src activation by β-adrenoreceptors is a key switch for tumour metastasis. Nat Commun. (2013) ;4: :1403. doi: 10.1038/ncomms2413 |
[28] | Sadasivam G , Willmann R , Lin S , Erb-Vögtli S , Kong XC , Rüegg MA , Fuhrer C Src-family kinases stabilize the neuromuscular synapse in vivo via protein interactions, phosphorylation, and cytoskeletal linkage of acetylcholine receptors. J Neurosci. (2005) ;25: (45):10479–93. |
[29] | Smith CL , Mittaud P , Prescott ED , Fuhrer C , Burden SJ Src, Fyn, and Yes are not required for neuromuscular synapse formation but are necessary for stabilization of agrin-induced clusters of acetylcholine receptors. J Neurosci. (2001) ;21: (9):3151–60. |
[30] | Röder IV , Petersen Y , Choi KR , Witzemann V , Hammer JA3rd , Rudolf R Role of Myosin Va in the plasticity of the vertebrate neuromuscular junction in vivo. PLoS One. (2008) ;3: (12):e3871. doi: 10.1371/journal.pone.0003871 |
[31] | Rudolf R , Bittins CM , Gerdes HH The role of myosin V in exocytosis and synaptic plasticity. J Neurochem. (2011) ;116: (2):177–91. doi: 10.1111/j.1471-4159.2010.07110.x |
[32] | Choi KR , Berrera M , Reischl M , Strack S , Albrizio M , Röder IV , et al. Rapsyn mediates subsynaptic anchoring of PKA type I and stabilisation of acetylcholine receptor in vivo. J Cell Sci. (2012) ;125: (Pt 3):714–23. doi: 10.1242/jcs.092361 |
[33] | Kim CH , Xiong WC , Mei L Inhibition of MuSK expression by CREB interacting with a CRE-like element and MyoD. Mol Cell Biol. (2005) ;25: (13):5329–38. |


