
Sil1-Mutant Mice Elucidate Chaperone Function in Neurological Disorders
Abstract
Chaperone dysfunction leading to the build-up of misfolded proteins could frequently be linked to clinical manifestations also affecting the nervous system and the skeletal muscle. In addition, increase in chaperone function is beneficial to antagonize protein aggregation and thus represents a promising target for therapeutic concepts for many genetic and acquired chaperonopathies. However, little is known on the precise molecular mechanisms defining the cell and tissue abnormalities in the case of impaired chaperone function as well as on underlying effects in the case of compensatory up-regulation of chaperones. This scarcity of knowledge often arises from a lack of appropriate animal models that mimic closely the human molecular, cellular, and histological characteristics. Here, we introduce the Sil1-mutant woozy mouse as a suitable model to investigate molecular and cellular mechanisms of impaired ER-chaperone function affecting the integrity of nervous system and skeletal muscle. The overlapping clinical findings in man and mouse indicate that woozy is a good copy of a human phenotype called Marinesco-Sjögren syndrome. We confirm the presence of ER-stress and expand the biochemical knowledge of altered nuclear envelope in muscle, a hallmark of SIL1-disease. In addition, our data suggest that impaired excitation-contraction coupling might be part of the SIL1-pathophysiology. Our results moreover indicate that divergent expression of pro- and anti-survival proteins is decisive for Purkinje cell survival. By summarizing the current knowledge of woozy, we focus on the suitability of this animal model to study neuroprotective co-chaperone function and to investigate the involvement of co-chaperones in the predisposition of other disorders such as diabetic neuropathy.
THEORETICAL BACKGROUND
SIL1 and Marinesco-Sjögren syndrome
Marinesco-Sjögren syndrome (MSS, MIM 248800) is a rare multisystemic disorder with autosomal recessive inheritance and infantile onset. So far, mutations of the SIL1 gene, located on chromosome 5q31.2, are the only identified genetic cause [1–5]. Reported mutations in the SIL1 gene include homozygous or compound heterozygous point mutations as well as larger genomic deletions leading to a loss of protein in MSS-patients [3, 6]. The ubiquitously expressed SIL1 protein functions as a nucleotide exchange factor (NEF) for the endoplasmic reticulum (ER) chaperone BiP, also known as GRP78. Quantification of the SIL1-BiP machinery including GRP170 as an alternative co-chaperone (Fig. 1) in human myoblastic cells revealed a molecular ratio of ∼1: 0.1: 0.01 (BiP-GRP170-SIL1) [7]. A comparison with the ratio of this complex in canine pancreas (∼1: 0.1: 0.001) [8] reveals a SIL1-increase in muscle cells and is thus indicative for a particular function of SIL1 in muscle tissue. BiP is a key regulator of several ER-related processes such as folding of nascent proteins, translocation of proteins across the ER membrane, modulation of the unfolded protein response (UPR) and of the ER-associated degradation (ERAD) pathway as well as regulation of calcium homeostasis. Due to its role as a co-chaperone SIL1 is involved in these functions of BiP (Fig. 1).
The initial clinical descriptions defined the triad of ataxia, bilateral cataracts and mental retardation as decisive for the diagnosis of MSS. Over time, further aspects were found to be characteristic, including vacuolar myopathy [9, 10] and cerebellar atrophy [11]. Thus the clinical hallmarks characteristic for MSS are cerebellar ataxia, infantile or congenital bilateral cataracts, progressive vacuolar myopathy and mental retardation. However, a recent study pointed out that mental retardation is present at a highly variable degree ranging from normal mental capacity to severe mental impairment. Additionally, further symptoms like hypogonadism, skeletal abnormalities, short stature [12, 13], strabismus and nystagmus [14] as well as pyramidal tract signs [3] were reported in patients harbouring pathogenic SIL1 mutations. Byrne and colleagues reported on a SIL1-related MSS-patient who presented with motor neuronopathy and a bradykinetic movement disorder which preceded the onset of ataxia [15]. Recently, two patients were described carrying compound heterozygous SIL1 missense mutations and suffering from cerebellar ataxia and atrophy without any other clinical hallmarks of MSS [16]. Still, all other MSS cases reported so far presented with mutations leading to a (premature) stop-codon and at least show progressive vacuolar myopathy and cataracts in combination with the cerebellar signs. This was also observed for cases presenting with larger genomic aberrations affecting the SIL1 gene [3, 5]. Despite these rather severe manifestations of SIL1-deficiency, life span of MSS-patients seems to be normal.
Even though MSS is a rare syndrome, investigations of its pathogenesis using patient-derived material and appropriate animal models such as the woozy mouse (Sil1-mutant; see below) appear to be of general interest due to its co-chaperone function of BiP for a number of reasons: (i) BiP is the major chaperone of the endoplasmic reticulum, thus controlling diverse cellular mechanisms (see above), (ii) impaired function of chaperones – leading to so called chaperonopathies [17] – is a pathophysiological hallmark of various disorders including neuromuscular diseases [17] and major neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease and Amyotrophic Lateral Sclerosis (ALS) [18], and (iii) examinations of various tissues will allow an improved biological understanding of chaperone function in MSS and related neuromuscular and neurodegenerative disorders and may lead to new therapeutic concepts. In this context, it is advantageous that suitable mouse models have been generated that can be used in longitudinal studies.
Sil1 mouse models
In 2005, Zhao and co-workers [19] published the woozy mouse model suffering from cerebellar ataxia at about 3 to 4 month of age in a homozygous setting. The authors could link homozygous mutations of the Sil1 gene (localized on mouse chromosome 18) to the cerebellar degeneration. They identified truncated Sil1 mRNA, which contains only exon 1-7. The Sil1 transcript splices from exon 7 into an ETn retrotransposon leading to an in-frame stop codon after 96 nucleotides of the ETn sequence. Due to this abbreviated mRNA the corresponding protein would lack the C-terminus. The same group made use of a gene-trap embryonic cell line in which the β-geo gene-trap construct pGT2Mpfa had integrated into the Sil1 gene after exon 7, in order to achieve a truncation of the Sil1 transcript. This way they generated the Sil1Gt mouse model which shows the same pathology as the woozy mutants, thus confirming the phenotype of Sil1 mutation.
Using the woozy model, several studies dealing with the examination of the SIL1-BiP pathophysiology have been published on tissues vulnerable in MSS over the last decade. Moreover, studies focusing on tissues that were not affected clinically were performed to achieve a more profound understanding of pathophysiological mechanisms and possible compensatory strategies upon SIL1 deficiency, which could be therapeutically relevant.
EXPERIMENTAL PROCEDURE
Immunohistochemistry, immunoblot (Table 1) as well as electron microscopic studies were carried as described before [20]. All described procedures were approved by the UK Aachen Institutional Animal Care and Use Committee and conducted in compliance with the Guide for the Care and Use of Laboratory Animals. Sil1 (woozy) mutants were obtained from Jackson Laboratories (strain name: CXB5/By-Sil1wz/J; stock number 003777).
RESULTS
Immunohistochemistry studies of woozy cerebellum
In order to obtain first insights in mechanisms decisive for the live-death response of Purkinje cells (PC) in SIL1-pathophysiology, H&E staining as well as immunohistochemistry studies of degenerating neocerebellar and surviving vestibulocerebellar PC were carried out. The H&E staining revealed neocerebellar PC degeneration and thus confirmed previous descriptions of this pathology [19, 20]. Via immunohistochemistry, we focused on the expression of PARP1 and BCL2, two functionally connected proteins involved in cell death (PARP1) and survival (BCL2) [21]. BCL2 is enriched in PC dendrites of wildtype animals (Fig. 2E). While this protein is decreased in the dying neurons of the neocerebellum, increased levels were detectable throughout the disease-resistant PC of the vestibulocerebellum (Fig. 2F, H). Notably, the opposite can be observed for the nuclear enrichment of PARP1 (Fig. 2I–L).
Woozy mouse muscle pathology and quantitative analysis of woozy mouse body weight
Investigation of woozy muscles using macroscopic inspection and light microscopy (H&E staining) revealed vacuolar myopathy with variable muscle fibre calibres and several non-subsarcolemmal myonuclei (Fig. 3) and thus confirmed the presence of myopathy upon Sil1 mutation. Quantitative analysis of wildtype and woozy mouse body weight (26-week-old) was performed in order to further study the SIL1-related muscle wasting. Woozy animals show a significant reduction in body weight (app. 20%).
Morphological studies of MSS-patient and woozy skeletal muscle
Electron microscopic studies of skeletal muscle derived from MSS-patients and Sil1-mutant muscle fibres were carried out in order to demonstrate similar myopathological changes defining woozy as a suitable phenocopy of the human disorder and to confirm these changes prior further immunohistochemistry and immunoblot investigations. Transmission electron microscopic studies showed pyknotic degenerating myonuclei with massive perturbations of nuclear envelope integrity, proliferated sarcoplasmic reticulum, presence of autophagic material in man and mouse with SIL1/Sil1 mutation (Fig. 4). In many affected fibres, the nuclear envelope together with the lamina fibrosa was lifted off from the chromatin, corresponding to the perinuclear membranous structures characteristic for the myopathy in MSS-patients [10, 20] (Fig. 4). Furthermore, enlarged mitochondria with disrupted cristae as well as accumulation of autophagic material were observed in human and murine muscle biopsy specimen (Fig. 4).
Table 2 provides a comparison of clinical and morphological findings described in MSS-patients and found in woozy animals. This comparison reveals that while cataracts and growth retardation (occasionally observed in patients) are not present in Sil1-mutant mice, cerebellar and muscular pathology are very well reflected in the animal model. As the presence of cataracts is part of the defined MSS symptom trias in man, absence of this hallmark in woozy is of particular interest. However, systematic studies dealing with the function of SIL1 in lens crystallina are still lacking but might have the potential to identify compensatory mechanisms when using woozy derived lenses.
Immunohistochemistry and immunoblot studies of woozy quadriceps muscle
Further biochemical studies including immunohistochemistry and immunoblot were carried out in order to gain further insights into the pathogenesis of SIL1-reated myopathy. Immunohistochemistry studies revealed a moderate increase of muscle creatine kinase in muscle fibres derived from woozy animals (Fig. 5A, B). In addition, BiP and DJ-1, two stress related proteins, and TDP-43 an aggregation-prone protein were increased in woozy muscle fibres (Fig. 5C–L). Thereby, TDP-43 shows an enhanced intermyofibrillar punctuate immunoreactivity which is accompanied by prominent labelling of the perinuclear region and the nuclear membrane. Further immunohistochemical studies of nuclear envelope proteins showed prominent LAP2-reactivity of the pathological vacuoles (Fig. 5M–P). Investigation of Emerin revealed irregular immunoreactivity of the sub-sarcolemmal region in Sil1-mutant muscle fibres (Fig. 5Q–T). Immunohistochemistry studies of Lamin A/C showed altered staining pattern with prominent reactivity adjacent to myonuclei in woozy (Fig. 5U–X).
Immunoblot studies of RYR1 and DHPR, two proteins involved in the Ca2+-dependent excitation-contraction (EC) coupling were performed in quadriceps muscles derived from 16, 26, 52 and 84-weeks old woozy animals and respective wildtype controls. These studies were prompted by (i) the known changes in triad morphology in SIL1-diseased muscle [2], (ii) the known role of BiP as a key player in Ca2+ homeostasis [22] and the (iii) alteration of proteins involved in Ca2+ homeostasis in SIL1-depleted HEK293 cells [23]. These immunoblot studies revealed a decrease of both EC coupling proteins in woozy muscle fibres (Fig. 6). GAPDH was used as a loading control and showed no changes in protein abundance upon Sil1 mutation (Fig. 6).
DISCUSSION AND REVIEW OF THE CURRENT LITERATURE
Cerebellar degeneration in MSS-patients and woozy mice
Zhao and co-workers identified loss of Purkinje cells (PC) within the lobules I-VIII (neocerebellum) as the substrate of cerebellar atrophy and the same finding could be confirmed within our studies (Fig. 2). Notably, PC of lobules IX-X (vestibulocerebellum) were spared from death. MRI of MSS-patients also revealed cerebellar atrophy. Electron microscopy of PC of woozy mice revealed condensed nuclear chromatin, multilamellar membranous structures with autophagic vacuoles as well as electron-dense globular structures reminiscent of protein inclusions in the perinuclear region and in the nucleus. These structures might indicate that the function of BiP is impaired which was supported by the finding that these aggregates were immunoreactive for ubiquitin and co-localized with several UPR proteins. The PC loss could have the two following reasons: (i) accumulation of proteins could lead to loss of their function or even to a toxic effect in the ER or in the nucleus, (ii) prolonged ER stress and loss of calcium homeostasis could result in PC death. The findings obtained thus far indicate that the functional SIL1-BiP machinery is necessary for PC maintenance and survival; at the same time UPR can be activated within this vulnerable neuronal cell population even when one co-chaperone of BiP is lost. In this context it is important to note that in ancillary studies it was shown that an alternative NEF for BiP called GRP170 (HYOU1) can rescue, when overexpressed in woozy mice, PC degeneration and therefore prevent cerebellar atrophy. Further genetic manipulation of woozy mice, leading additional heterozygous loss of GRP170, accelerates PC loss and also affects the vestibulocerebellum. Accordingly, genetic manipulation leading to loss of DNAJC3, another BiP co-chaperone promoting its ATPase activity (Fig. 1), partially suppresses ER stress in SIL1-deficient PC and suppresses their degeneration. These studies clearly demonstrate a link between ER stress intensity and time point of cell death in PC [24]. Moreover, mice with a conditional knockout of BiP, selectively in PC, show apoptosis and ataxia by 5 weeks of age. Interestingly, in thesemice PC degeneration occurs in all ten lobules of the cerebellum and is complete by 3 months [25]. This is different from woozy mice in which degeneration starts later and does not include vestibulocerebellum (see above) [19]. Taken together these studies demonstrate that the phenotype of PC loss, cerebellar atrophy and ataxia is caused by loss of function rather than loss of protein itself. This is in turn supported by the recent clinical and genetic findings published by Noreau and co-corkers [see above; 36]. However, it was still an enigma why particular PC populations are vulnerable while others are disease-resistant. Our immunohistochemistry findings (Fig. 2) indicate that the divergent expression of pro- (BCL2) and anti-survival (PARP1) proteins is involved in the live-death response and thus cellular vulnerability in SIL1-pathophysiology. Interestingly, BCL2 increase is known to promote PC survival [26].
Myopathic changes in MSS-patients and woozy mice
Severe progressive vacuolar myopathy is another hallmark of MSS [3, 10]. We recently investigated the myopathic changes in patients suffering from MSS and compared those with the progressive myopathic alterations in the quadriceps muscle of woozy mice [20]. Muscle biopsies of MSS-patients showed atrophic, rounded fibres and a considerable number of non-subsarcolemmal nuclei. Moreover, autophagic vacuoles were observed especially in the perinuclear sarcoplasm. A similar progressive vacuolar myopathy was observed in woozy mice. However, as in the MSS-patient muscle biopsies, acute muscle fibre necrosis and myophagic reactions were absent in the muscle of woozy mice [20]. In accordance with a progressive muscular atrophy (and cerebellar degeneration), woozy animals presented with a significant loss of weight of more than 20% (Fig. 3). A moderate intercellular increase of muscle creatine kinase is detectable during the course of the myopathy (Fig. 5). Up-regulation of the creatine synthetic pathway has been already described in the mdx mouse model [27]. In this context, increased creatine kinase expression most likely serves towards the maintenance of creatine levels in “leaky” damaged skeletal muscle fibres.
With regard to the known function of the SIL1-BiP machinery, increase of BiP as the major chaperone of the sarcoplasmic reticulum along with DJ-1, a well-known stress marker, were indicative for impaired protein quality control leading to ER-stress in diseased muscle fibres (Fig. 5). Notably, concomitant increase of DJ-1 and GRP170 under stress conditions has already been described [28]. By electron microscopy, we observed a focally widened proliferating sarcoplasmic reticulum especially in the perinuclear region of subsarcolemmal myonuclei sometimes associated with vacuoles (Fig. 4). These morphological findings confirm the biochemical results and thus suggest the SIL1 co-chaperone is essential for proper protein quality control in the sarcoplasmic reticulum. Myonuclei itself were frequently degenerated with condensed and fragmented shape thus indicating initiation of muscle fibre degeneration. This is in accordance with the reduced muscle bulk overserved on the macroscopic level and the reduced body weight in Sil1-mutant animals (Fig. 3). Perturbations of NE integrity associated with the nuclear lamina proliferation and a mislocalisation of NE resident proteins such as Emerin, LAP2 and Lamin A/C (Figs. 4 and 5) indicate that the SIL1-BiP machinery serves important functions within the NE. Indeed, enrichment of BiP within the NE of muscle fibres could repeatedly be demonstrated (Fig. 5) [20, 29].
Proliferated sarcoplasmic reticulum along with increase of ER-stress proteins (Fig. 4) [20] and the presence of intermyofibrillar autophagic vacuoles which are immunoreactive for TDP-43 (Fig. 5) (a well-known protein aggregation marker) indicate that loss of SIL1 activates mechanisms of cellular stress response (UPR and ERAD) and of proteolysis. Notably, these mechanisms depend on proper BiP function which in turn is controlled by SIL1. Although both processes were activated in a SIL1-independent manner, a proper degradation of misfolded proteins is obviously not possible. In this context, one might hypothesize that SIL1 represents a key co-chaperone of BiP in muscle fibres, loss of which can only partially be compensated. This assumption is supported by the fact the forced increase of GRP170 in vivo leads to severe myopathological alterations [30], thus declaring this alternative BiP co-chaperone to an unsuitable candidate to compensate SIL1 loss. Hence, woozy represents a promising animal model to study chaperone function in muscle fibre pathology including NE integrity.
Notably, BiP has also a key role in the regulation of Ca2+ storage, a cellular process decisive for EC coupling in muscle fibres. In addition, previous studies not only showed the Sil1-mutant muscles present with changes of the triadic system on the morphological level [20] but also that SIL1-depletion causes changes in protein abundances of other Ca2+ related proteins in vitro [23]. This prompted us to investigate level of RYR1 and DHPR, two EC coupling related transmembrane proteins which are also potential substrates of the SIL1-BiP machinery. Both proteins showed decreased level indicating that morphological alteration of the triadic system [20] is accompanied by decrease of functional-related proteins.
Suitability of woozy mice to study the neuroprotective function of SIL1
Based on the function of SIL1 as a NEF for BiP, perturbed function of the chaperone machinery due to loss of the NEF most likely results in impaired protein processing, thus causing the build-up of (toxic) protein aggregates. This assumption has been supported by studies showing that loss of SIL1 function causes activation of UPR, ERAD and proteolysis as well as of pro-survival mechanisms [20, 23, 31]. Therefore, the question was asked whether SIL1 itself has a role in handling the stress response through unfolded proteins and therefore has a protective function within the cell. This hypothesis was recently addressed by Filézac de L’Etang and co-workers [32]. They showed that SIL1 is expressed at lower levels in the low-excitable fast-fatigable (FF) motor neurons, which are much more vulnerable against ALS, than in the highly excitable slow-fatigable (S) motor neurons, which do not degenerate until the end stage of disease. Reduction of SIL1 expression induced ER stress in motor neurons. To investigate the role of SIL1 in ALS, the authors made use of SOD1-G93A-s mice as a fALS model and crossed these animals with mice either heterozygous or homozygous for woozy mutation to reduce SIL1 levels by degrees. By obtaining a reduction of functional SIL1, progression of the disease was highly accelerated in the double-mutant mouse model of ALS and also normally not affected S motor neurons show characteristics of the disease. Elevated expression of SIL1 in mouse models of ALS protected the cell of ER stress and reduced disease manifestations. This study showed that SIL1 influences the expression and function of several chaperones and influence cellular homeostasis. In addition, molecular genetic SNP-studies of SIL1 revealed a correlation between sALS and SIL1 genotype. Pyramidal tract signs in patients also suggested an involvement of motor neurones in MSS pathology [3, 15]. In order to address this question on the biochemical level, in a recent study, we combined proteomic analyses of SIL1-depleted cells with studies of the spinal cord of woozy animals. Immunohistochemistry showed that proteins which modulate ER stress and proteolysis as well as neuroprotective factors are increased in the spinal cord of Sil1-mutant mice [23]. VAPB, an ALS-related vesicular protein, was decreased. In a prolonged stress situation this could lead to apoptosis and therefore to cell death. These mechanisms are already known in several neurodegenerative disorders [23, 32]. In summary, woozy mice are a promising animal model for the study of neuroprotection in neurological disorders such as ALS.
Suitability of woozy mice to study the role of SIL1 in antibody production
In secretory cells of immune system, loss of SIL1 function could be expected to cause an impairment of function. However, such phenotypic alterations have thus far not been reported in MSS-patients. Studies dealing with these tissues in woozy animals aimed to get a further understanding of the role of SIL1 within these organs and of potential compensatory mechanisms, preventing clinical manifestation in the case of SIL1 deficiency. In this context the interaction with BiP is especially interesting. Immunoglobulins are very well investigated substrates of BiP [33–36]. Thus the mechanism dealing with the antibody formation and secretion was of particular interest and studied under the condition of SIL1 deficiency in mice (and lymphoblastoid cells derived from MSS-patients) by Ichhaporia and colleagues [37]. Remarkably, the population of immunoreactive cells was not reduced in Sil1Gt mice. Moreover, there was no reduction in the production and secretion of antibodies after immunisation with T-dependent antigens or in LPS stimulated B-cells. Therefore, SIL1 is obviously not required for antibody assembly or secretion. In B-cells no activation of an alternative chaperone system was detectable and GRP170, the alternative NEF, was not expressed at higher levels. These latter results obtained in woozy mice were also confirmed in lymphoblastoid cells of MSS-patients. Moreover, the authors found no indication of a reduced immune response in woozy mice as well as in MSS-patients [37].
Suitability of woozy mice to study the role of SIL1 in endocrine pathways
Because of its vast secretory activity and the high expression level on SIL1, the pancreas is a further interesting organ for the investigation of SIL1 function. Although BiP function is modulated by SIL1, it is interesting that MSS-patients do not show symptoms of impaired secretory function of the pancreas, suggesting mechanisms that successfully compensating for loss of SIL1 function. Ittner and his co-workers showed that beta cell number and cell mass were reduced in woozy mice [38]. Moreover, proinsulin as well as insulin levels were lower in Sil1-mutant animals compared to wild type littermates. Insulin is a major regulator of fuel metabolism in muscle fibres and also stimulates mitochondrial oxidative phosphorylation in skeletal muscle along with the expression of mitochondrial proteins [39]. In line with this, SIL1 deficient muscle fibres (and SIL1-depleted HEK293 cells [23]) present with severe mitochondrial perturbations on both, the morphological and biochemical level [23] (Figs. 4 and 5). Nevertheless, further precise biochemical and functional studies are needed in order to link these perturbations to altered insulin levels. Although ER stress was observed within the beta cells, no apoptotic cells were present. Based on the reduced levels of insulin, the authors concluded that SIL1 is required for proper insulin processing and secretion in (mouse) beta cells leading to reduced glucose tolerance in woozy animals. Further experiments confirmed that SIL1 deficient cells were more vulnerable to stressors like streptozotocin (STZ), which induced type 1 diabetes in woozy mice. Moreover, marked signs of apoptosis were detectable after STZ treatment [38].
In conclusion, woozy mice with a loss of functional SIL1 show remarkable similarities to the neurodegenerative and neuromuscular phenotype of MSS-patients: Cerebellar atrophy and progressive vacuolar myopathy with myonuclear envelope changes mimic the human pathology. Therefore, woozy mice appear to be a suitable model to investigate SIL1 function in vivo. Moreover, woozy mice provide an exceeding suitable tool to study the role of the SIL1-BiP machinery not only in skeletal muscle and nervous system and especially in neuroprotection but also in physiological cascades such as production of antibodies and insulin. These aspects also suggest that this in vivo model should be useful to investigate the pathophysiology of other chaperonopathies and neurodegenerative as well as neuromuscular disorders.
CONFLICT OF INTEREST
The authors declare that they do not have a conflict of interest. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ACKNOWLEDGMENTS
This work has been supported by a grant from START program of RWTH Aachen University (to A.R.; grant No. 41/12) and by the German Society for Muscular Disorders (Deutsche Gesellschaft für Muskelkranke; DGM).
REFERENCES
[1] | Anttonen AK , Mahjneh I , Hamalainen RH , Lagier-Tourenne C , Kopra O , Waris L , et al . The gene disrupted in Marinesco-Sjogren syndrome encodes SIL1, an HSPA5 cochaperone. Nature Genetics. (2005) ;37: (12):1309–11. PubMed PMID: 16282978. |
[2] | Senderek J , Krieger M , Stendel C , Bergmann C , Moser M , Breitbach-Faller N , et al . Mutations in SIL1 cause Marinesco-Sjogren syndrome, a cerebellar ataxia with cataract and myopathy. Nature Genetics. (2005) ;37: (12):1312–4. PubMed PMID: 16282977. Epub 2005/11/12. eng. |
[3] | Krieger M , Roos A , Stendel C , Claeys KG , Sonmez FM , Baudis M , et al . SIL1 mutations and clinical spectrum in patients with Marinesco-Sjogren syndrome. Brain: A Journal of Neurology. (2013) ;136: (Pt 12):3634–44.PubMed 572 PMID: 24176978. |
[4] | Jauhari P , Sahu JK , Roos A , Senderek J , Vyas S , Singhi P . SIL1-negative Marinesco-Sjogren syndrome: First report of two sibs from India. Journal of Pediatric Neurosciences. (2014) ;9: (3):291–2.PubMed PMID: 25624944. Pubmed Central PMCID: 4302561. |
[5] | Goto M , Okada M , Komaki H , Sugai K , Sasaki M , Noguchi S , et al . A nationwide survey on Marinesco-Sjogren syndrome in Japan. Orphanet Journal of Rare Diseases. (2014) ;9: , 58. PubMed PMID: 24755310. Pubmed Central PMCID: 4021608. |
[6] | Takahata T , Yamada K , Yamada Y , Ono S , Kinoshita A , Matsuzaka T , et al . Novel mutations in the SIL1 gene in a Japanese pedigree with the Marinesco-Sjogren syndrome. Journal of Human Genetics. (2010) ;55: (3):142–6.PubMed PMID: 20111056. |
[7] | Kollipara L , Buchkremer S , Weis J , Brauers E , Hoss M , Rutten S , et al . Proteome profiling and ultrastructural characterization of the human RCMH cell line: Myoblastic properties and suitability for myopathological studies. Journal of Proteome Research 2016.PubMed PMID: 26781476. |
[8] | Zahedi RP , Volzing C , Schmitt A , Frien M , Jung M , Dudek J , et al . Analysis of the membrane proteome of canine pancreatic rough microsomes identifies a novel Hsp40, termed ERj7. Proteomics. (2009) ;9: (13):3463–73.PubMed PMID: 19579229. |
[9] | Herva R , von Wendt L , von Wendt G , Saukkonen AL , Leisti J , Dubowitz V . A syndrome with juvenile cataract, cerebellar atrophy, mental retardation and myopathy. Neuropediatrics. (1987) ;18: (3):164–9. PubMed PMID: 3683758. |
[10] | Sewry CA , Voit T , Dubowitz V . Myopathy with unique ultrastructural feature in Marinesco-Sjogren syndrome. Annals of Neurology. (1988) ;24: (4):576–80. PubMed PMID: 3239958. |
[11] | Todorov A . Marinesco-Sjogren syndrome. 1st anatomo-clinical study. Journal De Genetique Humaine. (1965) ;14: (2):197–233. PubMed PMID: 5849252. Le syndrome de Marinesco-Sjogren premiere etude anatomo-clinique. |
[12] | Skre H , Berg K . Linkage studies on Marinesco-Sjogren syndrome and hypergonadotropic hypogonadism. Clinical Genetics. (1977) ;11: (1):57–66. PubMed PMID: 830450. |
[13] | Brogdon BG , Snow RD , Williams JP . Skeletal findings in Marinesco-Sjogren syndrome. Skeletal Radiology. (1996) ;25: (5):461–5. PubMed PMID: 8837278. |
[14] | Lagier-Tourenne C , Chaigne D , Gong J , Flori J , Mohr M , Ruh D , et al . Linkage to 18qter differentiates two clinically overlapping syndromes: Congenital cataracts-facial dysmorphism-neuropathy (CCFDN) syndrome and Marinesco-Sjogren syndrome. Journal of Medical Genetics. (2002) ;39: (11):838–43. PubMed PMID: 12414825. Pubmed Central PMCID: 1735003. |
[15] | Byrne S , Dlamini N , Lumsden D , Pitt M , Zaharieva I , Muntoni F , et al . SIL1-related Marinesco-Sjoegren syndrome (MSS) with associated motor neuronopathy and bradykinetic movement disorder. Neuromuscular Disorders: NMD. (2015) ;25: (7):585–8. PubMed PMID: 25958341. |
[16] | Noreau A , La Piana R , Marcoux C , Canada F , Dion PA , Brais B , et al . Novel SIL1 mutations cause cerebellar ataxia and atrophy in a French-Canadian family. Neurogenetics. (2015) ;16: (4):315–8. PubMed PMID: 26260654. |
[17] | Macario AJ , Conway de Macario E . Chaperonopathies and chaperonotherapy. FEBS Letters. (2007) ;581: (19):3681–8. PubMed PMID: 17475257. |
[18] | Doyle KM , Kennedy D , Gorman AM , Gupta S , Healy SJ , Samali A . Unfolded proteins and endoplasmic reticulum stress in neurodegenerative disorders. Journal of Cellular and Molecular Medicine. (2011) ;15: (10):2025–39.PubMed PMID: 21722302. Pubmed Central PMCID: 4394214. |
[19] | Zhao L , Longo-Guess C , Harris BS , Lee JW , Ackerman SL . Protein accumulation and neurodegeneration in the woozy mutant mouse is caused by disruption of SIL1, a cochaperone of BiP. Nature Genetics. (2005) ;37: (9):974–9.PubMed PMID: 16116427. |
[20] | Roos A , Buchkremer S , Kollipara L , Labisch T , Gatz C , Zitzelsberger M , et al . Myopathy in Marinesco-Sjogren syndrome links endoplasmic reticulum chaperone dysfunction to nuclear envelope pathology. Acta Neuropathologica. (2014) ;127: (5):761–77.PubMed PMID: 24362440. |
[21] | Dutta C , Day T , Kopp N , van Bodegom D , Davids MS , Ryan J , et al . BCL2 suppresses PARP1 function and nonapoptotic cell death. Cancer Research. (2012) ;72: (16):4193–203. PubMed PMID: 22689920. Pubmed Central PMCID: 4075432. |
[22] | Dudek J , Benedix J , Cappel S , Greiner M , Jalal C , Muller L , et al . Functions and pathologies of BiP and its interaction partners. Cellular and Molecular Life Sciences: CMLS. (2009) ;66: (9):1556–69.PubMed PMID: 19151922. |
[23] | Roos A , Kollipara L , Buchkremer S , Labisch T , Brauers E , Gatz C , et al . Cellular Signature of SIL1 Depletion: Disease Pathogenesis due to Alterations in Protein Composition Beyond the ER Machinery. Molecular Neurobiology 2015. PubMed PMID: 26468156. |
[24] | Zhao L , Rosales C , Seburn K , Ron D , Ackerman SL . Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjogren syndrome. Human Molecular Genetics. (2010) ;19: (1):25–35.PubMed PMID: 19801575. Pubmed Central PMCID: 2792147. |
[25] | Wang M , Ye R , Barron E , Baumeister P , Mao C , Luo S , et al . Essential role of the unfolded protein response regulator GRP78/BiP in protection from neuronal apoptosis. Cell Death and Differentiation. (2010) ;17: (3):488–98.PubMed PMID: 19816510. Pubmed Central PMCID: 2822118. |
[26] | Ghoumari AM , Wehrle R , Bernard O , Sotelo C , Dusart I . Implication of Bcl-2 and Caspase-3 in age-related Purkinje cell death in murine organotypic culture: An in vitro model to study apoptosis. The European journal of Neuroscience. (2000) ;12: (8):2935–49.PubMed PMID: 10971635. |
[27] | McClure WC , Rabon RE , Ogawa H , Tseng BS . Upregulation of the creatine synthetic pathway in skeletal muscles of mature mdx mice. Neuromuscular Disorders: NMD. (2007) ;17: (8):639–50.PubMed PMID: 17588756. Pubmed Central PMCID: 2706264. 686 |
[28] | Eltoweissy M , Muller GA , Bibi A , Nguye PV , Dihazi GH , Muller CA , et al . Proteomics analysis identifies PARK7 as an important player for renal cell resistance and survival under oxidative stress. Molecular Bio Systems. (2011) ;7: (4):1277–88.PubMed PMID: 21308111. |
[29] | Villa A , Podini P , Panzeri MC , Soling HD , Volpe P , Meldolesi J . The endoplasmic-sarcoplasmic reticulum of smooth muscle: Immunocytochemistry of vas deferens fibers reveals specialized subcompartments differently equipped for the control of Ca2+ homeostasis. The Journal of Cell Biology. (1993) ;121: (5):1041–51.PubMed PMID: 8388876. Pubmed Central PMCID: 2119688. |
[30] | Kobayashi T , Takita Y , Suzuki A , Katsu Y , Iguchi T , Ohta Y . Vacuolar degeneration of skeletal muscle in transgenic mice overexpressing ORP150. The Journal of Veterinary Medical Science/the Japanese Society of Veterinary Science. (2008) ;70: (1):115–8. PubMed PMID: 18250584. |
[31] | Howes J , Shimizu Y , Feige MJ , Hendershot LM . C-terminal mutations destabilize SIL1/BAP and can cause Marinesco-Sjogren syndrome. The Journal of Biological Chemistry. (2012) ;287: (11):8552–60.PubMed PMID: 22219183. Pubmed Central PMCID: 3318681. |
[32] | Filezac de L’Etang A , Maharjan N , Cordeiro Brana M , Ruegsegger C , Rehmann R , Goswami A , et al . Marinesco-Sjogren syndrome protein SIL1 regulates motor neuron subtype-selective ER stress in ALS. Nature Neuroscience. (2015) ;18: (2):227–38.PubMed PMID: 25559081. |
[33] | Haas IG , Wabl M . Immunoglobulin heavy chain binding protein. Nature. 306. (1983) )387–9.PubMed PMID: 6417546. |
[34] | Bole DG , Hendershot LM , Kearney JF . Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. The Journal of Cell Biology. (1986) ;102: (5):1558–66.PubMed PMID: 3084497. Pubmed Central PMCID: 2114236. |
[35] | Hendershot L , Bole D , Kohler G , Kearney JF . Assembly and secretion of heavy chains that do not associate posttranslationally with immunoglobulin heavy chain-binding protein. The Journal of Cell Biology. (1987) ;104: (3):761–7.PubMed PMID: 3102505. Pubmed Central PMCID: 2114523. |
[36] | Lee YK , Brewer JW , Hellman R , Hendershot LM . BiP and immunoglobulin light chain cooperate to control the folding of heavy chain and ensure the fidelity of immunoglobulin assembly. Molecular Biology of the Cell. (1999) ;10: (7):2209–19.PubMed PMID: 10397760. Pubmed Central PMCID: 25436. |
[37] | Ichhaporia VP , Sanford T , Howes J , Marion TN , Hendershot LM . Sil1, a nucleotide exchange factor for BiP, is not required for antibody assembly or secretion. Molecular Biology of the Cell. (2015) ;26: (3):420–9.PubMed PMID: 25473114. Pubmed Central PMCID: 4310734. |
[38] | Ittner AA , Bertz J , Chan TY , van Eersel J , Polly P , Ittner LM . The nucleotide exchange factor SIL1 is required for glucose-stimulated insulin secretion from mouse pancreatic beta cells in vivo . Diabetologia. (2014) ;57: (7):1410–9.PubMed PMID: 24733160. |
[39] | Stump CS , Short KR , Bigelow ML , Schimke JM , Nair KS . Effect of insulin on human skeletal muscle mitochondrial ATP production, protein synthesis, and mRNA transcripts. Proceedings of the National Academy of Sciences of the United States of America. (2003) ;100: (13):7996–8001.PubMed PMID: 12808136. Pubmed Central PMCID: 164701. |
Figures and Tables
Fig.1
Schematic overview of impaired BiP function due to loss of functional SIL1. The cellular processes highlighted in the respective boxes are potentially affected by loss of (functional) SIL1.
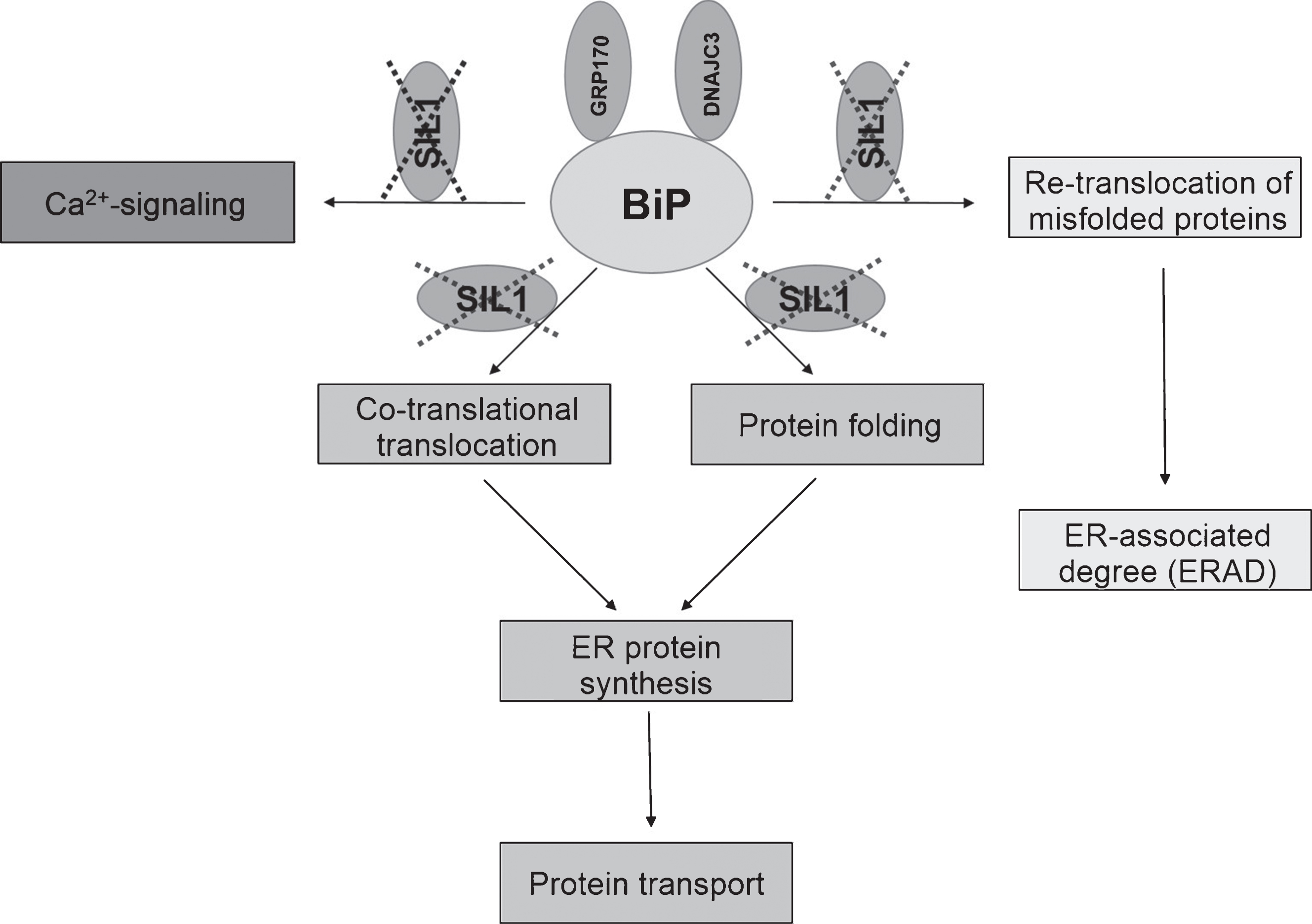
Fig.2
Cerebellar alterations in woozy mice. H&E: (A) Purkinje cell layer (white arrows) and absence of glial cell proliferation in a 6-week-old wild type littermate control. Scale bar = 20 μm. (B) Occasional mislocalisation of Purkinje-cells (white arrows) and proliferated (Bergmann) glial cells in a 6-week-old woozy mouse. Scale bar = 20 μm. (C) Purkinje-cell layer (white arrows) and absence of glial cell proliferation in a 26-week-old wild type littermate control. Scale bar = 20 μm. (D) Complete loss of Purkinje cells in a 26-week-old woozy animal. Scale bar = 15 μm. Immunohistochemistry: (E–L) Purkinje cells in the neo- and vestibulocerebellum of 6 weeks (E, G, I and K) and 26 weeks (F, H, J and L) old wildtype animals show enrichment of BCL2 in dendrites (E–H). Sil1-mutant Purkinje cells show decreased immunoreactivity in the neocerebellum (degenerating cells, G), but increased reactivity in Purkinje cells of the non-vulnerable vestibulocerebellum (surviving cells, H). (I–L) Investigation of PARP1 in Purkinje cells of woozy revealed increased immunoreactivity in the nuclei of neocerebellar Purkinje cells (K) but not in vestibulocerebellar Purkinje cells (L) compared to wildtype animals (I, J).
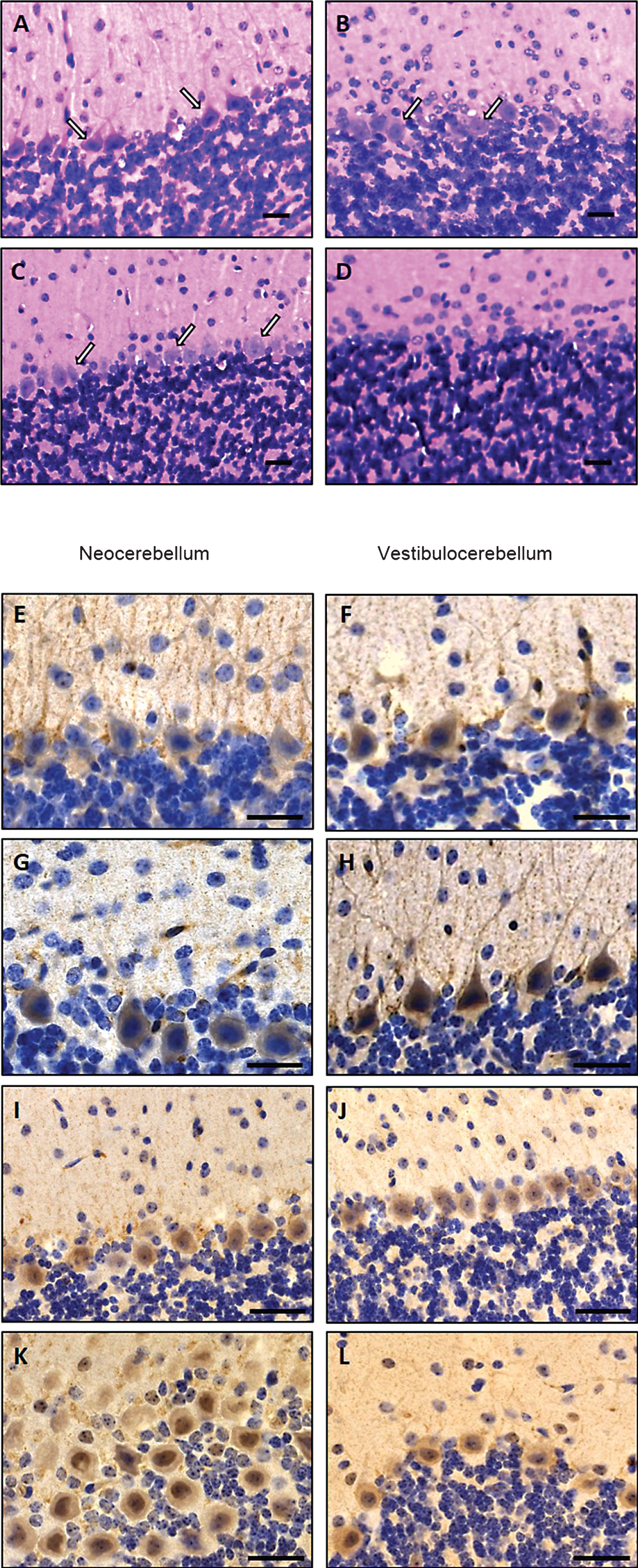
Fig.3
Pathology of Sil1-mutant mouse muscle: (A) Normal macroscopic appearance of hind limb muscle in a 26-week-old wild type mouse. (B) Reduced muscle bulk and in a woozy littermate. (C) Largely normal light microscopical appearance of wild type mouse quadriceps muscle at age 26 weeks. H&E. Scale bar = 25 μm. (D) Vacuolar myopathy with variable muscle fibre calibres and several non-subsarcolemmal myonuclei in a woozy littermate mouse; H&E. scale bar = 25 μm. (E) Quantitative analysis of wild type and woozy mouse body weight (26-week-old). Woozy animals show a significant reduction in body weight which is in line with muscle wasting in these animals.

Fig.4
Ultrastructural alterations in woozy mouse quadriceps muscle at age of 26 weeks (A–C) and in genetically proven MSS-patients (D–G) analysed by transmission electron microscopy. (A) Disrupted intermyofibrillar mitochondria. Scale bar = 1.4 μm. (B) Pyknotic, degenerating myonucleus showing lift-off of the nuclear envelope (white arrows) specific for SIL1 deficiency. Scale bar = 1.3 μm. (C) Prominently widened and proliferated SR (white arrow) associated with autophagic material adjacent to a centralised myonucleus as well as accumulation of autophagic, myelin-like material in the sub-sarcolemmal space. Scale bar = 2.5 μm. (D) Disrupted intermyofibrillar mitochondria. Scale bar = 1 μm. (E) Degenerating myonucleus showing lift-off of the nuclear envelope surrounded by electron-dense myelin-like material most likely corresponding to protein aggregates. Scale bar = 1.7 μm. (F) Magnification of E highlighting the lamina fibrosa alteration specific for SIL1-deficiency (white arrows). Scale bar = 0.5 μm. (G) Pyknotic subsarcolemmal myonucleus with massive lift-off of the nuclear envelope (white arrow). Electron-dense myelin-like and granular material is detectable adjacent to the diseased myonucleus as well as within the irregular nuclear envelope loop. Scale bar = 1 μm.
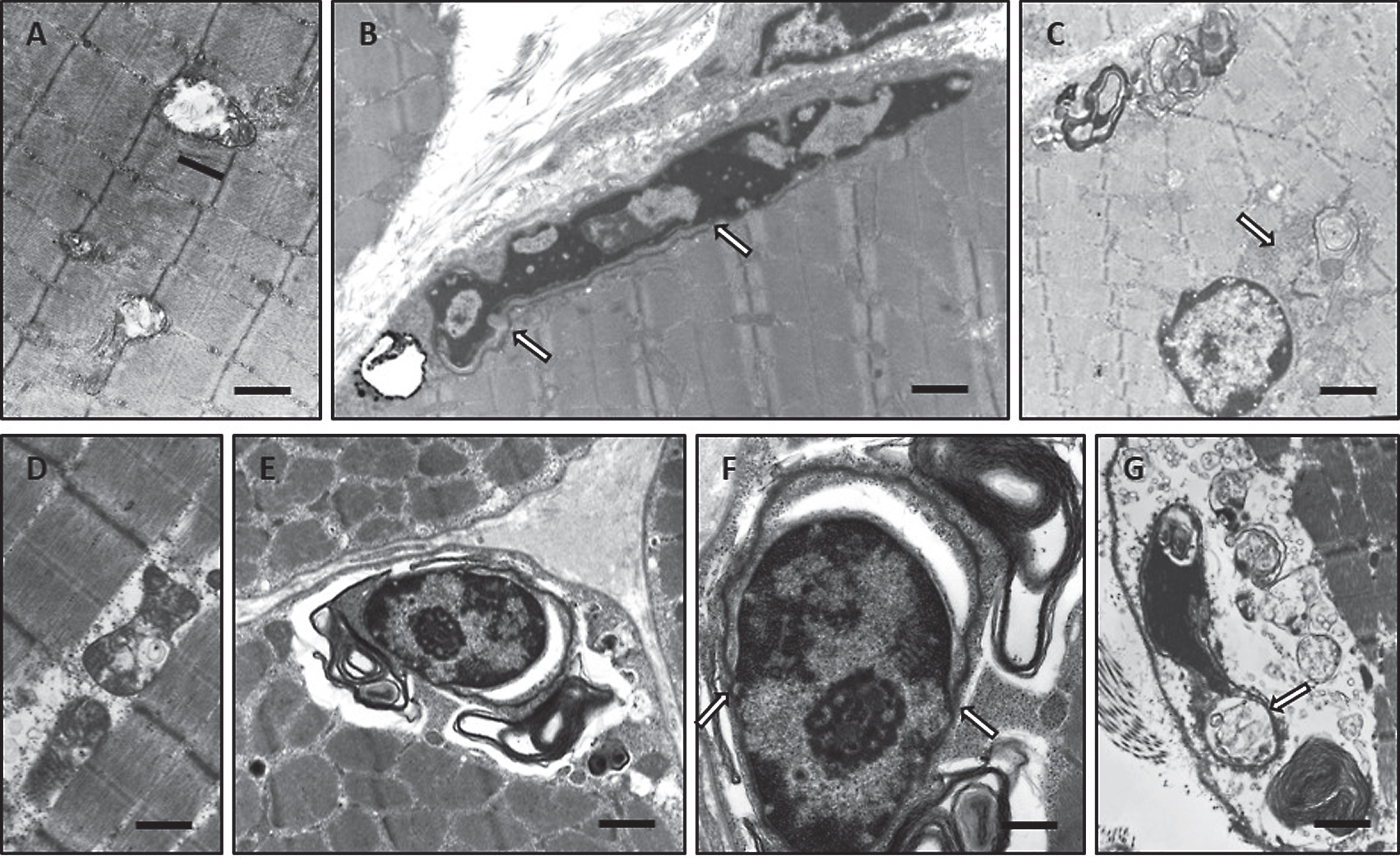
Fig.5
Immunohistochemistry of paraffin sections of paraformaldehyde-fixed quadriceps muscle specimens of 26-week-old wild type (A, C, E, F, I, M, Q and U) and woozy (B, D, G, H, J–L, N–P, R–T, V–X) animals: (A) weak creatine kinase (muscular) immunoreactivity of the sarcoplasm in wild type mouse muscle fibres. Scale bar = 22 μm. (B) Muscle fibres of normal size and especially partially atrophic muscle fibres showing increased muscle creatine kinase immunoreactivity. Scale bar = 19 μm. (C) Minor DJ-1 immunoreactivity of wild type mouse muscle and enhanced diffuse as well as perinuclear (black arrows) and vacuole-associated (white arrows) DJ-1 immunoreactivity in Sil1-mutant muscle fibres (D). Scale bars = 20 μm. (E) Minor diffuse immunoreactivity for the SIL1 binding partner BiP in wild type mouse muscle fibres is showing a sarcomeric pattern which probably corresponds to a labelling of the SR. Scale bar = 22 μm. (F) BiP immunoreactivity associated with myonuclei/ nuclear envelope (black arrows) in mouse wild type muscle fibres. Scale bar = 10 μm. (G and H) Partially atrophic and atrophic muscle fibres of a woozy animal show strong BiP immunoreactivity of subsarcolemmal and intermyofibrillar deposits, some of which are associated with vacuoles and myonuclei. Scale bars = 22 μm. (I) Diffuse immunoreactivity of TDP-43 within the sarcoplasm and occasionally in myonuclei (inset in I) of muscle fibres derived from wild type animals. Scale bar = 22 μm; in inset 10 μm. (J–L) In this woozy mouse muscle specimen, enhanced intermyofibrillär punctuate TDP-43 immunoreactivity is accompanied by prominent labelling of the perinuclear region and nuclear membrane (inset in J and L). Scale bars in J and K = 22 μm; inset in J = 10 μm and L = 15 μm. (M) Nuclear labelling after incubation with the LAP2 antibody in wild type mouse muscle. Scale bars in M = 22 μm, in inset = 10 μm. (N–P) Prominent LAP2 immunoreactivity of the abnormal vacuoles (white arrows) and of the sarcoplasm of woozy muscle fibres. Scale bars in N = 25 μm, in O = 22 μm, in P = 19 μm. (Q) Myonuclei with weak Emerin immunoreactivity in wild type muscle fibres. Scale bar = 10 μm. (R, S) Prominent immunoreactivity for Emerin in dystrophic muscle fibres (sarcoplasmic staining) and adjacent to myonuclei (white arrows). Scale bars = 19 μm. (T) Irregular Emerin immunoreactivity of the sub-sarcolemmal region. Scale bars in R and S = 20 μm, in T = 17 μm. (U) Myonuclei with very weak Lamin A/C immunoreactivity in wild type muscle fibres. Scale bar = 6 μm. (V) Prominent immunoreactivity for Lamin A/C adjacent to myonuclei and (W, X) in lesioned muscle fibres (sarcoplasmic staining). Scale bars in V = 6 μm, in W and X = 25 μm.
Fig.6
Immunoblot studies of total quadriceps muscle lysates from 16-, 26-, 52- and 84-week old woozy animals and wildtype controls. Compared to protein abundances in wildtype littermates, RYR1 and DHPR show decrease upon disease progression. GAPDH which was used as loading control did not show altered abundances in Sil1-mutant muscles compared to respective control samples.
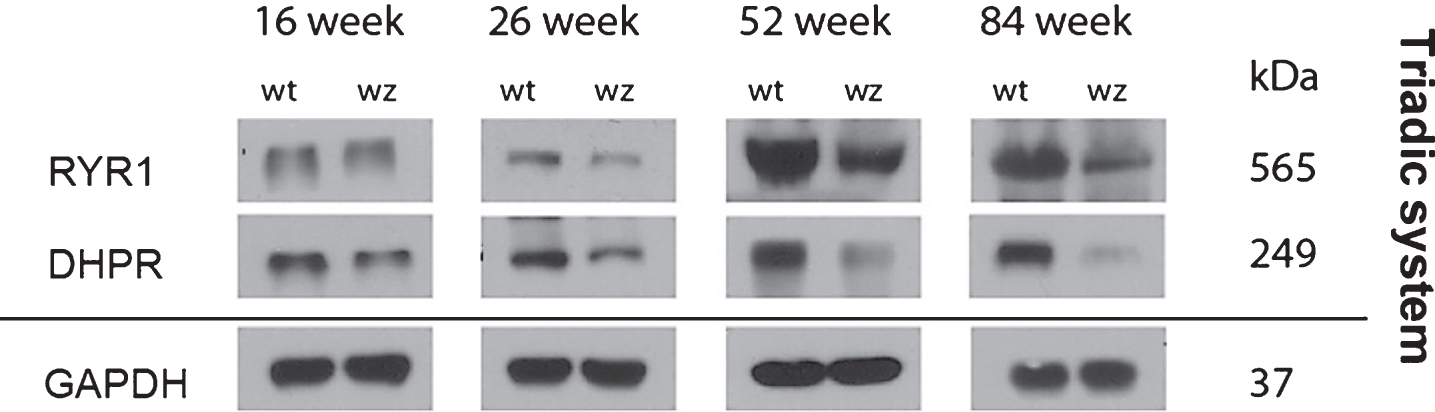
Table 1
List of antibodies used for the immunohistochemistry studies
| Antigene | Source | Dilution | Company |
| BCL2 | rabbit | 1:100 | Genetex |
| BiP | rabbit | 1:100 | Genetex |
| Creatine kinase (muscluar) | rabbit | 1:100 | Genetex |
| DHPR | rabbit | 1:1000 | Abcam |
| Dj-1 | rabbit | 1:100 | Santa Cruz |
| Emerin | mouse | 1:50 | Novocastra |
| GAPDH | rabbit | 1:1000 | Genetex |
| Lamin A/C | mouse | 1:50 | Vector Laboratories |
| LAP2 | rabbit | 1:50 | Millipore |
| PARP1 | rabbit | 1:100 | Genetex |
| RYR1 | rabbit | 1:1000 | Abcam |
| TDP-43 | mouse | 1:100 | Anova |
Table 2
Comparison of clinical and morphological findings in man and mouse with SIL1/Sil1-mutations. The description of the SIL1-related Marinesco-Sjögren syndrome phenotype is based on the clinical findings summarized/described in [3, 5, 15]
| Clinical features | MSS-patients | Woozy mice |
| Mental retardation | Varying degree | Not investigated |
| Cerebellar signs | ataxia | ataxia |
| Cerebellar atrophy | Present | Present |
| Purkinje cell loss | Not investigated | In neocerebellum but not in vestibulocerebellum |
| Nystagmus | Occasionally | Not present |
| Myopathological changes | ||
| Muscle atrophy | Present | Present |
| Autophagic vacuoles | Present | Present |
| Degenerating mitochondria in muscle fibres | Present | Present |
| Degeneration myonuclei | Present | Present |
| Proliferated SR | Present | Present |
| NE perturbations with electron-dense membranes | Present | Present |
| Altered triadic system | Present | Present |
| Elevated creatine kinase levels | Mild to moderate increase | Elevated level of CK protein in muscle fibres |
| Cataracts | Congenital or infantile | Not present |
| Growth | ||
| Short stature | Occasionally | Not present |
| Microcephaly | Rare | Not present |


