
ICNMD 2016: Abstract Book for the 14th International Congress on Neuromuscular Diseases, July 5–9, 2016 Toronto, Canada
CONTENTS
Plenary Sessions
PL 1: Plenary Lecture - Genetics ........................................................................................................S3
PL 2: Plenary Lecture - Hot Topics .....................................................................................................S4
PL 3: Plenary Lecture - Muscular Dystrophy ......................................................................................S8
PL 4: Plenary Lecture - Motor Neuron Disease ................................................................................S10
Late Breaking News Sessions
LB 1: Late Breaking News Session ..................................................................................................S15
Workshop Sessions
WS 1: Workshop Sessions ...............................................................................................................S19
WS 2: Workshop Sessions ...............................................................................................................S22
WS 3: Workshop Sessions ...............................................................................................................S28
WS 4: Workshop Sessions ...............................................................................................................S32
WS 5: Workshop Sessions ...............................................................................................................S37
WS 6: Workshop Sessions ...............................................................................................................S41
WS 7: Workshop Sessions ...............................................................................................................S45
WS 8: Workshop Sessions ...............................................................................................................S50
Poster Sessions
PS 1: Poster Sessions - Group 2......................................................................................................S55
PS 1: Poster Sessions - Group 4......................................................................................................S61
PS 1: Poster Sessions - Group 6......................................................................................................S91
PS 1: Poster Sessions - Group 8....................................................................................................S103
PS 2: Poster Sessions - Group 1....................................................................................................S132
PS 2: Poster Sessions - Group 3....................................................................................................S185
PS 2: Poster Sessions - Group 5....................................................................................................S204
PS 2: Poster Sessions - Group 7....................................................................................................S209
Author index .................................................................................................................................. 217
Plenary Sessions
PL 1.1 / #21GENOMIC APPROACHES TO DIAGNOSIS OF RARE MUSCLE DISEASE
Topic: Plenary Lecture - Genetics
Daniel MacArthur
Medical And Population Genetics, Broad Institute of Harvard and MIT, Cambridge, MA, US
Abstract: Genomic technologies have profoundly changed our ability to uncover the genes underlying a wide range of rare Mendelian diseases. Here I describe three major technological advances in genomic approaches to rare disease diagnosis, and their application to neuromuscular disease. Firstly, I discuss the development of a massive reference panel of “healthy” exomes, the Exome Aggregation Consortium (ExAC) and demonstrate how ExAC data can be used to more effectively filter the variants identified in rare disease patients. Secondly, I outline the value of whole-genome sequencing in the discovery of causal variants missed through exome sequencing. Finally, I describe a pilot study on the application of muscle transcriptome sequencing (RNA-seq) on a set of over 40 exome-unsolved muscle disease cases, and the high resulting diagnostic yield from discovery of splice-disrupting and expression-altering variants. Finally, I outline several unresolved challenges of genomic diagnosis in rare disease cases.
PL 1.2 / #22GENE DISCOVERY IN CHARCOT-MARIE-TOOTH NEUROPATHIES
Topic: Plenary label - Genetics
Stephan Züchner
Human Genetics, University of Miami, Miami, FL, US
Abstract: Next-generation sequencing has significantly enhanced the pace of gene discovery in peripheral neuropathies (also known as Charcot-Marie-Tooth neuropathies or CMT) and neuromuscular disorders. As gene panels and whole exomes sequencing become routine, the delineation and precise description of phenotypic spectra of disease genes has become a new frontier. The increasingly observed genotypic/phenotypic overlap of clinically distinct entities is a challenge to the existing classification of disorders. Finally, the long-standing clinical notion of modifiers of disease, environmental and genetic, has caught renewed attention, as dramatically improved genetic tools offer new analytical strategies. Over the last seven years, the CMT community has developed an infrastructure that has enabled the assimilation of clinical data from thousands of patients seen by CMT experts from around the world, along with their DNA, all in ways that de-identify their personal information. This is largely driven by the Inherited Neuropathy Consortium and the CMT-International Database. Key elements include a standardized approach to phenotyping, digital formatting of the clinical date that enables querying together with genetic data, and efficient processing of patient DNA samples. Defining the genetic basis of inherited peripheral neuropathies, also known as Charcot-Marie-Tooth disease (CMT), has progressed rapidly in the past five years. Beyond individual studies, the innovative sharing of genomic data in real time at any scale has fostered a collaborative spirit in the CMT and neighboring fields not seen before. Through these efforts, more than 30 novel genes have been published since 2012. Still more than 50% of axonal CMT patients go without diagnosis, thus whole genome sequencing will likely open new opportunities. This presentation will provide an overview and specific examples of recent success and challenges in the field.
PL 1.3 / #23RNA SEQUENCE AND RNA ANALYSIS
Topic: Plenary Lecture - Genetics
James J. Dowling
Neurology, Hospital for Sick Children, Toronto, ON, CA
Abstract: The application of next generation sequencing has ushered in a revolution in mutation discovery and clinical genetic diagnostics. However, despite its now widespread use in patients, the cause of disease remains unsolved in nearly 50% of cases of genetic neuromuscular disease. This lack of knowledge creates significant barriers to diagnostics, clinical care, and therapy development. One potential explanation for this knowledge gap is that many mutations reside in the non-coding genome. Such mutations are not assessed by current testing modalities such as gene panels and whole exome sequencing. We believe that RNA analysis (via RNA sequencing or RNAseq) is the ideal modality for overcoming these challenges, for identifying and interpreting non-coding mutations, and thus for bridging the current diagnostic gap. In this study, I will describe our efforts at mutation discovery using RNAseq as a diagnostic modality in childhood muscle disease, and will discuss the broader applicability of RNAseq to all genetic disorders of the peripheral nervous system.
PL 2.1 / #24STEM CELL THERAPY IN ALS
Topic: Plenary Lecture - Hot Topics
Jonathan D. Glass1, Nicholas M. Boulis1, Parag G. Patil2, Nazem Atassi3, Merit Cudkowicz4, Karl Johe5, Stephen A. Goutman2, Eva L. Feldman6
1Emory University, Atlanta, GA, US;
2University of Michigan, Ann Arbor, MI, US;
3Massachusetts General Hospital, Boston, MA, US;
4Neurology, MGH, Boston, MA, US;
5Neuralstem, Inc., Germantown, MD, US;
6Department Of Neurology, University of Michigan, Ann Arbor, MI, US
Abstract: Cellular therapies offer multiple benefits to combat the complex pathogenesis of amyotrophic lateral sclerosis (ALS), a fatal disease characterized by progressive motor neuron degeneration. While several cell types and delivery strategies have been examined, our experience developing an intraspinal transplantation strategy employing human spinal stem cells (HSSCs) has led to two first-in-human FDA-approved clinical trials. The Phase 1 trial followed a risk-escalation design whereby 15 ALS patients were subjected to increased risk across 5 cohorts based on their level of disability at the time of surgery and the number and placement of injections. Final doses ranged from 500,000 to 1 million cells and were delivered in 5 unilateral or 10 bilateral injections targeting the lumbar and/or cervical spinal cord. Results demonstrated that the intraspinal stem cell transplantation strategy was safe, feasible, and well-tolerated, and monitoring of clinical progression further revealed preliminary insight into potential windows of stem cell biological activity. In the subsequent Phase 2a trial, the safety of increasing stem cell doses ranging from 2 to 16 million cells, which were achieved via increased concentrations per injection and numbers of injections, was assessed. Analyses of the results are ongoing and will be presented; however, the approach was well-tolerated across all cohorts, including the final cohort which received 8 million cells over 20 injections in both the cervical and lumbar spinal cord regions. Comparison of patient functional outcome measures following the procedure with historical control groups also revealed that the therapy did not accelerate disease progression, further verifying safety, and potential windows of biological activity were again observed. Moreover, insight into potential outcome measures that correlate with ALSFRS-R scores were examined to inform future trial phase endpoints, and the Phase 2a trial was expanded to include 3 surgical centers, supporting the feasibility of future larger-scale trials. Overall, our progress to date supports continued examination of this therapeutic strategy, and future trial phases assessing efficacy are being planned. Funding support provided by: ALS Association, National Institutes of Health (R01 NS077982), and Neuralstem, Inc.
PL 2.2 / #25RESULTS OF THE THYMECTOMY TRIAL IN MYASTHENIA GRAVIS
Topic: Plenary Lecture - Hot Topics
Gil I. Wolfe1, Henry J. Kaminski2, Inmaculada B. Aban3, Gary R. Cutter3, Plus Mgtx Study Group4
1Dept Of Neurology, Univ at Buffalo/SUNY, Buffalo, NY, US;
2Dept Of Neurology, George Washington University, Washington, US;
3Univ. of Alabama-Birmingham, Birmingham, AL, US;
4Univ. at Buffalo, Buffalo, NY, US
Abstract: BACKGROUND: Since first utilized 75 years ago, a benefit from thymectomy in non-thymomatous myasthenia gravis (MG) has been claimed by numerous studies. In 2000, an evidence-based review summarized outcomes from 21 controlled, non-randomized MG cohorts, pointed out methodological flaws preventing definite conclusions regarding the procedure’s benefit, but concluded that thymectomy should be a treatment option. In concert with calls dating back a half century, the authors recommended a controlled, randomized study that employed a standardized medical therapy. METHODS: MGTX was organized as a five-continent, international, rater-blinded, randomized trial with the aim to answer three questions: Does extended transsternal thymectomy (ETTX) combined with a strictly defined prednisone protocol, when compared with the prednisone protocol alone, after 3 years (1) result in a greater improvement in myasthenic weakness, (2) result in a lower total dose of prednisone, and (3) enhance the quality of life by reduction of adverse events? Inclusion criteria were MG Foundation of America Clinical Classification 2 to 4, elevated acetylcholine receptor (AChR) antibody levels, age >18.0 and <65.0 years, and disease history <5 years. Patients were followed in rater-blind fashion for a minimum of 3 years. RESULTS: A total of 126 patients were randomized between 2006 and 2012 across 36 sites. In the two-stage primary analysis, patients who underwent ETTX showed a significantly improved clinical status based on the mean of the area under the Quantitative MG score time curve at month 36. Likewise, the area under the dose time curve for prednisone use was significantly lower for the ETTX group. Further details will be provided at the meeting. CONCLUSIONS: MGTX provides Class I evidence that ETTX has a favorable impact in MG, based on both clinical outcomes and reduced requirements for immunotherapeutic agents. This randomized, single-blinded, controlled study provides the strongest evidence to date to support the longstanding practice of recommending thymectomy as an intervention to improve outcomes in non-thymomatous MG.
PL 2.3 / #416REGAIN: A RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED MULTI-CENTER PHASE 3 STUDY OF THE SAFETY AND EFFICACY OF ECULIZUMAB IN SUBJECTS WITH REFRACTORY GENERALIZED MYASTHENIA GRAVIS
Topic: Plenary Lecture - Hot Topics
James F. Howard, Jr.1, Kimiaki Utsugisawa2, Michael Benatar3, Hiroyuki Murai4, Richard J. Barohn5, Isabel Illa Sendra6, Saiju Jacob7, John Vissing8, Ted M. Burns9, Carlos Casasnovas Pons10, Jan De Bleecker11, John T. Kissel12, Srikanth Muppidi13, Richard Nowak14, Tuan Vu15, Gary R. Cutter16, Fanny O’Brien17, Jing Jing Wang17, Renato Mantegazza18
1Department Of Neurology, The University of North Carolina, Chapel Hill, NC, US;
2Department Of Neurology, Hanamaki General Hospital, Hanamaki, JP;
3Neurology, University of Miami, Miami, FL, US;
4department Of Neurological Therapeutics, Kyushu University, Fukuoka Prefecture, JP;
5Neurology, The University of Kansas Medical Center, Kansas City, KS, US;
6Hospital Sant Pau, Universitat Autònoma, Barcelona, ES;
7Queen Elizabeth Neuroscience Centre, University Hospitals, Birmingham, Birmingham, GB;
8Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK;
9University of Virginia Health System, Charlottesville, VA, US;
10Hospital Universitari de Bellvitge, Barcelona, ES;
11AZ Sint-Lucas - Campus Sint-Lucas, Ghent, BE;
12Department Of Neurology, Ohio State University, Columbus, OH, US;
13Stanford University School of Medicine, Stanford, CA, US;
14Neurology, Yale School of Medicine, New Haven, CT, US;
15Neurology, University of South Florida, Tampa, FL, US;
16Univ. of Alabama-Birmingham, Birmingham, AL, US;
17Alexion Pharmaceuticals, Cheshire, CT, US;
18Neuromuscular Diseases And Neuroimmunology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, IT
Abstract: Background: Acetylcholine receptor (AChR) antibody-positive myasthenia gravis (MG), constituting ~80% of MG cases, is a rare, complement-mediated autoimmune disorder characterized by fluctuating and fatigable muscle weakness. Involvement of respiratory or oropharyngeal muscles may be life-threatening. Despite a wide range of available therapies (including cholinesterase inhibitors, corticosteroids, B−cell- and T−cell-directed immunosuppressive therapies, and thymectomy), some patients remain refractory to treatment due to inadequate response or inability to tolerate side effects.1 Moreover, no current treatments target the complement component of MG pathophysiology, which is thought to contribute substantially to loss of AChR function at the neuromuscular junction. Eculizumab is a humanized monoclonal antibody that inhibits the cleavage of C5 to C5a and C5b, thereby protecting the neuromuscular junction from the destructive effects of antibody-mediated complement activation.1 In a prior phase 2 randomized, double-blind, placebo-controlled crossover study, eculizumab-treated subjects with refractory MG demonstrated significant, clinically meaningful symptomatic improvement, with 86% experiencing a ≥3-point reduction, and 57% a ≥8-point reduction, in the Quantitative Myasthenia Gravis (QMG) score, compared with 57% and 14% of placebo-treated subjects, respectively.1 Eculizumab was also well tolerated in all treated MG patients.1 Methods: We conducted a phase 3 randomized, double-blind, placebo-controlled, multicenter study to evaluate the safety and efficacy of eculizumab in patients with refractory generalized MG. Subjects ≥18 years of age with confirmed anti-AChR antibody-positive generalized MG (Myasthenia Gravis Foundation of America Class II-IV), with a Myasthenia Gravis Activities of Daily Living profile (MG-ADL) screening/baseline score of ≥6 were eligible if they had failed to respond adequately to at least 2 immunosuppressive therapies (or at least 1 immunosuppressive therapy and required chronic intravenous immunoglobulin therapy or plasma exchange). Subjects were randomized 1:1 to receive intravenous infusion of eculizumab for 26 weeks (900 mg/week for 4 weeks, followed by 1200 mg/every 2 weeks) or matching placebo. Rescue therapy (high-dose corticosteroids, plasma exchange, or intravenous immunoglobulin therapy) was permitted at the discretion of the treating physician. The primary efficacy endpoint was change from baseline to week 26 in the MG-ADL total score; safety evaluations included adverse events, vital signs, electrocardiography, and laboratory assessments. Results and Discussion: Final results from the study will be presented and discussed. This study was sponsored by Alexion Pharmaceuticals (New Haven, CT, USA).
PL 2.4 / #26APPROACH TO PATIENT-CENTERED OUTCOMES RESEARCH
Topic: Plenary Lecture - Hot Topics
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: In recent years there has been a great effort to get patients, families, community and patient advisory groups more involved in the clinical research process. When developing an idea for a clinical research project we get patients involved to identify their needs and what areas they believe we should focus on. To do this we have had focus groups with neutral moderators. We then place patients and others on the project steering committees, communication committees and Data Safety Monitoring Boards (DSMB). Outcome measures are increasingly becoming patient driven and the field of patient reported outcomes measures (PROM) is extremely important. We often use various patient reported outcomes in our trials as either primary or secondary outcomes. To enable many of these concepts, the Patient-Centered Outcomes Research Institute (PCORI) was created. PCORI is an independent research organization authorized by US Congress as part of the 2010 Patient Protection and Affordable Care Act. The goal is to provide information to patients or those who care for them in ways that they can understand. This is accomplished by funding patient-centered comparative clinical effectiveness research (CER), identify critical research questions and disseminate results in ways the patients and clinicians find useful and valuable. There are many types of PCORI funding. One such funding mechanism is the Assessment of Prevention, Diagnosis and Treatment Options. The goal of this mechanism is to compare the effectiveness of two or more interventions or approaches to health care, examining their risks and benefits – CER. We have been successful in obtaining one of these awards to conduct a comparative effectiveness study of drugs for management of pain in cryptogenic sensory polyneuropathy (CSPN). The trial is called Patient Assisted Intervention for Neuropathy: Comparison of Treatment in Real Life Situations (PAINSCONTRoLS). We are comparing duloxetine, pregabalin, nortriptyline and mexiletine at about 40 sites in the USA and Canada. Another idea we have for a PCORI grant of this type including finding the best drug for saliva control in ALS. PCORI also funds an infrastructure called PCORnet: the National Patient-Centered Clinical Research Network. The goal is to foster a wide range of experimental and observational patient-centered studies. This network is broken down to Clinical Data Research Networks (CDRNs) and Patient-Powered Research Networks (PPRNs). Patients and other stakeholders are participating in the development and implementation of network activities in both the CDRNs and PPRNs. University of Kansas Medical Center has a CDRN called the Greater Plains Collaborative (GPC). The GPC disease areas we are currently focusing on initially involved Breast Cancer, Obesity and Amyotrophic Lateral Sclerosis. The GPC includes 12 academic centers in the Midwest and Texas from Milwaukee and Minneapolis to Dallas and San Antonio. The PPRN received a cooperative agreement award. The goals of the PPRN is to increase size and diversity of patient membership, willingness to build standardized database of patient-reported data and the willingness to explore collection of electronic clinical data. The idea is that this data would be suitable for sharing with other infrastructure members.
PL 2.5 / #27DO WE STILL NEED MUSCLE BIOPSY IN THE ERA OF ULTRASOUND?
Topic: Plenary Lecture - Hot Topics
Carsten Bonnemann
Neuromuscular And Neurogenetic Disorders Of Childhood Section, National Institutes of Health, Bethesda, MD, US
Abstract: not received.
PL 2.6 / #28THERAPEUTIC APPROACHES TO INCLUSION BODY MYOSITIS
Topic: Plenary Lecture - Hot Topics
Mazen Dimachkie
Neurology, The University of Kansas Medical Center, Kansas City, KS, US
Abstract: Sporadic inclusion body myositis (IBM) is the most common idiopathic inflammatory myopathy after the age of 40 to 50 years. IBM typically presents with chronic insidious mildly asymmetric proximal leg and distal arm muscle weakness. Based on histopathologic evidence of endomysial inflammation, IBM was originally believed to be a primary inflammatory myopathy. However, there are sarcoplasmic aggregates of p62 and TDP-43 redistribution in support of neurodegenerative etiology. While the exact contribution of these two pathways to the pathogenesis of IBM remains unknown, IBM is refractory to immunotherapies. Research is needed in this field and at least three major clinical trials are currently at different stages in an attempt to identify an effective therapy for people with IBM. Two of these are ongoing studies that aim to increase muscle size strength and function using different approaches. In the follistatin gene transfer therapy, follistatin gene carried by adeno-associated virus is injected into the thigh muscle of IBM and Becker muscular dystrophy patients. In a large multicenter blinded trial, Novartis is investigating the efficacy, safety and tolerability of bimagrumab, which inhibits the Activin II receptor and thereby interferes with the myostatin signaling cascade. A third approach is to target the protein homeostasis. We conducted a randomized double-blind placebo-controlled pilot trial which demonstrated arimoclomol to be safe and well tolerated in IBM. A large multicenter clinical trial powered for efficacy will be starting this year and aims to establish whether arimoclomol can slow down disease progression in IBM. These studies, amongst others, create hope and excitement in the field, both for people with IBM and their neuromuscular clinicians.
PL 2.7 / #29TREATMENT OF AMYLOID NEUROPATHY
Topic: Plenary Lecture - Hot Topics
David Adams
Neurology, APHP/INSERM U1195/NNERF/FILNEMUS/, Le Kremlin Bicêtre, FR
Abstract: Amyloid neuropathies are rare diseases. They represent the most disabling polyneuropathy in the adult and are fatal multisystemic diseases involving also heart, kidneys or sometimes eyes. They may be hereditary (Familial Amyloid Polyneuropathy) usually due to transthyretin (TTR) gene mutation or acquired in Light Chain (AL) amyloidosis. Treatment includes i) anti-amyloid therapy which depends of the biochemical nature of amyloidosis, ii) treatment of end-stage organ failure and iii) symptomatic therapy. They require a multidisciplinary management. TTR-FAP have 2 main patterns : the first one is associated with the Val30Met variant with an early onset (EO<50 yo) a length-dependent predominant small fibre polyneuropathy, autonomic dysfunction and weight loss ; it concerns restricted endemic areas in Portugal, Japan. The second pattern worldwide presents as a predominant large fiber polyneuropathy with usually sporadic presentation in late onset Val30Met people with early rapid walking difficulties, an higher Neuropathic Impairment Score (NIS) and a shorter survival of 7 versus 12 years. In TTR-FAP, anti-amyloid therapy include liver transplantation (LT) which improves long-term survival in EO Met30 TTR-FAP and may stop progression of the disease in early stages. Five pejorative factors have been identified for selection of candidates to LT: orthostatic hypotension, aid for walking, QRS complex >120, thickness of IVW, NYHA >1 and a online calculator is available to define 5 year survival (http://www.nnerf.fr/attrtransplantscore). Combined heart and liver transplantation should be discussed in patients with moderate neuropathy and an endstage cardiomyopathy in patients. Anti-amyloid drugs have emerged during the last 10 years : TTR kinetic stabilizers tafamidis and diflunisal, they showed in a phase III controlled study their ability to slow FAP progression. Only the first one obtained marketing authorization in EU in 2011 for stage 1 (walking unaided) of the disease. Other strategy for TTR-FAP include TTR gene silencing with small interfering RNA (SiRNA), antisense oligonucleotide (ASO) to knockdown both mutant and wild type TTR in the serum. An ongoing Open Label Extension study with RNAi therapy shows a good safety and tolerability of RNAi therapy after 750 infusions, a major knockdown of mutant and wild type TTR by more than 80 percent in the serum lasting for at least 18 months and a stability of a composite neuropathic score mNIS+7. Two multicentric randomized placebo control phase III studies are ongoing with RNAi therapy (Apollo, ClinicalTrials.gov Identifier: NCT01960348) and OAS (ClinicalTrials.gov Identifier: NCT01737398). Both had ended enrollment and definite results will be available at the end of 2017. For light chain neuropathy, Stem cell transplantation or alkeran-dexamethasone may be proposed. On univariate analysis, among patients with AL neuropathy who present with autonomic neuropathy (AN), cardiac involvement, AN, glomerular filtration rate, number of organs involved, and NT-pro-BNP all had an impact on median overall survival (OS). On multivariate analysis, AN retained an independent adverse impact on OS.
PL 3.1 / #30GENE THERAPY FOR MUSCULAR DYSTROPHY
Topic: Plenary Lecture - Muscular Dystrophy
Dongsheng Duan
Molecular Microbiology And Immunology, University of Missouri, Columbia, MO, US
Abstract: Duchenne muscular dystrophy (DMD) is caused by dystrophin gene mutation. Over the last three decades, there has been tremendous effort in the development of gene replacement therapy. Early proof-of-principle studies have used vectors based on plasmids, retrovirus, adenovirus and herpes simplex virus. Unfortunately, these vectors are either very inefficient or associated with significant toxicity. Adeno-associated virus (AAV) is now the leading vector. AAV is a single stranded DNA virus. It is the only viral vector capable of bodywide muscle transduction through circulation. However, AAV can only carry a 5-kb genome. The full-length dystrophin coding sequence is 11.2-kb. To overcome this hurdle, investigators have developed highly abbreviated synthetic mini- and micro-dystrophin genes. On the other hands, dual and tri-AAV vectors have also been engineered to expand AAV packaging capacity. The mini-dystrophin genes are 6 to 8-kb in length. They evolve from a 6.2-kb naturally occurring minigene that results in ambulation to the age 61. For AAV delivery, the minigene cassette is split to two parts and individually packaged in an AAV vector. Reconstitution is achieved in muscle through engineered recombination signals. Dual AAV minigene delivery has resulted in saturated transduction following direct muscle injection and widespread transduction after intravenous injection in mouse models of DMD. In an unpublished study, we also achieved efficient local dual AAV delivery in the canine DMD model. Tri-AAV vectors have the capacity to deliver full-length dystrophin but currently its efficiency is too low to yield a therapeutic benefit. Micro-dystrophin carries only one-third of dystrophin coding sequence. More than 40 microgenes have been evaluated in mice. Some are highly functional and some are no function at all. Dystrophin has four major domains including the N-terminal (NT), rod, cysteine-rich (CR) and C-terminal domains. The rod domain consists of 24 spectrin-like repeats and 4 hinges. All micro-dystrophins that are currently in the preclinical pipeline contain the NT and CR domains and a shortened rod domain with 4 or 5 repeats and 2 or 3 hinges. A major function of dystrophin is to anchor neuronal nitric oxide synthase (nNOS) to the sarcolemma. We recently identified repeats 16/17 as the nNOS-binding domain. Inclusion of these two repeats in micro-dystrophin results in significantly better disease rescue in mouse models. Advances in our understanding on dystrophin biology are expected to produce more powerful microgenes in the future. A major hurdle in the development of an experimental therapy is to scale up from rodent models to large mammals. Since AAV-mediated micro-dystrophin therapy has resulted in marvelous results in mouse models, verification of therapeutic efficacy in the canine model has become the focus of current research. We recently demonstrated that local AAV microgene therapy not only ameliorated histopathology but also improved muscle function. Since the ultimate goal is to treat all muscles in the body, we tested systemic AAV microgene therapy in affected dogs. A single intravenous injection resulted in efficient microgene delivery to whole body muscle and amelioration of histological lesions. These results have set the foundation for bodywide therapy in DMD patients.
PL 3.1 / #498GENE THERAPY FOR MUSCULAR DYSTROPHY (CLINICAL)
Topic: Plenary Lecture - Muscular Dystrophy
Dana Martin
Sarepta Therapeutics, Cambridge, MA, US
Abstract: not received.
PL 3.2 / #31ANTISENSE THERAPY FOR MYOTONIC DYSTROPHY
Topic: Plenary Lecture - Muscular Dystrophy
Charles Thornton
Department Of Neurology, University of Rochester, Rochester, NY, US
Abstract: Myotonic dystrophy (DM) stands at the crossroads of human genetics, neuromuscular medicine, RNA biology, and antisense therapeutics. There is still much to learn about how an expanded CTG repeat in the DMPK gene causes a remarkably diverse set of clinical findings. A major thrust of DM research over the past 15 years indicates that RNA toxicity – a concept that never existed before it emerged as a central mechanism for myotonic dystrophy – underlies most of the symptoms. Studies have shown that RNA from the DM gene is toxic because it contains a long segment of expanded repeats. This repetitive segment affects cellular function because a select group of proteins bind to it extensively and lose their activity. This presentation will focus on the connections between repetitive RNA and symptoms of myotonic dystrophy, and current efforts to harness gene silencing technologies, such as, antisense oligonucleotides, to eliminate toxic RNA and treat the disease.
PL 3.3 / #32CRISPR BASED GENE EDITING FOR MUSCULAR DYSTROPHY
Topic: Plenary Lecture - Muscular Dystrophy
Ronald D. Cohn
Clinical & Metabolic Genetics, The Hospital for Sick Children, Toronto, ON, CA
Abstract: Clustered regularly interspaced short palindromic repeat (CRISPR) has arisen as a frontrunner for efficient genome engineering. However, the potentially broad therapeutic implications are largely unexplored. Here, to investigate the therapeutic potential of CRISPR/Cas9 in a diverse set of genetic disorders, we establish a pipeline that uses readily obtainable cells from affected individuals. We show that an adapted version of CRISPR/Cas9 increases the amount of utrophin, a known disease modifier in Duchenne muscular dystrophy (DMD). Furthermore, we demonstrate preferential elimination of the dominant-negative FGFR3 c.1138G>A allele in fibroblasts of an individual affected by achondroplasia. Using a previously undescribed approach involving single guide RNA, we successfully removed large genome rearrangement in primary cells of an individual with an X chromosome duplication including MECP2. Moreover, removal of a duplication of DMD exons 18–30 in myotubes of an individual affected by DMD produced full-length dystrophin. Our findings establish the far-reaching therapeutic utility of CRISPR/Cas9, which can be tailored to target numerous inherited disorders.
PL 4.1 / #33ALS THERAPY DEVELOPMENT: CHALLENGES AND OPPORTUNITIES
Topic: Plenary Lecture - Motor Neuron Disease
Michael Benatar
Neurology, University of Miami, Miami, FL, US
Abstract: Despite decades of research and dozens of clinical trials, few therapies are proven to have benefit for patients with ALS. This realization has prompted an introspective look at the many and complex reasons why clinical trials have been so singularly unsuccessful, and provides an opportunity to identify opportunities to change our approach to therapy development. Recognition that ALS is an etiologically and biologically heterogeneous disorder raises the possibility that clinical trials should endeavor to focus on subsets of the ALS patient population that have in common shared etiology or biology. Limitations of existing animal models have prompted development of a more diverse array of pre-clinical models and greater attention to methodological rigor in pre-clinical drug development. Recognition of the tremendous phenotypic heterogeneity of disease, along with the realization that clinical trials utilizing traditional clinical outcome measures require that large numbers of patients be followed for prolonged periods and at great expense, has prompting a renewed vigor to develop biomarkers relevant to therapy development. These include prognostic biomarkers that help to define more homogeneous subsets of patients; predictive biomarkers that may identify subsets of patients most likely to respond to particular therapeutics; and pharmacodynamic biomarkers (including biomarkers of disease progression) that may facilitate demonstration of target engagement or biological effect of an experimental therapeutic. Growing insight into factors that motivate or discourage patients from participating in research studies is also yielding opportunities for greater degrees of patient engagement in the research process. Finally, the lingering concern that therapeutic interventions are being brought to bear too late in the course of disease is prompting an increased focus on the early pre-symptomatic stage of ALS, along with efforts to reduce the diagnostic delay and latency to initiation of an experimental therapeutic. The hope is that recognizing these challenges and seizing upon the opportunities that are emerging, will enable us to define a strategy for more efficiently developing effective treatments for ALS patients.
PL 4.2 / #34BIOLOGY OF C9ORF72 DISEASE
Topic: Plenary Lecture - Motor Neuron Disease
Leonard Petrucelli
Neurology, Mayo Clinic, Jacksonville, AL, US
Abstract: not received.
PL 4.3 / #480ANTISENSE THERAPY FOR SPINAL MUSCULAR ATROPHY
Topic: Plenary Lecture - Motor Neuron Disease
John T. Kissel
Neurology, Pediatrics & Neuroscience, Ohio State University Wexner Medical Center, Columbus, OH, US
Abstract: Spinal muscular atrophy (SMA) is an autosomal recessive neuronopathy that causes degeneration of anterior horn cells and resulting muscle weakness. It is the most common fatal genetic disorder of infants, but can affect individuals at any age. Advances in molecular genetics and biochemistry over the past three decades have led to the identification of the causative gene (SMN1) for SMA, the recognition of a nearly identical homologue gene (SMN2) that serves a rescue function and explains in large part the phenotypic heterogeneity of the disorder, the development of a simple gene-based test to diagnose SMA, the generation of several animal models for the disease, and the realization that SMA is one of a number of neurological disorders associated with disruption of RNA metabolism. Many questions remain to be answered for a full understanding of the pathogenesis of SMA, including the precise site of action and function of SMN, which downstream proteins are affected, to what extent possible modifier genes alter phenotypic expression of the disorder, and how much increase in full length SMA is required to correct the phenotype. Current knowledge however has identified a number of exciting possibilities for therapeutic interventions for SMA with multiple targets for both pharmacologic and genetic based therapies. Most notable are recent trials involving antisense oligonucleotide and small molecule based therapies designed to bind to the SMN2 pre-mRNA and promote inclusion of exon 7 with resultant increased expression of full length message and normal SMN protein. Several clinical trials are in various stages of completion or development based on these strategies; most promising are antisense oligonucleotide and total gene replacement therapies, both of which are being employed in current therapeutic trials. Although there are many practical, medical, regulatory and ethical issues that still need to be addressed going forward, the SMA field is entering a new era of therapeutic possibilities for this devastating disorder. The cumulative focused and energetic efforts of basic scientists, clinician scientists, and lay advocacy groups have transformed SMA from a neglected orphan disease into a prototypic genetic disorder amenable to “bench to bedside to bench” translational investigations aimed at developing safe and effective treatments for SMA patients.
Late Breaking News Sessions
LB 1.1 / #378A PHASE 2 TRIAL OF RITUXIMAB IN MYASTHENIA GRAVIS: STUDY UPDATE
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Richard Nowak1, Christopher Coffey2, Jonathan Goldstein3, Mazen Dimachkie4, Michael Benatar5, Safawa Huq6, Brenda Pearson2, Kevin O’Connor1, Robin Conwit7, John Kissel8, David Hafler1, Richard Barohn9, Merit Cudkowicz6
1Neurology, Yale School of Medicine, New Haven, CT, US;
2University of Iowa, Iowa City, IA, US;
3Neurology, HSS, New York, NY, US;
4Neurology, The University of Kansas Medical Center, Kansas City, KS, US;
5Neurology, University of Miami, Miami, FL, US;
6Neurology, MGH, Boston, MA, US;
7Ninds, National Institutes of Health, Bethesda, MD, US;
8Neurology, Ohio State University Wexner Medical Center, Columbus, US;
9University of Kansas Medical Center, Kansas City, KS, US
Abstract: The specific primary objective of the B Cell Targeted Treatment in Myasthenia Gravis (BeatMG) study is to determine whether rituximab is a safe and beneficial therapeutic for MG that warrants further study in a phase 3 efficacy trial. The study is coordinated by NeuroNEXT (the Network for Excellence in Neuroscience Clinical Trials) with support and funding from the National Institute of Neurological Disorders and Stroke (NINDS). The BeatMG study is a multicenter randomized, double-blind, placebo controlled phase 2 clinical trial utilizing a futility design and includes participants with acetylcholine receptor (AChR) antibody positive generalized MG on at least 15 mg of prednisone per day. We plan to enroll a total of 50 participants (25 treatment, 25 placebo) with a planned follow-up duration of 52 weeks. Participants begin a forced steroid taper at week 8, based on stability or improvement in their MG signs/symptoms. The primary outcome measure is steroid sparing effect, defined as the proportion of participants achieving a ≥ 75% reduction in mean daily prednisone dose in the 4 weeks prior to week 52 and with clinical improvement or no significant worsening of symptoms. Secondary outcomes focus on whether there is a trend toward clinical benefit as measured by the MG Composite (MGC) and the Quantitative MG (QMG) scores. This study also affords a unique opportunity to study both drug and disease mechanisms. Exploratory biomarker outcomes include studies focused on autoantibody-producing B cells and antigen-specific T cells. This work will further our understanding of MG immunopathology and it represents the first step toward gaining a more complete understanding of the immune mechanisms underlying treatment of MG with rituximab, hopefully leading to new ways to treat the disease. Biomarker work has received support from the Myasthenia Gravis Foundation of America and the National Institute of Allergy and Infectious Diseases. Since the study opened to enrollment in May 2014, 60 participants have been screened and 47 randomized as of February 2016. Enrollment will likely be completed over the next few months with baseline data available soon after and final results anticipated approximately one year later.
LB 1.2 / #500NEUROLOGICAL COMPLICATIONS ON ZIKA VIRUS
Topic: Late Breaking News
John D. England
Neurology, LSUHSC School of Medicine, New Orleans, LA, US
Abstract: not received.
LB 1.3 / #499GUILLAIN-BARRE SYNDROME AND VARIANTS ASSOCIATED WITH ZIKA VIRUS OUTBREAKS
Topic: Late Breaking News
Osvaldo J. Nascimento
Neurology, Federal Fluminense University, Rio de Janeiro, BR
Abstract: The Zika virus (ZIKV) was named after the Zika forest, located in Uganda. It was first isolated in April 1947 from specimens collected from a sentinel rhesus monkey. ZIKV is now considered an emerging flavivirosis, with a first large outbreak registered in the Yap Islands in 2007. In 2013, a new large outbreak was reported in the French Polynesia referring the association of ZIKV and neurological complications, including 70 cases. Among these, there were 38 cases of Guillain-Barré syndrome (GBS) and 25 cases of different neurological complications (encephalitis, meningoencephalitis, paresthesias, facial paralysis and myelitis). ZIKV was first detected in the Americas in 2015, where it is now spreading exponentially. The incidence of GBS in Brazil whitening increased after the ZIKV outbreak, although it is not clear yet the real impact of ZIKV in the occurrence of new cases of GBS, as this syndrome can be secondary to other infectious or immune events. It is considered that the incidence of such neurological condition has risen by five times in Brazil. ZIKV disease, transmitted by Aedes aegyptimosquitoes, often presents with mild or non-specific symptoms, similar to other prevalent viral diseases in Brazil, such as Dengue and Chikungunya, hampering attempts to perform an accurate diagnosis based solely on clinical grounds. The disease is usually characterized by an acute onset of fever, non-purulent conjunctivitis, headache, arthralgia, myalgia, asthenia and a maculo-papular rash. The disease course is self-limited, normally lasting 4-7 days. Currently, the most accurate diagnostic assay for ZIKV is the isolation of viral RNA. Even though its viremic period is not well described, the initial 3-5 days from the onset of symptoms is most likely the ideal timeframe to detect the virus. Serologic tests for specific IgM/IgG are available, but the cross-reactivity with other flaviviruses, such as dengue and West Nile virus, remains a challenge. Several acute cases presenting with painful symptoms in the first days of the acute ZIKV infection represents an infectious neuritis as seen in many other infectious diseases. Most patients in Rio de Janeiro presented with classical GBS, AMAN, and AMSAN less than three weeks after experimenting ZIKV symptoms. GBS variants as Miller-Fisher syndrome, paraparetic, pharyngeal-cervical-brachial syndrome, bifacial weakness with paraesthesias, and Bickerstaff’s brainstem encephalitis were also seen. Electrophysiological axonal compromise has being more common in ZIKV patients. Some patients worsened for more than 2 months, evoking CIDP presenting as GBS. Surprisingly, MRI neuroaxis scans has disclosed hyperintense signal not only in spinal roots and sensory ganglia, but also in the anterior horns, spinal cord and other neuroaxis segments showing a more spread demyelinating process in some patients, suggesting encephalomyelitis, ADEM. The protracted course of GBS and its variants in some patients with suspected ZIKV infection evokes possible hypothesis on its pathogenesis, such as an immune mediated neuronal injury or a viral persistence with direct lesions to the nervous system. Most patients responded to IVIG and/or plasma exchange.
Workshop Sessions
WS 1.1.1 / #36MUSCLE ULTRASOUND
Topic: Imaging of Muscle
Carsten Bonnemann
Neuromuscular And Neurogenetic Disorders Of Childhood Section, National Institutes of Health, Bethesda, MD, US
Abstract: not received.
WS 1.1.2 / #466THE APPLICATION OF MRI IN MUSCLE DISEASE
Topic: Imaging of Muscle
Volker Straub
Newcastle University, John Walton Muscular Dystrophy Research Centre, Newcastle upon Tyne, GB
Abstract: Diagnosis and therapy development for neuromuscular diseases has rapidly expanded in recent years and there is an urgent need to develop objective, non-invasive outcome measures to monitor disease progression and treatment effect. The use of novel magnetic resonance imaging techniques (MRI) applied to genetic muscle disease is showing increasing promise in this regard and is helped by sharing expertise and data, validating protocols across platforms and exploring the potential of MRI as a helpful diagnostic tool and a quantitative outcome measure in clinical trials. When muscle MRI is used for diagnostic purposes, the recognition of a selective pattern of pathology is the main objective of the application, whereas in clinical trials MRI is applied to quantify muscle pathology. These are two very different objectives of the application of muscle MRI. It is only very recently that muscle MRI has become part of the diagnostic algorithm for a few neuromuscular diseases, where it is already apparent that it adds diagnostic value, discriminating between patients with different diseases who are phenotypically very similar. In a number of muscle diseases the pattern of selective muscle involvement detected by MRI can help guide genetic testing, target the optimal muscle for biopsy, and explore pathomechanisms. It is already known that MRI can be very helpful to guide genetic testing in patients with myofibrillar myopathies, collagen VI related myopathies and RYR1-associated myopathies. Most publications that describe muscle pathology by MRI used T1 weighted images in small cohorts of neuromuscular patients with similar phenotypes. The regions of interest that are imaged are normally the pelvic girdle and lower limb muscles. There is still fairly little experience in describing muscle pathology by whole-body MRI or MRI of the truncal and upper limb muscles, although several centres are now starting to explore these applications more systematically. In natural history studies MRI techniques are used to describe the effects of disease on muscle morphology, signs of inflammation and the replacement of muscle tissue by fat and connective tissue. Knowledge about the onset of muscle pathology detected by imaging, about the degree of progression and about modifiers of these parameters is still very sparse for the majority of genetic muscle diseases, but increasing the collection of MR imaging data will help to establish MRI as an accepted objective, non-invasive outcome measure for interventional clinical trials.
WS 1.2.1 / #38SYMPTOMATIC TREATMENT OF ALS
Topic: Management of ALS Patients
Stacy Rudnicki1, Christen Shoesmith2
1Cytokinetics, Cytokinetics, South San Francisco, CA, US;
2Clincial Neurological Sciences, London Health Sciences Centre, London, ON, CA
Abstract: This workshop will cover the management of symptoms in patients with amyotrophic lateral sclerosis (ALS). Patients may report wide ranging symptoms, including those that are directly related to the disease (e.g. spasticity), a downstream effect (e.g. fatigue), and those that may be a consequence of immobility (e.g. pain). There are a number of available symptomatic treatments that have improved the care of patients with ALS. In the case of non-invasive ventilation, symptomatic treatment of dyspnea has also been shown to provide a survival benefit. For some medications and interventions, there are rigorous trials that provide evidence for use, while in other circumstances, treatment is primarily based upon personal experience, small open label trials, or anecdotal reports. Options for how to manage the protean symptoms associated with ALS will be covered. A case based format will be used.
WS 1.2.2 / #39END OF LIFE ISSUES IN ALS
Topic: Management of ALS Patients
Christen Shoesmith1, Stacy Rudnicki2
1Clinical Neurological Sciences, London Health Sciences Centre, London, ON, CA;
2Cytokinetics, Cytokinetics, South San Francisco, CA, US
Abstract: ALS is a devastating neurological disease which is ultimately fatal. Care of patients with ALS can be complex and is best managed with a multidisciplinary approach which includes anticipatory care and symptom focused care. As the disease progresses towards end of life, management typically shifts to palliative care. End of life care in patients with ALS has unique aspects which make palliative care of ALS patients different from palliative care of patients dying from other diseases. This workshop will involve case based discussions about symptom management and end of life care in ALS. It will build on the symptom management discussion from the previous workshop led by Dr. Rudnicki. The cases discussed will include managing ALS symptoms at the end of life and we will also reflect on caregiver support issues. We will discuss palliative care in ALS, including intensive palliative care of patients who wish to discontinue their BIPAP. Organ donation from patients with ALS through DCD (donation after cardiopulmonary death) will also be touched on.
WS 1.3.1 / #482EVALUATION AND TREATMENT OF POMPE DISEASE
Topic: Metabolic Myopathy
Mark Tarnopolsky
Pediatrics, McMaster University, Hamilton, ON, CA
Abstract: Pompe disease (PD) is a genetic myopathy/lysosomal storage disease LSD) due to mutations in the glucosidase, alpha; acid (GAA) gene. This leads to a loss of GAA enzyme activity causing a progressive accumulation of intra-lysosomal glycogen. Infantile onset PD (IOPD) is characterized by severe hypotonia and cardiomyopathy and late onset PD (LOPD) leads to a progressive myopathy. Enzyme replacement therapy (ERT) with recombinant GAA is the current standard of care and studies have shown dramatic benefits for the cardiomyopathy in IOPD and an attenuation of the decline in respiratory function and modest walking benefits in LOPD. The skeletal muscle benefits are limited by the relative low abundance of mannose-6-phosphate receptors, especially in type II muscle fibers. General supportive therapies include; exercise training (resistance and endurance), respiratory muscle training, optimizing nutrition (swallowing studies in children, identification and replacement of deficiencies (i.e., vitamin D), a higher protein intake), physiotherapy/bracing as needed, gait assistive devices, etc. Several strategies have been suggested based upon pre-clinical studies showing benefits from; clenbuterol (increase mannose-6-phosphate receptors), carbohydrate re-modelled ERT (to increase binding to the mannose-6-phosphate receptor), glycosylation independant lysosomal targetting (GILT-tagged, to take advantage of IGF-1 receptor mediated ERT uptake), and molecular chaperones (duvoglustat HCl, to stabilize ERT and enhance native GAA activity). Some of these strategies have undergone phase II clinical trials and show promise. Exosomes are small (30 - 130 nm) microparticles that are formed by inward budding of the endosome and carry proteins, mRNA and miRNA. The exosomes are released from the cell and/or enter the lysosome. We have developed a novel exosome-based ERT and GAA-mRNA delivery strategy that has shown close to 100 % restoration of GAA enzyme activity and function in the GAA-/- mice. The exosome delivery system shows robust uptake into both type I and II skeletal muscle fibers, heart and brain. The latter observation opens up the potential for LSDs that affect the brain such as NCL, NPC, Krabbe, etc. Studies have shown the long-term safety of eoxome-mRNA and exosome-ERT therapy and clinical studies are proposed.
WS 1.3.2 / #41DIETARY AND OTHER THERAPIES IN MUSCLE GLYCOGENOSES AND DISORDERS OF MUSCLE LIPID OXIDATION
Topic: Metabolic Myopathy
John Vissing
Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK
Abstract: Metabolic myopathies are divided into disorders of muscle carbohydrate metabolism (muscle glycogenoses, GSD) and disorders of muscle fat oxidation (FAOD). Each group can be divided into two main clinical phenotypes; those with static symptoms related to the development of fixed muscle weakness and atrophy, and those with dynamic, exercise-related symptoms that are brought about by a deficient supply of ATP. A little more than 20 disorders affecting muscle have been described, and for many of them there is an overlap between the dynamic and fixed phenotypes. Metabolic myopathies are amendable to experimental treatment strategies used in other hereditary myopathies, such as gene and stem cell therapies, and treatments that boost muscle regenerative capacity, but are also unique because they unlike other muscle diseases often can be treated with changes in diet, especially with supplements that bypass the metabolic block. Furthermore, enzyme replacement has been developed for one muscle glycogenosis, type II (Pompe disease). The talk will review fuel supplementation strategies in GSDs and FAODs, enzyme replacement therapy, and touch on strategies to increase gene expression of the affected protein by valproic acid in McArdle disease and by bezafibrate or reservatrol in FAODs. In McArdle disease (GSD V), supplementations with branched-chain amino acids, creatine, B6 vitamin, and a protein-rich diet have proved to be ineffective. Oral glucose shortly before exercise dramatically improves exercise performance, but should be restricted due to the high caloric intake, and risk of weight gain. Maintaining a diet high in carbohydrate to ensure sufficient levels of liver glycogen, which is an important source of glucose for exercising myophosphorylase deficient muscles, has been shown to improve exercise tolerance in McArdle disease. To compensate for impaired oxidation of muscle glycogen in McArdle disease, patients burn more fat, but fat oxidation is limited by the lack of pyruvate delivery to spark the tricarboxylic cycle. Similar dietary treatment can possibly work in other muscle glycogenoses in which the combustion of extra-cellular glucose is not blocked. Results will be shown for debrancher, phosphoglucomutase 1 and glycogenin 1 deficiencies (GSDs III, XIV and XV) to support this notion. Currently, dietary supplement with triheptanoin, which exerts an anaplerotic effect on the tricarboxylic cycle, is tested in McArdle disease and FAODs. Carnitine treatment is pivotal in primary carnitine deficiency, but effectiveness has never been proven for other FAODs. Likewise, riboflavin has been proven to help most cases of multiple acyl-CoA dehydrogenase deficiency patients. Avoidance of fasting, mental stress, cold shivering, prolonged exercise and fever is a mainstay in the treatment of FAODs. Maintenance of sufficient hepatic glycogen levels has been proven to help exercise tolerance. The effect of ingesting oral glucose, which is helpful in a number of muscle glycogenoses, is less certain in FAODs.
WS 1.4.1 / #42NEUROMUSCULAR DATABASES
Topic: Neuromuscular Databases
Lawrence Korngut
Department of Clinical Neurosciences, Hotchkiss Brain Institute, University of Calgary, Calgary, AB, CA
Abstract: not received.
WS 1.4.2 / #43TREAT NMD
Topic: Neuromuscular Databases
Kevin Flanigan
Neurology, The Research Institute at Nationwide Children’s Hospital, Columbus, OH, US
Abstract: not received.
WS 1.5.1 / #44GENERAL TREATMENT APPROACHES
Topic: Treatment of Myasthenia Gravis
Gil I. Wolfe
Neurology, Univ. at Buffalo/SUNY, Buffalo, NY, US
Abstract: Myasthenia gravis (MG) is the most common disorder of the neuromuscular junction (NMJ), with an estimated prevalence between 25 and 142 per million. It characteristically presents with fatigable weakness, often initially involving the ocular muscles and manifesting as intermittent ptosis and diplopia. Ultimately, the disease generalizes in two-thirds of patients, leading to weakness of bulbar, neck, limb, and respiratory muscles. The majority of patients with generalized MG and roughly half of patients with purely ocular disease harbor antibodies to skeletal muscle nicotinic acetylcholine receptors. A subset of patients with generalized disease have antibodies to muscle-specific receptor tyrosine kinase (MuSK). Acetylcholinesterase inhibitors are often the first modality of therapy for MG. As an immune-mediated disorder, MG can respond to a number of immunosuppressive agents such as corticosteroids, azathioprine, mycophenolate mofetil, cyclosporine, and tacrolimus. Thymectomy is a key component of management in appropriately chosen MG patients and those with thymoma. Newer or alternative immunotherapies including rituximab and complement inhibiting agents are under active investigation in Phase 2 to 3 studies. An update will be provided on the status of these studies. In addition, the overall management strategy for MG will be discussed in the context of recently established international consensus treatment recommendations for the disease.
WS 1.5.2 / #45TREATMENT OF MG IN THE PAEDIATRIC POPULATION
Topic: Treatment of Myasthenia Gravis
Susan Iannaccone
Pediatrics, Univerity of Texas Southwestern Medical Center, Dallas, TX, US
Abstract: While the incidence of myasthenia gravis in the pediatric population is less than in adults, it is not insignificant and may be growing. Onset may occur as early as the first year of life and management is in general similar to that of adults. However, there are age and developmental specific aspects to the work up and treatment. It is important to rule out genetic etiologies in patients who are antibody negative. Congenital myasthenic syndromes require specific diagnosis and treatment and are beyond the scope of this discussion.
WS 2.1.1 / #46EMERGING CONCEPTS IN THE PATHOBIOLOGY OF DEGENERATIVE CERVICAL MYELOPATHY, EPIDEMIOLOGY AND CLINICAL PRESENTATION
Topic: Emerging Concepts in the Pathology and Clinical Management of Degenerative Cervical Myelopathy (DCM)
Michael G. Fehlings
Neurosurgery, University Health Network, Toronto, ON, CA
Abstract: Workshop Title: Emerging concepts in the pathology and clinical management of degenerative cervical myelopathy (DCM). Presentation Synopsis: In most developed countries degenerative cervical myelopathy (DCM) is the most common cause of spinal cord dysfunction and it is expected that with population aging it’s incidence in future decades will increase dramatically. The incidence of spinal cord dysfunction due to degenerative spine problems has the potential to exceed the incidence of traumatic SCI (tSCI) and already it is having an impact on acute hospitals and specialist spinal rehabilitation units. It is recognized that DCM differs from tSCI with respect to the temporal profile and pathology of the spinal cord damage, the clinical presentation, which ultimately affects the treatment options and clinical management, as well as outcomes. In this workshop we focus on DCM and aim to identify the differences and similarities of DCM and tSCI by describing the pathobiology, epidemiology, clinical presentation, advanced assessment techniques and current treatments for SCI related to degenerative spinal cord compression. Learning Objectives: At the end of the session participants will be able to: Understand DCM with respect to pathophysiology, epidemiology, and clinical presentation (neural adaptation), including implications for future research. Understand how the methods of assessment for DCM differ from tSCI · Appreciate to a greater degree the differences in clinical management and outcomes of DCM in comparison to tSCI Presenters: 15 min Emerging Concepts in the pathobiology of Degenerative Cervical Myelopathy, epidemiology and clinical presentation -M.G. Fehlings 15 min Clinical Implications, Outcomes and Rehabilitation Pathways -A. Burns 15 min Understanding disease severity through Novel surrogate measurement approaches in NTSCI -S. Kalsi-Ryan 15 min Advanced Techniques in Imaging specific to degenerative myelopathy -J. Cohen-Adad 30 min Panel Discussion regarding the clinical relevance and impact of DCM on the field of SCI, how it can be managed and what we need to learn -All Speakers
WS 2.1.2 / #47CLINICAL IMPLICATIONS, OUTCOMES AND REHABILITATION PATHWAYS
Topic: Emerging Concepts in the Pathology and Clinical Management of Degenerative Cervical Myelopathy (DCM)
Anthony Burns
Brain and Spinal Cord Rehabilitation Program, University Health Network - Toronto Rehabilitation Institute, Toronto, ON, CA
Abstract: not received.
WS 2.1.3 / #48UNDERSTANDING DISEASE SEVERITY THROUGH NOVEL SURROGATE MEASUREMENT APPROACHES IN NTSCI
Topic: Emerging Concepts in the Pathology and Clinical Management of Degenerative Cervical Myelopathy (DCM)
Sukhvinder Kalsi-Ryan1, Michael Fehlings2, Anthony Burns3, Julien Cohen-Adad4
1Neurosurgery, University Health Network, Toronto, ON, CA;
2Neurosurgery, University Health Network, Toronto, CA;
3Spinal Cord Injury, Toronto Rehab, Toronto, AB, CA;
4Physics, University of Montreal, Montreal, CA
Abstract: Workshop Title: Emerging concepts in the pathology and clinical management of degenerative cervical myelopathy (DCM). Presentation Synopsis: In most developed countries degenerative cervical myelopathy (DCM) is the most common cause of spinal cord dysfunction and it is expected that with population aging it’s incidence in future decades will increase dramatically. The incidence of spinal cord dysfunction due to degenerative spine problems has the potential to exceed the incidence of traumatic SCI (tSCI) and already it is having an impact on acute hospitals and specialist spinal rehabilitation units. It is recognized that DCM differs from tSCI with respect to the temporal profile and pathology of the spinal cord damage, the clinical presentation, which ultimately affects the treatment options and clinical management, as well as outcomes. In this workshop we focus on DCM and aim to identify the differences and similarities of DCM and tSCI by describing the pathobiology, epidemiology, clinical presentation, advanced assessment techniques and current treatments for SCI related to degenerative spinal cord compression. Learning Objectives: At the end of the session participants will be able to: · Understand DCM with respect to pathophysiology, epidemiology, and clinical presentation (neural adaptation), including implications for future research · Understand how the methods of assessment for DCM differ from tSCI · Appreciate to a greater degree the differences in clinical management and outcomes of DCM in comparison to tSCI Presenters: min Emerging Concepts in the pathobiology of Degenerative Cervical Myelopathy, epidemiology and clinical presentation -M.G. Fehlings min Clinical Implications, Outcomes and Rehabilitation Pathways -A. Burns min Understanding disease severity through Novel surrogate measurement approaches in NTSCI -S. Kalsi-Ryan min Advanced Techniques in Imaging specific to degenerative myelopathy -J. Cohen-Adad 30 min Panel Discussion regarding the clinical relevance and impact of DCM on the field of SCI, how it can be managed and what we need to learn -All Speakers
WS 2.1.4 / #49ADVANCED TECHNIQUES IN IMAGING SPECIFIC TO DEGENERATIVE MYELOPATHY
Topic: Emerging Concepts in the Pathology and Clinical Management of Degenerative Cervical Myelopathy (DCM)
Julien Cohen-Adad
Biomedical Engineering, Ecole Polytechnique, University of Montreal, Montreal, QC, CA
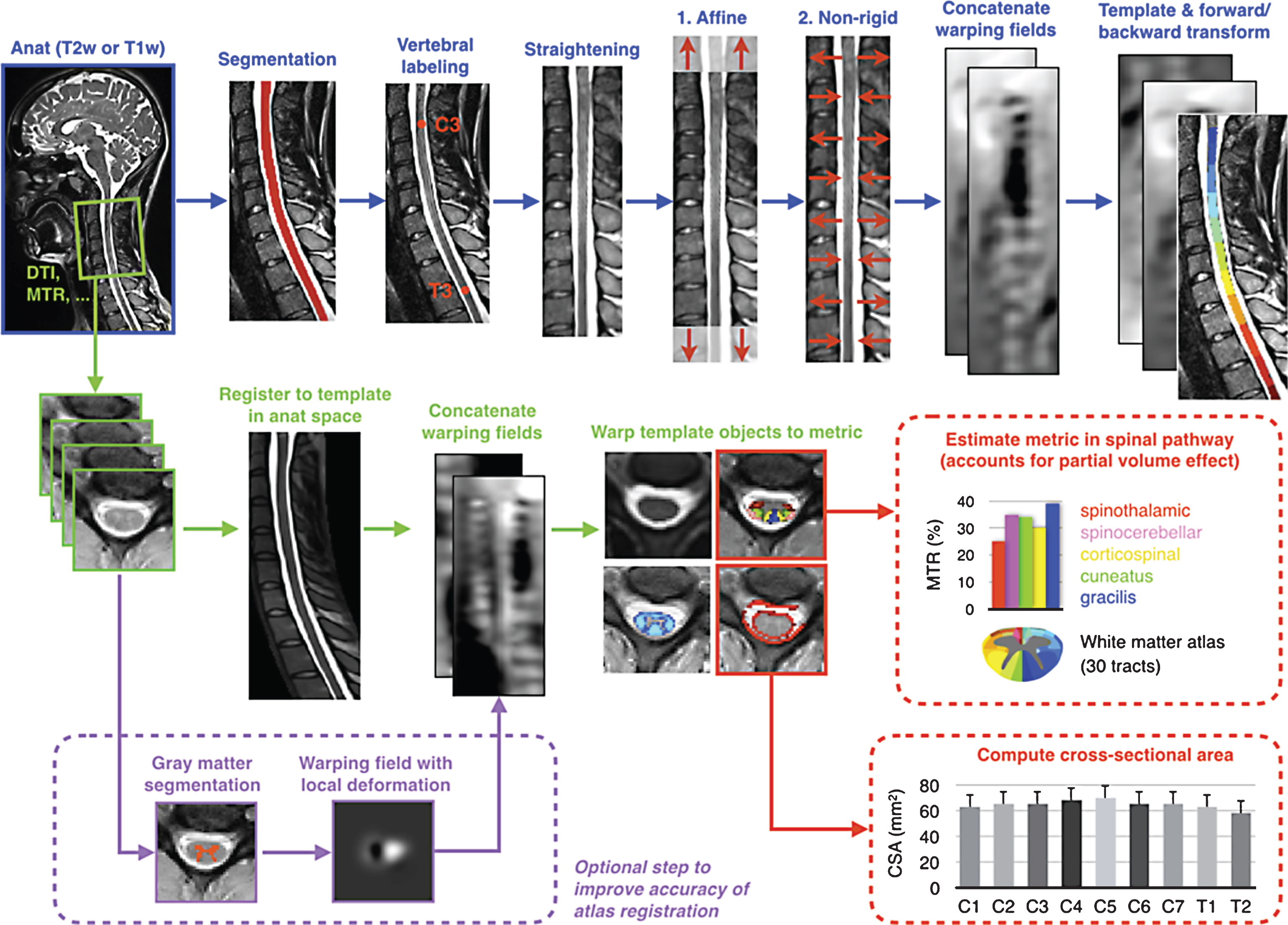
Abstract: While multi-parametric MRI (mpMRI, includes functional MRI, diffusion tensor imaging, etc.) has become popular for brain imaging, it is still difficult to apply these techniques to the spinal cord because of complex issues related to acquisition and processing of the data. In this talk we will examine state-of-the-art methods for doing mpMRI in the spinal cord in the context of degenerative myelopathy– discussing their present status, unresolved issues, and future directions. In particular, we will introduce the Spinal Cord Toolbox (SCT), a novel MRI data analysis toolbox dedicated to the spinal cord, and show its application in CSM patients.
Template and atlases included in SCT: T2-weighted template with vertebral levels and spinal-cord segmentation (top left), probabilistic atlases of white/gray-matter (top center), probabilistic map of spinal levels according to vertebral levels (top right) and white-matter atlas (bottom).
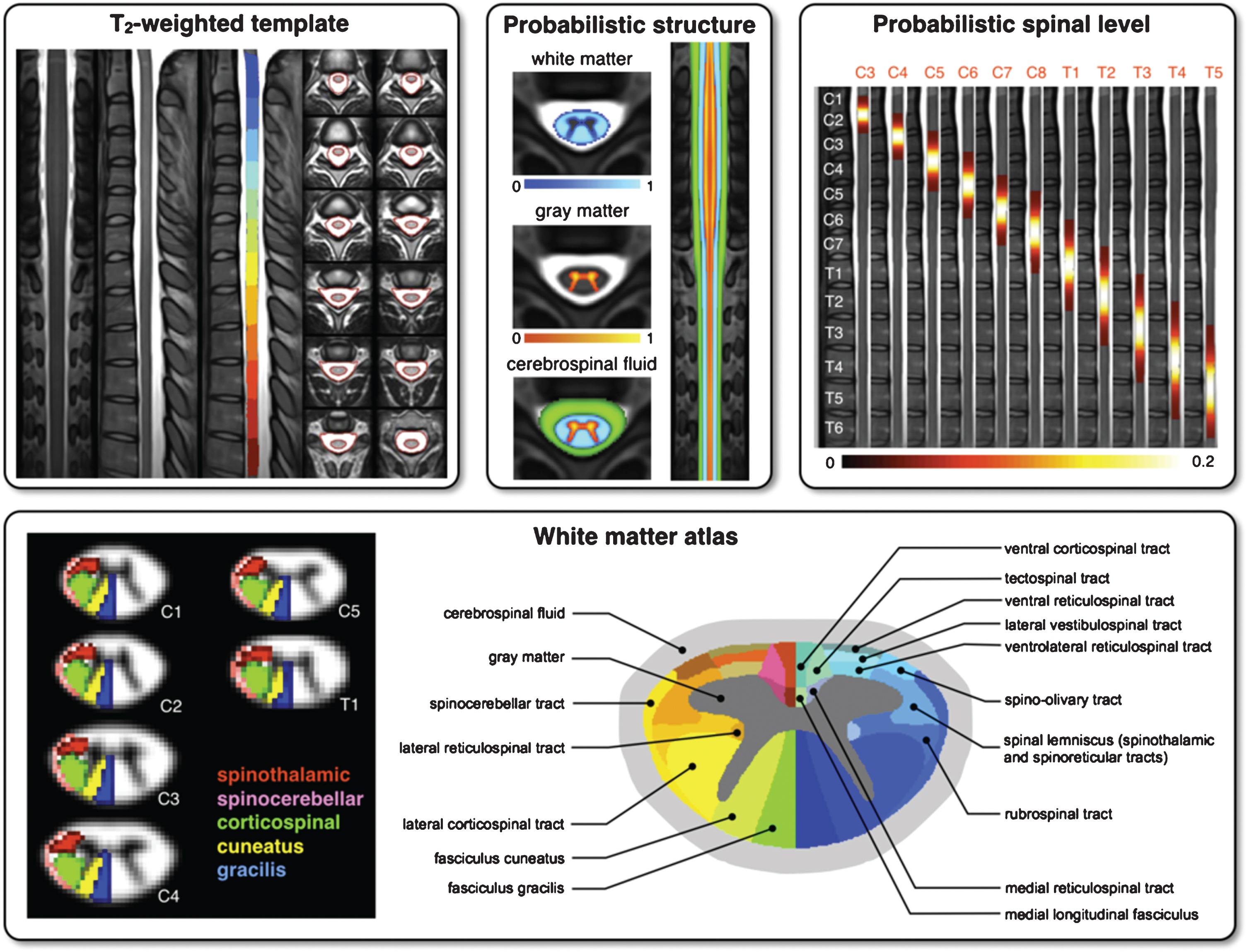
Overview of the template-based analysis pipeline. Anatomical data are fi rst registered to the template (blue arrows). Additional mpMRI data acquired during the same scan session are registered, then template objects arewarped to the multi-parametric data (green arrows). Subsequently, mpMRI metrics are quantifi ed within the spinal cord or spinal tracts at specifi c vertebral levels (red arrows). Cord and gray matter cross-sectional-area can be computed.
WS 2.2.1 / #50NOVEL PROCESSING METHODS FOR PERIPHERAL NERVE IMAGING
Topic: MRI Studies in Peripheral Nerve Disease
Jennifer Kollmer
Department Of Neuroradiology, University of Heidelberg, Heidelberg, DE
Abstract: Traditionally, diseases of the peripheral nervous system were diagnosed, based on a combination of detailed clinical and electrophysiological measurements, sometimes accompanied by electromyography. Magnetic Resonance Imaging (MRI) was predominantly included in the diagnostic work-up to rule out any underlying mass lesion within or adjacent to a peripheral nerve. After first experimental studies around 25 years ago, MRI has been further developed for peripheral nerve imaging, by increasing the structural resolution and contrast, and was therefore termed “MR-Neurography” (MRN)1, 2. Since that time, MRN has become a valuable tool in the diagnostic assessment of peripheral nerve lesions as it can determine the exact lesion localization, spatial lesion dispersion and lesion extension in-vivo3. T2-weighted (Tw) sequences with fat saturation were found to be most useful for the imaging of peripheral nerves. In these sequences, normal nerve tissue has a relatively similar signal compared to the surrounding muscles, while a marked signal increase can be seen in case of nerve impairment4, 5. The T2w signal increase of nerve tissue is unspecific, and can be found e.g. in entrapment neuropathies, nerve injuries, metabolic, hereditary or inflammatory neuropathies. Nevertheless, it has a high sensitivity and can help to clearly differentiate between demyelination and axonal nerve lesions, and in traumatic nerve injury it can distinguish between axonotmetic and neurotmetic injury. Additional measurement of the nerve caliber has also proven its validity, in that nerve impairment is often associated with an increase of nerve diameter. The development of high-resolution MRN is far from being completed, and more and more new sequences emerged over the last years in order to achieve signal quantification, higher sensitivity, and especially higher specificity. One possible sequence is the Diffusion Tensor Imaging (DTI), a method that is based on the direction-dependent diffusion in anisotropic structures, and which is a quantitative estimate of nerve anisotropy6. Further signal quantification can be achieved by calculating the proton spin density and the apparent T2 relaxation time, which requires the recording of the MR signal at different echo times7. Both parameters are known to be in-vivo markers of nerve tissue integrity and they depend on tissue density as well as on the macromolecular composition and the extracellular content of protein and lipid3. Another new method is the Magnetization Transfer (MT) imaging, where the MT Ratio (MTR) is computed, which can enhance the knowledge about the interaction between free water protons and protons that are associated with macromolecules or bound water, and therewith about the macromolecular tissue content8. Usefulness, validity and clinical relavence of MRN in general and of new diagnostic markers in particular will be demonstrated in non-focal (e.g. polyneuropathies) and focal neuropathies (different nerve entrapement syndroms).
WS 2.2.2 / #51CHALLENGES IN MRI STUDIES OF PERIPHERAL NERVES
Topic: MRI Studies in Peripheral Nerve Disease
Ali Naraghi
Department Of Medical Imaging, Toronto Western Hospital, Toronto, ON, CA
Abstract: With technical advances in MRI hardware and development of faster, higher resolution pulse sequences, MR imaging has been increasingly used to evaluate peripheral nerve pathologies. Available techniques include T2 based MR neurography utilized for detection of morphological changes and diffusion tensor imaging for detection of functional as well as anatomical changes. However, these techniques pose certain challenges. Some of these challenges are inherent to the technique whereas other challenges vary from case to case. Technical challenges include balancing spatial resolution and coverage and management of artifacts. In addition many pathologies can present with similar morphological changes on MR imaging and as such this technique cannot be used in isolation. Although MRI techniques can demonstrate peripheral nerve abnormalities, the accuracy of the technique and its role in the diagnostic pathway remains to be determined for various pathologies. These challenges will be addressed during the presentation.
WS 2.3.1 / #52OUTCOMES IN CMT
Topic: Outcomes in Hereditary Neuropathy
Mary M. Reilly
Mrc Centre For Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, London, GB
Abstract: Charcot Marie Tooth disease is genetically and clinically a heterogeneous group of hereditary neuropathies characterised by slowly progressive distal wasting, weakness and sensory loss. Although there are more than 80 causative genes described, the first described mutation i.e. the chromosome 17 duplication, which causes CMT1A is still the most common accounting for more than 50% of all cases of CMT in most populations. There are no drug therapies for CMT but in the last decade there have been major advances in understanding the pathogenesis of a number of forms of CMT and drug trials have both being completed and are ongoing in the common form, CMT1A. A major challenge for performing trials in CMT is the lack of valid, sensitive and responsive outcome measures. This is even more a challenge in CMT1A, which is the most slowly progressive form of CMT. Patients usually present in the first or second decade and have a slowly progressive length dependent motor and sensory neuropathy with a normal lifespan and usually remaining ambulant although often with aids to walk. Another challenge for trials in CMT is the lack of natural history studies in the various subtypes that could be used to inform the development of outcome measures. In CMT1A, the first trials were a number of trials of ascorbic acid (AA) in CMT1A including 2 large two year trials. Before these trials there had been some limited natural history data available in CMT1A and also a CMT specific composite outcome measure, the CMT Neuropathy score (CMTNS) had been developed and published in 2005. None of the AA trials showed any benefit from AA but worryingly none of the outcome measures used were sensitive enough to change over 2 years. This included the primary outcome which was the CMTNS, and all the secondary outcomes including myometry, the 9 hole peg test, the 10 meter timed walk, the overall neuropathy limitations scale, the SF-36 and various visual analogue scales. Professor Shy in the next talk with do an update on what has happened in all these areas to try to develop more sensitive and responsive outcome measures for phase 3 clinical trials in CMT1A. Another challenge is the need for responsive biomarkers especially for early phase trials. In the AA CMT1A trials, PMP22 levels have not been shown to be a responsive biomarker in either of the 2 year studies. Pilot data from both a CMT1A rat model and CMT1A patients have shown that cutaneous messenger RNA levels of glutathione S-transferase theta 2 and Cathepsin A may reflect disease severity and are further being investigated as potential disease biomarkers in CMT1A. In our group we have shown in a pilot study of CMT1A that calf level all muscle fat fraction as measured by 3 point MRI Dixon fat fraction quantification, increased significantly over a 12 month period in CMT1A patients with no significant changes seen in the control group. This is a promising outcome measure and is therefore potentially a useful biomarker for CMT1A trials.
WS 2.3.2 / #408MONITORING HEREDITARY NEUROPATHIES IN CLINICAL TRIALS
Topic: Outcomes in Hereditary Neuropathy
Michael Shy
Department Of Neurology, University of Iowa, Iowa City, IA, US
Abstract: We are currently in an era where rational therapies for inherited neuropathies are possible and practical. However, clinical trials to date have been limited by the lack of clinical outcome measures that can detect change in slowly progressive forms of Charcot Marie Tooth disease (CMT) such as CMT1A, the most common form. Moreover, there has been a dearth of CMT patient reported outcomes (PRO), disease specific quality of life (QoL) instruments, pediatric scales, and biomarker studies. The Inherited Neuropathy Consortium (INC) is a member of the Rare Disease Clinical Research Network (RDCRN) and was created, in part, to develop these outcome measures to enable natural history studies and clinical trials. Funded by the NINDS/NCATS, MDA, and CMTA the INC has recruited over 8,000 participants into protocols including >3,500 participants for natural history studies over the past 7 years. We have developed a Rasch modified CMT neuropathy score (CMTNS-Rasch) and a Rasch based pediatric scale (CMT-PedS), both of which detect change over two years in patients with CMT1A. We have also developed both adult and childhood CMT specific QoL scales and the PRO Disability Severity Index (DSI). Colleagues at the MRC Neuromuscular Center in the UK have demonstrated progression of the free fat (ff) fraction in calf muscles of patients with CMT1A that progresses within 12 months. These studies are now being extended to include sites within the INC. Taken together these results suggest that clinical trials in CMT1A will become increasingly possible and practical as therapies develop.
WS 2.4.1 / #54ROLE OF SKIN PUNCH BIOPSY IN CLINICAL PRACTICE
Topic: Role of Skin Punch Biopsy
David Saperstein
Neurology, Phoenix Neurological Associates, Phoenix, AZ, US
Abstract: Skin punch biopsy as a means to identify small fiber neuropathy (SFN) has moved from a research tool to a test readily available to the clinician. Although SFN can be clinically diagnosed in many patients, the ability to measure intraepidermal nerve fiber density (IENFD) can improve diagnostic certainty. More importantly, a broader use of IENFD testing has led to the appreciation of clinical patterns not previously attributed to SFN (such as intermittent, multifocal symptoms). This course will review the technique of skin punch biopsy and IENFD assessment. Discussion will include ways in which this testing can be useful in clinical practice as well as its limitations and pitfalls.
WS 2.4.2 / #55ROLE OF SKIN PUNCH BIOPSY AS A RESEARCH OUTCOME MEASURE
Topic: Role of Skin Punch Biopsy
Michael James Polydefkis
Neurology - Neuromuscular Medicine, Johns Hopkins Bayview Medical Center, Baltimore, MD, US
Abstract: not received.
WS 2.5.1 / #56PRACTICAL DEMONSTRATION AND DISCUSSION
Topic: Ultrasound in Peripheral Nerve Disease, Upper Limb
Francis Walker
Neurology, Wake Forest University School of Medicine, Winston-Salem, NC, US
Abstract: Neuromuscular ultrasound is an emerging subspecialty area of clinical neurophysiology and neuromuscular medicine. The diagnostic value of ultrasound for muscle disease was apparent as early as 1979, but its recognition as a tool useful for studying nerve required the evolution of high frequency transducers, which were not widely available until after 2000. Since then, a number of investigators have demonstrated multiple ways in which this technology can be applied to clinical management. In the upper extremities, ultrasound has a strong evidence base for accuracy in the diagnosis of carpal tunnel syndrome and is likely to prove of similar value in ulnar neuropathy at the elbow. Key findings in entrapment neuropathies include focal enlargement of nerve cross sectional area, loss of echogenicity, increased vascularity by power Doppler, and sometimes, changes in nerve mobility with joint flexion and extension. In hereditary demyelinating neuropathies, ultrasound demonstrates nerve enlargement which is more prominent proximally than distally. In inflammatory neuropathies, ultrasound has more complex patterns of abnormality which vary with time and severity of the underlying problem. Ultrasound can demonstrate other findings as well, including bifid nerves, persistent median arteries, cervical ribs, variant nerve branching or tendon anatomy. Pathologic structures such as aneurysms, arteriovenous malformations, benign tumors, neuromas, malignant tumors, enlarged lymph nodes, bone fractures, and cysts are also readily visualized. Finally, ultrasound can be helpful in guiding interventions, including nerve and muscle biopsies, steroid injections for carpal tunnel syndrome, and botulinum injections for muscle and phenol injections for nerve. The purpose of the practical demonstration and hands-on activities is to acquaint participants with how to recognize and follow the brachial plexus, and the entire course of major upper extremity nerves, muscles and tendons as well as common locations of where to look for pathologic changes in nerve, muscle or surrounding structures.
WS 3.1.1 / #57CHALLENGES FOR INVESTIGATOR INITIATED TRIALS AND FOR CONDUCTING MULTICENTER TRIALS
Topic: Challenges in Design of Investigator-Initiated Research
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: There are many challenges and barriers for conducting investigator initiated clinical trials. The ten barriers are: ideas, interest/desire, talent, training, time, team, regulatory, space, money and subjects. We will explore the barriers that can potentially hinder investigator initiated trials. Barriers 1-5: ideas, interest, talent, time and training: It can be difficult convincing young healthcare professionals to pursue a career path in clinical research. The time to interest potential researchers is in medical school, residency, fellowship or as junior faculty. The longer you wait, the harder it is to have the interest and develop an idea and make a commitment. Training options in clinical research are available at all stages. However young faculty members face difficulty being released from clinical and teaching duties to get training and conduct clinical research. The NIH is now strongly suggesting Good Clinical Practice training for all involved in clinical trials. Barrier 6: team approach: To perform multi-center trials, there is a substantial set of team members needed to carry out the study. The team members range from the investigators, project managers, study coordinators, biostatisticians, research pharmacy, budgets and contracts personnel, safety monitors, to patients, and finding sites that can conduct the studies. In many cases, you need multiple sites to conduct studies even for condition/disease that is common. Selecting sites and then motivating and encouraging sites to recruit is a challenge. Barrier 7: regulatory: Time to obtain Interval Review Board (IRB) approvals, and contracts slows down a trial. There is a movement now for a single IRB for multicenter studies – several models exist. Local IRBs need to be educated in these models. Knowing if you need to file an Investigation New Drug Application or not is another regulatory challenge. Sometimes the funding agency you are attempting to obtain funds from requires it. If you obtain it, then you have to maintain that IND status with yearly reports. Monitoring single or multi-center investigator-initiated trials is another challenge. A FDA White paper in 2007 (OEI-01-00160) discusses that monitoring of investigator initiated trials is a significant problem. Unfortunately in most investigator initiated trials, there is limited money and resources to do monitoring well. Barriers 8, 9, 10: Finding space to do clinical research is always a challenge. Sometimes this is done in defined clinical research units but funding of these is problematic. Performing a clinical trial, particularly a multicenter trial, is expensive. Several sources of grant funding are available (NIH,PCORI, Industry, Foundations). We have come up with a number of ways to do a multicenter trial of about 50 patients on budgets of about $200,000 annually but this is challenging. Finally, recruitment is one of the most difficult barriers to conducting successful research. Various techniques have been developed to overcome this challenge.
WS 3.1.2 / #58TRANSATLANTIC CHALLENGES
Topic: Challenges in Design of Investigator-Initiated Research
Richard J. Barohn
Neurology, The University of Kansas Medical Center, Kansas City, KS, US
Abstract: not received.
WS 3.2.1 / #59HOW TO TREAT MYOTONIC DYSTROPHY
Topic: Diagnosis and Treatment of Myotonic Dystrophy
Charles Thornton
Department Of Neurology, University of Rochester, Rochester, NY, US
Abstract: This workshop is focused on different aspects of treating myotonic dystrophy, including myotonic dystrophy type 1 (DM1) and type 2 (DM2). As yet there is no treatment that stops progression or fundamentally alters the trajectory of these diseases. Few randomized controlled trials have been completed. The most important life sustaining treatments in DM1 are to recognize heart block, nocturnal hypoventilation, and anesthetic risk. These can be effectively managed before major arrhythmia, hypoxemia, or perioperative complications may occur. The indications and benefits of anti-myotonia treatment will be reviewed. While CNS effects are quite variable, hypersomnolence can be one of the most troublesome features, and usually will respond to stimulant medications. Supportive care for other aspects of progressive myopathy is reminiscent of other paralytic disorders. There is a growing appreciation of the need for regular exercise and social engagement. Progress toward developing more definitive treatments are discussed elsewhere at the meeting, but will be briefly summarized in this workshop.
WS 3.2.2 / #60CURRENT KNOWLEDGE OF DISEASE PROGRESSION IN MYOTONIC DYSTROPHY
Topic: Diagnosis and Treatment of Myotonic Dystrophy
Richard T. Moxley, III
Neurology, University of Rochester Medical Center, Rochester, NY, US
Abstract: This portion of the workshop focuses on our current knowledge of the rate of progression of the major disease manifestations in myotonic dystrophy type 1 (DM1) and type 2 (DM2) and current methods to assess this progression. Identifying and enhancing our clinical and research capabilities to measure the impact and rate of change of disease manifestations is becoming increasingly important. Preclinical discoveries in the mouse model of DM1 and recent studies in humans, especially related to the use of antisense oligonucleotide treatment to remove mutant expanded RNA, give evidence that trials of disease specific treatments are eminent and underway. No currently available treatment stops progression or significantly alters the course of DM1 or DM2. However, progress has occurred to identify potential measures that may prove useful clinically in assessing our current symptomatic treatments and in evaluating patients participating in upcoming trials of treatment. We will review patient reported outcome measures along with measures of skeletal muscle strength and function, including different methods to assess myotonia, currently being used in selected research protocols. These include the patient reported instrument, The Myotonic Dystrophy Health Index for DM1, and information from the Patient-Reported Impact of Symptoms in DM2. We will also review findings of assessments of manual muscle strength testing, quantitative myometry, and maximum isometric grip evoked grip myotonia in 60 DM1 patients tested serially for 36 months. In the final portion we list some of the challenges we face in assessing the clinical progression of disease manifestations in DM1 and DM2 and highlight some of the opportunities we have as clinicians and researchers to meet these challenges.
WS 3.3.1 / #153MOLECULAR DIAGNOSTICS IN THE NEUROMUSCULAR CLINIC
Topic: Modern Concepts in Genetics
Grace Yoon
Clinical And Metabolic Genetics, Sickkids, Toronto, ON, CA
Abstract: Genetic testing for neuromuscular disorders has undergone enormous changes over the past several years. Implementation of advanced genetic testing technologies into routine clinical practice is becoming more common for the diagnosis and management of patients with suspected inherited neuromuscular disorders. Interpretation of genetic test results is not always straightforward, and careful integration of clinical and genetic information is critical to establishing the diagnosis. In this workshop, three clinical case vignettes will be presented and for each, the molecular techniques which enabled the genetic diagnoses will be discussed. The objectives of this workshop are: 1) To develop an appreciation of the diverse molecular mechanisms which cause neuromuscular disorders 2) To develop an appreciation of molecular techniques utilized for diagnosis of neuromuscular disorders
WS 3.3.2 / #62TBD
Topic: Modern Concepts in Genetics
Kevin Flanigan
Neurology, The Research Institute at Nationwide Children’s Hospital, Columbus, OH, US
Abstract: not received.
WS 3.4.1 / #63AMYLOID NEUROPATHY AS A MODEL OF SMALL FIBER NEUROPATHY
Topic: Small Fibre Neuropathy
David Adams
Neurology, APHP/INSERM U1195/NNERF/FILNEMUS/, Le Kremlin Bicêtre, FR
Abstract: Amyloid neuropathies (AN) are rare diseases due to endoneurial amyloid deposits leading to a progressive peripheral and autonomic neuropathy associated with multisystemic involvement. Natural course is progressive disabling and irreversible, usually fatal within 3 to 12 years. They may be hereditary or acquired. Familial Amyloid Polyneuropathy (FAP) have autosomal dominant transmission and are usually due to a point mutation of Transthyretin (TTR) gene.They may be acquired in 15% of AL amyloidosis and in recipients of Domino liver transplant but not in secondary amyloidosis (AA). Classical diagnostic tools were until recently nerve biopsy to detect amyloid deposit in the endoneurium and biological markers to specify biochemical nature : mainly TTR gene testing in endemic areas, free light chains dosing in the serum. Diagnosis is usually delayed by 1 to 3 years, facilitated by a positive family history. FAP are usually due to mutation of transthyretin gene.The most common variant is Val30Met with endemic areas including Portugal, Japan, Sweden. The age of onset is early before 50 years. Small fiber neuropathy is common and a hallmark of the disease. Inaugural manifestations are distal paresthesia, digestive complaints, erectile dysfunction and weight loss. Early diagnosis is difficult during the first months due to an insidious onset. On examination, there is a predominant involvement of pain and thermal sensations in a length-dependent distribution and NCS are normal at that time, during a 2 years period. Making diagnosis is challenging at that stage using varied testing to assess small fibres. The tools in FAP are many and time-consuming: skin biopsy,Quantitative Sensory Testing (QST) measurement of cold and warm detection thresholds (CWT), cortical laser-evoked potentials (LEP) to dorsal foot stimulation latency (LEP N2), plantar sympathetic skin response (SSR) amplitude, electrochemical skin conductance (ESC) which measures autonomic sudomotor glands, Skin denervation is common in Val30Met TTR and other variants. Degeneration of cutaneous nerve terminals is correlated with the severity of clinical phenotypes and the level of CSF protein in Ala97Ser. Congo positive amyloid deposits are present in dermis in 67% patients including in 100% of EO Val30Met suggesting a toxic role of amyloid fibrils on the nerve terminals. There is also pathological evidence of sudomotor denervation in FAP with functional correlation with autonomic symptoms, autonomic tests, ambulation status, and progression of disability. Skin denervation seems to be an early event as indicated with the reduction of Intra Epidermal Nerve Fiber Density (IENFD) at distal leg in 59 % of carriers of TTR mutation.Cardiovascular autonomic testing for heart rate variability, Other methods include 123I-meta-iodobenzylguanidine (MIBG) cardiac scintigraphy, gastric emptying scintigraphy. The diagnostic accuracy of (SSR) amplitude and cortical LEP N2 latency is similar with very high specificity (94 to 97%) but low sensitivity (22 to 32%) in distinguishing controls from carriers and early-symptomatic patients. In our experience, for monitoring carriers of TTR mutations Scale to assess small fiber : abnormal questionnaire of SFN Symptom Inventory Questionnaire (SFN-SIQ), measure of IENFD and amyloid deposit finding are the key stools to define onset of FAP disease and start an anti-amyloid therapy.
WS 3.4.2 / #493DIAGNOSIS OF SMALL FIBRE NEUROPATHY
Topic: Small Fibre Neuropathy
Giuseppe Lauria
Neuroalgology, IRCCS “Carlo Besta” Neurological Institute, Milan, IT
Abstract: SFN represents a distinct condition encountered in patients with different acquired and genetic disorders. Clinically based diagnostic criteria for SFN have been proposed and reliably supported by the recent availability of age- and sex-adjusted normative values for intraepidermal nerve fibre density. Besides skin biopsy, corneal confocal microscopy and nociceptive evoked potentials have been implemented to investigate SFN of different causes, and correlated with skin biopsy findings, especially in diabetic patients. The association between SFN and several metabolic and immune-mediated systemic diseases, and drugs toxic to this subset of peripheral nerve fibres has been reported. Moreover, besides the classical length-dependent presentation, small nerve fibre damage has been identified in patients complaining of widespread pain like fibromyalgia and neurodegenerative disorders like Parkinson disease and amyotrophic lateral sclerosis. An exciting advance in the field has been the identification of variants in genes encoding for various sodium channels expressed in the peripheral nociceptive pathway that led to the definition of a new syndrome within the spectrum of pain disorders. These improvements in the definition and pathogenesis of SFN allow clinicians to reliably meet the diagnosis, identify the underlying cause, and prescribe appropriate treatments for neuropathic pain, and researchers to adapt the design of new symptomatic and disease-modifying clinical trials.
WS 3.5.1 / #65PRACTICAL DEMONSTRATION AND DISCUSSION
Topic: Ultrasound in Peripheral Nerve Disease, Lower Limb
Francis Walker
Neurology, Wake Forest University School of Medicine, Winston-Salem, NC, US
Abstract: Neuromuscular ultrasound is an emerging subspecialty area in clinical neurophysiology that is increasingly being adopted by electrodiagnostic laboratories across the world. Whereas EMG and NCS can localize pathology to segments of nerve, ultrasound can localize it at the millimeter level, evaluate pathology involving nearby non-nervous tissue, provide useful information about blood flow and kinesiology, distinguish nerve transection from nerve crush, and it can do this both painlessly and non-invasively. Furthermore, the technology can be used to guide needle placement for EMG or botulinum toxin, identify areas of nerve or muscle for biopsy, and help with steroid and phenol injections. Ultrasound is rarely compromised in hostile electrical environments, and, in select circumstances, can be used intra-operatively, or via endoscopic or endovascular approaches. It is ideal for pediatric patients and for the occasional adult intolerant of needle examination or electrical stimulation. The demonstration/hands-on portion of this workshop is designed to acquaint participants with the examination of common nerves in the lower extremity, demonstrate areas where nerves are difficult to evaluate, review muscle and tendon anatomy, and indicate where to look for pathology during the examination. Advantages and limitations of the technique will be discussed as will potential future applications and its correlation with electrodiagnostic findings.
WS 4.1.1 / #66TECHNOLOGY PLATFORMS FOR COLLABORATIONS IN CLINICAL RESEARCH
Topic: Bioinformatics and Clinical Research
Alexander Sherman
Neurological Clinical Research Institute, Massachusetts General Hospital, Boston, MA, US
Abstract: OBJECTIVE: Implement collaborative patient-centered environment, methodology, processes and distributed infrastructure for creating “Big Data” in rare diseases. BACKGROUND: Large datasets are critical for identifying statistically significant and biologically relevant observations. Heterogeneity of many diseases complicates disease modeling and potential therapies development, making clinical trials challenging. Proposed approach and platform for patient-centered clinical and research information aggregation (including biobanks, image and GWAS repositories), collaboration and sharing allow rare diseases to enter the Age of “Big Data”. DESIGN/METHODS: Patient-centered approach requires unique patient identification across clinical research continuum. Global Unique Identifiers (GUIDs) allow patients’ clinical and research information aggregation and linking phenotypical data to biospecimens, patient-reported outcomes, genetic files and images across clinical visits and research projects over time, regardless where/when information was collected. Such approach gives researchers access to patient information collected elsewhere. Stakeholders include clinicians, research and commercial biobanks, academic consortia, patient organizations, foundations, industry and patients. Collaboration models for information sharing are essential. RESULTS: Distributed “virtual” biobanks are connected to health records and research data, image banks and GWAS files, linked and identified by GUIDs. Patients enter outcomes and priorities through Patient Portal. NeuroBANK™, a patient-centered accelerated clinical research platform, is in the center of this clinical research universe. Standard procedures, common consent language and technology unify multiple participants of clinical care and research (physicians, researchers, industry, patients and non-profits) and informational components (clinical data captured at bedside and from health records, research data, disease-specific consortia; patient-reported outcomes, and data from completed clinical trials) into single disease-specific research continuum. Several disease networks, such as ALD Connect and NEALS ALS consortia, successfully implemented this approach. CONCLUSIONS: Extremely powerful concept of Patient Centricity in clinical research paired with modern technology like NeuroBANK™ Accelerated Research Environment platform and insentives to collaborate create clinical research continuum from bedside to clinical trials and back to research and clinical care.
WS 4.1.2 / #67USING THE ELECTRONIC MEDICAL RECORD FOR RESEARCH
Topic: Bioinformatics and Clinical Research
Jon Katz
Neurology, California Pacific Medical Center, San Francisco, AL, US
Abstract: not received.
WS 4.2.1 / #68NERVE BIOPSY ARE RARELY NEEDED
Topic: Controversies Over Large Nerve Biopsy
Anthony A. Amato
Neurology, Brigham and Women’s Hospital, Boston, AL, US
Abstract: Nerve biopsy interpretation requires correlation of histological changes with clinical information, including the results of electrophysiological studies. Nerve biopsies are generally less useful than muscle biopsies because the pathologic abnormalities are often nonspecific and frequently do not help distinguish one form of peripheral neuropathy from another. In addition, there is increased morbidity associated with the removal of a segment of sensory nerve, which leads to permanent numbness in the corresponding cutaneous distribution. Also, nerve biopsies can be complicated by significant neuropathic pain in the distribution of the nerve for several months and the potential for growth of painful neuromas. More importantly, the cause of a polyneuropathy can be made in most cases on clinical grounds backed by electrophysiology and laboratory results. Additionally, with increasing availability and reduction of costs of genetic testing, the need for nerve biopsies has diminished. Therefore, I do not recommend nerve biopsies just because a patient has a generalized neuropathy of undetermined etiology despite an extensive laboratory evaluation- which makes up a large percentage of the patients who are referred to me for consideration of a nerve biopsy. This is not to say that nerve biopsies are not indicated or valuable in certain situations. Suspected amyloidosis and vasculitis are the main indications for nerve biopsy. Additional, nerve biopsy may be helpful in other autoimmune inflammatory conditions (e.g., sarcoidosis), infectious processes (e.g., leprosy), and tumor infiltration (e.g., lymphoma and leukemia). Also, a nerve biopsy is typically required for diagnosis of a tumor of the peripheral nerve (e.g., perineurioma). This said, in most of the above cases, a good clinician usually has a strong suspicion of the cause of the neuropathy before the biopsy and the likelihood that the results of a nerve biopsy on the majority of cases will change clinical management. For example, if a patient has painful, multiple mononeuropathies, elevated ESR, and ANCA antibody, are you not going to treat a patient for vasculitis if the biopsies does not demonstrate diagnostic features?
WS 4.2.2 / #69IT IS VALUABLE
Topic: Controversies Over Large Nerve Biopsy
P. James B. Dyck
Neurology, Mayo Clinic, Rochester, MN, US
Abstract: THERE IS STILL A PLACE FOR NERVE BIOPSY IN MODERN CLINICAL PRACTICE P. James B. Dyck Nerve biopsy is a useful tool for the diagnosis and treatment of peripheral neuropathies but the need for its continued use has been questioned. With improvement in laboratory, radiographic and electrophysiological evaluations, along with the introduction of quantitative autonomic and sensory testing, the need for nerve biopsy has been on the decline. With better understanding of the pathophysiology, some classes of neuropathy no longer need to be biopsied (inherited) whereas others can be recognized by the disease pattern (CIDP). These observations have led experts to question whether nerve biopsy is needed at all. When deciding to do a nerve biopsy, there are some features that are helpful. A thorough neurological evaluation should be completed before nerve biopsy is performed. One should select patients with rapidly worsening, progressive neuropathies who have large clinical deficits. Traditionally, whole distal cutaneous nerve biopsies are performed. These are taken from sensory nerves (e.g. sural, superficial peroneal, saphenous, superficial radial, etc.) and so are only useful in sensorimotor or pure sensory neuropathies and usually are best in length-dependent neuropathies. A key point to remember is that the distribution of the nerve needs to be clinically affected in order for a nerve biopsy to show findings. Laboratories that process nerve biopsies should do a wide variety of preparations including teased fibers, paraffin and epoxy sections, immunohistochemistry and electron microscopy. Reports on the diagnostic yield of whole cutaneous nerve biopsy vary. The conclusions from these reports are hard to judge because the quality of the evaluations, indication for biopsy, and quality of pathological specimens vary. In my experience, nerve biopsies from carefully selected patients frequently show important findings and change management. For motor predominant, focal or proximal neuropathies the traditional distal cutaneous biopsy may be inadequate. With improved resolution of MRI, focal lesions along the length of the nerve can now be identified and biopsied. We have extensive experience with targeted fascicular nerve biopsies and find that about 84% of these targeted biopsies reveal meaningful pathology from which 76% are potentially treatable. Such cases usually are focal or multifocal, proximal or motor predominant. The causes include inflammatory demyelinating, vasculitis, sarcoidosis, amyloidosis, leprosy, lymphoma, metastatic tumors, vascular malformations and others. In conclusion, there is strong evidence that both whole distal cutaneous and targeted fascicular nerve biopsy should still be performed in modern neurological practice. For most typical length-dependent neuropathies whole distal cutaneous biopsy is the procedure of choice. In contrast, for focal motor predominant or proximal neuropathies targeted fascicular nerve biopsy is a superior approach. Patients should be carefully screened and have a thorough evaluation and only patients with severe neuropathies that have a high likelihood of being treatable should be biopsied to offset potential side effects. Biopsies should be performed at centers with expertise in peripheral nerve diseases with specialized neurologists, radiologists, peripheral nerve surgeons and pathologists and laboratories that offer a variety of nerve preparations to ensure maximal benefit with minimal morbidity.
WS 4.3.1 / #70OVERVIEW OF GENETICS OF HEREDITARY POLYNEUROPATHY
Topic: Genetics of Hereditary Polyneuropathy
Stephan Züchner
Human Genetics, University of Miami, Miami, FL, US
Abstract: Next-generation sequencing has significantly enhanced the pace of gene discovery in peripheral neuropathies (also known as Charcot-Marie-Tooth neuropathies or CMT) and neuromuscular disorders. As gene panels and whole exomes sequencing become routine, the delineation and precise description of phenotypic spectra of disease genes has become a new frontier. The increasingly observed genotypic/phenotypic overlap of clinically distinct entities is a challenge to the existing classification of disorders. Finally, the long-standing clinical notion of modifiers of disease, environmental and genetic, has caught renewed attention, as dramatically improved genetic tools offer new analytical strategies. Over the last seven years, the CMT community has developed an infrastructure that has enabled the assimilation of clinical data from thousands of patients seen by CMT experts from around the world, along with their DNA, all in ways that de-identify their personal information. This is largely driven by the Inherited Neuropathy Consortium and the CMT-International Database. Key elements include a standardized approach to phenotyping, digital formatting of the clinical date that enables querying together with genetic data, and efficient processing of patient DNA samples. Defining the genetic basis of inherited peripheral neuropathies, also known as Charcot-Marie-Tooth disease (CMT), has progressed rapidly in the past five years. Beyond individual studies, the innovative sharing of genomic data in real time at any scale has fostered a collaborative spirit in the CMT and neighboring fields not seen before. Through these efforts, more than 30 novel genes have been published since 2012. Still more than 50% of axonal CMT patients go without diagnosis, thus whole genome sequencing will likely open new opportunities. This presentation will provide an overview and specific examples of recent success and challenges in the field.
WS 4.3.2 / #71CELLULAR REPROGRAMMING AND INHERITED PERIPHERAL NEUROPATHIES: PERSPECTIVES AND CHALLENGES
Topic: Genetics of Hereditary Polyneuropathy
Mario Saporta
Neurology, University of Miami, Miami, FL, US
Abstract: Inherited peripheral neuropathies (or Charcot-Marie-Tooth disease, CMT) are a phenotypically and genetically heterogeneous group of disorders, which are currently untreatable. They are the most common inherited neuromuscular disorder, affecting around 1 in every 2500 people. From a biological standpoint, they are associated with mutations in over 70 different genes, affecting Schwann cell development and myelination or peripheral axon physiology. To date, over 70 genes have been associated with a CMT phenotype, making CMT an attractive natural model to study peripheral nervous system biology. Despite significant advances made in our knowledge of diseases mechanisms in CMT, findings from animal models have so far translated poorly in clinical trials, underscoring the need for innovative methods to investigate the pathophysiology of these human disorders. Induced pluripotent stem cells (iPSC) offer an unlimited source of patient specific, disease-relevant cell lines that can be used as a platform for identification of disease mechanisms, discovery of molecular targets and development of phenotypic screens for drug discovery. iPSC-based models of neuromuscular disorders, including Amyotrophic Lateral Sclerosis (ALS), Spinal Muscular Atrophy (SMA) and inherited peripheral neuropathies, have successfully reproduced pathophysiological findings from previous animal and cellular models and have also identified new disease mechanisms with potential therapeutical implications. We have recently established an iPSC bank from 14 patients with multiple forms of inherited peripheral neuropathies as a human platform to study CMT. We have also derived motor neuron lines from two patients with early onset axonal forms of CMT associated with MFN2 (CMT2A) and NEFL (CMT2E) point mutations and used them to study disease mechanisms in CMT2. Immunostaining for intermediate filaments revealed an increased content of neurofilaments in the neuronal body of CMT2E spinal cord motor neurons. Reduced mitochondrial mobility was found in axons from human CMT2E neurons, compared to age-matched controls, confirming previous data suggesting that mutant neurofilaments can interfere with normal mitochondrial trafficking along axons. The same phenomenon, albeit in a lesser degree, was observed in CMT2A motor neurons. CMT2A and CMT2E iPS-derived motor neurons also generated action potentials at lower membrane potentials compared to control motor neurons, suggesting that these cells are hyperexcitable. Analysis of ion channel properties by voltage clamp demonstrated an increased density of sodium channels in CMT2A motor neurons, which also inactivated less efficiently, when compared to controls. Both CMT2A and CMT2E motor neurons also demonstrated an impaired inactivation of calcium channels. Taken together, this data suggest that ion channel dysfunction may play a role in the pathogenesis of axonal CMT, providing novel therapeutical targets for inherited neuropathies. Cellular reprogramming offers, therefore, a new approach for the study of inherited peripheral neuropathies by enabling the study of human, patient-derived cell types directly related to their pathophysiology (neurons and glia) in vitro. Many challenges remain for investigators in the field, including developing subtype-specific differentiation protocols and co-culture systems to study myelination and neuromuscular junction pathology in vitro. Cell replacement therapy using patient-derived cells corrected by gene editing techniques may be a feasible treatment strategy especially for demyelinating forms of CMT.
WS 4.4.1 / #72WHAT THE PERINOMS STUDY TAUGHT US
Topic: Outcome Measures in Inflammatory Neuropathy
Ingemar Merkies
Neurology, Academic Hospital Maastricht, Maastricht, NL
Abstract: not received.
WS 4.4.2 / #244HOW WE SHOULD ASSESS INFLAMMATORY NEUROPATHY
Topic: Outcome Measures in Inflammatory Neuropathy
Jean-Marc Léger
National Referral Center For Neuromuscular Diseases, University Hospital Pitié-Salpêtrière, Paris, FR
Abstract: The selection of outcome measures for use in clinical trials and clinical practice is a challenge for inflammatory neuropathy. The outcome measures should cover deficit, disability, impairment, and quality of life. They should be simple, valid, reliable, and responsive, and correlate with disease severity. A workshop organized by the European Neuromuscular Centre was held in 2013 and edited recommendations for Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), monoclonal gammopathy of undetermined significance (MGUS)-related polyneuropathy, and multifocal motor neuropathy (MMN), most of them being selected from the Peripheral Neuropathy Outcome Measurement Standardization (PeriNoms) study. It pointed out a better evaluation of data analysis with the Rasch method, which enables a transformation of ordinal data into interval metric data, increasing the level of precision in assessment. The Inflammatory Rasch-built Overall Disability scale offers the promise of being a more sensitive measure and is highly recommended in future trials involving patients with GBS and CIDP. However, further developements are needed in MMN and MGUS-related polyneuropathy, mainly anti-myelin-associated-glycoprotein (MAG) neuropathy. References JM Léger, R Guimaraes Costa, C Muntean. Immunotherapy in Peripheral Neuropathies. Neurotherapeutics 2016;13: 96-107 Vanhoutte EK, Faber CG, Merkies IS. PeriNoms study group. 196th ENMC International Workshop: outcome measures in inflammatory peripheral neuropathies 8-10 February, Naarden, The Netherlands. Neuromusc Disord 2013; 23: 924-933
WS 4.5.1 / #74EVALUATION OF VARIANTS OF UNKNOWN SIGNIFICANCE
Topic: Primer for Genetic Testing
Mary M. Reilly
MRC Centre For Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, London, GB
Abstract: Charcot Marie Tooth disease (CMT) and the related neuropathies, the distal hereditary motor neuropathies (HMN) and the hereditary sensory neuropathies (HSN) are genetically and clinically a heterogeneous group of neuropathies characterised by slowly progressive distal wasting, weakness and / or distal sensory loss. Like many other neuromuscular diseases, major advances have occurred in identifying the causative genes over the last 25 years and especially over the last 5 years in the era of next generation sequencing (NGS) such that there are now more than 80 identified causative genes for the inherited neuropathies. The major challenge for clinicians now is interpreting the results they receive. Most diagnostic laboratories are using next generation sequencing techniques (NGS) and have developed disease-specific, or multi-gene, testing panels which employ NGS to regions of the exome that contain known inherited neuropathy genes. These disease-specific panels are currently the best method for simultaneously screening a large number of inherited neuropathy genes although WES (whole exome sequencing) is increasingly being used diagnostically and WGS (whole genome sequencing) to a lesser extent. All of the above techniques result in multiple potentially pathogenic variants being identified in an individual patient and the challenge is to identify the causative variant in an individual patient. Normal practise is to look at the known inherited neuropathy genes first but even this often results in a number of potentially pathogenic variants being identified. Careful phenotyping in an individual (which may include new phenotyping methods such as neuromuscular MRI) is still an extremely important tool in validating a variant as in certain circumstances the phenotype is tightly associated with a particular variant e.g. CMT1A phenotype with the chromosome 17 duplication. The next step is segregation of a variant within a family which is a powerful tool but multiple family members are not always available. Predictive tools are useful but they are limited by current knowledge of an individual genes function. The increasing availability of population specific control data from NGS studies is proving very useful in predictive programmes. In rare forms of inherited neuropathies such as deoxysphingolipid levels (DSBs) in HSN 1 due to SPTLC1 / 2 mutations, biomarkers can be useful in validating the pathogenicity of a variant. In this talk, I will outline an approach to interpreting the results of NGS testing in the inherited neuropathies with an emphasis in making it useful for practising clinicians.
WS 4.5.1 / #75EVALUATION OF VARIANTS OF UNKNOWN SIGNIFICANCE
Topic: Primer for Genetic Testing
James J. Dowling
Neurology, Hospital for Sick Children, Toronto, ON, CA
Abstract: Next generation sequencing technology has led to a dramatic improvement in clinical genetic diagnostics, and the cause of disease is now established in an ever increasing subset of patients. Next gen sequencing platforms include gene panels and whole exome/genome sequencing. While these platforms provide comprehensive genetic information, they also provide an incredible amount of sequence data, a lot of which is difficult to understand and/or interpret. In this workshop, I will discuss the important issue of sequence variants that are not obvious mutations, or “variants of unknown significance”. I will outline the problems and concerns related to these variants, and discuss novel strategies that we and others have developed to try and analyze them. I will particularly focus on the application of these strategies to childhood muscle disease, though will center this with a broader discussion applicable to all genetic neuromuscular disorders.
WS 4.5.2 / #469CLINICAL WHOLE EXOME SEQUENCING
Topic: Primer for Genetic Testing
Livija Medne
Genetics & Neurology, The Children’s Hospital of Philadelphia, Philadelphia, PA, US
Abstract: not received.
WS 4.5.2 / #470GENE PANELS
Topic: Primer for Genetic Testing
Kimberly Amburgey1, James Dowling2
1Neurology, The Hospital for Sick Kids, Toronto, ON, CA;
2Neurology, The Hospital for Sick Kids, Toronto, ON, CA
Abstract: Genetic confirmation of neuromuscular diseases has greatly improved with the clinical availability of genetic testing panels using next generation sequencing. With the multitude of tests available, it can be difficult to select the most appropriate test. This session will cover the following topics:
• Review the clinically available genetic testing panels for neuromuscular diseases
• Discuss the testing methodologies and limitations of the technology
• Review the considerations when selecting a panel
• Present case reports demonstrating the best use of single gene, panel and exome testing
WS 5.1.1 / #467THE CLINICIANS APPROACH TO LIMB GIRDLE MUSCULAR DYSTROPHY
Topic: Approach to Muscular Dystrophies
Volker Straub
Newcastle University, John Walton Muscular Dystrophy Research Centre, Newcastle upon Tyne, GB
Abstract: The limb-girdle muscular dystrophies (LGMD) are a heterogeneous group of rare autosomal recessive and dominant diseases that clinically present with progressive weakness and wasting of shoulder and pelvic-girdle muscles. Over the last 20 years, the underlying genetic defects for many of the LGMDs have been identified and insight into pathomechanisms has been gained. Since we have entered an era of translational research for some of the more common forms of LGMD, the need for precise molecular diagnoses, a thorough understanding of the natural history of the diseases and guidelines for standardized assessments of the patients become even more relevant. In this context, our team has started a project called MYO-SEQ, that focuses on the application of next generation sequencing, in particular whole exome sequencing (WES), in a large cohort of patients with unexplained limb-girdle weakness. Focusing on undiagnosed patients with a clearly defined clinical phenotype enables increased diagnostic rates for known genes in this cohort, while the use of WES provides scope both for new gene discovery and for additional research into disease modifiers and genotype-phenotype correlation with substantial cost effectiveness. The MYO-SEQ project is exploring 1000 exomes of patients that were aged 10 years and above and presented with unexplained limb girdle weakness and an elevated serum CK activity. Patients were recruited from more than 50 sites across Europe. Results from the project will be presented and it will be discussed how the project has helped to approach patients with limb girdle weakness of unknown origin from a clinicians point of view.
WS 5.1.1 / #468THE CLINICIANS APPROACH TO LIMB GIRDLE MUSCULAR DYSTROPHIES
Topic: Approach to Muscular Dystrophies
Carsten Bonnemann
Neuromuscular And Neurogenetic Disorders Of Childhood Section, National Institutes of Health, Bethesda, MD, US
Abstract: not received.
WS 5.2.1 / #250INTERESTING NEUROMUSCULAR CASES
Topic: Interesting Neuromuscular Cases
Hans Katzberg
Neurology, University of Toronto, Toronto, ON, CA
Abstract: not received.
WS 5.2.1 / #129INTERESTING NEUROMUSCULAR CASES
Topic: Interesting Neuromuscular Cases
Aaron Izenberg
Dept Of Medicine, Division Of Neurology, University of Toronto, Toronto, ON, CA
Abstract: not received.
WS 5.3.1 / #78DIAGNOSIS AND TREATMENT OF DIABETIC NEUROPATHY
Topic: Diagnosis and Treatment of Diabetic Neuropathy
Bruce A. Perkins
Division Of Endocrinology, University of Toronto, Toronto, ON, CA
Abstract: Diabetic sensorimotor polyneuropathy (DSP) involves progressive, diffuse and length-dependent injury to peripheral nerves and affects up to 50% of people with diabetes. Its underlying causes are complex and include, but are not limited to, chronic hyperglycemia. The progression of DSP is associated with significant morbidity and costs. It can cause pain, imbalance, and foot deformity; in later stages it can result in infection, ulceration, and amputation. In view of a long subclinical phase there is an urgent need for a biomarker of early DSP for use in clinical practice and implementation in future trials of therapies aimed at preventing DSP onset and progression. This workshop will engage participants into discussion surrounding the clinical context for screening, diagnosis, and treatment of DSP, including discussion of the symptomatic therapy for neuropathic pain in diabetes and new research avenues for screening and diagnosis. The primary objective is to permit the neurology specialist view the context of diabetic neuropathy management in the general clinical practice environment from which they receive referrals. In that light, the key objectives are to help develop a practical approach to making a working diagnosis of diabetic sensorimotor polyneuropathy in general and diabetes practice, consideration of key alternative diagnoses to exclude, and to understand the therapeutic options for the treatment of pain. From a research perspective and frequently in clinical practice, the diagnosis of DSP requires confirmation by electrodiagnostic nerve conduction studies (NCS). However, NCS may have limitations in detecting early, pre-clinical stages in which abnormalities in small nerve fibre function or structure are more prominent than those in large nerve fibres. Simple screening tests - such as use of the monofilament score - have sufficient concurrent validity for identification of DSP and even sufficient predictive validity for the reasonable prediction of future neuropathy onset. Diagnostic tests currently being explored in research protocols include examination of corneal nerve fibres through the method of In-Vivo Corneal Confocal Microscopy, a method that could in future harmonize with annual eye specialist screening for retinopathy. After identification of the presence of polyneuropathy, clinicians can implement a simple protocol for reasonably excluding major non-diabetes causes and initiate intensification in metabolic control and, if needed, therapy for neuropathic pain. Approaches for pain include the anticonvulsant and antidepressant drug classes, as well as topical and non-pharmacological approaches. Guidance for initiation, titration, and consideration of side effects and costs will be discussed for these therapies.
WS 5.3.2 / #79THE USE OF OMEGA-3 SUPPLEMENTATION FOR MANAGING DIABETIC NEUROPATHY: RESULTS FROM A CLINICAL PILOT TRIAL
Topic: Diagnosis and Treatment of Diabetic Neuropathy
Evan Lewis1, Bruce Perkins2, Richard Bazinet1, Thomas Wolever1, Vera Bril3
1Nutritional Sciences, University of Toronto, Toronto, ON, CA;
2Leadership Sinai Centre For Diabetes, Mount Sinai Hospital, Toronto, ON, CA;
3Division Of Neurology, University Health Network, Toronto, ON, CA
Abstract: Background: Diabetic sensorimotor peripheral neuropathy (DSP) is the leading complication in diabetes mellitus (DM). Omega-3 polyunsaturated fatty acids (N-3 PUFA) are integral to the development and maintenance of healthy nerve tissue but have not yet been investigated for their ability to maintain nerve structure and function in DSP. Methods: Individuals with type 1 (T1DM) and evidence of DSP as determined by a Toronto Clinical Neuropathy Score (TCNS) ≥1 were recruited to participate in an open-label trial of mammalian N-3 PUFA supplementation (10 mL/d; 750 mg EPA, 560 mg DPA and 1020 mg DHA; Auum Inc.) for 1-year (NCT02034266). The primary outcome is the 1-year change in corneal nerve fibre length (CNFL) measured by in vivo corneal confocal microscopy (IVCCM). Secondary outcomes include nerve conduction studies (NCS) and quantitative sensory testing. Results: 40 participants (53% female), aged 48 (SD 14), BMI 28.1 (SD 5.8) with diabetes duration of 27 (SD 18) years were enrolled between March 2014 and June 2015. The baseline TCNS showed participants have a broad range of DSP from no to moderate neuropathy. Mean IVCCM CNFL was 12.0±5.2 mm/mm2, for which ≥14.9 mm/mm2 is considered normal. Median sural and peroneal nerve conduction velocities were 43.8±11.0 and 38.8±6.6 m/sec. Conclusion: In baseline data analysis, we could demonstrate a profile of small and large fibre structure and function that will delineate the effect of omega-3 fatty acid supplementation on these parameters in patients with established neuropathy, and a mixed group of patients without neuropathy at both low and high risk of future onset. Trial results and clinical implications will be presented at ICNMD 2016.
WS 5.4.1 / #80THE ROLE OF GUIDELINES IN DECISIONS ON TREATMENT
Topic: Neuropathic Pain
John D. England
Neurology, LSUHSC School of Medicine, New Orleans, LA, US
Abstract: Guidelines serve an important role in deciding the currently best available treatments for patients with neuropathic pain. The most appropriate way to incorporate guidelines in clinical decision making is to use them in the context of best practices. Best practices incorporate all of the available scientific evidence and fit this to the best and most practical recommendations for the individual patient. As such, guidelines are not a replacement for solid clinical experience and judgment, but serve as an integral part of the decision making process. Even the best guidelines have limitations and do not provide recommendations to fit every patient and every situation. To understand these limitations, one has to understand the process involved in developing guidelines. The best guidelines start with a comprehensive review and rating of the evidence (systematic evidence review). Based upon the quality of evidence and best practices, the evidence is then translated into specific recommendations. Because high quality scientific evidence is always limited, guideline recommendations are hardly ever proscriptive for a particular patient. For the treatment of neuropathic pain, most of the high quality evidence is limited to the treatment of painful diabetic neuropathy. The recommendations from these guidelines are solid, but of limited generalizability to other painful neuropathic conditions. In this presentation, the results of these published guidelines will be presented, and their limitations and future needs will be discussed in detail.
WS 5.4.2 / #81UPDATE ON TREATMENT OF NEUROPATHIC PAIN
Topic: Neuropathic Pain
Jaya Trivedi
University of Texas Southwestern Medical Center, Dallas, TX, US
Abstract: Neuropathic pain is a common symptom and a major source of morbidity in patients with peripheral neuropathy. The mechanisms underlying the generation of neuropathic pain are not fully understood but likely involve a variety of pathophysiologic processes. Numerous clinical trials evaluating the efficacy of agents used in the treatment of neuropathic pain have been performed but are fraught with challenges and methodological shortcomings. However, there is good evidence for the use of tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitors, a number of anticonvulsants, and several other medications in the treatment of neuropathic pain. Of these, duloxetine and pregabalin are the only agents approved for treatment of neuropathic pain in peripheral neuropathy. Treatment must be individualized to a particular patient taking comorbidities and concurrent medication use into account.
WS 5.5.1 / #82 MYASTHENIA GRAVIS IMPAIRMENT INDEX
Topic: Outcome Scales in Myasthenia Gravis
Carolina Barnett Tapia1, Vera Bril2, Moira Kapral1, Abhaya Kulkarni3, Aileen Davis4
1Medicine (neurology), University of Toronto, Toronto, ON, CA;
2University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, ON, CA;
3Neurosurgery, Hospital for Sick Children, Toronto, ON, CA;
4Health Outcomes Research, Toronto Western Research Institute, Toronto, ON, CA
Abstract: Objectives: To develop a measure of the impairments in patients with Myasthenia Gravis (MG) that would quantify disease severity and detect change in clinical trials and practice. Methods: These included a qualitative study of patients experiences to develop a conceptual framework of disease severity in MG. Additionally, items were obtained from the literature or developed de novo and input from clinicians and MG experts was obtained for a final draft of the measure. The measure was field-tested in a broad population of MG patients to determine feasibility, reliability, construct validity and responsiveness. Results: A conceptual framework of MG-related impairments was developed where both the severity of the impairments and fatigability defined overall disease severity. A first draft was assessed by 18 patients through cognitive debriefing and by 78 international specialists through a survey. The second draft had 7 examination and 22 patient-reported items. Field-testing included 200 MG patients with 54 of these included in reliability evaluation and 50 in the responsiveness studies. All items had <7% missing responses. Test-retest reliability for the total score was excellent (ICC: 0.92, 95% CI: 0.79-0.94), as was inter-rater reliability for the examination component (ICC:0.91, 95% CI: 0.88-0.96). The correlations of total scores with other impairment measures were within hypothesized ranges (r: 0.69-0.91, p < 0.0001). Treated patients had significantly greater improvement in scores than controls (ANOVA p < 0.001), with an effect size of 0.40 and standardized response mean of 0.80. The measure was able to detect patient meaningful change and was more efficient than other MG-specific measures in terms of sample size requirements for comparative studies. Conclusions: The Myasthenia Gravis Impairment Index (MGII) has been developed following current recommendations using a patient-centred approach. It has demonstrated construct validity, reliability and responsiveness.
WS 5.5.2 / #83REVIEW OF CURRENT MG SCALES
Topic: Outcome Scales in Myasthenia Gravis
Ted Burns
Department Of Neurology, University of Virginia, Charlottesville, VA, US
Abstract: not received.
WS 6.1.1 / #84GENETIC ASPECTS OF ALS OVERLAP SYNDROMES
Topic: ALS Overlap Syndromes
Ekaterina Rogaeva
Department Of Medicine, University of Toronto, Toronto, ON, CA
Abstract: not received.
WS 6.1.2 / #85CLINICAL ASPECTS OF ALS OVERLAP SYNDROMES
Topic: ALS Overlap Syndromes
Maria Carmela Tartaglia
Neurology, Centre for Research in Neurodegenerative Diseases, Toronto, ON, CA
Abstract: not received.
WS 6.2.1 / #251TREATMENT OF MUSCLE CRAMPS
Topic: Cramps in Neuromuscular Disease
Hans Katzberg
Neurology, University of Toronto, Toronto, ON, CA
Abstract: Initial management of muscle cramps centers around treating the underlying cause for symptoms, such as elimination of underlying causes and triggers for muscle cramps such as correction of vitamin or electrolyte deficiencies. In the setting of cirrhosis, albumin infusions used to help treat contracted extracellular volume have been shown in a single study to be beneficial for cramps and is often tried. For patients with hemodialysis induced cramps, changing the dialysate has been shown to reduce the frequency of cramps. Non-pharmacological interventions include stretching can help avoid muscle cramps not only prior to exercise, but has been shown to prevent nocturnal muscle cramps in a sham-controlled clinical study. Ensuring adequate hydration can also help prevent muscle cramps. If these interventions fail, pharmacological interventions are often needed to control symptoms. In 2010, the American Academy of Neurology issued guidelines on the pharmacological and non-pharmacological treatment of patients with muscle cramps. Although quinine sulphate had the most evidence (Class A) for efficacy, because of safety concerns, it was recommended for use in muscle cramps only when first line therapies have failed and if cramps and prominent and disabling. Other identified agents included calcium channel blockers (class B) and B-complex vitamins (class B). Natidrofuryl which is usually used to treat arterial insufficiency had Class II evidence for use but is not available for use in the US and not routinely used in clinical practice for treatment of cramps. Other medications which are used for muscle cramps which have limited evidence in various patient populations include mexiletine, carbamazepine, baclofen and leveteracitam. Although cramp frequency has been used most commonly as a primary outcome measure in muscle cramp trials, a validated assessment scale which factors in not only cramp frequency but duration and severity may be a more optimal way to assess muscle cramps and measure clinically relevant change. Therapeutic and epidemiological research dealing with muscle cramps lack an established measure of functional impairment, that represents disease severity, which is also sensitive to change, and can be easily administered.
WS 6.2.2 / #156ASSESSMENT OF MUSCLE CRAMPS
Topic: Cramps in Neuromuscular Disease
Nicholas Silvestri
Neurology, SUNY at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY, US
Abstract: Muscle cramps are defined as a painful, sustained contraction of a muscle or muscle group and are frequently referred to by patients as “charley horses.” Most people have experienced a muscle cramp at some point in their lifetime, and the prevalence ranges from 37-95% depending on the population studied. Cramps are most often a neurogenic phenomenon and are due to high frequency firing of motor neurons, seen on electromyography as spontaneous 50-150 Hz discharges, leading to a coordinated contraction of muscle. There is data supporting both a central and a peripheral pathophysiological origin. Potential central factors leading to muscle cramps include amplification of inward sensory input at the spinal level, or disruption of sensory afferent input. Theories of a peripheral origin of cramps include anterior horn cell damage, aberrant ephaptic transmission, and nerve terminal hyper-excitability. The differential diagnosis of persistent muscle cramps include physiologic cramps (e.g. heat or exercise-induced), idiopathic cramps, and cramps secondary to metabolic derangements, certain medications, and neuromuscular disorders such as motor neuron disorders and disorders of peripheral nerve hyper-excitability. Cramps are also common in pregnancy. The initial work-up of such patients includes complete blood count, comprehensive metabolic profile, creatine kinase, thyroid function studies, as well as consideration of electromyography and nerve conduction studies with repetitive nerve stimulation. In selected cases, magnetic resonance imaging of the spine and evaluation for voltage-gated potassium channel antibodies may also be considered. The treatment of muscle cramps is covered in another presentation.
WS 6.3.1 / #88EPIDEMIOLOGY AND PATHOPHYSIOLOGY OF DIABETIC NEUROPATHY
Topic: Diabetic Neuropathy
James W. Russell
Neurology, University of Maryland School of Medicine, Baltimore, MD, US
Abstract: The geographic distribution of diabetic neuropathy continues to change across the world. It is estimated that there are in excess of 400 million diabetics in the world and up to 50% of those diabetics will develop clinical neuropathy during their lifetime. The largest populations of diabetics are in the Western Pacific and South-East Asia. Most of the countries with the highest prevalence of diabetes (12% or higher) are in the Western Pacific or Middle East/North Africa. Over the next 20 years, the largest percent increase in newly diagnosed diabetics will occur in Africa, the Middle East and North Africa, and South-East Asia. In contrast, the rate of growth will be much slower in Europe and North America. In the United States, there are at least 1.25 million adults and children with type 1 diabetes mellitus, 29 million diagnosed type 2 diabetics, 8 million undiagnosed type 2 diabetics, and 86 million Americans with prediabetes. There are 1.4 million new cases diagnosed with diabetes every year and diabetes is the 7th leading cause of death. The direct medical costs for diabetes are $176 billion and $69 billion in reduced productivity. In the United States, there is encouraging evidence that the rate of development of diabetes is slowing possibly due to public awareness of risk factors. There is also evidence in the US that overall management of diabetes and diabetic complications is resulting in improved complication rates and severity. A complex constellation of pathways are directly or indirectly affected in diabetic neuropathy. Dissecting out these pathways can lead to development of new approaches to reverse neuropathy and treat neuropathic pain. Despite the focus on glycemic control in diabetes, the evidence from randomized clinical trials that improved glycemic control reduces the prevalence of neuropathy in the majority of diabetics is unclear. The evidence for type I diabetes is strong but weak or absent for type 2 diabetes mellitus. Abnormal lipid metabolism, or increased oxidation of lipids, may play an important role in the pathogenesis of neuropathy, particularly in type 2 diabetes. Increased oxidative injury and impairment of mitochondrial function affect metabolic processes and signaling pathways that are crucial for function of neurons, glial cells and axons in the peripheral nervous system. For example, generation of reactive oxygen species (ROS) activates several other pathways that can lead to neuronal or axonal injury. In turn, these pathways can increase generation of ROS. Unfortunately, despite considerable evidence that there is an increase in ROS, treatment trials with one antioxidant, thioctic acid (alpha lipoic acid), have had only a modest effect. However, combination approaches with nutraceuticals that increase normal mitochondrial efficiency and reduce oxidative stress pathways have shown some benefit both in pre clinical and clinical studies. For example, combination therapy with L-methyl folate, pyridoxyl 5 phosphate and methylcobalamine reduces endothelial oxidative stress, inhibits endothelial nitric oxide synthetase, and replenishes reduced glutathione. Thus, understanding the critical mechanisms that lead to diabetic neuropathy may result in improved therapy and management of neuropathic symptoms.
WS 6.3.2 / #89TREATMENT OF DIABETIC NEUROPATHY
Topic: Diabetic Neuropathy
Vera Bril
Medicine (neurology), University Health Network, University of Toronto, Toronto, ON, CA
Abstract: Diabetic sensorimotor polyneuropathy (DSP) is the most common complication of diabetes. Reversal of this chronic axonopathy is not possible at this time. In type I diabetes, intensive glycemic control reduces the prevalence of DSP. Attention to modifiable risk factors such as hypertension, smoking and hyperlipidemia may further ameliorate neuropathy. In type II diabetes, the results of improved glycemic control are less evident with only minimal changes after 5-8 years of intervention. Symptomatic treatment of painful symptoms of DSP can be achieved with various interventions including adjuvant analgesic therapy. It is crucial to determine that neuropathy in diabetes patients is, in fact, related to diabetes and not representative of another process such as nutritional deficiency, monoclonal gammopathy, or chronic inflammatory demyelinating polyneuropathy (CIDP). CIDP is more 9 times more prevalent in patients with diabetes and responds similarly to treatment with immunomodulatory or immunosuppressive interventions, but is offered to patients with CIDP + diabetes less frequently than to those with CIDP alone.
WS 6.4.1 / #279LYMPHOMA AND OTHER PERIPHERAL NERVE TUMORS
Topic: Peripheral Nerve Tumors
Wolfgang Grisold1, Anna Grisold2
1Neurology, Sozialmedizinisches Zentrum Süd, Wien, AT;
2Neurology, Med Uni Wien, Wien, AT
Abstract: Peripheral nerve tumors usually present with a slowly progressive mononeuropathy. Clinically paresthesia, pain, followed by motor or sensory loss and sometimes neuropathic pain occurs. The tumors may be seen, palpated or displayed in imaging. Often mechanically evoked factors (e.g. sitting , stretching the sciatic nerve, walking if tumor is on the foot) can exacerbate pain or paresthesia. Tinel sign may be present on palpation. Tumors of the peripheral nerves are rare and in addition to intrinsic nerve tumors can be caused by several malignant mechanisms as cancer, lymphoma and leukemia. Tumors can occur in all parts of the peripheral nervous system as the extracavitary portions of cranial nerves, nerve roots, nerve plexus and in peripheral nerves. The involvement of the meningeal space by tumors is not considered here, although synchronous. combinations of meningeal and extrameningeal involvement have been observed. Cancer: Direct involvement of peripheral nerves by cancer is clinically observed at several sites as nerve roots and nerve plexus. Direct metastatic deposition in individual nerves is rare. However spread along peripheral nerves and cranial nerves either in retro- and anterograde pattern, often causing marked nerve thickening has been observed in several types of cancer. The different types of nerve infiltration have been described as: compression, invasion, “engulfing” and diffuse infiltration. In the region of the base of the skull, the territory of the trigeminal and facial nerve are most frequently involved. Lymphoma and leukemia: Diffuse involvement of peripheral nerves is termed neurolymphomatosis and neuroleukemiosis. Also intravascular lymphoma and rarely combinations of neurolymphomatosis and intravascular lymphoma were observed. Clinically this presentation is usually symmetric, but also focal limb presentations were described. Tumefactive nerve enlargement has been described in lymphoma and leukemia. Focal malignant tumors in peripheral nerves in extranodal lymphoma are rare, can be the first manifestation of lymphoma and have been described in several peripheral nerves, most frequently in the sciatic nerve. Rarely focal lymphoma can mimic an entrapment syndrome. Solid focal depositions of leukemia, termed chloroma or myelosarcoma have been described in cranial nerves, the cauda equine and peripheral nerves, also indicating relapse. Differential diagnosis of the nerve tumor includes inflammatory lesions, diffuse infiltrative lymphocytosis syndrome, hypertrophic neuritis, perineuroma, pseudotumors, and amyloidoma among others. In cancer patients with preradiated tissue in addition to scarring also radiation induced tumors must be considered. The diagnosis depends on imaging either by MRI or increasingly ultrasound. Combined MR/PET studies are recommended to detect enhanced metabolism. Biopsy is still the most important diagnostic tool, and will be performed either as tumor resection, or fascicular biopsy, depending on the site and functional property of the nerve. Surgical therapy depends on the site and relation of the affected nerve towards it surroundings, as well as the aim to preserve function. Depending on the histologic pathology local RT, chemotherapy or other anti tumor therapies will be used.
WS 6.4.2 / #91NEUROFIBROMATOSIS 1 AND MALIGNANT TRANSFORMATION OF PERIPHERAL NERVE SHEATH TUMORS
Topic: Peripheral Nerve Tumors
Gelareh Zadeh
Surgergy, University Health Network, Toronto, ON, CA
Abstract: not received.
WS 6.5.1 / #92NEUROMUSCULAR PHYSICIANS SHOULD PERFORM NM ULTRASOUND
Topic: Ultrasound of Muscle and Nerve
Steven Shook
Neuromuscular Center, Neurological Insitute, Cleveland Clinic, Cleveland, OH, US
Abstract: Neuromuscular Physicians Should Perform NM Ultrasound This presentation will highlight the pros and cons of neurologists, physiatrists, and other neuromuscular clinicians performing ultrasound, and will emphasize the importance of accurate image interpretation and appropriate selection of ancillary testing to optimize patient care. The unique experience neuromuscular physicians bring to the field will be presented, including: familiarity with surface landmarks and anatomy associated with electrodiagnostic testing, as well as the knowledge required to place ultrasound findings in the appropriate clinical context. Common situations in which ultrasound can enhance neuromuscular patient care will be presented, including ultrasound as an adjunct to electrodiagnostic testing, point-of-care decision making in patients with nerve injury, post-operative management of nerve surgery patients, and ultrasound-guided EMG of the diaphragm. The session will conclude with a demonstration of a collaborative effort to accurately diagnose a neuromuscular process and with emphasis on the value of collaboration between neuromuscular specialists and radiologists.
WS 6.5.2 / #93RADIOLOGISTS SHOULD PERFORM NM ULTRASOUND
Topic: Ultrasound of Muscle and Nerve
Linda Probyn
Medical Imaging, University of Toronto, Toronto, ON, CA
Abstract: Radiologists Should Perform Neuromuscular Ultrasound The presentation will highlight both the pros and cons of Radiologists performing neuromuscular ultrasound and will emphasize the importance of a collaborative effort that is frequently required so that accurate ultrasound image interpretation can be performed to ensure the best possible patient care. A description of required ultrasound training requirements for Radiologists will be provided including teaching of physics, knowledge of artifacts and quality assurance activities necessary for optimizing imaging quality and to aid in the appropriate interpretation of findings. Required picture archiving and report generation for accurate communication and accountability of results from radiologists to clinicians will be reviewed. Common requests that radiologists receive for neuromuscular ultrasound and sample pathologies will be shown. These include a demonstration of real time dynamic assessment of pathologies to reproduce symptoms and demonstrate pathology. Cases will be illustrated whereby other pathologies (i.e. extrinsic masses) cause neurologic symptoms and the value of a radiologist’s interpretation is emphasized. In addition, the importance of other imaging modalities (i.e. MRI) to assist in the accurate interpretation of some neuromuscular pathologies with be reviewed. The session will conclude with a demonstration of a collaborative effort to accurately diagnose a neuromuscular process and will emphasis the value of a Radiologist’s involvement in neuromuscular ultrasound.
WS 7.1.1 / #94IGG4 AUTOANTIBODIES RELATED TO NEUROMUSCULAR DISEASES: THERAPEUTIC IMPLICATIONS
Topic: Autoantibodies in Neuromuscular Disease
Luis Querol, Isabel Illa
Neuromuscular Diseases Unit, Hospital de la Santa Creu i Sant Pau, Barcelona, ES
Abstract: Pathogenic antibodies of the IgG4 isotype have been described in different autoimmune diseases, including diverse rheumatologic, cutaneous and neurological diseases. IgG4 isotype antibodies are a rare antibody subtype with anti-inflammatory properties: they do not activate complement or bind to Fcgamma immunoglobulin receptors and are bispecific. They are synthesized by specific B-cell subsets and, most importantly, IgG4-mediated diseases respond very well to B-cell depleting therapies. Antibodies targetting the muscle-specific kinase (MusK) in myasthenia gravis and contactin-1 or neurofascin-155, two proteins of the paranode of Ranvier, in chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) are also of the IgG4 isotype. The IgG4 nature of these antibodies likely reflects fundamental differences in the immunological environment leading to antibody synthesis compared to classical IgG1-3 mediated immune responses and, thus, diverse triggers, genetic backgrounds and pathogenesis. The discovery of IgG4-mediated diseases highlights the need to define the target antigens in autoimmune diseases as the critical step to stablish, within heterogenoeus syndromes, clinical subsets. This ultimately leads to the possibility of tayloring the theraputic regimens according to serological status and changing prognosis dramatically.
WS 7.1.2 / #95IS IT ALL ABOUT THE ANTIBODIES
Topic: Autoantibodies in Neuromuscular Disease
Andrew Mammen
National Institute Of Arthritis And Musculoskeletal And Skin Diseases, National Institutes of Health, Bethesda, MD, US
Abstract: not received.
WS 7.2.1 / #96NERVE
Topic: Evaluation of Variants of Unknown Significance
Mary M. Reilly
MRC Centre For Neuromuscular Diseases, National Hospital for Neurology and Neurosurgery, London, GB
Abstract: Charcot Marie Tooth disease (CMT) and the related neuropathies, the distal hereditary motor neuropathies (HMN) and the hereditary sensory neuropathies (HSN) are genetically and clinically a heterogeneous group of neuropathies characterised by slowly progressive distal wasting, weakness and / or distal sensory loss. Like many other neuromuscular diseases, major advances have occurred in identifying the causative genes over the last 25 years and especially over the last 5 years in the era of next generation sequencing (NGS) such that there are now more than 80 identified causative genes for the inherited neuropathies. The major challenge for clinicians now is interpreting the results they receive. Most diagnostic laboratories are using next generation sequencing techniques (NGS) and have developed disease-specific, or multi-gene, testing panels which employ NGS to regions of the exome that contain known inherited neuropathy genes. These disease-specific panels are currently the best method for simultaneously screening a large number of inherited neuropathy genes although WES (whole exome sequencing) is increasingly being used diagnostically and WGS (whole genome sequencing) to a lesser extent. All of the above techniques result in multiple potentially pathogenic variants being identified in an individual patient and the challenge is to identify the causative variant in an individual patient. Normal practise is to look at the known inherited neuropathy genes first but even this often results in a number of potentially pathogenic variants being identified. Careful phenotyping in an individual (which may include new phenotyping methods such as neuromuscular MRI) is still an extremely important tool in validating a variant as in certain circumstances the phenotype is tightly associated with a particular variant e.g. CMT1A phenotype with the chromosome 17 duplication. The next step is segregation of a variant within a family which is a powerful tool but multiple family members are not always available. Predictive tools are useful but they are limited by current knowledge of an individual genes function. The increasing availability of population specific control data from NGS studies is proving very useful in predictive programmes. In rare forms of inherited neuropathies such as deoxysphingolipid levels (DSBs) in HSN 1 due to SPTLC1 / 2 mutations, biomarkers can be useful in validating the pathogenicity of a variant. In this talk, I will outline an approach to interpreting the results of NGS testing in the inherited neuropathies with an emphasis in making it useful for practising clinicians.
WS 7.2.2 / #97MUSCLE
Topic: Evaluation of Variants of Unknown Significance
Raveen Basran
Neurology, Hospital for Sick Children (SickKids), Toronto, ON, CA
Abstract: not received.
WS 7.3.1 / #98GENERAL TREATMENT APPROACHES
Topic: Inclusion Body Myopathy
Mazen Dimachkie
Neurology, The University of Kansas Medical Center, Kansas City, KS, US
Abstract: Sporadic inclusion body myositis (IBM) is the most common idiopathic inflammatory myopathy after age 40 to 50. It commonly presents with chronic insidious proximal leg and distal arm asymmetric muscle weakness. Some IBM cases may have an atypical presentation such as quadriceps sparing weakness, foot drop or even dysphagia. Besides clinical evaluation, laboratory testing is important to exclude mimics of IBM and muscle biopsy is central to the diagnosis of IBM. Muscle histopathology demonstrates endomysial inflammatory exudates surrounding and invading non-necrotic muscle fibers often times accompanied by rimmed vacuoles and protein aggregates. There is a debate about the specificity of a novel autoantibody to cytosolic 5′-nucleotidase 1A that is detectable in 33% to 70% of IBM cases. IBM causes significant morbidity during its relentless progression to disability. We are beginning to identify genetic modifiers that influence the age of IBM onset and perhaps the phenotype. While clinical trials of immunosuppressive therapies have been largely negative, it is suggested that mild intensity exercise may slow down the rate functional decline. Finally, much research is needed and studies are currently ongoing in an attempt to identify novel and effective therapies for people with IBM.
WS 7.3.2 / #99ONGOING DEVELOPMENTS IN IBM
Topic: Inclusion Body Myopathy
Anthony A. Amato
Neurology, Brigham and Women’s Hospital, Boston, MA, US
Abstract: From a clinical perspective, an important finding was the demonstration of cytosolic 5’-nucleotidase 1A antibodies in approximately two-thirds of IBM patients. Antibody testing can help diagnose IBM, particularly when not all the characteristic clinical or histopathological features of IBM are present. Also, in regard to diagnosis, Lloyd et al compared 24 published IBM diagnostic categories. The ENMC 2011 “probable IBM” category performed the best and probably should be used in future clinical trials. A pilot study of bimagrumab, a monoclonal antibody inhibiting activin receptors in 14 subjects with sIBM using one dose of bimagrumab (n = 11) or placebo (n = 3) revealed increase in thigh muscle volume and lean body mass in bimagrumab-treated subjects. In a subsequent 16-week observation phase involving bimagrumab and 2 placebo-treated subjects 6-minute walking distance improved in the Bimagrumab-treated subjects. A large, phase III trial of Bimagrumab was conducted(results pending). Various histopathological studies have demonstrated sarcoplasmic aggregation and myonuclear depletion of the nucleic acid binding proteins, including TDP-43 and nuclear heterogeneous nuclear ribonucleoproteins (hnRNPA1 and hnRNPA2B1). Perhaps abnormal RNA processing plays a role in the pathogenesis of IBM as speculated to occur in the “multisystem proteinopathies”, including forms of familial ALS and frontotemporal dementia that can occur in combination with hereditary inclusion body myopathy. IBM might, in part, be caused by abnormal RNA metabolism, resulting in overassembly of hnRNP granules mediated by prion-like domains in hnRNPs, which evolve into pathological aggregates. This could also lead to abnormal translation of mRNA to aberrant proteins. Studies involving RNA sequencing combined with proteomic studies of muscle biopsy tissue should lead to intriguing findings. Viral infections have been speculated in the pathogenesis of IBM. Patients with retroviral infections (HIV and HTLV-1) can develop IBM. Uruha and colleagues recently assessed the prevalence of HCV infection in 114 patients in Japan with IBM. They found a significantly higher number of patients with IBM (28%) had anti-HCV antibodies as compared with patients with polymyositis (4.5%) and the general Japanese population in their 60s (3.4%). Might anti-viral therapies be studied in IBM? Greenberg and colleagues recently published their prospectively study of 38 IBM patients looking for the presence of expanded large granular lymphocyte (LGL) populations. Most IBM patients (22/38) had aberrant populations of LGL meeting diagnostic criteria for T-cell large granular lymphocytic leukemia. These T cell populations were clonal and stably present on follow-up testing. T cell aberrant loss of CD5 or gain of expression of CD16 and CD94 were common (19/42). In comparison, only 2/15 age-matched patients with other forms of myositis and 0/20 age-matched healthy controls had large granular lymphocyte expansions, with none of these patients having T cell aberrant expression of CD5, CD16, or CD94. The authors speculated that the autoimmune T cell expansion may evolve into a neoplastic-like or overtly neoplastic disorder in IBM, perhaps contributing to its relative refractoriness to immune-directed therapies. Furthermore, the presence of LGL correlated with lower CD4/CD8 ratio and in and of itself may be a helpful diagnostic marker. Novel treatment targeting LGLs might be beneficial.
WS 7.4.1 / #100SMA TODAY
Topic: Modern Concepts in Spinal Muscular Atrophy
Susan Iannaccone
Pediatrics, Univerity of Texas Southwestern Medical Center, Dallas, TX, US
Abstract: Spinal muscular atrophy (SMA) is an autosomal recessive neuronopathy that causes degeneration of anterior horn cells and resulting muscle weakness. It is the most common fatal genetic disorder of infants, but can affect individuals at any age. For almost a century after its initial descriptions, it was considered the typical rare neurodegenerative disorder that could be clinically diagnosed and classified but not treated or understood on a molecular level. Advances in molecular genetics and biochemistry over the past three decades have led to the identification of the causative gene (SMN1) for SMA, the recognition of a nearly identical homologue gene (SMN2) that serves a rescue function and explains in large part the phenotypic heterogeneity of the disorder, the generation of several animal models for the disease, and the realization that SMA is one of a number of neurological disorders associated with disruption of RNA metabolism. These discoveries have in turn led to the development of a simple and accurate diagnostic test for SMA that has fundamentally changed how we diagnose these patients. Although SMA is currently considered untreatable in the usual sense, clinical investigators have developed patient standards of care (currently being revised and updated) that have vastly prolonged survival and improved function in patients with SMA. This clinical work coupled with already completed therapeutic trials has also been instrumental in identifying valid, reproducible, and meaningful outcome measures for clinical trials. This information is vitally important since the new genetic and biochemical data has identified a number of exciting possibilities for therapeutic intervention and multiple targets for both pharmacologic and genetic based therapies. Several clinical trials are in various stages of completion or development based on these strategies; most promising are antisense oligonucleotide and gene replacement therapies, both of which are being employed in therapeutic trials. The two lectures in this workshop will provide a brief yet comprehensive update and state-of-the-art review of the diagnosis, clinical management, and outlook for definitive therapies in this challenging disorder. Attention will focus on meaningful interventions that can be made to prolong and improve quality of life in even the most severely affected patients.
WS 7.4.2 / #481UPDATE ON SPINAL MUSCULAR ATROPHY
Topic: Modern Concepts in Spinal Muscular Atrophy
John T. Kissel
Neurology, Pediatrics & Neuroscience, Ohio State University Wexner Medical Center, Columbus, OH, US
Abstract: Spinal muscular atrophy (SMA) is an autosomal recessive neuronopathy that causes degeneration of anterior horn cells and resulting muscle weakness. It is the most common fatal genetic disorder of infants, but can affect individuals at any age. For almost a century after its initial descriptions, it was considered the typical rare neurodegenerative disorder that could be clinically diagnosed and classified but not treated or understood on a molecular level. Advances in molecular genetics and biochemistry over the past three decades have led to the identification of the causative gene (SMN1) for SMA, the recognition of a nearly identical homologue gene (SMN2) that serves a rescue function and explains in large part the phenotypic heterogeneity of the disorder, the generation of several animal models for the disease, and the realization that SMA is one of a number of neurological disorders associated with disruption of RNA metabolism. These discoveries have in turn led to the development of a simple and accurate diagnostic test for SMA that has fundamentally changed how we diagnose these patients. Although SMA is currently considered untreatable in the usual sense, clinical investigators have developed patient standards of care (currently being revised and updated) that have vastly prolonged survival and improved function in patients with SMA. This clinical work coupled with already completed therapeutic trials has also been instrumental in identifying valid, reproducible, and meaningful outcome measures for clinical trials. This information is vitally important since the new genetic and biochemical data has identified a number of exciting possibilities for therapeutic intervention and multiple targets for both pharmacologic and genetic based therapies. Several clinical trials are in various stages of completion or development based on these strategies; most promising are antisense oligonucleotide and gene replacement therapies, both of which are being employed in therapeutic trials. The two lectures in this workshop will provide a brief yet comprehensive update and state-of-the-art review of the diagnosis, clinical management, and outlook for definitive therapies in this challenging disorder. Attention will focus on meaningful interventions that can be made to prolong and improve quality of life in even the most severely affected patients.
WS 7.5.1 / #102RECENT CONCEPTS IN FSHD
Topic: Update on FSHD
Rabi Tawil
Department Of Neurology, University of Rochester Medical Center, Rochester, AL, US
Abstract: Facioscapulohumeral muscular dystrophy (FSHD) is the most common adult-onset dystrophy after myotonic dystrophy. The association of FSHD, in its most common form, with a contraction of a repetitive DNA sequence (D4Z4 repeats) on chromosome 4q35 was established in 1991. However, how this repeat contraction resulted in FSHD remained a mystery until recently. There is now consensus that repeat contraction to 1-10 repeats (normal 11-100), results in opening of the chromatin structure allowing the expression of DUX4, transcriptional regulator gene that is normally expressed only in the germline. DUX4 protein in turn is toxic to muscle fibers. Confirmation of this epigenetic mechanism for FSHD soon followed by the discovery of a FSHD phenotype not associated with D4Z4 repeat contraction. FSHD2, which accounts for about 5% of all FSHD, is the result of a mutation in the SMCHD1 gene on chromosome 18. SMCHD1 is a chromatin regulator and mutation in the gene results in loss of methylation and a more permissive chromatin structure on 4q35 resulting in inappropriate DUX4 expression. The discovery of a unifying mechanism has paved the way for rational, targeted drug development in FSHD.
WS 7.5.2 / #103CLINICAL PRESENTATION IN FSHD
Topic: Update on FSHD
Jeffrey M. Statland
Neurology, University of Kansas Medical Center, Kansas city, KS, US
Abstract: In this session I will present an update on the clinical features of facioscapulohumeral muscular dystrophy (FSHD). FSHD is one of the most prevalent muscular dystrophies (1 in 15,000). The most common form FSHD type 1 (~95% of individuals) is inherited in an autosomal dominant fashion due to deletion of repeated elements on the long arm of chromosome 4q (3.3 kb repeats in the D4Z4 region). Patients with FSHD have between 1-10 residual D4Z4 units; whereas normal individuals have >10. An additional 5% termed FDHD type 2 have disease through a deletion-independent mechanism, about 80% of whom will have a mutation on a gene SMCHD1, which has a role in DNA methylation. FSHD type 2 is a digenic disease with more complicated inheritance patterns. Both FSHD types 1 and 2 converge on a common downstream mechanism of aberrant expression of the normally repressed gene DUX4. Clinically patients with FSHD types 1 and 2 are similar. FSHD can be diagnosed over the course of a lifespan, from the very young to old age, with a typical age of diagnosis in the second or third decade. Penetrance in women is believed to be lower than in men, and on average women are diagnosed at older age, and are often less severely affected There are no genetic racial or ethnic predispositions to developing FSHD, and large case series have been reported in Asia, Europe, and the United States. FSHD has a distinct initial pattern of muscle involvement, often affecting the facial muscles, shoulder girdle, and upper arms, followed by weakness of the trunk, distal lower extremity and more proximal muscles later in the disease course. However in FSHD the exception is often the rule – as there is a great deal of variability in muscle involvement between patients, from one side of the body to the other in the same patient, and between individuals with the same mutation in single families. Patients can have debilitating truncal weakness. Although FSHD does not typically shorten the life span, it can result in significant morbidity, loss of the ability to earn a living, and approximately 1 in 5 may require the use of a wheelchair. Mainly atrial cardiac arrhythmias have been described in FSHD, but these are almost entirely not symptomatic. Restrictive respiratory involvement can be seen and typically follows weakness, more common once individuals are wheelchair bound, or when they demonstrate marked paraspinal weakness. There are rare but important extra-muscular manifestations which require screening: 1) symptomatic retinal vascular disease called Coat’s syndrome which is potentially treatable with laser therapy; and 2) hearing loss that if missed in a young child can impact language and learning. The symptomatic extra muscular complications are almost entirely seen in patients with the smallest number of remaining D4Z4 units (1-3 units), and has not been reported in FSHD type 2. There are no registered treatments for FSHD, so treatment is largely supportive. The AAN has published care guidelines which will be reviewed here.
WS 8.1.1 / #104EXERCISE THERAPY IN MITOCHONDRIAL DISORDERS
Topic: Exercise Therapy for Metabolic Myopathies
Ronni Haller
Neurology, University of Texas Southwestern Medical School, Dallas, TX, US
Abstract: not received.
WS 8.1.2 / #105EXERCISE TRAINING AND PATHOPHYSIOLOGY OF EXERCISE IN METABOLIC MYOPATHIES
Topic: Exercise Therapy for Metabolic Myopathies
John Vissing
Neurology, Copenhagen Neuromuscular Center, Section 6921nm, Rigshospitalet, Copenhagen, DK
Abstract: Metabolic myopathies encompass muscle glycogenoses (GSD) and disorders of muscle fat oxidation (FAOD). FAODs and GSDs can be divided into two main clinical phenotypes; those with static symptoms related to fixed muscle weakness and atrophy, and those with dynamic, exercise-related symptoms that are brought about by a deficient supply of ATP. Together with mitochondrial myopathies, metabolic myopathies are unique among muscle diseases, as the limitation in exercise performance is not solely caused by structural damage of muscle, but also or exclusively relate to energy deficiency. ATP consumption can increase 50-100-fold in contracting, healthy muscle from rest to exercise, and testing patients with exercise is therefore an appropriate approach to disclose limitations in work capacity and endurance in metabolic myopathies. Muscles rely almost exclusively on muscle glycogen in the initial stages of exercise and at high work intensities. Thus, patients with GSDs typically have symptoms early in exercise, have low peak work capacities and develop painful contractures in exercised muscles. Muscle relies on fat oxidation at rest and to a great extent during prolonged exercise, and therefore, patients with FAODs typically develop symptoms later in exercise than patients with GSDs. Due to the exercise-related symptoms in metabolic myopathies, patients have generally been advised to shun physical training. However, immobility is associated with multiple health issues, and may even cause unwanted metabolic adaptations, such as increased dependence on glycogen use and a reduced capacity for fatty acid oxidation, which is detrimental in GSDs. Training has not been studied systematically in any FAOD and in just a few GSDs. However, studies on single bouts of exercise in most metabolic myopathies show that particularly moderate-intensity aerobic exercise is well tolerated in these conditions. Even low-intensity, resistance training of short duration is tolerated in McArdle disease. The talk will focus on the particular pathophysiology associated with exercise in different metabolic myopathies, and the advice and recommendations for exercise training in these conditions will be reviewed.
WS 8.2.1 / #106WHY PATIENT-CENTERED OUTCOMES RESEARCH?
Topic: How To Do Investigator-Initiated Trials; PCORI
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: In recent years there has been a great effort to get patients, families, community and patient advisory groups more involved in the clinical research process. When developing an idea for a clinical research project we get patients involved to identify their needs and what areas they believe we should focus on. We then place patients and others on the project steering committees, communication committees and Data Safety Monitoring Boards (DSMB). Outcome measures are increasingly becoming patient driven and the field of patient reported outcomes measures (PROM) is extremely important. We use various patient reported outcomes as either primary or secondary outcomes. There are many types of PCORI funding. One such funding mechanism is the Assessment of Prevention, Diagnosis and Treatment Options. The goal is to compare the effectiveness of two or more interventions or approaches to health care. One way this is can be accomplished is by comparing interventions by answering questions important to patients and other stakeholders. We have been successful in obtaining one of these awards to conduct a comparative effectiveness study of drugs for management of pain in cryptogenic sensory polyneuropathy (CSPN) called Patient Assisted Intervention for Neuropathy: Comparison of Treatment in Real Life Situations (PAINSCONTRoLS). We are comparing four medications at about 40 sites in the USA and Canada. We have submitted 2 additional studies for possible funding, one to PCORI and one to the NIH. Both applications utilized patients and their caregivers in developing the concept and preparing the grant application. The application to PCORI is a Comparative Effectiveness Study of Sialorrhea in Patients with ALS. In that study, we are looking at which of the 3 medications might be the best in controlling the sialorrhea in patients with ALS. The study submitted to the NIH is a study looking at drug combinations (somewhat similar to the approach used in HIV). Early on PCORI created an award that provides infrastructure in which the PCORI grants can operate through. This infrastructure called(PCORnet, the National Patient-Centered Clinical Research Network) goal is to foster a wide range of experimental and observational patient-centered studies. This network is broken down to Clinical Data Research Networks (CDRNs). University of Kansas Medical Center has a CDRN called the Greater Plains Collaborative (GPC). One of the first accomplishments by the GPC was to deploy the development of the Healthcare Enterprise Repository for Ontological Narration (HERON) system at KUMC. This is an i2B2 software program that allows use of electronic medical records to survey for potential research subjects. One of the first projects was to develop a survey and utilizing the sites EMR, identify patients and utilize this list to send out the survey. We took an existing functional status form (the Amyotrophic Lateral Sclerosis Functional Rating Scale- Revised (ALSFRS-R)) and asked patients what they wanted to change or what additional questions they wanted to see poised to them regarding their functional status. We then modified the form and then submitted a protocol to be able to send out this survey.
WS 8.2.2 / #107INVESTIGATOR-INITIATED CLINICAL TRIALS
Topic: How To Do Investigator-Initiated Trials; PCORI
Richard J. Barohn
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: not received.
WS 8.3.1 / #108NEUROPATHY IN PRE-DIABETES & THE METABOLIC SYNDROME
Topic: Metabolic Neuropathies
A. Gordon Smith
Neurology, University of Utah, Salt Lake City, UT, US
Abstract: Peripheral neuropathy is a major cause for patient morbidity due to pain, gait instability and elevated fall risk. The most common cause for neuropathy after diabetes is idiopathic or cryptogenic sensory peripheral neuropathy (CSPN). CSPN patients have a significantly elevated risk of having both prediabetes (particularly impaired glucose tolerance) and metabolic syndrome. Obesity and dyslipidemia are most closely linked with neuropathy risk. Evolving data similarly indicate patients with metabolic syndrome and prediabetes have an elevated risk of neuropathy. It is probable that CSPN associated with obesity shares common mechanisms with diabetic neuropathy. Diet and exercise based lifestyle interventions have shown promise, likely via positive effects on peripheral nerve regenerative capacity. Novel therapeutic strategies include a focus on reducing sedentary behavior and medical therapy aimed at improving insulin sensitivity and reducing weight.
WS 8.3.2 / #109NEUROPATHY DUE TO SYSTEMIC DISEASE
Topic: Metabolic Neuropathies
Mamatha Pasnoor
Neurology, University of Kansas Medical Center, Kansas City, KS, US
Abstract: Neuropathies can be associated with systemic diseases including disorders of the kidney, liver, lung as well as critical illness in the setting of sepsis and multiple organ failure. It is usually difficult to separate neuropathies developing as consequence of systemic disease from those conditions that affect these systems and the nerve together as seen with alcoholism, sarcoidosis and other conditions. Autonomic and somatic polyneuropathies are seen with chronic liver disease. Acute inflammatory demyelinating polyradiculoneuropathy is seen with viral hepatitis. Porphyric neuropathy presents with an axonal neuropathy, prominent autonomic dysfunction along with psychosis and visceral symptoms. Other liver conditions that can cause either sensory, motor or sensorimotor neuropathies include vasculitis with hepatitis C and cryoglobulinemia, but also primary biliary cirrhosis and cholestatic liver disease. Navajo neuropathy is a progressive autosomal recessive, severe sensory and motor polyneuropathy associated with progressive liver disease. Fabry disease (α-galactosidase A deficiency) is an X-linked genetic disorder in which young men complain of lower extremity pain and is responsive to enzyme replacement therapy. Several reports noted a relationship between the duration of hypoxia and findings of polyneuropathy in chronic obstructive polyneuropathy. There are few reports of median mononeuropathy associated with obstructive sleep apnea and nocturnal hypoxemia. There has been extensive literature showing the relationship between renal disease and neuropathy at serum creatinine of 5mg/dl or higher. Early hemodialysis and aggressive transplantation has lessened the prevalence of uremic polyneuropathy. In addition to polyneuropathies, ischemic monomelic neuropathies have been reported with renal failure and A-V fistula placement. Polyneuropathy associated with sepsis and multiple organ failure has various proposed mechanisms. Early aggressive management of systemic disease and avoidance of triggers are important in preventing the development of neuropathies in some of these conditions.
WS 8.4.1 / #110BEST OUTCOME MEASURES TO USE FOR NM PATIENTS
Topic: Outcome Measures in Neuromuscular Disorders
Linda Lowes
Clinical Therapies Research, Nationwide Children’s Hospital, Columbus, OH, US
Abstract: not received.
WS 8.4.2 / #111OUTCOME MEASURES IN MUSCULAR DYSTROPHY
Topic: Outcome Measures in Neuromuscular Disorders
Craig Mcdonald
Physical Medicine And Rehabilitation, Center for Healthcare Policy and Research, UC Davis Children’s Hospital, Sacramento, CA, US
Abstract: not received.
WS 8.5.1 / #112GENE-DIRECTED TREATMENT OF MUSCULAR DYSTROPHY
Topic: Treatment of Muscular Dystrophy
Kevin Flanigan
Neurology, The Research Institute at Nationwide Children’s Hospital, Columbus, OH, US
Abstract: not received.
WS 8.5.2 / #160NON-GENE DIRECTED
Topic: Treatment of Muscular Dystrophy
Craig Campbell
Pediatrics (pediatric Neurology), London Health Sciences Centre, London, ON, CA
Abstract: not received.
Poster Sessions
PS1Group2-001 / #409STATIN-INDUCED NECROTIZING AUTOIMMUNE MYOPATHY. RECURRENCE WITH FIBRATE USE
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Mario Fuentealba1, Jorge Bevilacqua2
1Neurology, University of Concepcion, concepcion, CL;
2Departamento De Neurología Y Neurocirugía, Hospital Clinico de la Universidad de Chile, Santiago, CL
Abstract: We report a 65-year-old woman with medical history of diyslipemia, and no family hystory of neuromuscular disease or consanguinity. She was treated during one year with atorvastatine 40 mg/day and presented sub acute onset (1 month) of myalgia, fatigability and weakness in the lower limbs, with difficulties to run and climb stairs. Physical examination demonstrated positive Gowers sign and proximal lower limb weakness (M3+ MRC scale) and proximal upper limb weakness (M4). Electromyography revealed myopathic changes in the lower and upper limbs (small polyphasic and short duration motor units), and resting spontaneous activity (fibrillations, positive sharp waves). The serum creatine kinase (CK) was 10284 IU/Land was the only abnormal finding in the routine laboratory assessment. Para-neoplastic and autoimmunity serologic screening including viral hepatitis B and C and VIH were all normal or negative. A left deltoid muscle biopsy was consistent with a necrotizing myopathy with isolated atrophic fibers and several necrotic fibers without inflammatory infiltrates, and immunohistochemistry ruled out muscular dystrophy. Oral prednisone was initiated and titrated up to a daily dose of 80 mg in June 2012, resulting in a significant reduction of myalgia and weakness. In December 2012 the physical examination revealed normal muscle strength (M5 MRC) and serum CK levels were normal (116 UI/l). During 2013 the corticotherapy was therefore reduced and additional metrotrexate was initiated with persistence of the clinical improvement. In May 2013 the patient stopped the treatment. In November 2013 she remained asymptomatic with normal serum CK. In March 2014, treatment for dyslipemia was re-initiated with oral gemfibrozyl 600 a day mg). After 2 months she presented recurrence of myopathic symptoms and objetve clinical worsening (myalgia and fatigability) and a significant increase of CK level (6500 UI/L) Oral gemfibrozyl was withdrawn and oral prednisone was reestablished with a rapid disappereance of symptoms and normalization of CK ( 168 UI/L) after, 1 month of treatment. Two months later, prednisone was tapered and additional teraphy with azathioprine (100 mg a day) was initiated
PS1Group2-002 / #406STUDY OF HYALURONIDASE-FACILITATED SCIG IN CHRONIC INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHY (CIDP)
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Claudia Sommer1, John D. England2, Johannes Jakobsen3, Russell Reeve4, David Gelmont5
1Neurologische Klinik, Universitätsklinikum Würzburg, Wurzburg, DE;
2Neurology, LSUHSC School of Medicine, New Orleans, LA, US;
3Department Of Neurology, Rigshospitalet, Copenhagen, DK;
4Quintiles Inc, Durham, NC, US;
5Clinical Development, Baxalta US Inc, Westlake Village, CA, US
Abstract: Background: CIDP is an acquired, immune-mediated, progressive or relapsing peripheral neuropathological condition with significant disease burden. Corticosteroids, plasma exchange, and intravenously-administered immunoglobulin (IVIG) are current treatment options with limitations (eg, adverse events; time commitment). IGHy (HYQVIA®) is a subcutaneously-administered immunoglobulin that may be self-administered at rates, volumes, and frequencies similar to IVIG but with better systemic tolerability. The design of a Phase III, prospective, multicenter study for the evaluation of the efficacy, safety, and tolerability of IGHy as maintenance therapy to prevent relapse of neuromuscular disability and impairment in patients with CIDP is presented. Methods: Adults (N=174) with typical or atypical CIDP receiving stable IVIG for ≥3 months prior to screening will be randomized 1:1 to IGHy or placebo with rHuPH20 for 6 months. Treatment will be administered subcutaneously every 2, 3, and 4 weeks at the same monthly immunoglobulin dose (IGHy) (or matching infusion volume for placebo group) as the subject’s pre-enrollment IVIG treatment. Subjects who relapse during SC treatment will be provided IVIG to restore functional ability. The primary efficacy outcome measure is worsening of functional disability at study completion or last study visit, relative to pre-treatment baseline. Secondary/exploratory outcome measures include time to relapse, activities of daily living, hand grip strength, muscle strength, quality of life, health resource utilization, and treatment satisfaction. Results: Non-applicable Conclusions: IGHy may provide an alternative maintenance treatment option enabling self-administration of a full therapeutic dose every 2–4 weeks in patients with CIDP. Enrollment of patients into this Phase III study ongoing
PS1Group2-003 / #405MECHANISM OF HYALURONIDASE-FACILITATED SCIG ALLOWING INVESTIGATIONS IN NEUROMUSCULAR DISEASE
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Christopher J. Rabbat1, Tobin Chettiath2, Martin Noel3, Robert Peterman4, Todd Berner5
1Medical Affairs, Baxalta US Inc, Bannockburn, IL, US;
2Medical Affairs, Baxalta US, Westlake Village, CA, US;
3Medical Affairs, Baxalta Canada, Mississauga, ON, CA;
4Medical Affairs, Baxalta, Vienna, AT;
5Medical Affairs, Baxalta US Inc, Bannockburn, US
Abstract: Intravenously-administered immunoglobulin (IVIG) is used to treat neuromuscular disorders (NMDs), such as multifocal motor neuropathy (MMN), chronic inflammatory demyelinating polyradiculoneuropathy (CIDP), and Guillian-Barré Syndrome (GBS). From the patient’s perspective, IVIG has limitations such as of the risk for systemic adverse reaction [AR], need for venous access, requirement of a healthcare professional to perform the infusion, and time commitments for infusions, impacting the overall patient burden of treatment. Studies found the use of patient self-administered subcutaneous immunoglobulin therapy (SCIG) was as efficacious as IVIG in patients with neuromuscular disorders (NMDs). In spite of the advantages of self-administered SCIG (e.g., autonomy, flexibility to schedule, lower risk for systemic ARs), SCIG is not widely utilized to treat NMDs. While the doses for the treatment of primary immunodeficiency (PIDD) range from 0.3–0.6 g/kg/month, dosing for MMN are much larger (range 0.5–2.4 g/kg/month). The limitation associated with administering larger doses of SCIG per site is related to hyaluronan, which fills the space between the structural components of the extracellular matrix of the subcutaneous (SC) tissue causing resistance to bulk fluid flow restricting the volume of Ig that can be administered in a single site to 20–30 mL. This limitation necessitates multiple infusion sites and an increased frequency of administration in order to achieve therapeutic dosage requirements. For example, an 80 kg patient receiving 1 g/kg of SCIG 20% would require a volume of 400 mL necessitating a patient to endure 16 needle sticks and weekly infusions rather than the monthly frequency of IVIG. This number of needle sticks and frequency of infusions may not be practical or desired by many patients. HYQVIA® (IGHy) is a hyaluronidase-facilitated SCIG that may be self-administered at rates, volumes, and frequencies similar to IVIG. The enzyme rHuPH20 (recombinant human hyaluronidase) acts similarly to naturally occurring hyaluronidase causing local and transient increases in dispersion and absorption of SC-administered fluids and drugs. Volumes up to 600 mL can be delivered into a single SC infusion site. IGHy allows for infusion rates up to 300 mL/hour/site that are approximately 10- to 15-fold higher and volumes/site of 20- to 30-fold larger than conventional SCIG. As a result, many NMD patients could self-administer their full monthly dose of Ig therapy with just 1 or 2 infusion sites. Skin structure for patients with NMD is similar to other populations within which HYQVIA has been extensively studied (e.g., PIDD); therefore, the ability to infuse large volumes of Ig using hyaluronidase facilitation would translate to use in patients with NMD. Hyaluronan is rapidly resynthesized and tissue permeability is completely restored within 24–48 hours. As such, repeated large volume infusions of IGHy have demonstrated no long-term effects on skin integrity. Conclusion: IGHy may present an attractive alternative treatment option for NMDs relative to conventional SCIG, enabling self-administration of a full monthly therapeutic dose in 1 or 2 infusion sites. IGHy is being investigated in patients with CIDP within a phase 3 clinical trial to evaluate the efficacy, safety, and tolerability of IGHy co
PS1Group2-004 / #393RATIONALE FOR TOLL-LIKE RECEPTOR ANTAGONISM AS A POTENTIAL NOVEL THERAPEUTIC APPROACH FOR DERMATOMYOSITIS
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Kirsten Gruis1, Kanneboyina Nagaraju2, Tahseen Mozaffar3, Anthony Amato4, Julie Brevard5, Lindsey Granlund5, Joanna Horobin5
1Senior Medical Director, Idera Pharmaceuticals, Cambridge, MA, US;
2Center For Genetic Medicine Research, Children’s National Medical Center, Washington, DC, US;
3Department Of Neurology, University of California, Irvine, Orange, CA, US;
4Department Of Neurology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA, US;
5Idera Pharmaceuticals, Cambridge, MA, US
Abstract: Background: Dermatomyositis (DM) is a rare idiopathic inflammatory myopathy characterized by an auto-inflammatory immune response in muscle and skin. Toll-like receptors (TLRs) are key components of the innate immune system, and are involved in regulating inflammation and adaptive immunity. In DM, cell death can release damage associated molecular patterns (DAMPs), which activate TLR signaling leading to induction of the inflammatory cascade. Consequently, a self-sustaining autoinflammatory response secondary to persistent TLR activation contributes to chronic inflammation in affected tissues. Targeting TLRs to block inflammation is a novel therapeutic approach for DM that has the potential to reduce chronic inflammation and improve clinical symptoms. Here we propose the therapeutic rationale for TLR antagonism in DM and review the design of a recently initiated Phase 2 clinical trial of the TLR antagonist drug candidate IMO-8400 in DM. Results: Multiple lines of evidence have demonstrated the role of TLRs in the pathogenesis of DM. A retrospective study evaluating muscle biopsy samples showed that TLR9, 4 and 2 were significantly over-expressed in skeletal muscle and infiltrating cells in DM subjects compared to controls. Types I and II interferon and other cytokines were over-expressed. Expression of certain cytokines also had significant positive correlations with expression of TLR9 and 4. In preclinical studies in mouse models of skin inflammation, a TLR antagonist candidate blocked IL-17, IL-6, IFN-γ and IL-1. In a clinical study in patients with psoriasis, an autoimmune disease in which TLRs are implicated, the TLR7, 8 and 9 antagonist drug candidate IMO-8400 was generally well tolerated and demonstrated clinical activity. Conclusions: DM is a severe and chronic inflammatory myopathy. TLRs are key drivers of inflammation and have been shown to be significantly over-expressed in DM muscle. TLR antagonist drug candidates have demonstrated activity in preclinical studies and a proof-of-concept trial in patients with an immune-mediated inflammatory disease. Based on these data, we initiated and are actively enrolling a double-blind, placebo-controlled, 24-week Phase 2 clinical trial of IMO-8400 in DM patients (NCT02612857). Key entry criteria include adults with DM aged 18-75 years, active skin and muscle disease, and abnormal serum CK or ALD, EMG, muscle biopsy or MRI. The primary endpoint is the change from baseline on the Cutaneous Dermatomyositis Disease Area and Severity Index (CDASI). Additional efficacy outcome measures include MMT-8, timed function tests, and exploratory biomarkers of disease activity. References: Rayavarapu S, et al. Idiopathic inflammatory myopathies: pathogenic mechanisms of muscle weakness. Skeletal Muscle. 2013 Jun 7;3(1):13.Kim GT, et al. Expression of TLR2, TLR4, and TLR9 in dermatomyositis and polymyositis. Clin Rheumatol. 2010 Mar;29(3):273-9. Brunn A, et al. Toll-like receptors promote inflammation in idiopathic inflammatory myopathies. J Neuropathol Exp Neurol. 2012 Oct;71(10):855-67. Cappelletti C, et al. Type 1 interferon and Toll-like receptor expression characterizes inflammatory myopathies. Neurology. 2011 Jun 14;76(24):2079-88. Balak, DMW, et al. Results from a randomized, double-blind, placebo-controlled, monotherapy trial of IMO-8400 demonstrate clinical proof-of-concept for Toll-like receptor 7, 8, and 9 antagonism in psoriasis. American Academy of Dermatology Annual Meeting. San Francisco, California. March 2015.
PS1Group2-005 / #391INFLAMMATORY MYOPATHIES: NEEDLE ELECTROMYOGRAPHY CHARACTERISTCS IN A SERIE OF CASES
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Rosana H. Scola, Paulo J. Lorenzoni, Claudia S.k. Kay, Renata D. Ducci, Paula Rodrigues, Lineu C. Werneck
Neuromuscular Service, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR
Abstract: Objective: The aim of our study was analyze the EMG characteristics in a series of patients with Inflammatory Myopathy (IM). Background: Clinical data, laboratorial analysis, electrophysiological studies, MRI and muscle biopsy are crucial in the diagnostic evaluation of IM. Needle electromyography (EMG) can reveals myopathic features (small, polyphasic and short motor unit potentials) and spontaneous (irritative) activity of muscle fibers (active myopathy) which are strongly associated with IM. Methods: We retrospectively analyzed EMG, laboratory, muscle biopsy and clinical data of patients with IM that was confirmed to muscle biopsy performed between 2010 and 2014. Results: We analyzed 67 patients with confirmed IM. EMG showed myopathic findings in 38 patients (56,70%), neurogenic findings in 12 (17,91%), mixed features (myopathic and neurogenic) in 2 (2,98%) and was normal in 15 (22,38%). Biceps brachii and tibialis anterior muscle revealed more abnormal EMG findings than deltoids and vastus lateralis muscles. Fibrillation potential and positive sharp waves were found only in few muscles (34,0% and 19,4%, respectively). Other spontaneous muscle fiber activity (myotonic discharges, neuromyotonia, CRD, Rippling) was found only in 8,95% of the patients. Serum creatine quinase range from normal to 62.3 times increased above normal limit. Previous therapy to IM was done before EMG in 75% of patients who 50% had more than 3 months of therapy. The time of disease therapy before EMG be performed ranged from 15 days to 96 months. Conclusions: Our study showed few myopathic features and spontaneous muscle fiber activity in the EMG of IM. The long time of disease onset as well as IM therapy before EMG was associated with decreased frequency of abnormalities in the EMG.
PS1Group2-006 / #324NECROTIZING MYOPATHY ASSOCIATED TO HIV: CASE REPORT
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Renata D. Ducci1, Francisco B. Magalhães2, Daniel Collares1, Monica M. Gomes-Da-Silva2, Paulo J. Lorenzoni1, Claudia S.K. Kay1, Mauricio Carvalho3, Lineu C. Werneck1, Rosana H. Scola1
1Neuromuscular Service, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR;
2Infectology Service, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR;
3Medicine Interne, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR
Abstract: Introduction: Necrotizing myopathy (NM) is an inflammatory myopathy, which diagnosis is performed by histopathologic analysis, showing myocyte necrosis without significant inflammation. NM can occur after drug abuse, use of toxic agents, due to autoimmune mechanisms and may be associated with neoplasia. NM associated to HIV has rarely been documented. The aim of this report is demonstrate the association between NM and HIV. Case report: A 20-year-old man presented with proximal progressive muscle weakness and intense and diffuse myalgia. On examination: lymphadenopathy, diffuse edema and proximal weakness (grade 3). Laboratorial tests showed: CK 26,167U/L (Normal:30-200U/L), positive HIV serology, CD4 count: 300/UL, viral load: 50,027 copies. Conduction velocity was normal and needle electromyography with myopathic pattern. Muscle biopsy revealed necrotizing myopathy. The diagnosis of NM with HIV was made and management with antiretroviral and immunosuppressive therapy was done. Follow-up after one month showed: CK 139U/L, CD4 count 1,179/UL and viral load was undetectable. Discussion: Myalgia and weakness are common in HIV. The critical test for the diagnosis in this case was the muscle biopsy. Treatment of NM associated to HIV remains empirical with no randomized controlled trial and there is little information about the importance of HIV therapy and about the prognosis.
PS1Group2-007 / #189SRP ANTIBODY ASSOCIATED NECROTIZING MYOPATHY MIMICKED LGMD: A CASE REPORT
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Pariwat Thaisetthawatkul1, Rodney Mccomb2
1Neurological Sciences, University of Nebraska Medical Center, Omaha, NE, US;
2Pathology, University of Nebraska Medical Center, Omaha, NE, US
Abstract: Objective To report a case of signal recognition particle antibody associated necrotizing myopathy (SRPNM) presenting atypically as a slowly progressive myopathy mimicking limb girdle muscular dystrophy(LGMD) Background: SRPNM is an inflammatory myopathy in which the pathological features are myofiber necrosis without endomysial or perimysial inflammation. The typical course is that of subacute or rapidly progressive proximal weakness and atrophy associated with very high CPK. SRPNM presenting as very slowly progressive proximal weakness and atrophy similar to LGMD is rare. Design/methods A case report Results A 42-year-old African American female patient presented with severe progressive proximal weakness and atrophy 11 months before evaluation. Physical exam showed marked proximal weakness and atrophy but she was able to walk without assistance. Her CPK was 963 iu/L. Electrophysiologic studies showed diffuse myopathy with fibrillations. A muscle biopsy showed mild variation of fiber sizes, rare myofiber necrosis and very mild inflammation. She was treated with oral corticosteroid and azathioprine for 4 months but did not improve and her CPK increased to 3703 iu/L. She was suspected to have LGMD and immunotherapy was stopped. She remained stable for 8 years with minimal progression until she developed chest pain, severe truncal and limb weakness and was not able to sit or stand. Her CPK was 2980 iu/L. Another muscle biopsy showed severe chronic myopathy with marked variation in fiber size, severe fiber atrophy and hypertrophy, extensive myofiber necrosis and increased fatty and fibrous infiltration. SRP antibody was positive. She was treated with rituximab and intravenous corticosteroid. Her condition improved significantly. Her CPK came down to 467 iu/L and she walked with assistance. Conclusions SRPNM can present with very slowly progressive chronic myopathy and stepwise progression. A serologic test for myositis specific antibody can lead to correct diagnosis when the clinical course and muscle biopsy findings are atypical.
PS1Group2-008 / #157MYASTHENIA GRAVIS AND POLYMYOSITIS PRESENTED SIMULTANEOUSLY
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.1 Inflammatory / Dysimmune Myopathies
Florentina Berianu
Rheumatology, Mayo clinic, jacksonville, US
Abstract: Background: Myasthenia gravis (MG) and Polymyositis (PM) has been found to be present simultaneously. Objectives: to present two new cases seen in our clinic with concomitant onset of MG and PM. Methods: we reviewed the electronic medical records of the patients diagnosed with MG and PM. Results: Two male patients, 50 and respectively 66 year-old presented to our clinic with significant muscle weakness. Both patients had described dysphagia for solids and liquids and neck weakness. Muscle enzymes were highly elevated in both and EMG showed myopathic features. Patient has limited improvement with pulse therapy with steroids and on further interview we learned that one patient had experienced fatigable muscle weakness involving proximal as well as distal muscle and the other one had experienced diplopia. These features are more typical for MG and the specialized EMG testing were suggestive of MG along with positive anti-ACh antibodies. The muscle biopsy was supportive of polymyositis. Fortunately the patients responded to Prednisone 1 mg /kg, IVIG and acetylcholinesterase inhibitors. Conclusion: There are more than 20 cases described in the literature of simultaneous presentation of MG and PM. Physician should consider this possibility when patients have atypical presentation for PM with distal weakness, muscular fatigability, diplopia and dysphagia or do not respond well to pulse doses steroids. Specialized EMG helps with the diagnosis. References: 1. M. Seton, C.C. Wu, A. Louissaint Jr. Case records of the Massachusetts General Hospital. Case 26-2013. A 46-year-old woman with muscle pain and swelling. N Engl J Med, 369 (2013), pp. 764–773 2. Paik JJ1, Corse AM2, Mammen AL. The co-existence of myasthenia gravis in patients with myositis: a case series. Semin Arthritis Rheum. 2014 Jun;43(6):792-6.
PS1Group2-009 / #362FOLLISTATIN GENE THERAPY IMPROVES SIX MINUTE WALK DISTANCE IN SPORADIC INCLUSION BODY MYOSITIS (SIBM)
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.2 Inclusion Body Myositis
Jerry R. Mendell1, Z Sahenk2, Mark J. Hogan2, Samiah Al-Zaidy3, Kevin Flanigan1, Louise Rodino-Klapac1, Markus Mccolly1, Kathleen Church2, S Lewis2, Linda P. Lowes4, Lindsay N. Alfano4, Katherine M. Berry4, Natalie F. Miller4, Igor Dvorchik5, Melissa Moore-Clingenpeel5, Brian K. Kaspar1
1Neurology, The Research Institute at Nationwide Children’s Hospital, Columbus, US;
2Nationwide Children’s Hospital, Columbus, OH, US;
3Neurology, Nationwide Children’s Hospital, Columbus, OH, US;
4Center For Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, US;
5Biostatistics, Nationwide Children’s Hospital, Columbus, OH, US
Abstract: Sporadic inclusion body myositis (sIBM) is a progressive myopathy generally developing after the age of 50 years. Early atrophy of the quadriceps muscles is a clinical hallmark of the disease, with progressive muscle weakness leading to impaired mobility, increased susceptibility to falls, and loss of independence. The cause of this disease is unknown, and although considered to be an inflammatory myopathy, it demonstrates resistance to anti-inflammatory and immunosuppressive agents. sIBM muscle biopsies show vacuolated muscle fibers, widespread inflammation, and intracellular amyloid deposits. Follistatin is a potent inhibitor of the myostatin pathway and its potential as a therapeutic vehicle is enhanced by a pathway independent of the activin IIB receptor. Safety and efficacy following direct intramuscular injection of follistatin in the quadriceps muscle has been demonstrated in a previously reported gene therapy trial in Becker muscular dystrophy (Mendell JR, et al. Mol Ther 2015). No off target effects of an alternatively spliced follistatin isoform, FS344, were encountered. Enrollment in the current gene therapy trial included 6 subjects with either definite or possible sIBM (Griggs RC, et al. Ann Neurol 1995). MRI guided direct intramuscular injection of AAV1.CMV.FS344 in 12 to 14 sites in the quadriceps muscle delivered a total of 1.2×1012 vg/kg. Deliberate efforts to circumvent fibrotic regions of muscle were improved by ultrasound and EMG. The 6-minute walk test (6MWT) was completed at baseline and every follow up visit up to 2 years post-gene transfer. A concurrent control sIBM group (n=20) was prospectively studied by performance of the 6MWT with follow up from 9-28 months. sIBM patients treated with gene therapy increased the 6MWT distance by 46 meters (457 to 503, p=0.001). Untreated sIBM controls lost 38.5 meters over a similar time period resulting in net difference of 85 meters between groups (p=0.0007). To validate findings, a subgroup of untreated sIBM controls (n=8), matched for age, gender, and 6MWT at baseline was compared. Matched controls lost 39 meters (p=0.0036) in the 6MWT, a virtually identical loss to the larger control group. The results of this study demonstrate that patients with sIBM can benefit from follistatin gene therapy based on improvement in distance walked in the 6MWT. It should also be emphasized that the most robust responses to follistatin were seen in patients who actively exercised during the trial. In this study, gene delivery was limited to the quadriceps muscle, but in future trials more widespread delivery could be more effective.
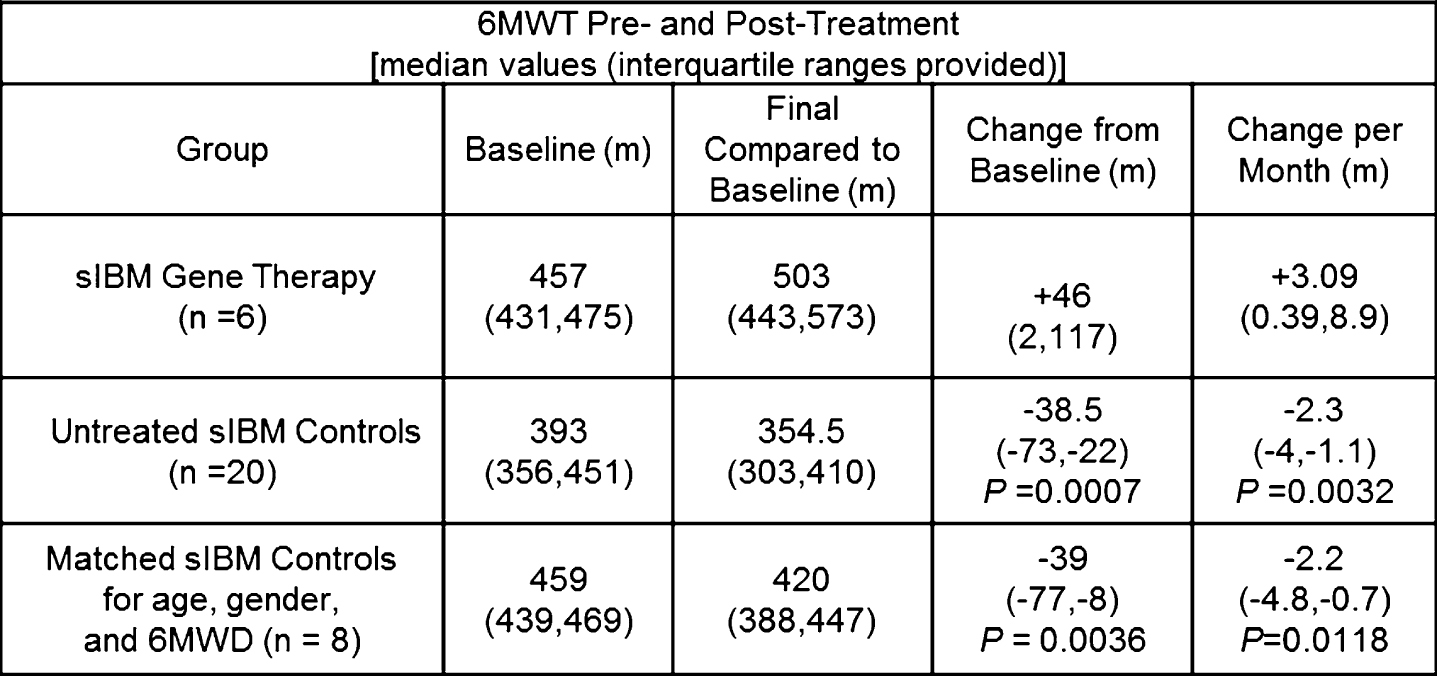
PS1Group2-010 / #305WHOLE-BODY MRI IN AMYOPLASIA CONGENITA
Topic: Group 2 – Acquired Myopathies: Clinical Features, Pathophysiology, Therapy / 2.3 Toxic / Endocrine / Other Acquired Myopathies
Cam-Tu Emilie Nguyen, Sharan Goobie, Craig Campbell
Pediatrics (pediatric Neurology), London Health Sciences Centre, London, ON, CA
Abstract: Background: Amyoplasia congenita is a sporadically occurring condition characterized by congenital underdevelopment of muscles and replacement by fibrous-fatty tissue. The affected children present at birth with arthrogryposis multiplex congenita, markedly decreased muscle bulk and shortened limbs. The pathophysiology has not yet been elucidated, but the leading hypothesis proposes that the developing fetus’ anterior horn cells suffer a vascular insult. There is very little literature describing muscle MRI (magnetic resonance imaging) data on these patients. Objective: To describe the whole-body MRI appearance of amyoplasia congenita. Methods: We present the clinical findings, longitudinal electrophysiological studies and whole-body MRI results of three children with amyoplasia congenita. Results: Three children, two boys and one girl, had classical clinical features of amyoplasia congenita. At birth, they had severely decreased muscle mass, marked limb weakness and hypotonia, and diffuse joint contractures with shoulders in internal rotation, elbows extended, wrists flexed, hips in abduction or adduction, knees in flexion and feet in equinovarus. Nerve conduction studies showed small amplitudes of motor responses and relative preservation of sensory responses in all patients. In two patients, needle electromyography showed large and polyphasic motor units, without any significant spontaneous activity. Electrodiagnostic studies were thus suggestive of a chronic motor neuronopathy. Whole-body MRIs were performed in the neonatal or infantile period and showed generalized atrophy and fatty replacement of both proximal and distal limb muscles, with normal appearance of the axial musculature. Two out of three patients had milder involvement of some muscle groups, including hip extensors, hip adductors, knee flexors and gastrocnemius. One patient had asymmetrical involvement, with smaller muscle bulk of the right thigh and the left forearm when compared to the other side. Nerve biopsy was performed in one patient and showed normal findings. We are currently awaiting the result of whole exome sequencing on these patients and their families. Conclusion: Whole-body MRIs of three patients with amyoplasia congenita confirms the presence of generalized underdevelopment of the appendicular musculature. However, it appears that not all limb muscles are involved to the same degree and that an asymmetrical pattern is possible. Since these children very often require multiple orthopedic surgeries to optimize limb function, these MR images are crucial to identify relatively spared muscles and help plan these complex procedures.
PS1Group4-001 / #461RANDOMIZED, DOUBLE-BLIND, PLACEBO-CONTROLLED STUDY TO INVESTIGATE THE EFFICACY, SAFETY AND TOLERABILITY OF TWO DIFFERENT DOSES OF IGPRO20 (SUBCUTANEOUS IMMUNOGLOBULIN) FOR THE TREATMENT OF CIDP - IGG DEPENDENCY AND RESTABILIZATION PHASE
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Ivo N. Van Schaik1, Vera Bril2, Nan Van Geloven3, Hans-Peter Hartung4, Richard A. Lewis5, G. Sobue6, Billie Durn7, John-Philip Lawo4, Orell Mielke7, David Cornblath8, Ingemar S.j. Merkies9, on On Behalf Of The Path Study Group1
1Neurology, Academic medical Center, University of Amsterdam, Amsterdam, NL;
2University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, CA;
3Department Of Biostatistics And Bioinformatics, Leiden University Medical Center, Leiden, NL;
4Neurology, Heinrich Heine University, Dusseldorf, DE;
5Department Of Neurology, Cedars-Sinai, Los Angeles, CA, US;
6Neurology, Nagoya University Graduate School of Medicine, Nagoya, JP;
7CSL Behring Biotherapies for Life™, Marburg, DE;
8Neurology, Johns Hopkins University School of Medicine, Baltimore, MD, US;
9Neurology, Maastricht University Medical Center, Maastricht, NL
Abstract: Background: Subcutaneous immunoglobulin Ig (SCIg) has gained popularity as an alternative route of Ig administration in CIDP, but has never been rigorously examined for efficacy. Methods: The PATH trial is a randomized, multi-center, double-blind trial investigating two different weekly doses of SCIg IgPro20 versus placebo for maintenance treatment in CIDP. Adult patients with definite or probable CIDP, according to the EFNS/PNS guideline, are eligible. All patients progress through two study periods before randomization. In the first, patients are tested for Ig dependency. Subjects receive non-study IVIg at prescribed dose and subsequent administration is delayed up to maximal 12 weeks until Ig dependency is confirmed. Patients are assessed using three instruments: INCAT every 2 weeks, I-RODS and Grip strength by daily self-assessments and confirmatory visits. Patients who deteriorate by ≥1 point on the INCAT, by ≥4 points on the I-RODS or by ≥8 kPa grip strength in one hand are determined to be Ig dependent and enroll into the IVIg re-stabilization period (10 to 13 weeks). Only patients whose INCAT score improves during IVIg re-stabilization to at least the score at screening and who maintain a stable INCAT during the last 3 weeks of this period are eligible for randomization. This abstract reports on these two pre-randomization periods. Preliminary results: After screening, 248 patients were enrolled. Thirty-one (12,5%) were withdrawn because they were not Ig dependent. 208 patients entered the IVIg re-stabilization phase. Most patients were re-stabilized, 35 have been withdrawn and 5 are still active. Additional details will be presented. Conclusions: The number of subjects not needing IVIg is within the expected range. This study design was safe as deterioration could be sensitively picked up by meaningful measures, and most patients were re-stabilized quickly back to baseline. The study design was beneficial in selecting patients in need of IgG therapy.
PS1Group4-002 / #449SWITCHING PATTERNS IN PATIENTS WITH ICD-9 DIAGNOSED CIDP INITIATING IVIG TREATMENT
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Jeffrey T. Guptil1, Jeffrey A. Allen2, Micheal C. Runken3, Josh M. Noone4, Emily Zacherle5, Chris M. Blanchette5
1Neurology, Duke University, Durham, NC, US;
2Department Of Neurology, University of Minnesota, Minneapolis, MN, US;
3Heor, Grifols SSNA, Raleigh, NC, US;
4The University of North Carolina at Charlotte, Charlotte, NC, US;
5The University of North Carolina at Charlotte, Charlotte, US
Abstract: Introduction: Current medical literature often portrays immunoglobulins (IG) as being clinically interchangeable. However, anecdotal reports from patients, pharmacists, and infusion nurses describe clear differences between infused IG products. Real-world data from large sample populations comparing the different product characteristics of the available intravenous immunoglobulin (IVIG) therapies are limited. This study describes the use of available IVIG products among insured CIDP patients in the United States. Methods: Using 2010-2013 data from the Marketscan US commercial claims database, covering more than 200 million unique patients since 1995, we identified a cohort of treatment naïve chronic inflammatory demyelinating polyneuropathy (CIDP) patients. These patients were required to have at least two ICD-9 diagnosis codes (357.81) for CIDP at least 3 months apart as well as 1 year pre and 2 years post period enrollment to be included in the study. All patients were indexed on their first dose of IVIG therapy and divided into those who switched therapy after the index administration and those who did not. To better understand if comorbidities may affect the choice of IVIG product, Charlson Comorbidity Indices (CCI) were calculated from baseline claims data prior to diagnosis for each treatment group. Results: 151 CIDP patients initiating IVIG treatment were identified in the database. Mean age was 49.2 years with 53% of patients being male. Product specific comparisons of CCI scores for Gammagard and Gamunex were 1.68 and 1.64, respectively, while Privigen, Carimune and Flebogamma scores were all 1.0. Gamunex was initiated most often (37%), followed by Gammagard (33%), Privigen (14%) and Flebogamma (7%). Forty patients (26%) switched therapy during the two year follow-up period. Product-specific switch rates showed Gamunex having a significantly lower proportion (p<0.001) of switchers at 9%, followed by Flebogamma (18%), Privigen (24%), and Gammagard (34%). Additionally, Gamunex along with Privigen had the highest prescription fill rates. Analyses also showed that the majority (54%) of those patients who switched from their index medication, switched to Gamunex during the follow-up period. Conclusion: This real world US payer claims analysis assessing switch rates in naïve IVIG-treated CIDP patients showed that Gamunex had a significantly lower switch rate than all other IVIG products. Further analyses need to be conducted to 1) explore the tolerability and outcomes among the different immunoglobulins; and 2) determine which factors contribute to patients switching IVIG therapies.
PS1Group4-003 / #442SYSTEMIC LUPUS ERYTHEMATOSUS PRESENTING WITH AUTONOMIC AND SOMATIC SMALL FIBER NEUROPATHY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Oscar Trujillo1, Juan Idiaquez2, Ricardo Fadic1
1Departamento De Neurología, Pontificia Universidad Católica de Chile, Santiago, CL;
2Neurología, Universidad de Valparaíso, Santiago, CL
Abstract: Subacute and chronic autonomic dysfunctions, usually subclinical, including sympathetic, parasympathetic and enteric denervation may occur in systemic lupus erythematosus (SLE). Case: 27 years old woman with subacute severe pain and allodynia in both feet. Examination: Bilateral mydriasis. Normal strength and reflexes, sensory: severe allodynia. Tests: Normal motor and sensory nerve conduction. Normal EMG. Autonomic test function: normal blood pressure change on standing. Bilateral pupillary parasympathetic denervation (dilute Pilocarpine 0.005%). Serological tests showed antinuclear antibody 1:1280, positive: Ro/SSA, Anti SM, Anti RNP, low complement. Patient was treated with prednisone and pregabalina. After a month she noticed improvement of pain. This case show neuropathic pain and pupillary dysfunction as a presenting form of SLE.
PS1Group4-004 / #404INCIDENCE OF GUILLAIN-BARRE SYNDROME IN IRANIAN CHILDREN UNDER FIFTEEN YEARS OLD; NATIONAL AFP SURVEILLANCE REPORT (2008-2014).
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Seyed Hassan Tonekaboni1, Habibeh Nejad Biglari2
1Pediatric Neurology, Shahid Beheshti Medical University, Tehran, IR;
2Pediatric Neurology, Kerman Medical University, Tehran, IR
Abstract: The aim of this study was to investigate the incidence of Guillain-Barre syndrome in Iranian children under fifteen years old based on Acute flaccid paralysis (AFP) surveillance program. This program was designed by WHO for countries which are Polio virus free for many years but are located geographically near countries where Poliomyelitis is still endemic like Pakistan and Afghanistan. Iranian Ministry of Health is responsible for performing the task, which consist of detection and report of every case of Acute Flaccid Paralysis (AFP) all around the country. This surveillance system can evaluate the vigilance of our health system to detect any new case of Poliomyelitis. As more than half of our cases had Guillain-Barre syndrome (GBS) and we found this fact an opportunity to find the incidence of GBS in Iranian children. This is a cross-sectional descriptive, analytic study that was performed between 2008 and 2014. The incidence was 1.65 Per 100,000 in 2008, 1.64 Per 100,000 in 2009 , 1.67 Per 100,000 in 2010, 1.72 Per 100,000 in 2011 , 1.93 Per 100,000 in 2012 and 1.68 in 2013 in children under 15 years. There was no seasonal pattern for the illness.
PS1Group4-005 / #386POLYNEUROPATHY IN THE LIMELIGHT: A CASE
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Sandya Tirupathi1, Matthew Sayers1, John Mcconville2, K Pang3, Marie-Louise Kane3
1Paediatric Neurology, Royal Belfast Hospital for Sick Children, BTBA, GB;
2Adult Neurology, Royal Victoria Hospital, BTBA, GB;
3Neurophysiology, Royal Victoria Hospital, BTBA, GB
Abstract: Objective: To discuss the differentials in a case of childhood mononeuritis multiplex Method: We report a case of a 13 year old presenting with parasthesia, and asymmetric weakness in his limbs. Result: A 13 year old boy of Chinese origin presented with a six week history of abnormal sensation in his legs, which progressed to difficulty climbing stairs and bilateral leg weakness. He also developed intermittent finger numbness and weakness of his right hand, lasting for around two weeks before fully resolving. There was associated weight loss and flu like symptoms. There was no past medical history, family history or foreign travel of note. On examination he had reduced power in his right wrist and finger muscles. He had bilaterally reduced power on foot dorsiflexion and eversion, absent knee and ankle jerks and he had a high stepping gait and bilateral foot drop. He had absent vibration, joint position sense and reduced sharp touch sensation on the lateral dorsum of both legs right more than left. Motor nerve conduction studies were suspicious of conduction block in many nerves with focal slowing, with difficult to mark F waves and satisfactory distal motor latencies. Sensory nerve conduction studies showed absent right sural amplitudes and borderline right peroneal amplitudes. There was also a small response on the right ulnar nerve and the median nerve was attenuated on both sides. Lumbar puncture showed an elevated protein 1.48g/dl with normal cell counts. MRI Spine showed mild pial enhancement overlying the conus and also of cauda equina nerve roots with no nerve root thickening or clumping. The child was commenced on IV immunoglobulin 2g/kg over four days, with marked clinical improvement in his clinical condition, particularly in ankle power. After the first course his strength made 50-60% recovery. He received further 2 courses of IVIG with a 6 week interval between them at the end of which he made a full functional recovery. The reflexes never recovered. Repeat nerve conductions 6 months and a year later showed only some improvement. 2½ years later he presents acutely with a partial isolated left 3rd nerve palsy sparing the pupils. Discussion: A wide differential diagnosis was considered, including Mononeuritis Multiplex, Acute/Chronic Inflammatory Polyneuropathy and Hereditory Neuropathy with liability to Pressure Palsies and Neuroborreliosis. His symptoms were asymmetrical and affected multiple nerves, both upper and lower limb. They were very responsive to immunoglobulins. His condition was felt most in keeping with the Lewis Summer Variant of Chronic Inflammatory Polyneuropathy: Multifocal Acquired Demyelinating Sensory and Motor Neuropathy. This usually presents with an asymmetrical involvement of the upper limbs with distal sensorimotor deficit in median or ulnar territories. Cranial nerve involvement is seen in 20% of cases, which has recently been observed in this patient with a 3rd nerve palsy.
PS1Group4-006 / #349VASCULITIC NEUROPATHY COMPLICATED BY ANTERIOR SPINAL ARTERY SYNDROME
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Michael Ackerl, Wolfgang Grisold
Department Of Neurology, Kaiser Franz Josef Hospital, Vienna, AT
Abstract: Anterior spinal artery syndrome as complication of vasculitic neuropathy is rare. The objective is to provide single patient experience in a patient with anterior spinal artery syndrome in vasculitic neuropathy. This work is a single patient observation. Standard methods for blood tests, nerve conduction velocities (NCV), ultrasound, MRI, CSF studies, muscle and nerve biopsy were used. Anti-PR3 ANCA was used for measuring disease activity. A 56-year-old female patient was admitted with a progressive sensorimotor neuropathy. Guillain-Barré syndrome was suspected and IVIG treatment with 0.4g/kg body weight for 5 days was performed. Following a short period of improvement she experienced severe neuropathic pain radiating unilaterally from the brachial plexus and asymmetrically in both legs. Although the rather symmetric motor dysfunction was unusual for vasculitic neuropathy, the neuropathic pain syndromes were suggestive. Anti-PR3 ANCA was highly elevated with 608 U/ml (<5 U/ml). The sural nerve biopsy showed inflammatory lymphocytic infiltrates but did not allow any further classification. Apart from reduced renal function, no other organ involvement could be detected. NCV now showed asymmetric axonal nerve lesions, compatible with a multifocal neuropathy. Ultrasound demonstrated short segmental thickening of several peripheral nerves (median, ulnar, peroneal, tibial and sciatic). Perception of joint movement of the upper and lower extremities was severely reduced, walking felt like “walking on clouds”. Steroid therapy was initiated but the disease progression could not be halted. The patient experienced a painless sigma perforation and hemicolectomy was performed. Prednisolone therapy was drastically reduced to allow wound healing. 4 weeks later she reported dramatic and painful worsening of her condition. She experienced further pain in both hands and legs as well as an autonomic dysregulation with hypotonia. The coordination of both hands fingers was further reduced, lower extremities were paraplegic, she had a possible sensory level at TH6, and light touch on lower extremities had an allodynic spread. MRI studies of the cervical and thoracic spine were repeated which showed a ventral medullar signal alteration from C5 to TH3 resembling the vascular distribution of the spinal anterior artery. In addition to steroids now several cycles of rituximab 375 mg/m2 once weekly were given and further long term treatment with azathioprine was initiated. This therapy had an effect on the anti-PR3 ANCA which was declining (110 U/ml). Still she has remained bedbound with proximal muscle activity but distally absent nerve conduction velocities in upper and lower extremities. This case provides several differential diagnostic difficulties: 1) the rather symmetric onset of an acute neuropathy resembling GBS, 2) the pattern of vasculitis not allowing any further histological classification, 3) the painless perforation of the sigma as possible sign of autonomic involvement and 4) a rare anterior spinal cord infarction in a patient with severe neuropathy, which was difficult to asses, as the vasculitic neuropathy concealed the symptoms of spinal cord infarction. Acute anterior spinal infarction appearing in vasculitic neuropathy is rare and is difficult to detect in a severe case of neuropathy.
PS1Group4-007 / #335CLINICAL AND ELECTROPHYSIOLOGICAL CHARACTERISTICS OF CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY IN KOREA
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Seol-Hee Baek1, Jun-Soon Kim1, Bongje Kim1, So Hyun Ahn2, Kyomin Choi1, Seok-Jin Choi1, Jung-Joon Sung1, Yoon-Ho Hong3
1Department Of Neurology, Seoul National University Hospital, Seoul, KR;
2Neurology, Seoul National University Hospital, Seoul, KR;
3Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, KR
Abstract: Background: Chronic inflammatory demyelinating polyneuropathy (CIDP) is an immune-mediated neuropathy with insidious onset, progressive symmetric or asymmetric polyneuropathy. CIDP has a heterogeneous clinical manifestation, classified into ‘typical’ CIDP and ‘atypical’ subtypes including multifocal acquired demyelinating sensory and motor neuropathy (MADSAM), Distal acquired demyelinating symmetric (DADS) polyneuropathy, and pure sensory or motor CIDP. Objective: The aim of this study is to investigate the frequency of CIDP subtypes in Korea, and to elucidate clinical and electrophysiological characteristics in each subtypes. Methods: We reviewed medical records of 67 patients with CIDP fulfilling EFNS/PNS criteria who visited Seoul National University Hospital or Seoul Metropolitan Government Boramae Medical Center between January 2004 and August 2015. We enrolled patients aged over 18 years and fulfilling EFNS/PNS criteria for definite CIDP. We collected the following data: demographics, clinical data, laboratory data, and electrophysiological data. ‘Typical’ CIDP was defined as symmetric sensory and motor polyneuropathy involving proximal and distal, or proximal part. In contrast, DADS type is restricted to a distal, symmetrical distribution. The diagnosis of MADSAM was defined as asymmetry of symptoms, which was determined as differences in muscle strength by one or more Medical Research Council scale in homonymous muscles. Results: A 67 patients (44 men, 24 women) were classified as having typical CIDP (n=36, 54%), MADSAM (n=14, 21%), DADS (n=12, 18%), pure sensory CIDP (n=4, 6%) and pure motor CIDP (n=1, 1.5%). The age ranged from 21 to 85 years (mean ± standard deviation, 42.75 ± 7.12 years). All patients were initially treated with high-dose prednisolone, intravenous steroid pulse therapy, or intravenous immunoglobulin (IVIG). 38 patients (56.7%) were improved after immunomodulatory therapy, whereas 10 patients (14.9%) were worse even after treatment. Patients with typical CIDP had more frequently ataxia (63.9% vs. 28.6%; p=0.026), whereas patients with MADSAM had more frequently muscle atrophy (27.8% vs. 64.3%; p=0.02). Clinical outcome at the end of follow-up was favorable in 66.7% of patients with typical CIDP, whereas 42.9% of patients with MADSAM. However, there was no significance between two groups (p=0.11). Patients with DADS had more mild disability at the first visit compared with patients with typical CIDP (p=0.018). Clinical outcome at the end of follow-up was favorable in both groups (typical CIDP was 66.7%; DADS was 83.3%; p=0.27). The patients with DADS showed longer distal latency compared with patients with typical CIDP or MADSAM. However, the patterns of distribution of demyelination according to EFNS/PNS electrodiagnostic criteria were no difference among 3 groups. Conclusion: Although typical CIDP is most common phenotypes in Korean patients with CIDP, only 54% of patients with CIDP are belonged to this group. The others are atypical CIDP with heterogeneous features. Most patients with CIDP have favorable outcome. There are some difference of clinical manifestations and prognosis between typical and atypical CIDP. In conclusion, it might be assumed that each subtypes of CIDP have discrepancies in clinical features, electrophysiological findings, and prognosis.
PS1Group4-008 / #308NON-TRAUMATIC PLEXOPATHIES AND RADICULOPATHIES IN CHILDREN
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Cam-Tu Emilie Nguyen1, Craig Campbell1, Hugh Mcmillan2, Chantal Poulin3, Michel Vanasse4, Jiri Vajsar5
1Pediatrics (pediatric Neurology), London Health Sciences Centre, London, ON, CA;
2CHEO, Ottawa, ON, CA;
3McGill University Health Centre, Montreal, QC, CA;
4CHU Sainte-Justine, Montreal, QC, CA;
5Neurology, SickKids, toronto, ON, CA
Abstract: Background: Non-traumatic plexopathies and radiculopathies in children are rare and often puzzling conditions with a myriad of possible causes. There are very few epidemiological studies on the subject, and none that focuses on pediatric non traumatic plexopathies and radiculopathies. Consequently, little is known on the clinical presentation, electrophysiology and epidemiology of pediatric non-traumatic plexopathies or radiculopathies. In addition, most studies do not report genetic testing for conditions such as hereditary neuropathy with liability to pressure palsies (HNPP) or hereditary neuralgic amyotrophy (HNA) which predisposes individuals to developing plexopathies. Objective & Methods: Describe the clinical presentation and course, electrophysiology, investigation, etiology, treatment and epidemiology of the pediatric non-traumatic and non-obstetrical plexopathy and radiculopathy cases seen at 5 Canadian University Hospitals in a retrospective cohort study. Results: So far, we have identified ten pediatric patients with non-traumatic and non-obstetrical plexopathy or radiculopathy. Our cohort is composed of five boys and five girls. The age at onset ranges from 2 to 14 years of age. In all but one patient, negative motor signs and symptoms predominated. Only two patients had prominent sensory symptoms, and only two had prominent pain. Neurological evaluation and electrodiagnostic and imaging studies localized the lesion to the brachial plexus in seven patients, the lumbosacral plexus in two patients and to the right C7 nerve root in one patient. Final diagnoses were as follows: neuritis in four patients, structural in three patients (one with plexiform neurofibroma, one with fibrous band compression and one with osteoid osteoma), genetic in two patients (one with HNPP and one with HNA), and unknown in one patient (who was awaiting biopsy of the plexus). In four patients, a short course of corticosteroids was attempted. This resulted in no change in two patients (brachial neuritis and plexiform neurofibroma), mild improvement in one patient (brachial neuritis) and coincided with marked improvement in the patient with HNPP. At the latest follow-up (data available for seven patients), the only two patients who have shown marked improvement or complete recovery are the patient with HNPP and the patient with HNA. Discussion: Based on the adult literature on non-traumatic plexopathies, we expected that many patients would have a diagnosis of brachial or lumbosacral neuritis. In addition, our cohort, albeit small, highlights that structural and genetic causes should be carefully sought in children presenting with non-traumatic plexopathies or radiculopathies, as these etiologies have crucial treatment and prognostic implications. Interestingly, all four patients who were diagnosed with idiopathic brachial or lumbosacral neuritis had a painless presentation. The only two patients who had pain at presentation were the patient with HNA and the patient with a vertebral osteoid osteoma causing C7 radiculopathy symptoms. Conclusions: In children presenting with non-traumatic plexopathy or radiculopathy, imaging should be performed to look for a possible structural cause. Moreover, a detailed family history should be done and genetic testing for conditions predisposing to plexopathy, such as HNPP and HNA, should be strongly considered.
PS1Group4-009 / #281PERIPHERAL T CELL LYMPHOMA PRESENTING AS MILLER FISHER’S SYNDROME
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
So Hyun Ahn1, Seol-Hee Baek1, Jun-Soon Kim1, Kyomin Choi1, Seok-Jin Choi1, Yoon-Ho Hong2, Jung-Joon Sung3
1Department Of Neurology, Seoul National University Hospital, Seoul, KR;
2Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, KR;
3Neurology, Seoul National University Hospital, Seoul, KR
Abstract: Background: Miller Fisher’s syndrome (MFS) is a rare variant of Guillain-Barre’s syndrome and characterized by the clinical triad of ophthalmoplegia, ataxia, and reduced or absent deep tendon reflexes. MFS generally is self-limited and has an excellent prognosis. Here, we report a patient with Miller Fisher’s syndrome in whom detailed clinical investigations disclosed a peripheral T cell lymphoma. Case: A 67-year-old man presented with diplopia and disequilibrium since 5 days ago. He had no history of vaccination, viral infection, or travel abroad. He was no history of illness except anemia which was diagnosed 3 month ago. A neurologic examination revealed the weakness of lateral rectus on left. In the lower extremities, the proximal muscle strength was MRC Gr. IV+, and the distal muscle strength was MRC Gr. III. Deep tendon reflexes were hypoactive or absent at all joints. He had positive Romberg sign and impaired tandem gait. He had wide base and ataxic gait. The cerebrospinal fluid (CSF) examination showed elevated protein concentration without cell (protein 122mg/dl; WBC 0/μl). The CSF cytology was negative for malignant cells. Serologies for anti-Hu, anti-Yo, anti-Ri, and anti-ganglioside (anti-GM1, anti-GD1b, and anti-GQ1b) antibodies were negative. Nerve conduction studies were suggestive of an early stage of polyneuropathy. On the basis of his clinical, neurological, and electrophysiological findings, he was diagnosed with MFS, and received intravenous immunoglobulin (IVIG) 400mg/kg/day for 5 days. However, his symptoms were not improved after IVIG therapy. Laboratory investigation revealed a pancytopenia (Hb was 9.7, Hct was 27.5%, WBC was 2570 and platelet was 61000), and mild elevated hepatic enzyme (ALT was 65, ALP 231, LDH 429). So, we had detailed investigation for hematologic etiology including malignancy. Computerized tomographic scan of the abdomen and pelvis showed mild splenomegaly, and the chest revealed bilateral axillary, mediastinal lymph nodes of variable size. Position emission tomography scan showed suggestive of hematologic malignancy involving in the whole bone marrow, spleen and bones. Left axillary lymph node was biopsied, and microscopic and immunohistochemical findings were consistent with a diagnosis of peripheral T cell lymphoma. He underwent the standard chemotherapy that included cyclophosphamide, doxorubicin, vincristine, prednisone (CHOP), and intrathecal methotrexate. After chemotherapy, his neurologic symptoms including ophthalmoplegia, weakness, and ataxia were much improved. However, he have not achieved remission state and have still been under treatment. Conclusion: It is extremely rare event that the neuropathy appears the initial symptom of underlying malignant. The neuropathy associated with lymphoma could be occurred by direct infiltration or compression, metabolic and infectious process, vascular impairment, or immunologic/paraneoplastic mechanism. Our patient with peripheral T cell lymphoma initially presented as MFS improved his neurologic symptoms after chemotherapy. Therefore, our case might be supported that MFS could be occurred by the paraneoplastic etiology.
PS1Group4-010 / #219A COMPARATIVE, DOUBLE-BLIND, RANDOMIZED, MULTICENTRE CLINICAL TRIAL TO ACCESS THE EFFICACY AND SAFETY OF CLAIRYG VS TEGELINE IN MAINTENANCE TREATMENT OF CHRONIC INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHY (CIDP)
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Claude Desnuelle1, Jean Pouget2, Jean Christophe Antoine3, Arnaud Lacour4, Jerome De Seze5, Christophe Vial6, Anne-Laure Bedat Millet7, Julien Cassereau8, David Adams9, Guilhem Sole10, Yann Pereon11, Philippe Corcia12, Thibault Moreau13, Steve Genestet14, Rabye Ouaja15, Anne Hufschmitt16, Chrystelle Mercier16
1Neuro-muscular Diseases Department, University Hospital L’Archet, NICE, FR;
2Neuro-muscular Diseases Department, University Hospital La Timone, MARSEILLE, FR;
3Neurology Department, Saint- Etienne University Hospital, ST ETIENNE, FR;
4Neurology Department, Lille University Hospital, LILLE, FR;
5Neurology Department, Strasbourg University Hospital, STRASBOURG, FR;
6Neurology Department, Lyon University Hospital, LYON, FR;
7Neurology Departement, Rouen University Hospital, ROUEN, FR;
8Neurology Department, Angers University Hospital, ANGERS, FR;
9Neurology Department, Le Kremlin-Bicêtre University Hospital, LE KREMLIN BICETRE, FR;
10Neuro-muscular Department, Pellegrin University Hospital, BORDEAUX, FR;
11Neurology Department, Nantes University Hospital, NANTES, FR;
12Neurology Department, Tours University Hospital, TOURS, FR;
13Neurology Department, Dijon University Hospital, DIJON, FR;
14Neurology Department, Brest University Hospital, BREST, FR;
15Global Medical Departement, LFB, LES ULIS, FR;
16Clinical Research And Development, LFB, LES ULIS, FR
Abstract: Introduction The European Federation of Neurological Societies (EFNS)/Peripheral Nerve Society (PNS) guidelines(1) recommend the use of intravenous immunoglobulin (IVIg) or corticosteroids as first line treatment in CIDP. The published clinical trials reporting the efficacy and safety of IVIg in CIDP showed heterogeneity concerning the number of included patients (from 7 to 118), the study population, the primary efficacy endpoint, the duration of exposure to IVIg and IVIg regimen. LFB Biotechnologies has conduct a clinical trial in CIDP comparing a new 5% liquid IVIg (ClairYg®)(2), with Tegeline®(3), another IVIg for which the efficacy in CIDP has already been demonstrated. The results will be published soon. TREATMENTS ClairYg® is the IVIg for wich the efficacy and safety is investigated in CIDP. It is compared to Tegeline®, the French referent IVIg in CIDP (Market authorization in 2009). The characteristics of ClairYg® and Tegeline® are:
• • 5% ready-to-use liquid for ClairYg® and 5% freeze-dried for Tegeline®
• • Highly purified with nanofiltration (20 nm for ClairYg® and 35 nm for Tegeline®)
• • Low levels of anti-A and anti-B haemagglutinins for both (specific step: affinity chromatography for ClairYg®)
• • Low activated factor XI and kallikrein for both (process to eliminate the procoagulant factors approved by French Health Agency for both)
• • All IgG functionalities preserved (ethanolic process for Tegeline® and caprylic process for ClairYg®).
endpoints EFFICACY PRIMARY ENDPOINTS
• • Proportion of patients with no relapse during the 6-month follow-up i.e. whose adjusted INCAT disability score:
∘ remains at the same baseline level or improves or
∘ increases by one point without change in CIDP treatment schedule
EFFICACY SECONDARY ENDPOINTS
• Proportion of patients whose adjusted INCAT disability score remains at the same level or improves during the 6-month follow-up
• Change in the patient’s CIDP treatment schedule, as defined in primary endpoint section
• Mean changes in INCAT disability score
• Mean changes in INCAT Sensory Sumscore (ISS)
• Mean changes in Medical Research Council (MRC)-sumscore
• Mean changes in grip strength with the Martin Vigorimeter in the dominant hand.
safety ENDPOINTs
• Number of treatment-emergent adverse events and the number of patients in both groups
• Changes in vital signs from before infusions to 20 minutes after infusions.
• Changes in laboratory parameters from before to after IMP courses.
conclusion This is the first clinical trial comparing the efficacy and safety of two IVIg in CIDP, ClairYg® to Tegeline®, in patients on maintenance treatment for at least 6 months, and for whom the minimal efficient treatment schedule has been individually determined.

PS1Group4-011 / #208AN INTERNATIONAL, MULTICENTRE, EFFICACY AND SAFETY STUDY OF I10E/ IQYMUNE IN INITIAL AND MAINTENANCE TREATMENT OF PATIENTS WITH CHRONIC INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHY (CIDP)
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Eduardo Nobile-Orazio1, Richard Hughes2, Isabel Illa3, Jean Marc Leger4, S.J.Ingemar Merkies5, Luca Padua6, Rabye Ouaja7, Witold Malyszczak8, Sophie Puget9
1Humanitas Clinical And Research Center, Milan University, MILAN, IT;
2Mrc Centre For Neuromuscular Disease, National Hospital for Neurology and Neurosurgery, LONDON, GB;
3Hospital Sant Pau, Universitat Autònoma Barcelona, BARCELONA, ES;
4National Referral Center For Neuromuscular Diseases, University Hospital Pitié-Salpétrière, Paris, PARIS, FR;
5Maastricht University Medical Center, Maastricht & Spaarne Hospital, Hoofddorg, NL;
6Neurology Departement, Policlinico Universitario Agostino Gemelli, ROMA, IT;
7Global Medical Department, LFB, LES ULIS, FR;
8Clinical Research And Development, LFB, LES ULIS, FR;
9International Scientific Affairs Unit, LFB, LES ULIS, FR
Abstract: INTRODUCTION: The EFNS/PNS Guideline 20101 recommends the use of IVIg (level A recommendation) or corticosteroids (level C recommendation) as first a line treatment in CIDP. For the maintenance treatment, if IVIg is effective in first-line, they can be continued until the maximum benefit has been achieved then the dose should be the lowest effective maintenance dose. LFB BIOTECHNOLOGIES is currently conducting a clinical trial in CIDP with their new 10% liquid IVIg (I10E). The objective of this study is to provide confirmatory data on the efficacy and safety of I10E in the initial and maintenance treatment of CIDP over 21 weeks. An extension study (PRISM-2) is also planned for accessing the efficacy of I10E administered at a reduced maintenance dose in sustaining CIDP response after an initial 6-month treatment.ENDPOINTS EFFICACY ENDPOINTS
• Primary:
Responder rate at End of Study (EOS) visit (responders are defined as patients with a decrease ≥ 1 point in the adjusted INCAT disability score between baseline and EOS visit
• Secondary:
∘ Responder rate at 12 weeks
∘ Time to response
∘ Percentage of patients at 12 weeks and EOS visit with no change in CIDP treatment
∘ Adjusted INCAT disability score
∘ Grip strength with the Martin dynamometer in both hand
∘ Rasch-built Overall Disability Scale (R-ODS)
∘ Patient and Investigator Clinical Global Impression (CGI)
∘ MRC 12 muscles sum score (0 to 5) and Rasch-modified MRC (0 to 3)
EXPLORATORY ENDPOINTS
• Biomarker study:
∘ Anti-contactin 1 and anti-neurofascin 155 antibodies titers
∘ FcγRIIB B cells marker levels
∘ B cell activating factor (BAFF) and complement components (C3 and C4 antigens, CH50)
∘ Serum total IgG trough levels
SAFETY ENDPOINTS
• Treatment-emergent adverse events including Serious Adverse events
• Adverse events temporally associated to infusion
CONCLUSION This prospective open label study plan to assess the efficacy and safety of I10/ IQYMUNE in the initial and maintenance treatment (during 18 months) in patients with CIDP. It will also investigate for the presence of possible biomakers of response to therapy and the utilty of ancillatory investigation such as nerve ultrasonography and electrophysiology to assess response to therapy.

PS1Group4-012 / #207NEUROMUSCULAR COMPLICATIONS ARE NOT RARE IN MIDDLE EAST RESPIRATORY SYNDROME
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Jee-Eun Kim1, Su-Yeon Park2, Jae-Hyeok Heo1, Hye-Ok Kim3, Sook-Hee Song3, Sang-Soon Park1, Tai-Hwan Park1, Jin-Young Ahn1, Min-Ky Kim1, Jae-Phil Choi3
1Department Of Neurology, Seoul Medical Center, Seoul, KR;
2Department Of Neurology, Korea cancer center hospital, Seoul, KR;
3Department Of Internal Medicine, Seoul Medical Center, Seoul, KR
Abstract: Middle East respiratory syndrome (MERS) has a high mortality rate and pandemic potential. However, very little information has become available on this syndrome since it first erupted in 2012. This study aimed to evaluate the incidence of neurological complications and their clinical presentations in MERS. We reviewed the medical records of all patients who were diagnosed with laboratory-confirmed MERS coronavirus (CoV) infections and subsequently admitted to the single main nationally designated MERS treatment center during the 2015 MERS outbreak in the Republic of Korea. 17.4% of the patient reported neurological symptoms during or after MERS-CoV infection. The potential diagnoses for these cases were Bickerstaff’s encephalitis overlapping with Guillain-Barré syndrome, critical illness polyneuropathy or other toxic or infectious neuropathies. Neurological complications did not appear concomitantly with respiratory symptoms, but were instead delayed by 2-3 weeks. Neuromuscular complications were frequent in MERS-CoV-infected patients, and they may have previously been underdiagnosed. Understanding neurological manifestations is important in an infectious disease like MERS, because evaluation is frequently limited during treatment, but it can interfere with prognosis and sometimes require modification of treatment.
PS1Group4-013 / #206NOVEL ANTIGEN-SPECIFIC TREATMENT FOR ANTI-MYELIN-ASSOCIATED GLYCOPROTEIN NEUROPATHY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Ruben Herrendorff1, Pascal Haenggi1, Hélène Pfister1, Andreas Steck2, Beat Ernst1
1Department Of Pharmaceutical Sciences, University of Basel, Basel, CH;
2Department Of Neurology, University Hospital Basel, Basel, CH
Abstract: Anti-myelin-associated glycoprotein (MAG) neuropathy is a disabling antibody-mediated dysimmune peripheral neuropathy. Pathogenic monoclonal IgM autoantibodies are thought to be the culprit of the disease. These autoantibodies recognize the human natural killer-1 (HNK-1) carbohydrate epitope, highly expressed in myelin. The self-attack of the anti-glycan antibodies causes demyelination of peripheral nerves and neuropathy. In clinical studies, it has been shown that the reduction of the anti-MAG IgM titer leads to clinical improvement of patients. Furthermore, the therapeutic response is correlated to the level of the autoantibody reduction. Therefore, agents that selectively block and deplete the pathogenic autoantibodies are of substantial clinical interest for patients suffering from anti-MAG neuropathy. We designed carbohydrate ligands that block the IgM binding sites by mimicking the natural HNK-1 epitope. A mimetic of the glycoepitope, consisting of a sulfated saccharide, was synthesized and multiple copies were linked to a biodegradable poly-L-lysine backbone. The high density multivalent presentation of the mimetic dramatically increased the affinity to pathogenic IgM from patients’ sera. With the glycopolymer, MAG-binding by pathogenic autoantibodies could be inhibited at low nanomolar concentrations in a competitive binding assay. We generated a mouse model for anti-MAG IgM antibodies by immunizing BALB/c mice against the HNK-1 bearing glycolipid sulfoglucuronyl paragloboside. When the mice reached sufficiently high IgM titers, they were treated intravenously with 10mg/kg glycopolymer in saline solution. In this pilot study, the anti-HNK-1/MAG IgM antibody levels were decreased by more than 93% compared to pre-treatment levels, 3 hours after treatment. Even 7 days after the single injection of the glycopolymer in immunized mice, the IgM titers were still substantially decreased. Based on these data, we are currently investigating the in vivo effect of different dose levels as well as multiple dosages. Furthermore, the potential immunogenicity of the compound is studied in mice. Overall, we can conclude that the glycopolymer has the potential to deplete circulating pathogenic antibodies and potentially down-regulate their production without non-selective immunosuppression in anti-MAG neuropathy patients.
PS1Group4-014 / #203A EUROPEAN, RANDOMISED, DOUBLE-BLIND, CROSS-OVER STUDY OF A NEW 10% HUMAN INTRAVENOUS IMMUNOGLOBULIN VERSUS OTHER IVIG IN PATIENTS WITH MULTIFOCAL MOTOR NEUROPATHY - LIME STUDY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Jean Marc Leger1, Richard Hugues2, S.J.Ingemar Merkies3, Eduardo Nobile-Orazio4, Rabye Ouaja5, Witold Malyszczak6, Sophie Puget7
1National Referral Center For Neuromuscular Diseases, University Hospital Pitié-Salpétrière, Paris, PARIS, FR;
2Mrc Centre For Neuromuscular Disease, National Hospital for Neurology and Neurosurgery, LONDON, GB;
3Maastricht University Medical Center, Maastricht & Spaarne Hospital, Hoofddorg, NL;
4Humanitas Clinical And Research Center, Milan University, MILAN, IT;
5Global Medical Department, LFB, LES LUIS, FR;
6Clinical Research And Development, LFB, LES ULIS, FR;
7International Scientific Affairs Unit, LFB, LES LUIS, FR
Abstract: Intravenous immunoglobulin (IVIg) is the standard (Level A for EFNS/PNS Guidelines 20101 and B for American Academy of Neurology 20122). In order to register their new 10% liquid IVIg (I10E) in MMN, LFB BIOTECHNOLOGIES is conducting a clinical trial comparing two IVIg (I10E versus Kiovi) in the maintenance treatment of MMN. METHODS: This study is a phase III, European, multicenter, randomized, double-blind, active-comparator-controlled, cross-over, non-inferiority, efficacy and safety study (NCT01951924)3. I10E and Kiovig® are administered at the dose of 1 g/kg for 1-3 days up to 2g/kg for 2-5 days every 4 to 8 weeks (±7 days), according to the scheme of the standard of care of MMN1 EFFICACY ENDPOINTS:
• Primary: Difference between I10E and Kiovig4 in the mean assessments of MMRC 10E sum score5
• Secondary: Difference between I10E and Kiovig® in :
∘ MMRC 10 new sum score
∘ Rasch built MMRC sum score (based on the 10 muscles in the MMRC sum score)
∘ Grip strength with dynamometer in the most affected hand
EXPLORATORY ENDPOINTS
• Description of differences between I10E and Kiovig® :
∘ Change in MMN-Rasch-built Overall Disability Scale (R-ODS)
∘ Total serum IgG trough levels
∘ Drop in IgM anti-GM1 and anti-GM2 antibodies levels
∘ Change in hand-grip scores in the most affected hand...
SAFETY ENDPOINTS:
• Adverse events (AEs), including serious adverse events (SAEs)
• Clinically significant changes from baseline in vital signs and laboratory tests
CONCLUSION This is the first clinical trial comparing the efficacy and the safety of two IVIg in MMN (I10 versus Kiovig®), in patients on maintenance treatment. The exploratory study aims to address questions on additional outcomes (MMN R-ODS, SES), and the relevance of measuring serum IgG levels and IgM anti-GM1 and anti-GM2 antibody titers in the follow-up of MMN patients.
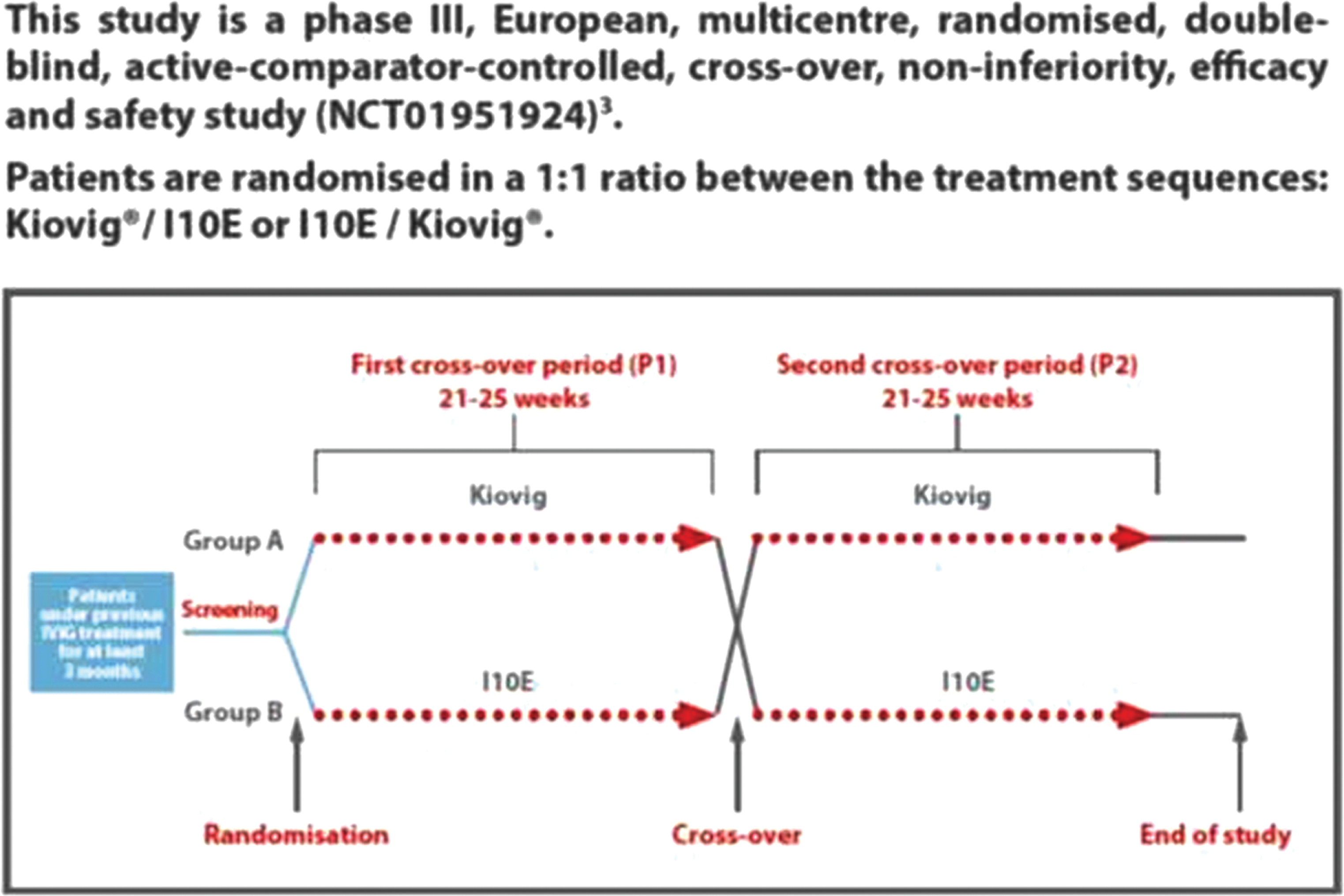
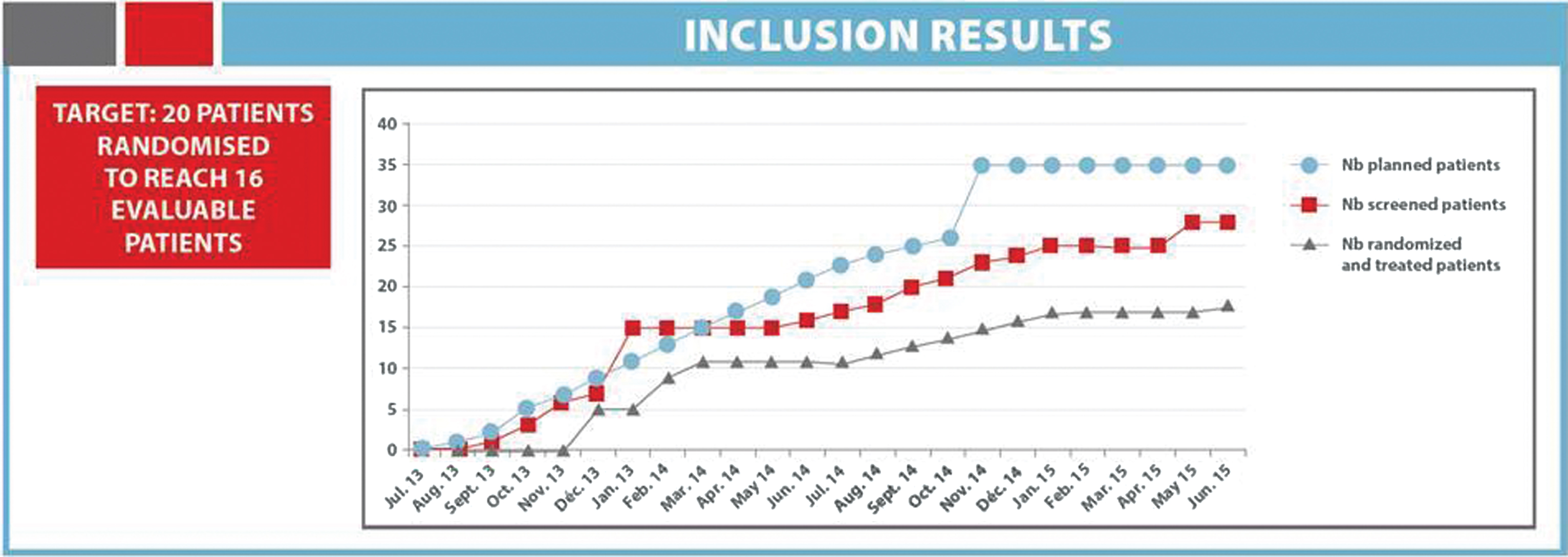
PS1Group4-015 / #200CORRELATION BETWEEN IGM PARAPROTEINEMIA AND MORPHOMETRIC PARAMETERS OF SURAL NERVE IN ANTI-MAG/SGGL NEUROPATHY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Kon Ping Lin1, Hua Chuan Chao2, Cheng Ta Chou2, Yi-Chung Lee1
1Neurology, Taipei Veterans General Hospital, Taipei, TW;
2Neurology, Taipei-Veterans General Hospital, Taipei, TW
Abstract: Objects: To know the correlation between the levels of IgM paraproteinemia/titers of anti-SGGL (sulfoglucuronyl glycolipid) antibody and morphometric parameters of sural nerve in anti-myelin associated glycoprotein (MAG)/SGGL neuropathy Methods: We collected 4 patients with anti-MAG/SGGL neuropathy from archive files. They are 4 men and age from 63 to 77 years old (71±6.24). Two of them showed demyelinating ataxic neuropathies and two mild demyelinating sensory neuropathies. Their terminal latency index (TLI) were median nerve 0.16 to 0.38 (0.28- 0.49) and ulnar nerve 0.23 to 0.34 (0.37-0.66). The immunoglobulin levels of IgM are 462 to 6430 mg/dl (55-300). Electrophoresis (EP) and immunoelectrophoresis (IEP) of serum disclosed IgM paraproteinemia. The anti-SGGL antibody revealed from 1600 to 25600 (<1600) by ELISA method. The western blotting analysis of serum anti-MAG activities was done in patient 1 and 3. The fiber densities of sural nerve were from 3295 to 4877 fibers/mm sq. (4927-9907). The cluster ratio (cluster numbers per 1000 myelinated fibers) was from 0 to 8.03 (<8). The percentage of onion formations were from 1.56% to 3.75% (<0.03%). Results: The levels of IgM paraproteinemia were negative correlation to fiber densities (r = –0.69, P = 0.31) and cluster ratio (r = –0.69, p = 0.0.31) but positive correlation to onion bulb formation (r = 0.96, p = 0.03). The titers of anti-SGGL antibody were no correlation to fiber densities (r = -0.40, p = 0.60) but negative correlation to cluster ratio (r = –0.69, p = 0.31) and positive correlation to onion bulb formation (r = 0.98, p = 0.02). Conclusions: The levels of IgM paraproteinemia evenly affected the severity of the morphometric parameters of histopathological findings in this work, but the titers of anti-SGGL antibody did not have the same even effects on those parameters. The levels of IgM paraproteinemia were the major roles to affect the pathological changes in anti-MAG/SGGL neuropathy.
PS1Group4-016 / #187INFLAMMATORY DIABETIC NEUROPATHY: HELPFUL DIAGNOSTIC PARAMETERS
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.1 Inflammatory / Dysimmune / Associated with Monoclonal Gammopathy/Paraneoplastic
Pariwat Thaisetthawatkul1, J Americo Fernandes, Jr1, Ezequiel Piccione1, Laetitia Truong2, P. James B. Dyck3
1Neurological Sciences, University of Nebraska Medical Center, Omaha, NE, US;
2Creighton University, Creighton University, Omaha, NE, US;
3Neurology, Mayo Clinic, Rochester, MN, US
Abstract: Introduction An inflammatory diabetic neuropathy (IDN) is difficult to diagnose due to overlapping features in clinical presentation, electrodiagnostic studies or blood tests with diabetic sensorimotor neuropathy (DPN). Objectives To determine if IDN can be differentiated from DPN by clinical features, electrodiagnostic studies or laboratory tests. Methods Medical records of patients with IDN and DPN followed up at the University of Nebraska Medical Center between 2004 to 2014 were reviewed. IDN is defined by subacute progressive sensory and/or motor symptoms with electrophysiologic changes of neuropathy in diabetic patients and good response to immunotherapy. Demographic, clinical, electrodiagnostic, laboratory, and nerve biopsy data were extracted. Descriptive and analytical statistics were used. Results 27 patients (16 IDN, 11 DPN) were identified. There were 17 males. Mean age 61 years. Mean duration of diabetes and neurological symptoms were 143 and 26 months respectively. IDN has significantly higher prevalence of motor weakness in the legs and arms, walking difficulty, and electrophysiologic abnormality of demyelination. Absence of peroneal motor, median, ulnar and radial sensory and at least 2 sensory nerve responses in the upper limbs were more frequent in IDN. Sensory nerve biopsies in 10 IND patients showed perivascular inflammatory infiltrates in 7 and decreased fiber density in all (7 generalized, 3 multifocal). Teased fibers showed increased demyelination in 2 and axonal degeneration in 4. All IND improved with immunotherapy. Summary Clinical features, electrodiagnostic abnormalities and response to immunotherapy are the main differentiating features of IDN from DPN. A sensory nerve biopsy can show supporting diagnostic evidence.
PS1Group4-017 / #491EGR2 MUTATION ENHANCE PHENOTYPE SPECTRUM OF DEJERINE-SOTTAS SYNDROME
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Elena Gargaun1, Andreea M. Seferian2, Ruxanda Cardas2, Anne Gaelle Lemoing2, Juliette Nectoux3, Catherine Delanoe4, Gisèle Bonne5, Anne Boland5, Jean-Francois Deleuze6, Cécile Masson7, Laurent Servais1, Teresa Gidaro2
1I-motion, Institute of myology, PARIS, FR;
2Institute of myology, PARIS, FR;
3Molecular Genetics And Biochemistry Unit, HUPC Cochin Hospital, Paris, FR;
4Neurophysiologic Department, Robert Debré Hospital, PARIS, FR;
5Center For Research In Myology, UPMC, PARIS, FR;
6National Center Of Genotyping, Institute of Genomics, Evry, FR;
7Bioinformatics Unit, Necker Hospital, Paris, FR
Abstract: Bulbospinal atrophy is a rare neuromuscular disorder characterized by degeneration of nervous cells localized in the medulla and bulbar region of the spinal cord. It is clinically characterized by facial weakness, swallowing difficulties, drooling and lingual fasciculation. Bulbospinal atrophy has been reported in several syndromes with phenotypic overlap such as Fazio-Londe, Madras or Nathalie syndrome and severe progressive polyneuropathy like Dejerine Sottas syndrome. We report a 5-year old patient who presented with normal motor development until the age of 2, then with progressive proximal, axial and facial weakness leading to complete bulbar palsy and tongue fasciculations. After sequencing SLC52A2, SLC52A3 and SMN1 genes, we performed the whole exome sequencing for our patient and its parents and we found a de novo pathogenic mutation in EGR2 (early growth response 2) gene c.1075C>T, p.Arg359Trp. The knowledge of the progressive bulbar syndromes has significantly increased in recent years due to advances in next generation sequencing. This group of heterogeneous diseases can occur in children or adults and form a spectrum of severity, based around the common phenotype of bulbar and motor deficit. This report underlines EGR2 gene as a potential cause of progressive severe facial weakness with tongue fasciculations.
PS1Group4-018 / #389DEMYELINATING FEATURES IN NEUROPHYSIOLOGICAL STUDY OF TRANSTHYRETIN FAMILIAL AMYLOID POLYNEUROPATHY DUE TO VAL30MET MUTATION IN A PORTUGUESE POPULATION
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Marcio Cardoso1, Ana P. Sousa2, Katia Valdrez3, Teresa Coelho3
1Unidade Corino De Andrade And Serviço De Neurofisiologia, Hospital Santo António, Porto, PT;
2Neurophysiology, Centro Hospitalar do Porto, Porto, PT;
3Unidade Corino De Andrade E Serviço De Neurofisiologia, Hospital Santo António, Porto, PT
Abstract: Introduction Transthyretin Familial Amyloid Polyneuropathy due to Val30Met mutation (TTR-Val30Met-FAP) is almost the only variant found in Portugal, where most of families have early-onset symptoms, although late-onset cases are also commonly seen. Typically, TTR-Val30Met-FAP presents as a length-dependent small-fibre sensory-motor polyneuropathy with autonomic dysfunction. However, many atypical demyelinating features have been described in recent literature. Objective To search for and characterise demyelinating neurophysiological features in TTR-Val30Met-FAP patients followed up in our centre in recent years. Methods We have analysed 2302 nerve conduction studies from 650 TTR-Val30Met-FAP patients, performed from 2006 to 2015, searching for demyelinating features, namely significant motor velocity reduction, distal motor latency increase and minimum F-wave latency increase, excluding common compression sites, in ulnar, median, peroneal and tibial nerves), according to the medical literature. Demyelinating features were adapted to the respective compound motor action potential (CMAP), as recommended by the criteria. Results There was no nerve conduction study with definitive criteria for demyelination. However, we have identified 124 patients (male: 78) with at least one demyelinating feature (one single feature in 77 patients), regardless CMAP value. The median of the disease time-progression, in each study, was 6.3 years. Conclusion Although it is possible do detect demyelinating features in TTR-VAL30MET-FAP patients followed in our centre, no definitive criteria for a demyelinating polyneuropathy could be matched. Those particular demyelinating features that were found could be a consequence of the severity of the axonal polyneuropathy itself, usually seen in TTR-Val30Met-FAP. More studies, including pathologic findings, may provide a better understanding of this phenomenon.
PS1Group4-019 / #434TRANSTHYRETIN-RELATED HEREDITARY AMYLOIDOSIS IN AN ARGENTINE FAMILY WITH TTR TYR114CYS MUTATION
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Marcelo F. Rugiero, Marcelo Chaves, Mariela Bettini, Maria Ines Araoz, Maria S. Saez, Patricia Sorroche, Edgardo Cristiano
Neurology, hospital italiano de buenos aires, buenos aires, AR
Abstract: Transthyretin-related hereditary amyloidosis is an autosomal dominant inherited disease caused for mutations in the transthyretin (TTR) gen. Corresponding to the various transthyretin gene mutations and a wide range of geographical distribution this disorder presents diverse characteristics in genotype-phenotype correlation. Familial Amyloid Polyneuropathy (FAP) is the more common clinical presentation and the Val30Met its more frequent mutation described Objetive/Method: we describe de clinical characteristics of an Argentine family affected by transthyretin-related amyloidosis with TTR Tyr114Cys mutation Methods/Patients: we present a 65-year-old female patient who has a history of 3 years of recurrent episodes of perspiration and dizziness followed by nausea, vomiting and syncope. She also developed distal paraesthesia and progressive autonomic dysfunction: orthostatic hypertension, decreased libido and alternating diarrhea and constipation. She lost weight and had distal dysesthesias and muscle atrophy. Hypoesthesia in stocking-glove distribution and arreflexia was found on physical examination. She did not showed symptoms or signs of CNS involvement. Her oldest sister died for biopsy proven amyloid leptomeningitis and her 52-year-old brother has the similar clinical picture. Amyloid deposition was found in sural nerve biopsy. MRI showed no leptomeningeal lesion, cardiac evaluation and ocular examination were unremarkable TTR gene sequencing showed the Tyr114Cys mutation Conclusion: we present an argentine family with TTR amyloidosis with a Tyr114Cys mutation, which is extremely rare and is the first reported in our country.
PS1Group4-020 / #343NOVEL INF2 GENE MUTATIONS IN CZECH PATIENTS WITH SPORADIC HMSN
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Pavel Seeman1, Petra Lassuthova2, Dana Šafka Brožková2, Jana Neupauerová2, Marcela Krůtová2, Jana Šoukalová2, Zdeněk Kalina2, Dagmar Grečmalová4, Jana Haberlová5
1Dept Of Child Neurology, Dna Laboratory, Charles University in Prague and University Hospital Motol, Praha, CZ;
2Dept Of Child Neurology, Dna Laboratory, Charles University in Prague and University Hospital Motol, Praha, CZ;
3Dept Of Medical Genetics, University Hospital Brno, Brno, CZ;
4Dept Of Medical Genetics, University Hospital Ostrava, Ostrava, CZ;
5Dept Of Child Neurology, Charles University in Prague and University Hospital Motol, Praha, CZ
Abstract: Mutations in the INF2 gene (inverted formin 2) cause about 15 % of autosomal dominant focal segmental glomerulosclerosis (FSGS) and about 75 % of cases with Charcot-Marie-Tooth disease (CMT or HMSN) associated with glomerulopathy. All yet known pathogenic INF2 mutations are clustered in the DID domain coded by exons 2 and 3. Nineteen different mutations (mostly missense) in INF2 were yet reported as the cause of CMT with FSGS. Majority of reported cases seems to be sporadic. We report first three Czech unrelated patients with sporadic hereditary motor and sensory neuropathy who were referred for DNA testing for CMT without the information about their kidney disease and before the INF2 mutations were discovered as the cause of CMT with FSGS. All three patients presented a typical CMT phenotype with a childhood onset of peroneal weakness and foot deformities of variable severity and a diffuse polyneuropathy with signs of axonal loss and slowing of nerve conduction velocity (NCV) compatible with intermediate type of neuropathy. Patient 1, 2 and 3 are now 39, 16 and 25 years old respectively. All were initially tested for several relevant causes of CMT as CMT1A/HNPP rearrangement, MPZ, GJB1, SH3TC2, MFN2, GDAP1 gene by Sanger sequencing. Finally we used massively parallel sequencing (MPS) of a diagnostic gene panel containing all 64 - 93 genes yet associated with hereditary neuropathy including the INF2 gene using the HaloPlex method (Agilent). Three different mutations, all located in exon 2 were detected and confirmed by Sanger sequencing in patients, but not in available parents . One mutation was previously reported (p.Leu128Pro – patient 1) and two are novel (p.Leu78Pro – patient 2 and an in-frame deletion of 4 aminoacid residues p.Lys55_Glu58del – patient 3). De-novo origin was confirmed only for the p.Lys55_Glu58del mutations were both healthy parents were available for testing. In the remaining two mutations the de-novo origin is only assumed from the family history and the mutation was not detected in the only one living or available parent. Both missense mutations affect a conserved aminoacid and have deleterious/disease causing predictions. Only after we detected these mutations in patients, additional targeted questioning about nephropathy revealed that patient 1 underwent a liver transplantation at the age of 27 years after a kidney failure and hypertension crisis two years before and patient 3 is followed for proteinuria with edemas and hypertension suspected from glomerulonefritis. Patient 2 have not responded our report about the mutation finding yet. All these cases show that there is a lack of knowledge about the possible causal association of unexplained CMT and nephropathy. The information about the signs of glomerulopathy or nephropathy could crucially shorten and make effective the diagnostic DNA testing when only 2 exons of the INF2 gene could have been tested instead of a whole panel of nearly 80 genes or more as in our patients. Early clarification of the disease cause is crucial not only for genetic counseling and prevention, but especially for effective early nephrological intervention. Supported by AZV 15-33041A and AZV 16-31173A
PS1Group4-021 / #193PREDICTION OF NERVE CONDUCTION STUDIES OUTCOMES IN PATIENTS WITH FAMILIAL AMYLOIDOTIC POLYNEUROPATHY RECEIVING TAFAMIDIS THERAPY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Sandra Sousa1, Katia Valdrez2, Isabel Fonseca3, Teresa Coelho2
1Neurology, Hospital de Cascais, Cascais, PT;
2Unidade Corino De Andrade E Serviço De Neurofisiologia, Hospital Santo António, Porto, PT;
3Unidade Corino De Andrade, Hospital Santo António, Porto, PT
Abstract: Introduction: The most frequent clinical expression of hereditary transthyretin amyloidosis is a severe progressive sensory, motor and autonomic neuropathy also known as Familial Amyloid Polyneuropathy (FAP). The disease is particularly frequent in certain regions of Portugal where an oral treatment, Tafamidis, became available to treat neuropathy in stage 1 patients (able to walk without assistance) in July 2012. The objective of this study was to determine the evolution of nerve conduction studies after 12 and 24 months of treatment with tafamidis 20mg once daily. Methods: We retrospectively identified all FAP patients treated with tafamidis for 24 months. Patients had nerve conduction studies at baseline and every year. Compound motor action potential (CMAP) amplitudes from unilateral cubital, peroneal and tibial nerves were summated as well as sensory nerve action potential (SNAP) amplitudes from unilateral cubital, sural and superficial peroneal nerves. We considered that a 20% decrease in the summated value corresponded to a meaningful worsening of the nerve conduction studies. The following demographic and clinical variables were recorded: age, gender, time of onset symptoms, time from symptoms onset to baseline, degree of amyloid deposition salivary glands, quantitative sensory testing (QST), heart rate response to deep breathing (HRDB) and sympathetic skin response (SSR). Determinants of NCS evolution were tested with univariate and multivariate regression analysis (95% confidence intervals); p value <0.05 was considered significant. Results: We identified 129 patients (male gender 52,7%, n=68). The mean age of onset symptoms was 37,1±11,83 years (range20-77); 83,7% (n=108) of them considered early onset (< 50 years). The means time from symptoms onset to baseline was 34,50±24,85 months (range 5-192). Baseline NCS were abnormal in 57,4%(n= 74) patients. At 12 months, 39,5%(n=47) and 26,3%(n=34) patients showed worsening of sensory and motor studies, respectively. Predictors of CMAP amplitudes and SNAP amplitudes worsenning were baseline abnormal NCS (OR21,54; p=0,00), baseline sympathetic dysfunction (OR20,1; p=0,00) and baseline autonomic-sensitive-motor polyneuropathy (OR20,9; p=0,00). In multivariate analysis, abnormal baseline NCS is an independent predictor of NCS variation at 12 months (OR16,4; p=0,00). At 24 months, CMAP amplitudes and SNAP amplitudes worsening was detected in 44,2% (N=57) and 47,9% (n=57) patients, respectively. Male gender (OR18,3; p<0,01), abnormal baseline NCS (OR24,9; p=0,01), baseline parasympathetic dysfunction (OR17,6; p=0,01), baseline sympathetic dysfunction (OR23,5; p=0,00) are associated with worsening of CMAP amplitudes. Male gender is associated to worsenning of SNAP amplitudes, (OR31,2; p=0,00). In multivariate regression, male gender is an independent predictor of CMAP amplitudes (OR14,5; p=0,02) and SNAP amplitudes (OR40,2; p=0,01) worsenning; abnormal baseline NCS (OR 23,0;p=0,00) and age of symptoms onset (OR -0,5; p=0,046) are independent predictor of CMAP amplitudes outcomes at 24months. Conclusions: Our results indicate that after 24month treatment CMAP amplitudes and SNAP amplitudes worsenning was present in 44,2% and 47,9% patients, respectively. The independent predictors for these results are baseline abnormal NCS, male gender and early age of symptom onset. These results emphasize the importance of early treatment.
PS1Group4-022 / #280HEREDITARY MOTOR AND SENSORY NEUROPATHY WITH PYRAMIDAL SIGNS CAUSED BY NEFL GENE MUTATION
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Akihiro Hashiguchi1, Akiko Yoshimura2, Yujiro Higuchi2, Tomonori Nakamura1, Junhui Yuan2, Eiji Matsuura3, Hiroshi Takashima3
1Neurology, Neurological Disease Center, Kagoshima University Medical and Dental Hospital, Kagoshima, JP;
2Kagoshima University Graduate School, Kagoshima, JP;
3Neurology And Geriatrics, Kagoshima University Graduate School, Kagoshima city, JP
Abstract: To identify novel mutations causing hereditary motor and sensory neuropathy (HMSN) with pyramidal signs, a variant of Charcot–Marie–Tooth disease (CMT), we screened CMT and related genes in members of a Japanese family affected with HMSN with pyramidal signs. Clinical features, peripheral nerve conduction properties, and magnetic resonance imaging (MRI) findings were examined in 4 patients from a Japanese family to confirm the presence of HMSN with pyramidal signs. We then screened 28 CMT and related genes using a custom microarray chip. Clinical features included mild weakness of distal lower limb muscles, foot deformity, mild sensory loss, and late onset of progressive spastic paraparesis. Electrophysiological studies revealed slower sensory and motor nerve conduction and small sensory and muscle compound action potentials in multiple limb nerves, indicating widespread neuropathy. Electron microscopic analysis showed abnormal mitochondria and mitochondrial accumulation in the neurons and Schwann cells. Brain MRI revealed an abnormally thin corpus callosum. In all 4 patients, microarrays detected a novel heterozygous missense mutation c.1166A>G (p.Y389C), in the gene encoding the light-chain neurofilament protein (NEFL). All members of the Japanese family affected by motor and sensory neuropathy with pyramidal signs revealed the same novel NEFL mutation, indicating that NEFL mutations can result in a HMSN with pyramidal signs phenotype.
PS1Group4-023 / #204ATYPICAL CIDP OR CMT IN THE ELDERLY? A CASE REPORT
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Min-Xia Wang1, Penny Spring2, John Pollard1, Judy Spies1
1Neurology, University of Sydney and RPAH, Sydney, NSW, AU;
2Neurology, Concord Hospital, Concord, AU
Abstract: An 88-year-old Greek Jehovah’s Witness lady presented in November 2015 with a 5-month history of progressive numbness and severe painful paraesthesia in both hands, as well as unexplained falls and increasing gait ataxia. She had a previous history of breast cancer with right lumpectomy, and radiotherapy (2008), followed by right mastectomy and axillary node clearance for local recurrence (2014). Recent breast clinic review had excluded recurrence. Examination revealed generally wasted, severely weak, erythematous hands with finger hyperalgesia (other modalities intact), and to lesser extent feet. There was mild distal upper limb weakness, with 4 +/5 in wrist flexion and all fingers, 4/5 power in hips proximally, with other muscle groups intact. Reflexes were reduced in the upper and absent in the lower limbs, with downgoing plantar responses. Romberg test was positive and there were no cerebellar or cranial nerve signs. Blood investigations demonstrated ANA 1:40 homogeneous, ANCA uninterpretable, CRP 13, mildly impaired renal function. Other autoantibody and blood tests were unremarkable. CSF examination showed an elevated protein of 0.9g/L, no cells. CT chest/abdomen/pelvis and whole body PET scan showed no evidence of tumour recurrence. Nerve conduction studies were consistent with a symmetrical sensorimotor demyelinating polyneuropathy. Sural nerve biopsy showed a hypertrophic neuropathy with marked variability in the extent of onion bulb formation. Some fascicles showed striking onion bulbs surrounding most remaining myelinated fibres, with varying myelin sheath thickness. There were subperineurial, epineurial, and endoneurial perivascular infiltrates composed of mostly CD4 and CD8 positive T lymphocytes, suggesting CIDP.
These pathological and electrophysiological features and the age of presentation strongly suggest CIDP despite the unusual presentation affecting mainly the hands. After sural nerve biopsy, 1mg/kg daily prednisone was commenced with denosumab injections. Initial treatment with pregabalin was minimally effective. With slowly weaning steroids, introduction of azathioprine and physical therapy, by 2 months, she had a definite partial clinical improvement in all deficits and gait, improved muscle bulk and painless hands, and was mobilizing short distances without assistance. This is a case with marked onion bulb formation on biopsy that would be unusual for CIDP, and would favour Charcot-Marie-Tooth disease (CMT). However, the inflammatory infiltrates and her response to immunosuppression would be out of keeping with CMT. Could this be CIDP with an atypical presentation and uncertain disease duration in the elderly, an inflammatory process superimposed on CMT, or another diagnosis? Additional tests have been arranged for CMT/PMP22 genetic analysis, Refsum/phytanate, and paranodal antibodies.
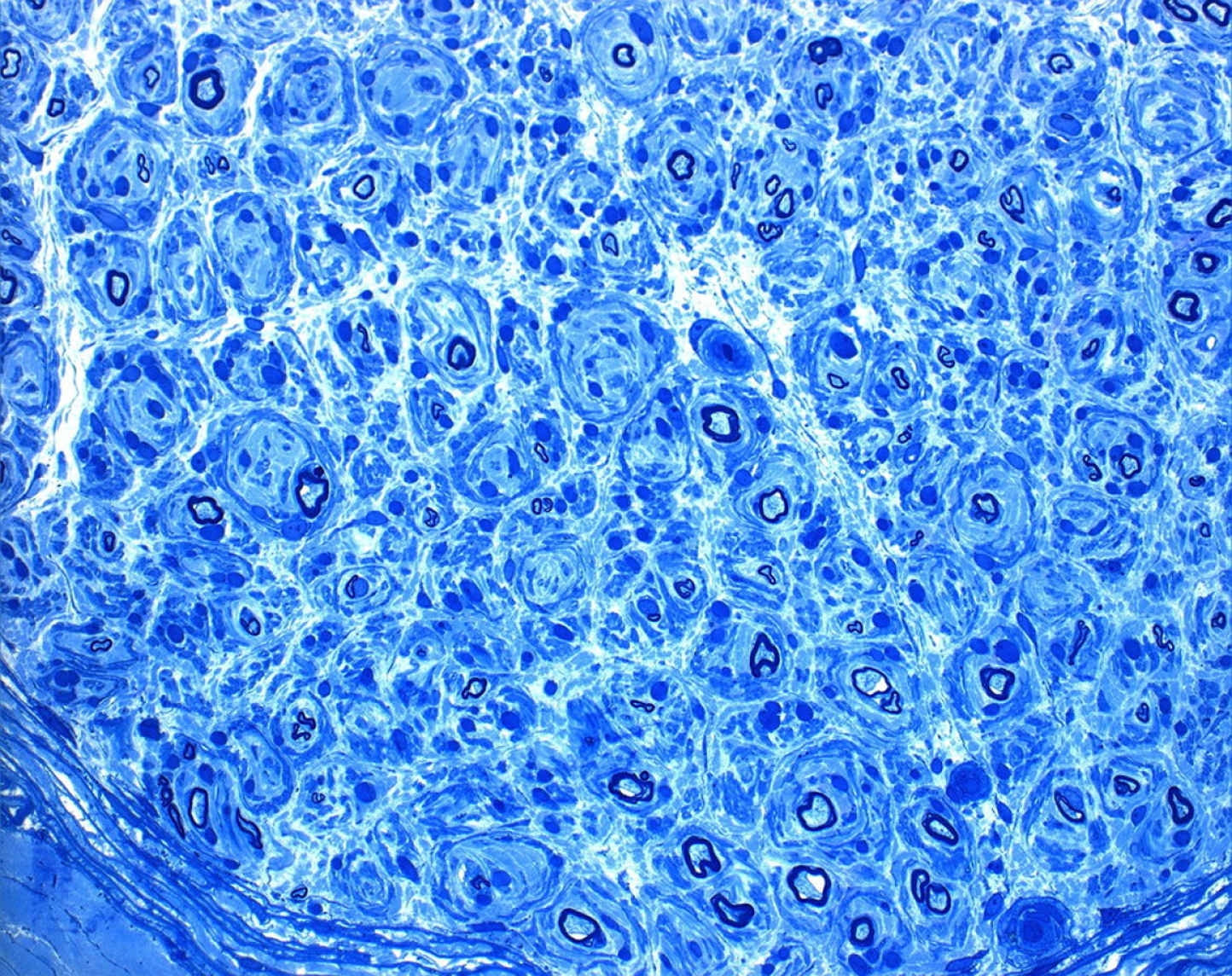
PS1Group4-024 / #273ANTI-GRAVITY AEROBIC TRAINING IN PATIENTS WITH CHARCOT-MARIE-TOOTH DISEASE TYPES 1A AND 1X
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Kirsten L. Knak, Linda K. Andersen, John Vissing
Neurology, Copenhagen Neuromuscular Center, Section 6921nm, Rigshospitalet, Copenhagen, DK
Abstract: Anti-gravity aerobic training in patients with Charcot-Marie-Tooth disease types 1A and X Kirsten Lykke Knak*, PT, MSc, Linda Kahr Andersen*, PT, MSc, John Vissing, MD, PhD Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark *These authors are co-authors on this work. Background Charcot-Marie-Tooth disease (CMT) types 1A and X are neuromuscular diseases, characterized by distal muscle weakness affecting walking ability. There is no cure for CMT, but studies indicate that exercise tends to be safe and beneficial for patients with CMT. However, no studies have investigated the effect of walking on an anti-gravity treadmill. Objective The objective was to investigate if more severely affected patients with CMT 1A and X have beneficial effects of aerobic anti-gravity training. Methods Four adult patients with CMT 1A and X performed a 10-week moderate aerobic training on an anti-gravity treadmill for 30 minutes, 3/week. The study is ongoing, aiming at a sample size of 8 patients according to a priori sample size calculation. The primary outcome is the 6-minute walk test (6MWT), and secondary outcomes are balance (Berg Balance Scale, Biosway), lower extremity muscle strength (Biodex), and fitness. Results Preliminary results show a tendency towards improvement in balance, lower extremity muscle strength, 6MWT and fitness, although findings were not statistically significant. Conclusions Preliminary data show that anti-gravity training tends to improve walking ability, balance, muscle strength and fitness in patients with CMT 1A and X. More patients are needed to conclude whether or not anti-gravity training has an effect in patients with CMT 1A and X.
PS1Group4-025 / #259COEXISTENCE OF CHARCOT MARIE TOOTH DISEASE TYPE 1A AND DIABETES: A CLINICOPATHOLOGICAL STUDY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Hua Chuan Chao1, Kon Ping Lin2
1The Neurological Institute, Taipei Veterans General Hospital, Taipei, TW;
2Neurology, Taipei Veterans General Hospital, Taipei, TW
Abstract: Charcot Marie Tooth disease type 1A (CMT1A) is the most commonly inherited demyelinating polyneuropathy with variable phenotypes, affected by several comorbidities, especially diabetes mellitus (DM). Previous studies showed that DM exacerbates the clinical manifestations of CMT1A. We retrospectively evaluated patients with CMT1A in our hospital, and identified three groups among 12 cases, which comprised four patients with CMT1A, four with CMT1A + DM, and four with DM. We reviewed the CMT neuropathy score (CMTNS), electrophysiological data, and histomorphological parameters of the sural nerve, including fiber density, myelin thickness, axon diameter, g-ratio, regenerative clusters, and regeneration ratio. The CMTNS was significantly higher in patients with CMT1A + DM (21.5 ± 2.52) than in those with CMT1A only (10.8 ± 4.4; p = 0.03). Pathological findings in patients with CMT1A + DM included a significant decrease of myelinated fiber density (p = 0.02) and reduction in the regenerative ratio (p = 0.01), indicating severe degeneration with impaired regeneration. In non-parametric analyses, DM was found to play a more important role than CMT1A in influencing nerve degeneration and regeneration. In patients with CMT1A, DM exacerbated clinical and pathological manifestations including increased loss of myelinated fibers, abnormal axon–myelin interaction, and impaired nerve regeneration.
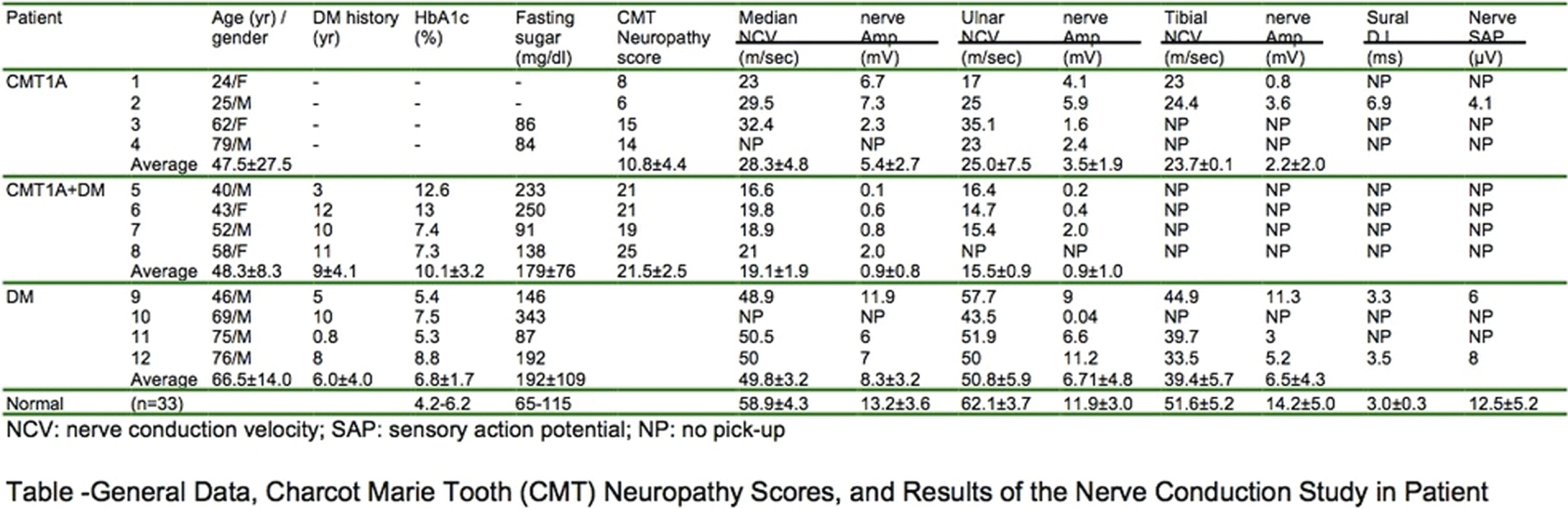
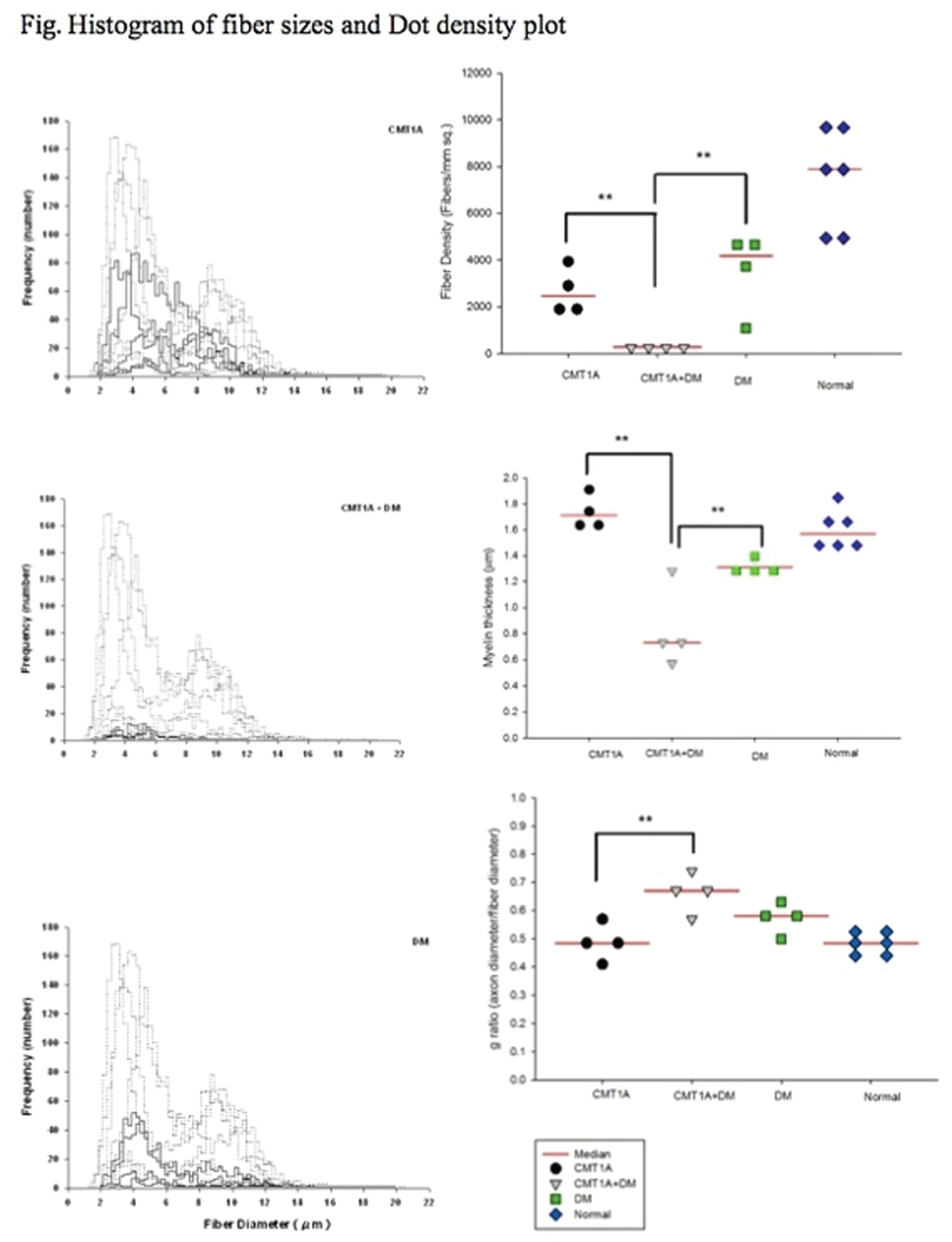
PS1Group4-026 / #215IN VIVO FUNCTIONAL ANALYSIS OF THE NOVEL BSCL2 P.R96H MUTATION RESULTING IN HEREDITARY MOTOR NEUROPATHY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Cheng-Tsung Hsiao1, Pei-Chien Tsai1, Yi-Chu Liao2, Kon Ping Lin2, Yi-Chung Lee1
1Department Of Neurology, Taipei Veterans General Hospital, The Neurological Institute, Taipei, TW;
2Neurology, Taipei Veterans General Hospital, Taipei, TW
Abstract: Background A small group of patients with inherited neuropathy that has been shown to be caused by mutations in the BSCL2 gene. We identified a novel BSCL2 mutation, p.R96H, in a patient with distal hereditary motor neuropathy (dHMN) and a reported BSCL2mutation, p.S90L, in a patient with hereditory motor and sensory neuropathy. The pathogenesis of the p.S90L BSCL2 mutation has had been documented well. However, the cellular effect of the mutation remains elusive. The aim of this study is to investigate the impacts of the p.R96H mutation on the expression, intracellular localization and solubility of seipin protein, and also cellular viability. Method The wild-type (WT) and mutant human seipin (S90L and R96H BSCL2 mutations)-expressing plasmids were constructed and transfected into HEK293 cells, respectively. BSCL2 mRNA and seipin protein expressions were analyzed by real-time PCR and western blot analysis. Seipin protein solubility and stability were also investigated. Immunofluorescence analyses were utilized to characterize the intracellular localization of the WT and mutant seipin. ER stress was evaluated by analyzing the Bip and CHOP mRNA expression. Cellular viability was assessed by Cell counting kit-8 (CCK-8) colorimetric assay. Results The pedigrees of the families carring BSCL2 mutations, and the characteristics of the novel p.R96HBSCL2 mutation were showed.(Figure 1) The BSCL2 p.R96H mutation results in a profoundly decreased soluble level of seipin protein due to decreased BSCL2 mRNA expression and increased propensity to form insoluble aggregates. Immunofluorescence analyses showed that both the R96H and S90L mutant seipin formed cytoplasmic aggregates around cellular nuclei. Expression of the R96H and S90L seipin reduced cellular viability, although ER stress was not observed in HEK293 cells transfected with R96H mutant.(Figure 2)
Conclusion This study demonstrated the cellular deleterious effect of the BSCL2 p.R96H mutation and supports its pathogenic role in inherited neuropathy.
Fig. 1. The pedigrees and sequencing data of the two families carrying BSCL2 mutations, and the characteristics of the novel p. R96H mutation.
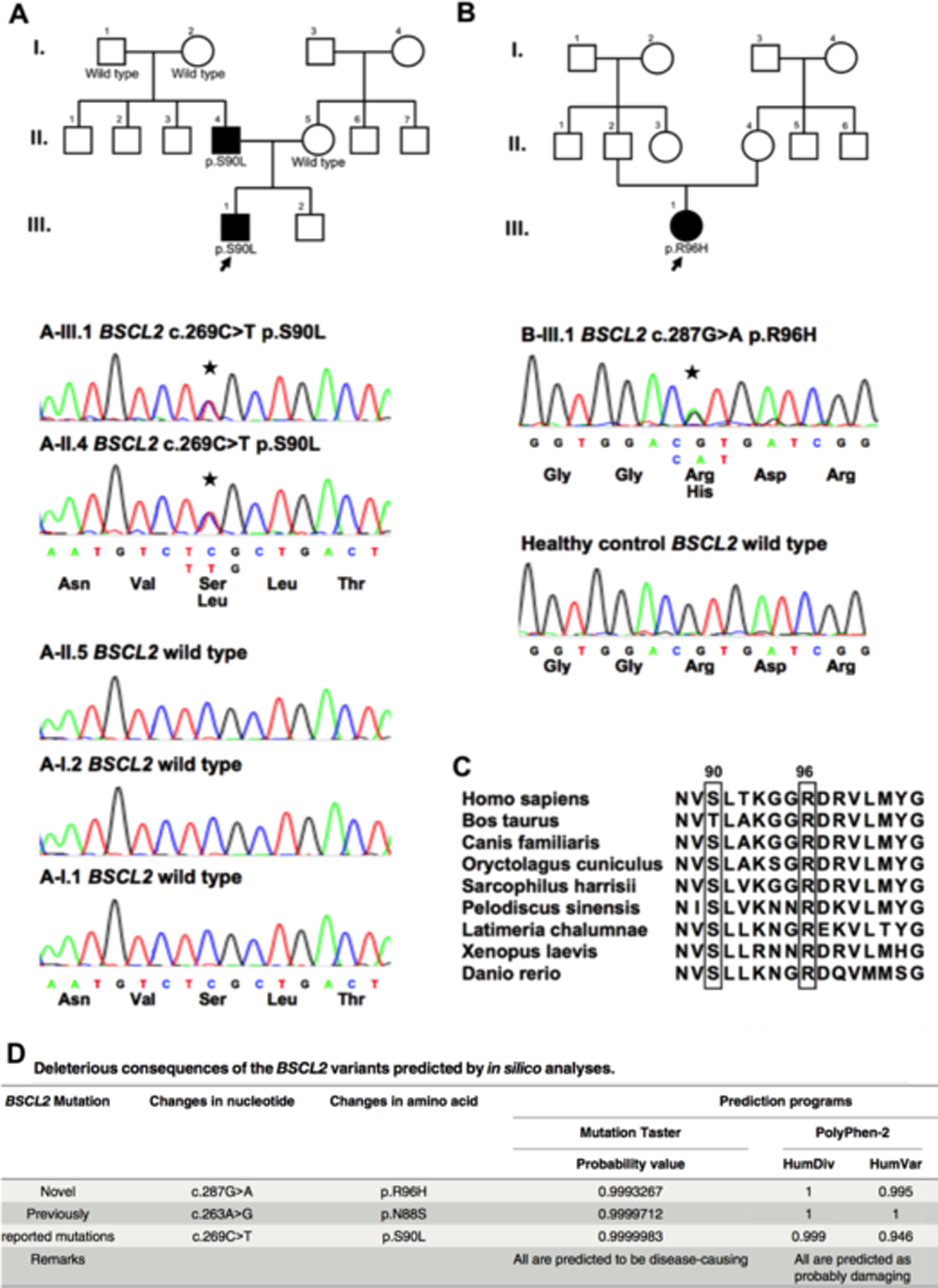
Fig. 2. In vitro expression of BSCL2 and the characterization of wild-type (WT) and mutant seipin proteins in HEK293T cells.
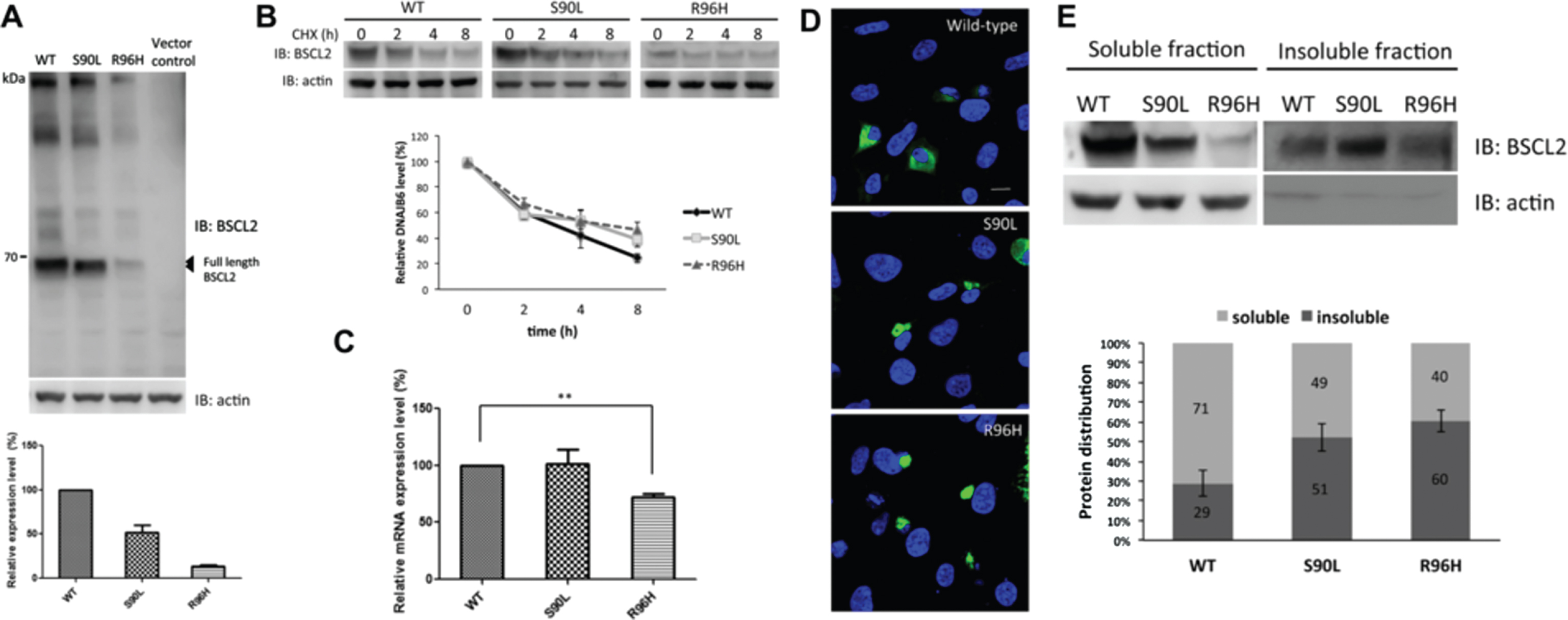
PS1Group4-027 / #142TWO NOVEL DE NOVO GARS MUTATIONS CAUSE EARLY-ONSET AXONAL CHARCOT-MARIE-TOOTH DISEASE
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Yi-Chu Liao, Yo-Tsen Liu, Pei-Chien Tsai, Bing-Wen Soong, Yi-Chung Lee
Neurology, Taipei Veterans General Hospital, Taipei, TW
Abstract: Background Mutations in the GARS gene have been identified in a small number of patients with Charcot-Marie-Tooth disease (CMT) type 2D or distal spinal muscular atrophy type V, for whom disease onset typically occurs during adolescence or young adulthood, initially manifesting as weakness and atrophy of the hand muscles. The role of GARS mutations in patients with inherited neuropathies in Taiwan remains elusive. Methodology and Principal Findings Mutational analyses of the coding regions of GARS were performed using targeted sequencing of 54 patients with molecularly unassigned axonal CMT, who were selected from 340 unrelated CMT patients. Two heterozygous mutations in GARS, p.Asp146Tyr and p.Met238Arg, were identified; one in each patient. Both are novel de novo mutations. The p.Asp146Tyr mutation is associated with a severe infantile-onset neuropathy and the p.Met238Arg mutation results in childhood-onset disability. Conclusion GARS mutations are an uncommon cause of CMT in Taiwan. The p.Asp146Tyr and p.Met238Arg mutations are associated with early-onset axonal CMT. These findings broaden the mutational spectrum of GARS and also highlight the importance of considering GARS mutations as a disease cause in patients with early-onset neuropathies.
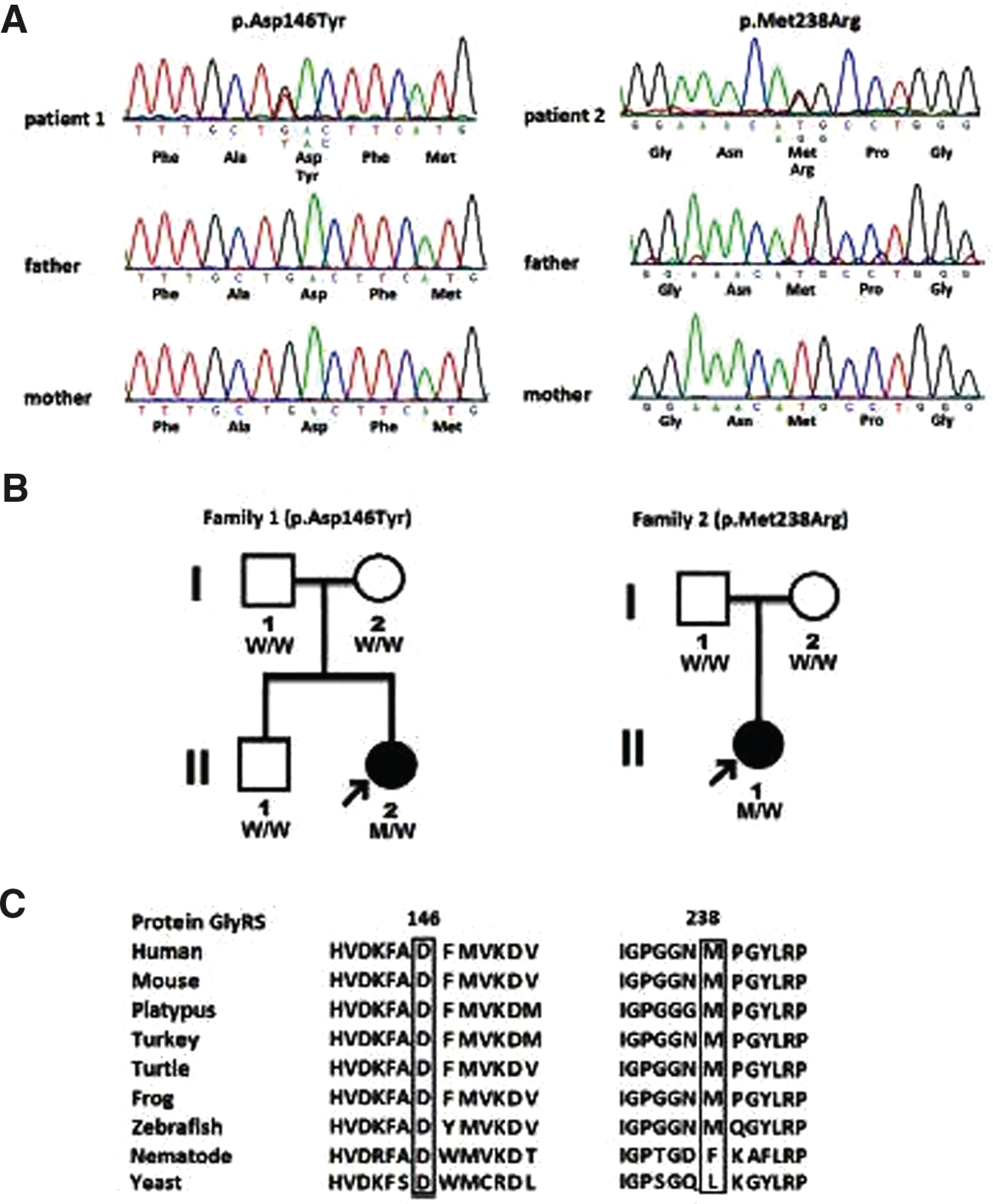
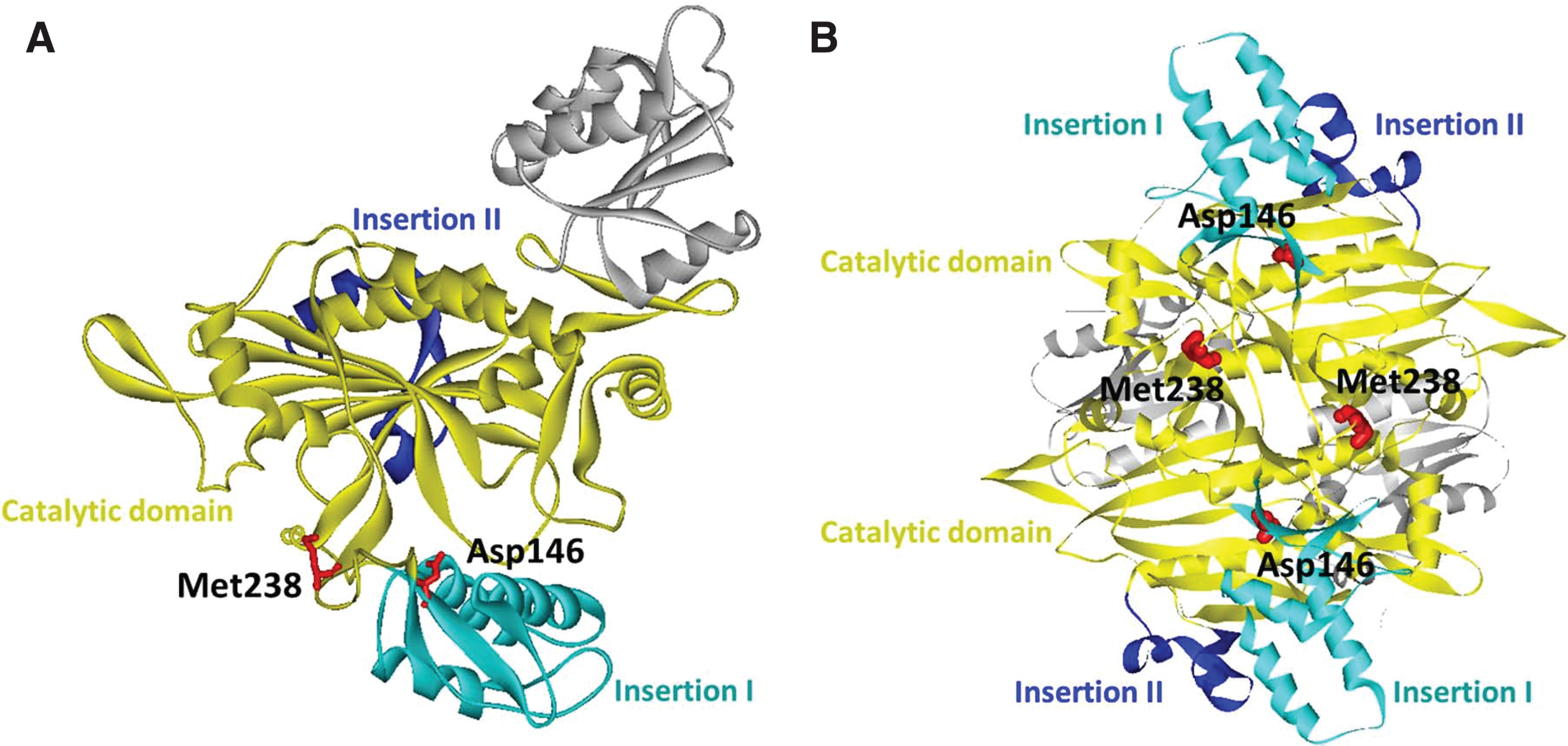
PS1Group4-028 / #141BIOPHYSICAL CHARACTERISTICS AND CLINICAL CORRELATION OF GJB1 MUTATIONS IN CHARCOT-MARIE-TOOTH DISEASE TYPE X1Â
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.2 Hereditary Peripheral Neuropathy
Pei-Chien Tsai, Yi-Chu Liao, Kon Ping Lin, Yo-Tsen Liu, Yi-Chung Lee
Neurology, Taipei Veterans General Hospital, Taipei, TW
Abstract: Objective Charcot-Marie-Tooth disease (CMT) type X1 (CMTX1) caused by mutations in the GJB1 gene is the second most common form of CMT. GJB1 encodes the gap junction (GJ) beta-1 protein (GJB1), which is expressed by Schwann cells in peripheral nerves to form GJs within the myelin sheath. How GJB1 mutants cause neuropathy is still not fully elucidated. The aim of this study was to characterize the biophysical characteristics of the GJB1 mutants and their correlation with the clinical features of patients with CMTX1. Methods All patients with a validated GJB1 mutation were assessed by the Charcot–Marie–Tooth disease neuropathy score version 2 (CMTNS). The impacts of the mutations on the biophysical functions of GJB1 were characterized by intracellular localization, expression pattern and the GJ Ca2+permeability. Results Nineteen GJB1 mutations were identified in 24 patients with the clinical diagnosis of CMT. Six of the mutations are first reported in patients with classic CMT: p.L6S, p.I20F, p.I101Rfs*8, p.F153L, p.R215P and p.D278V. Diverse pathological effects of the mutations were demonstrated, including reduced expression, intracellular mislocalization and altered GJ function. The GJB1 mutations causing failure in GJB1 expression or loss of the GJ permeability were associated with earlier onset and faster progression of the symptoms.Conclusion This study demonstrated that altered expression, abnormal intracellular trafficking and impaired GJ functions were the fundamentally biophysical dysfunctions of GJB1 mutations and the GJ functions might influence the clinical features of CMTX1. Our results are helpful in further understanding of the mechanisms underlying CMTX1.
PS1Group4-029 / #445A NICOTINAMIDE ADENINE NUCLEOTIDE (NAD+) PRECURSOR IS A POTENTIAL THERAPY FOR DIABETIC NEUROPATHY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.3 Metabolic / Toxic
Krish Chandrasekaran, Chen Chen, Avinash R. Sagi, James W. Russell
Neurology, University of Maryland School of Medicine, Baltimore, MD, US
Abstract: Introduction: NAD+ is a critical metabolite in energy metabolism and mitochondrial (Mt) electron transfer. The SIRT1/PGC-1alpha pathway is regulated by NAD+ and is associated with neuronal protection. We tested if treatment with a precursor of NAD+, Nicotinamide Mono Nucleotide (NMN), or increasing the expression of SIRT1 in neurons would reverse diabetic neuropathy in a mouse model of type 2 diabetes mellitus (T2DM). Methods: Adult C57BL6 mice were fed a high fat diet (HFD: Bio Serv, F3282; 36% fat, 60% calories from fat) for two months until they developed neuropathy. Then, 100 mg/kg NMN was administered subcutaneously on alternative days for 2 months (optimal NMN dose based on previous experiments). Control animals were fed a Harlan-Teklad diet (2018; 6.2% fat, 18% calories from fat). Diets had similar protein and carbohydrate contents. At 2 months, blood glucose levels in HFD-fed mice were >200 mg/dL. In a separate experiment, A doxycycline (DOX)-inducible neuron-specific SIRT1 over expression (SIRT1OE) C57BL6 mouse was generated. In SIRT1OE mice, SIRT1 expression was shut off by feeding DOX (200mg/kg) in the diet. Mice were then fed with a HFD for 2 months during which they developed neuropathy. SIRT1 was then activated (by stopping DOX) for a further two months, then neuropathy was measured. Neuropathy endpoints were motor sciatic-fibular nerve conduction velocities (MCV), mechanical allodynia (MA), and intraepidermal nerve fiber density (IENFD). In the dorsal root ganglion neurons (DRG) neurons, NAD+ levels were quantified by HPLC. SIRT1 protein levels and activity were quantified. Mt respiration was measured using the Seahorse XFe24. Results: Both MA and MCV improve in HFD mice with NMN treatment, or when SIRT1 is activated (P<0.001 mice at 4 months compared to the 2 month time point). There was no change in control diet (CD) animals. HFD animals continued to develop neuropathy over the 4 mo. period. At 4 mo., the IENFD was decreased in the HFD but not the NMN or the SIRT1OE group (P<0.001 HFD mice at 4 months vs baseline). NMN treatment and SIRT1OE did not significantly affect glucose, insulin, or lipid measurements in HFD animals. In diabetes, there was a parallel decrease in the NAD+ level, in SIRT1 activity, and in PGC-1 alpha levels in DRG. NMN or SIRT1OE normalized these measurements. Mt respiration spare reserve capacity was reduced by high glucose (approximately 400%). Treatment with NMN or SIRT1 activation preserved the spare reserve capacity under high glucose conditions. SIRT1 inhibition prevented NMN improvement in Mt function Conclusion: (1) Therapies that are precursors for NAD+ can reverse neuropathy in a model of T2DM (2) Importantly, inhibition of the SIRT1-PGC-1alpha pathway blocked NMN mediated improvement in Mt function. Supported in part by NIH NIDDK, VA 101RX001030, Diabetes Action Research and Education Foundation.
PS1Group4-030 / #186FREQUENT LABORATORY TESTS ABNORMALITIES IN PERIPHERAL NEUROPATHY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.3 Metabolic / Toxic
Alon Abraham1, Majed Majed Alabdali2, Abdulla Alsulaiman2, Hana Albulaihe2, Ari Breiner3, Carolon Barnett3, Hans Katzberg3, Danah Aljaafari2, Leif Lovblom2, Bruce Perkins2, Vera Bril2
1Neurology, Toronto General Hospital, Toronto, AB, CA;
2Toronto General Hospital, Toronto, CA;
3Neurology, Toronto General Hospital, Toronto, CA
Abstract: Introduction: Laboratory tests are commonly performed for patients with polyneuropathy, as screening tests for specific etiologies. Objectives: Explore the frequency and relevance of various laboratory tests abnormalities in common types of peripheral neuropathy. Methods: Patients with polyneuropathy, attending the neuromuscular clinic from 01/2013 to 09/2015 were evaluated. Clinical, electrophysiological and laboratory tests results were extracted from patients’ charts. Results: Abnormal laboratory tests results were found in 91% of the total cohort, with an average of 2.3 abnormal tests results per patient. Abnormal glucose handling tests were the most common, followed by paraproteinemia, anemia, and various additional hematological, biochemical, immunological and endocrinal abnormalities. Discussion: The vast majority of patients with peripheral neuropathy have laboratory test abnormalities. Although only a limited number of laboratory tests is relevant for the common types of peripheral neuropathy, broader screening might be warranted due to the high rate of comorbidities. The frequency of paraproteinemia was significantly higher in our cohort than previously reported, and the implication of this finding needs to be explored in future studies.
PS1Group4-031 / #327CLINICAL AND LABORATORY FEATURES OF SMALL FIBER NEUROPATHIES (SFN) WITH IGM VS TS-HDS
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
1Neurology, Saint Louis University, Saint Louis, US;
2Neurology, Saint Louis University, Saint Louis, MO, US
Abstract: Objective: Four case studies describing the characteristics of Small fiber neuropathy (SFN) associated with IgM vs TS-HDS. Background: SFN is a type of peripheral neuropathy that selectively affects the axons of small and poorly myelinated/unmyelinated fibers. Serum IgM binding to IdoA2S-GlcNS-6S, a trisulfated heparin disaccharide (TS-HDS) [normal <10,000], is associated with sensory-motor polyneuropathies. This antibody is associated with impairment of vascular endothelial growth factor with findings of capillary pathology such as thickening walls, circumferential enlargement and C5b9 complement deposits in the basal lamina. Methods: Case studies. Results: 1)16 year old healthy male presented with several years of diffuse intermittent paresthesia. Symptoms began in the left upper extremity which became generalized in few months. Exam showed decreased sensation to pin-prick on distal lower extremities in a symmetric stocking-glove distribution. NCS/EMG was unremarkable. IgM vs TS-HDS was upper normal limit at 16,000. Skin biopsy of the left thigh and calf showed low INFD with normal sweat gland nerve fiber density (SGNFD). 2)16 year old healthy male presented with acute onset of diffuse pain. Symptoms began as viral-like syndrome with bilateral lower extremity pain. This progressed to generalized pain, dry eyes/mouth and urinary urgency in few weeks. Exam showed decreased sensation to pain and temperature distally, and widespread areas of dysesthesia and allodynia from neck down. CK and Aldolase were elevated. IgM vs TS-HDS was abnormally elevated at 23,000. MR L-spine/thighs and NCS/EMG were unremarkable. Skin biopsy of the right thigh and calf showed low INFD with normal SGNFD. 3)15 year old healthy female presented with sharp abdominal pain one month after contracting infectious mononucleosis. This progressed to headaches and intermittent numbness/tingling on her bilateral distal extremities as well as autonomic instability in one month. Exam showed decreased sensation to light touch in patchy distribution all over her body. IgM vs TS-HDS was abnormally elevated at 18,000. MR T/L spines and NCS/EMG were unremarkable. Skin biopsy of the left thigh and calf showed low INFD with normal SGNFD. 4)16 year old female presented with back pain, leg pain/paresthesia as well dry eyes and lightheadedness. On exam, sensation was decreased to pain and temperature distally. IgM vs TS-HDS was abnormally elevated at 26,000. NCS/EMG was unremarkable. MR T/L spines were unremarkable. Skin biopsy of the left thigh and calf showed low INFD with normal SGNFD. Conclusions: All four patients presented with acute onset of progressive painful neuropathy which was confirmed to be SFN with abnormally low INFD. Among them, noticeable characteristics include non-length dependency (Case 1), high CK (Case 2), onset in one extremity (Cases 1 and 4), 50% male (Case 1 and 2) and one preceding event (Case 3). Despite there were autonomic dysfunction in the majority (Case 2,3 and 4), SGNFD was normal in all cases. IgM vs TS-HDS may enhance diagnostic yield for acute onset of SFN. Quantification of INFD seems to be correlated with intensity of the pain. Larger sample is needed to conclude clinical manifestation of the symptoms in relation to quantitative titers of IgM vs TS-HDS.
PS1Group4-032 / #462AN INTERESTING CASE OF SCIATIC NEUROPATHY.
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
Jason Lazarou
Neurology, University of Toronto, Toronto, ON, CA
Abstract: A 68-year-old man with a past medical history of type 2 diabetes presented with severe pain, rapidly progressive weakness and numbness in the left leg. Three months after the onset of symptoms power was zero at the ankle and knee flexion was weak consistent with a severe sciatic neuropathy. Nerve conduction studies confirmed the presence of a very severe sciatic neuropathy as well as a polyneuropathy thought likely to be related to his diabetes. MRI neuropathy revealed an enlarged and enhancing sciatic nerve that was thought to represent isolated inflammatory neuritis. Lymphoma was considered less likely. CT imaging of the chest, abdomen and pelvis was negative. CSF analysis revealed a mild lymphocytic pleocytosis and flow cytometry was did not reveal evidence of a lymphoproliferative disorder. The patient was treated with prednisone that resulted in a rapid improvement in pain, but little clinical or electrodiagnostic improvement. A sural nerve biopsy was obtained and revealed a non-specific axonal neuropathy. One and a half years after initial presentation he developed an acutely edematous right leg. CT imaging revealed massive lymphadenopathy within the abdomen and pelvis and masses within the liver and pancreas. The findings were considered highly suggestive of lymphoma. A percutaneous biopsy was organized and is pending. This case represents a rare but serious cause of sciatic neuropathy: neurolymphomatosis.
PS1Group4-033 / #394SUBACUTE BRACHIAL PLEXOPATHY ASSOCIATED WITH CYSTIC SUBCORACOID BURSITIS
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
Suk-Won Ahn1, Dae-Woong Kang2, Myung-Jin Kim3, Jung-Joon Sung4, Yoon-Ho Hong5, Chang-Seop Kim6
1Neurology, Chung-Ang University Hospital, seoul, KR;
2Chung-Ang University Hospital, Seoul, KR;
3Chung-Ang University Hospiatl, seoul, KR;
4Department Of Neurology, Seoul National University Hospital, Seoul, KR;
5Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, KR;
6Chung-Ang-University Hospital, seoul, KR
Abstract: Case: A 65-year-old female presented with weakness of the right arm and hand a few days ago. Neurological examination revealed weakness of shoulder abduction and arm extension, and wrist and finger drop. Also sensory loss was noted in the lateral arm, posterior arm and radial dorsal hand with radiating pain on right scapular. Nerve conduction study revealed brachial plexopathy involving posterior cord. Shoulder magnetic resonance imaging showed 2x3.4cm sized cystic subcoracoid bursitis adjacent to axillary artery and brachial plexus. Conclusively, the patient’s brachial plexopathy involving posterior cord was resulted from subacute entrapment by the cystic subcoracoid bursitis.
PS1Group4-034 / #383A CASE OF NEUROMYOTONIA ASSOCIATED WITH A CHRONIC POLYRADICULONEUROPATHY
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
Anna Paula P.M. Covaleski1, Vanessa V.D.L. Mota1, Otávio G. Lins2, Wilson Marques3
1Corpo Clínico, AACD - Recife, Recife, BR;
2Neurofisiologia, Hospital das Clínicas da UFPE, Recife, BR;
3Neuromuscular, Hospital das Clinicas da FMRP-USP, Ribeirão Preto, BR
Abstract: Introduction Neuromyotonia is a rare disease characterized by spontaneous and continuous muscle activity due to a disorder of the peripheral nerve hyperexcitability. Patients develop persistent muscle contraction, classically worsening after exercise. Electromyography is characterized by neuromyotonic discharges and about 40% of patients have detectable voltage-gated potassium-channel (VGKC) antibodies. The association with autoimmune diseases and neoplasms can be observed. Initial treatment is symptomatic, but immune therapy is often needed. Objective To describe the case of a patient with Neuromyotonia associated with Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP) and review the literature. Case report A 8-year-old boy, from nonconsanguineos parents, with no significant family history, presented with muscle stiffness, repeated falls and hyperhidrosis. Physical examination revealed distal muscle weakness, a myotonic-like slow relaxation of muscles after voluntary contraction, myokymias and abolished tendon reflex response. Electroneuromyography revealed a chronic demyelinating polyradiculoneuropathy and needle electromyography showed neuromyotonic discharges. A diagnostic search for cancer was negative. Laboratorial tests to diagnose autoimmune disorders revealed only increased serum thyroperoxidase antibodies level, with normal thyroid function Tests. Treatment was initiated with carbamazepine with good response. Because the patient developed Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS) syndrome , withdrawal of carbamazepine was necessary with later introduction of intravenous immunoglobulin, with no improvement of muscle stiffness and weakness. The next drugs tried were sodium valproate and acetazolamide, with no response, and currently mexiletine with partial response. Discussion Neuromyotonia is a treatable condition. The diagnosis is accomplished primarily through clinical and electrophysiological findings. The association with autoimmune diseases can be observed; however, the association of Neuromyotonia with CIDP is rarely reported in the literature. Initial treatment is symptomatic, but immune therapy is often needed.
PS1Group4-035 / #372QUALITY OF LIFE IN PATIENTS WITH DIABETIC PERIPHERAL NEUROPATHY: A LITERATURE REVIEWÂ
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
Semra Aciksoz
School Of Nursing, Gulhane Military Medical Academy, Ankara, TR
Abstract: Background: Diabetic peripheral neuropathy is the most common type of neuropathy in diabetic patients and leads to the greatest morbidity and mortality. It is closely associated with foot ulcers, amputations, falls and pain, which profoundly affect the quality of life. Objective: The aim of this literature review is to investigate research that evaluated the results of quality of life in patients with diabetic peripheral neuropathy in Turkey, and to determine the requirements and priorities of research in this area. Methods: In this literature analysis, ULAKBIM Turkish Medical Literature, google academic, YOK theses search, pubmed, google scholar, Ebsco HOST were scanned. A combination of key words such as “diabetic peripheral neuropathy”, “quality of life” and “Turkey” along with the prefered time limit of “2006 onwards” were used to search the literature. The results of 34 national and international publications suitable for the purposes of this analysis were included. Results and Discussion: The studies have shown that diabetic patients with peripheral neuropathy reported lower levels of quality of life in comparison to other patients with uncomplicated diabetes. Diabetic peripheral neuropathy affects multiple aspects of a patient’s life. Several studies have shown that foot-related complications in diabetic patients have a significant impact on their quality of life. Qualitative studies reported that diabetic foot ulcers have negative psychological and social effect. Painful diabetic neuropathy interfere with general activity, mobility and enjoyment of life and the negative impact is higher in patients with greater pain severity. Conclusion: Understanding the quality of life in patients with diabetic peripheral neuropathy is essential to the development of treatment strategies and is also helpful for delivering the optimal patient care. Data on the incidence of neuropathic pain and other complications in Turkey, its consequences for the quality of life are still lacking. Disease-specific studies in this area need to be further enhanced.
PS1Group4-036 / #164SEASONAL VARIATION OF BELL’S PALSY: A HOSPITAL BASED RETROSPECTIVE STUDY OVER 9 YEARS
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
Byung-Nam Yoon1, Jung-Joon Sung2, Suk-Won Ahn3, Ji-Eun Kim4, Yoon-Ho Hong5
1Neurology, InHa University Hospital, Incheon, KR;
2Neurology, Seoul National University Hospital, Seoul, KR;
3Neurology, Chung-Ang University Hospital, Seoul, KR;
4Neurology, Seoul Medical Center, Seoul, KR;
5Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, KR
Abstract: Epidemiological data on the incidence of Bell’s palsy (BP) are conflicting. Herpes simplex 1 was suggested as a cause of BP. The aim of this retrospective study was to evaluate the variation of cases of BP and herpes simplex infection (HSI) in our department over 9 years. The study population comprised all patients with idiopathic peripheral facial nerve palsy, who presented for acute neurological evaluation and all patients with herpes simplex infection in our hospital during the period from January 1, 1995, until December 31, 2013. We examined the monthly and seasonal distribution of BP and HIS, and compared it with the assumed equal distribution of cases over the year. Winter lasts from 12, 1, 2, spring from 3, 4, 5, summer from 6, 7, 8, autumn from 9, 10, 11. A total of 1,389 patients with BP and 10,883 patients with HSI were presented in our hospital. The monthly distribution of BPs shows a decrease during summer and especially during June. The monthly distribution of HSI shows no definite variations. A decline during the summer was observed, in contrast to a peak documented during the spring and winter. March and December were the months with the highest frequency of BP, and June was lowest frequency. The frequency of herpes simplex infection was evenly distributed around years. A markedly different pattern has been described between BP and HIS. These results will be useful to demenstrate whether the herpes simplex virus can be a pathogen of BP.
PS1Group4-037 / #171THE USE OF OMEGA-3 SUPPLEMENTATION FOR MANAGING DIABETIC NEUROPATHY: RESULTS FROM A CLINICAL PILOT TRIAL
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
Evan Lewis1, Bruce Perkins2, Richard Bazinet1, Thomas Wolever1, Vera Bril3
1Nutritional Sciences, University of Toronto, Toronto, ON, CA;
2Leadership Sinai Centre For Diabetes, Mount Sinai Hospital, Toronto, ON, CA;
3University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, CA
Abstract: Background: Diabetic sensorimotor peripheral neuropathy (DSP) is the leading complication in diabetes mellitus (DM). Omega-3 polyunsaturated fatty acids (N-3 PUFA) are integral to the development and maintenance of healthy nerve tissue but have not yet been investigated for their ability to maintain nerve structure and function in DSP. Methods: Individuals with type 1 (T1DM) and evidence of DSP as determined by a Toronto Clinical Neuropathy Score (TCNS) ≥1 were recruited to participate in an open-label trial of mammalian N-3 PUFA supplementation (10 mL.d-1; 750 mg EPA, 560 mg DPA and 1020 mg DHA; Auum Inc.) for 1-year (NCT02034266). The primary outcome is the 1-year change in corneal nerve fibre length (CNFL) measured by in vivo corneal confocal microscopy (IVCCM). Secondary outcomes include nerve conduction studies (NCS) and quantitative sensory testing. Results: 40 participants (53% female), aged 48 (SD 14), BMI 28.1 (SD 5.8) with diabetes duration of 27 (SD 18) years were enrolled between March 2014 and June 2015. The baseline TCNS showed participants have a broad range of DSP from no to moderate neuropathy. Mean IVCCM CNFL was 12.0 ± 5.2 mm/mm2. Median sural and peroneal nerve conduction velocities were 43.8±11.0 and 38.8±6.6 m/sec. Conclusion: The baseline characteristics of the 40 participants enrolled in this trial show that they have a broad spectrum of DSP and display evidence of both small and large fibre nerve dysfunction. Trial results and clinical implications will be presented at ICNMD 2016.
PS1Group4-038 / #188B12 DEFICIENCY IS A CAUSE OF REVERSIBLE AUTONOMIC FAILURE: A CASE REPORT
Topic: Group 4 – Peripheral Neuropathy: Clinical Features, Pathophysiology, Therapy / 4.5 Others
Pariwat Thaisetthawatkul
Neurological Sciences, University of Nebraska Medical Center, Omaha, NE, US
Abstract: OBJECTIVE To report a case of reversible autonomic failure (AF) associated with B12 deficiency having comprehensive autonomic evaluation BACKGROUND AF associated with B12 deficiency is an uncommon cause of orthostatic hypotension (OH) and falls. A case report with comprehensive autonomic evaluation and autonomic reflex screening test (ARS) before and after the treatment with B12 supplement was not found. DESIGNS/METHODS A case report RESULTS A 66-year-old female patient presented with recurrent orthostatic lightheadedness, syncope and falls while walking for 7 months prior to evaluation. She had well-controlled type 2 diabetes mellitus for 10 years. Physical exam was unremarkable. Blood tests revealed serum B12 <50 pg/ml, elevated serum methylmalonic acid, small amount of IgG kappa, normal hemoglobin and MCV, negative parietal cell antibody, negative paraneoplastic antibodies including acetylcholine ganglionic antibody and HgA1C 5.7%. Electrophysiologic studies were normal. ARS showed severe generalized autonomic failure affecting postganglionic sudomotor, cardiovagal and adrenergic functions associated with OH on head-up tilt (total CASS=9). Supine and upright serum norepinephrine was 1224 and 2504 pg/ml respectively. 24-hour urine sodium was 63 mmol. She was treated with daily followed by monthly intramuscular B12 injection, midodrine and fludrocortisone and became asymptomatic. Midodrine and fludrocortisone were stopped once her serum B12 level normalized. Follow-up ARS 10 and 14 months after the start of B12 supplement and the patient not taking midodrine or fludrocortisone were normal. Last serum B12 was 662 pg/ml. Hematologic evaluation showed that her monoclonal protein was benign monoclonal gammopathy. Gastrointestinal evaluation showed that intestinal bacterial overgrowth was the cause of B12 deficiency. The patient remained asymptomatic at last follow-up. CONCLUSIONS B12 deficiency is a cause of reversible AF. AF associated with B12 deficiency can present without peripheral neuropathy or macrocytic anemia. It is potentially treatable and important to recognize to prevent further injury from falls related to OH.
PS1Group6-002 / #179UNUSUAL CAUSE OF NOCTURNAL HAND PAIN- Â 3 CASES DIAGNOSED BY POCUS AFTER NORMAL EMG
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.1 Ultrasound
Abraham Chaiton
Rheumatology, University of Toronto, Toronto, ON, CA
Abstract: Abstract Three cases of hand pain in which the typical triad of nocturnal hand pain, acute point tenderness and cold sensitivity are presented. These represent an uncommon cause of hand pain, a glomus tumour. Point of care ultrasonography (POCUS) is the most sensitive method of imaging these small and highly vascular tumours. Surgical excision is curative. Glomus tumour of the finger, may be confirmed promptly at point of care with clinical strategies alone (case 1) and with supplemental imaging (cases 2&3). A 33 year old female lawyer, presented for the evaluation of hand pain with a 2 year history of aching, tenderness, and throbbing of third digit of her left hand. Throbbing was worse at night and when exposed to colder temperatures. Night splint gave no relief. Tinel’s sign was absent, Phalen’s test was negative, and the patient showed no clinical synovitis or tenosynovitis. Examination revealed an acutely tender 2 by 3 mm bluish splinter-like lesion under the nail (Fig. 1A), near the nail plate of the symptomatic 3rd digit. EDX studies were normal. No other investigations were performed, although an US examination was requested. Based on the history and reproducible clinical findings the surgeon agreed with the referral diagnosis of a glomus tumour. The patient underwent a successful surgical excision. A 46 year old medical assistant with psoriasis presented with a 12 year history of pain in the 4th right digit. She reported worse pain at night and when exposed to cold. The nail bed showed some discolouration and acute point tenderness at the site . There was no distal IP joint tenderness or swelling, and no psoriatic nail changes. X-ray revealed an erosion of the ulnar portion of the distal phalanx, under the site of discolouration (Fig. 1B). POCUS examination allowed the detection of a round shaped 5mm lesion under the nail bed of the 4th digit where there was discontinuity of the distal phalanx cortex and an intense colour Doppler signal consistent with a focal vascular lesion (Fig. 1E and F). At surgery, a glomus tumour was excised. A 33 year old office manager, presented with a 2 year history of pain in the left hand, and was referred for evaluation for possible median nerve entrapment at the wrist. EDX studies and ultrasound of the carpal tunnel were normal. Her symptoms were most intense at the 4th finger nail bed where she displayed acute tenderness to palpation and to pressure from the ultrasound probe. Discoloration under the nail was apparent. POCUS was performed at the symptomatic site, using a high-resolution 22MHz ultrasound transducer (Fig. 1 C and D). In all 3 cases a 22 MHz linear probe was used. A vascular malformation consistent with a 2mm glomus tumour was suspected. The ultrasound study guided the follow up MRI examination, confirming an isolated glomus tumour (Fig. 2A and B). The lesion was surgically excised. When faced with symptoms of paroxysmal pain, cold sensitivity, and acute tenderness, there are clinical and imaging strategies that can be employed to identify glomus tumours with greater certainty.
PS1Group6-003 / #422IMPACT OF DRISAPERSEN ON APPARENT FAT FRACTION IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.2 MRI
Courtney Bishop1, Rexford Newbould2, Zhengning Lin3, Robert Janiczek4, Susanne Wang3
1Centre For Imaging Sciences, Imanova Limited, Hammersmith Hospital, London, GB;
2Imanova Limited, Hammersmith Hospital, London, CA, GB;
3BioMarin Pharmaceutical Inc, Novato, CA, US;
4GlaxoSmithKline, Middlesex, GB
Abstract: The apparent fat fraction (AFF), the ratio of fat to total signal in a muscle detected by MRI, has been proposed as a biomarker of the health status of skeletal muscles and is linked to functional tests (e.g., 6 minute walk distance test, 6MWD). Thirty-four out of 51 subjects from a double-blind, randomized, placebo-controlled Phase 2 study of Duchenne Muscular Dystrophy patients amenable to exon 51 skipping therapy (NCT01462292) attended at least one scanning session. Data from the 18 boys who were imaged at baseline, week 24 and week 48 are presented here. Subjects were treated weekly for 24 weeks with either 3 or 6 mg/kg drisapersen, or placebo, then followed for 24 additional weeks. Only the 6 mg/kg and placebo groups are presented here due to small sample size in the 3 mg/kg group. T1-weighted FSE and IDEAL sequences were used to detect fat infiltration in 6 muscles within the thigh including the rectus femoris, vastus lateralis/medialis/intermedius, semitendinosus, and the biceps femoris. Six 6 mg/kg and 5 placebo subjects passed QC at baseline and at least one follow up visit to allow change-from-baseline analysis. Regions of interests (ROIs) were manually drawn by a blinded experienced reader around each muscle on the central five (mid-thigh) imaging slices in the T1w FSE at baseline, and five imaging slices that best matched those of the baseline scan at each follow-up, using Analyze 11.0 software, and applied to the co-registered IDEAL scans. Two independent sets of software were developed based on the same iterative decomposition (IDEAL) principle - one modelling a 7-peak fat spectrum (Method 1), the other utilizing a single fat peak with T2* correction (Method 2) - to calculate the AFF within each muscle. This analysis focused on the AFF results for the two software readings. Treatment difference was evaluated based on mixed effect model repeat measurement adjusting for baseline. The individual patient baseline AFF ranged from 0.07 to 0.61 across the 6 muscles. Using Method 1, the drisapersen group had lower average AFF of 6 thigh muscles at Week 24 (–1.33; p=0.41) than the placebo group. This difference increased further at Week 48 (–3.86; p=0.05). A similar trend was observed using the Method 2: Week 24 (–1.24; p=0.23), Week 48 (–2.99; p=0.10). The two software methods exhibited consistent results as the overall average AFF of 6 thigh muscles of both methods in favor of 6 mg/kg drisapersen treatment. The delayed onset of drisapersen’s effect in muscle structure may be explained by the time (36 weeks) required to achieve steady state in muscle. These results are in line with the positive trend in the clinical outcome (6MWD) of the overall study population favoring the drisapersen 6 mg/kg treatment group.
PS1Group6-004 / #419USEFULNESS OF MRI IN CASES OF HYPERCKEMIA
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.2 MRI
Pilar Marti1, Nuria Muelas2, Jordi Diaz-Manera3, Juan J. Vilchez2
1Neuromusucular, hospital la fe, Valencia, ES;
2Neurologia, Hospital la Fe, Valencia, ES;
3Neurologia, Hospital Santa Creu i Sant Pau, Barcelona, ES
Abstract: Muscle MRI (mMRI) is an important tool for diagnosis and management in myopathies. However, its utility has not been comprehensively assessed on the study of asymptomatic or paucisymptomatic hyperCKemia (HCK). The objective is to evaluate the mMRI diagnostic performance and its predictive capacity in a large series of patients with HCK. We perform total body mMRI in 142 patients (80% male) diagnosed of asymptomatic or paucisymptomatic HCK (CK>250 IU/L). T1 and STIR sequences were obtained on the axial plane from 6 segments (neck, shoulder girdle, trunk, pelvic, thighs and legs). Fat replacement was graded according to Mercuri scale (grades 1-4). All patients were studied using a protocol including blood enzymatic studies, muscle biopsy and genetic tests including dystrophin MLPA and iontorrent LGMD genepanel. Diagnoses were as follow: 7 dystrophinopathies, 37 LGMDs, 5 inflammatory myopathies, 6 glycogenosis, 6 neurogenic, 7 myofibrillar myopathies and 70 idiopathic. mMRI alterations were found in 67% of the patients, 34% of them showed a generalized muscle involvement and 33% a focal pattern. The most marked changes were located in pelvic, thigh and leg muscles. A specific mMRI signature of HCK was not found but a few profiles oriented to some diagnoses (dystrophin, LGMDs and glycogenosis). The extent of mMRI involvement was not length dependent but higher degree of mMRI involvement was detected in patients who developed long-term weakness. MRI can help to the diagnosis of HCK and can give information about prognosis. Its inclusion in an HCK algorithm investigation may help to optimize the diagnostic process.
PS1Group6-005 / #302LOWER LIMB MUSCLE VOLUME TEST/RE-TEST VARIABILITY USING MRI
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.2 MRI
Hui Jing Yu1, Thomas Fuerst2, Randall Stoltz3, Juan Chavez4
1Medical Affairs, Bioclinica, Inc, Princeton, NJ, US;
2Bioclinica, Inc, Newark, CA, US;
3Covance, Indianapolis, IN, US;
4Biogen, Inc, Cambridge, MA, US
Abstract: Changes in lower limb muscle cross-sectional area (CSA) as measured by T1-weighted Magnetic Resonance Imaging (T1W-MRI) are quantitative endpoints in the evaluation of treatments of neuromuscular diseases. However, physiologic variations, the imaging technique or the measurement method can induce some variability. This study aimed at assessing the degree of muscle cross-sectional area and volume variability on successive imaging examinations while controlling for time, physiologic variations and measurement method, to determine whether variability due to acquisition significantly affects trial endpoints. Before knee-joint immobilization, 30 healthy male volunteers were enrolled in a disuse muscle atrophy clinical trial and underwent MRI scans with the same imaging protocol two weeks prior to immobilization (Day 1) and on the day of immobilization (Day 14). Half of the subjects received iv infusion of study treatment on Day 1 and Day 14. Right thigh and calf muscle contours were semi-automatically delineated on axial T1W-MRI by experienced technicians. Total volume was defined as the sum of CSAs on all five axial slices for thigh and calf separately. The root mean square coefficient of variation (RMSECV) and Pearson’s correlation were computed. Mean differences between time points were assessed using a paired t-test. Variability ranged from 0.0% to 4.8% (RMSECV=1.7%) for thigh muscle volume and from 0.2% to 5.5% (RMSECV=2.2%) for calf muscle volume. Measurements were highly correlated (r2>0.95). No statistical significant differences were found between Day 1 and Day 14 (p >0.01). The lack of difference in means between the two time points suggest that there was unlikely a pharmacological effect on MRI muscle volume within the first two weeks prior to immobilization. If time, physiologic variation and measurement method are controlled for, then variability in acquisition appears to have an insignificant effect on the assessments of neuromuscular trial efficacy endpoints of muscle atrophy.
PS1Group6-006 / #328VITAMIN DEFICIENCIES IN PATIENTS WITH VARIOUS MYOPATHIES AND OTHER NEUROMUSCULAR CONDITIONS-PILOT STUDY
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.3 Other Biomarkers
Dubravka Dodig1, Mark Tarnopolsky2
1Medicine (neurology), UHN (University of Toronto), Toronto, ON, CA;
2Neurology, McMaster University, Hamilton, CA
Abstract: INTRODUCTION: Vitamins are organic micronutrients that are essential to normal growth and metabolism. Vitamin deficiency disorders have been major causes of neurological morbidity and mortality throughout the world in the past. We suspect that hypovitaminosis D , frequent in Canadian population, may be a significant comorbidity in various types of myopathies and other neuromuscular conditions. METHODS: We have conducted a pilot retrospective analysis of serum Vitamin levels in patients with various neuromuscular diagnoses seen in the neuromuscular tertiary referral clinic. Data was collected from 1852 patients (905 men and 947 women) from July 1, 1996 to June 15, 2001. We included serological results for patients with at least one vitamin level obtained including vitamins: D (25-OH), B12, E, A, and RBC folate. We analyzed data sets available for each Vitamin Deficiency and Type of Diagnosis separately. We compared the results with results for vitamin level D deficiencies available for general population in Canada. RESULTS AND DISCUSSION: For vitamin D, 124 patients (6.7% ) were tested; 74 (59.7%) were deficient and 34 (27.4%) were insufficient. For vitamin B12, 730 patients (39.4%) were tested, and 104 (14.2%) were deficient. For RBC folate, 551 (29.8%) were tested; 35 (1.9%) were deficient. For vitamin A, 29 were tested (1.6% ); 29 were deficient (100%). For vitamin E, 97 (2.1%) were tested; 2 (0.1%) were deficient. In the subgroup of myopathies (including metabolic, inflammatory ,toxic congenital, and channelopathies), 454 patients was tested for various vitamin levels. Vitamin D levels were available for 31 patients; 19 were deficient and 9 were insufficient (61% ). Statistical significance has not been reached due to small sample size. Overall, 57 (13%) of 454 patients with various myopathies had some sort of vitamin deficiency, reaching statistical significance. CONCLUSIONS: Vitamin deficiencies may be an important unrecognized contributing aetiology in neuromuscular disorders including various myopathies. Vitamin B12 and Vitamin D deficiencies are most frequently identified in neuromuscular patients. Vitamin D deficiency in myopathies seems to exceed the reported frequency in general population in Canada. We are currently analyzing Vitamin D levels in patients with myopathies seen from 2010-2015.
PS1Group6-007 / #143POTENTIOALLY CONFOUNDING VARIABLES OF GDF-15: NEW BIOMARKER OF MITOCHONDRIA DISEASES
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.3 Other Biomarkers
Akiko Ishii1, Seitaro Nohara1, Fumiko Yamamoto1, Shuichi Yatsuga2, Makoto Terada1, Tetushi Aizawa1, Tetsuto Yamaguchi1, Kumi Yanagiha1, Tetsuya Moriyama1, Naoki Touzaka1, Zenshi Miyake1, Hiroshi Tsuji1, Yasushi Tomidokoro1, Kiyotaka Nakamagoe1, Kazuhiro Ishii1, Masahiko Watanabe1, Yasutoshi Koga2, Akira Tamaoka1
1Neurology, University of Tsukuba, Tsukuba, JP;
2Kurume University, Kurume, JP
Abstract: GDF-15 (Growth Differentiation Factor 15) is a member protein of transforming growth factor beta superfamily and regulate inflammatory and apoptotic pathways in various disease, such as heart failure, kidney dysfunction, and tumor. Recently, Yatsuga et al. reported that GDF-15 was a new biomarker of mitochondria diseases. The aim of this study is to clarify potentially confounding variables of GDF-15 and demonstrate as mitochondria biomarker. Patients and methods We used serum from 16 patients of Mitochondria diseases (14 MELAS and 2 CPEO, average of age is 44), 15 patients of limbic encephalitis (LE, average of age is 47), and 10 patient of multiple sclerosis (MS, average of age is 44). GDF-15 and FGF-21 are measured using ELISA. Clinical features of patients are also evaluated retrospectively. Results Average of GDF-15 and FGF-21 in mitochondria diseases are 2860.7 pg/ml and 855 pg/ml, respectively. Average of GDF-15 and FGF-21 in LE are 15786.2 pg/ml and 238.2 pg/ml, respectively. GDF-15 and FGF-21 in MS are 575.4 pg/ml and 384.0 pg/ml, respectively. GDF-15 and FGF-21 are significantly elevated in mitochondria diseases. There is no correlation between GDF-15 and clinical features, such as ADL level, number of cells in cerebrospinal fluid (CSF), and brain MRI findings. No GDF-15 elevation is found in three patients having cancer. There are tendency of correlation between GDF-15 and protein concentration of CSF in LE patients. GDF-15 shows positive correlation between EDSS in MS patients (Pearson correlation coefficient 0.74). Conclusion GDF-15 is a useful biomarker of mitochondria disease and potentially influenced by severity of diseases.
PS1Group6-008 / #228A NOVEL ASSESSMENT OF BAROREFLEX ACTIVITY BY PHOTOPLETHYSMOGRAPHY AND TERNARY ARITHMETIC CODING IN A RAT MODEL
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.4 Electrodiagnosis
An-Bang Liu1, Hsien-Tsai Wu2, Chun-Keng Lin2
1Neurology, Buddhist Tzu Chi General Hospital, Hualien, Hualien, TW;
2Department Of Electrical Engineering, National Dong Hwa University, Shoufeng, TW
Abstract: Baroreflex sensitivity (BRS) has been applied to assess autonomic nervous system activity in many clinical trials. This indicator has been proposed to relate to many cardiovascular diseases, such as hypertension, myocardial infarction, stroke, etc. Traditionally, BRS is defined as the relationship between changes of blood pressure and heart beat interval (indicated by ECG R-R interval) after intravenous injection of vasoactive agents such as phenylephrine, sodium nitroprusside and so on. There are some commercialized instruments used to assess BRS by a noninvasive method to measure blood pressure. These instruments are expansive and need professionals to operate. Therefore, we attempted to use volume pulse acquired by infrared sensor photoplethysmography (PPG) to replace blood pressure. In the study, BRS, estimated by spontaneous sequence, driven by PPG signals was compared with those estimated by blood pressure. Reproducibility of these two indicators is good and validity is also good as compared with the traditional pharmacological intervention. Furthermore, we used ternary arithmetic coding to quantify the oscillation of blood pressure and heart rates. The similarity between these two signals was proposed to be used as a new indicator of baroreflex activity, named ternary arithmetic coding probability (TACpr). We acquire blood pressure, ECG and PPG signals from 11 Wistar Kyoto (WKY) rats to analyze BRSs and TACpr, and then verify the reproducibility and validity. Our results showed that BRS and TACpr had very good reproducibility. The validity of these two parameters was also good and acceptable. Meanwhile, we also found that pulse-pulse interval (PPI) could substitute for ECG R-R interval (RRI) to be used in assessing BRS in either method. Lastly, we demonstrated that TACpr was significantly attenuated as BRS was in streptozocin-induced diabetic rats. In summary, volume pulse acquired by PPG could replace BP in the assessment of BRS, and TACpr might be a better method than the traditional one for measuring BRS in clinical studies.
PS1Group6-009 / #293AGREEMENT BETWEEN AUTOMATED AND MANUAL QUANTIFICATION OF CORNEAL NERVE FIBER LENGTH: IMPLICATIONS FOR DIABETIC NEUROPATHY RESEARCH
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.5 Small Nerve Fibre Evaluation
Daniel Scarr1, Cesar M. Falappa1, Ilia Ostrovski1, Leif E. Lovblom1, Mohammed A.M. Farooqi1, Dylan Kelly1, Tong Wu1, Elise M. Halpern1, Mylan Ngo2, Eduardo Ng2, Andrej Orszag1, Vera Bril3, Bruce A. Perkins4
1Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, ON, CA;
2mUniversity Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, ON, CA;
3University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, CA;
4Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, CA
Abstract: Aim: Quantification of corneal nerve fiber length (CNFL) by in vivo corneal confocal microscopy represents a promising diabetic neuropathy biomarker, but applicability is limited by resource-intensive image analysis. We evaluated agreement between the established manual analysis protocol and a novel automated protocol. Methods: Sixty-eight controls, 139 participants with type 1 diabetes, and 249 participants with type 2 diabetes underwent CNFL measurement (N=456). Neuropathy severity was determined by clinical and electrophysiological criteria. CNFL was determined by manual (CNFLManual, reference standard) and automated (CNFLAuto) protocols, and results were compared for correlation and agreement using Spearman coefficients and the method of Bland and Altman. Associations with neuropathy severity were evaluated by analysis-of-variance. Results: Participants demonstrated broad variability in clinical characteristics associated with neuropathy. The mean age, diabetes duration, and HbA1c were 53±18 years, 15.9±12.6 years, and 7.4±1.7%, respectively, and 218(56%) individuals with diabetes had neuropathy. Mean CNFLManual was 15.1±4.9 mm/mm2, and mean CNFLAuto was 10.5±3.7 mm/mm2 (CNFLAuto underestimation bias, -4.6±2.6 mm/mm2 corresponding to –29±17%). Percent bias was similar across non-diabetic controls (-33±12%), type 1 (–30±20%), and type 2 diabetes (-28±16%) subgroups (ANOVA p=0.07). Levels of CNFL Auto and CNFLManual were both inversely associated with neuropathy severity (ANOVA p<0.0001). Conclusions: Although CNFLAuto substantially underestimated CNFLManual, its bias was non-differential between diverse patient groups and its relationship with neuropathy severity was preserved. Determination of diagnostic thresholds specific to CNFLAuto should be pursued in diagnostic studies of diabetic neuropathy.
PS1Group6-010 / #300USE OF CORNEAL NERVE FIBRE LENGTH (CNFL) FOR DIABETIC NEUROPATHY IDENTIFICATION IN OLDER PATIENTS WITH LONGSTANDING TYPE 1 DIABETES
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.5 Small Nerve Fibre Evaluation
Mohammed A.M. Farooqi1, Leif E. Lovblom1, Daniel Scarr1, Julie A. Lovshin1, Yuliya Lytvyn2, Genevieve Boulet1, Alanna Weisman1, Hillary A. Keenan3, Michael H. Brent4, Narinder Paul5, Vera Bril6, David Z. Cherney2, Bruce A. Perkins1
1Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, ON, CA;
2Division Of Nephrology, Department Of Medicine, University of Toronto, Toronto, ON, CA;
3Research Division, Joslin Diabetes Center, Boston, MA, US;
4Department Of Ophthalmology And Vision Sciences, University of Toronto, Toronto, ON, CA;
5Joint Department Of Medical Imaging, Division Of Cardiothoracic Radiology, University Health Network, University of Toronto, Toronto, ON, CA;
6University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, CA
Abstract: Aim: CNFL using in-vivo corneal confocal microscopy is a valid screening tool for diabetic neuropathy, but it has not been systematically evaluated in older adults with longstanding diabetes. We aimed to explore the diagnostic performance of CNFL in a cohort with ≥50 years of type 1 diabetes (T1D). Methods: As part of the Canadian Study of Longevity in Diabetes in which 150 subjects will undergo deep-phenotyping procedures, to date 48 T1D and 46 age- and gender-matched non-diabetic controls underwent clinical evaluation for symptoms, signs, and nerve conduction studies to define neuropathic status according to the 2010 Toronto consensus criteria. CNFL was determined by confocal microscopy and diagnostic performance for neuropathy was determined by Receiver Operating Characteristic (ROC) curves. Results: T1D participants were mean age 66±7 years, had median diabetes duration 54 [IQR 52-59] years, 28(58%) female; the non-diabetic controls were mean age 65±8 years and 32(70%) female. 41(85%) of the T1D participants met the criteria for neuropathy. Mean sural nerve amplitude for non-diabetic controls (9.5±5.5 μV) was similar to T1D without neuropathy (8.4±2.8 μV, p=0.83), while it was lowest for T1D with neuropathy (3.4±1.6 μV, p<0.001 for both comparisons). Mean CNFL for non-diabetic controls (20.0±5.7 mm/mm2) was similar to T1D without neuropathy (18.8±8.9 mm/mm2; p=0.74) and was lower for T1D with neuropathy (9.5±5.4 mm/mm2; p<0.001 for both comparisons). Among T1D subjects, area under the ROC curve for CNFL in identifying neuropathy was 0.81 with optimal threshold 13.7 mm/mm2, sensitivity 78%, specificity 86%. Conclusion: The diagnostic performance – even the optimal threshold value – for CNFL in older adults with longstanding T1D appears to parallel that observed in studies of younger patients with shorter duration. This implies that potential age-related changes in CNFL do not impair diagnostic validity and that neuropathy screening protocols using CNFL may be applied to broad T1D populations.
PS1Group6-011 / #298VALIDITY OF AN AUTOMATED PROTOCOL OF IN VIVO CORNEAL CONFOCAL MICROSCOPY FOR DIABETIC SENSORIMOTOR POLYNEUROPATHY DETECTION IN TYPE 1 DIABETES
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.5 Small Nerve Fibre Evaluation
Daniel Scarr1, Nancy Cardinez1, Ilia Ostrovski1, Tong Wu1, Mohammed A.M. Farooqi1, Leif E. Lovblom1, Elise M. Halpern1, Ausma Ahmed1, Mylan Ngo2, Eduardo Ng2, Andrej Orszag1, Vera Bril3, Bruce A. Perkins4
1Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, ON, CA;
2University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, ON, CA;
3University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, CA;
4Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, CA
Abstract: Aim: In vivo corneal confocal microscopy (IVCCM) has been validated as a diagnostic tool for diabetic sensorimotor polyneuropathy (DSP), with manual assessment of corneal nerve fibre length (CNFL) showing best operating characteristics. We aimed to determine and compare the diagnostic validity of CNFL when measured using an automated analysis protocol. Methods: IVCCM was performed on 89 type 1 diabetes participants and 71 healthy volunteers concurrent with clinical and electrophysiological examinations. Post-examination, CNFL was determined using both a manual and automated analysis protocol. CNFL from both methods was compared between healthy volunteers, DSP controls, and DSP cases using ANOVA. Receiver operating characteristic (ROC) curves for the identification of DSP were generated for both methods, and results were compared. Results: Mean age of the 71 healthy volunteers, 50(56%) DSP controls, and 39(44%) DSP cases was 40±17, 34±15, and 49±14y respectively (p<0.0001). CNFLManual was 18.6±4.5, 17.2±4.2, and 11.4±3.7 mm/mm2 for the healthy volunteers, DSP controls, and DSP cases, respectively (p<.0001), and CNFLAutomated was 12.4±3.8, 11.8±3.2, and 8.3±2.8 mm/mm2 (p<0.0001). Area under the ROC curve was 0.87 for CNFLManual and 0.80 for CNFLAutomated (p=0.0002). The optimal diagnostic threshold was 14.0 mm/mm2for CNFLManual 11.3 mm/mm2 for CNFLAutomated. Conclusions: Though lower when compared to the manual method, CNFL measured by the automated protocol retains its diagnostic validity for identifying DSP in type 1 diabetes. This suggests that automating the image analysis process can improve efficiency and generalizability of IVCCM without sacrificing diagnostic capability. Further work must be done to validate diagnostic thresholds.
PS1Group6-012 / #248VALIDATION OF COOLING DETECTION THRESHOLD AS A MARKER OF SENSORIMOTOR POLYNEUROPATHY IN TYPE 2 DIABETES
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.5 Small Nerve Fibre Evaluation
Mohammed A.M. Farooqi1, Andrej Orszag1, Zoe Lysy1, Leif E. Lovblom1, Elise M. Halpern1, Mylan Ngo2, Eduardo Ng2, Ari Breiner2, Vera Bril3, Bruce A. Perkins1
1Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, ON, CA;
2University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, ON, CA;
3University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, CA
Abstract: Aim: The measurement of cooling detection thresholds (CDT) has been established in previous cross-sectional studies as a valid test for diabetic sensorimotor polyneuropathy (DSP) in type 1 diabetes. We aimed to validate its diagnostic performance in type 2 diabetes (T2D). Methods: 220 T2D subjects from a larger cohort underwent clinical and electrophysiological examinations including 3 small-fiber function tests: CDT, heart rate variability (HRV) and laser Doppler imaging of axon-mediated neurogenic flare responses to cutaneous heating (LDIFLARE), along with the Toronto Clinical Neuropathy Score (TCNS). Clinical DSP was defined by consensus criteria whereas preclinical DSP was defined by at least one electrophysiological abnormality. Area under the curve (AUC) and optimal thresholds were determined by receiver operating characteristic (ROC) curves. Results: Subjects were aged 63±11 years with mean HbA1c of 7.5±1.6%. The 139(63%) clinical DSP cases had mean CDT value 18.3±8.9 °C; the 52(24%) preclinical DSP had 25.3±3.5 °C; and the 29(13%) controls had 27.1±3.8 °C; (p-value <0.02 for all three comparisons). For identification of clinical DSP AUCCDT was 0.79, which exceeded AUCHRV (0.60, p=<0.0001), AUCLDI FLARE (0.69, p=0.0003) and AUCTCNS (0.73, p=0.03). Optimal threshold for clinical DSP identification was <22.8 °C (64% sensitivity and 83% specificity). For Preclinical DSP, AUCCDT was 0.80, and the optimal threshold was ≤27.5 °C (83% sensitivity and 72% specificity). Conclusions: Akin to studies of T1D, CDT has acceptable diagnostic performance for the identification of both clinical and preclinical neuropathy in patients with T2D. Application of CDT as a non-invasive tool for systematic screening of early neuropathy in diabetes clinics should be considered.
PS1Group6-013 / #294HEREDITARY NEUROPATHIES: THE ROLE OF COPY NUMBER VARIATIONS (CNVS) IN THE NGS TARGETED GENE PANEL DIAGNOSTIC TESTING
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.6 Biochemical and Molecular Techniques
Petra Lassuthova1, Jana Neupauerová1, Simona Marková1, Marcela Krůtová1, Radim Mazanec2, Dana Š. Brožková1, Pavel Seeman1
11- department Of Paediatric Neurology, Dna Laboratory, 2nd Faculty of Medicine, Charles University in Prague and University Hospital Motol, Prague, CZ;
22- department Of Neurology, Dna Laboratory, 2nd Faculty of Medicine, Charles University in Prague and University Hospital Motol, Prague, CZ
Abstract: Introduction: Inherited peripheral neuropathies (IPN) are an extremely heterogeneous group of disorders, both clinically and genetically. Mutations of more than 90 genes are associated with IPN. Copy number variations (CNVs) are an important mechanism in hereditary neuropathies, as the most common type CMT1A/HNPP is caused by duplications/deletions of the PMP22 gene. Methods: One hundred and ninety-eight patients were tested using massively parallel sequencing (MPS) with a targeted gene panel with custom designed HaloPlex kits (Agilent Technologies, CA, USA). Causative point mutations or small indels were found in fifty-one of them (26%). Data for remaining one hundred and forty-seven patients were further tested with other algorithms with the aim of finding the causal mutations. Copy number variation analysis was performed with three different approaches – firstly, SureCall analysis (Agilent Technologies, CA, USA) predicts CNVs based on log ratio of the normalized sample read depth to the reference sample read depth. Secondly, NextGene (SoftGenetics LLC., PA, USA) analysis based on hidden Markov model (HMM) and lastly, NextGene analysis based on data normalization (beta testing version). Results: Large deletion of the GJB1 gene was found in one patient with progressive peripheral motor and sensory neuropathy. The deletion of the entire GJB1was later also confirmed by Multiplex Ligation-dependent Probe Amplification (MLPA, MRC-Holland, NL). The SALSA MLPA P129-B1 GJB1 probemix was used. Large duplication of the SEPT9 gene was identified in one patient with HNA with autosomal dominant inheritance and without point mutations in this gene. This duplication was confirmed with MLPA (SALSA MLPA P307 SEPT09 probemix) and a microarray technique (HumanOmni BeadChip Kit, Illumina Inc., CA, USA). The duplication was further detected in the similarly affected father of the patient with both methods (MLPA and the microarray). Duplications of the SEPT9 gene have already been described as the cause of HNA in a few patients. Furthermore, more CNVs were detected, however, confirmation with second method is in process (CNVs in these genes were identified with software analysis: FGD4, MARS, ATL1, SPTLC2, SPTLC1, FIG4, GARS, BICD2). For three patients also smaller duplications in the SEPT9 gene were detected with data analysis, however these were later not confirmed with MLPA (SALSA MLPA P307 SEPT09probemix). Smaller CNVs (encompassing only one or few exons) thus seem to be mostly false positives. Conclusions: Copy number variations affecting entire gene are an important mechanism causing IPN and algorithms for CNVs detection should be included in evaluation process of data from MPS. Algorithm based on HMM is the most reliable according to our experience. Sensitivity depends on coverage and we have observed indirect relationship between the reliability and the length of the CNVs. There is a need for reliable and simple methods for confirmation of such suspected CNVs. Supported by Ministry of Health of the Czech Republic, grant nr. AZV 16-30206A.
PS1Group6-014 / #235HOW MULTI-GENE PANELS CAN CHANGE THE LANDSCAPE OF DIAGNOSING NEUROMUSCULAR DISORDERS
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.6 Biochemical and Molecular Techniques
Margaret Bradbury1, Amanda Lindy1, Amy Decker1, Deborah Copenheaver2, Sharon Suchy1
1Neurogenetics, GeneDx, Gaithersburg, MD, US;
2Exome, GeneDx, Gaithersburg, MD, US
Abstract: OBJECTIVE: To assess the clinical utility of a multi-gene panel for diagnosing neuromuscular disorders (NMD). BACKGROUND: NMD encompass over 200 clinically and genetically heterogeneous disorders that primarily affect the peripheral nervous and musculoskeletal systems. Overlapping clinical features can make a clinical diagnosis challenging and may lead to a diagnostic odyssey while potential health risks associated with the condition go unmonitored. Molecular testing using a large multi-gene next generation sequencing (NGS) panel is a newly available diagnostic tool for providing a genetic diagnosis. METHODS: In this retrospective study, molecular testing was performed on 288 individuals with NMD using NGS and exon-level copy number analysis of up to 76 NMD-related genes. Confirmed test results and clinical information were evaluated to assess the positive rate and concordance with the patient’s suspected diagnosis. Positive results were defined as one pathogenic or likely pathogenic variant in an autosomal dominant or X-linked disorder, or two pathogenic or likely pathogenic variants in an autosomal recessive disorder. RESULTS: The positive diagnostic rate for the panel was 26.4% (76/288). We found that 52.6% (40/76) of positive cases were concordant with the clinical diagnosis at the time of testing. However, molecular testing provided an alternative diagnosis in 39.5% (30/76) of individuals. One example was a clinical diagnosis of limb girdle muscular dystrophy (LGMD) with an identified molecular diagnosis of hypokalemic periodic paralysis, type 1, and another example was a clinical diagnosis of FSHD with a molecular diagnosis of a dystrophinopathy. In 7.9% (6/76) of cases no prior clinical diagnosis was provided. Individuals under 18 years of age were more likely (55.6%; 15/27) to receive a diagnosis that differed from the clinical suspicion than those over 18 years of age (34.9%; 15/43). This may suggest that atypical or overlapping clinical presentations may pose an increased diagnostic dilemma in children. The genes with highest test sensitivity on the NMD panel were DMD (9/76) and RYR1 (9/76). These genes were associated with a high frequency of ‘alternative diagnoses’ for which the molecular diagnosis differed from the suspected clinical diagnosis; however, the molecular diagnosis was consistent with the clinical presentation (7/9 DMD; 4/9 RYR1). Additionally, LGMD2L (5/6) and myotonia congenita (7/9) were the most commonly concordant diagnoses. Of the positive cases, 50.0% (38/76) had pathogenic variants in genes associated with LGMD, and 31.6% (24/76) in genes associated with congenital muscular dystrophies, consistent with the prevalence of these disorders. CONCLUSION: Our results demonstrate that multi-gene panels for NMD are an effective diagnostic tool to concurrently evaluate many genes related to the same or overlapping phenotypes. In this series the panel provided a diagnosis that differed from the clinical suspicion in approximately one-third of cases, most often in individuals under the age of 18. Multi-gene panels can provide a molecular diagnosis quickly, which is necessary for many therapeutic interventions and is required for proper surveillance and family planning, as well as entering clinical trials.
PS1Group6-015 / #263SENSITIVITY AND SPECIFICITY OF DR1 BISULFITE SEQUENCING IN DETECTING SMCHD1 MUTATION IN A COHORT OF FSHD1 AND FSHD-LIKE PATIENTS
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.6 Biochemical and Molecular Techniques
Audrey Briand1, Christian Baudoin2, Nadira Lagha2, Pilvi Nigumann2, Francoise Chapon3, Tania Stojkovic7, Christophe Vial5, Francoise Bouhour5, Elena Pegoraro6, Philippe Petiot7, Antony Behin7, Bruno Eymard7, Pascal Laforêt7, Leonardo Salviati6, Marc Jeanpierre1, Michel Vidaud1, Claude Desnuelle8, Gael Cristofari2, Sabrina Sacconi8
1Biochemistry And Molecular Genetics, University Hopital Cochin, Paris, FR;
2IRCAN INSERM U1081 CNRS UMR 7284 University of Nice, Nice, FR;
3University Hospital of Caen - anatomopathology laboratory and neuropathology, CAEN, FR;
4Centre De Référence Pathologie Neuromusculaire Paris Est, Hôpital Pitié-Salpêtrière, Paris, FR;
5Neurology Department, Lyon University Hospital, BRON, FR;
6Azienda ospedaliera di Padova, Padova, IT;
7Croix Rousse Hospital - Neurological Functional Explorations, LYON, FR;
8Neurology, University Hospital of Nice, Nice, FR
Abstract: Facioscapulohumeral muscular dystrophy (FSHD) is characterized by typical, progressive and asymmetric weakness of selective muscle groups. The variability of clinical expression and the existence of myopathies mimicking FSHD clinical phenotype may complicate diagnosis. We can distinguish autosomal dominant FSHD1, associated with a contracted and permissive D4Z4 repeat array on chromosome 4 (≥1, <11 repeated units), from FSHD2, linked to pathogenic dominant mutations in the SMCHD1 gene on chromosome 18 and non-contracted allele on chromosome 4 (>11 repeated units). FSHD2 patients display hypomethylation of the D4Z4 repeats both on chromosomes 4 and 10, while in FSHD1 patients, only the pathogenic contracted allele is hypomethylated. We recently described patients with severe clinical phenotype carrying both FSHD1 and FSHD2 mutations. This multicentric study assessed the sensitivity and specificity of DR1 methylation analysis by bisulfite sequencing (MABS) of blood DNA in detecting SMCHD1 pathogenic mutations. We analyzed 68 FSHD1 patients and 42 patients with typical FSHD clinical features and no contraction of the D4Z4 repeats on chromosome 4. All patients underwent clinical examination, haplotyping of 4q allele, targeted sequencing of the SMCHD1 gene by NGS and DR1 MABS (threshold level for DR1 hypomethylation ≤30%). The pathogenicity of SMCHD1 mutations was computationally predicted and additional functional analyses were performed for new mutations. Among FSHD1 patients, we found 25 patients with DR1 methylation levels below the threshold, 6 of them carrying pathogenic mutations in SMCHD1 and a permissive 4Q allele, while 5 of them carry rare polymorphisms. The sensitivity of DR1 MABS in detecting patients with pathogenic SMCHD1 mutation was 100 %, while specificity was 69% Among 42 FSHD-like patients, we found 32 patients with DR1 hypomethylation, 29 of them carrying SMCHD1 pathogenic mutations and a permissive 4Q allele, 1 carries a rare polymorphism. For 6 other patients, an alternative diagnosis was confirmed and none shows DR1 hypomethylation. In this cohort, the sensitivity of DR1 MABS in predicting SMCHD1 pathogenic mutations was 96% and specificity was 75%. In conclusion, DR1 MABS is highly sensitive to identify patients with SMCHD1 mutations in FSHD1 and FSHD-like cohorts, but has moderate specificity. This may be improved by accurate sizing of chromosome 4 and 10 D4Z4 loci.
PS1Group6-016 / #318ROBUST GENOTYPING IN THE DIAGNOSTICS OF LIMB GIRDLE MUSCULAR DYSTROPHIES
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.6 Biochemical and Molecular Techniques
Baiba Lace1, Jurgis Strautmanis2, Ieva Micule3, Maruta S. Naudina4, Loreta Cimbalistiene5, Algirdas Utkus5, Birute Burnyte5, Janis Stavusis6, Inna Inashkina7
1Medical Genetics, CHUQ, Quebec, QC, CA;
2Childrens’ University hospital, RIga, LV;
3Medical Genetics, Childrens’ University hospital, RIga, LV;
4Pauls Stradins Clinical University hospital, RIga, LV;
5Department of Human and Medical Genetics, Faculty of Medicine, Vilnius, LT;
6Mgmrg, Biomedical Research and Study centre, RIga, LV;
7Mgmrg, 1Biomedical Research and Study centre, RIga, LV
Abstract: Limb-girdle muscular dystrophies (LGMD) are characterized by the predominant involvement of the shoulder and pelvic girdle and trunk muscle groups. Diagnosing of specific types of LGMD is a challenging process due to the many genes involved, including unknown genes within the identified chromosomal loci. Although common mutations have been identified for some causative genes a lot of private mutations are identified in patients. Confirming the diagnosis of LGMD, in addition to subtyping, is usually done using molecular techniques. The aim of this study was to develop a mutation screening technique for molecular diagnostics and evaluate it in the context of the routine diagnostics. Objects. We investigated 94 patients from Latvia and Lithuania with clinical symptoms of LGMD, along with 565 healthy unrelated controls from general and ethnic populations, using our developed test kit. Methods. We developed a test kit for LGMD-2 based on the Illumina VeraCode GoldenGate genotyping platform that analyzes 77 mutations, including insertions/deletions and single-nucleotide polymorphisms, within different LGMD-related genes with proven reproducibility. We Sanger sequenced the coding region and exon/intron boundaries of CAV3 and FKRP genes. The mutation c.191dupA in ANO5 gene was also identified by direct sequencing. The commercially available AmpliSeq Inherited Disease Panel (Life Technologies) that covers 325 gene exons and intron-exon boundaries of most common inherited diseases, including genes of LGMD and other muscle dystrophies, was used for the patients, who remained undiagnosed. Results. Twenty of the tested mutations demonstrated superior performance in the assay. Analysis revealed a homozygous CAPN3 c.550delA mutation in 14 patients and three heterozygous single-nucleotide polymorphisms in controls: DYSF c.5028delG, CAPN3 c.2288A>G, and rs2287717. ANO5 dup191A was not present in either population. FKRP gene analysis revealed one c.286C>A homozygous and two compound heterozygous mutations within the coding region of FKRP gene: c.826C>A/ c.404_405insT; c.826C>A/ c.204_206delCTC. CAV3 gene analysis revealed three novel sequence variations (c.183C>G, p.S61R; c.220C>A, p.R74S; c.220C>T, p.R74C) and found evidence that one was associated with hyper-CK-emia. Additionally, next generation sequencing using AmpliSeq Inherited Disease Panel was performed on four patient samples. These patients had no diagnosis found by other methods described. One heterozygous mutation within chloride voltage-gated channel 1 (CLCN1) gene – c. 2680C>T p.894R>X was identified. Patient was an eight years old girl with CK level (up to 30 000 U/L), proximal muscle weakness and progressive disease course. Muscle biopsy was inconclusive, electromyography confirmed severe muscle damage and myotonia. Steroid therapy was initiated and CK level decreased to almost normal. She developed complications of the long term steroid use, but currently she is physically active and disease remission was reached. Conclusions. Selective mutation analysis for the patients are justified in the populations with prevalent common mutations, like c.del550A (CAPN3 gene) in the population of Lithuania (15% of patients diagnosed), however the best results we reached by Sanger sequencing of the selected genes (additionally 9% of patients diagnosed). Nextgen sequencing is powerful tool, yet we have insufficient knowledge about the effect of mutations, it can reveal to us.
PS1Group6-017 / #274PROFLECT: A USER-FRIENDLY TOOL TO DETECT COPY NUMBER VARIATION (CNV) AMONG AMPLICON SEQUENCING DATA
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.6 Biochemical and Molecular Techniques
Paco Derouault1, Claire-Cécile Barrot1, Rémi Moulinas2, Franck Sturtz1, Stéphane Merillou3, Anne-Sophie Lia1
1Ea6309 - Faculty Of Medicine, University of Limoges, Limoges, FR;
2Umr-s1092 - Faculty Of Medicine, University of Limoges, Limoges, FR;
3Xlim, University of Limoges, Limoges, FR
Abstract: Next Generation Sequencing is a major evolution in the diagnosis of diseases exhibiting a large genetic heterogeneity because all the genes involved in them can be tested at once for one or several patients. Amplicon sequencing using the Polymerase Chain Reaction (PCR) amplification of a limited number of the genomic regions of interest followed by high-throughput sequencing is very appropriate for diagnosis since it is fast and not too expensive. It also exhibits a very nice coverage of the targeted amplicon sequences (more than 99%), a good read-depth and a variant calling allowing being confident of the results. However, while several tools exist for Copy Number Variation (CNV) for Whole Genome Sequencing (WGS) or Whole Exome Sequencing (WES), no suitable tool was available for amplicon sequencing. Here, we present a novel method to detect deletion and duplication events from sequencing data generated by this amplicon sequencing for eight or more patients simultaneously by applying several normalization steps to the Raw Read Count (RRC). These normalization steps allow comparing the amplicon normalized values of all the patients in order to identify amplicons which are duplicated or deleted. First, we correct the value of RRC by the library size. Secondly, we apply two successive normalizations in order to correct the GC content bias and the bias due to the length of the amplicon. The regions rich or poor in GC are significantly less amplified than the others and smaller amplicons generates more reads than the larger ones due to a more efficient PCR amplification. Thirdly, we use a normalization step similar to that of a housekeeping gene in RT-PCR. We choose amplicons that are equally sequenced in our patients and we normalize our data by the median of these amplicons. Fourthly, for each amplicon, the median value of all patients of a run is calculated and the value of each amplicon is divided by this median. This step allows obtaining a final value of 1 if there are two alleles on the analyzed amplicon, 0.5 if only one copy, 1.5 for three copies and so on. We validated our method on several datasets containing positive controls (duplication and deletion of the PMP22 gene involved in Charcot-Marie-Tooth (CMT) disease) previously diagnosed by MLPA. In addition, our bioinformatics approach permitted the detection of new CNVs different from the PMP22 duplication, in CMT patients. We confirmed these CNVs by quantitative PCR and/or CGH-array. The pathogenicity of these CNVs is now under investigation. Finally, we implemented our method in a user-friendly tool, ProfLect, freely-available, which visualizes the read depth values, cleans the data, and applies the different normalization steps. This tool, developed in Java, is available for the main operating systems.
PS1Group6-018 / #234GENETIC SEQUENCING OF PATIENTS WITH LIMB GIRDLE MUSCLE WEAKNESS USING AN NGS PANEL
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.6 Biochemical and Molecular Techniques
Elaine Lee1, Madhuri R. Hegde2, Hillarie P. Windish3, Babi R. Nallamilli2, Laura Rufibach1
1Jain Foundation, Seattle, WA, US;
2Emory Genetics Laboratory, Emory University, Decatur, US;
3Jain Foundation, Jain Foundation, Seattle, WA, US
Abstract: Molecular diagnosis of limb girdle muscular dystrophies (LGMDs) is challenging because there are over 20 different types of LGMD, each caused by mutations in a different gene. Diagnosis is further complicated by the fact that the symptoms of LGMDs overlap with other hereditary muscle-wasting conditions including Becker muscular dystrophy, Pompe, FSHD (Facioscapulohumeral muscular dystrophy), Miyoshi myopathy, Emery-Dreifuss muscular dystrophy, Bethlem myopathy and Nonaka/HIBM (Hereditary inclusion body myopathies). To help physicians diagnose LGMDs and other diseases with similar symptoms, we recently developed a genetic diagnostic program that is free for patients living in the United States. There are several ways for both patients and physicians to access this program. Individuals with muscle weakness who do not have a genetic diagnosis can take a short online quiz (www.lgmd-diagnosis.org) to determine whether they are eligible. Alternatively, physicians can apply on behalf of their patients using a free online diagnostic algorithm called the Automated LGMD Diagnostic Assistant (ALDA) (www.jain-foundation.org/alda) or by contacting the Jain Foundation ([email protected]) directly to set up a diagnosis drive in their clinic. Eligible individuals who consent to participate in the free diagnostic program send a saliva sample for analysis to Emory Genetics Laboratory. The genetic analysis consists of a next-generation sequencing gene panel that evaluates 35 genes associated with 23 LGMDs and 8 other muscular dystrophies with similar symptoms. Samples from over 600 individuals have already been analyzed and about 85% have had at least one variant in one of the included genes. Around 20% of the reports provide a confirmed diagnosis (i.e. 2 pathogenic mutations for recessive diseases or one pathogenic mutation for dominant diseases), 25% of the reports provide a likely diagnosis (i.e. 2 variants of unknown significance identified in the same gene) and 15% of the reports rule out pathogenic variants in all 35 genes. After their DNA sample has been analyzed, participants and physicians receive a genetic report, as well as supplemental information that can assist them with proper disease management and provides places to contact for disease specific information. We are in the process of evaluating an additional 400 patient samples and hundreds more have begun the process and will soon submit samples.
PS1Group8-001 / #426A PHASE III DOUBLE-BLIND, RANDOMIZED, PLACEBO-CONTROLLED STUDY (SIDEROS) ASSESSING THE EFFICACY OF IDEBENONE IN SLOWING THE RATE OF RESPIRATORY FUNCTION LOSS IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY RECEIVING GLUCOCORTICOID STEROIDS
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Gunnar Buyse1, Oscar H. Mayer2, R. Donisa-Dreghici3, F. Couttet4, Jodi Wolff5, Nick Coppard4
1Organ Systems, University Hospitals Leuven, Leuven, BE;
2The Children’s Hospital of Philadelphia, Philadephia, PA, US;
3Santhera Pharmaceuticals, Santhera Pharmaceuticals, Liestal, CH;
4Santhera Pharmaceuticals, Liestal, CH;
5Santhera Pharmaceuticals, Santhera Pharmaceuticals, Tucson, US
Abstract: In DMD, progressive weakness of respiratory muscles leads to sleep disordered breathing, nocturnal hypoventilation and ineffective cough leading to inability to clear lung secretions and to life-threatening pulmonary infections. Respiratory failure is commonly observed during the second or third decade of life. In a Phase III randomized, placebo-controlled clinical trial (DELOS trial; Buyse et al, 2015), in DMD patients 10-18 years of age not taking concomitant glucocorticoid steroids (GCs), idebenone slowed the rate of decline in respiratory function over the 1-year study period. An earlier Phase II randomized, placebo-controlled pilot trial (DELPHI Buyse et al., 2011) in DMD patients 8-16 years of age demonstrated that idebenone slowed the rate of decline in respiratory function in GC non-using patients whilst GC-using patients were predominantly not in the decline phase of their disease. The SIDEROS trial is a multi-center trial of the efficacy of idebenone in slowing the rate of respiratory function decline in 266 GC-using DMD patients in the respiratory decline phase of their disease (30% ≤ Forced Vital Capacity percent predicted (FVC%p) ≤80%). The study will be conducted in the U.S. and Europe and patients will be randomized in a 1:1 ratio to 900mg idebenone (two 150 mg tablets to be taken three times a day with meals), or to matching placebo for a duration of 78 weeks. Patients must be able to provide reliable and reproducible FVC assessments and must have been on a stable GC regimen (intermittent or continuous) for at least 12 months prior to Baseline, without any dose adjustments on a mg/kg basis during the last 6 months. The primary endpoint is the change from Baseline to Week 78 in FVC%p assessed using hospital-based spirometry. Secondary endpoints include other measures of expiratory and inspiratory functions assessed using hospital-based spirometry as well as a portable hand-held device designed for regular use at home.
PS1Group8-002 / #463REDUCTION OF ISOAGGLUTININS IN IVIG BY ANTI-A DONOR SCREENING REDUCES THE RISK OF HEMOLYTIC EVENTS.Â
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Ayman R. Kafal
Medical Affairs, CSL Behring Canada, Montreal, QC, CA
Abstract: Background: Hemolytic events after the administration of high dose intravenous immunoglobulin (IVIG) have been linked to anti-blood group A and B antibodies (isoagglutinins) in the product. Isoagglutinins can be reduced by anti-A donor screening and exclusion of plasma form high titer donors, or by a specific immunoaffinity chromatography step. However, the impact of such isoagglutinin reduction measures on the risk of hemolysis is not known. Methods: We analyzed the impact of an anti-A donor screening program on spontaneous reports of hemolytic events with an IVIG product (Privigen, CSL Behring). From July 2013 to December 2015, Privigen was produced from plasma form which high titer donors (about 5 % of donors) was excluded. This measure produced a decrease in anti-A and anti-B antibodies by one titer step (measured by indirect agglutination test). We compared the number of hemolytic events reported to the CSL Behring Pharmacovigilance department from 2008 to 2013 (before donor screening) to 2013 to 2015 (during donor screening). In order to account for the different amounts of Privigen administered during these 2 periods, we calculated the relative rate of hemolytic event reporting. Results: In the period before donor screening, 265 hemolytic events were reported, in the period during donor screening, there were 107[HAC1] . Taking the amount of Privigen used during the periods into account, the decrease in the reporting rate was 41.5 %. Discussion: This investigation suggests that with an anti-A donor screening program producing a modest reductions in anti-A and anti-B antibodies, the risk of hemolysis after high dose IVIG therapy can be reduced to a relevant extent. Limitations of this research include possible confounders between the 2 evaluation periods (as the use of IVIG may differ) and the reliance on spontaneous reports. Form October 2015 onwards, anti-A donor screening is progressively replaced with a specific immunoaffinity chromatography step in the Privigen production processes (implementation depending on the registration status in the respective countries), The immunoaffinity chromatography reduces isoagglutinins by 2 to 3 titer steps, with a possible further reduction of the risk of hemolysis.
PS1Group8-003 / #433DEVELOPMENT OF A NOVEL TOOL FOR ASSESSMENT OF CRAMP SEVERITY: THE TORONTO CLINICAL CRAMP INDEX (TCCI)
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Hans Katzberg1, Vera Bril2, Carolina Barnett-Tapia3
1Neurology, Toronto General Hospital, Toronto, ON, CA;
2Division Of Neurology, University Health Network, Toronto, CA;
3Toronto General Hospital, Toronto, ON, CA
Abstract: BACKGROUND: Although cramp frequency has been used most commonly as a primary outcome measure in muscle cramp trials, a validated assessment scale which evaluates all aspects of clinically relevant change important to patients is lacking. DESIGN/METHODS: Patients over the age of 18 evaluated at the Prosserman Family Neuromuscular Clinic (Toronto, Ontario) experiencing muscle cramps across different medical conditions including polyneuropathy, motor neuron disease, pregnancy-induced and exercise induced cramps, cramps related to cirrhosis and hemodialysis and idiopathic nocturnal cramps were recruited. Semi-structured interviews were taped, transcribed and analyzed. Using data saturation technique, a conceptual framework of cramp severity based on qualitative data of patients’ experience was created. RESULTS: Fifteen patients, 8 males and 7 females ranging from 28 to 65 years of age representing all intended cramp etiologies were included in the qualitative assessments. Average duration of the interviews was 27 minutes. Two major domains identified during the interviews were issues relating to cramp severity and cramp impact on daily life. Within cramp severity, cramp duration, intensity, frequency and location were common sub-domains. Within the cramp impact domain, issues related to effects on daytimes activities, impact on sleep, daytime sleepiness and harm experienced from muscle cramps were identified as key features. CONCLUSIONS: Initial qualitative analysis of patients’ cramps experience demonstrates that cramp severity including assessment of cramp intensity, duration, frequency and location as well as cramp impact including daytime impairment, sleep impairment and harm from muscle cramps are clinically significant to patients. The cramp experience appears to be consistent across disease states.
PS1Group8-004 / #353RELIABILITY AND VALIDITY OF THE 100 METER TIMED TEST AS AN OUTCOME MEASURE IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Lindsay N. Alfano1, Natalie F. Miller1, Katherine M. Berry1, Kevin Flanigan2, Linda H. Cripe3, Jerry R. Mendell2, Linda P. Lowes1
1Center For Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, US;
2Neurology, The Research Institute at Nationwide Children’s Hospital, Columbus, US;
3Cardiology, Nationwide Children’s Hospital, Columbus, OH, US
Abstract: Duchenne muscular dystrophy (DMD) is a rare disorder leading to impaired cognitive and language development, as well as progressive skeletal and cardiac muscle weakness due to a genetic mutation in the DMD gene. The rarity of this disorder coupled with mutation-specific therapies reduces potential subject pools for current and future trials in this disorder. The success of clinical trials will be dependent upon selection of appropriate outcome measures that minimize variability in this pediatric population. The 6 minute walk test (6MWT) has been modified for use in the DMD population, but can be problematic in this cohort due to children’s difficulty in understanding the abstract concept of time, especially those with cognitive impairments. The 100 meter timed test (100m) has been proposed as an outcome measure in the DMD population to overcome the limitations seen in the 6MWT(Alfano 2013). The 100m requires a child to complete 2 laps around cones placed 25 meters apart as fast as safely possible. Unlike the 6MWT, the boys are encouraged to run if able, thus encouraging maximal performance and potentially eliminating any ceiling effect of the assessment. The concrete distance allows children to visualize the finish line which could improve consistency of performance compared to 6MWT. The purpose of this study was to determine the construct validity and test-retest reliability of the 100m in the DMD population. A cohort of boys with DMD (n=30) completed the 100m and common functional assessments (i.e. 6MWT, NSAA, time to rise, 10 m run, and 4 stair climb) at one study visit. The assessments were performed in random order to decrease order bias. A subset of 9 boys repeated the 100 m in the same day and an additional subset of 7 boys repeated 100 m the following day. Repeated 100 m tests within the same day were separated from any other testing by at least 30 minutes to minimize fatigue effects. The 100m timed test had a stronger correlation to all of the tests (r = 0.623 – 0.915, P<0.001) when compared to the 6MWT (r = 0.467 – 0.849, P<0.001; except P=0.01 time to rise) excluding the timed stair tests (6MWT: r=0.76; 100 m: r=0.74), though the correlations were still large. The 100 m shows exceptional ICC values for inter-day reliability (ICC=0.99, P<0.001). The intra-day reliability was very good (but slightly lower) (ICC=0.87, P=0.004). We attribute this difference to fatigue when completed twice in the same day as the average change between attempts was an increase of 9.7 ± 15.1 seconds (range = –3.0 to +44.5 seconds) within the same day compared to 0.0 ± 4.7 seconds (range = –4.5 to +8.3) across days. These pilot data provide evidence of the validity and excellent test-retest reliability of the 100 m in DMD and should be considered as an outcome measure for this population. The limited inter-day variability seen in the 100 m provides evidence that this test measures maximal ambulatory ability rather than more variable self-selected walking speeds seen in other timed walking tests.
PS1Group8-005 / #163INTRAVENOUS IMMUNOGLOBULIN “WEAR-OFF EFFECT” IN CIDP: STUDY DESIGN AND PROGRESS UPDATE
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Jeffrey A. Allen1, Jeffrey A. Allen2, Mamatha Pasnoor3, Ted Burns4, Senda Ajroud-Driss2, John P. Ney5, Albert A. Cook6, Thomas H. Brannagan, Iii7, John T. Kissel8, Kenneth C. Gorson9, Richard A. Lewis10, Melvin Berger11, Patty Riley12, David Schaefer13, Timothy Walton13
1Department Of Neurology, University of Minnesota, Minneapolis, MN, US;
2Department Of Neurology, Northwestern University, Chicago, IL, US;
3Department Of Neurology, Kansas University Medical Center, Kansas City, KS, US;
4Department Of Neurology, University of Virginia, Charlottesville, VA, US;
5Department Of Neurology, University of Washington, Seattle, WA, US;
6Department Of Neurology, Neurology and Johns Creek, Johns Creek, GA, US;
7Department Of Neurology, Columbia University Medical Center, New York, NY, US;
8Department Of Neurology, Ohio State University, Columbus, OH, US;
9Department Of Neurology, St. Elizabeth’s Medical Center/Tufts University School of Medicine, Boston, MA, US;
10Department Of Neurology, Cedars-Sinai, Los Angeles, CA, US;
11Medical Research Strategy, CSL Behring, King of Prussia, PA, US;
12Medical Affairs, CSL Behring, King of Prussia, PA, US;
13Clinical Research, AxelaCare Health Solutions, Lenexa, KS, US
Abstract: Background: IVIg randomized controlled trials (PRIMA and ICE trials) have demonstrated safety and efficacy in the treatment of CIDP. Despite these critical landmark studies, there are no IVIg dosing studies and no evidence based dose optimization guidelines. The degree to which “wear off” or other treatment related fluctuations impacts IVIg dosing or treatment optimization is unknown. Objectives: We describe an investigator-initiated, multi-center study (Intravenous Immunoglobulin (IVIg) Treatment-Related Fluctuations in Chronic Inflammatory Demyelinating Polyneuropathy (CIDP) Patients Using Daily Grip Strength Measurements (GRIPPER)) that prospectively evaluates treatment related fluctuations in between IVIg cycles. We specifically aim to determine the extent and frequency of strength and disability fluctuations near the end of each IVIg dosing interval as well as the degree to which serum IgG levels correlate with the collected clinical assessments. We intend to use this data as a foundation by which to better develop evidence-based IVIg dose optimization strategies. Methods:This is a prospective observational study of 30 adult CIDP patients who receive home IVIg infusions. Subjects perform and document daily grip strength (GS) measurements for 6-months. Weekly visits to the subjects home by a trained nurse additionally capture Rasch-built Overall Disability Scale (R-ODS) score, Timed Up and Go Test (TUGs), Overall Neuropathy Limitations Scale (ONLS), Modified Fatigue Severity Scale (mFSS), and Visual Analog Pain Severity Scale (VAS). QOL Short Form Physical Component Summary, Version 2 (SF-36v2®) is collected at baseline, week 12, and week 24. Serum IgG levels are collected by the home study nurse at 3 time-points surrounding the first 4 IVIg infusions (peak, trough, and mid-cycle). To determine “wear-off” frequency, we will analyze the proportion of subjects with any given degree of GS and R-ODS intracycle fluctuation and the proportion of cycles in which GS and R-ODS fluctuation occurs. To determine the extent of “wear-off” the degree of difference between maximum and minimum GS, R-ODS, TUGs, ONLS, and VAS scores will be analyzed. Results: Six out of the 7 projected sites are currently open to enrollment. Preliminary representative data is presented for a limited number of subjects, demonstrating observed IVIg “wear-off” effects on GS. Conclusion: This ongoing study seeks to help clinicians better understand the extent and frequency of IVIg treatment-related fluctuations. Obtaining a greater understanding of IVIg effect should provide clinicians with a framework to develop evidence based dose optimizing strategies. We expect that this information will be extremely important in forming hypotheses to be tested in future studies (for example, comparing different dosage intervals, optimal IVIg taper guidelines, or assessing the long-term outcome of short-term cycle to cycle clinical fluctuations).
PS1Group8-006 / #346IDEBENONE REDUCES RESPIRATORY COMPLICATIONS IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Craig M. Mcdonald1, Thomas Meier2, Thomas Voit3, Ulrike Schara4, Chiara S. Straathof5, Maria Grazia D’Angelo6, Günther Bernert7, Jean-Marie Cuisset8, Richard S. Finkel9, Nathalie M. Goemans10, Christian Rummey11, Mika Leinonen11, Paolo Spagnolo12, Gunnar Buyse13
1University of California Davis Medical Center, Sacramento, CA, US;
2Santhera Pharmaceuticals, Santhera Pharmaceuticals, Liestal, CH;
3Nihr Biomedical Research Centre, Institute Of Child Health, University College London, London, GB;
4Universitatsklinikum Essen, Essen, DE;
5Leiden University Medical Center, Leiden, NL;
6Istituto di Ricovero e Cura a Carattere SEM, Lecco, IT;
7GVP Kinderspital, Vienna, AT;
8CHRU de Lille, Lille, FR;
9Nemours Children’s Hospital, Orlando, FL, US;
10Child Neurology, University Hospitals Leuven, Leuven, BE;
114Pharma, Liestal, CH;
12University of Padua, Padova, IT;
13Organ Systems, University Hospitals Leuven, Leuven, BE
Abstract: In DMD, progressive loss of respiratory function leads to restrictive pulmonary disease that evolves into severe respiratory complications. Of particular concern are ineffective cough, secretion retention and recurrent respiratory tract infections. In a Phase 3 randomized controlled clinical trial (DELOS; Buyse et al., 2015), idebenone reduced the loss of respiratory function in DMD patients 10-18 years of age and not taking concomitant glucocorticoid steroids (GCs) over a 1-year study period. In a post-hoc analysis of DELOS, “bronchopulmonary adverse events” (BAEs) were defined by a study-independent physician as bronchitis, pneumonia, upper respiratory tract infection, influenza and/or viral infection with respiratory symptoms, laryngitis, respiratory failure, acute respiratory failure, cough and dyspnea. More patients in the placebo group than in the idebenone group reported BAEs (placebo: 17 of 33 patients, 28 events; idebenone: 6 of 31 patients, 7 events). The Hazard ratios (HR) calculated “by patient” (HR 0.33, p = 0.0187) and for “all BAEs” (HR 0.28, p=0.0026) indicated a clear idebenone treatment effect. The overall duration of BAEs was 222 days (placebo) vs. 82 days (idebenone). In addition, there was also a difference in the use of systemic antibiotics typically utilized for the treatment of BAEs. In the placebo group, 13 patients (39.4%) reported 17 episodes of antibiotic use compared to 7 patients (22.6%) reporting 8 episodes of antibiotic use in the idebenone group. Furthermore, patients in the placebo group used systemic antibiotics for longer (105 days) compared to patients in the idebenone group (65 days). This post-hoc analysis of DELOS indicates that idebenone reduces the risk of bronchopulmonary adverse events and reduced the need for systemic antibiotics.
PS1Group8-007 / #342TREATMENT EFFECT OF IDEBENONE ON INSPIRATORY FUNCTION IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Gunnar Buyse1, Thomas Voit2, Ulrike Schara3, Chiara S. Straathof4, Maria Grazia D’Angelo5, Günther Bernert6, Jean-Marie Cuisset7, Richard S. Finkel8, Nathalie M. Goemans9, Christian Rummey10, Mika Leinonen11, Oscar H. Mayer12, Paolo Spagnolo13, Thomas Meier14, Craig M. Mcdonald15
1Organ Systems, University Hospitals Leuven, Leuven, BE;
2Nihr Biomedical Research Centre, Institute Of Child Health, University College London, London, GB;
3Universitatsklinikum Essen, Essen, DE;
4Leiden University Medical Center, Leiden, NL;
5Istituto di Ricovero e Cura a Carattere SEM, Lecco, IT;
6GVP Kinderspital, Vienna, AT;
7CHRU de Lille, Lille, FR;
8Nemours Children’s Hospital, Orlando, FL, US;
9Child Neurology, University Hospitals Leuven, Leuven, BE;
104Pharma, Liestal, CH;
114Pharma, Stockholm, SE;
12The Children’s Hospital of Philadelphia, Philadephia, PA, US;
13University of Padua, Padova, IT;
14Santhera Pharmaceuticals, Liestal, CH;
15UCD Medical Center, Sacramento, CA, US
Abstract: Assessment of dynamic inspiratory function may provide valuable information about the degree and progression of pulmonary function decline in patients with Duchenne muscular dystrophy (DMD). The aims of this study were to characterize inspiratory function and to assess the efficacy of idebenone on this pulmonary function outcome in a large and well-characterized cohort of 10-18 year old DMD patients not taking glucocorticoid steroids (GCs) enrolled in the randomized controlled DELOS trial (Buyse et al., 2015). We evaluated the effect of idebenone on the highest flow generated during an inspiratory FVC maneuver (maximum inspiratory flow; V’I,max(FVC)) and the ratio between the largest inspiratory flow during tidal breathing (tidal inspiratory flow; V’I,max(t)) and the V’I,max(FVC). The fraction of the maximum flow that is not used during tidal breathing has been termed Inspiratory Flow Reserve (IFR). DMD patients in both treatment groups of DELOS (idebenone, n=31; placebo: n=33) had comparable and abnormally low V’I,max(FVC) at baseline. During the study period, V’I,max(FVC) further declined by -0.29 L/s in patients on placebo (95% CI: -0.51, -0.08; p=0.008 at Week 52), whereas it remained stable in patients on idebenone (change from baseline to Week 52: 0.01 L/s; 95% CI: -0.22, 0.24; p=0.950). The between-group difference favoring idebenone was 0.27 L/s (p=0.043) at Week 26 and 0.30 L/s (p=0.061) at Week 52. In addition, during the study period, the IFR improved by 2.8% in patients receiving idebenone and worsened by -3.0% among patients on placebo (between-group difference 5.8% at Week 52; p=0.040). This study suggests that idebenone preserved inspiratory muscle function as assessed by V’I,max(FVC) and IFR in patients with DMD.
PS1Group8-008 / #338REFERENCE VALUES FOR THE THREE-MINUTE WALK TEST, NORTH STAR AMBULATORY ASSESSMENT AND TIMED TESTS IN TYPICALLY DEVELOPING BOYS AGED 2.5-5 YEARS
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Katrijn Klingels1, Jasmine Hoskens1, Lise Van Verdegem1, Marleen Van Den Hauwe2, Gunnar Buyse3, Nathalie M. Goemans2
1Rehabilitation Sciences, KU Leuven, Leuven, BE;
2Child Neurology, University Hospitals Leuven, Leuven, BE;
3Organ Systems, University Hospitals Leuven, Leuven, BE
Abstract: Background: The 6-minute walk test is used to evaluate functional capacity in ambulant boys with Duchenne muscular dystrophy (DMD) and accepted as primary endpoint in registration-directed therapeutic studies. The feasibility of this test may be tackled by short attention span or developmental delay in younger children. Therefore, a shorter 3-minute walk test (3MWT) may potentially be a useful tool for assessing functional capacity in this age group. Objective: 1) To generate reference values for the 3MWT, North Star Ambulatory Assessment (NSAA) and timed tests in typically developing boys aged between 2 years 6 months and 5 years. 2) To describe the relation between the functional tests and anthropometric variables. Methods: A total of 114 typically developing boys (mean 4 years 2 months) were recruited across four age subcategories (2.5-<3 years; 3-<4 years; 4-<5 years; 5-<6 years). Results: The 3MWT was feasible for all participants, although more encouragements were needed to keep the attention of the very young ones. The three-minute walk distance (3MWD) increased significantly with age, from 160.4m ± 18.8m at 2.5 years to 209.7m ± 19.1m at 5 years. Median score (interquartile range, IQR) on the NSAA increased with age from 25 (23-27) at 2.5 years to 33 (33-34) at 5 years. For the timed tests (Gower’s test, 10 m run and climb and descend four stairs), median values (IQR) for the total group were 2.6 (2.2-3.3), 4.1 (3.7-4.7), 2.3 (1.9-3.0) and 2.6 (2.1-3.7) respectively. The correlation coefficient between 3MWT and NSAA was 0.57 and high correlations were found between all timed tests. Correlations with age, height and weight varied between 0.56 and 0.79. Conclusion: These reference values of the 3MWT, NSAA and timed tests according to age and height provide a useful tool to assess functional capacity in boys aged 2 years 6 months to 5 years.
PS1Group8-009 / #432NEUROMUSCULAR JUNCTION IN EXPERIMENTAL AUTOIMMUNE ENCEPHALOMYELITIS: A HISTOPATHOLOGICAL ANALYSIS
Topic: Group 8 – Miscellaneous / 8.2 Biomarkers in Neuromuscular Disorders
Thalita Rocha1, Jetro G.D. Sguarezi1, Sara A. Ferreira1, Rodolfo Thomé2, Liana Verinaud2, Catarina Rapôso2
1Multidisciplinary Laboratory, São Francisco University, Bragança Paulista, BR;
2Department Of Biochemistry And Tissue Biology, Institute of Biology, State University of Campinas (UNICAMP), Campinas, BR
Abstract: Experimental Autoimmune Encephalomyelitis (EAE) is a good model to study Multiple Sclerosis (MS), which is a chronic autoimmune and inflammatory disease of the central nervous system (CNS), leading to demyelination of neurons and psychomotor impairment. Regarding the peripheral nerve system (PNS), no morphological changes were observed in skeletal muscles, as myonecrosis or fibrosis, but a connective tissue inflammation and demyelination of peripheral nerves were observed and evidenced by an increase of TNFα and F4/80 expression, and by a decrease in expression of S100 protein, respectively. Current treatments aim to reduce neuroinflammation and can act directly in the prevention of demyelination. Searching for new drugs, studies in EAE have proposed that the Sildenafil® (Viagra - Pfizer) could act as a possible adjunctive therapy, based on its anti-inflammatory effect in the CNS. Considering these information, the present study investigated, by immunofluorescence (IF), the expression of some neuromuscular junction (NMJ) proteins to identify possible damages to this structure. Therefore, EAE was induced in C57BL6 mice (n = 5) by immunization with100 μg myelin oligodendrocyte glycoprotein (MOG35–55) peptide, emulsified with Complete Freunds Adjuvant. 200 ηg Pertussis toxin was administrated via i.p. at 0 and 48 h after MOG35–55 inoculation. The animals were submitted (EAESC group) or not (EAE group) to intraperitoneal injections of 0.2 mL of Sildenafil®, each 8 hours for 15 days. A group treated only with Sildenafil® and a naïve group (without any treatment) were also studied. Weight changes and clinical signs were followed and graded daily according to a score method, where 0: no sign, 1: flaccid tail, 2: hind limbs weakness, 3: hind limbs paralysis, 4: hind paralysis and fore limbs weakness, 5: full paralysis/dead. Gastrocnemius muscles were dissected to cryomicrotomy and submitted to IF. Clinical analysis revealed that EAE animals suffered from motor limitations, such as tremors, and abnormal walking and posture. However, the animals from EAESC group exhibited normal walking and posture, and tremors were either mild or nonexistent. The Sildenafil® presented protective action to neuroinflammation, restraining and/or reversing the demyelination. The expression of synaptophysin, synaptobrevin and syntaxin proteins was maintained positive in all groups, with an increase of synaptophysin and syntaxin expression after Sildenafil® treatment, indicating no changes in synaptic vesicle-forming machines. However, a decrease in the expression of SNAP25 protein in EAE and EAESC groups was observed, indicating a possible change in the anchoring mechanism of synaptic vesicles into the presynaptic membrane. The αBgTx reaction was positive in all groups, confirming that there are no changes in acetylcholine receptors into the postsynaptic membrane. Statistical analysis revealed that there are no significant differences between groups, leading us to conclude that, considering synaptophysin, synaptobrevin, syntaxin and SNAP25 proteins there is no damage into the neuromuscular junction in EAE.
PS1Group8-010 / #286WHEN SHOULD WE TREAT HYPERCKEMIA?
Topic: Group 8 – Miscellaneous / 8.2 Biomarkers in Neuromuscular Disorders
Astrid Emilie Buch1, Karen B.H. Pedersen1, Sofie T. Ostergaard1, Jesper Q. Thomassen2, Ruth Frikke-Schmidt2, Nanna Witting1, John Vissing1
1Copenhagen Neuromuscular Center, Rigshospitalet, University Hospital of Copenhagen, Copenhagen E, DK;
2Department Of Clinical Biochemistry, Rigshospitalet, University Hospital of Copenhagen, Copenhagen E, DK
Abstract: High concentrations of creatine kinase (hyperCKemia) and myoglobin in blood can cause kidney failure. Therefore, it is standard care to treat hyperCKemia with forced diuresis. Generally, treatment starts when concentration of creatine kinase in plasma (p-CK) exceeds 3,000 U/L. However, the level of hyperCKemia that causes acute renal failure (AKI) has never been investigated. We hypothesize that the current treatment threshold is too low. We therefore investigated the frequency of AKI in patients with hyperCKemia with and without treatment, in this ongoing study. Study design was retrospective, with analysis of register-based cases admitted to a hospital in the Capital Region of Denmark between 2009 and 2014 with p-CK>3,000 U/L and at least two repeated measurements of p-creatinine. Patients with other medical reasons for AKI, such as sepsis, heart failure, preexisting kidney disease, hypothermia or hypotension and children under the age of 18 were excluded. So far, we have reviewed 890 cases of which 723 were excluded. 167 patients were found eligible of whom 22 did not receive any treatment (17 males, age 22-84 years) 136 were treated with forced diuresis (87 males, age 21–93 years) and 9 received dialysis (6 males, age 31-79 years). Primary outcome was AKI evaluated by change in p-creatinine according to the RIFLE criteria. Average peak p-CK did not differ significantly between untreated and treated patients (p-CK=7,551 U/L, range: 3,152-17,854 vs. p-CK=10,350 U/L, range: 3,022-25,300 U/L), but tended to be lower in untreated (p=0.056). Average peak p-CK in the dialysis group was markedly higher compared to the two other groups (p-CK=21,628 U/L, range: 14,400-25,300; p<0.001). Classifying patients according to peak p-CK (see figure), no patient with peak p-CK <13,000 U/L received dialysis (n=119, encompassing 91% of the untreated and 72% of those treated with forced diuresis). Of the 119, only 3 patients met the RIFLE criteria for stage 1 AKI (2.5%), with peak levels of p-CK of 3,404, 6,107 and 6,521 U/L, respectively. All 3 patients were from the group treated with forced diuresis. Overtreatment is a serious issue, increasing the duration of hospitalization and the risk of nosocomial diseases, as well as being a socioeconomic burden to the society. Our preliminary findings suggest that treating low to moderate hyperCKemia with an exploratory threshold of peak p-CK of 13,000 U/L with forced diuresis has no effect on the risk of developing AKI. Prospective studies are needed to establish the actual threshold for when to initiate treatment.
PS1Group8-011 / #285VALIDATION OF PROTEIN BIOMARKERS FOR DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 8 – Miscellaneous / 8.2 Biomarkers in Neuromuscular Disorders
Kristin Strandberg1, Burcu Ayoglu2, Mojgan Reza3, Diana Johnsson4, Joana Pisco Domingos4, Alison Blain3, Erik Niks5, Mathias Uhlén2, Annemieke Aartsma-Rus6, Hanns Lochmuller3, Francesco Muntoni4, Peter Nilsson2, Pietro Spitali6, Cristina Al-Khalili Szigyarto1
1Divison Of Proteomics And Nanobiotechnology, KTH-Royal Institute of Technology, Stockholm, SE;
2Scilife Lab Stockholm, School Of Biotechnology, KTH-Royal Institute of Technology, Solna, SE;
3Institute Of Genetic Medicine, Newcastle University, Newcastle upon Tyne, GB;
4Dubowitz Neuromuscular Centre And Mrc Centre For Neuromuscular Diseases, UCL Institute of Child Health, London, GB;
5Department Of Neurology, Leiden University Medical Centre, Leiden, NL;
6Department Of Human Genetics, Leiden University Medical Centre, Leiden, NL
Abstract: Although the number of identified biomarkers is increasing rapidly, only a few biomarkers are approved for regulatory use each year. To facilitate the use of biomarkers in a clinical setting; to monitor disease progression or to use as a surrogate endpoint, validation strategies need to be developed. In this study we show a multianalyte and multicenter approach for validation of Duchenne muscular dystrophy biomarkers, using a high-throughput suspension bead array platform. Duchenne muscular dystrophy is a progressive, childhood onset muscle degenerative disease. Many therapeutic options are being tested in clinical trials, using functional outcome measures as primary endpoints. Affected patients would benefit of validated, reliable biomarkers in easily accessed serum or plasma, that can be used to predict disease severity, monitor disease progression, and measure treatment outcome in clinical trials and for clinical management of the disorder. Recent studies have identified potential biomarkers for Duchenne muscular dystrophy in serum and plasma using different types of assays, immuno-based and aptamer-based assays as well mass spectrometry. In this study an antibody suspension bead array, containing 254 antibodies, was utilized to validate protein biomarkers using a total of 493 longitudinal patient samples (gathered from 285 Duchenne muscular dystrophy patients) and 67 control samples, which were collected at three different locations. Myosin light chain 3, Electron transfer flavoprotein alpha, Malate dehydrogenase 2 and Carbonic anhydrase 3 were validated from previous findings as disease progression markers with high significance (p-value <0.001). Myosin light chain 3, Electron transfer flavoprotein alpha and Malate dehydrogenase 2 show a fast decrease in protein level in patients younger than 12 years and from 12 years and on the decrease stagnate, while Carbonic anhydrase 3 shows a more constant decrease in protein level with increasing age. Studies on the association of these protein profiles with clinical parameters in ambulant and non-ambulant patients are ongoing.
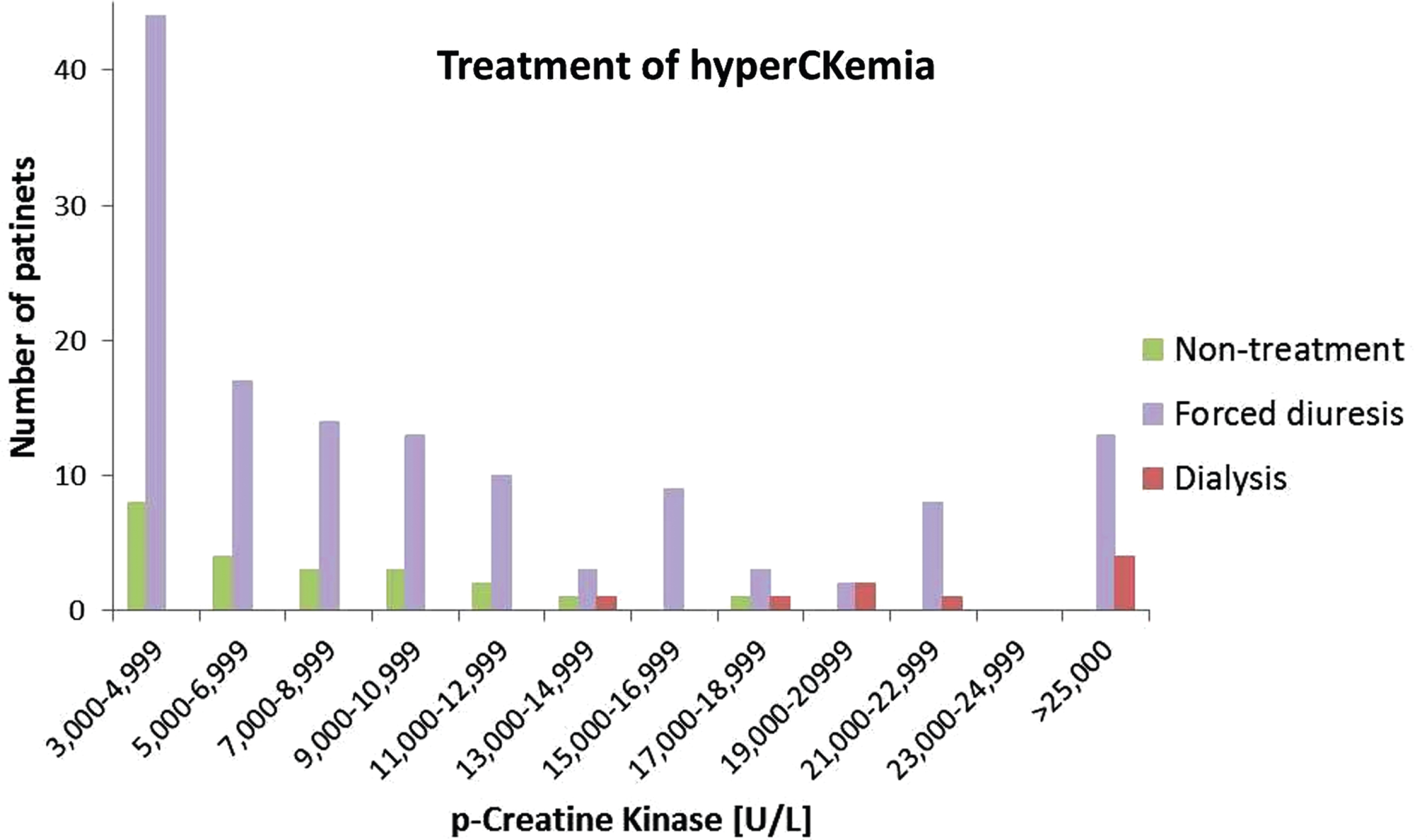
PS1Group8-012 / #267SURVEY ON USAGE OF TELECOMMUNICATION TERMINALS IN JAPANESE PATIENTS WITH NEUROMUSCULAR DISEASES
Topic: Group 8 – Miscellaneous / 8.3 Home Care / Social Programs in Neuromuscular Diseases
Katsuhisa Ogata1, Mikiya Suzuki1, Kana Yatabe1, Kazunari Momma1, Yuzo Tanaka1, Ikuya Nonaka1, Takuhisa Tamura1, Mitsuru Kawai1, Toshiaki Takahashi2
1Institute Of Clinical Research / Department Of Neurology, National Hospital Organization Higashisaitama Hospital, Hasuda, Saitama, JP;
2Neurology, National Hospital Organization Sendai-Nishitaga National Hospital, Sendai, JP
Abstract: Background. Patient registries for rare diseases have founded to accelerate development of therapies; some of them were designed feasible for self-registration by patients on the Web. Many of medical information have uploaded to the Web. Internet usage might be effective for either daily medical care or development of therapies. Objective. Investigating popularization of the Internet and telecommunication terminals among Japanese patients with neuromuscular diseases (NMD). Methods. We carried out a questionnaire targeting patients with neuromuscular disorders, about usage of telecommunication terminals; personal computer (PC), smartphone and feature phone. Results. We obtained 128 answers; mean age of patients was 40.1+-19.1; 90 males, 38 females; 12 with spinal muscular atrophy including amyotrophic lateral sclerosis (G12 of ICD-10), five with disorders of the peripheral nervous system (G60-G65), 111 with diseases of neuromuscular junction and muscle (G70-G73). Patients who owned any telecommunication terminals were 100. PC owners were 73; smartphone, 33; feature phone, 47. Dual owners of PC and smartphone were 26; 24 of them were previous owner of feature phone. Feature phone owners without PC and smartphone were 20. Twenty-eight patients owned neither PC nor smartphone nor feature phone; four of them had no family who owned any telecommunication terminals. Conclusion. Approximately 80% of Japanese patients with NMD are accessible to the Internet with their own devices. PC is most popular terminal; smart phone is emerging. Feature phone is still popular; 16% of the patients are accessible to the Internet only with feature phone. Some patients don’t have any devices to access the Internet. For effective provision of disease information and patient registration, multiple telecommunication channels should be prepared.
PS1Group8-013 / #227IS HOME TREATMENT IN AUTO-IMMUNE DISEASE PATIENTS TREATED BY IVIG SAFE?
Topic: Group 8 – Miscellaneous / 8.3 Home Care / Social Programs in Neuromuscular Diseases
Guilhem Sole1, Claude Desnuelle2, Jean-Philippe Azulay3, Gérard Besson4, Jean Christophe Antoine5, Francoise Buhour6, Alain Creange7, Gwendal Le Masson1, Laurent Magy8, Sebastien Marcel8, Jean-Michel Paquet10, Francois Rouhart11, Rabye Ouaja12, Marc Gauthier-Darnis13, Sophie Puget14
1Neuro-muscular Department, Pellegrin University Hospital, BORDEAUX, FR;
2Neuro-muscular Diseases Department, University Hospital L’Archet, NICE, FR;
3Neurology Department, University Hospital La Timone, MARSEILLE, FR;
4Neurology Department, Grenoble University Hospital, GRENOBLE, FR;
5Neurology Department, Saint- Etienne University Hospital, ST ETIENNE, FR;
6Neuro-muscular Diseases Department, Lyon University Hospital, LYON, FR;
7Neurology Department, University Hospital Henri Mondor, CRETEIL, FR;
8Neurology Department, Limoges University Hospital, LIMOGES, FR;
9Neurology Department, Chambery Hospital, CHAMBERY, FR;
10Neurology Department, LAVAL HOSPITAL, LAVAL, FR;
11Neurology Department, Brest University Hospital, BREST, FR;
12Global Medical Department, LFB, LES ULIS, FR;
13Market Access And Pricing Department, LFB, LES ULIS, FR;
14International Scientific Affairs Unit, LFB, LES ULIS, FR
Abstract: Intravenous immunoglobulins (IVIg) are widely used especially in auto-immune diseases (AID). IVIg infusions are most often done in hospital however it can be costly, time-consuming and detrimental to patient’s QoL. A retrospective study was conducted in 22 French centers to evaluate the feasibility and the safety of TEGELINE® (5% IVIg, LFB Biomedicaments), when infused at home. The 46 included patients were suffering from AID, mostly dysimmune neuropathies (71.7%) and treated with at least 1 cycle of TEGELINE® at home after receiving 3 consecutive cycles of TEGELINE® at hospital. A total of 138 and 323 cycles of TEGELINE® were performed at hospital and home respectively. The mean posology was 1.6±0.4 g/kg at hospital and 1.57±0.4 g/kg at home. In 28 patients (60.9%), at least one risk factor of IVIg-related adverse events (AEs) occurred. For 15 patients, no precautionary measures were used and in 23 patients, preventive measures were similar in both places. At home, 45 TEGELINE®-related AEs occurred in 17 patients compared to 24 AEs occurring in 15 patients at hospital. All at-home AEs were resolved. No correlation between the place of TEGELINE® infusion with either the maximum AEs intensity (p=0.339) or the number of TEGELINE®--related AEs occurrences (p=0.56) was found. At the end of the study, 45 patients continued their IVIg treatment (including 39 (84.8%) at home) and one stopped following recovery. The study results show the feasibility and the good safety profile of TEGELINE® when administered at home in selected patients.
PS1Group8-014 / #226AT HOME VERSUS HOSPITAL IVIG FOR THE TREATMENT OF MULTIFOCAL MOTOR NEUROPATHY (MMN), CHRONIC INFLAMMATORY DEMYELINATING POLYRADICULONEUROPATHY (CIDP) AND LEWIS SUMNER SYNDROME (LSS): A COST OF ILLNESS STUDY
Topic: Group 8 – Miscellaneous / 8.3 Home Care / Social Programs in Neuromuscular Diseases
Emilen Delmont1, Claude Desnuelle2, Guilhem Sole3, Isabelle Durand-Zaleski4, Marc Gauthier-Darnis5, Rabye Ouaja6, Sophie Puget7
1Neuro-muscular Diseases Department, University Hospital La Timone, MARSEILLE, FR;
2Neuro-muscular Diseases Department, University Hospital L’Archet, NICE, FR;
3Neuro-muscular Department, Pellegrin University Hospital, BORDEAUX, FR;
44public Health, University Hospital Henri Mondor, CRETEIL, FR;
5Market Access And Pricing Department, LFB, LES ULIS, FR;
6Global Medical Departement, LFB, LES ULIS, FR;
7International Scientific Affairs Unit, LFB, LES ULIS, FR
Abstract: Intravenous immunoglobulins (IVIg) are widely used especially in auto-immune diseases, such as dysimmune neuropathies. IVIg infusions are usually made at hospital however at-home treatment could save healthcare resources and costs. Previous studies showed that home-based IVIg treatment can be effective, safe and less costly than hospital-based IVIg infusions. A retro-prospective study was conducted in 2 French centers to evaluate, from a national payer point of view, the direct and indirect costs related to TEGELINE® administration (5% IVIg, LFB Biomedicaments) when infused at hospital versus at home (fig. 1). The 24 patients enrolled were aged of 52.5 years (median) and suffered from Multifocal Motor Neuropathy (MMN) (37.5%), Chronic Inflammatory Demyelinating Polyradiculoneuropathy (33.3%) and Lewis Summer Syndrome (29.2%). All patients were treated with TEGELINE® first at hospital (median posology: 1.51±0.43 g/kg), for at least 5 infusion cycles prior to switching to home treatment (1.52±0.4 g/kg), where they received at least 3 more TEGELINE® infusion cycles (fig.2). The main reasons reported for the switch were: “Good tolerance of TEGELINE® (87.5%)”, “Lack of AEs occurrence during the last 3 infusions at hospital (75%)”, “Good patient understanding on the pros and cons of home treatment (75%)”, “Patient willingness (62.5%)” and “Hospital organization constraints (54.2%)”. “Hospital costs burden” was reported in only 4.2%. The total median cost, per patient treated with TEGELINE® was estimated at €74,750.89 at hospital and €44,148.44 at home (p<0.001). The study results show that home-based IVIg (Tegeline®) maintenance treatment in selected patients with dysimmune neuropathies allows cost savings.
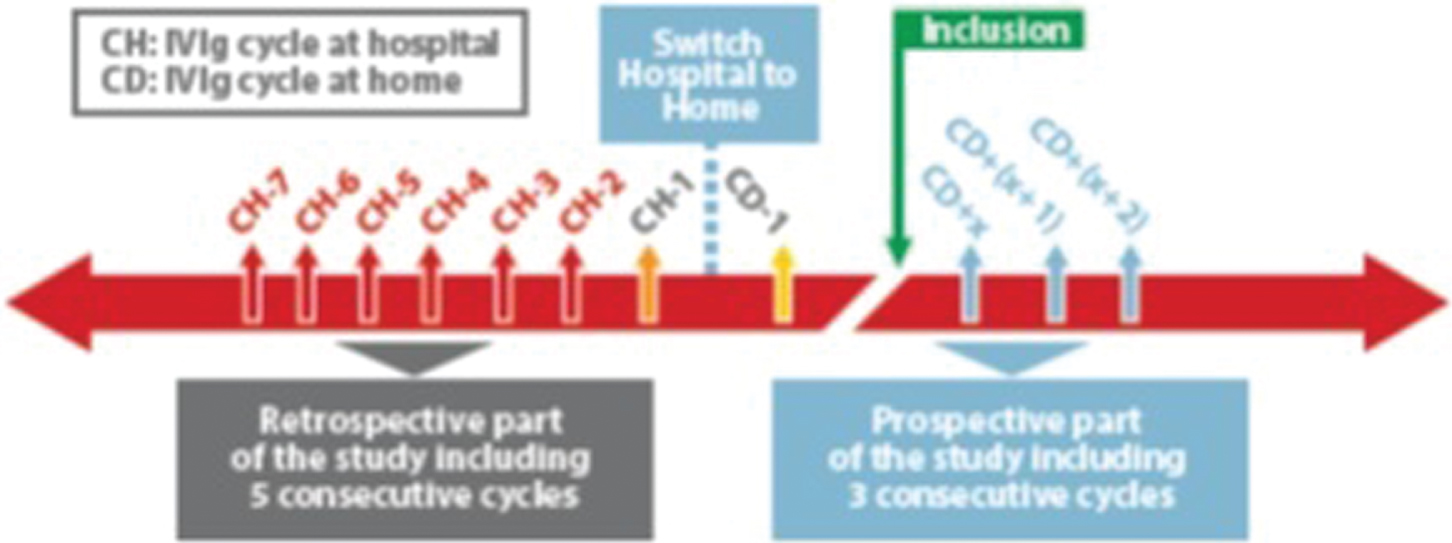
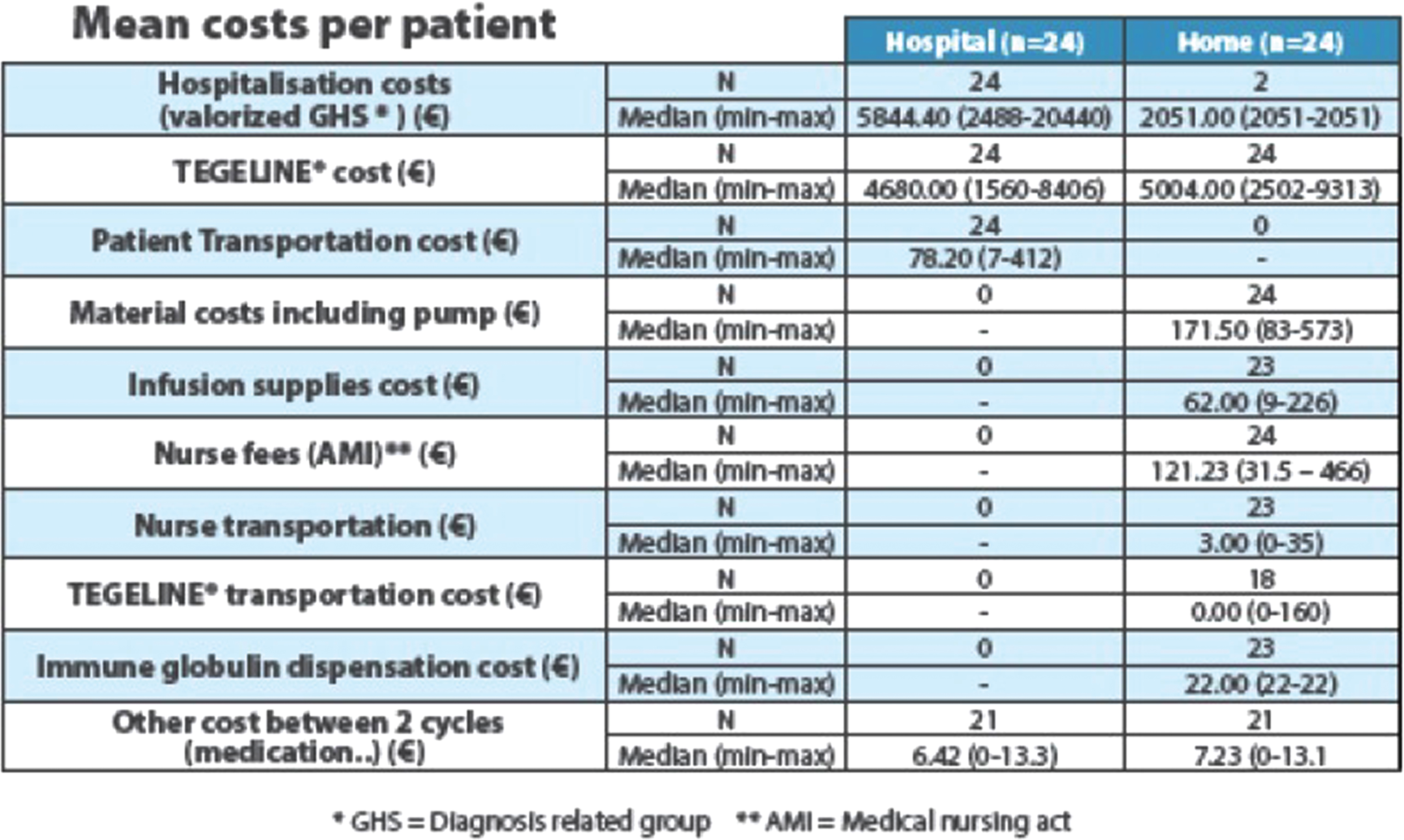
PS1Group8-015 / #301IMPACT OF DIABETIC NEUROPATHY ON DIABETES DISTRESS AND DEPRESSION IN LONGSTANDING T1DM: RESULTS FROM THE CANADIAN STUDY OF LONGEVITY IN TYPE 1 DIABETES
Topic: Group 8 – Miscellaneous / 8.4 Psychological and Neuropsychological Approaches in Neuromuscular Diseases
Johnny-Wei Bai1, Alanna Weisman1, Mohammed A.M. Farooqi1, Leif E. Lovblom1, Elise M. Halpern1, Genevieve Boulet1, Devrim Eldelekli1, Julie A. Lovshin1, Yuliya Lytvyn2, Hillary A. Keenan3, Michael H. Brent4, Narinder Paul5, Vera Bril6, David Z. Cherney7, Bruce A. Perkins8
1Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, ON, CA;
2Division Of Nephrology, Department Of Medicine, University of Toronto, Toronto, CA;
3Research Division, Joslin Diabetes Center, Boston, US;
4Department Of Ophthalmology And Vision Sciences, University of Toronto, Toronto, ON, CA;
5Joint Department Of Medical Imaging, Division Of Cardiothoracic Radiology, University Health Network, University of Toronto, Toronto, ON, CA;
6University Health Network, Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto, CA;
7Division Of Nephrology, Department Of Medicine, University of Toronto, Toronto, ON, CA;
8Division Of Endocrinology And Metabolism, Department Of Medicine, Mount Sinai Hospital, University of Toronto, Toronto, CA
Abstract: Aim: Older patients with longstanding type 1 diabetes (T1DM) are at risk of neurological and micro/macrovascular complications, which can negatively impact quality of life. We aimed to compare associations of various diabetes complications including neuropathy with depression and diabetes-related emotional distress. Methods: 323 Canadians with over 50 years of T1DM submitted a questionnaire and recent laboratory and retinopathy reports. Neuropathy status was determined using the Michigan Neuropathy Screening Instrument questionnaire potion (score≥3/15). Other complications were determined by eye-specialist reports, laboratory glomerular filtration rate<60mL/min/1.73m2 or urine albumin:creatinine ratio>2 mg/mmol if on a renin-angiotensin-system blocker and ≥3.4 mg/mmol if otherwise, self-reported cardiovascular disease (CVD), and self-reported peripheral vascular disease (PVD). Distress was measured using the Problem Areas in Diabetes (PAID) score, and depression was measured using the Geriatric Depression Scale (GDS). Factors independently associated with depression and distress were analyzed by ordinal logistic regression using separate models for each of the two outcomes. For analysis, PAID was divided into quartiles, and GDS into tertiles. Results: Among the 323 participants, 137(42.4%) had neuropathy, 212(70.2%) had retinopathy, 116(37.2%) had nephropathy, 110(33.8%) reported CVD, and 32(10.0%) reported PVD. In univariable regression analysis, presence of neuropathy was associated with higher PAID quartile (p<0.001) and GDS tertile (p=0.001), while presence of nephropathy was associated with higher PAID quartile (p=0.027), presence of PVD was associated with higher GDS tertile (p<0.001), and retinopathy and CVD were not associated with either score. In multivariable regression adjusting for each of the 5 complications, neuropathy was the only complication significantly associated with higher PAID quartile (OR 2.11, p=0.001) and the only complication significantly associated with higher GDS tertile (OR 2.95, p=0.008). In a separate multivariable regression modelling PAID and adjusting for possible confounders including age, age at onset of diabetes, sex, HbA1c, BMI, and self-reported physical activity, the presence of neuropathy was significantly associated with higher PAID quartile (OR 1.90,p=0.013). In a similar adjusted model, the presence of neuropathy was significantly associated with higher GDS tertile (OR 3.79, p=0.003). Those with a painful subtype of neuropathy had numerically higher PAID and GDS scores than those with non-painful neuropathy, but the differences were not statistically significant (median[IQR] PAID score 14.4[6.25,28.75] vs. 10.0[5.0,22.5], p=0.17; median GDS 2.5[1.0,5.0] vs. 2[1,4], p=0.44). Conclusion: In longstanding T1DM, presence of neuropathy was independently and consistently associated with emotional distress and depression and not explained by the presence of painful symptoms. Future research must explore causality and determine potential interventions to improve distress and depressive symptoms.
PS1Group8-016 / #415ORAL MOTOR COMMUNICATION INVENTORY FOR ALS: CONTENT VALIDATION
Topic: Group 8 – Miscellaneous / 8.6 Rehabilitation in Neuromuscular Diseases
Laura J. Ball1, Gar;y L. Pattee2, Jichuan Wang3
1Center For Translational Science, Children’s National Medical Center, Washington, DC, US;
2Neurology, University of Nebraska Medical Center, Lincoln, NE, US;
3Biostatistics, Children’s National Medical Center, Washington, DC, US
Abstract: The motor system is directly involved in speech production, with damage to any major division (i.e., final common pathway & cranial nerves; direct activation & cortico-bulbar/spinal pathways; indirect activation pathway & brainstem synapses; cerebellum & control circuit; basal ganglia) resulting in distinct neuromotor speech deficits(1). Speech is produced through movements triggered by nerves innervating speech subsystems: respiration (speech respiratory support), phonation (voice), resonance (velopharyngeal function), and articulation (mandible, lips, tongue)(2). Motor speech disorders resulting from amyotrophic lateral sclerosis (ALS) are classified as dysarthria (i.e., neurologic speech disorders of abnormal movement strength, speed, range, steadiness, tone, or accuracy). Dysarthria reflects damage to any of the major neurological divisions; most commonly classified as mixed spastic-flaccid in ALS(1). Reduced intelligibility is a consequence of dysarthria in ALS and has a multidimensional impact(4). Swallowing motor impairment may cause aspiration or catastrophic airway compromise(5). Dysphagia is associated with high morbidity and mortality rates and can lead to dehydration, malnutrition, or aspiration pneumonia; it has effects on social and psychological well-being(6). Reliable clinical assessment of swallowing is critical for management decisions and essential for research quantification. With progressive symptoms of bulbar impairment, people with ALS (PALS) develop complex communication needs. Strong evidence exists to support augmentative and alternative communication (AAC) interventions, with benefits including positive communication gains as well as significant decreases in communication-related frustration(7–12). Currently, no ALS-targeted comprehensive standardized protocols exist for the purpose of evaluating bulbar function through motor speech and oral-pharyngeal swallowing function. We hypothesize that disease status may be reflected in communication and swallowing functions and thereby characterized and tracked for change(13). The Oral Motor-Communication Inventory (OMCIALS) assesses bulbar domains described. Based on the ICF(15), items on the OMCIALS characterize oral motor speech, eating/swallowing, and communication . Procedures Clinical neurologists and speech language pathologists (N=7) with experience evaluating and treating PALS participated in a structured survey examining the appropriateness and relevance of OMCIALS items. Ratings of OMCIALS items were collected and content validity ratio (CVR) calculated. The content validity formula [CVR = (ne-N/2)/(N/2)] was calculated for each item, where ne represents the number of raters indicating ‘yes, the item is relevant and appropriate’, and N = total number of raters. CVR values range from +1 to –1 with positive values indicating at least half of the raters scored the item as ‘yes’. Conservatively, the minimum CVR selected to ensure that agreement was unlikely to be due to chance was 0.99. For calculations, 5-point Likert scale items were recoded as dichotomous (i.e., recoded 1 from extremely or veryappropriate, recoded 0 when rated neutral or lower). Results Experts rated domains for bulbar assessment based on the ICF. Agreement levels will be reported for key OMCIALS categories and individual tasks. Items in the majority of domains yielded high content validity; however not all items achieved CVR >0.99. Items with CVR less than the cutoff will be removed in next stage OMCIALS psychometric evaluation. Construct validity and reliability (test-retest, Chronbach’s alpha), will be evaluated during planned future clinical implementation.
PS1Group8-017 / #357REHABILITATION NURSING IN NEUROMUSCULAR DISEASES
Topic: Group 8 – Miscellaneous / 8.6 Rehabilitation in Neuromuscular Diseases
Tulay Basak
School Of Nursing, Gulhane Military Medical Academy, Ankara, TR
Abstract: REHABILITATION NURSING IN NEUROMUSCULAR DISEASES: A LITERATURE REVIEW Tulay BASAK RN, PhD Background: Most of neuromuscular diseases are slowly progressive and involve several impairments and disabilities. Rehabilitation approaches in these diseases are to protect the maximum functions as long as possible, to prevent the complications and to increase the health related quality of life. Rehabilitation nurses are part of the multidisciplinary rehabilitation team. The goal of rehabilitation nursing is to assist individuals with disability to attain and maintain maximum function by providing evidence-based nursing care. Objective. The aim of the literature review was to investigate studies that evaluated the rehabilitation in neuromuscular diseases, and to determine the research requirements of rehabilitation nursing. Methods:. Studies published were undertaken using the following databases: PubMed, Google Scholar, and EBSCO Host. Key words as “rehabilitation” “nursing” “neuromuscular diseases” along with unlimited time of were used to search the literature. Using the selected keywords, our searches identified 8 potentially relevant studies. After selection criteria of the study, a final total of 2 studies were included for the review. Results: Two studies addressing rehabilitation nursing in neuromuscular diseases. It was investigated psychosocial responses and quality of life among patient with amyotrohic lateral sclerosis and their caregivers in one of the studies. The other study present a healthcare management plan and explores an innovative to be used within a disease management program. Conclusion: There is no study evaluating the effectiveness of interventions of rehabilitation nurses for patients with neuromuscular diseases to provide evidence based nursing care. It is important for rehabilitation nurses to be aware of the result. More studies about this field are needed to determine rehabilitation nurses’ roles and responsibilities in neuromuscular diseases.
PS1Group8-018 / #361PROBLEMS OF FAMILIES LIVING WITH BOYS WITH DUCHENNE MUSCULAR DYSTROPHY (DMD) IN A DEVELOPING COUNTRY
Topic: Group 8 – Miscellaneous / 8.6 Rehabilitation in Neuromuscular Diseases
1Management, Association of Neuromuscular Disorders, İstanbul, TR;
2Management, Assocation of Neuromuscular Disorders, İstanbul, TR
Abstract: This presentation aims to discuss the problems of families living with their sons who have DMD at the distant corners of a developing country. Association of Neuromuscular Diseases of Turkey (NMD) prepared a documentary film (Kite in My Dream) taken in different 3 cities of South East Turkey, displaying the living conditions of these families and their various problems arising from living in rural and undeveloped area of the country. They suffer a lot and do not know how to handle their difficulties. They are not aware of concepts like rehabilitation, management, or improving quality of life. They need physiotherapy; they need transportation: and education. They desperately need sound and accurate information about the disease and genetic counseling. Unfortunately these services are not available to them. They have no idea about the standards of caring for DMD patients. They are not aware of the research about drug trials or exon skipping . They have no chance to reach a centre of excellence which could help them . There is a big gap between eastern and western part of Turkey.This is due to the privatization in every sector in the country. Modern centers are located in big cities. We estimate there about 1500-2000 DMD cases in Turkey. Our Association of NMD tries very hard to reach and support them all . A very hard working group of volunteers who are also patients of a neuromuscular disease try to get in touch with them and support them in different ways. Association of NMD does its best to help and support these volunteers. We know these are common problems at many other developing countries. We hope this presentation sheds a light into their challenges too.


PS1Group8-019 / #464REAL WORLD USE OF PRIVIGEN IN THE TREATMENT OF GBS AND CIDP: RESULTS OF A RETROSPECTIVE OBSERVATIONAL STUDY
Topic: Group 8 – Miscellaneous / 8.7 Others
Abstract: Objectives To better characterize the patient population treated with an IVIG product (Privigen, CSL Behring, Bern Switzerland) for GBS and CIDP, and to gain data on the doses used in clinical practice. Methods: An observational retrospective cohort study of in- and outpatients treated with Privigen for GBS and CIDP between 2008 and 2012 was conducted using a large US hospital database (Premier Research Services). Results At total of 646 patients received Privigen for GBS (991 treatment episodes) and 367 for CIDP (1663 episodes). Median patient age was 54 and 63 for GBS and CIDP, respectively. Frequent comorbidities were diabetes (23.3% and 34.3%), chronic pulmonary disease (18.6 and 19.1%), cardio- and cerebro-vascular disease (12.1 and 22.2%), neoplasia (9.1 and 12.1%), liver disease (6.8 and 5.2%), and renal disease (6.3 and 8.6%). Median Privigen dose per episode was higher in GBS (1.57 g/kg) than in CIDP (0.65 g/kg). Discussion: These results indicate that the patient population treated with IVIG (Privigen) consists mainly of adults, including the elderly and those with frequent significant comorbidities. As expected, IVIG doses were higher in GBS (high dose acute treatment only) than in CIDP (high dose initiation and lower dose maintenance therapy).
PS1Group8-020 / #455REFRACTORY POSTURAL ORTHOSTATIC TACHYCARDIA SYNDROME: EFFICACY AND SAFETY OF WEEKLY ALBUMIN INFUSIONS
Topic: Group 8 – Miscellaneous / 8.7 Others
Zaeem A. Siddiqi1, Aimee Soloway2, Derrick Blackmore3
1Neurology, University of Alberta Hospital, Edmonton, CA;
2Neurology, University of Alberta Hospital, Edmonton, AB, CA;
3University of Alberta Hospital, Edmonton, CA
Abstract: Background: Postural Orthostatic Tachycardia Syndrome (POTS) is characterized by excessive heart rate increase on assuming an upright posture, orthostatic light-headedness and syncope. Other disabling symptoms can include fatigue, exercise intolerance, chronic headaches, palpitations, nausea and abdominal discomfort, diminished concentration and sleep difficulties, tremulousness and anxiety, chest pain, and limb paresthesias. Management includes a combination of exercise, plasma volume expansion, alpha agonists and beta-blockers. About 25% have a highly disabling and resistant form of POTS that is nonresponsive to all treatments. Methods: We performed weekly infusions of 500cc of 5% albumin in normal saline in six patients with severe and longstanding POTS. All patients had previously failed a number of treatment modalities including saline infusions. All were markedly disabled and had severe restriction of routine daily activities (RDA). Results: Average observation period was nine months (range 6 to 12 months). Five of six patients reported dramatic improvement within few weeks of initiation of albumin, while one did not. Most robust improvement occurred in orthostatic light-headedness, fatigue, nausea and acral paresthesias. Brain fog and headache also showed appreciable improvement. Abdominal symptoms were least responsive. All five patients were able to resume RDA. One patient with long standing amenorrhea had resumption of monthly cycles. No major systemic or hematological adverse effects were noted. Conclusions: This is the first report of efficacy of weekly albumin infusions in intractable POTS. Most patients in our cohort showed robust improvement in a wide array of symptoms. Larger and controlled studies are needed to confirm our results.
PS1Group8-021 / #439RD-CONNECT: DATA SHARING AND ANALYSIS FOR RARE DISEASE RESEARCH WITHIN THE INTEGRATED PLATFORM AND THROUGH GA4GH BEACON AND MATCHMAKER EXCHANGE
Topic: Group 8 – Miscellaneous / 8.7 Others
Andreas Roos1, Sergi Beltran2, Davide Piscia2, Steven Laurie2, Joan Protasio2, Anastasios Papakonstantinou2, Andrés Cañada3, Jose Maria Fernández3, Mark Thompson4, Rajaram Kaliyaperumal4, Séverine Lair5, Pedro Sernadela6, Marta Girdea7, Michael Brudno7, André Blavier5, Rachel Thompson1, Volker Straub1, Matthew Bellgard8, Justin Paschall9, Marco Roos4, Peter A.C. ‘T Hoen4, Alfonso Valencia3, David Salgado10, Christophe Béroud11, Ivo Glynne Gut2, Hanns Lochmüller1
1John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, Newcastle, GB;
2Centro Nacional De Análisis Genómico (cnag-crg), Center For Genomic Regulation, Institute of Science and Technology (BIST), Barcelona, ES;
3Centro Nacional de Investigaciones Oncológicas (CNIO), Madrid, ES;
4Department Of Human Genetics, Leiden University Medical Center, Leiden, NL;
5Interactive Biosoftware, Rouen, FR;
6Deti/ieeta, University of Aveiro, Aveiro, PT;
7Centre For Computational Medicine, Hospital for Sick Children and University of Toronto, Toronto, AB, CA;
8Centre For Comparative Genomics, Murdoch University, Perth, Perth, ACT, AU;
9European Molecular Biology Laboratory,, European Bioinformatics Institute (EMBL-EBI), Cambridge, GB;
10Inserm, Umr_s 910, Aix-Marseille Université, Marseille, FR;
11Laboratoire De Génétique Moléculaire, APHM, Hôpital TIMONE Enfants, Marseille, FR
Abstract: RD-Connect is an EU-funded project building an integrated platform to narrow the gaps in rare disease research, where patient populations, clinical expertise and research communities are small in number and highly fragmented. Guided by the needs of rare disease researchers and with neuromuscular and neurodegenerative researchers as its original collaborators, the RD-Connect platform securely integrates multiple types of omics data (genomics, proteomics and transcriptomics) with biosample and clinical information – at the level of an individual patient, a family or a whole cohort, providing not only a centralized data repository but also a sophisticated and user-friendly online analysis system. Whole-genome, exome or gene panel NGS datasets from individuals with rare diseases and family members are deposited at the European Genome-phenome Archive, a longstanding archiving system designed for long-term storage of these large datasets. The raw data is then processed by RD-Connect’s standardised analysis and annotation pipeline to make data from different sequencing providers more comparable. The corresponding clinical information from each individual is recorded in a connected PhenoTips instance, a software solution that simplifies the capture of clinical data using the Human Phenotype Ontology, OMIM and Orpha codes. Results are made available to authorised users through the highly configurable online platform (platform.rd-connect.eu), which runs on a Hadoop cluster and uses ElasticSearch – technologies designed to handle big data at high speeds. The user-friendly interface enables filtering and prioritization of variants using the most common quality, genomic location, effect, pathogenicity and population frequency annotations, enabling users from clinical labs without extensive bioinformatics support to do their primary genomic analysis of their own patients online and compare them with other submitted cohorts. Additional tools, such as Exomiser, DiseaseCard, Alamut Functional Annotation (ALFA) and UMD Predictor (umd-predictor.eu) are integrated at several levels. The RD-Connect platform is designed to enable data sharing at various levels depending on user permissions. At the most basic level (“does this specific variant exist in any individual in this cohort?”) it has lit a Beacon within the Global Alliance for Genomics and Health’s Beacon Network (www.beacon-network.org). At the next stage of sharing – finding similarities between patients in different databases with a matching phenotype and a candidate variant in the same gene – it is actively involved in the development of Matchmaker Exchange (www.matchmakerexchange.org), allowing users of different systems to securely exchange information to find confirmatory cases. Finally, since all patients within the system have been consented for data sharing, users of the system, after validation and authorization, are able to access datasets from other centres, providing an instant means of gathering cohorts for cross-validation and further study. Although open to any rare disease, the platform is currently enriched for neuromuscular and neurodegenerative phenotypes and includes almost 1000 genomic datasets from the NeurOmics project (www.rd-neuromics.eu) with several other contributions in the pipeline, including 1000 limb-girdle muscular dystrophy index cases from the Myo-Seq project (www.myo-seq.org) and more. The platform is free of charge to use and is open for contributions of NGS and phenotypic data from research labs worldwide via [email protected]
PS1Group8-022 / #438CASE REPORT OF RECURRENT MENINGITIS SECONDARY TO CSF RHINORRHEA
Topic: Group 8 – Miscellaneous / 8.7 Others
Aleena F. Soomro
Neurology, ZIAUDDIN MEDICAL UNIVERSITY AND TEACHING HOSPITA;, KARACHI, PK
Abstract: Recurrent Bacterial meningitis is a severe, potentially life-threatening infection that is associated with high rates of morbidity and significant disability in survivors with overall mortality rates around 20% to 25% .Potential long-term neurological sequelae include cranial nerve palsies, hemiparesis, hydrocephalus, and seizures as well as visual and hearing impairment. Recurrent bacterial meningitis is a much less common phenomenon but generally poses a considerable diagnostic challenge. The early diagnosis of any underlying pathology is crucial to prevent further episodes and improve the overall outcome for the affected individual.CSF rhinorrhea leads to disruption of the barriers between the sinonasal cavity and the anterior and middle cranial fossae which is the underlying factor leading to the discharge of CSF into the nasal cavity. The resulting communication with the CNS can result in a multitude of recurrent bacterial meningitis complications that impart significant morbidity and potentially disastrous long-term deficits for the patient. this case report highlightsan 18 year old male witha history of road traffic accident 8 years back leading to defect in cribriform plate of ethmoid bone resulting in csf rhinorrea and presenting with recurrent episodes of bacterial meninngitis diagnosed with csf studies and radilologic evidence.
PS1Group8-023 / #437CLINICAL PRESENTATION OF ANTI-NMDA ENCEPHALITIS
Topic: Group 8 – Miscellaneous / 8.7 Others
Rabail Karim, Bashir Soomro
Neurology, ziauddin hospital, karachi, PK
Abstract: ABSTRACT Anti-NMDA Encephalitis is an immune mediated encephalitis associated with antibodies against NR1-NR2 heteromers of the NMDA receptor. This encephalitis is a severe, curable and potentially reversible disorder that presents as a multistage illness proceeding from psychosis, memory loss, seizures and language disintegration into a state of unresponsiveness with dystonia and autonomic dysfunction. Mostly affects young females with increased incidence in those having ovarian teratoma. Early diagnosis is essential since outcome depends on timely immunotherapy and in paraneoplastic cases, resection of tumor. As its one of the rarity, I decided to report a case of it illustrating clinical presentation, so that anti-NMDA encephalitis should be kept in differential diagnosis. This case report highlights a 35 years old female initially managed as Meningoencephalitis got readmitted with status epilepticus, psychiatric symptoms and dystonia, was diagnosed as anti-NMDA encephalitis on clinical and radiological evidence, responded well to steroids.
PS1Group8-024 / #427NUDT15 VARIANT IS THE MOST COMMON VARIANT ASSOCIATED WITH THIOPURINE-INDUCED EARLY LEUKOPENIA AND ALOPECIA IN KOREAN PATIENTS WITH VARIOUS NEUROLOGICAL DISEASES
Topic: Group 8 – Miscellaneous / 8.7 Others
Sun-Young Kim1, Dae-Seong Kim2, Jin-Hong Shin2, Jin-Sung Park3, Sa-Yoon Kang4, Ki-Jong Park5, Tai-Seung Nam6, So-Young Huh7, Jong-Kuk Kim8
1Neurology, Ulsan Univerisity Hospital, Ulsan, KR;
2Pusan National Universit Hospital, Yangsan, KR;
3Kyungpook National Univerisity Hospital, Daegu, KR;
4Cheju National University Hospital, Jeju, KR;
5Gyeongsang National University Hospital, Jinju, KR;
6Chonnam National University Hospital, Gwangju, KR;
7Kosin University Hospital, Pusan, KR;
8Dong-A University Hospital, Pusan, KR
Abstract: (Background & Significance) Azathioprine (AZT), commonly used in various neurological diseases, could be complicated by life-threating leukopenia. This leukopenia has been known to be associated with genetic variation in TPMT (thiopurine S-methyltransferase). In recent study, NUDT15 R139C was strongly associated with AZT induced leukopenia in Korean and Caucasian population with inflammatory bowel disease. We performed an association study to investigate and replicate the association of TPMT/NUDT15 with AZT induced leukopenia in Korean patients with various neurological diseases. (Methods) A total of 85 patients with neurological diseases in whom AZT treatment was indicated were enrolled from multi center. DNA sequencing for exons of TPMT/NUDT15 were performed by Sanger method. Clinical data for these patients were analyzed from medical records with specific focus in leukopenia (early vs. late, defined as early leukopenia cases who developed leukopenia within 8 weeks of AZT therapy). (Results) Of the 85 patients, 20 patients (23.5%) patients showed leukopenia (7 patients with early leukopenia, 19 patients with late leukopenia). Known intronic and exonic TPMT polymorphisms do not correlate with AZT induced leukopenia. Five patients developed early leukopenia with severe grade (white blood cells<1,000mm-3) and severe alopecia. All of them having both of early severe leukopenia and alopecia had homozygous (T/T) for R139C. Patients with TT genotype were exquisitely sensitive to AZT, compared with those with T/C and C/C genotype, who tolerated 80% and 75% with a maintenance dose (50~100 mg). The interval from the onset of AZT therapy to the development was shorter and the grade of observed leukopenia was more severe in patients with T/T genotype compared with those with C/T genotype. (p=0.001, p=0.000, respectively) Patients with C/T genotype showed a significant high risk of early leukopenia compared with those with C/C genotype. Overall, in our study, the association of R139C with early leukopenia was replicated in our Korean patients with various neurological diseases. (OR 48.9, p=7.61 × 10-5) However, we could not confirm the association with late leukopenia in our patients. Mean maintenance dose (mg) of AZT and discontinuation rate (%) were not significantly different between patients with the C/T genotype and C/C genotype. (68.75±45.81 vs. 70.42±40.26, 25% vs. 20%). (Conclusions) We verified the previously reported association of R139C with early leukopenia in Korean patients with various neurological diseases. The treatment with AZT should be avoided for patients with the T/T genotype. However, late leukopenia could not be predicted by R139C genotypes..
PS1Group8-025 / #412DATABASE OF NEUROMUSCULAR DISEASES IN REGION OF CONCEPCION IN CHILE
Topic: Group 8 – Miscellaneous / 8.7 Others
Mario Fuentealba
Neurology, University of Concepcion, concepcion, CL
Abstract: Introduction: There are few epidemiological studies in the world about the group of neuromuscular diseases, and there are none in our country. The creation of clinical databases that allow the appropriate recruitment of patients for epidemiological studies may be the first step in reversing this situation. In this context we decided to review the experience of the Neuromuscular Disease Clinic of the Neurology Department at the Regional Hospital of Concepción. Method: 106 electronic medical records were reviewed from the second semester of 2014 to the first semester of 2015, creating a database that considers the neuromuscular diagnosis and epidemiological data. The patients that met the previously defined inclusion criteria were included in the registry until reaching 100 patients. Each case was registered in three neuromuscular diagnostic levels: topographic, etiopathogenic and specific. Results and Conclusions: At the topographic diagnosis level, significant statistical differences were observed in the type of diagnosis presented by the patients, with a higher frequency for diseases that affect the peripheral nerve (40%), followed by the neuromuscular junction (32%) and muscle (22%), being the motor neuron the least frequent (6%). At the etiopathogenic diagnosis level, similar relative frequencies were observed of autoimmune myasthenia gravis (32% ), acquired polineuropathies (30%) and hereditary neuromuscular diseases (22%). In both these levels there is consistency with the results of epidemiological studies of neuromuscular diseases in adult patients. At the specific diagnoses, an unusual high frequency of patients with ocular myasthenia (13%) is seen in our series, noting that its age average is significantly higher than that of patients with generalized myasthenia .
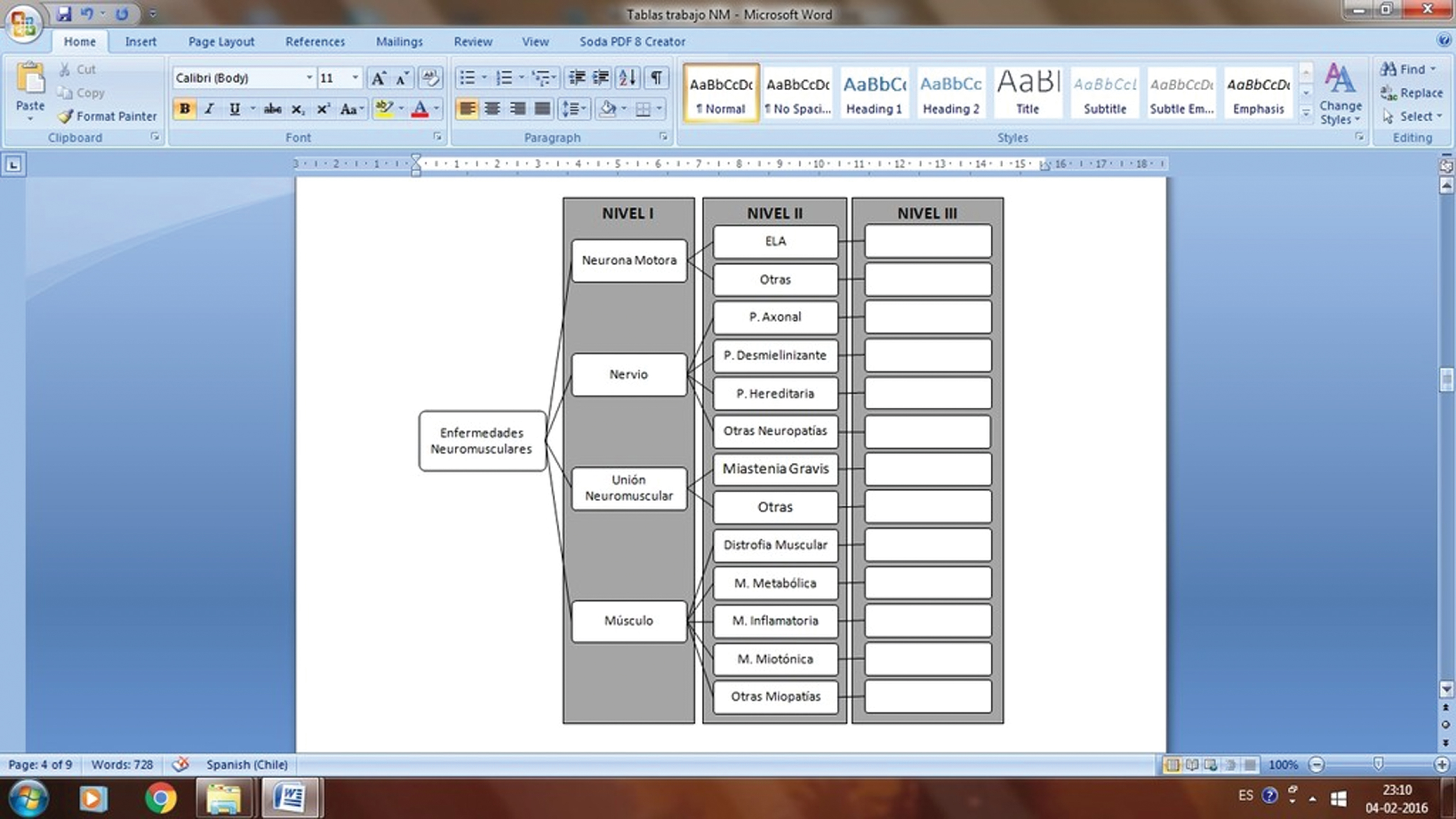
PS1Group8-026 / #255SEROPREVALENCE OF HUMAN T-LYMPHOTROPIC VIRUS TYPE 1 IN PATIENTS WITH SURGICAL HISTORY IN KAGOSHIMA, SOUTHERN JAPAN
Topic: Group 8 – Miscellaneous / 8.7 Others
Yuichi Tashiro1, Eiji Matsuura1, Satoshi Nozuma1, Akihiro Hashiguchi1, Osamu Watanabe1, Hiroshi Takashima2
1Neurology And Geriatrics, Kagoshima University Graduate School of Medical and Dental Sciences, Kagoshima city, JP;
2Neurology And Geriatrics, Kagoshima University Graduate School, Kagoshima city, JP
Abstract: [Introduction] Human T-lymphotropic virus type 1 (HTLV-1) is known as causal virus of adult T-cell leukemia/lymphoma(ATLL), HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) and HTLV-1-associated Uveitis(HAU). It has been suggested that HTLV-1 associates with some other diseases such as Sjögren syndrome, polymyositis, polyneuropathy, and infectious dermatitis. Because HTLV-1 infection rate is different by generation and region, actual HTLV-1 prevalence is required for elucidating the causal connection between HTLV-1 and the diseases in a region. We report seroprevalence of HTLV-1 by generation during 15 years in Kagoshima, southern Japan, as known as an HTLV-1 endemic area. [Method] 23,710 patients (11,069 males and 12,641 females) with surgical history in Kagoshima University Hospital since January 2001 to December 2014, are screened for the presence of serum anti-HTLV-1 antibody. We calculated HTLV-1 seropositivity rates by genders, generations, and periods (2001-2005, 2006–2010, 2011–2014). [Result] HTLV-1 seropositivity rates in patients of the 10s, 50s, and 80s are 1.1%, 9.8%, and 17.6%, respectively. Higher seropositive rates were noted in elder groups. While both of male and female seropositivity are equally about 10% (male 9.0%, female 10.5%) in the 50s, the seroprevalence of female are higher than that of male in the 60s and elder ages. The difference in ratio between the sexes tends to increase by ages. Peaks of HTLV-1 seropositivity are 15.1% in the 70s of male and 22.0% in the 80s of female. The seropositivity rates of any groups classified by age of birth are almost as same as the rate after 10 years. [Discussion] The current HTLV-1 prevalence remains markedly high in the people older than 50 years old in Kagoshima. Our results suggest that high HTLV-1 seropositivity rate in elder female patients is attributed to any factor of those days, other than horizontal transmission including sexual transmission.
PS1Group8-027 / #395HIGH RISK BREAST CANCER SCREENING IN WOMEN WITH NEUROFIBROMATOSIS TYPE 1
Topic: Group 8 – Miscellaneous / 8.7 Others
Jeanna Mccuaig1, Shelley Westergard1, Catherine Maurice2, Paul Kongkham3, Galareh Zadeh3, Carolina Barnett-Tapia4, Vera Bril4, Raymond Kim1
1Princess Margaret Cancer Centre, Toronto, ON, CA;
2Neurology, Princess Margaret Hospital, Toronto, ON, CA;
3Toronto Western Hospital, Toronto, ON, CA;
4Toronto General Hospital, Toronto, ON, CA
Abstract: Women with neurofibromatosis type 1 (NF1) have an estimated 4- to 5-fold increased risk of breast cancer, particularly under age 50. In the province of Ontario, women between age 30 and 69, who have a genetic or familial breast cancer risk estimated to be a ≥25% lifetime risk, may have annual mammograms and breast MRIs arranged through the Ontario Breast Screening Program High Risk screening program (OBSPHR). To determine the feasibility and outcomes of high-risk breast screening for women with NF1, a review of patients seen in the Elisabeth Raab Neurofibromatosis Clinic (ERNFC) in Toronto between May 2015 and March 2016 was conducted. A date of May 2015 was selected as genetic services and regular referrals to the OBSPHR began at this time. Thirty women with a known mutation in the NF1 gene were identified and 14 (46.7%) were referred to the OBSPHR. Of the 16 who were not referred, 15 were excluded from referral based upon their age; 14 (3.3%) were under age 30 and 1 (3.3%) was over age 69. An additional woman was excluded based on a previous bilateral mastectomy at age 52 following a diagnosis of bilateral breast cancer. A second woman had a history of breast cancer at age 45; however, she did not undergo bilateral mastectomies and was referred to the OBSPHR. Of the 14 women referred to OBSPHR, 3 have received high-risk screening at the Princess Margaret Cancer Centre, 8 have upcoming appointments, two have not yet booked an appointment, and one is being screened at an outside hospital. In the first round of screening, 2 breast cancers were identified; the ages at diagnosis were 44 and 45. Considering those over age 30, 4 women (25%) followed in the ERNFC have a personal history of breast cancer, which is consistent with the elevated risk of breast cancer reported in women with NF1. Referral for annual high risk breast screening is acceptable to eligible patients, with the majority scheduling appointments. The combination of mammogram and breast MRI is effective in identifying breast cancer in high risk women, including women with NF1.
PS1Group8-029 / #365UNDERSTANDING THE CANADIAN NEUROMUSCULAR DISEASE RESEARCH LANDSCAPE
Topic: Group 8 – Miscellaneous / 8.7 Others
Megan Johnston1, Christopher Macdonald1, Jeff Dilworth2, Hans Katzberg3, Jean K. Mah4, Lawrence Korngut1
1Department Of Clinical Neurosciences, Hotchkiss Brain Institute, University of Calgary, Calgary, AB, CA;
2Cellular And Molecular Medicine, University of Ottawa, Ottawa, AB, CA;
3Neurology, Toronto General Hospital, Toronto, CA;
4Alberta Children’s Hospital, University of Calgary, Calgary, AB, CA
Abstract: BACKGROUND: The Canadian Neuromuscular Diseases Network (CAN-NMD) is a national network of healthcare providers, scientists and researchers, patients, families and other stakeholders with an interest in neuromuscular disease (NMD) and therapy. Funded by the Canadian Institutes of Health Research and Muscular Dystrophy Canada through a Network Catalyst grant obtained in April of 2014, the network aims to improve clinical care for Canadian NMD patients, enhance capacity for research, promote collaborative work, and increase education and training opportunities in NMD in Canada. Until recently, the lack of a pan-Canadian network for NMD research has limited collaboration among researchers; they must rely on their local connectivity or other national or international network to develop collaborations. In particular, collaboration between basic science and clinical research experts, an essential ingredient for successful translational research, is extremely difficult to establish as NMD and professional events rarely invite both types of experts. There is a substantial opportunity to facilitate enhanced interactions and collaboration across the Canadian research landscape through CAN-NMD. As an initial step towards understanding how best to accomplish enhanced collaboration, the CAN-NMD Research Task Force undertook a web-based survey to assess the Canadian landscape. The survey aimed to broadly engage NMD stakeholders, to appreciate the scope of work in the Canadian NMD research community, and to identify perceived needs and potential opportunities. METHODS: A 40-question survey was created by members of the CAN-NMD Research Task Force between March and May 2015. The survey was developed after examining the literature for similar efforts to characterize a research community in other countries or disciplines. The survey was translated into French by a certified translator based in Canada. The bilingual survey was mounted on the Survey Gizmo platform (, Boulder, CO, USA). Links to the survey were distributed by email to CAN-NMD members and known NMD researchers across Canada. A link to the survey was also publicly available on the CAN-NMD website. RESULTS: 32 survey respondents represented 5 provinces and several research disciplines across 12 academic institutions, with an equal number of MD and PhD holders. Respondents were mainly mid-career, and an encouraging 29% of respondents were early career. Overall, CAN-NMD has identified 137 researchers across the country, and 122 (89%) have become members of the Network. Key services and added value for the Network included: assisting with identification of funding opportunities, enabling collaboration through meetings and networking, and creating an interactive research road map. CONCLUSIONS: The Canadian research community is vibrant; the members are motivated to increase their connectivity and to further productivity in NMD research. CAN-NMD is providing a unique opportunity to enhance and facilitate collaborations across the Canadian research landscape.
PS1Group8-030 / #317CANADIAN NEUROMUSCULAR DISEASES NETWORK (CAN-NMD) THE DEVELOPMENT & IMPLEMENTATION OF A WEB-BASED KNOWLEDGE SHARING AND EXCHANGE PLATFORM
Topic: Group 8 – Miscellaneous / 8.7 Others
Gracia Mabaya1, Craig Campbell1, Cynthia Gagnon2, Megan Johnston3, Laura Mcadam4, Jeremy Dixon5, Kelvin Jones6, Charles Kassardjian7, Aneal Khan3, Jane Mitchell8, Annie Plourde9, Maryam Oskoui10, Lawrence Korngut3
1Department Of Paediatric Neurology, Children’s Hospital London Health Sciences Centre, London, ON, CA;
2School Of Rehabilitation, Faculty Of Medicine, University of Sherbrooke, Jonquière, QC, CA;
3Department Of Clinical Neurosciences, Hotchkiss Brain Institute, University of Calgary, Calgary, AB, CA;
4Department Of Paediatrics, Univeristy Of Toronto, Bloorview Research Institute, Holland Bloorview Kids Rehabilitation Hospital, Toronto, ON, CA;
5Board Of Directors, Muscular Dystrophy Canada, Calgary, AB, CA;
6Department Of Engineering, Faculty Of Physical Education And Recreation, University of Alberta, Edmonton, AB, CA;
7Division Of Neurology, St. Michael’s Hospital, University of Toronto, Toronto, ON, CA;
8Department Of Pharmacology And Toxicology, University of Toronto, Toronto, ON, CA;
9Groupe De Recherche Interdisciplinaire Sur Les Maladies Neuromusculaires, CRDP Le Parcours, Jonquière, QC, CA;
10Montreal Children’s Hospital Divison Of Pediatric Neurology, McGill University Health Centre, Montreal, QC, CA
Abstract: BACKGROUND: To date in Canada, despite a highly collaborative neuromuscular disease (NMD) community, there has been no formalized effort to promote collaboration between NMD stakeholders nation-wide. Collaboration and knowledge sharing are further limited by the limited size of individual teams at the local level and their geographic separation across regions and the nation overall. In September 2015, the Network released its KT Framework aiming to provide a roadmap through which Network-wide KT activities can be planned, implemented and evaluated. As a next step in knowledge translation enhancement the Network is launching a web-based platform to facilitate enhanced collaboration and knowledge sharing beyond geographical and temporal constraints. METHODS: The CAN-NMD features a Knowledge Translation (KT) Task Force, which supports all knowledge-related activities initiated by the Network’s four other task forces. Knowledge brokering has been adopted as a role-based knowledge translation strategy with the ultimate goal of enhancing NMD clinical practice, research and education through knowledge sharing and exchange, and increasing collaborations among NMD healthcare providers, researchers, patients, families and other stakeholders. During the development phase of the KT web platform, the KT task force has employed two needs assessment strategies. The first consisted of the completion of a 15-minute online survey disseminated to various stakeholders across Canada. The second strategy consists of consultations with neuromuscular clinical care teams across Canada, conducted both in person and via videoconference, facilitated by the Network’s knowledge broker. RESULTS: The Network’s KT task force is in the process of implementing a KT web platform. This platform will serve to connect professionals and increase their ability to share best practice experiences in relation to NMD clinical care, research and education. The platform will initially target two audiences: neuromuscular healthcare providers (clinicians and allied health staff) and NMD researchers (both clinical and basic scientists). The short-term goals of the platform are: 1) Enabling collaborations and relationship building between the target audiences; 2) Enabling knowledge sharing and exchange between the target audiences through file sharing and the ability to store data; 3) Promoting the use of research evidence by healthcare providers; and 4) Providing educational opportunities through lunch & learns, cases of the month and other similar experiences. In the long-term the platform will assist the Network in transforming gaps identified in clinical practice. CONCLUSION: The implementation of the KT web platform represents an important and unique opportunity to increase collaborations and best practice sharing among NMD professionals across Canada. It is hoped that the platform will become a critical component in enhancing the delivery of evidence informed clinical care for Canadian NMD patients by facilitating rapid knowledge and best-practice sharing amongst researchers and healthcare providers across the country.
PS1Group8-031 / #315UNDERSTANDING DECISION NEEDS FOR RESPIRATORY INTERVENTIONS IN PAEDIATRIC NEUROMUSCULAR DISORDERS FROM THE PERSPECTIVE OF HEALTHCARE PROVIDERS
Topic: Group 8 – Miscellaneous / 8.7 Others
Gracia Mabaya1, Sherri Katz2, Margaret L. Lawson3, April Price4, Dhenuka Radhakrishnan2, Jean K. Mah5, Lawrence Korngut6, Hugh Mcmillan7, Cheryl Scholtes8, Allyson Shephard9, Melissa Heletea2, Lynda Hoey2, Craig Campbell1
1Department Of Paediatric Neurology, Children’s Hospital London Health Sciences Centre, London, ON, CA;
2Division Of Paediatric Respirology, Children’s Hospital of Eastern Ontario, Ottawa, ON, CA;
3Division Of Endocrinology And Metabolism, Children’s Hospital of Eastern Ontario, Ottawa, ON, CA;
4Paediatric Respirology, Children’s Hospital London Health Sciences Centre, London, ON, CA;
5Departments Of Pediatrics And Clinical Neurosciences, University of Calgary, Calgary, AB, CA;
6Division Of Neurology, Department Of Paediatrics, Hotchkiss Brain Institute, University of Calgary, Calgary, AB, CA;
7Department Of Paediatric Neurology, Children’s Hospital of Eastern Ontario, Ottawa, ON, CA;
8Department Of Physiotherapy, Thames Valley Children’s Centre, London, ON, CA;
9Family Decision Services, Children’s Hospital of Eastern Ontario, Ottawa, ON, CA
Abstract: Background: Paediatric neuromuscular disorders (NMD), including Duchenne muscular dystrophy (DMD) and spinal muscular atrophy (SMA), are associated with progressive muscle weakness leading to respiratory failure. Non-invasive ventilation (NIV) and lung volume recruitment (LVR) are therapies used to mitigate respiratory complications. Shared decision-making (SDM) is a collaborative model of health decision making (DM), where healthcare providers (HCP) and patients participate in an exchange of information to integrate the best available evidence with patient values and preferences, in order to make a shared decision. Objectives: Following the assembling of a steering group (including physicians, registered nurses and allied healthcare professionals from Neurology, Respirology and SDM) to refine the scope of the study study objectives were derived. The study goal was to conduct a thorough needs assessment to identify decision-making needs related to LVR and NIV by engaging DMD and SMA patient advocacy groups, patients/caregivers, and HCPs. Decision needs identified within the HCP participant group will be the focus of this report. Methods: A focus group was held with 4 HCPs (all physicians) in September 2015. Findings guided the development of a questionnaire that was then disseminated online to 30 NMD HCPs across Canada. The objective of the questionnaire was to understand patients DM processes, the types of support they receive when making their decision, the certainty of their decision, their use of decision aid tool(s) when making the decision, their knowledge about the decision as well as their choices, and their decision satisfaction from a HCP’s perspective. Results: Findings obtained through the focus group highlighted HCPs’ concerns about the different institutional philosophies that exist around treatment options given to patients across healthcare facilities in Canada as well as differences between DMD and SMA disease trajectories. Data obtained via the online survey thus far shows that for both DMD and SMA, all HCPs primarily provide the option to use NIV using a bilevel positive airway pressure (BiPAP) machine, less commonly recommending LVR using either a cough assist machine or an Ambu bag. HCPs affirm that their role is to provide support or advice, enabling the patient and their family to make the decision on their own. Half of the time conflicts arise because patients are lacking information on the probability of benefits and harms for various treatment options. Conclusion: These findings validate the strong need for SDM to provide patients and families with evidence-based probabilities on benefits and harms of different treatment options for respiratory care and account for the varying disease trajectories in DMD and SMA. There is particularly a need to establish a clinical environment conducive to SDM to support patients in making decisions that are informed and values-based and able to mitigate the internal conflict that may be experienced by HCPs when a patient’s decision does not reflect the best option from the HCP’s perspective. We anticipate that the data from all collection methods will further provide results to support the development of a sound SDM environment as well as innovative and evidence-informed decision support tools for respiratory interventions.
PS1Group8-032 / #331A PROPOSAL: ISAACS SYNDROME (ACQUIRED NEUROMYOTONIA) DIAGNOSTIC CRITERIA
Topic: Group 8 – Miscellaneous / 8.7 Others
Osamu Watanabe1, Kimiyoshi Arimura2, Hiroshi Takashima3
1Neurology, Kagoshima University Hospital, Kagoshima, JP;
2Neurology, Ookatsu Hospital, Kagoshima, JP;
3Neurology And Geriatrics, Kagoshima University Graduate School, Kagoshima, JP
Abstract: Acquired neuromyotonia, also called Isaacs’ syndrome has characteristic features of the clinical manifestations and electrophysiological findings. VGKC complex antibodies are well known as its biomarker, but the positive rate is around 30%. In Japan, the number of patients of the Isaacs’ syndrome is estimated to be at least around 100 cases in Japan than the number of VGKC complex antibody screening requests to our laboratory. We made provisional diagnostic criteria (the following) and gathered the opinion through the announcement in the Japanese associated societies. A. Major symptoms/signs: 1. neeuromyotonia (indispensable: grip myotonia without percussion myotonia) 2. continuous muscle cramp or stiffness. Peripheral Nerves Hyperexcitability (ex. myokymic discharges, meuromyotonic dischartes) on EMG. 3. anti-VGKC complex antibodies positive 4. effectiveness of immunemodulation/immunesuppression therapy (ex. Steroids, Plasma exchanges, others) B. Minor (supporting) symtoms/signs: 1. hyperhidrosis. 2. numbness or pain of limbs 3. thymoma 4. skin color changes 5. other autoantibodies (anti-AChR antibodies, ANA, TPO, etc.) Definite: all of A. Probable (1 or 2 of A, more than 2 points of B). Possible (1 or 2 of A, one point of B).
PS1Group8-033 / #258UNDERSTANDING THE PERSPECTIVES OF YOUNG ADULTS WITH DUCHENNE MUSCULAR DYSTROPHY AS THEY TRANSITION TO ADULTHOOD AND ADULT HEALTH CARE
Topic: Group 8 – Miscellaneous / 8.7 Others
Sally Lindsay1, Laura Mcadam2, Tania Mahenderin3
1Department Of Occupational Science & Occupational Therapy, Univeristy Of Toronto, Bloorview Research Institute, Holland Bloorview Kids Rehabilitation Hospital, Toronto, ON, CA;
2Department Of Paediatrics, Univeristy Of Toronto, Bloorview Research Institute, Holland Bloorview Kids Rehabilitation Hospital, Toronto, ON, CA;
3Holland Bloorview Kids Rehabilitation Hospital, Bloorview Research Institute, Toronto, ON, CA
Abstract: The majority of children with Duchenne Muscular Dystrophy now live into adulthood. Such young adults will have specialized health care needs that are unfamiliar to most adult health care providers. There are often several challenges encountered in the process of transitioning to adult health care including lack of readiness and lack of specialized health care providers. It is particularly critical to ensure that the proper mechanisms are in place as youth transition to adult care so that their health and well-being are not impacted. Adolescents and young adults with disabilities are often significantly under-serviced in health and social care and many ‘fall through the cracks’ as they transition to adulthood. Such gaps in the system combined with inadequate transition arrangements are particularly concerning because they are linked with poorer long-term health outcomes, increased hospitalizations and reduced opportunities to participate in the community. Although there is a growing literature on transitions to adult care, youth with Duchenne Muscular Dystrophy (DMD) have largely been neglected. Exploring this population is vital because they are now living well into adulthood and have different needs than other clinical groups. DMD is often seen as a pediatric disease associated with a corresponding lack of transition planning. Thus, youth with DMD may not have been empowered or supported in decision-making about their care and other aspects of their lives. It is critical, therefore, to understand youths’ needs to inform the development of an effective transition pathway. Objective: to explore clinician, youth and parent experiences of transitioning from an adult DMD clinic within a pediatric hospital to an adult health care facility. Methods: This study involves a retrospective qualitative design (16 semi-structured interviews) with 9 recently transitioned young adults and/ or their parents and 7 clinicians involved in the transition. The interview guide was based on key transition principles. An open-coding, constant-comparison thematic approach was used to analyze the data. Results: Clinicians, young men with DMD and their caregivers reported several enablers and barriers in transitioning from the adult DMD clinic within the pediatric hospital to the adult health care facility. Enablers included structural factors (leadership and advocacy), availability of care (inter-agency partnerships, teamwork and specialists knowledgable about DMD), and relational factors (effective communication, development of trust and rapport and family involvement). Barriers that influenced the transition included lack of clarity around the timing of the transition and lack of funding at the adult health care center to take on additional clients. Youth and parents noted differences in the models of care between the pediatric and adult health care centre. Finally, there were difficulties at the indidiviaul readiness of youth and connecting them to other community resources.
PS1Group8-034 / #233CHARACTERISTICS DEVELOPMENT OF A NEW IVIG (I10) Â “ THE QUALITY BY DESIGN APPROACH (QBD)
Topic: Group 8 – Miscellaneous / 8.7 Others
Philippe Paolantonacci1, Catherine Decoupade1, Philippe Appourchaux2, Catherine Michalski2, Rabye Ouaja3, Ousmane Alfa Cisse3, Ludovic Burlot1
1Biopharmaceutical Development Department, LFB, LES ULIS, FR;
2Industrial Department, LFB, LES ULIS, FR;
3Global Medical Department, LFB, LES ULIS, FR
Abstract: QbD in pharmaceuticals involves designing and developing pharmaceutical formulation and manufacturing process to help ensure product quality1. It is the latest standard of quality recommended by the FDA and the EMA since 20092. This approach was adopted by the LFB in order to ensure a better quality of his IvIG for the patients and to offer a product that meets current regulatory requirements The process development of our IvIG was focused on obtaining a high purity product with a favorable safety profile by removing targeted unwanted accompanying proteins. More precisely, the development of I10 purification process was mainly driven by five key objectives which were part of I10 Quality Target Product Profile (QTPP):
• Removal of activated coagulation factors to reduce the risk of thrombogenic adverse events,
• Removal of IgA to avoid immune responses in patients deficient in IgA,
• Removal of Anti-A and Anti-B haemagglutinins to avoid adverse events on patients of blood groups A, B or AB,
• Reduction of aggregates to avoid adverse events through complement activation,
• Reduction of potential adventitious and non-adventitious agents.
Three purification steps by precipitation or chromatography and two dedicated viral removal steps were specifically developed to reach these objectives. The sequence of these unit operations forms the core I10 purification process, called IGNG (ImmunoGlobulin New Generation) Process. This process is also used for the 5% liquid human normal immunoglobulin for intravenous administration of LFB BIOMEDICAMENTS marketed in France since 2010. This product has the same purity profile as I10, only product concentration and formulation differ.
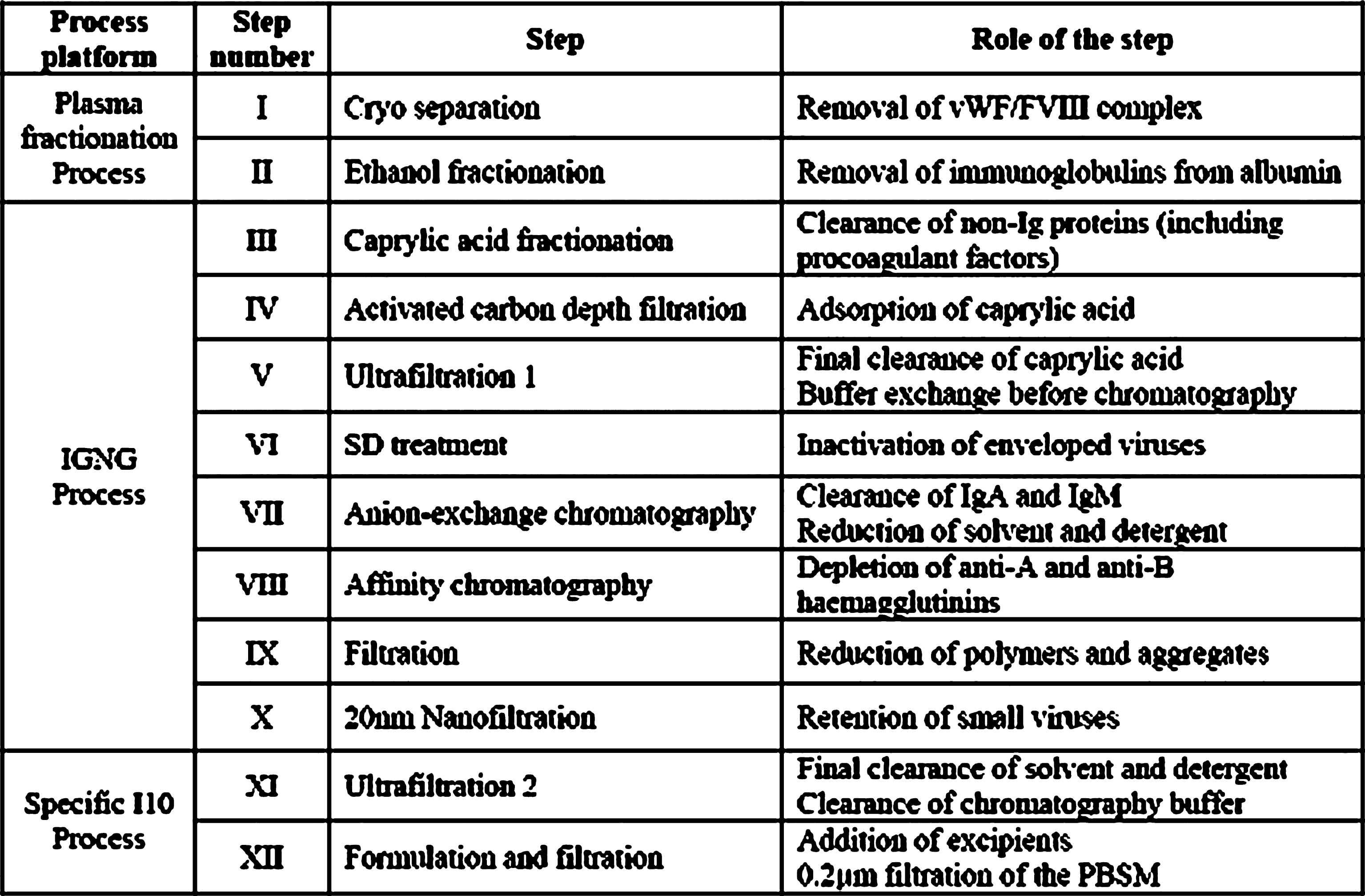
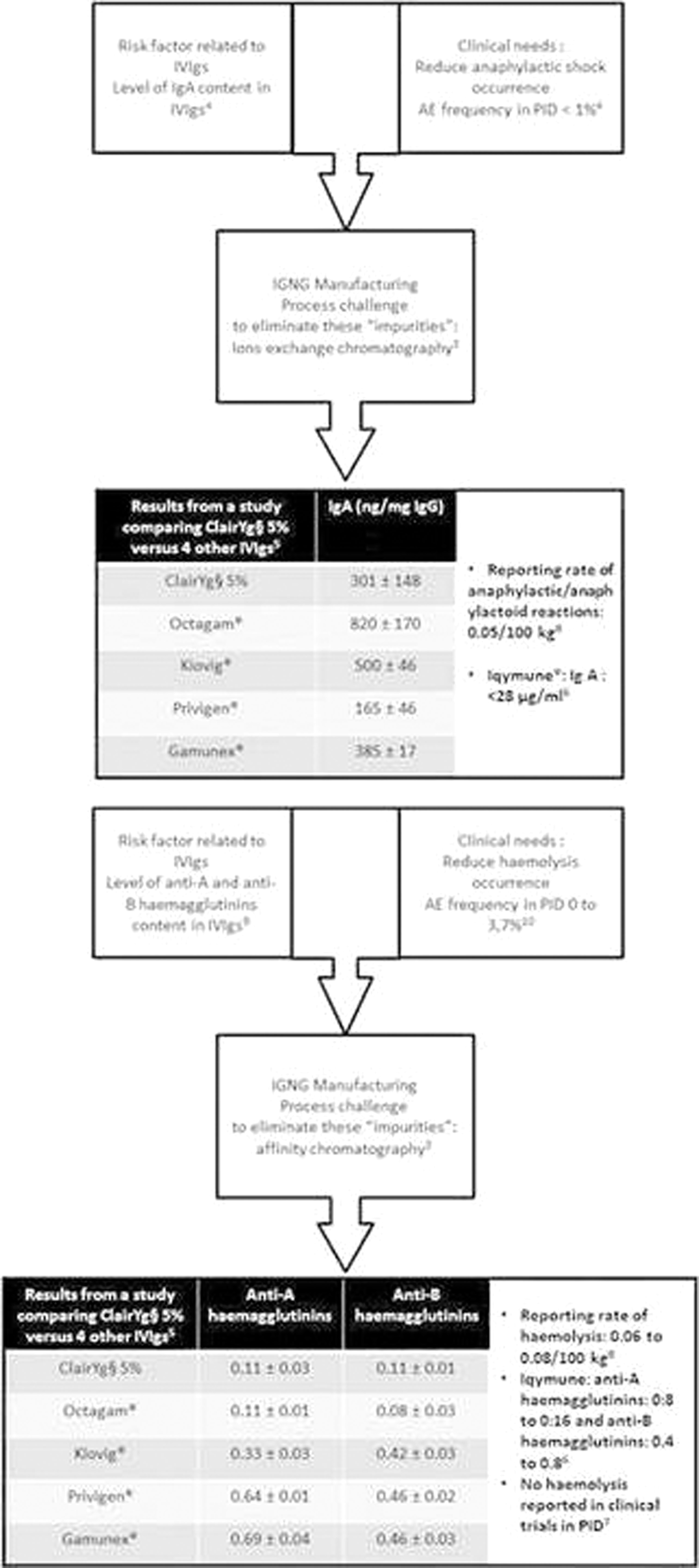
PS1Group8-035 / #231PURIFICATION OF IVIG (INTRAVENOUS IMMUNOGLOBULIN) FROM IGNG MANUFACTURING PROCESS TO OPTIMIZE PRODUCT TOLERABILITY PROFILE: EXAMPLE OF HUMAN NORMAL IMMUNOGLOBULIN (IQYMUNE® 100 MG/ML, SOLUTION FOR INFUSION)
Topic: Group 8 – Miscellaneous / 8.7 Others
Philippe Paolantonacci1, Catherine Decoupade1, Philippe Appourchaux2, Catherine Michalski2, Rabye Ouaja3, Sophie Puget4, Ludovic Burlot1
1Biopharmaceutical Development Department, LFB, LES ULIS, FR;
2Industrial Department, LFB, LES ULIS, FR;
3Global Medical Departement, LFB, LES ULIS, FR;
4International Scientific Affairs Unit, LFB, LES ULIS, FR
Abstract: Introduction: During IVIg replacement therapy in primary immunodeficiency patients (PID), minor adverse events such as chills and fever occur frequently whilst major adverse events (AEs) such as anaphylactic shock, thrombosis or haemolysis are rare (Ref. 1). These associated risks were reported to be related to impurities found in each product (Ref. 1), and therefore vary, depending upon each IVIg’s purification process which is unique and specific Methods: LFB has designed a novel purification process (the IGNG platform) for eliminating or reducing impurities in order to reduce the occurrence of adverse events, whilst maintaining the structural and functional integrity of IgG, and ensuring a constant quality of product batches. ClairYg®, 5% liquid preparation licensed in France and other international markets, was the first product developed from IGNG platform and based on this experience a 10% liquid IVIg, Iqymune® has also now been developed. ClairYg® and Iqymune® share the same manufacturing process with a different formulation (5% versus 10%) Results :
• 5% ready-to-use liquid preparation for ClairYg® and 10% ready-to-use liquid IVIg preparation for Iqymune®
• Low level of IgA for both preparations
• Low levels of anti-A and anti-B haemagglutinins for both
• Low levels of activated factor XI and kallikrein content for both
• Saccharose- and maltose-free for both
ClairYg® and Iqymune® (Ref. 2, 3, 4) Conclusion: IGNG purification process, based on LFB’s experience with ClairYg®, has been designed to produce an immunoglobulin (Iqymune®) with a consistently low level of impurities, theoretically leading to improved tolerability (Ref.2, 6).
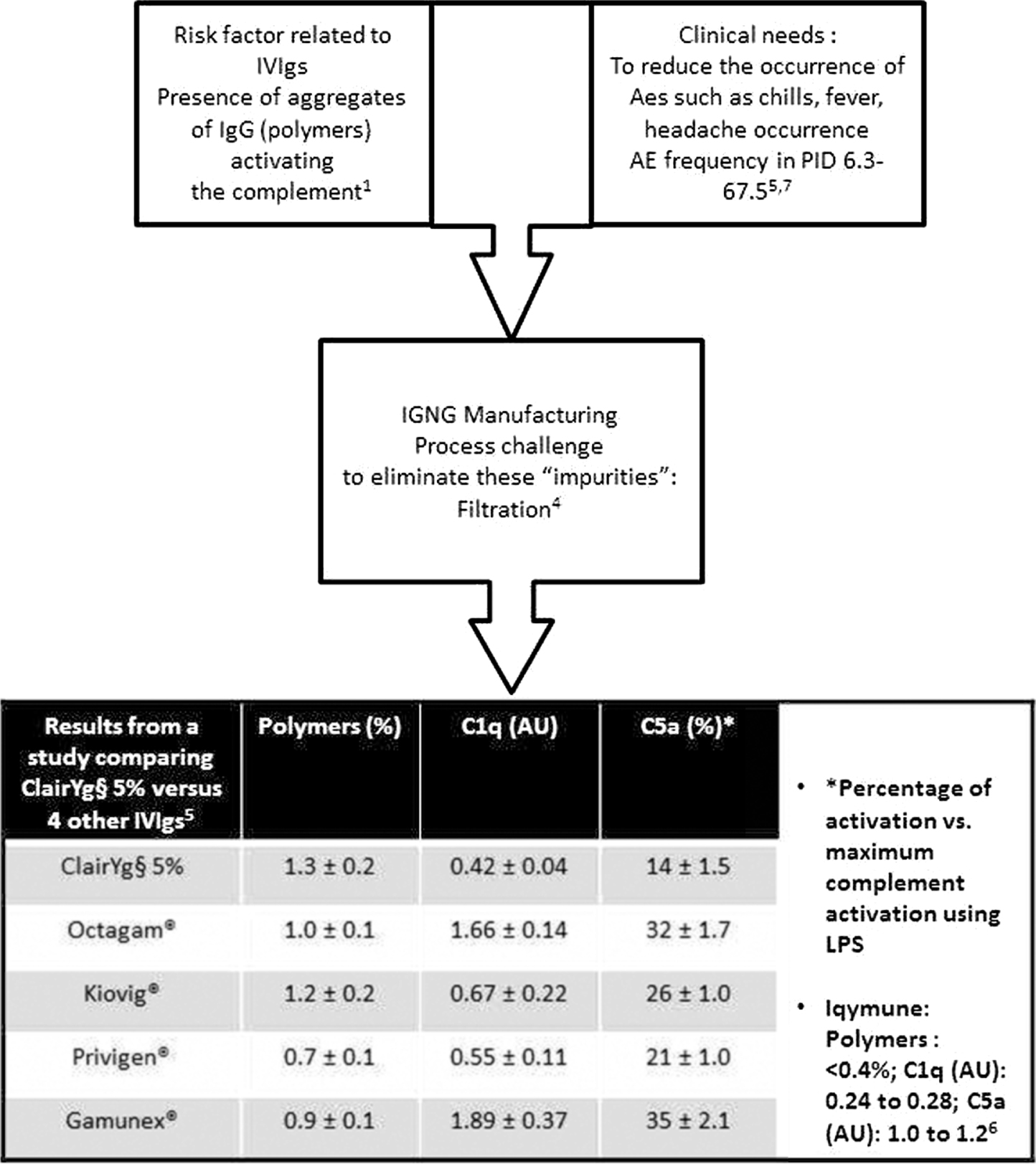
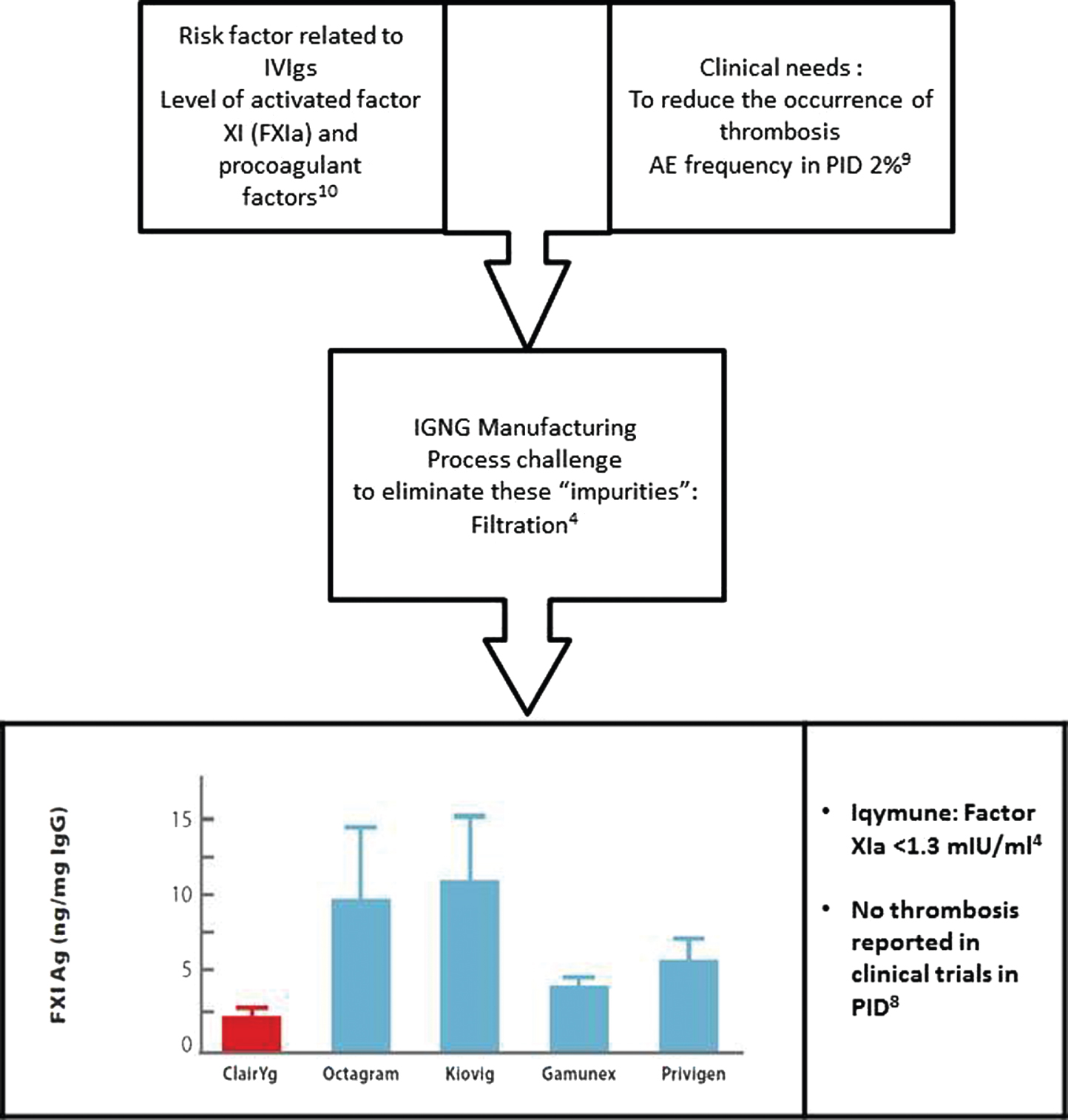
PS1Group8-036 / #196HEREDITARY MUSCLE DISORDERS IN MIDDLE EUROPE: DATA FROM HOSPITAL REGISTRY
Topic: Group 8 – Miscellaneous / 8.7 Others
Stan Vohanka1, Josef Bednarik1, Olesja Parmova1, Magda Chmelikova1, Lenka Fajkusova2
1Neurology, University Hospital, Brno, CZ;
2Centre For Molecular Biology, University Hospital Brno, Brno, CZ
Abstract: Aim of the study. Neuromuscular Centre University Hospital Brno is one of two largest neuromuscular centres in Czech Republic. The aim of the study is to analyse data from a local hospital registry of neuromuscular disorders and present a spectrum of hereditary muscle disorders. The registry may reflect the spectrum of the hereditary muscle disorders in the local population of South Moravia (the part of Czech Republic with approx. 2 mil. inhabitants). Patients and methods. Local registry of neuromuscular disorders is based on IBM notes/domino environment database system. It has been used at University Hospital more than 15 years as the office software package for inner communication (databases, mail, to do lists etc.). This collaborative workgroup software enables the personalized access and different roles for each user (neurologists, paediatricians, pathologists, and molecular biology specialists). Results. The registry was established in 2003 and to the end of 2015 286 files have been identified. The largest group consists of patients suffering from myotonic dystrophy type 2 (105) and myotonic dystrophy type 1 (46). Limb girdle muscular dystrophy pattern was identified in 56 cases. Eight patients have mutation of calpain 3 gene, 7 dystrophinopathy, and 6 suffer from deficit of anoctamin 5. Despite of the new generation of sequencing methods, the gene defect was not discovered in 25 cases (45%). Third most frequent group are patients with facioscapulohumeral muscular dystrophy (38 persons). Oculopharyngeal muscular dystrophy was diagnosed in 5 cases and McArdle disease (GSD V) in 3 patients. Emery Dreifuss muscular dystrophy was found in 3 cases. Conclusion. The most frequent muscular dystrophy in Middle European Region is myotonic dystrophy type 2. Among limb girdle muscular dystrophies the common mutations are calpain 3 and anoctamin 5. FSHD is the third most frequent form of muscular dystrophy in Central Europe.
PS1Group8-037 / #496ANTINOCICEPTIVE AND ANTI-INFLAMMATORY EFFECTS OF COMBINED ADMINISTRATION OF VITAMIN B12 AND KETOROLAC IN RATS
Topic: Group 8 – Miscellaneous / 8.7 Others
Md Mizanur Rahman
Physiology, Enam Medical College & Hospital, Dhaka, BD
Abstract: Background: Effects of vitamin B12 on nociceptive pain, inflammatory pain and inflammation have been demonstrated in animal and human studies. But comparison of these effects with similar effects of ketorolac tromethamine (KT) and their combination (vitamin B12 + KT) has not been established. Objective: To assess the effects of KT against nociceptive and inflammatory pain after single administration in rat and compare them with those of combination of vitamin B12 with KT. Methods: An experimental study was conducted in Department of Physiology, BSMMU, Dhaka on Fifteen healthy Long Evans rats of both sexes. On the basis of drug and vitamin administration, the rats were divided into 3 groups (5 rats/group). Control group received normal saline (5 ml/kg body weight), ketorolac treated group received KT (10 mg/kg body weight), combined treated group received vitamin B12 (15 mg/kg body weight) and KT (10 mg/kg body weight) intraperitoneally one hour before the tests. Thereafter, all the rats underwent formalin test and formalin induced paw edema test. Data were expressed as mean ± SEM and statistically analysis was done by ANOVA followed by Bonferroni’s post hoc test. Afterwards, the rats were deeply anesthetized followed by euthanasia. Results: Almost all the variables for nociceptive pain (p≤0.001), inflammatory pain (p≤0.001), inflammation (p≤0.01) were reduced by combined administration of vitamin B12 and KT than KT alone. Conclusion: Single dose administration of combined vitamin B12 and KT may be more effective in reducing pain and inflammation than the individual administration of KT.
PS1Group8-038 / #364MEASURING PRIORITIES AND GOALS OF CHILDREN WITH DUCHNNE MUSCULAR DYSTROPHY TO DEVELOP A MEANINGFUL PATIENT REPORTED OUTCOME MEASURE
Topic: Group 8 – Miscellaneous / 8.1 Outcome Measures in Clinical Trials
Roni Propp1, Sarah Buttle2, Shannon Weir3, Clarissa Encisa3, Aileen Davis4, Laura Mcadam5, Nancy M. Salbach6, Unni G. Narayanan7
1Rehabilitation Sciences Institute, University of Toronto, Toronto, ON, CA;
2Medicine, University of Ottawa, Ottawa, ON, CA;
3Child Health Evaluative Sciences, SickKids Research Insitute, Toronto, ON, CA;
4Krembil Research Institute, University Health Network, Toronto, ON, CA;
5Department Of Paediatrics, Univeristy Of Toronto, Bloorview Research Institute, Holland Bloorview Kids Rehabilitation Hospital, Toronto, ON, CA;
6Department Of Physical Therapy, University of Toronto, Toronto, ON, CA;
7Orthopaedic Surgery, The Hospital for Sick Children, Toronto, ON, CA
Abstract: Background: Duchenne muscular dystrophy (DMD) is among the most common and severe childhood onset neuromuscular disorders. DMD has a significant impact on all aspects of health related quality of life (HRQOL). In the absence of a cure, interventions aim to improve health, prolong life, and increase quality of life of children with DMD. It is imperative to measure whether these interventions address the goals of children with DMD and their parents. Although there are patient reported outcome measures specifically used for children with DMD, these do not adequately reflect the priorities (concerns, wishes and expectations) of children with DMD. The Caregiver Priorities and Child Health Index of Life with Disabilities (CPCHILD) questionnaire is a multidimensional HRQOL measure that was developed specifically for children with severe disabilities and has been validated for severe (non-ambulant) cerebral palsy. Pilot testing of the CPCHILD in children with DMD and their parents has shown that the items and domains are important and relevant to them, further justifying the investigation of the utility of the measure for this population. Objective: To present a proposal outlining the evaluation of the suitability and validity of the CPCHILD as an outcome measure for children with DMD, utilizing the ICNMD as a forum to solicit feedback and participation of international experts for development of the adapted version (CPCHILD-DMD). Methods: Cognitive interviews with a purposive sample of children with DMD and their parents will be conducted to evaluate the suitability of content and format of the CPCHILD. This iterative process will inform the retention, modification, elimination or addition of items to the CPCHILD. A multidisciplinary group of healthcare professionals with expertise in this population will be e-surveyed for their perspectives on the suitability of the CPCHILD and their recommendations will be incorporated. The resulting CPCHILD-DMD will be administered alongside other measures to a cohort of children with DMD and their parents to analyze the psychometric properties of the questionnaire, including test-retest reliability, construct validity, and differences between the child and parent scores, and ultimately sensitivity to change. Results: In pilot testing, the CPCHILD was completed by children with DMD (n=13) and parents (n=22), who also rated the importance of each item on an ordinal scale from 0 (Not at all important) to 5 (Most Important). The mean importance rating of each item ranged from 3 (Fairly important) to 5 (Most important). The overall mean (SD) importance rating of the items was 4.11 (±0.36) and 3.88 (±0.38), reported by the children and parents, respectively. This suggests that the items of the CPCHILD are important and relevant to this population, justifying further evaluation and adaptation followed by rigorous psychometric evaluation of the new outcome measure. Discussion and Conclusion: A comprehensive condition-specific priority based outcome measure for DMD will serve as a much needed tool to guide decision-making and evaluate effectiveness of interventions consistent with the goals of children with DMD and their parents. This is especially relevant given the development of promising new treatments for children with DMD.
PS2Group1-001 / #436DYSFELINOPATHIES IN BURKINA FASO: A CASE REPORT
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Anselme Dabilgou, Christian Napon, Julie M A Kyelem, Alassane Drave, Anila Bhunnoo, Jean Kabore
Neurology, CHU Yalgado Ouedraogo, Ouagadougou, BF
Abstract: Introduction Dysferlinopathies encompass a large variety of neuromuscular diseases characterized by the absence of dysferlin in skeletal muscle and an autosomal recessive mode of inheritance. So far, three main phenotypes have been reported: Miyoshi myopathy (MM), limb girdle muscular dystrophy type 2B (LGMD 2B), and distal myopathy with anterior tibial onset (DMAT). Although rare, dysferlinopathies occur frequently in the Middle East and the Indian subcontinent. Limb girdle muscular dystrophy is rare in Africa, specially in Burkina Faso. The objectives of this study is to report a case of dysferlinopathy occur in Burkina Faso. Clinical observation It is a case of a burkinabe patient, a teacher, born on 1984 who has consulted on March 2009 in neurology. He was suffering from walk troubles with falls, difficulties to be up, and muscular cramps with a progressive evolution since one year. In the past medical history, three cases of walk troubles have been registred. The neurogical examination noticed a tretraparesy with a proximal predominance. The achilleen and osteo-tentinous kneejerk were abolished, so are the idiomuscular reflexes. Serum creatine kinase (CK) was elevated, 24 800UI/l. Electromyography (EMG) has not been done due to diverse reasons. The muscular biopsy showed muscles abnormalities. The clinical outcome was effective under a corticotherapy. In biological level, a regressive progression of the CPK rate of 7 518 UI/1 in 2014, was noticed. Conclusion It is the first case of dysferlinopathy occurs in Africa and clinical examination noticed proximal dystrophy.
PS2Group1-002 / #471THE QUALITY OF LIFE IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY, IRANIAN EXPERIENCE
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Gholamreza Zamani, Morteza Heidari, Mahshid Mehdizadeh
Neurology, Tehran University of Medical Sciences,Children’s Medical Center, Tehran, IR
Abstract: Introduction Duchenne Muscular Dystrophy is the most common hereditary progressive muscular disease in children with a prevalence of 1 in every 3500 boys . Despite the advances in diagnosis and therapeutic modalities , less attention is still offered to their psychological and environmental aspects of life in daily practice. Very few studies have compared the perception of the children with DMD and their parents in this regard and no study has reported the health related quality of life of children with DMD from Iran. Materials and Methods This study was performed on 85 patients aged 8-18 years living in Tehran ,with definite diagnosis of DMD biopsy or genetic testing confirming definite diagnosis of DMD and 136 healthy case controls, those with intellectual disability were not enrolled in this study. The patients and one of their parents completed the 27-item Persian version of KIDSCREEN questionnaire (child and adolescent version and parent version) separately. That covers 5 major subclasses of Physical activities and health , General mood and feelings , Family and free time , Friends , School and learning. Results Comparative analysis of the health related quality of life in healthy and affected children showed from the perspective of the children and adolescents, significant lower scores were observed in “physical activity and health” (p<0.001) and “friends” (p=0.005) subclasses of the patients group but other subclasses of the quality of life were similar between two groups. From the perspective of the parents, the quality of life of the patients was significantly lower in the subclasses of “physical activity and health” (p<0.001), “general mood and feelings” (p<0.001), and “friends” (p<0.001) than that of the control group while the quality of life was similar in the subclasses of “family and free time” (p=0.94) and “school and learning” (p=0.07) 4. Discussion The present study showed that from the perspective of children and adolescents, the quality of life was similar in boys with DMD when compared to healthy controls except for the subclasses of “physical activity and health” and “friends”. The subclasses of “general mood and feeling”, “family and free time”, and “school and learning” were similar in patients and controls, indicating that these subclasses were not affected by DMD. In our study, with increasing age, the QOL decreased significantly only in the subclasses of “physical activity and health” and “friends”. . Therefore, social networking should develop to support the patients’ families. Furthermore, the parents should be made aware of the opinion differences that they have in contrast to their children in order to feel more relaxed and confident and continue their children’s supportive and therapeutic measures with more hope.Conclusion: The above-mentioned findings suggest that boys with DMD have a positive attitude toward life despite the progressive nature of the disease and limitations in physical activities and relationship with friends. However more happier and promising quality of life can be reachable for them through improvement of social supports services and promotion of their parents knowledge in concern to their children’s positive perspective of life.
PS2Group1-003 / #428CURRENT STATUS OF DYSTROPHINOPATHY NATIONAL REGISTRY IN JAPAN
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
En Kimura1, Madoka Mori-Yoshimura2, Satomi Mitsuhashi3, Fumi Takeuchi4, Harumasa Nakamura4, Hirohumi Komaki5, Ichizo Nishino6, Mitsuru Kawai7, Shin’Ichi Takeda8
1Translational medical center, National Center of Neurology and Psychiatry, Kodaira, Japan, JP;
2Department Of Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira,Tokyo, JP;
3Department Of Neuromuscular Research, Institute of Neuroscience, National Center of Neurology and Psychiatr, Kodaira, Tokyo, JP;
4Translational Medical Center, National Center of Neurology and Psychiatry, Kodaira, Tokyo, JP;
5Department Of Child Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira, Tokyo, JP;
6Department Of Neuromuscular Research, Institute of Neuroscience, National Center of Neurology and Psychiatry, Kodaira, Tokyo, JP;
7Institute Of Clinical Research / Department Of Neurology, National Hospital Organization Higashisaitama Hospital, Hasuda, Saitama, JP;
8Director, Institute of Neuroscience, National Center of Neurology and Psychiatr, Kodaira, Tokyo, JP
Abstract: Background Rare disease registries are recognized as an important tool of clinical research, such as the Global dystrophinopathy patient registry, a harmonized registry from over 30 national registries (http://www.umd.be/TREAT_DMD/). An achievement made possible through collaboration with TREAT-NMD, which was established to accelerate the international clinical developments in neuromuscular diseases. Remudy was established in collaboration with national neuromuscular experts, patient advocacy groups, and the National Center of Neurology and Psychiatry in 2009. The launch featured the Japanese national Dystrophinopathy registry which was followed by a national GNE myopathy registry in 2012, a national Myotonic Dystrophy (DM) registry in 2014, and some registries for other diseases just in preparation.Objective To present current status of Japanese dystrophinopathy national registry with international collaboration with TREAT-NMD global registry, and discuss its utility for clinical development and necessity of continuation. Methods Remudy took 1) Patient-reported system, 2) Genetic and Clinical curators, 3) Information committee to judge enquiry from third parties, 4) Following the Charter for TREAT-NMD Patient Database/Registry, and 5) Co-working with MDCTN (Muscular Dystrophy Clinical Trial Network). Here we present detailed clinical and genetic data in present Remudy dystrophinopathy database. Results By February 2016, total registrants were 1,497: 1,377 completed and 120 during process. Clinical classifications were DMD: 1,091, IMD: 35 and BMD: 252. Most individuals aged less than 20 years. In terms of genetic mutations, exonic deletion was 60.5%, followed by exonic duplication, 12.6%, nonsense mutations, 12.6%, frame shift mutations, 7.3%, intronic mutations, 4.4%, missense mutations, 1.1%, others, 0.2%, coexistence of exonic deletion and duplication, 0.2%, and 1.1% were not detected. Clinically, 39.2% of DMD, 39.4% of IMD and 74.6% of BMD were ambulant, 45.8% of DMD, 36.4% of IMD and 8.4% of BMD were treated with steroids, 38.4% of DMD, 45.5% of IMD and 30.0% of BMD were diagnosed having cardiac dysfunction, and 26.8% of DMD, 33.3% of IMD and 4.6% of BMD were under the ventilation support. Remudy contributed to feasibility studies and trial recruitments with TREAT-NMD or domestic groups 25 times. Furthermore, Remudy shared management know-how with other diseases in domestic and the Chinese national neuromuscular disease registry launched in 2012. Conclusion Remudy demonstrated utility in clinical research feild and standardization of patients care for dystrophinopathy in Japan, as well as informed a promising style of patient registry system in other rare diseases with international collaborations.
PS2Group1-004 / #407CHARACTERISTICS OF JAPANESE PATIENTS WITH BECKER MUSCULAR DYSTROPHY IN A JAPANESE NATIONAL REGISTRY OF MUSCULAR DYSTROPHY (REMUDY): HETEROGENEITY AND CLINICAL VARIATION
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Madoka Mori-Yoshimura1, Satomi Mitsuhashi2, Hirohumi Komaki3, Harumasa Nakamura4, Naohiro Yonemoto5, Fumi Takeuchi6, Yukiko K. Hayashi7, Miho Murata1, Ichizo Nishino2, Shin’Ichi Takeda8, En Kimura9
1Department Of Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Tokyo, JP;
2Department Of Neuromuscular Research, Institute of Neuroscience, National Center of Neurology and Psychiatry, Tokyo, JP;
3Department Of Child Neurology, National Center Hospital, National Center of Neurology and Psychiatry, Tokyo, JP;
4Department Of Promoting Clinical Trial And Translational Medicine, National Center Hospital, National Center of Neurology and Psychiatry, Kodaira, Tokyo, JP;
5Department Of Neuropsychopharmacology, National Institute of Mental Health, National Center of Neurology and Psychiatr, Tokyo, JP;
6Translational Medical Center, National Center of Neurology and Psychiatry, Kodaira, Tokyo, JP;
7Department Of Neurophysiology, Tokyo Medical University, Tokyo, JP;
8Director, Institute of Neuroscience, National Center of Neurology and Psychiatr, Tokyo, JP;
9Translational medical center, National Center of Neurology and Psychiatry, Kodaira, JP
Abstract: Obtaining an adequate number of patients to conduct a natural history study for rare diseases such as Becker muscular dystrophy (BMD) is difficult. The present study used data from Remudy, a national registry for neuromuscular diseases in Japan, to conduct a phenotypic analysis of BMD. We analyzed Remudy data of patients with dystrophinopathy. All patients who were either ambulant at age 13 or predicted to be ambulant by age 13 and who were included in the initial analysis were included as the BMD subpopulation for further analysis of age at ambulation, cardiopulmonary function, and genotype. For clinical comparisons, patients who were administered steroids were excluded. As of September 2015, 279 BMD patients had registered with Remudy. Mean patient age was 27.6±15.9 (range, 2-78) years, and 65.2% of patients were ambulant. Patients with cardiomyopathy and respiratory failure comprised 27% and 27% of this population, respectively, and 33 patients required a ventilator. The most frequent genetic mutations were exon deletions (70%), followed by point mutations (20%) and duplications (10%). The most frequent mutation was ex45_ex47del (55 patients). Patients with the ex45-49del or ex3_ex7del mutation lost their ambulation earlier than those with the ex45_ex47del mutation. In total, there were 81 combinations of exon deletions and duplications in the study population. In conclusion, patient registries can be useful tools for clinical research. Cooperation with international registries may allow for a detailed analysis of genotype-phenotype relationships in BMD.
PS2Group1-005 / #400META-ANALYSES OF ATALUREN IN PATIENTS WITH NONSENSE MUTATION DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Craig Campbell1, Francesco Muntoni2, Eugenio Mercuri3, Xiaohui Luo4, Gary Elfring4, Hans Kroger4, Peter Riebling4, Tuyen Ong4, Robert Spiegel4, Stuart W. Peltz4, Craig M. Mcdonald5
1Pediatric Neurology, London Health Sciences Centre and Western University, London, ON, CA;
2Developmental Neuroscience, UCL Institute of Child Health, London, GB;
3Paediatric Neurology, Catholic University, Rome, IT;
4Statistics, PTC Therapeutics, South Plainfield, NJ, US;
5University of California Davis Medical Center, Sacramento, CA, US
Abstract: For the Ataluren DMD Study Steering Committee Background: Ataluren (Translarna) is an orally bioavailable drug approved in Europe, Israel and South Korea for treating nonsense mutation (nm) Duchenne muscular dystrophy (DMD) in ambulatory patients aged 5 years and older. Ataluren promotes ribosomal readthrough of a premature stop codon, enabling the formation of full-length, functional dystrophin protein; 10-15% of DMD boys have nonsense mutations. The phase 2b (Study 007) and phase 3 (ACT DMD) clinical trials of ataluren are the largest, randomized, double-blind, placebo-controlled studies in nonsense mutation DMD to date, focusing on a subset of an already rare disorder. To assess the totality of the evidence regarding the efficacy of ataluren in patients with nmDMD using the 6-minute walk test and timed function tests, a meta-analysis of patients pooled from both trials was prespecified in the ACT DMD statistical analysis plan. Objective: To evaluate the efficacy of ataluren in a meta-analysis of 2 clinical trials in patients with nmDMD. Methods: For this analysis, only patients in Study 007 who met the ACT DMD inclusion criteria were combined with patients in ACT DMD. Males 7–16 years with nmDMD, baseline 6-minute walk distance (6MWD) of ≥150 meters and ≤80% of predicted, and ≥6 months of steroid use received ataluren 10, 10, 20 mg/kg or placebo administered orally 3 times daily for 48 weeks. The primary endpoint in both trials was change in 6MWD at Week 48. Changes in timed function tests at Week 48 were also assessed in both trials. The meta-analysis was repeated using all patients in Study 007 (>5 years old with baseline 6MWD of > 75m) and ACT DMD. Results: Overall, 291 patients were included in this analysis (ACT DMD: ataluren, n=114; placebo, n=114; Ph2b: ataluren, n=32; placebo, n=31). A statistically significant 21.1-meter benefit in 6MWD (p=0.0193) was observed in patients who received ataluren compared with placebo. Other timed function tests also showed statistically significant improvement with ataluren over placebo, including time to walk/run 10 meters (-1.4 seconds; p=0.0251), time to climb 4 stairs (-1.6 seconds; p=0.0184), and time to descend 4 stairs (-2.0 seconds; p=0.0044). When the meta-analysis was repeated using all patients in Study 007 and ACT DMD, similar results were obtained (p<0.05 for all endpoints). Discussion/Conclusions: This meta-analysis of 291 patients with nmDMD across 2 clinical trials demonstrated that patients who received ataluren over 48 weeks obtained a statistically significant clinical benefit as measured by 6MWD and timed function tests compared with placebo. No other dystrophin restoration therapy for DMD has resulted in statistically significant benefits vs. placebo at 48 weeks of treatment using pre-specified analyses across both the 6MWT and timed function test endpoints. The relative benefit-risk profile of ataluren confirms its utility in ambulatory nmDMD. Study Supported By: PTC Therapeutics Inc.
PS2Group1-006 / #369ETEPLIRSEN FOR DUCHENNE MUSCULAR DYSTROPHY (DMD): CLINICAL AND BIOCHEMICAL RESULTS WITH LONGITUDINAL COMPARISON TO EXTERNAL CONTROLS ON SIX-MINUTE WALK TEST (6MWT)
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
J R. Mendell1, Nathalie M. Goemans2, Louise Rodino-Klapac3, Z Sahenk1, Linda P. Lowes1, Lindsay N. Alfano1, Katherine M. Berry1, E Peterson1, S Lewis1, K Shontz1, P Duda4, C Donoghue4, J Dworzak4, B Wentworth4, E M. Kaye5, Eugenio Mercuri6, Dmd Italian Network7
1Nationwide Children’s Hospital, Columbus, OH, US;
2Child Neurology, University Hospitals Leuven, Leuven, BE;
3Center For Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, US;
4Sarepta Therapeutics, Cambridge, MA, US;
5Medical, Sarepta Therapeutics, Cambridge, MA, US;
6Dep. Of Child Neurology, POLICLINICO GEMELLI, ROME, IT;
7Fondazione Telethon DMD Italian Network, Milano, IT
Abstract: OBJECTIVE: DMD, a rare, degenerative, X-linked genetic disease occurring in ~1:3500-5000 males worldwide, results in progressive muscle loss and premature death. DMD is primarily caused by whole-exon deletions in the DMD gene that disrupt the RNA reading frame and prevent production of dystrophin protein. Eteplirsen, a phosphorodiamidate morpholino oligomer (PMO), is designed to restore the reading frame and induce production of internally-shortened dystrophin in the subset of patients who are amenable to exon 51-skipping.METHODS: An analysis of 6MWT performance over 4 years compared boys treated with 30/50 mg/kg eteplirsen weekly IV (N=12) versus a cohort of untreated patients (N=13) who were comparable based on age, corticosteroid use, and genotype and therefore served as an external control (EC). Muscle biopsies of eteplirsen-treated boys were analyzed for exon skipping and dystrophin expression. RESULTS: At Year 4, a statistically and clinically significant treatment benefit of 162 meters on 6MWT was observed in eteplirsen-treated patients compared with EC (p=0.002). Furthermore, 2/12 (17%) eteplirsen patients lost ambulation by Year 1 with no additional losses observed, compared with 10/13 EC at Year 4 (77% by raw calculation, or 90% by Kaplan-Meier Analysis with p=0.004). Muscle biopsy analysis demonstrated exon 51-skipping in consenting eteplirsen-treated patients (N=11) by RT-PCR and statistically significant increases (p<0.001) of dystrophin intensity and % dystrophin-positive fibers by immunohistochemistry over untreated DMD controls (N=9). Western blot confirmed dystrophin production in 9/11 patients. No deaths, discontinuations due to AEs, or treatment-related SAEs were observed. LVEF on ECHO was stable over 4 years. AEs were generally mild and unrelated to study-drug. CONCLUSIONS: After 4 years of eteplirsen-treatment, DMD patients walked an average of 162 m further on 6MWT than the comparable external cohort (p=0.002) and fewer experienced loss of ambulation (17% vs. 90%, p=0.004). Eteplirsen’s mechanism of action was confirmed by detection of exon 51-skipped mRNA by RT-PCR/sequencing and by increased expression of de novo dystrophin as demonstrated by 3 complementary methods in nearly all eteplirsen-treated patients.
PS2Group1-007 / #385LONGITUDINAL EFFECT OF DRISAPERSEN VERSUS HISTORICAL CONTROLS ON AMBULATION IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Nathalie M. Goemans1, Már Tulinius2, Anna-Karin Kroksmark2, Marleen Van Den Hauwe1, Zhengning Lin3, Susanne Wang4, Giles Campion3
1Child Neurology, University Hospitals Leuven, Leuven, BE;
2Queen Silvia Children’s Hospital, Gothenburg, SE;
3BioMarin Pharmaceutical Inc, Novato, CA, US;
4Clinical Sciences, BioMarin Pharmaceutical Inc, Novato, CA, US
Abstract: The objective of this analysis was to evaluate long-term efficacy and safety of drisapersen, an antisense oligonucleotide inducing specific skipping of exon 51 during pre-mRNA splicing, designed to treat patients with Duchenne muscular dystrophy (DMD). This analysis compared disease progression in subjects treated with drisapersen with matched natural historical controls (NHC) amenable to exon 51 skipping. 12 DMD subjects were enrolled in PRO051-02 (NCT01910649), an open-label extension of the dose-escalation study; median (mean [standard deviation; range] change from baseline to week 177 in 6-minute walking distance (6MWD) was 8 m (–24.5 [161; –263, 163] for subjects (n=10) able to complete the test at baseline. All 12 subjects were treated with drisapersen at 6mg/kg/week, and maintained on corticosteroids, with 9 patients aged ≥7 yrs, able to complete a baseline 6MWD. Because of the absence of a control group, these subjects were compared to NHC from 2 DMD natural history cohorts: the Leuven Neuromuscular Reference Center (LNMRC) and the Italian Telethon registry. These NHC have prospective 6MWD assessments on a substantial number of subjects for 36 months or longer1,2. The 12 drisapersen treated subjects were also treated at one of the clinical centers as the NHC, thereby controlling for variability in standard of care across centers. The NHC were matched with the 9 subjects ≥7 yrs, based on age, corticosteroid use, and exon 51 skippable genotype. Longitudinal 6MWD outcomes from these subjects were used in a comparative analysis with the NHC. Mean age was 9.9 yrs and mean baseline 6MWD was 388 m in the 9 drisapersen subjects. 13 NHC had a mean age of 9.4 yrs and baseline 6MWD of 360 m, indicating that these subjects were well matched according to age and 6MWD. The 9 drisapersen subjects maintained stable 6MWD after 12 months of treatment, while 6MWD declined by 79.5 meters in the NHC at Month 12 (Figure 1). At 36 months, drisapersen-treated subjects (n=9) demonstrated a statistically significant difference of 240m (p < 0.01) in 6MWD change from baseline in comparison to NHC (n = 11). There was a slightly lower incidence of drisapersen subjects losing ambulation (1/9) compared to NHC (2/13) over this 36 month period. After 3 years of follow-up, drisapersen-treated, DMD subjects showed stable ambulation as assessed by 6MWD compared to NHC with the exon 51 skippable genotype. The treatment effect of drisapersen was observed within 12 months, and was maintained at 36 months. These findings support the efficacy of drisapersen in patients with DMD ≥7 years of age. 1Goemans N, van den Hauwe M, Wilson R, et al. Ambulatory capacity and disease progression as measured by the 6-minutewalk-distance in Duchenne muscular dystrophy subjects on daily corticosteroids. Neuromuscul Disord 2013;23:618–623. 2Pane M, Mazzone ES, Sivo S, et al. Long term natural history data in ambulant boys with Duchenne muscular dystrophy: 36-month changes. PLoS One 2014;9:e108205.
PS2Group1-008 / #377IMPACT OF MUSCLE FUNCTION, NUTRITIONAL STATE AND SYSTEMIC INFLAMMATION, ON BONE MINERAL DENSITY IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Oriana D.R. Cruz1, Maricela Rodriguez-Cruz2, Carlos Wong-Baeza1, Salvador Atilano-Miguel2, Tomas Almeida-Becerril2
1Bioquímica, Laboratorio Biomembrana, Escuela Nacional de Ciencias Biológicas, IPN, Mexico, D.F, MX;
2Unidad De Investigación Médica En Nutrición, Laboratorio De Biología Molecular, Centro Medico Nacional Siglo XXI, IMSS, D.F, MX
Abstract: Duchenne Muscular Dystrophy (DMD) is a hereditary disease that affects the skeletal muscle; some clinical characteristic are the lost of muscle mass, obesity, chronic inflammation and low bone mineral density (BMD). It has been proposed that BMD is affected by different factors such as the muscle mass, nutritional state and the systemic inflammation, nevertheless the impact of these factors on the bone health in DMD boys it’s unknown. Thus, the aim of this study was to evaluate the impact of these factors in the bone mineral density in DMD patients. We studied 63 glucocorticoid-naive DMD boys. The systemic inflammation was evaluated in serum by measuring cytokines (IL-1, IL-6, TNF-α), C-reactive protein (CRP), and creatine kinase (CK). Muscle function was evaluated using Vignos Scale and categorized in groups: Vignos A, the patients with better function, Vignos B; patients who lost the ability to walk with out any help, and Vignos C; patients who lost the total ability to walk. Weight and height were recorded to calculate body mass index (BMI), the body composition; BMD and body mineral content (BMC) were measured by absorptiometry dual-energy X-ray. Patients were categorized into three groups according their body mass index (BMI), expressed as percentiles. Children with BMI ≤5th percentile were underweight; BMI >5th but <85th percentile as normal weight; BMI ≥85th but <95th percentile as overweight, and BMI ≥95th percentile as obese, according with criteria established by the Centers for Disease Control and Prevention. Data was analyzed using SPSS, the confidence intervals were 95% with an alpha value (p<0.05). ANOVA, Bonferroni post test and T3-Dunett or statistics Kruskal-Wallis and Mann-Whitney U tests were applied, and for the correlation Sperman or Pearson were applied. Patients with less muscle function had the highest lost of BMD compared with patients who are still capable to walk and with the better muscle function. Moreover the BMC was lower than normal values acording to the age in the patients with less muscle function (z-score of -2.10 ± 1.23), also this group present a negative correlation between the muscle function with the BMD (r = -0,422, p <0.05). Additionally the CK marker of muscle damage showed a positive correlation with BMD (r = 0.412, p <0.05). Furthermore the adipose mass (% and Kg) has a negative correlation (r=-.250, p <0.05) with BMD, although no differences were observed in the BMD in the groups with different levels of nutritional status (underweight, normal weight, overweight/obese). The CRP, a circulation inflammation marker, shows a negative correlation with BMD z-score (r = -0. 279, p <0.01), while the others cytokines didn’t show any significant correlation with BMD. These results show that loss of function muscle and the increased adipose mass has an impact in the bone health in the DMD. However in the other hand the nutritional status and the systemic inflammation does not affect the BMD in patients with DMD. Based on these results, therapies focused on the muscle function and the degree of adiposity can be proposed to improve the loss of BMD.
PS2Group1-009 / #283MIBG THERAPY FOR AN INOPERABLE PARAGANGLIOMA IN DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Denis Duboc1, Marine Paul2, Laurie Fanon1, Karim Wahbi1, Marco Alifano3, Florence Tenenbaum4, Laurence Guignat2
1Cardiologie, Hôpital Cochin, paris, FR;
2Endocrinologie, Hôpital Cochin, paris, FR;
3Chirurgie Thoracique, Hôpital Cochin, paris, FR;
4Médecine Nucléaire, Hôpital Cochin, paris, FR
Abstract: The authors present a case report of a 16 year-old adolescent associating thoracic paraganglioma (PGL), a rare neuroendocrine neoplasm of the autonomic nervous system, in relation to SDHB mutation, a mutation of the subunit B of the mitochondrial succinate dehydrogenase complex, transmitted by his father and a Duchenne muscular dystrophy (DMD) inherited from his mother. He received a cardioprotective treatment that combines beta-blockers and ACE-inhibitors (angiotensin-converting enzyme inhibitors) since 10 years of age. By the age of 14, he complained of episodes of dizziness, palpitations, nauseas, sweats, pallor, and paroxysmal hypertension. Twenty-four hour urinary normetanephrine was 16647 nmol/24 hour (normal: 500–2400 nmol/24 hour), urine norepinephrine was 5099 nmol/24 hour (normal: 70-500) and urine metanephrine and epinephrine were within normal limits. Computed tomography found a left paravertebral tumor (size 25×29×40 mm) opposite the 4th and the 5th thoracic vertebrae. The adrenal scintigraphy with 123I-MIBG (Iodine-123-labelled meta-iodobenzylguanidine) described PGL left paravertebral at the top of the aortic arch. Radiopharmaceutical high uptake was only present in this localization. The patient underwent testing for susceptibility genes for pheochromocytoma/PGL, which revealed a SDHB mutation. We performed PET-TDM FDG, because this gene mutation predisposes to develop malignant and metastatic paraganglioma. The exam confirmed the unique localization. The only curative treatment (surgery) was unfeasible in this case. Embolization of this hypervascularized tumor with multiple anastomosis with medullar arteries could cause medullar ischemia and a potential lesion of the Adamkiewicz artery, near the tumor, increased the risk of post-surgery paraplegia. Furthermore, the preexistent restrictive respiratory insufficiency would increase the risk of general anesthesia. MIBG is a radioactive analogue of norepinephrine. When it is labeled with 131Iodine, it is usually used for treatment of metastatic PGLs and refractory neuroblastomas. The patient received 5 doses of 131I-MIBG from April 2013 to January 2016 from 740 MBq to 3700 MBq. The treatment was initiated with low doses to evaluate the safety, in the event of a release of catecholamines, which could cause high blood pressure crisis, arrhythmia and adrenergic cardiopathy caused by radionecrosis. We observed spacing out between crises from 3 per months to 2 in 6 months. These crises became shorter and the patient reports no more palpitations, nausea or sweats. We noticed a biochemical response with a decrease of urinary normetanephrine from 7 ULN to 1.2 ULN. The tumor size was stable. No metastasis was discovered. This treatment was well tolerated and without life-threatening adverse events (cardiological, hematological), as documented by clinical laboratory tests and vital signs, in the patient nor in his parents who attended physically all the treatment sessions. 131I-MIBG radiotherapy seems to be an alternative when surgery is unfeasible in non-metastatic paraganglioma.
PS2Group1-010 / #354EFFECT OF METFORMIN ON IN VIVO AND EX VIVO PATHOLOGY SIGNS IN EXERCISED DYSTROPHIC MDX MICE
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Roberta F. Capogrosso1, Anna Cozzoli1, Arcangela Giustino2, Paola Mantuano1, Francesca Sanarica1, Michela De Bellis1, Annamaria De Luca1
1Unit Of Pharmacology, Department Of Pharmacy And Drug Sciences, University of Bari “A. Moro”, BARI, IT;
2Department Of Biomedical Sciences And Human Oncology, University of Bari, Medical School, BARI, IT
Abstract: The progressive degeneration and myofibers fragility during contractile activity in Duchenne muscular dystrophy (DMD) are caused by the complex cascade of events triggered by the absence of dystrophin, a protein with a key role for a proper mechano-transduction (Hoffman & Dressman, Trends Pharmacol. Sci., 2001). We recently have shown that, in the mdx mouse, the most widely used animal model for DMD, the protocol of chronic treadmill exercise leads to a failing mechanical-metabolic coupling. In particular, genes of protective metabolic pathways such as Sirt-1/Pgc1-alpha, Ppar-gamma, adiponectin, Bnip-3 are severely down-regulated and are likely unable to contrast the high-expression of damage-related genes (NADPH-oxidase 2, TGF-beta1, TNF-alpha, c-Src tyrosine kinase), then accounting for muscle damage and dysfunction (Camerino et al., Hum Mol Genet., 2014). In line with this, various studies showed that metabolic modulators lead to beneficial effects on pathology-related signs of mdx mouse. The aim of this study was to evaluate the effect of chronic treatment with metformin hydrochloride, currently in clinical trial in DMD boys (NCT01995032.clinicaltrials.gov), able to indirectly activate AMPK by modulating mitochondrial activity and cellular energetic state. Mdx mice underwent a long protocol of exercise (24-28 weeks) to exacerbate metabolic sufferance and were in parallel treated with metformin (200 mg/kg/days orally). A multidisciplinary approach in vivo and ex vivo was used according to standard operating procedures to assess the impact of drug treatment on primary readouts. In vivo, metformin significantly increased mouse strength, with normalized forelimb force values of 5.66 ± 0.16 (n=7) vs. 4.66 ± 0.01 (n=6; p<0.001) of untreated mice, but did not improve exercise performance. Metoformin did not ameliorate twitch and tetanic force values of extensor digitorum longus (EDL) muscle, while a slight, but not significant increase, was observed in diaphragm (+54% and +35% for twitch and tetanic force, respectively, versus untreated mdx mice). Moreover no protection of the treatment was observed on force drop of mdx EDL muscle during eccentric contraction. Interestingly, metformin ameliorated histopathology of mdx gastrocnemius muscle with a significative reduction of the total area of damage, paralleled by a clear trend of reduction in plasma level of matrix metalloprotease 9. By contrast the treatment did not counteract the high plasma level of creatine kinase and lactate dehydrogenase. Then the present results show a partial efficacy of metformin on pathology signs of exercised mdx mice. Further molecular biology analysis will help to gain a better insight in its overall efficacy and mechanism of action in dystrophic muscle (Supported by Prin-MIUR n °20108YB5W3_004).
PS2Group1-011 / #340DEVELOPMENT OF A PATIENT-REPORTED OUTCOME MEASURE FOR UPPER LIMB FUNCTION IN DUCHENNE MUSCULAR DYSTROPHY (DMD-UPPER LIMB PROM)
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Katrijn Klingels1, Anna Mayhew2, Elena S. Mazzone3, Michelle Eagle2, Tina Duong4, Valérie Decostre5, Marion Main6, Marleen Van Den Hauwe7, Ulla Werlauff8, Imelda De Groot9, Sonia Messina10, Valeria Ricotti6, Giles Campion11, Laurent Servais5, Elizabeth Vroom12, Eugenio Mercuri3, Nathalie M. Goemans7
1Rehabilitation Sciences, KU Leuven, Leuven, BE;
2Institute Of Genetics Medicine, The John Walton Muscular Dystrophy Research Centre, Newcastle upon Tyne, GB;
3Paediatric Neurology, Catholic University, Rome, IT;
4Neurology, Stanford University School of Medicine, Stanford, US;
5Gh Pitié Salpêtrière, Institut de Myologie, Paris, FR;
6Ucl Institute Of Child Health, Dubowitz Neuromuscular Centre, London, GB;
7Child Neurology, University Hospitals Leuven, Leuven, BE;
8The Danish National Rehabilitation Centre for Neuromuscular Diseases, Aarhus, DK;
9Radboud University Medical Centre, Nijmegen, NL;
10Neurosciences, University of Messina, Messina, IT;
11Biomarin, Leiden, NL;
12Duchenne Parent Project, Veenendaal, NL
Abstract: Background: A multidisciplinary international Clinical Outcomes group consisting of clinicians, scientists, industries, patients and their advocacy groups designed the Performance of Upper Limb module (PUL) for Duchenne Muscular Dystrophy (DMD), according to a contextual framework of upper limb function in both ambulant and non-ambulant individuals with DMD. This group also identified the need to develop in parallel a patient-reported outcome measure (PROM) to evaluate upper limb function related to activities of daily living (ADL) that cannot be observed in a clinical setting. Objective: To develop a patient-reported outcome measure (PROM) assessing upper limb function related to activities of daily living (ADL), specifically for patients with Duchenne Muscular Dystrophy (DMD) across a wide age range and applicable in the different disease stages. Methods: The developmental process was based on US Food and Drug Administration guidelines. This included item generation from a systematic review of existing tools and expert opinion on task difficulty and relevance, involving individuals with DMD. Cultural aspects affecting ADLs were taken into consideration to make this tool applicable to the broad DMD community. Items were selected in relation to a conceptual framework reflecting disease progression covering the full range of upper limb function across different domains of ADL. Results: After pilot testing followed by a preliminary Rasch analysis in 88 boys with DMD, redundant or clinically irrelevant items were removed. A second Rasch analysis in a larger dataset of 357 boys with DMD collected within an international multicenter collaboration resulted in further modifications of the UL-PROM. The questionnaire consists of 32 items covering four domains of ADL (Food 7 items, Self-care 8 items, Household and environment 6 items, Leisure and communication 11 items) and is recommended to be used in boys from 7 years of age. Conclusion: A DMD specific Upper Limb PROM was developed based on clinical relevance of the items. Psychometric robustness and test-retest reliability was confirmed. The main purpose is to document the patient self-reported natural history of DMD and assess the efficacy of interventions in patients’ daily life.
PS2Group1-012 / #322GENOTYPE PHENOTYPE ANALYSIS OF MULTIPLEX LIGATION DEPENDENT PROBE AMPLIFICATION (MLPA) POSITIVE DUCHENNE/BECKER MUSCULAR DYSTROPHY (DMD/BMD) PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Seena Vengalil1, Kiran Polavarapu2, Veeramani Preethish-Kumar2, Atchayaram Nalini1, Meera Purushottam3, Deepha Sekar3
1Neurology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
2Clinical Neurosciences, National Institute of Mental Health and Neurosciences, Bangalore, IN;
3Molecular Genetics, National Institute of Mental Health and Neurosciences, Bangalore, IN
Abstract: BACKGROUND: Lack of a large scale Indian study to analyse the mutation characteristics of dystrophin gene and the relation between the reading frame and phenotype METHOD : Clinical and genetic data of MLPA positive cases of Duchenne/ Becker muscular dystrophy patients attending neuromuscular clinic in a tertiary care center in South India between 2010-2013 was collected RESULTS: Out of 317 MLPA positive cases , 279 had DMD, 32 had BMD and 4 were intermediate dystrophinopathies . Positive family history was found in 41 out of 317 (12.9%) of cases . Duplications constituted 10.3% (29/279) of the cases of DMD and deletions constituted 89.7%. Among BMD cases , only one was caused due to a duplication involving 4-6 and 61-69 exon. Single exon deletions were the most frequent constituting 24.92% of the cases , most common deletion being exon 45, followed by 3 exon deletions (14.19%) , 5 exon deletions (8.2%) , two exon deletions (7.5%) , 6 exons (7.2%) and 4 exons (4.1%). Other large deletions constituted 24.2% of the cases. Exon 50-55 was the most frequent duplicated one. In BMD, most common deletion was 45-47 exon . Reading frame was out of frame in 255 patients (91.3%) of DMD and 3 patients of BMD (9.3%). Cardiomyopathy was found in 5 patients of BMD (15.6%) CONCLUSION: Better understanding of DMD and BMD genetics is needed for future research and advances in therapeutics
PS2Group1-013 / #314CLINICAL AND MUSCLE BIOPSY CHARACTERISTICS OF A COHORT OF CHILDREN UNDER TWO YEARS OF AGE WITH DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Ana P. Sousa1, Elisa Costa2, Ricardo Taipa2, Melo Pires2, Manuela Santos3
1Neurophysiology, Centro Hospitalar do Porto, Porto, PT;
2Neuropathology, Centro Hospitalar do Porto, Porto, PT;
3Neuromuscular - Pediatric Neurology, Centro Hospitalar do Porto, Porto, PT
Abstract: Introduction: Duchenne muscular dystrophy (DMD) is a severe and progressive disease usually diagnosed around 3-5 years of age, only when the physical ability of the affected boys diverges from their peers. The objective of this work is to report the structural changes in the preclinical stages of DMD. Methods: In a cohort of families with DMD followed in our centre, we retrieved the patients younger than 2 years of age with muscular biopsy performed in our Neuropathology Unit. Clinical and molecular characteristics were retrospectively reviewed. Muscle biopsies were re-analyzed blinded to the demographic and clinical features. Results: Ten muscle biopsies obtained in the first two years of life were available. One of them was a fetus with 16 weeks from a patient with a gene dystrophin mutation. The others were performed between 3 and 22 months due to raised creatine kinase. Five children were already able to walk at the time of biopsy. The muscle biopsies revealed marked variability in fiber size diameter, with atrophy and hypertrophy, necrotic and basophilic regenerating fibers. Endomysial fibrosis was present in all biopsies being more prominent in older patients. Only one case (19 months) had discrete adipose tissue substitution. Immunohistochemistry revealed absent dystrophin staining in all cases but one, presenting discrete and irregular immunoreactivity for Dys2. Three cases showed revertant fibers. The sixteen week-old fetus muscle was negative for dystrophin staining. Genetic studies were available in 8 cases revealing dystrophin gene deletions in 6 and point mutations in 2 patients. Conclusion: This study indicates that muscle show severe pathological changes at preclinical stages of DMD, raising questions of whether gene therapy should be considered at younger age, even before the clinical stage.
PS2Group1-014 / #304NOVEL MOUSE MODEL OF DUCHENNE MUSCULAR DYSTROPHY WITH DELETION OF EXONS 8-34
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Tatiana Dimitrieva1, Alexey Deikin1, Denis A. Reshetov2, Dmitry V. Vlodavets3, Eugeniia Zotova1
1Marlin Biotech LLC, Moscow, RU;
2Research Department, Marlin Biotech LLC, Moscow, RU;
3Russian Children Neuromuscular Center, Pirogov Russian National Research Medical University, Moscow, RU
Abstract: We present a novel mouse model of Duchenne muscular dystrophy with deletion of exons 8-34 generated with CRISPR/Cas9 technology. We have selected a set of gRNAs to target introns 7th and 35th. Then we assessed their activity with in vitro assay. Two gRNAs that showed the best cleavage rate were selected for injections into zygote with homemade Cas9 protein. After a series of microinjection and embryo transfer we got a male mouse with whole 8-34 exons region deleted. This male carrier of 8-34 exons deletion was backcrossed to C57/BL6 lane to establish colony. Mice from this colony display features of muscle degeneration as do classic mdx model. This novel model could be used for assessing therapeutic strategies for frame correction for mutations starting in the beginning of the DMD gene.
PS2Group1-015 / #309THE NEED FOR TRANSITION THE NEED FOR TRANSITION. WORKSHOP TC10.2
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Jiri Vajsar
Neurology, SickKids, toronto, ON, CA
Abstract: Improvements in the management and new treatments extend the lifespan of patients with neuromuscular diseases. For example, boys with Duchenne Muscular Dystrophy are now living longer, many of them into their 30s and even 40s. Successful adults with DMD need to acquire the necessary educational, vocational and psychosocial skills and transition to adult services including health care. Several obstacles to successful transition have been described, including a lack of transition programs which focus on education, vocation, relationships and independence and there continues to be gaps in the timing and availability of services. And as DMD was previously a pediatric disease there are a few professionals who are comfortable and knowledgeable about the needs of adults with DMD. This presentation will review the current practices, needs, and barriers to transition patients with chronic neuromuscular diseases from children’s to adult services. A recent survey to capture the current approach for transition in Canada was conducted among the members of the Canadian Pediatric Neuromuscular Group and included 18 centers that follow DMD patients: 11/18 centers indicated that they transition youth with DMD. Transition practices varied widely across Canada and included the process of preparing adolescents for adulthood, independence skills to be developed prior to transition, discussing legal documents and the process to transition them from pediatric to adult services: 11/11 have transition discussions; 9/11 assist with social and healthcare benefit applications; 7/11 have programs to prepare adolescents for transition; 6/11 provide developmentally appropriate care; 5/11 transition occurs at youths pace and only 4/11 provide vocational advice. It is obvious that transition approaches and comprehensiveness of programs to prepare adolescents to transition to adult services vary and this presentation will: 1/ list existing knowledge of physical, social and psychological aspects of the transition care; 2/ review the different national guidelines and discuss organizational solutions to transition care; 3/ outline approaches to transition care such as use of educational programs, effective use of technology, joint pediatric/adult clinics, specific young adult clinics or specialized neuromuscular clinics dealing with both pediatric and adult patients, and, outline directions for the development of Standards of Care guidelines for patients transitioning to adult care, similar to those available for pediatric populations (DMD, Congenital Myopathies and SMA). This presentation will help professionals identify factors that are likely to have an impact on successful transition to adult services. Every transfer should be a culmination of a period of planned transition and the approaches need to be tailored to the individual and need to be developmentally appropriate. Stability in care, the adolescent’s motivation and parent readiness, support of advocacy groups and integration of social issues are all important for successful transition.
PS2Group1-016 / #307A CASE REPORT OF A 10 YEAR OLD BOY WITH COMBINATION OF DMD AND DOWN SYNDROME
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Dmitry V. Vlodavets1, Marina I. Komarova2, Denis A. Reshetov3
1Russian Children Neuromuscular Center, Pirogov Russian National Research Medical University, Moscow, RU;
2Center Of Curative Pedagogics, Center of Curative Pedagogics, Moscow, RU;
3Research Department, Marlin Biotech LLC, Moscow, RU
Abstract: We present a case report of a 10 year old Russian boy with DMD combined with Down syndrome. The evidence of Down syndrome was seen at birth. Parents abandoned the child, and he was sent to an orphanage. He was born with a floppy infant signs and had a motor delay at first year of life. His karyotype was assessed (47,XY+21) and Down syndrome was confirmed. Apart from mental retardation patient also expressed motor function problems. He started to walk with support only at the age of 7. The orphan center staff was very surprised that a child with Down syndrome had such a profound motor function problems. Blood biochemistry done as a part of routine examination revealed abnormally high levels of CK – 4267, 7750 U/l (normal range is 190 U/l). At the age of 8 the boy visited Russian Children Neuromuscular Center and was diagnosed with Duchenne muscular dystrophy. EMG test revealed myopathic changes. The boy had gait disturbances and muscular wasting. He used Govers maneuvers to get up from the floor and it was impossible for him to ascent or descent the stairs. Muscle tone was decreased. DTR were absent. He had ankle contractions and calf pseudohypertrophy. We performed a genetic testing and MLPA revealed a duplication of 3 and 4 exons of DMD gene, which led to the diagnosis of a combined Down syndrome and Duchenne muscular dystrophy. The duplication of 3-4 exons in DMD gene could lead to restoration of reading frame in DMD gene and looks like Becker muscular dystrophy, but duplication effects may be complex and reading frame rule is not always applicable. The main issue is that exons 2 to 9 of DMD gene are responsible for special NH2 terminus that binds to F-actin in skeletal and cardiac muscles. Abnormal configuration of NH2 terminus which include 2 extra exons are not suitable to anchor F-actin domain and leads to a decrease or absence of dystrophin protein. We hypothesize that his evident motor delay and motor function problems were caused by DMD, and Down syndrome was responsible for his prominent mental retardation. On the other hand most of the DMD patients are ambulant before 8-12 years of live whereas our patient just started to walk at 7 years of age. We postulate that the upregulation of DMD gene and other regulatory genes could also be the reason for the motor function delay. About 30% of DMD patients report mental retardation. In our patient two disorders are combined which lead to an unusual clinical phenotype. It is of a great interest to explore the distortion of DMD gene expression in this patient. We have obtained a primary fibroblast cell culture from the patient and can transform it iPS cells and then to neurons. We are interested in a collaborative research on this matter.
PS2Group1-017 / #287PROGNOSTIC MODEL FOR 1-YEAR CHANGE IN 6-MINUTE WALK DISTANCE (6MWD) IN PATIENTS WITH DUCHENNE MUSCULAR DYSTROPHY (DMD)
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Nathalie M. Goemans1, James Signorovitch2, Elyse Swallow3, Jinlin Song3, Susan J. Ward4
1Child Neurology, University Hospitals Leuven, Leuven, BE;
2Analysis Group,Inc, Boston, US;
3Analysis Group,Inc, Boston, MA, US;
4TAP Collaboration, Cambridge, MA, US
Abstract: Background: Disease progression is inherently heterogeneous among individuals with DMD. The resulting variation in outcome measures can complicate clinical trial design and potentially cloud the interpretation of results. Objective: To develop a prognostic model for 1-year change in 6MWD among DMD patients, and to assess the additional predictive value of the model compared to commonly used factors (i.e., age, baseline 6MWD, and steroid use). Methods: Natural history data were collected from DMD patients approximately every 6 months at routine follow up visits over the course of 2 to 5 years. At each visit, patient demographics, treatment experience and ambulatory outcomes were recorded. Annualized changes in 6MWD were studied between all pairs of visits that were separated by ~1 year (8-16 months). Prediction models were developed using multivariable regression for repeated measures, and were evaluated using cross-validation. Results: A total of n=191 ~1-year follow-up intervals from n=39 boys were included. Mean age was 9.4 years and mean 6MWD was 351.8 meters at the start of these intervals; 74.9% of the patients had been using steroids for at least a year. The mean annualized change in 6MWD was -37.0 meters with a standard deviation (SD) of 93.7 meters during the subsequent ~1 year. Predictions based on age, baseline 6MWD, and steroid use explained 28% of the variation in annualized 6MWD changes (R-squared = 0.28, residual SD=79.4 meters). A broadened prognostic model, incorporating timed 10-meter walk/run, 4-stair climb, and rise from supine, as well as height and weight, significantly improved prediction, explaining 59% of the variation in annualized 6MWD changes after cross-validation (R-squared=0.59, residual SD=59.7 meters). Conclusion: Incorporating timed function tests into the prognostic model significantly improved the prediction of 1-year changes in 6MWD. Explained variation was more than doubled compared to predictions based only on age, baseline 6MWD, and steroid use, indicating significant potential for broader prognostic models to inform clinical trial design and interpretation in DMD.
PS2Group1-018 / #282DIAGNOSIS OF DUCHENNE MUSCULAR DYSTROPHY IN ITALY: CRITICAL ISSUES AND AREAS FOR IMPROVEMENTS.
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Adele D’Amico1, Michela Catteruccia2, Marika Pane3, Giovanni Baranello4, Alessandra Govoni5, Sonia Messina6, Maria Grazia D’Angelo7, Luisa Politano8, Ksenjia Gorni9, Stefano Carlo Previtali10, Antonella Pini11, Roberta Battini12, Angela Berardinelli13, Federica Ricci14, Elena Pegoraro15, Claudio Bruno16, Federica Trucco16, Barbara Panasisi4, Giuseppe Vita6, Tiziana Mongini14, Maurizio Moggio17, Giacomo Pietro Comi5, Eugenio Mercuri3, Enrico Bertini1
1Unit Of Neuromuscular And Neurodegenerative Disorders, Dep Of Neurosciences, Bambino Gesu’ Children’s Research Hospital, Rome, IT;
2Unit Of Neuromuscular And Neurodegenerative Diseases, Department Of Neurosciences, BAMBINO GESU’ HOSPITAL, ROME, IT;
3Dep. Of Child Neurology, POLICLINICO GEMELLI, ROME, IT;
4Developmental Neurology, Child Neurology, And Neuromuscular Diseases And Neuroimmunology Units, Istituto Neurologico “Besta”, MILAN, IT;
5Neuroscience Section, Department Of Pathophysiology And Transplantation (dept), UNIVERSITY OF MILAN, MILAN, IT;
6Department Of Clinical And Experimental Medicine And Nemo Sud Clinical Centre, UNIVERSITY OF MESSINA, MESSINA, IT;
7Neuromuscular Disorders Unit, MEDEA INSTITUTE, BOSISIO PARINI, IT;
8Dipartimento Di Medicina Sperimentale, UNIVERSITY OF NAPLES, NAPLES, IT;
9Neuromuscular Omnicentre (nemo), Fondazione Serena Onlus, HOSPITAL NIGUARDA, MILAN, IT;
10Inspe And Division Of Neuroscience, SAN RAFFAELE HOSPITAL, MILAN, IT;
11Child Neurology And Psychiatry Uni, NEUROLOGICA SCIENCES INSTITUTE, BOLOGNE, IT;
12Department Of Developmental Neuroscience, STELLA MARE iNSTITUTE, PISA, IT;
13mChild Neurology And Psychiatry Unit, “C. Mondino” Foundation, PAVIA, IT;
14Department Of Neurosciences “rita Levi Montalcini”,, University of Turin, TURIN, IT;
15Department Of Neurosciences, UNIVERSITY OF PADUA, PADUA, IT;
16Neuromuscular Disease Unit, G.GASLINI INSTITUTE, GENOA, IT;
17Neuromuscular And Rare Diseases Unit, Department Of Neuroscience, UNVERSITY OF MILAN, MILAN, IT
Abstract: The mean age at diagnosis of Duchenne Muscular Dystrophy (MD) is around 4.3-5 years all over the world. Early diagnosis has several implications including a timely access to Standards of Care and a prompt genetic counselling. Moreover, if novel treatments will be approved they could be started in the early phase of the disease. Here we report findings on a retrospective multicenter study that explores the age at DMD diagnosis in Italy in the past 10 years. Results: We identified 384 Italian boys who were diagnosed with DMD from 2005 to 2014.The mean age of first suspect was 31 months and the mean age at diagnosis was 41 months. The main finding that brought to suspect a DMD was raised CK or transaminases (53% of cases) followed by motor delay (16%), and muscle weakness (14%), Initial concerns about DMD were raised by general pediatricians (30%), by specialists of tertiary centers and first level hospitals (22% and 38% respectively) or parents (10%). Children with an incidental finding of high CK reached the diagnosis first whereas the most delayed diagnosis occurred in those patients who did not manifest any developmental delay. Conclusions: the mean age at diagnosis, over the last decade, in Italy is about 10-12 months less than other country had reported. . The detection of raised CK is the factor that mostly reduces the time for diagnosis. We believe that all male children should be screened in early infancy by a CK assessment even in absence of a neurodevelopmental delay.
PS2Group1-019 / #242Â THE PROPHYLACTIC USE OF PAMIDRONATE ON GLUCOCORTICOID-INDUCED BONE LOSS IN THE MDX MOUSE MODEL OF DUCHENNE MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Sung-Hee S. Yoon1, Jinghan J. Chen2, Marc Grynpas3, Jane Mitchell2
1Pharmacology And Toxicology, Lunenfeld-tanenbaum research institute, Toronto, ON, CA;
2Pharmacology And Toxicology, University of Toronto, Toronto, ON, CA;
3Laboratory Medicine And Pathobiology, lunenfeld-tanenbaum research institute, Toronto, ON, CA
Abstract: Duchenne Muscular Dystrophy (DMD) results from genetic mutations on dystrophin-encoding gene on X chromosome1. Dystrophin is critical in stabilizing the link between the actin fibers in muscle and the extracellular matrix during muscle contractions2. Besides muscular dystrophy, DMD patients also show decreased bone mineral density and higher incidence of fracture due to reduced mechanical stimuli on bone and chronic inflammation of dystrophic muscle, which negatively affect bone health3,4. Most DMD patients receive long-term glucocorticoid (GC) treatment beginning around 5 years of age for beneficial effects on dystrophic muscles, despite the numerous side effects, including GC-induced osteoporosis. Thus, additional therapeutic agents to protect the skeletal system are required. This study examined the effects of the prophylactic, short-term use of bisphosphonates (BPs), anti-resorptive drugs to protect bone from GC-induced osteoporosis, on dystrophic muscle and growing bone in a DMD mouse model (Mdx mice) receiving GCs during growth. For the long-term GC treatment, prednisone pellets releasing 5mg over 60 days were implanted in Mdx mice at 5 weeks of age. Some of the mice received S.C. injections of pamidronate at the initiation of GC treatment for 2 weeks, at a dose of 4mg/kg body weight per week. The mice were sacrificed at 13 weeks of age, after rapid bone growth, and their bone and muscle phenotypes were analyzed in comparison with placebo treated Mdx mice, and placebo treated wild-type mice (C57BL10). Treatment of mdx mice with prednisone had positive effects on dystrophic muscle. GC treatment significantly improved grip strength and decreased the percentage of central nuclei containing fibers in diaphragm and tibialis anterior muscle. GC treated Mdx mice also showed significantly reduced serum creatine kinase (CK) levels and lower Igf-2 and collagen1a1 mRNA levels. The beneficial effects of GCs on dystrophic muscle were maintained with the additional BP treatment. Mdx mice treated with both GCs and BPs showed comparable grip strength, percentage of central nuclei in tibialis anterior, and Igf-2 and collagen1a1 mRNA levels compared to GC treated Mdx group, while serum CK level was further reduced in co-treated Mdx mice. As anticipated, prednisone treatment had negative effects on bone growth and strength. Prednisone treatment significantly decreased cortical bone, which was ameriolated by the additional treatment with pamidronate for the first two weeks of glucocorticoid exposure. Although trabecular bone did not show GC-induced bone loss, the early, short-term treatment with pamidronate significantly increased trabecular bone content. This effect appeared to be mediated by the anti-resorptive action of bisphosphonates, resulting in unresorbed calcified cartilage inside trabeculae. In summary, the early administration of pamidronate at initiation of GC exposure had protective effects on growing bone after 8 weeks of glucocorticoid treatment in a mouse model of Duchenne muscular dystrophy. Bisphosphonate treatment did not interfere with the positive effects of prednisone on dystrophic muscle suggesting that prophylactic use of these drugs early in the course of GC treatment may be helpful in protecting boys with DMD from GC-induced osteoporosis.
PS2Group1-020 / #224TRPV2 INHIBITION THERAPY CAN BE EFFECTIVE FOR CARDIOMYOPATHY OF MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.1 Dystrophinopathy
Tsuyoshi Matsumura1, Misa Matsui1, Yuko Iwata2, Masanori Asakura3, Toshio Saito1, Harutoshi Fujimura1, Saburo Sakoda1
1Neurology, National Hospital Organization Toneyama National Hospital, Toyonaka, JP;
2Molecular Physiology, National Cerebral and Cardiovascular Center Research Institute, Suita, JP;
3Clinical Research And Development, National Cerebral and Cardiovascular Center, Suita, JP
Abstract: Heart failure is the most serious complication in muscular dystrophy. Cardioprotective therapies had shown certain effects however the situation is still unsatisfactory. Transient receptor potential cation channel, subfamily V, member 2 (TRPV2) is a stretch sensitive calcium channel. It normally localizes in the intracellular membrane compartments but translocates to the cytoplasmic membrane in damaged myocytes or cardiomyocytes and enhances influx of calcium, witch trigger cell damage process. In various animal models, TRPV2 inhibition improved cardiac function and/or muscle damages. Tranilast, which has already been approved as anti-allergy drug, has TRPV2 inhibitory effect and may improve cardiac condition of muscular dystrophy. We made a pilot study of tranilast in two muscular dystrophy patients with advanced stage of heart failure. Subjects were two patients with muscular dystrophy. Both patients presented severe heart failure despite aggressive treatments including cardioprotective therapy, diuretics, antiarrhythmic agents and mechanical ventilation. Their left ventricular diastolic dimension (LVDd) exceeded 60mm, fractional shortenings were below 10%, and plasma levels of brain natriuretic peptide (BNP) were over 100 pg/ml. They took tranilast orally at 300 mg/day for three months. Ultrasound cardiogram (UCG), Holter electrocardiogram, BNP, serum level of cystatin C (CysC) and cardiac troponin T (cTnT), expression of TRPV2 in peripheral blood mononuclear cells, circulating microRNA profiles were assessed at before, one month and three months after initiation of tranilast. There were no apparent changes in LVDd and FS of UCG and cTnT levels in both patients. In the first case, BNP decreased from 144.2 pg/ml to 119.7 pg/ml at one month, and was 99.8 pg/ml at three months. He had taken warfarin and had to decrease it on eighth days, since international normalized ratio of prothrombin time elevated from 2.25 to 3.64. He complained mild elevation in pulse rate. Holter ECG also showed increase in mean heart rate (from 75bpm to 88bpm at one month, 87bpm at three months) and premature ventricular contractions (from 2891 beats to 7712 beats at one month, 8445 beats at three months). No other clinically-relevant changes were observed. The second case showed recurrent infections and general condition was unstable. Nonetheless, BNP decreased from 164.3 pg/ml to 102.6pg/ml at one month. He stopped taking tranilast at seven weeks, because CysC was elevated from 0.92 mg/L to 2.86 mg/L. Then BNP increased from 64.5 pg/ml to 228.3 pg/ml in two weeks. After restart of tranilast, BNP decreased to 114.6 pg/ml in ten days. The expression of TRPV2 at plasma membrane were reduced in both patients. In microRNA profiles analyses, some heart related miRNA (miR-208-a-5p, miR-223) were elevated in both patients at the beginning and reduced to control level after initiating tranilast. These findings suggested that TRPV2 inhibition can be also effective for human cardiomyopathy of muscular dystrophy. Further studies are needed to certify the effects and safety of it.
PS2Group1-021 / #381A MISSENSE MUTATION IN THE PUTATIVE SARCOPLASMIC RETICULUM TRANSMEMBRANE PROTEIN DCST2 CAUSES THE STRONGMAN SYNDROME.
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.10 Other Myopathies Including GNE – Hereditary Inclusion Body Myopathy
Talita C. Conte1, Martine Tétreault11, Marie-Josée Dicaire1, Sylvie Provost2, Najwa Al-Bustani1, Marie-Pierre Dubé2, Véronique Bolduc1, Myriam Srour1, Erin K. O’Ferrall1, Jean-Pierre Bouchard3, Gina Ravenscroft4, Russell Hepple5, Tanja Taivassalo6, Nigel Laing4, Phillipa Lamont7, Jean Mathieu8, Bernard Brais1
1Department Of Neurology And Neurosurgery, Montreal Neurological Institute, Montreal, QC, CA;
2Statistical Genetics, Montreal Heart Institute Research Center, Montreal, QC, CA;
35. department Of Neurological Sciences, Hôpital de l’Enfant-Jésus, Quebec, QC, CA;
4Centre For Medical Researc, Western Australian Institute for Medical Research, Nedlands, WA, AU;
5Department Of Kinesiology Centre For Translational Biology, University of McGill, Montreal, QC, CA;
6Department Of Kinesiology, University of McGill, Montreal, QC, CA;
7Department Of Neurology, Royal Perth Hospital, Perth, WA, AU;
810. cliniques Des Maladies Neuromusculaires, CSSS de Jonquière, Jonquiere, QC, CA
Abstract: Muscle mass and strength are variable traits in humans. Individual French Canadians such as Louis Cyr (1863-1912) became international celebrities because of their exceptional strength. This study was based on cohort of 117 cases belonging to 64 families suffering from a heterogeneous dominant condition that we refer to as: “Strongman syndrome” (SM). In all families, the most affected cases suffer from incapacitating myalgias with variable degree of muscle cramps and fatigue, and have a personal and familial history of above average strength. All of the affected individuals had extensive workups, including reported normal muscle biopsies and EMGs, and were found to have no mutations in the following genes that can be associated with cramps and muscles hypertrophy: ANO5, CLCN1, SCN4A, ATP2A1, CAV3, RYR1, MSTN(myostatin), PLIN1 and LMNA. On examination, these patients have large well-defined muscles, above average strength, non-electric prolonged muscle contractions and progressive weakness on repeated contractions. To search for a first SM gene, we focused our efforts on a large French-Canadian (FC) family (42 participants, 20 affected) from the Saguenay-Lac St-Jean region of Quebec. A linkage analysis mapped the mutated gene to chromosome 1q21.3-q23.3 (Maximum multipoint LOD score of 8.1). Whole exome sequencing identified a single rare segregating variant predicted to be damaging in the DCST2 gene in the candidate region. The DCST2 gene is expressed in skeletal muscle and is predicted to code for a six transmembrane domain protein. COS7 cells transfected with a tagged DCST2 protein shows its localization in the ER, whereas mutated DCST2 carrying the SM mutation leads to a different localization and morphological changes that influence overall mitochondrial and ER distribution suggesting that indeed the mutation has a physiological effect that is likely related to its ER function. We also found that DCST2 is not expressed in human myoblasts, nor it is in any of the most used cell lines (HEK293, HeLa, fibroblasts), but once myoblasts start to differentiate into myotubes, DCST2 is highly expressed. DCST2 was found to be mostly expressed in type I myofibers and localizes in close proximity to SERCA2 suggesting that it may play a role in the modulation of intracellular calcium homeostasis. We hypothesize that DCST2 might modify calcium homeostasis in skeletal muscle leading to prolonged intracellular calcium availability and/or sarcoplasmic reticulum store depletion, causing longer contractions in patients which in time will lead to an incapacitating myalgia and fatigue. The uncovering of the function of DCST2 will contribute to a better understanding of pathways important for muscle hypertrophy and strength needed for athletic power performance; moreover it may lead to novel targets for the development of new strategies to treat muscle pain, fatigue and weakness in SM, other neuromuscular diseases and conditions in which muscle atrophy is present, such as aging.
PS2Group1-022 / #194HEREDITARY MYOPATHY WITH EARLY RESPIRATORY FAILURE
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.10 Other Myopathies Including GNE – Hereditary Inclusion Body Myopathy
Sandra Sousa1, Jorge Oliveira2, Emília Vieira2, Teresa Coelho3, Manuela Santos4, Marcio Cardoso3, Ricardo Taipa5, Melo Pires5, Rosário Santos2
1Neurology, Hospital de Cascais, Cascais, PT;
2Unidade De Genética Molecular, Centro De Genética Medica, Centro Hospitalar do Porto, Porto, PT;
3Unidade Corino De Andrade E Serviço De Neurofisiologia, Hospital Santo António, Porto, PT;
4Neurologia Pediátrica, Centro Hospitalar do Porto, Porto, PT;
5Neuropathology, Centro Hospitalar do Porto, Porto, PT
Abstract: Introduction: Different clinical phenotypes have been associated with pathogenic variants in TTN (titin) such as Udd distal myopathy, limb-girdle muscular dystrophy type 2J, hypertrophic and dilated cardiomyopathy, Salih myopathy and hereditary myopathy with early respiratory failure (HMERF). HMERF is a slowly progressive myopathy that typically begins in the third to fifth decades of life. Most common clinical findings are gait disturbance related to distal weakness or nocturnal respiratory failure due to muscle weakness. Phenotype is variable, even within the same family: clinical findings may be distal, proximal or respiratory muscle weakness presenting either alone or in different combinations. The prevalence of HMERF is unknown, but it’s probably under-recognized not only because of the broad phenotypic spectrum but also because of the underlying genetic defect was recently discovered. Muscle biopsy can be useful to the diagnosis but very often the pathological changes are nonspecific, such as eosinophilic cytoplasmic inclusions, rimmed vacuoles and abnormal myofibrillar aggregates. The definitive diagnosis of HMERF is established by the presence of a pathogenic variant in the region of TTN that encodes the 119th fibronectin-3 domain of titin. Clinical Cases: Index Case - female, no relevant past history, admitted to the Intensive Care Unit in 2005 because of respiratory failure of unknown etiology. She had a family history of gait disturbance and respiratory problems. During work-up investigation the muscle biopsy showed myopathic changes with cytoplasmic inclusions localised in subsarcolemmal regions. During the follow-up, she developed the need for noninvasive ventilation and in 2011 the gait began to deteriorate. Case 1 - Sister. At the age of 45 years old, she slowly developed a progressive lower limbs weakness affecting the thigh muscles. 12 years later she suffered respiratory failure and needed noninvasive ventilation. The muscle biopsy showed rimmed vacuoles. Case 2 - First degree cousin. Clinical history of lower limb proximal weakness followed by restrictive respiratory syndrome at 50 years old. Case 3 - First degree cousin. The muscle biopsy demonstrated abundant cytoplasmic body inclusions and myofibrillar aggregates. Case 4 – Second degree cousin. Clinical picture of lower limb distal weakness and simultaneous restrictive respiratory syndrome at 40 years old. Muscle biopsy showed cytoplasmic body inclusions, rimmed vacuoles and abnormal accumulations of desmin. In the index case, we sequenced the exon 343 of TTN gene (based on ENST00000589042), which encodes the fibronectin-3 (FN3) 119 domain of the A-band and is a mutational hot spot for HMERF. We identified a heterozygous mutation NM_001267550.1:c.95134T>C (p.Cys31712Arg), previously reported in European, Indian and Japanese families. Conclusion: We present a family with MHIRP showing different clinical manifestations, inherited in an autosomal dominant manner. These cases highlight the crucial role of a combined correlation between the clinical phenotype and the muscle pathology findings in order to guide some genetic studies of the TTN gene, since the whole TTN gene analysis it’s impossible due to the size and complexity of this gene.
PS2Group1-023 / #345GNE MYOPATHY: MILESTONES AND DISEASE PROGRESSION BASED ON PATIENT SELF-REPORTED DATA COLLECTED THROUGH THE GLOBAL PATIENT REGISTRY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.10 Other Myopathies Including GNE – Hereditary Inclusion Body Myopathy
Oksana Pogoryelova1, Phillip Cammish1, Supriya Rao2, Ed Conner2, Alison Skrinar2, Hanns Lochmüller1
1Newcastle University, The John Walton Muscular Dystrophy Research Centre, Newcastle Upon Tyne,, GB;
2Ultragenyx Pharmaceutical, Ultragenyx Pharmaceutical, Novato, US
Abstract: GNE myopathy is an ultra-rare autosomal recessive distal myopathy caused by mutations in the GNE gene resulting in reduced sialic acid synthesis. Clinical presentation varies from almost asymptomatic carriers to severely debilitating forms. It is considered that this is a relatively slowly progressing disease and takes on average one to two decades to progress from the onset of symptoms to non-ambulant state. In this study we investigate the timeline between the milestones by looking at the mobility level and use of assistive devices over time. We analyzed self-reported data obtained from 120 genetically confirmed GNE patients. Questionnaires were completed by patients through an online database-GNE myopathy registry (https://www.gnem-dmp.com/). The GNE myopathy registry is part of the GNE myopathy disease monitoring program which also includes the GNE myopathy natural history study conducted in selected expert centers worldwide. Mean age of the registry participants was 39.5 years [range 20.5 to 70.1]. Weakness in feet and legs was the reported as the earliest sign of the disease, starting on average at 28.5 years [range 15 to 50 years]. Difficulty walking and stumbles was reported on average at 30.3 years. At an average age of 31.5 years patients reported increasing tiredness, fatigue, difficulty climbing stairs, muscle spasms and twitching. Five years after the onset of first symptoms patients started to notice weakness in arms and hands and muscle pain (33.3 and 33.2 years respectively). On average patients then noticed difficulty moving (e.g. sit to stand or changing position in bed) and started using assistive devices for walking (34.1 and 34.2 years respectively). At the mean age of 35.3 sitting unaided was reported as difficult. First use of wheelchair or scooter occurred on average at 39.1 [range between 21 and over 50]. We estimate that on average the progression from the onset of symptoms to first use of wheelchair takes 10.6 years [range 4.5 to 20.0 years]. Forty one registry participants (34.1%) have stopped working because of the muscle weakness caused by GNE myopathy at the average age of 33.5 years [range 22.9 to 48.0 years] which coincides with the muscle weakness in upper extremity. The presented timeline based on the registry data confirms that the average progression with GNE myopathy takes place over years from first symptoms to substantial disability. This is important for counseling patients with this ultra-rare disease and to ensure the disease management and support is planned and organized efficiently. However, significant differences in speed of the progression highlight the importance of studying factors which can influence the severity of the disease and its course, e.g. concomitant diseases, lifestyle and underlying genetic variability. The above data also contributes to understanding of the health-economic burden of the disease. Future work is required in order to find associated factors contributing to GNE myopathy progression and markers of the severity of the disease.
PS2Group1-024 / #326MULTISYSTEM PROTEINOPATHY WITH MOTOR NERVE CONDUCTION BLOCKS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.10 Other Myopathies Including GNE – Hereditary Inclusion Body Myopathy
Oscar Trujillo1, Juan C. Casar1, Roger Gejman2, Ricardo Fadic1
1Departamento De Neurología, Pontificia Universidad Católica de Chile, Santiago, CL;
2Departamento De Anatomía Patológica, Pontificia Universidad Católica de Chile, Santiago, CL
Abstract: JMM was a 54-year-old right-handed man with history of hypertension and Paget disease. His father died aged 72 of a neurodegenerative disease described as “amyotrophic lateral sclerosis complicated with dementia”. He had a younger sister with Paget’s disease. He had a 4-month history of right foot dorsiflexion progressive weakness. Initial neurologic examination was significant for foot dorsiflexion paralysis on the right, and 3/5 weakness on the left side. He also had 4/5 weakness of the right biceps, deltoid and infraspinous muscles. He had hyperreflexia with a brisk jaw reflex. Plantar responses were indifferent. Muscle tone was mildly increased due to spasticity. Sensory exam was normal. Electromyography and nerve conduction studies showed proximal motor conduction blocks in the left tibial nerve and right cubital nerves. There was denervation and chronic reinnervation signs in muscles innervated by brainstem, cervical, thoracic, and lumbosacral spinal cord motor neurons. There were also small, polyphasic motor units in a few muscles. A muscle biopsy showed an increased muscle fiber sized variation with small and hypertrophic fibers. There were a few regenerating muscle fibers. Rimmed-vacuoles were present in the periphery of some muscle fibers. There were also hyaline zones with loss of the myofibrillar architecture. There were no inflammatory cells. Total creatine kinase (CK) levels were slightly increased or normal (198, 74, 150 U/L, normal values: 38-174). Cell blood count, biochemical profile, and cerebrospinal fluid analysis were normal. Patient’s DNA was sent to the MitoMed laboratory at the University of California Irvine for sequencing of the Valosin containing protein (VCP) gene. A heterozygous deleterious mutation was found at position c.476 G>A in exon 5 of the gene causing an amino acid Arginine/Histidine substitution at position p.R159H. This result is consistent with the diagnosis of Inclusion Body Myopathy associated with Paget’s disease of bone and Fronto-temporal Dementia/ALS (IBMPFD/ALS). He had a rapidly progressing muscle weakness being bed ridden 5 months alter our initial evaluation. At the time he had a feeding tube placed through a gastrostomy and he required noninvasive ventilatory support through Bipap. He died of a respiratory failure two months later. VCP interacts with a large number of ubiquitinated proteins to allow degradation or recycling and functions in multiple protein clearance pathways, including extracting misfolded proteins from the ER and sorting of endosomal proteins for proper trafficking. Depletion of VCP leads to accumulation of immature autophagosomes. Given the crucial role of VCP in maintaining cellular proteostasis, autosomal dominant mutations in the VCP gene lead to a multisystem degenerative disorder. Parkinsonism, ataxia, cataracts, dilated cardiomyopathy, hepatic fibrosis and hearing loss has been related to VCP gene mutations. The term ‘Multisystem Proteinopathy’ has been proposed as the nomenclature for an emerging family of genetic disorders that are unified by this characteristic variation in the penetrance of muscle, bone and CNS degenerative phenotypes. Motor nerve conduction blocks should be added to the continuosly growing Multisystem Proteinopathy phenotypic presentation.
PS2Group1-025 / #202PHENOTYPIC CHARACTERIZATION AND PATTERN OF MUSCLE INVOLVEMENT IN GNE MYOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.10 Other Myopathies Including GNE – Hereditary Inclusion Body Myopathy
Veeramani Preethish-Kumar1, Oksana Pogoryelova2, Kiran Polavarapu1, Narayanappa Gayathri3, Seena Vengalil4, Judith Hudson5, Chandrajit Prasad6, Hanns Lochmüller7, Atchayaram Nalini4
1Clinical Neurosciences, National Institute of Mental Health and Neurosciences, Bangalore, IN;
2John Walton Muscular Dystrophy Research Centre, Mrc Centre For Neuromuscular Diseases, Institute of Genetic Medicine, Newcastle Upon Tyne, GB;
3Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
4Neurology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
5Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, GB;
6Neuroimaging And Interventional Radiology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
7John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, Newcastle, GB
Abstract: Background: GNE myopathy is an autosomal recessive disease caused by mutations in UDP N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene. The disorder is typically characterised by adult onset symmetrical/asymmetrical foot drop progressing to involve the proximal muscles. Quadriceps is generally spared even in advanced stages. Homozygous 712 (p.M712T) mutation is found in Middle Eastern families, whereas patients of other ethnicities are usually compound heterozygous or homozygous for different mutations. Methods: Genetic confirmation of 31 patients was done by direct sequencing of coding exons 1 to 13 and >10 nucleotides of 5’and 3’ intronic sequence. Alamut-Mutation software was used to predict pathogenicity of novel variants. 1.5T AERA MR Scanner was used for evaluating 37 muscles on each side.T1-W images were obtained to look for degree of fibro fatty replacement (Mercuri score) and T2-W STIR for myoedema (Borsato et al). Results: 31 patients were examined from 2007-2015 and all of them were ambulant at the time of evaluation. The mean duration of illness was 7.1 SD ± 5.9 years. The mean age at presentation was 32.9 SD ± 6.7 years and age at onset: 26.0 SD ± 5.4 years. All patients had symptom onset in the lower limbs [proximal- 5/31, distal – 25/31] except one with distal upper weakness as initial symptom. The onset was asymmetrical in 12/31 and symmetrical limbs in 19/31 (61.3%) patients. The time to involve the opposite limb was 1.4 SD ± 2.8 years. Three cases had rapid progression and the rest had a slowly progressive course. The distal upper limb muscles were involved in 20/ 31 (64.5%) and proximal in 7/31 (22.6%), while all had lower limb weakness. Consanguineous parentage (6/31) and family history of affected siblings was present in 7/31 cases. Foot deformities [Pes Cavus-15; Pes Planus-8], scapular winging-2/31 and spinal deformities-13/31 were observed. One patient had mild facial weakness. The classical pattern of muscle selectivity in hip muscles [Flexors weaker than extensors-22/31, adductors weaker than abductors–27/31] were present and knee extensors weaker than flexors was recorded in 28/31 cases. The mean creatine kinase levels were 467.5 SD ± 339.8 years. Electromyography done in 22 patients showed myopathic pattern in 15; neurogenic in 4; normal in 3. All underwent muscle biopsy of either anterior Tibialis / Biceps muscles. Histopathological features included presence of ‘rimmed’ vacuoles, variation in fiber size, atrophic fibers and grouping. Mutations (known and novel) in GNE gene were confirmed in all. MRI showed severe involvement of Biceps femoris (short head), Gluteus minimus, Tibialis anterior, Extensor hallucis and digitorum longus with moderate involvement of Adductors, Hamstrings, Sartorius, medial Gastrocnemius, Tensor fascia lata which was consistent even in early stages by MRI in typical and atypical presentations also. Conclusion: The clinical features of this large cohort of GNEpatients from India is similar to those reported elsewhere with different founder mutations. This suggests that GNE myopathy is a more homogenous disease unlike other distal myopathies. The study also provides the base for using MRI as important tool for diagnosis of clinically suspected cases.
PS2Group1-026 / #478CALPAINOPATHIES IN CHILE
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Jorge A. Bevilacqua1, Yves Mathieu2, Martin Krahn2, Marc Bartoli2, Claudia Castiglioni3, Karin Kleinsteuber4, Jorge Díaz5, Francesca Puppo2, Mathieu Cerino2, Sebastien Courrier2, Svetlana Gorokhova2, Alejandra Trangulao6, Natalia Miranda1, Patricio Gonzalez-Hormazabal6, María De Los Ángeles Avaria4, J. A. Urtizberea7, Pablo Caviedes6, Lilian Jara6, Nicolas Levy2
1Neurología Y Neurocirugía, Hospital Clínico Universidad de Chile, Santiago, CL;
2Inserm, Gmgf, Umr_s 910, Aix Marseille University, Marseille, FR;
3Unidad De Neuropediatria, Clinica Las Condes, Santiago, CL;
4Neuropediatría, Hospital Roberto Del Río, Universidad de Chile, Santiago, CL;
5Centro De Imagenología, Hospital Clínico Universidad de Chile, Santiago, CL;
6Icbm, Universidad de Chile, Santiago, CL;
7Unité Neuromusculaire., Hôpital Marin de Hendaye AP-HP, Hendaye, FR
Abstract: Limb girdle muscular dystrophy 2A (LGMD2A; MIM # 253600) is an autosomal recessive disorder caused by mutations of the CAPN3 gene, which encodes for calpain-3 (CAPN3), a muscle specific calcium-activated neutral protease involved in remodelling of the sarcomere. No patients with calpainopathy have been reported hitherto from Chile. Herein, we describe five patients belonging to four unrelated Chilean families harbouring mutations of the CAPN3 gene. Patient 1 is a 26-year-old female that presented with proximal lower limb weakness since she was 8 years old. She had severe bilateral Achilles tendon retraction that determined a tiptoe gait. She showed hyperlordosis and scapular winging. CK levels were elevated 45-fold. Patients 2 and 3 are two sisters born from a consanguineous marriage. The older sister (Patient 2) presented generalized weakness since she was 7 years old. She walked in tiptoes and underwent a left Achilles tenotomy due to a severe retraction. She showed severe proximal pelvic and shoulder girdles weakness, hyperlordosis and mild scapular winging. CK levels were within normal range. Her younger sister (Patient 3) complained of proximal lower limb weakness since she was 25-years old, and showed severe weakness of the pelvic girdle, associated with distal lower limb involvement and bilateral Achilles retraction. The shoulder girdle was less affected, but presented severe scapular winging. Serum CK levels showed a 5-fold increase. Patient 4 is a 21-year old male that presented delayed motor milestones and increased lower limb weakness since he was 12-years old. He showed a marked amyotrophy of the ischiotibial and adductor muscles on the thighs, with relative sparing of the quadriceps, and a severe impairment in the posterior leg compartments. He shows a scapular winging and anterior arm compartment involvement. CK levels were increased by 34-fold. Patient 5 is the only child of a non-consanguineous marriage, with a history of tiptoe walking associated with calf pain after exercise since he was 10-years old. He presented discrete scapular winging, diffuse amyotrophy, mild gastrocnemius hypertrophy and selective distal biceps hypotrophy. Muscle strength showed a predominantly proximal limb girdle weakness, with Achilles and elbow retractions. CK levels showed a 63-fold increase. The muscle biopsies of all patients showed a non-specific dystrophic pattern, with eosinophilic infiltrates in patient 5. None of the patients showed cardiac or respiratory compromise. Whole body muscle MRI performed to patients 1 to 4 showing a variable degree of fatty replacement, according to disease duration, following the pattern described for LGMD2A. Genetic screening for LGMD mutations was performed in the four patients on a NGS panel of 306 genes involved in neuromuscular diseases, using HaloPlex (Agilent TechnologiesTM) enrichment method and sequenced on the NextSeq500 (IlluminaTM) by HelixioTM (Biopôle Clermont-Limagne, France). The screening allowed the identification of the variant p.Arg788Serfs*14 of the CAPN3 gene (NM_000070.2) for patients 2 and 3; as well as novel mutation p.Gly36Valfs*21 found in a homozygous state for Patient 4 and in a compound heterozygous state, associated with variants p.Arg748Gln and p.Arg788Serfs*14 for patient 1 and 5 respectively. FONDECYT Grant 1151383.
PS2Group1-027 / #401THE FIRST FUNCTIONALLY MATURE HUMAN PRIMARY IN VITRO MUSCLE MODEL: A NEW PARADIGM TO EXPLORE MUSCLE PHYSIOPATHOLOGY AND ACCELERATE DRUG DISCOVERY FOR MUSCLE DISORDERS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Joris Michaud1, Mathieu Fernandes1, Eve Duchemin-Pelletier1, Pauline Poydenot1, Pauline Menager2
1R&d, CYTOO, Grenoble, FR;
2Bd, CYTOO, Bethesda, MD, US
Abstract: To date, a large range of muscle disorders suffers from the lack of treatment opportunities. This is mainly due to the absence of relevant and scalable in vitro models, i.e. mature human models in a miniaturized format combined with relevant readouts having a clear significance to diseases. In this context, we developed the first in vitro human skeletal muscle model compatible with High Content Screening, MyoScreenTM. Based on a tight guidance of myogenesis trough micropatterns, we demonstrated that normal human primary myotubes are fully mature and functional. They present striated sarcomeres, clusters of AChRs, and contract after ACh stimulation, recapitulating key muscle functions that are typically missing in existing in vitro models. Based on high content analysis, we demonstrated that MyoScreen model allows the detection of drug effects and the characterization of their mode of action by discriminating impacts on myogenesis, myotube maturation, and morphology. To demonstrate the benefits of our approach for muscle disorders, we established a model for muscle wasting correction. To do so, atrophy was induced on a healthy MyoScreenTM model using reference compounds known to lead to muscle loss or to impair its renewal. Then, we screened for compounds that could counteract this phenotype. Interestingly, IGF-1 and Trichostatin A were capable of robustly rescuing the induced atrophy showing the relevance of such approach. Altogether, we developed the first functionally mature and scalable in vitro human muscle model. By combining a higher relevance to in vivo situation with an access to phenotypic readouts, MyoScreen represents a new paradigm to improve our understanding of the molecular mechanisms driving muscle disorders and perform drug discovery screening campaigns.
PS2Group1-028 / #366DOMINANT TRUNCATING MUTATIONS IN THE A-BAND OF TTN ARE A CAUSE OF LIMB-GIRDLE MUSCULAR DYSTROPHY WITH CARDIOMYOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Jennifer Roggenbuck1, Ana Morales2, Ray E. Hershberger3, John T. Kissel4
1Neurology, Ohio State University Wexner Medical Center, Columbus, OH, US;
2Division Of Human Genetics, Ohio State University Wexner Medical Center, Columbus, OH, US;
3Divisions Of Human Genetics And Cardiovascular Medicine, Davis Heart And Lung Research Institute, Ohio State University Wexner Medical Center, Columbus, OH, US;
4Neurology, Nationwide Children’s Hospital, Columbus, OH, US
Abstract: Mutations in TTN, encoding the largest human protein, are known to cause dilated cardiomyopathy and several skeletal myopathies without cardiac involvement. Known skeletal myopathies associated with TTN include Udd distal myopathy (caused by dominant M line mutations), limb-girdle muscular dystrophy type 2J (caused by a homozygous M line deletion), hereditary myopathy with early respiratory failure (caused by dominant missense mutations in exon 344), and centronuclear myopathy (caused by recessive truncating or inframe deletions/duplications). Recently, recessive truncating TTN mutations have also been reported to cause several severe childhood onset phenotypes presenting with both skeletal and cardiac muscle disease, including early-onset myopathy with fatal cardiomyopathy (EOMFC). Although dominant truncating mutations in the A-band region of TTN are known to cause 15-20% of dilated cardiomyopathy cases, affected individuals have not been reported to have skeletal muscle involvement. We report three novel truncating mutations in the A-band of TTN, segregating in 3 families with variable dilated cardiomyopathy, variable muscle weakness in a predominantly limb-girdle pattern, and variably elevated creatine kinase (CK). In four cases, the skeletal muscle involvement preceded the cardiac symptoms, while in one case the cardiomyopathy occurred first. See Table. We conclude that, in addition to dilated cardiomyopathy, truncating mutations in the A-band of TTN may cause a variable, progressive skeletal myopathy with proximal muscle weakness, variably elevated CK, with onset ranging from childhood to adulthood. Truncating TTN mutations, while not uncommon in the general population, may be an under-recognized cause of limb-girdle muscular dystrophy, particularly in the context of dilated cardiomyopathy.
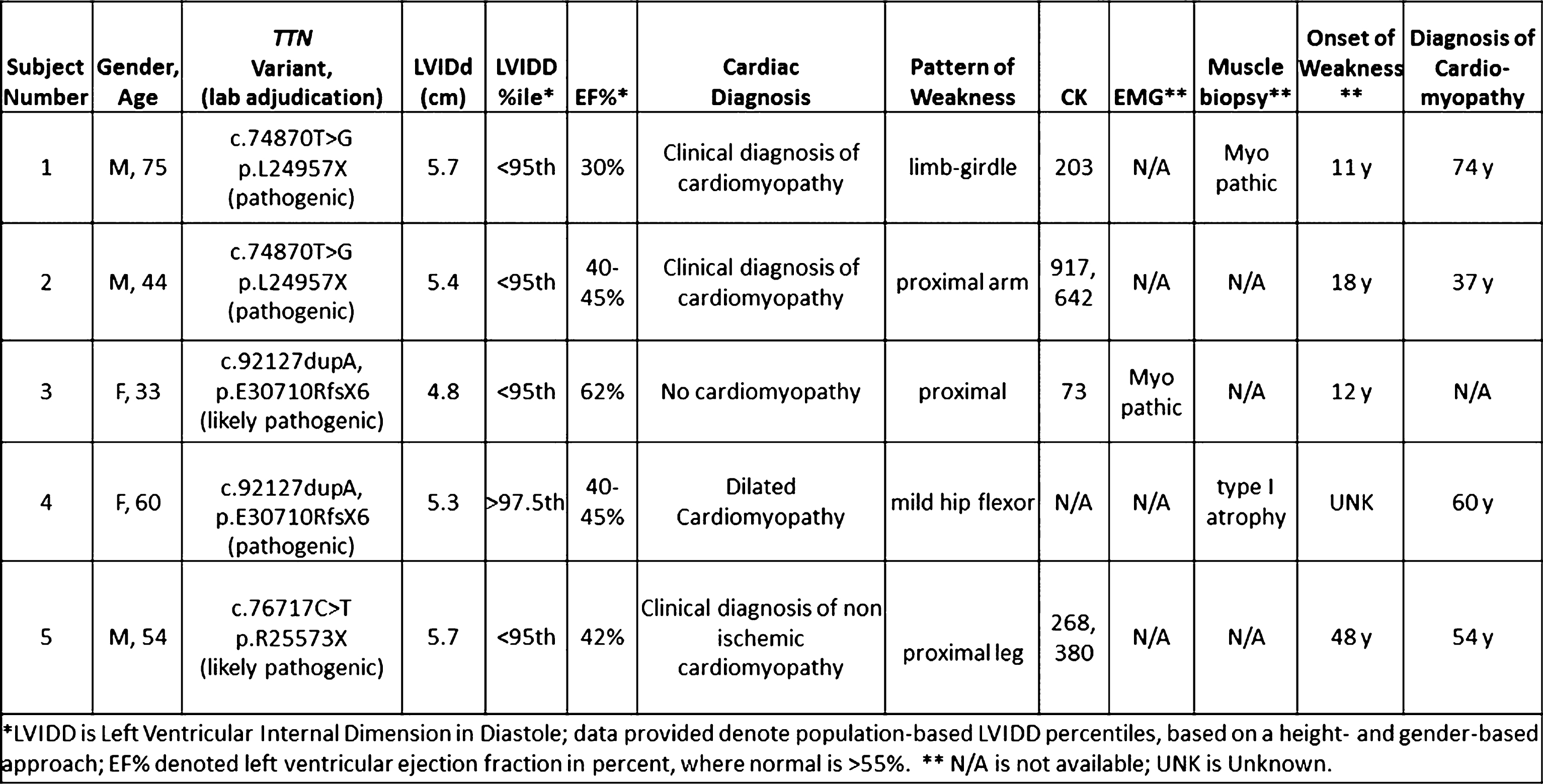
PS2Group1-029 / #363RESCUE OF FOLDING DEFECTIVE ALPHA-SARCOGLYCAN MUTANTS BY MEANS OF PROTEIN FOLDING CORRECTORS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Chiara Fecchio1, Marcello Carotti1, Elisa Bianchini1, Romeo Betto2, Roberta Sacchetto3, Dorianna Sandona1
1Biomedical Sciences, University of Padova, Padova, IT;
2Institute Of Neuroscience, National Research Council of Italy, Padova, IT;
3Department Of Comparative Biomedicine And Food Science, University of Padova, Legnaro (PD), IT
Abstract: Sarcoglycans (SG) are glycosylated proteins (alpha-, beta-, delta- or gamma-SG) forming a key structural complex, essential for the sarcolemma integrity of striated muscles during contraction. It is well known that defects in any one of the genes coding for sarcoglycans lead to the severe reduction or even the complete loss of SG-complex. Most of the mutations associated to sarcoglycanopathy are missense mutations and the disease severity is strictly related to the residual amount of sarcoglycans found at sarcolemma. We have recently proven that the primary event in these cases is the premature degradation of a folding-defective sarcoglycan, followed by the secondary loss of the wild-type partners, operated by the Endoplasmic Reticulum-Associated Degradation. We have also demonstrated that many missense mutants retain their function and that the entire complex can be properly rescued by targeting the degradative pathway. The knowledge of the pathogenic mechanism of sarcoglycanopathy has been also essential to design novel therapeutic strategies for this neglected and still incurable disease. These strategies intend not only to merely inhibit degradation of sarcoglycan mutants, but particularly to help their folding so that, structurally stabilized, mutants can skip disposal and traffic at the proper site of action. We tested several protein folding correctors, screened for the treatment of cystic fibrosis and called CFTR correctors. These small molecules were effective in recovering different mutants of alpha-sarcoglycan in cellular models and, notably, in primary myotubes from a patient suffering of alpha-sarcoglycanopathy. In the latter case, the whole sarcoglycan complex was properly rescued at the plasma membrane, suggesting that a sort of “protein repair strategy” can be adopted to treat sarcoglycanopathy. Although the mechanism by which CFTR correctors act on sarcoglycan mutants need to be clarified, these data represent the proof o principle of a novel pharmacological strategy aiming at correcting mutant folding by using well-known and available small molecules.
PS2Group1-030 / #355CLINICAL OUTCOME STUDY FOR DYSFERLINOPATHY: ONE-YEAR FOLLOW-UP
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Meredith James1, Ursula Moore1, Anna Mayhew1, Michelle Eagle1, Karen Bettinson1, Elena Pegoraro2, Kate Bushby1
1Institute Of Genetics Medicine, The John Walton Muscular Dystrophy Research Centre, Newcastle upon Tyne, GB;
2Department Of Neurosciences, University of Padova, Padova, IT
Abstract: We report on longitudinal clinical outcome data over the first year of a longer 3-year natural history study. An international cohort of 203 adults with dysferlinopathy have been recruited and are assessed 6 times (screen, baseline, 6 months and at 1, 2 and 3 years) gathering diagnostic and medical data, physiotherapy assessment, cardiac and muscle MRI data. A range of physiotherapy assessments are employed at the assessments to assess respiratory involvement (FVC in sitting), muscle strength (Manual muscle testing MMT and hand held Dyometry HHD), functional ability (a modified North Star Ambulatory Assessment, Brooke Score for upper limb, Jebsen Test, the Motor Function Measure (MFM-20). The Six Minute Walk and Timed Tests) (Rise from floor, 10 metre walk/run, four stair climb and descend, timed Up and Go are also performed. Here we examine physical data obtained from physiotherapy assessments at the baseline, 6 month and one year visits. Physical outcomes are reviewed for intrapersonal change over this period The strength assessments were able to detect statistically significant change over a one year period in multiple muscle groups. MMT data demonstrates significant change over 12 months with shoulder abduction, elbow flexion biceps, wrist flexion, hip adduction and hip flexion. The eleven point scale is more sensitive to change over this time period. Hand Held Dyometry (HHD) also indicated significant change for elbow flexion biceps and hip adduction. Additionally HHD identified change for elbow flexion brachioradialis, grip, pinch, knee extension, knee flexion, ankle PF knee bent. Of the Functional Tests MFM and NSAA both highlighted statistically significant change. Two items on the Jebsen (stacking cans and writing) showed significant change while other items did not. The Brooke did not identify change over one year in this cohort. Despite clinically small changes in times, statistical tests suggest that the timed tests are sensitive to change in this population over a year. Rise from floor, 10 metre walk/run, four stair climb and descend, timed Up and Go showed statistically significant change. The six minute walk showed little change in this population over one year. These data allow comparison of how these tests perform as outcome measures in this population in order to refine physical testing in Dysferlinopathy as the study progresses. This Study has been supported by the Jain Foundation
PS2Group1-031 / #347HISTOPATHOLOGICAL AND CLINICAL CHARACTERIZATION OF A SPORADIC TNPO3-MUTATED PATIENT
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Alessandra Ruggieri1, Sara Gibertini1, Barbara Pasanisi1, Vincenzo Nigro2, Marco Savarese2, Maurizio Moggio3, Corrado Angelini4, Renato Mantegazza1, Lorenzo Maggi1, Lucia Morandi1, Marina Mora1
1Neuromuscular Diseases And Neuroimmunology Unit, Fondazione IRCCS Istituto Neurologico C. Besta, Milan, IT;
2Dipartimento Di Biochimica, Biofisica E Patologia Generale, Seconda Università di Napoli, Naples, IT;
3Neuromuscular And Rare Diseases Unit, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, IT;
4Neurobiology Laboratory, Fondazione IRCCS Ospedale San Camillo, Venezia Lido, IT
Abstract: Background: In 2001 Gamez et al reported clinical and morphological phenotype of a novel dominant form of limb girdle muscular dystrophy (LGMD) affecting a large Spanish-Italian family classified as LGMD1F. LGMD1F is clinically characterized by pelvic and shoulder girdles and a wide variability in the age of onset, from 1 to 58 years. Individuals with juvenile onset present severe and rapid disease progression involving proximal and distal limb muscles and leading to early loss of autonomous walking. Patients with adult disease onset manifest slowly progressive limb myopathy and persistent ability to walk, associated, in some patients, with dysphagia, arachnodactyly, and dysarthria. Recently, the causative mutation of the LGMD1F was identified by whole genome sequencing in the transportin 3 (TNPO3) gene. So far only another single sporadic LGMD patient has been identified with a TNPO3 gene point mutation. Objective: We report histopathological and immunochemical findings and long term clinical and radiological follow-up of this sporadic patient. Methods Immunohistochemical evaluation using a panel of antibodies against autophagy related proteins (EEA1, LC3, LAMP2, P62, FK2, BAG3, SMI31) and dystrophin and dystrophin associated proteins, as well as electron microscopy was performed. The patient DNA was analyzed by next generation sequencing (NGS) using a custom panel of causative genes for muscular disorders, including TNPO3. TNPO3 protein level was assessed by Western Blot. Results: The patient was firstly seen at 38 years of age because of a slowly progressive difficulty in walking and climbing stairs in the last 3 years with no familiarity for neuromuscular disorders or parents’ consanguinity Neurological evaluation showed proximal limb muscle atrophy with weakness of shoulder girdle muscles. Finger flexors and extensors’ strength was normal, no face involvement and dysphagia were present. Respiratory and cardiac functions were normal. The symptoms progressively worsened. When last seen, almost 16 years after onset, he had lost his ability to climb stairs and presented hand atrophy and weakness of finger extensors. Muscle MRI revealed severe fibro-fatty substitution of bilateral anterior and posterior thigh muscles, with minimal preservation of rectus femoris and medial compartment of vastus intermedius. Histology showed fiber size variability, central nuclei, scattered degenerating fibers and no ragged red fibres. Immunohistochemical assessment failed to show autophagy activation. Western Blot showed a transportin 3 band of normal molecular weight and reduced intensity. NGS results pointed out a heterozygous G>A transition (c.G2453A) in exon 21 of the TNPO3 gene leading to substitution of the conserved residue arginine 818 to proline. Discussion In our sporadic case, the missense mutation causes a reduction in the level of the protein, which however is of normal size. The role of the conserved residue changed by the mutation is unknown. Differently from the familial cases the autophagy machinery appears unaltered. Clinically, the patient is similar to the adult onset familial cases, but does not manifest any of the additional symptoms described and shows some differences at muscle MRI.
PS2Group1-032 / #266ACE-083, A LOCALLY-ACTING MUSCLE AGENT, INCREASES MUSCLE VOLUME IN HEALTHY VOLUNTEERS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Kenneth M. Attie1, Chad E. Glasser1, Michael R. Gartner2, Brian L. Boes3, R S. Pearsall4, Xiaosha Zhang1, Jade Sun1, Brian Vidal1, Ashley Bellevue1, Monty Hankin4, Matthew L. Sherman1
1Medical Research, Acceleron Pharma, Cambridge, MA, US;
2Celerion, Lincoln, NE, US;
3Bryan Health, Lincoln, NE, US;
4Acceleron Pharma, Cambridge, MA, US
Abstract: Background: ACE-083 is an investigational protein therapeutic that acts as a localized ligand trap for myostatin (GDF8) and other negative regulators of muscle growth in the TGF-β superfamily. In wild-type mice, mdx model of Duchenne muscular dystrophy (DMD), and SOD1 model of amyotrophic lateral sclerosis (ALS), local injection of ACE-083 into a target muscle led to dose-dependent increases in muscle mass and force with no systemic pharmacodynamic (PD) effects. Methods: This is a single-center, double-blind, placebo-controlled, dose escalation study to evaluate the safety, tolerability, pharmacokinetics, and PD effects of ACE-083 in healthy, postmenopausal women. Five cohorts of 8 subjects each (Cohorts 1-5) were randomized to ACE-083 (n=6) or placebo (n=2), administered as 2-4 EMG-guided injections into the right rectus femoris (RF) muscle; two additional cohorts of 9 subjects each (Cohorts 6-7) were randomized to ACE-083 (n=6) or placebo (n=3), and received 4 EMG-guided injections into the right tibialis anterior. Dosing was as follows: Cohorts 1-3 (50, 100, 200 mg) on Day 1, Cohorts 4-5 (100, 200 mg) on Days 1 and 22, and Cohorts 6-7 (100, 150 mg) on Days 1 and 22. MRI to assess thigh or lower leg muscle volume was performed at baseline, and 3 and 8 weeks after last dose. Strength measurements of knee extension or dorsiflexion were evaluated during treatment and follow-up using both fixed and hand-held dynamometers. Results: Data were available for Cohorts 1-5 as of 26 Aug 2015. At the highest dose level, the mean percent change from baseline to 3 weeks after the last dose in muscle volume of the injected right RF was +14.5%. The mean difference in percent change from baseline in muscle volume between the injected right RF and the uninjected left RF was +0.6% in the placebo group, compared to +1.2%, +2.8%, +4.2%, +6.2%, and +13.2% in ACE-083-treated subjects from cohorts 1-5, respectively. In Cohorts 2-5, RF volume remained increased, though attenuated, at 8 weeks after the last dose. Changes in knee extension strength did not correlate with muscle volume increases in these healthy subjects. All AEs were grade 1-2 and reversible. The most frequent related AEs (≥15%) included injection site pain, muscle twitching, myalgia, and injection site reaction, with similar incidences in active- and placebo-treated groups. Conclusions: Local administration of ACE-083 into the RF muscle was well-tolerated and associated with dose-dependent increases in RF muscle volume. These observed increases in muscle mass compare favorably with systemic agents in development and support further studies of ACE-083 in diseases with focal muscle involvement, such as facioscapulohumeral muscular dystrophy (FSHD).
PS2Group1-033 / #292TITLE: CLINICAL OUTCOME STUDY FOR DYSFERLINOPATHY: CLINICAL DATA FROM BASELINE ASSESSMENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Elizabeth Harris1, Ursula Moore1, Catherine L. Bladen1, Anna Mayhew1, Meredith James1, Karen Bettinson1, Heather Hilsden1, Hillarie P. Windish2, The Jain Foundation Cos Consortium1, Kate Bushby1
1Institute Of Genetics Medicine, The John Walton Muscular Dystrophy Research Centre, Newcastle upon Tyne, GB;
2Jain Foundation, Jain Foundation, Seattle, WA, US
Abstract: Background: The Clinical Outcome Study for Dysferlinopathy is the first international multicentre study which aims to characterise the natural history of dysferlinopathy and develop validated functional outcome measures. This study includes patients from 15 sites in 8 different countries. Methods Here we describe the cohort and report genetic and clinical data from the first 193 patients with completed baseline assessments. Participants have a confirmed diagnosis of dysferlinopathy proven by: a) two predicted pathogenic dysferlin mutations (as predicted by the UMD predictor at Marseille) b) one predicted pathogenic dysferlin mutation and absent dysferlin protein on muscle immunoblot, or c) one predicted pathogenic dysferlin mutation and dysferlin protein level ≤20% of normal level determined by blood monocyte testing. Participants are assessed on six occasions over a 3 year period (at screening, baseline, 6 months, 1 year, 2 year and 3 years) gathering diagnostic and medical, physiotherapy assessment, cardiac and muscle MRI data. Biobanking samples are collected as an optional element. DNA and RNA are collected. Plasma and Serum are collected annually. 169 participants have consented to donate samples. Results: The cohort is 52% female, 48% male. 75% participants are ambulant and 25% non-ambulant. The age range is from 12- 88 years old, with a mean age of 40 years. Patients have been symptomatic for 3-52 years (median 17years.). Mean time from symptom onset to diagnosis was 6 years. The data indicates that time to diagnosis is improving. 175 different pathogenic mutations were identified. 112 mutations were unique to a single individual. 84% have 2 pathogenic mutations in DYSF. 3% have more than 2 mutations. 13% have only one known pathogenic mutation plus evidence of dysferlin absence or reduction. 32% have one known nonsense mutation.
PS2Group1-034 / #264MOLECULAR PATHOGENESIS OF CAVEOLIN-3-RELATED LIMB-GIRDLE-MUSCULAR-DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
José Andrés González Coraspe1, Denisa Hathazi2, Hanns Lochmüller3, René Zahedi4, Joachim Weis1, Andreas Roos3
1University Hospital Rwth Aachen, Institute of Neuropathology, Aachen, DE;
2Bioanalytics, Tissue Omics, Leibniz-Institut für Analytische Wissenschaften -ISAS- e.V., Dortmund, DE;
3John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, Newcastle, GB;
4Bioanalytics, Protein Dynamics, Leibniz-Institut für Analytische Wissenschaften -ISAS- e.V., Dortmund, DE
Abstract: Caveolin-3 is a muscle specific protein involved in caveolae-formation, signal transduction, lipid metabolism, cell growth, mechanoprotection and apoptotic cell death. This protein usually appears during the differentiation of myoblasts and it is localized to the sarcolemma, where it interacts with the dystroglycan complex establishing a connection between the extracellular matrix and cytoskeleton. Muscle diseases caused by mutations in the CAV3 gene are called Caveolinopathies. So far, more than 40 pathogenic CAV3 mutations related to the have been described leading different disease phenotypes including Limb Gridle Muscular dystrophy (LGMD), rippling muscle disease (RMD), distal myopathy (DM) and hyperCKemia (HCK). A transgenic animal model harboring a Pro104Leu missense mutation (Cav3P104L) was generated in order to study the nature of Caveolin-3-related LGMD. The phenotype of this animal model was already extensively examined within different studies declaring this model as a suitable phenocopy of the human disorder. To understand the molecular aspects of the skeletal muscle impaired by this mutation, we performed unbiased label-free quantitative LC-MS/MS investigations of quadriceps muscles. Our data revealed up-regulation of 130 and down-regulation of 43 proteins. Notably, further immunoblot and immunohistochemistry studies confirmed the proteomic findings and were also in accordance with electron microscopic findings. Interestingly localization studies of sarcolemmal proteins do not suggest a major contribution in the genesis of Caveolinopathy. To further elucidate the pathogenic character of Cav3P104L and to identify new binging partners of the wild-type protein, unbiased interaction screening (TAP-assay) was performed. These studies revealed a new binding partner for the wild-type protein which is involved in cytoskeleton and EGFR signaling. Interestingly, expression of this binding partner is altered by the presence of Cav3P104L. The combined data contribute to an improved understanding of the onset of the LGMD.
PS2Group1-035 / #168TRIM32 GENE MUTATIONS DETECTED BY NEXT GENERATION SEQUENCING
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Judith Hudson1, Eileen Graham1, Chiara Marini Bettolo2, Teresinha Evangelista2, Volker Straub3, Fiona Norwood4, Kate Bushby2, Rita Barresi5
1Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, GB;
2John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle upon Tyne, GB;
3Newcastle University, Institute of Genetic Medicine, Newcastle upon Tyne, GB;
4Kings College Hospital, London, GB;
5Muscle Immunoanalysis Unit, Newcastle upon Tyne Hospitals NHS Foundation Trust, Newcastle upon Tyne, GB
Abstract: The TRIM32 gene encodes the tripartite motif-containing protein 32, which is known to be associated with a range of diseases including LGMD2H, Bardot-Biedl syndrome and sarcotubular myopathies. LGMD2H is an extremely rare autosomal recessive, mild muscular dystrophy which is characterised by myopathic features. It is typically a late-onset condition with a highly variable phenotype. Initial reports identified a single homozygous mutation to be responsible for causing cases of LGMD2H in the inbred Manitoba Hutterite population. Subsequent studies have shown LGMD2H can be caused by a broader range of mutations including missense mutations, deletions and frameshift mutations. The precise mechanism by which TRIM32 gene mutations result in LGMD2H remains to be elucidated. We present a case report of a 41year old patient who presented with proximal lower limb weakness, atrophy of the quadriceps and calf hypertrophy. He first manifested clinical symptoms at age 30, with waddling gait and difficulties climbing stairs and walking uphill. Photographic evidence suggests calf hypertrophy was present in his teenage years. His serum CK activity was elevated up to 1844 IU/L. An EMG showed a myopathic pattern and muscle biopsy analysis was consistent with a muscular dystrophy, though no abnormalities were detected following immunohistochemistry and immunoblotting with a panel of antibodies for diagnosis of LGMDs. Muscle MRI showed selective involvement of the posterior compartment of the thigh. Molecular genetic testing was performed for a range of commonly mutated LGMD genes, all of which gave negative results. Next generations sequencing analysis was performed for a panel of 32 genes known to cause LGMD. The patient was found to be heterozygous for the novel pathogenic mutations c.691delG and c.1108delA in exon 2 of the TRIM32 gene. This case report highlights the value of utilising next generation sequencing as a diagnostic tool for rare forms of LGMDs.
PS2Group1-036 / #174MUSCLE INVOLVEMENT IN LIMB GIRDLE MUSCULAR DYSTROPHY WITH GMPPB DEFICIENCY (LGMD2T)
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Sofie T. Østergaard
Copenhagen Neuromuscular Center Department 3342, Rigshospitalet, Copenhagen, DK
Abstract: Objective: Limb girdle muscular dystrophy due to mutations in GMPPB has only been described in a few patients and the pattern of muscle involvement has not been studied in detail. In this study, muscle involvement was assessed by MRI and by levels of GMPPB and glycosylation of α-dystroglycan expression in muscle. Methods: Five patients with genetically verified mutations in GMPPB were recruited. In four of the subjects, T1-weighted images were evaluated. Subjects were asked about medical history and current symptoms. Muscle strength and potential involvement of extramuscular organs were examined. From muscle biopsies, Glycosylation of α-dystroglycan was studied and GMPPB expression by western blotting. Prevalence of LGMD2T was calculated from the total LGMD population, and GMPPB was sequenced in all unclassified cases. Results: One patient carried new mutations in GMPPB, which were deemed pathogenic. The other four carried previously described pathogenic mutations in GMPPB. MRI showed that the paraspinal muscles was the most affected muscle, followed by involvement of hamstrings. Our results showed that GMPPB deficiency led to a secondary loss of merosin expression and loss of glycosylation of α-dystroglycan when tested with the IIH6C-antibody, while glycosylation tested with the VIA4-antibody appeared normal. The prevalence of LGMD2T is 1.5%. Conclusion: The new findings of this study are 1) the consistent finding of a preferential affection of paraspinal and hamstring muscles in LGMD2T, 2) two new mutations in GMPPD, 3) specific loss of glycosylation to IIH6-, but not VIA4-antibody staining, and 4) a prevalence of LGMD2T of 1.5% in a well characterized LGMD cohort.
PS2Group1-037 / #149MUSCLE MRI CAN BE A POWERFUL TOOL TO DIAGNOSE LIMB GIRDLE MUSCULAR DYSTROPHY 2L
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Maria Elena Farrugia1, Cheryl Longman2, William Stewart3, Volker Straub4, Richard K. Petty1
1Neurology, Institute of Neurological Sciences, Glasgow, GB;
2Clinical Genetics, West of Scotland Regional Genetics Service, Glasgow, GB;
3Neuropathology, Neuropathology, Glasgow, GB;
4Newcastle University, Institute of Genetic Medicine, Newcastle upon Tyne, GB
Abstract: Attaining a molecular diagnosis in limb girdle muscular dystrophies (LGMD) can be challenging. In some cases, such as dysferlinopathies and LGMD caused by ANO5 mutations, patients may have high creatine kinase (CK) levels but be virtually asymptomatic or may simply complain of muscle pain and discomfort before developing muscle weakness. We present the case of a female patient now age 58 years who had normal motor developmental milestones but was labelled as being “lazy” all her life. Symptom onset started at the age of 43 (in 2000) with a flu-like illness. She was pyrexial, and developed a confluent rash over her trunk, arms and face with sparing of her lower limbs, but no anorexia or weight loss. She developed fulminant muscle pain with weakness and was bedbound for 5 days. She recovered, but thereafter complained of myalgia and was noted to have a hyperCKemia of up to 6000 U/l (normal range up to 200 U/l). Her EMG showed myopathic motor unit potentials but no spontaneous activity. She was commenced on corticosteroid treatment for a diagnosis of “polymyositis” despite her muscle pathology showing no evidence of inflammation or HLA upregulation. On assessment she was felt to have almost normal power. Her CK ranged around 1669 in 2008 and 610 in 2012. In 2005, corticosteroids were gradually weaned off and stopped and she felt that her myalgia worsened but examination remained unremarkable. She was started on statins in 2005 because of familial hypercholesterolemia but was intolerant of them with worsening myalgia. When assessed by a neurologist in 2009, she was still strong on examination and complaining bitterly of muscle pain. Her investigations excluded myotonic dystrophy type 1 and 2, common dystrophin mutations, FKRP and adult-onset Pompe disease. Over time, she developed a mild proximal myopathy, with myalgia remaining a significant feature. MR imaging of her thigh and calf muscles showed patchy involvement of the adductor magnus and striking involvement of the medial gastrocnemius muscles. The imaging findings were similar to another patient (male) who attends our muscle clinic with a confirmed pathogenic mutation in ANO5, and also with the pattern reported in the literature. Genetic studies confirmed that she was heterozygous for the ANO5 splice site mutation c.1898+1G>A in intron 17, which was previously reported as pathogenic to the Leiden muscular dystrophy database, consistent with limb girdle muscular dystrophy type 2L. She was also found to be a putative homozygote for the missense mutation c.2018A>G in exon 18 that has also been reported as pathogenic to the Leiden Muscular Dystrophy database. LGMD2L may cause late-onset proximal muscle weakness, often asymmetric, or less commonly Miyoshi muscular dystrophy 3 with early adult-onset calf distal myopathy. The CK is markedly elevated. An important differential diagnosis is LGMD associated with dysferlin mutations but the imaging findings are distinct. Muscle MRI can guide the neuromuscular physician in furthering the molecular diagnosis of a patient with LGMD.
PS2Group1-038 / #139MOLECULAR DIAGNOSIS OF BETA-SARCOGLYCANOPATHY: REPORTING TWO NOVEL MUTATIONS IN IRAN
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.2 Muscle Dystrophies (Non-Dystrophinopathy)
Marzieh Mojbafan1, Sirous Zeinali2, Seyed Hasan Tonekaboni3, Abdolazim Nejati Zadeh4, Yalda Nili Pour5
1Department Of Molecular Medicine, Pasteur Institute of Iran-Tehran university of medical sciences, Tehran, IR;
2Department Of Molecular Medicine, Biotechnology Research Center, Pasteur Institute of Iran, Tehran, IR;
3Department of Pediatric Neurology, Shahid Beheshti University of Medical Sciences, Tehran, Iran, Tehran, IR;
4Research Center for Molecular Medicine, Hormozgan University of Medical Sciences, Bandar Abbas, Iran., Bandar Abbas,, IR;
5Pediatric Pathology research centre, Pathology Mofid hospital, Shariatist., Tehran, Iran, Tehran, IR
Abstract: Background: Autosomal recessive limb-girdle muscular dystrophies (LGMDs) are genetically heterogeneous muscular dystrophies distinguished by progressive proximal muscle weakness. Sarcoglycanopathies are a subgroup of this disorder which are caused by mutations in sarcoglycan (SGs) genes. SG proteins form a core complex consists of α, β, γ and δ sarcoglycans which are encoded by SGCA, SGCB, SGCG and SGCD genes respectively. A subgroup of autosomal recessive limb girdle muscular dystrophies (LGMDs) which are caused by mutations in sarcoglycan (SGs) genes called Sarcoglycanopathies. SG proteins form a core complex consists of α, β, γ and δ sarcoglycans which are encoded by SGCA, SGCB, SGCG and SGCD genes respectively. Muscle biopsy studies in patients with sarcoglycanopathies have indicated that loss of one SG subunit leads to instability of whole SG complex.This effect can be helpful in interpreting muscle biopsy results. Autozygosity mapping is a powerful gene mapping approach for rare recessive inherited disorders in consanguineous families. Methods: three unrelated Iranian families, having 10 affected patients, were investigated. All patients had raised CpK. EMG-NCV showed myopathic process and muscle immunohistochemistry in 9/10 severe case showed sarcoglycanopathy. Haplotype analysis using short tandem repeat (STR) flanking all SG genes was performed and all coding and non-coding exons and intron boundaries of the SGCB gene were sequenced. Results: Autozygosity mapping, using four STR markers for each of the SG genes, showed that the phenotype may segregate with SGCB gene. DNA sequencing identified 2 novel splicing mutation and one recurrent homozygous mutation c.-10_16dup26. Conclusions: Since two novel splicing mutations are at acceptor and donor sites, they cause abnormal splicing. The third mutation was observed in a large multifamily in which two crossing overs have been observed within SGCB gene during autozygosity mapping suggesting the mutation might be in the first two exons of SGCB gene. Mutation analysis showed a 26bp duplication (10 bp before the initiation codon till 13 bp after the ATG). This will cause a frameshift in protein synthesis.
PS2Group1-040 / #485A NOVEL PATHOGENIC MUTATION IN TPM3 GENE IN A 5 YEARS OLD IRANIAN PATIENT WITH AUTOSOMAL RECESSIVE NEMALINE MYOPATHY-1
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.4 Congenital Myopathies / Myopathies with Prominent Muscle Contractures
Saeid Morovvati1, Yashar Morovvati2
1Research Center for Human Genetics, Baqiyatallah University of Medical Sciences, Tehran, IR;
2Research Center For Human Genetics, Baqiyatallah University of Medical Sciences, Tehran, IR
Abstract: Introduction: Nemaline myopathy-1 (NEM1) is a disorder characterized by muscle weakness, usually beginning in early childhood. The severity and pattern of muscle weakness varies, but most affected individuals show mildly delayed motor development, hypotonia, generalized muscle weakness, and weakness of the proximal limb muscles and neck muscles, resulting in difficulty running and easy fatigability. Most patients have respiratory insufficiency due to muscle weakness. Other common features include myopathic facies, high-arched palate, and scoliosis. NEM1 is caused by heterozygous, homozygous, or compound heterozygous mutations in the alpha-tropomyosin-3 (TPM3) gene. NEM1 is a rare disorder with an estimated incidence of 1:50,000 live births. Materials and Methods: A 5 years old symptomatic male with muscular disease resulted from a consanguineous marriage was brought to our clinic for genetic testing. 53 genes involved in Muscular Diseases were analyzed in the patient by Next Generation Sequencing method. The identified variants and mutations were confirmed in the patient and his parents by Sanger Sequencing method. Results: A likely pathogenic mutation, c.79_80insGCAG (p.Glu27GlyfsX4), on TPM3 gene, was detected in the patient a homozygous state. The same mutation was detected in his parents in a heterozygous state. Discussion: Although this mutation has not been reported, its frequencies in normal population are very low. The frameshift mutation leads to early termination of the amino acid coding, which is expected to affect the protein’s function. The mutation c.79_80insGCAG (p.Glu27GlyfsX4) on TPM3 gene is possible the pathogenic mutation of the patient, which was consistent with the clinical diagnosis.
PS2Group1-041 / #414MTM1-RELATED MYOPATHY CARRIER FEMALES MANIFEST SIGNIFICANT ASYMMETRIES AND A SPECTRUM OF MUSCLE INVOLVEMENT
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.4 Congenital Myopathies / Myopathies with Prominent Muscle Contractures
Benjamin T. Cocanougher1, Pomi Yun2, Lauren Flynn3, Mina Jain4, Melissa Waite4, Ruhi Vasavada4, Jason Wittenbach1, Sabine De Chastonay5, Sandra Donkervoort2, A.R. Foley2, Carsten G. Bonnemann2
1Janelia Research Campus, Howard Hughes Medical Institute, Ashburn, VA, US;
2National Institutes Of Health, NINDS, Neuromuscular and Neurogenetic Disorders of Childhood Section, Bethesda, MD, US;
3National Institute Of Neurological Disorders And Stroke, National Institutes of Health, Bethesda, MD, US;
4Clinical Center, National Institutes of Health, Bethesda, MD, US;
5Congenital Muscular Dystrophy International Registry, Cure CMD, Torrance, CA, US
Abstract: X-linked myotubular myopathy (XLMTM) is a rare genetic disorder caused by mutations in the MTM1 gene. The disease manifests with severe congenital weakness and hypotonia in male infants. In the absence of supportive care, most males die in early infancy of respiratory insufficiency. Carrier females are less well characterized. Single cases from multiple reports exhibit a spectrum of disease in females, ranging from mild facial weakness to severe congenital weakness. We deeply phenotyped a cohort of ten females with causative mutations in MTM1 by critically evaluating the severity of disease expression based on muscle weakness, skeletal morphology, pulmonary function (spirometry and dynamic breathing MRI) and muscle imaging (ultrasound and MRI). Neuromuscular examination revealed consistent patterns of muscle involvement along a spectrum of weakness severity which we categorized as severe (non-ambulant) (n=1); moderate (assisted ambulation) (n=2); mild (independent ambulation but with clear evidence of muscle weakness) (n=5) and non-manifesting (no evidence of muscle weakness) (n=2). Both the severity of respiratory insufficiency and the degree of abnormal signaling on muscle imaging were directly correlated with the degree of muscle weakness. Interestingly, we noted significant and consistent left-to-right asymmetries evident in all ten females evaluated in our cohort, even in the absence of muscle weakness. Asymmetries of muscle strength and skeletal morphology appeared correlated with handedness, with weakness and atrophy of extremities evident contralateral to each individual’s dominant side (i.e. right-handed patients were weaker on the left). The degree of skeletal asymmetry did not necessarily correlate with the degree of weakness, however, potentially suggesting distinct pathways for MTM1-encoded myotubularin, a ubiquitously expressed phosphoinosotide phosphatase, in the development of skeletal morphology and skeletal muscles. Deep phenotyping of females with MTM1 mutations is particularly timely as gene therapy trials for infant males with XLMTM are being planned; the outcome of these trials will determine whether gene therapy may be extended to carrier females.
PS2Group1-042 / #295P4HA1 MUTATIONS CAUSE A UNIQUE CONGENITAL DISORDER OF CONNECTIVE TISSUE INVOLVING TENDON, BONE, MUSCLE AND THE EYE
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.4 Congenital Myopathies / Myopathies with Prominent Muscle Contractures
Sandra Donkervoort1, Yaqun Zou1, Antti M. Salo2, Aileen M. Barnes3, Ying Hu1, A. R. Foley1, Elena Makareeva4, Meganne E. Leach1, Wendy Dinonno5, Jahannaz Dastgir6, Ronald D. Cohn7, Sergey Leikin4, Joan C. Marini3, Johanna Myllyharju2, Carsten G. Bonnemann1
1National Institutes Of Health, NINDS, Neuromuscular and Neurogenetic Disorders of Childhood Section, Bethesda, MD, US;
2Faculty Of Biochemistry And Molecular Medicine, Oulu Center for Cell-Matrix Research, Biocenter Oulu, Oulu, FI;
3National Institutes Of Health, Bone and Extracellular Matrix Branch, Eunice Kennedy Shriver National Institute of Child Health and Human Development, Bethesda, MD, US;
4Section On Physical Biochemistry, Eunice Kennedy Shriver National Institute Of Child Health And Human Development, National Institutes of Health, Bethesda, MD, US;
5Eastern Virginia Medical School, Newport News, VA, US;
6Ninds, Neuromuscular And Neurogenetic Disorders Of Childhood Section, National Institutes of Health, Bethesda, MD, US;
7Division Of Clinical And Metabolic Genetics, Centre For Genetic Medicine, The Hospital for Sick Children, Toronto, ON, CA
Abstract: Collagen prolyl 4-hydroxylases (C-P4Hs) play a central role in the formation and stabilization of the collagenous triple helical domain that are the defining structural motif of collagens. P4HA1 encodes for one of the catalytic α units, and thus far no mutation in C-P4Hs has been reported to cause human disease. We report the first human P4HA1 mutations in a family with a congenital-onset disorder of connective tissue, manifesting as early-onset joint hypermobility, weakness and bone malformations as well as high myopia, with clinical improvement over time in the surviving patient. Similarly to P4ha1 null mice, which die prenatally, patients were found to have reduced collagen IV expression at the muscle basement membrane. Patients were compound heterozygous for nonsense and splice mutations leading to reduced P4HA1 protein level and C-P4H activity in dermal fibroblasts compared to age-matched control samples. Differential Scanning calorimetry assay revealed reduced thermal stability of collagen with a decrease in proline hydroxylation in patients versus age-matched controls. The uniqueP4HA1 mutations identified in our patients shed light on the normal function and alternative splicing of exons 9 and 10 splice forms in P4HA1 in different tissues during development. Mutations affecting C-P4Hs should be considered in patients presenting with connective tissue overlap disorders with features of Ehlers-Danlos Syndrome.
PS2Group1-043 / #270DE NOVO DOMINANT MOSAIC MUTATIONS IN COLLAGEN 6 GENES: UNCOMMON CAUSE OF BETHLEM AND ULLRICH MYOPATHIES THAT MAY BE MISSED BY SANGER SEQUENCING.
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.4 Congenital Myopathies / Myopathies with Prominent Muscle Contractures
Adele D’Amico1, Fabiana Fattori1, Francesca Gualandi2, Stefania Petrini1, Valentina Doria1, Giorgio Tasca1, Michela Catteruccia1, Marcello Niceta3, Marco Tartaglia3, Alessandra Ferlini2, Enrico Bertini1
1Unit Of Neuromuscular And Neurodegenerative Disorders, Dep Of Neurosciences, Bambino Gesu’ Children’s Research Hospital, Rome, IT;
2Department Of Reproduction And Growth And Department Of Medical Science, UOL of Medical Genetics, University of Ferrara, Ferrara, IT;
3Molecular Genetics And Genomics Unit, Division Of Genetics And Rare Diseases, Bambino Gesu’ Children’s Research Hospital, Rome, IT
Abstract: Collagen type VI-related dystrophies (COL6-RD) are a spectrum of conditions ranging from severe Ullrich congenital muscular dystrophy (UCMD) to intermediate phenotype and the milder Bethlem myopathy (BM). COL6-RD are caused by mutations in the three different genes, COL6A1, COL6A2 and COL6A3. COL6-RD are characterized by inter and intra-familial phenotypic variability. Somatic mosaicisms have been recently reported partially explaining the phenotypic heterogeneity in 4 families. We report on clinical, immunohistochemical, and genetic data about 3 unrelated patents affected by a COL6-RD who carried a de novo mosaic mutations in col VI genes. All patients had clinical, histochemical and myoimaging findings consistent with a diagnosis of COL6-RD, athough mutations in none of the three patients were detected by Sanger Sequencing. Whole Exome Sequencing allowed the identification of a de novo mosaic COL6A3 mutation in one patient who presented an intermediate phenotype and two de novo mosaic COL6A2 mutations in the additional 2 patients who manifested a Bethlem phenotype. This study highlights the importance of an extensive diagnostic workup when clinical and histological finding are consistent with a COL6-RD.
PS2Group1-044 / #213A PROSPECTIVE STUDY OF HISTOLOGY, PHENOTYPES AND GENETICS IN CONGENITAL MYOPATHY PATIENTS ABOVE 5 YEARS OF AGE IN DENMARK
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.4 Congenital Myopathies / Myopathies with Prominent Muscle Contractures
Nanna Witting1, Ulla Werlauff2, Morten Duno3, John Vissing1
1Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK;
2The Danish National Rehabilitation Centre for Neuromuscular Diseases, Aarhus, DK;
3Rigshospitalet, Copenhagen, DK
Abstract: Background: The entity congenital myopathy has been recognized for decades, but knowledge of overall prevalence and frequency of subtypes are still lacking. Moreover detailed systematic evaluation of phenotype characteristics for different genetic causes has not been reported. Methods: A national cohort of 107 (35 years old) patients with a diagnosis of congenital myopathy was assessed clinically and histologically, and based on these findings evaluated systematically for genetic cause. Results: Twenty-four were excluded due to features not typical for congenital myopathy or identification of aetiology other than congenital myopathy. The remaining 83 corresponded to a prevalence of 2:100,000. Histological exam revealed 14 (17%) with core disease, 15 (18%) with centronuclear myopathy, 12 (14%) with nemaline bodies, 28 (34%) with congenital fibre type disproportion or type I predominance, and 14 (17%) subjects with non-specific myopathic changes. A genetic diagnosis was reached in 45 patients (54.2%.). 4.8% had mutation in ACTA1, 2.4% in TPM2, 2.4% in TPM3, 7.2% in NEB, 14.5% were heterozygote for mutation in RYR1, and 6.0% were compound heterozygous for mutations in RYR1, 7.2% had mutation in DNM2, 3.6% in MTM1, 2.4% in TTN, and 2.4% in SC4NA. The detailed clinical exam found distinct clinical differences among genetic subtypes, with discrepancies in the pattern of involvement of proximal/distal extremities and respiratory and external ocular muscle affection. The success rate of identifying a genetic aetiology was highly dependent on histology; in the group with central cores, rods or centronuclear myopathy, 83% were genetically diagnosed, whereas the genetic cause only was identified in 29% of patients in the group with more unspecific histology; congenital fibre type disproportion, type 1 dominance and unspecific myopathic changes. Genetic aetiology was identified in 13 in the excluded cohort; 6 had mutations in COL6, 2 in DES, 2 had CMS and 3 LGMD (1 LGMD2L, 1 LGMD2A, 1 LGMD1C). Discussion: The study presents novel findings of a national prevalence and distribution of subtypes of congenital myopathy and contributes new phenotypic characteristics. This information can be used to navigate genetic test strategies and interpret results of NGS testing, and the results may also impact on clinical follow-up and counselling of patients.
PS2Group1-045 / #492IDENTYFYING AND CHARACTERIZING NOVEL MUTATIONS CAUSING ARTHROGRYPOSIS.
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.4 Congenital Myopathies / Myopathies with Prominent Muscle Contractures
Forough Noohi1, Martine Tétreault2, Jacek Majewski3, Bernard Brais4
1Human Genetics, Montreal Neurological Institute, Montreal, CA;
2McGill University and Genome Quebec Innovation Center, Montreal, CA;
3McGill University and Genome Quebec Innovation Center, Montreal, QC, CA;
4Department Of Neurology And Neurosurgery, Montreal Neurological Institute, Montreal, QC, CA
Abstract: Congenital Muscular Dystrophies (CMDs) are a heterogeneous group of inherited muscle disorders that comprise low muscle tone and poor movements as well as delayed motor development and joint contractures. On the other hand, Arthrogryposis, or arthrogryposis multiplex congenita (AMC), is a nonprogressive condition characterized by congenital multiple joint contractures. Arthrogryposis is often considered as a distinct entity which shares some genotypic and phenotypic overlap with CMDs. We have recruited a large consanguineous family from the Middle East with more than twenty two affected individuals diagnosed with a dominantly inherited distal Arthrogryposis. Clinical manifestations include joint contractures in both hands and feet. However, the diverse representation of the disease in the family ranges from severely affected with minimal muscle development throughout the entire body to very mild with only minor contractures in hands. The most severely affected individuals in the family exist mostly within the consanguinity loop, whose severe phenotypes might be explained by the inheritance of double mutation. We have performed Whole Exome Sequencing (WES) on four affected individuals from the family. We first searched for rare variants in genes known to cause CMDs, Myopathies, and distal Arthrogryposis. However, we did not find any promising candidate variants. Next, we looked for rare heterozygous variants consistent with an autosomal dominant inheritance and obtained a long list of candidate variants. We are in the process of validating the candidate variants through the use of Sanger sequencing and so far have uncovered a very promising candidate variant in TTN that segregates in fifteen individuals that we have collected DNA samples from. We are in the process of designing more functional analysis to determine the causative mutation and are confident that WES would uncover a novel mutation causing a new phenotype in this family.
PS2Group1-046 / #475DESMINOPATHY IN CHILE, TWO FIRST CASES REPORTED
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.5 Distal Myopathy / Myofibrillar Myopathies
Jorge A. Bevilacqua1, Lidia González-Quereda2, Ivonne Zamorano3, Claudia Castiglioni4, Lorena Acevedo1, Jorge Díaz5, María José Rodríguez6, Alejandra Trangulao7, Mario Rivera8, Pia Gallano2
1Neurología Y Neurocirugía, Hospital Clínico Universidad de Chile, Santiago, CL;
2Servicio De Genética U705 Ciberer, Hospital de Sant Pau, Barcelona, ES;
3Neurología Y Neurocirugía, Eduardo Schütz Schroeder en Puerto Montt, Puerto Montt, CL;
4Unidad De Neuropediatria, Clinica Las Condes, Santiago, CL;
5Centro De Imagenología, Hospital Clínico Universidad de Chile, Santiago, CL;
6Servicio De Genética, Hospital de Sant Pau, Barcelona, ES;
7Icbm, Universidad de Chile, Santiago, CL;
8Neurología Y Neurocirugía, Clínica Davila, Santiago, CL
Abstract: Mutations in the gene encoding for the muscle-specific intermediate filament desmin on chromosome 2q35, are the underlying cause of variable clinical phenotypes collectively referred as desminopathies. Patients with desminopathy may variably present as myofibrillar myopathy (DRM, MIM#601419), dilated cardiomyopathy (CMDII, MIM#604765) or autosomal dominant or recessive forms of limb girdle muscular dystrophy (LGMD1E, MIM#602067 and LGMD2R, MIM#615325). The incidence and prevalence of desmin myopathy and/or cardiomyopathy are unclear. In Chile, no cases with any form of desminopathy have been reported to date. Herein we describe three patients belonging to two unrelated Chilean families harbouring mutations of the desmin (DES) gene. Patient 1 is a 36-year-old man with a history of two years of progressive lower limb weakness. At first examination he presented distal anterior and posterior leg amyotrophy and bilateral “stepagge”. In the upper limbs, muscle strength was in normal range. CK plasma level was elevated 6-fold and an electromyogram showed a myopathic pattern with normal nerve conduction values. A left quadriceps muscle biopsy showed severe dystrophic changes, with normal immunostain for sarcolemmal proteins and no myofirillar aggregates. A whole body MRI revealed a moderate selective involvement of the deltoids in the shoulder girdle, and a severe fatty replacement affecting the anterior and posterior leg compartments and the thighs, with a selective more severe involvement of the semitendinosus muscle. This particular MRI involvement pattern prompted the sequencing of the DESgene (NM_001927.3) allowing the identification of the missense mutation p.Leu370Pro in exon 6 of the DES gene, in heterozygous state. Patient 2 is a 48 years-old sportive man that experienced progressive lower limb weakness that prevented him from running. Examination showed a slim phenotype, with a generalized weakness involving predominantly the right lower limb. He was able to stand up in his toes, but not in the heels. Osteotendinous reflexes were preserved. CK levels were slightly increased (0.5-fold). His three-years younger brother (Patient 3) had a history of lower limb weakness beginning when he was 40-years old. Impairment started at the anterior leg compartment and later progressed proximally. Electromyography was myogenic; CK levels were increased 8-fold, and a biopsy performed on his left gastrocnemius had been reported as inflammatory. At time of examination he was 45 years old, he was wheelchair-bound and a pacemaker was indicated due to an arrhythmia. One year later, Patient 2 underwent a quadriceps muscle biopsy that showed non-specific myopathic changes. The whole body muscle MRI revealed a selective involvement of the tibialis anterior muscles in the legs, and a selective fatty replacement of the semitendinosus in the thighs. The genetic screening allowed the identification in the two brothers, of the pathogenic variant p.Arg350Pro in exon 6 of the DES gene, in heterozygous state. These patients illustrate the utility of the MRI in myopathies, when clinical and biopsy findings are non-specific. In both families the particular pattern of MRI involvement guided the molecular diagnosis. The timely diagnosis of patients with desminopathy is essential to prevent and treat the known potentially fatal cardiac complications. FONDECYT Grant 1151383
PS2Group1-047 / #181MULTIPLE DELETIONS IN MITOCHONDRIAL DNA IN MYOFIBRILLAR MYOPATHY AND CENTRONUCLEAR MYOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.5 Distal Myopathy / Myofibrillar Myopathies
Jochen Schaefer, Heinz Reichmann, Sandra Jackson
Neurology, Uniklinikum C.G.Carus Dresden, Dresden, DE
Abstract: Background and aims: Multiple deletions in mitochondrial DNA (mtDNA) are associated with mutations in nuclear genes which encode proteins either directly or indirectly involved in the replication or maintenance of mtDNA. To date, mutations in twelve nuclear genes,POLG, POLG2, C10orf2, SLC25A4, RRM2B, TK2, MPV17, DGUOK, OPA1, MFN2, MGME1, and DNA2 have been identified in patients with multiple deletions in mtDNA. Mitochondrial changes have also been identified in skeletal muscle from patients with myofibrillar myopathy, with histochemical changes such as rubbed out fibres observed with COX and NADH staining, and focal clustering and depletion of mitochondria. Mitochondrial pathology, including COX negative and ragged red fibres, and paracrystalline inclusions have also been described in patients with centronuclear myopathy due to mutations in DNM2, which encodes Dynamin 2, a GTPase which is involved in membrane trafficking. Methods: We describe our clinical, histological and molecular findings in two patients with multiple mtDNA deletions. Results: In one patient , a 70 year old male who presented with a history of exercise induced muscle pain and distal myopathy we identified a novel pArg2364His mutation in the gene FLNC which encodes the actin binding protein filamin-C. In the second patient, a 40 year old male who presented at age 3 years with ataxia and later developed facial myopathy, proximal muscle weakness, and CPEO, histochemical investigation indicated centronuclear myopathy, and a p.Glu560Lys mutation in DNM2 was identified. Conclusion: Our findings suggest that FLNC and DNM2 should be added to the list of genes associated with multiple deletions in mtDNA.
PS2Group1-048 / #137THE UK MYOTONIC DYSTROPHY PATIENT REGISTRY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.6 Myotonic Myopathies
Nikoletta Nikolenko1, Libby Wood1, Chris Turner2, David Hilton-Jones3, Antonio Atalaia1, Chiara Marini-Bettolo1, Paul Maddison4, Margaret Philips5, Mark Roberts6, Mark Rogers7, Volker Straub1, Simon Hammans8, Hanns Lochmuller1
1John Walton Muscular Dystrophy Research Centre, Institute Of Genetic Medicine, University of Newcastle, Newcastle Upon Tyne, GB;
2Institute Of Neurology, UCL MRC Centre for Neuromuscular Diseases, London, GB;
3Department Of Clinical Neurology, John Radcliffe Hospital, Oxford, GB;
4Department Of Neurology, Queen’s Medical Centre, Nottingham, GB;
5Royal Derby Hospital, University of Nottingham, Derby, GB;
6Department Of Neurology, Salford Royal NHS Foundation Trust, Salford, GB;
7Institute Of Medical Genetics, University Hospital of Wales, Cardiff, GB;
8Wessex Neurological Centre, University Hospital of Southampton, Southampton, GB
Abstract: Background Myotonic Dystrophy type 1 (DM1) is the most common adult muscular dystrophy affecting an estimated 8,000 people in the UK. It is an autosomal dominant disorder caused by the triplet CTG repeat expansion in the 3’ untranslated region of the DMPK on chromosome 19. DM1 is associated with a progressive multisystemic disease and currently there is no cure or disease modifying treatment for this disease. The average age of death is 53 years from cardiac and respiratory complications that are usually preceded by decades of morbidity and reduced quality of life. The UK Myotonic Dystrophy Registry is a patient initiated online database collecting detailed clinical and genetic information about both DM1 and myotonic dystrophy type 2 (DM2). It is coordinated from the John Walton Muscular Dystrophy Research Centre (Newcastle University) and the data collected includes all items agreed at the 2009 TREAT-NMD and Marigold Foundation ENMC workshop. Results Of the 451 DM1 patients enrolled from May 2009 until March 2016, 386 (85.6%) patients reported a positive family history of the disease. An even distribution is seen between genders (Female: 236, Male: 215) and a broad range of ages is present from 9 to 81 years old (mean 46.56 ± 14.35). 290 (64.3%) patients are ambulatory-unassisted, 146 (32.4%) are ambulatory-assisted and 10 (2.2%) are non-ambulatory. The most common reported symptom in the Registry is fatigue, occurring in 355 (78.7%) patients of which 89 (25.1%) are taking medication to treat this condition. Myotonia occurs in 351 (77.8%) patients and dysphagia in 210 (46.6%) patients. Dysphagia occurs mostly in patients also reporting myotonia and a statistically significant association can be seen between the two with more severe myotonic occurring more often in those with dysphagia (p =<0.0001).92 (20.4%) patients are aware of having of a heart condition of which 30 (32.6%) have a cardiac implant. A non-invasive ventilation is used by 24 (5.3%) patients. 52 (11.5%) patients reported that they underwent cataract surgery in the past. Conclusion The UK Myotonic Dystrophy Patient registry is an example of a novel patient driven registry. Its success can be measured by its continuous growth and utilisation. The registry has successfully assisted the recruitment and planning of a number of commercial and academic studies, including EU funded project OPTIMISTIC and the Newcastle Upon Tyne Hospitals NHS Foundation Trust funded natural history study PhenoDM1. In addition to this purpose the registry also provides interesting and important data characterising the DM1 community in the United Kingdom.
PS2Group1-049 / #41318F-FDG-PET STUDY IN PATIENTS WITH MYOTONIC DYSTROPHY TYPE 1 AND 2
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.6 Myotonic Myopathies
Vidosava M. Rakocevic Stojanovic1, Stojan Peric1, Vera Ilic1, Aleksandra Parojcic1, Jovan Pesovic2, Dusanka Savic-Pavicevic2, Leposava Brajkovic3
1Neurology Clinic, Clinical Center Of Serbia, School of Medicine, University of Belgrade, Belgrade, RS;
2Center For Human Molecular Genetics, Faculty of Biology, Belgrade, RS;
3Center For Nuclear Medicine, Clinical Center Of Serbia, School of Medicine, University of Belgrade, Belgrade, RS
Abstract: Introduction: Different cognitive and behavioral changes have been described both in myotonic dystrophy type 1 (DM1) and type 2 (DM2). Positron emission tomography (PET) of the brain is considered a valuable neuroradiologic diagnostic tool which could provide us with detailed information about metabolic activity of different brain regions. The aim: To determine brain regions with glucose metabolism impairment in DM1 and DM2 subjects using 18F-FDG-PET, and to correlate these findings with the results of neuropsychological testing. Material and methods: This study included 16 DM1 (50% males, mean age 45.6 ± 9.6 years and mean disease duration 21.8 ± 8.3 years) and 13 DM2 patients (23% males, mean age 51.8 ± 8.4 years and mean disease duration 15.3 ± 10.5 years). 18F-FDG-PET and detailed neuropsychological testing were performed in all patients. Results: Brain PET scan has shown significant impairment of glucose metabolism in frontal (75% of patients) and temporal regions (62%) in patients with DM1. On the other hand, in DM2 patients the most affected regions were frontal (77%), temporal (77%) and parietal (69%). Total number of affected cortical regions was similar in both groups (2.1 ± 1.3 in DM1 vs. 2.8 ± 1.2 in DM2, p>0.05). Patients with DM1 have shown low performances on tests which examined visuospatial (60%), naming (67%) and executive abilities (40%). DM2 patients performed better than DM1 on cognitive testing, and they achieved bad results regarding executive (30%) and naming (30%) abilities. Conclusion: PET scans of the brain showed affection of the frontal and temporal regions in DM1 patiens, and frontal, temporal and parietal regions in DM2 patients. These results are generally consistent with findings obtained by neuropsychological testing.
PS2Group1-050 / #185ELEVATED PLASMA LEVELS OF CARDIAC TROPONIN-I PREDICT LEFT VENTRICULAR SYSTOLIC DYSFUNCTION IN PATIENTS WITH MYOTONIC DYSTROPHY TYPE 1: A COHORT FOLLOW-UP STUDY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.6 Myotonic Myopathies
Mark J. Hamilton1, Yvonne Robb2, Helen Gregory3, Sarah Cumming4, Berit Adam4, Josephine Mcghie4, Anneli Cooper5, Jillian M. Couto6, Alexis Duncan1, Monika Rahman1, Anne Mckeown1, Alison Wilcox1, Catherine Mcwilliam7, Maria Elena Farrugia8, Richard K. Petty8, Cheryl Longman1, Iain Findlay9, Alan Japp10, Darren G. Monckton4, Martin Denvir10
1West Of Scotland Clinical Genetics Service, Queen Elizabeth University Hospital, Glasgow, GB;
2Clinical Genetics Service, Western General Hospital, Edinburgh, GB;
3Department Of Medical Genetics, Grampian University Hospitals, Aberdeen, GB;
4Institute Of Molecular, Cell And Systems Biology, University of Glasgow, Glasgow, GB;
5Institute Of Biodiversity, Animal Health And Comparative Medicine, University of Glasgow, Glasgow, GB;
6School Of Engineering, University of Glasgow, Glasgow, GB;
7Department Of Clinical Genetics, Ninewells Hospital, Dundee, GB;
8Neurology, Institute of Neurological Sciences, Glasgow, GB;
9Inherited Cardiac Conditions Service, Queen Elizabeth University Hospital, Glasgow, GB;
10Centre For Cardiovascular Science, Queens Medical Research Institute, Edinburgh, GB
Abstract: BACKGROUND: Cardiac involvement in myotonic dystrophy type 1 (DM1) is common and potentially life threatening. The 12-lead electrocardiogram (ECG) is the main test currently used for cardiac surveillance. High sensitivity plasma cardiac troponin-I (cTnI) is emerging as a strong predictor of cardiac events in a variety of clinical settings. We have explored its utility in patients with DM1. METHODS: Informed consent to participate was obtained from 118 ambulatory patients with clinically and genetically defined DM1. A single blood sample was drawn at the time of a routine out-patient clinic review and cTnI was measured using a high sensitivity assay (Abbott Architect). Patients with a recent hospital admission and those currently experiencing angina or cardiac-type chest pains were excluded. Demographic, ECG, echocardiographic and relevant cardiac history data were obtained from electronic medical records. Patients were followed up for a mean of 25 months (range 7-32 months). Complete data was available for ECG in 110 patients and echocardiography in 52. Genetic data relating to CTG repeat expansion size were available for 111. Cardiac devices had been previously implanted in 21 patients. RESULTS: The mean age of participants was 47.6 years (range of 21-72) including 55 females and 63 males. cTnI levels above the 99th percentile of the range observed in the general population (used for diagnosis of myocardial infarction) occurred in 10 patients (8.5%; 3 male and 7 female). Echocardiographic data was available for five of these, with three showing left ventricular systolic dysfunction (LVSD) giving an extrapolated prevalence of LVSD in the elevated cTnI group of 3/10 (30%). By contrast, LVSD was present in only five of 47 of patients for whom echocardiography data was available in the normal cTnI group, giving an overall prevalence of 5/108 (4.6%). This difference was shown to be statistically significant (p = 0.02 in two-tailed Fisher’s exact test). There was no association between elevated cTnI and presence of a cardiac device (20% vs 17.6%) in elevated versus normal cTnI groups respectively. All five patients with PR > 240 ms had cTnI level less than 9 ng/L and of 19 patients with QRS duration of 120 ms or greater only one had an elevated cTnI. Eight patients died during follow-up. Two of the 10 patients with elevated cTnI died within the study period (20%), compared with six of 108 with normal-range cTnI (5.6%). This difference was not statistically significant (p = 0.14 in two-tailed Fisher’s exact test). CONCLUSION: Plasma cTnI in ambulatory patients with DM1 shows promise as a predictor of clinical events and appears to map closely with the presence of LVSD. However, further studies are needed with larger patient numbers and with longer term follow-up to assess its true clinical utility in this setting.
PS2Group1-051 / #443ONE-YEAR MRI-FOLLOW-UP IN 45 PATIENTS WITH FACIOSCAPULOHUMERAL MUSCULAR DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.7 Facioscapulohumeral Muscular Dystrophies / Oculopharyngeal Muscular Dystrophy
Grete Andersen1, Julia Dahlqvist2, Christoffer Vissing2, Karen B.H. Pedersen3, Carsten Thomsen2, John Vissing4
1Copenhagen Neuromuscular Center, Rigshospitalet, University of Copenhagen, Copenhagen, DK;
2Rigshospitalet, University of Copenhagen, Copenhagen, DK;
3Copenhagen Neuromuscular Center, Rigshospitalet, University Hospital of Copenhagen, Copenhagen E, DK;
4Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK
Abstract: Although the genetic cause of facioscapulohumeral muscular dystrophy (FSHD) was identified as early as in 1990, a consensus model of the pathophysiology has only recently been developed. This has opened up the possibility for therapeutic development, but also a need for validated outcome measures. MRI may be a useful tool in further understanding of the pathology and a potential outcome measure for future clinical trials in patients with FSHD. In this study we evaluate the disease progression in paraspinal, thigh and calf muscles using quantitative MRI Dixon and STIR techniques, comparing these findings with more classical follow-up tests (FSHD-clinical-score, muscle strength, 6-minute-walk-test, 14-step-stair-test, and 5-time-sit-to-stand-test). All patients were re-examined after one year. In 25 men and 20 women (age: 20-75 years, BMI: 18-37 kg/m2, FSHD-score: 0-12) muscle fat-fraction increased 4.2 % in mean. All measured muscle groups increased significant. Inflammatory sites were identified in the thigh of one-third of the patients, these sites had a higher progression of fat-infiltration (32 % vs. 13% in non-inflammatory sites). The disease severity score increased by 10 % (P=0.002), muscle strength decreased over the hip, neck and back (8 %, 8 % and 17 %, respectively, P<0.002) without correlations with the progression of fat-infiltration. MRI provides an objective measurement of disease progression. MRI detects progression before it can be identified by the clinical and functional tests. The STIR technic link inflammatory-sites with increased fat-infiltration. This indicates that quantitative MRI can be a strong end-point in follow-up and therapeutic trials.
PS2Group1-052 / #367OCULOPHARYNGODISTAL MYOPATHY , A CASE REPORT FROM INDIAÂ
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.7 Facioscapulohumeral Muscular Dystrophies / Oculopharyngeal Muscular Dystrophy
Seena Vengalil1, Atchayaram Nalini1, Veeramani Preethish-Kumar2, Kiran Polavarapu2, Narayanappa Gayathri3, Anita Mahadevan3
1Neurology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
2Clinical Neurosciences, National Institute of Mental Health and Neurosciences, Bangalore, IN;
3Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore, IN
Abstract: Background : Oculopharyngodistal myopathy ( OPDM ) is a rare adult onset muscle disease characterised by ocular , bulbar, distal limb weakness and histologically by rimmed vacuoles . The gene causing the defect is not known. Diagnosis is based upon characteristic clinical features along with histological and electron microscopy features . The largest case series reported probably is that of 47 patients from Turkey . Oculopharyngeal distal myopathy has not been reported from India before Case history : 43 year old man presented with hoarseness of voice and difficulty in swallowing since 12 years , drooping of eye lids since 6 years and distal weakness of lower limbs since 6 years . He was born of a non consanguineous parentage and no other family members were affected . He had undergone treatment for myasthenia elsewhere with no significant improvement .On examination he had severe hypophonia and hoarseness of voice , asymmetrical ptosis with lateral gaze restriction , mild facial wasting and bulbar weakness . He had distal weakness of lower limbs with mild proximal weakness of upper and lower limbs. Creatine kinase was 3250 IU/L. EMG showed myopathic potentials . Pulmonary function test showed severe restrictive abnormality worsening in supine position with a postural drop in forced vital capacity of more than 10 % indicative of diaphragmatic weakness . ENT evaluation showed bilateral adductor vocal cord palsy . Cardiac evaluation was normal. Muscle biopsy showed variation in fiber size, necrosis, myophagocytosis , regenerating fibres, hypertrophied fibres with internalised nuclei and splitting with increased endomysial collagen , numerous ragged red fibres on Modified Gomori trichrome stain , numerous ragged blue fibres on oxidative enzymes and several COX deficient fibres with large lipid aggregates and some atrophic fibres showing rimmed vacuoles suggestive of oculopharyngeal distal myopathy.Electron microscopy report is being awaited Discussion Oculopharyngodistal myopathy can have autosomal dominant or autosomal recessive pattern of inheritance. However our patient didnt have any family history of similar illness . Most common presenting complaint is asymmetrical ptosis . Our patient had bulbar weakness as the initial symptom and later developed asymmetrical ptosis and lateral gaze restriction as described in literature . He had distal weakness of ankle dorsiflexion and toe extension and mild proximal weakness of upper and lower limbs . Pulmonary function tests showed significant drop in forced vital capacity in supine position suggestive of diaphragmatic weakness . Other investigations and biopsy were confirmative of diagnosis as described in literature ONCLUSION Adequate knowledge about clinical feature and careful histopathological examination will help in diagnosis of this rare disorder and may help in preventing misdiagnosis and incorrect management . This may be the first reported case of oculopharyngeal distal myopathy from India
PS2Group1-053 / #260NEUROMUSCULAR ELECTRICAL STIMULATION TRAINING OF THE TIBIALIS ANTERIOR MUSCLE IN FSHD1 PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.7 Facioscapulohumeral Muscular Dystrophies / Oculopharyngeal Muscular Dystrophy
Jeremy Garcia1, Aude C. Doix2, Pauline Lahaut1, Véronique Tanant1, Manuella Fournier-Mehouas1, Serge Colson2, Claude Desnuelle1, Sabrina Sacconi1
1Neurology, University Hospital of Nice, Nice, FR;
2Laboratory Pf Human Motricity, University of Nice Sophia Antipolis, Nice, FR
Abstract: The purpose of this study was to investigate whether 8-week neuromuscular electrical stimulation (NMES) training of the tibialis anterior (TA) muscles in adults with facioscapulohumeral muscular dystrophy type 1 (FSHD1) would improve motor function, muscle strength and endurance. Eleven patients with FSHD1 and 10 age and gender matched healthy participants achieved a 8-week bilateral NMES training of the TA muscles 20 minutes per session, 3 sessions per week. Ankle dorsiflexion (DF) and plantar flexion (PF) maximal voluntary isometric contractions (MVC), a 2-minute sustained MVC ankle dorsiflexion with surface electromyography recordings (EMG) of the TA and the soleus (SOL) muscles were measured and functional tests were performed prior to and after the NMES training to disclose training effects. To assess the biological tolerance, plasma Creatine Kinase (CK) was measured before, at 4 weeks (W4), after the 8-week training (W8) and once randomly during the training. No training effect was found in any of the investigated variable for either group. Patients with FSHD showed lower MVC and lower maximal TA EMG amplitude during the DF MVCs. During the 2-minute sustained MVC, the percentage of force loss was lower for the FSHD patients. The percentage of TA EMG loss amplitude after the 2-minute MVC were found to be similar in both groups before and after the training but partly increased with training for the group of patients with FSHD1. Besides drastic differences between groups, none of the clinical motor function measures were improved with the training in patients with FSHD. CK did not change significantly during the NMES training period for both groups. Although the program was biologically tolerated, the NMES protocol was not strenuous enough and/ or parameters of stimulation were not adequate to improve ankle strength, muscle endurance and motor function for the group of patients with FSHD1. Additionally, the absence of training effects may be explained by the NMES protocol which was designed for the patients with FSHD1, likely not appropriate to induce strength gains in healthy participants. Finally, the group of patients with FSHD1 showed lower force losses during the 2-minute sustained MVC, suggesting that they were experiencing a lower amount of muscle fatigue compared to the HP group.
PS2Group1-054 / #192SPEECH IMPAIRMENT IS COMMON IN EARLY ONSET FACIOSCAPULOHUMERAL DYSTROPHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.7 Facioscapulohumeral Muscular Dystrophies / Oculopharyngeal Muscular Dystrophy
Megan Hodge1, Jia Feng2, Jean K. Mah3, Cooperative International Neuromuscular Research Group Investigators2
1Department Of Speech-language Pathology, University of Alberta, Edmonton, AB, CA;
2Children’s Research Institute, Children’s National Health System, Washington, DC, US;
3Departments Of Pediatrics And Clinical Neurosciences, University of Calgary, Calgary, AB, CA
Abstract: Background: Facioscapulohumeral dystrophy (FSHD) is one of the most common forms of muscular dystrophies worldwide. We described the prevalence and extent of speech impairment among participants enrolled in a prospective study of early onset FSHD. Methods:This multicenter study was conducted at participating Cooperative International Neuromuscular Research Group (CINRG) centers. Baseline vision, hearing, speech, cognition, and motor function assessments were performed as part of a standardized protocol. Speech evaluation included a self-reported questionnaire on current and past speech function, Maximum Performance Tasks (MPT), and a connected speech sample to evaluate intelligibility and related speech dimensions. MPT included prolongation of [a] and [mama] to yield maximum phonation duration (MPD), prolongation of [f], [s], and [z] to yield maximum fricative duration (MFD), repetition of the single syllables [pa], [ta], and [ka] to yield maximum repetition rate-monosyllabic (MRRmono), and repetition of the syllable sequence [pa-ta-ka] to yield maximum repetition rate-trisyllabic (MRRtri). Each assessment was administered and audio-recorded by a trained research assistant using the TOCS+MPT and TOCS+Recorder-Player software. Waveforms of the recordings were measured for each MPT and the results compared to age- and gender-matched norms. Experienced speech-language pathologists (SLP) rated the speech samples on intelligibility, articulation, resonance, voice quality, and rate using a 4-point ordinal scale. Results: 53 participants (41% males, mean age 23.1 (SD 14.6) years) were enrolled. 48/53 (90.6%) had speech questionnaire completed; 22 (45.8%) had past concerns about speech, 19 (39.6%) received speech therapy, and 10 (20.8%) had current speech concerns. 38/53 (71.7%) participants had full information from the questionnaire, MPT, and connected speech recordings. 26/38 (68%) had shorter than expected MPD. All 13 (34%) participants with slow MRRmono also had MPDs more than 1.5 SD below the reference mean. Results from MPT suggested that laryngeal-respiratory support was compromised more frequently than speed of articulator movements. Ratings of the speech samples supported this finding; more participants were judged to have laryngeal (27/38) than articulatory (17/38) and resonance (10/38) impairment. 32/38 (84%) of participants were judged as demonstrating speech impairment, which ranged from mild to profound. 22/32 (69%) of those judged to have speech impairment had MPD more than 1.5 SD below the reference mean. Participants judged to have the most severe speech impairments on the connected speech sample also had lower than expected MPD and MRRmono. Of the 10 participants judged to have speech impairment but had MPD and MRRmono within the expected range, 8 were identified with only mild or moderate impairment on the voice scale. Conclusion: Speech impairment is very common in early onset FSHD, with voice being affected most frequently, followed by articulation and resonance. These impairments were judged to reduce intelligibility in about 25% of participants, most often of mild-to-moderate severity. Maximum phonation duration (MPD) appeared to be the most sensitive MPT variable and the voice to be particularly at risk. This may reflect weakness in the laryngeal musculature, vocal strain and fatigue in compensation for limitations in other speech muscles, or both. Professional counseling in healthy voice practices is recommended for all affected individuals.
PS2Group1-055 / #120ANTISENSE TARGETING OF 3’END ELEMENTS INVOLVED IN DUX4 MRNA PROCESSING IS AN EFFICIENT THERAPEUTIC STRATEGY FOR FACIOSCAPULOHUMERAL DYSTROPHY: A NEW GENE SILENCING APPROACH.
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.7 Facioscapulohumeral Muscular Dystrophies / Oculopharyngeal Muscular Dystrophy
Anne-Charlotte Marsollier1, Lucasz Ciszewski2, Virginie Mariot3, Linda Popplewell2, Thomas Voit3, George Dickson2, Julie Dumonceaux3
1Inserm, Cnrs, Centre De Recherche En Myologie, Sorbonne Universités UPMC Univ Paris 06, Paris, FR;
2Centre Of Biomedical Sciences, School Of Biological Sciences, Royal Holloway University of London, London, GB;
3Inserm, Cnrs, Centre De Recherche En Myologie, Sorbonne Universités UPMC Univ Paris 06, Paris, FR
Abstract: Defects in mRNA 3’ end formation have been described to alter transcription termination, transport of the mRNA from the nucleus to the cytoplasm, stability of the mRNA and translation efficiency. Therefore, inhibition of polyadenylation may lead to gene silencing. Here, we choose FacioScapuloHumeral Dystrophy (FSHD) as a model to determine whether or not targeting key 3’end elements involved in mRNA processing using antisense oligonucleotide drugs can be used as a strategy for gene silencing within a potentially therapeutic context. FSHD is a gain-of-function disease characterized by the aberrant expression of the DUX4 transcription factor leading to altered pathogenic deregulation of multiple genes in muscles. Here we demonstrate that targeting either the mRNA polyadenylation signal and/ or cleavage site is an efficient strategy to downregulate DUX4 expression and to decrease the abnormally high pathological expression of genes downstream of DUX4. We conclude that targeting key functional 3’end elements involved in pre-mRNA to mRNA maturation with antisense drugs can lead to efficient gene silencing and is thus a potentially effective therapeutic strategy for at least FSHD. Moreover polyadenylation is a crucial step in the maturation of almost all eukaryotic mRNAs, and thus all mRNAs are virtually eligible for this antisense-mediated knockdown strategy.
PS2Group1-056 / #479TREATMENT RELATED EFFECTS OF ANTI-GAA ANTIBODIES IN LATE ONSET POMPE DISEASE
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Marie Wencel1, Claudia Shambaugh2, Namita Goyal1, Virginia Kimonis2, Tahseen Mozaffar1
1Neurology, University of California, Irvine, Orange, CA, US;
2Pediatrics, University of California, Irvine, Orange, CA, US
Abstract: Late onset Pompe disease (LOPD), a lysosomal storage disorder characterized by deficiency of the enzyme acid alpha-glucosidase (GAA), presents with diaphragm and skeletal muscle weakness with little or no cardiac involvement. The disease is progressive, and if left untreated, may result in significant motor disability and respiratory failure. Pompe disease is now considered treatable, with FDA-approved enzyme replacement therapy (ERT) Lumizyme®. Initial published data in LOPD showed improvement in forced vital capacity (FVC) and endurance, measured by 6-minute walk. However, subsequent publications have shown plateauing of this benefit and worsening of FVC may occur with time. In infantile cases of Pompe disease, development of IgG antibodies against GAA results in reduced treatment efficacy, especially in cross-reactive immunologic material negative (CRIM-) individuals. The role of these antibodies in neutralizing effects of treatment in LOPD is not clear since most individuals at this stage are CRIM+. We plan to present a retrospective analysis of our 9 patients who regularly follow with us, have been on uninterrupted enzyme replacement therapy and have been checked for these antibodies on a routine basis. We intend to correlate their treatment related adverse events, their treatment response, as measured by muscle function tests (manual muscle strength, 6-minute walk test), respiratory function trends (serial FVC, maximal inspiratory pressures (MIP) and sniff nasal inspiratory pressures (SNIP)), and quality of life data with the antibody titers. Furthermore we will correlate these data with their genotype results, and when available, pre-treatment GAA enzyme levels and urinary Hex4 results. Our hypothesis is that development of anti-GAA antibody titers has no effects on treatment efficacy in LOPD.
PS2Group1-057 / #417POMPE DISEASE IN AUSTRIA
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Wolfgang Löscher1, Thomas Stulnig2, Phillip Simschitz3, Michaela Brunner-Krainz4, Martina Huemer5, Stephan Iglseder6, Florian Lagler7, Herman Moser8, Stefan Quasthoff4, Julia Wanschitz1, Wolfgang Grisold9
1Neurology, Medical University Innsbruck, Innsbruck, AT;
2Clinical Division Of Endocrinology And Metabolism, Department Of Medicine Iii, Medical University of Vienna, Vienna, AT;
3Landeskrankenhaus Klagenfurt, Klagenfurt, AT;
4Neurology, Medical University Graz, Graz, AT;
5Landeskrankenhaus Bregenz, Bregenz, AT;
6Barmherzige Brüder Linz, Linz, AT;
7Paracelsus University Salzburg, Salzburg, AT;
8Neurologisches Therapiezentrum Gmundnerberg, Altmünster am Traunsee, AT;
9Kaiser Franz Josef Hospital, Vienna, Vienna, AT
Abstract: Pompe disease is a rare inherited glycogenosis (type 2) caused by mutations in GAA, which encodes acid α-1,4-glucosidase. Depending on the level of residual enzyme activity, disease onset varies from pre-/neonatal to adulthood. If untreated, the disease is lethal in 95~% of infantile cases. Early-onset Pompe disease is characterized by cardiomyopathy and generalized weakness while the heart is usually spared in later onset disease. In most cases respiratory muscles are affected at some stage of the disease. Here we review the key features of patients with Pompe disease in Austria and their current state of treatment. Cases were ascertained via Myozyme® prescription and personal contacts to various Pediatric and Neurological Departments in Austria. Currently 18 patients have been diagnosed with Pompe disease in Austria, 4 with early-onset and 14 with late-onset. Thus, the diagnostic prevalence of Pompe disease in Austria is 1:477.000. In early-onset Pompe, disease onset was between 2 m and 3.5 ys and a final diagnosis was reached after 1-18 m. These patients are currently 7-14 ys old and only 2 need at least intermittent non-invasive ventilation (NIV). Cardiomyopathy was present in the two patients with the earliest disease onset and improved during enzyme replacement therapy (ERT). The early-onset patients have been on ERT since 6.5-9 ys without development of neutralizing antibodies against glucosidase alfa. Mean disease onset in late-onset Pompe disease was 26.3 (11 – 47) ys and the diagnostic delay was 6.7 ± 9.7 ys, with a range of 0.5 – 34 ys. Disease duration currently is 15.3 ± 9.1 (range: 6-36) ys. The presenting symptom was an isolated limb girdle weakness in 10, isolated respiratory insufficiency in 2 and a combined weakness in 2 patients. All but 1 patient receive ERT and have been treated with ERT for 7.2 ± 2.2 ys (range: 2-10 ys). The patient without ERT chose to stop treatment due to his severe illness. Neutralizing antibodies were negative in the 3 patients tested. 9 of the 14 patients require NIV, 4 permanently, 5 during night-time only. 12 of these 14 patients remained ambulatory. This survey shows that in most cases late-onset Pompe disease presents with a limb-girdle phenotype. As the prevalence of Pompe disease in Europe is estimated to be 1:283.000, the present data suggest that several undiagnosed patients live in Austria. Therefore we recommend testing for Pompe’s disease in all patients with limb-girdle weakness also in the absence of respiratory insufficiency. All known Pompe patients in Austria currently have access to ERT. As expected, ERT is highly effective in early-onset Pompe disease dramatically improving survival and cardiomyopathy. In adults the efficacy is more difficult to assess but in addition to improved respiratory and ambulatory function achieved within the first 6 months of treatment the present data suggest, that ERT at least slows disease progression.
PS2Group1-058 / #356PHASE 1 SAFETY, PHARMACOKINETICS, AND EXPLORATORY EFFICACY OF NEOGAA, A NOVEL ENZYME REPLACEMENT THERAPY, IN TREATMENT-NAIVE AND ALGLUCOSIDASE ALFA-TREATED LATE-ONSET POMPE DISEASE PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Loren D.M. Pena1, Richard Barohn2, Barry Byrne3, Claude Desnuelle4, Ozlem Goker-Alpan5, Shafeeq Ladha6, Pascal Laforêt7, Eugen Mengel8, Alan Pestronk9, Benedikt Schoser10, Volker Straub11, Jaya Trivedi12, Philip Van Damme13, John Vissing14, Peter Young15, Beth L. Thurberg16, Raheel Shafi16, Kerry Culm-Merdek16, Gerard Short16, Ans Van Der Ploeg; For The Neo1 Investigator Group17
1Medical Center, Duke University, Durham, NC, US;
2University of Kansas Medical Center, Kansas City, KS, US;
3University of Florida, Jacksonville, FL, US;
4Neurology, University Hospital of Nice, Nice, FR;
5O and O Alpan LLC, Fairfax, VA, US;
6St Joseph’s Hospital & Medical Center, Phoenix, AZ, US;
7Paris-est Neuromuscular Center, Inserm U974, Université Pierre et Marie Curie Hôpital Pitié-Salpêtrière, AP-HP, Paris, FR;
8Universitätsklinikum Mainz, Mainz, DE;
9Washington University School of Medicine, St Louis, MO, US;
10Friedrich-Baur Institut München, München, DE;
11John Walton Muscular Dystrophy Research Centre, Newcastle upon Tyne, GB;
1213University of Texas Southwestern Medical Center, Dallas, TX, US;
13KU Leuven (University of Leuven) — Department of Neurosciences, VIB — Vesalius Research Center, and University Hospitals Leuven —, Leuven, BE;
14Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK;
15Universitätsklinikum Münster, Münster, DE;
16Sanofi Genzyme, Cambridge, MA, US;
17Erasmus MC, Rotterdam, NL
Abstract: This phase 1, open-label, ascending-dose study (NCT01898364) evaluated safety, tolerability, pharmacokinetics, pharmacodynamics, and exploratory efficacy of a second-generation recombinant acid α-glucosidase, neoGAA, in late-onset Pompe disease patients. neoGAA contains multiple copies of a synthetic bisM6P-containing hexasaccharide to improve muscle targeting. Patients were either treatment-naïve to alglucosidase alfa (Naïve Group) or had previously received alglucosidase alfa for ≥9 months (Switch Group). Inclusion criteria were: acid α-glucosidase deficiency; ≥18 years old; walk ≥50m independently; upright forced vital capacity (FVC) ≥50% predicted. Patients received intravenous neoGAA (5, 10, or 20 mg/kg qow) for 24 weeks. In the Naïve Group, 10 patients were treated; 9 completed (n=3 per dose level). In the Switch Group, 14 patients were treated; 12 completed (5 mg/kg, n=3; 10 mg/kg, n=4; 20 mg/kg, n=5). No deaths/life-threatening serious adverse events (SAEs) occurred. One Naïve Group patient experienced SAEs of respiratory distress and chest discomfort, which were considered study drug-related infusion-associated reactions (IARs); 1 Switch Group patient had 1 SAE unrelated to study drug. IARs affected 4/10 Naïve Group patients (11 events) and 4/14 Switch Group patients (14 events). Most (72.7%) treatment-emergent AEs were mild in intensity. In the Naïve Group, 9/10 patients developed anti-neoGAA antibodies, which inhibited uptake in 1 patient. In the Switch Group, 5/14 patients had anti-neoGAA antibodies at screening/Week 1, despite having only alglucosidase alfa exposure; 2 of the 9 patients without baseline anti-neoGAA seroconverted without uptake inhibition. No enzyme-inhibitory antibodies or IgE antibodies developed. For both groups, neoGAA plasma concentrations declined monoexponentially post-infusion (mean t1/2z ~1.0 h), and neoGAA pharmacokinetics were similar and unchanged over time. Following 24 weeks of neoGAA treatment, pulmonary function (upright % predicted FVC, maximum expiratory pressure [MEP], and maximum inspiratory pressure [MIP]) remained stable or improved in the Naïve Group and remained stable in the Switch Group (Table). Six-minute walk test (6MWT) distances were generally stable or increased irrespective of group or dose (Table). Quick Motor Function Test (QMFT) and lower-body Hand-Held Dynamometry (HHD) scores improved irrespective of group or dose. Gait, Stairs, Gowers, Chair ability and Gross Motor Function Measure-88 showed minimal changes across groups and doses. Quadriceps muscle biopsy glycogen levels were low (~6% of tissue surface area) in most patients in both groups at baseline and remained mostly unchanged. In summary, neoGAA had a well-tolerated safety profile in treatment-naïve and previously treated patients with late-onset Pompe disease. The exploratory efficacy results support further development of neoGAA for Pompe disease.
TABLE
Mean ± SD changes in pulmonary function, 6MWT, HHD, and QMFT at Week 25 relative to baseline
| Group | Naïve | Naïve | Naïve | Switch | Switch | Switch |
| neoGAA dose | 5 mg/kg | 10 mg/kg | 20 mg/kg | 5 mg/kg | 10 mg/kg | 20 mg/kg |
| Patients, n | 4 | 3 | 3 | 4 | 4 | 6 |
| FVC, % predicted | −2.7 ± 8.81 | 4.3 ± 4.90 | 6.2 ± 3.15 | −0.5 ± 4.31 | −2.0 ± 2.24 | 1.4 ± 5.71 |
| MEP, % predicted | 8.1 ± 2.79 | 16.5 ± 7.95 | 12.0 ± 4.05 | −3.5 ± 10.81 | 15.7 ± 38.35 | 6.0 ± 21.80 |
| MIP, % predicted | 6.1 ± 0.73 | 10.6 ± 4.91 | 7.9 ± 15.73 | 10.5 ± 7.26 | 4.2 ± 12.58 | −0.2 ± 6.85 |
| 6MWT, % predicted | 2.6 ± 3.89 | ™2.1 ± 2.19 | 3.9 ± 3.45 | ™1.2 ± 5.80 | 0.7 ± 1.25 | ™1.3 ± 8.94 |
| HHD (lower body), % | 11.6 ± 4.69 | 21.4 ± 10.31 | 14.2 ± 15.90 | −0.5 ± 13.07 | 14.3 ± 27.32 | −14.5 ± 42.23 |
| QMFT, total score | 0.7 ± 4.93 | 1.7 ± 2.31 | 3.0 ± 2.65 | −1.5 ± 2.65 | 3.0 ± 1.63 | 1.2 ± 1.92 |
PS2Group1-059 / #276FAT OXIDATION IS LIMITED IN MADD DURING EXERCISE, BUT GLUCOSE INFUSION IMPROVES EXERCISE CAPACITY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Karen L. Madsen1, Nicolai Preisler1, Astrid Emilie Buch1, Mads Stemmerik1, Pascal Laforêt2, John Vissing1
1Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK;
2Centre De Référence Pathologie Neuromusculaire Paris Est, Hôpital Pitié-Salpêtrière, Paris, FR
Abstract: We have studied whole-body metabolism and exercise performance in patients with Multiple Acyl-CoA Dehydrogenase deficiency (MADD) and the effect of intravenous glucose supplementation. MADD is an inborn lipid storage myopathy caused by mutations in the electron transfer flavoprotein (ETF) or ETF dehydrogenase, which transfer high-energy electrons produced in beta-oxidation to the respiratory chain. This leads to dysfunction of the acyl-CoA dehydrogenases reducing flux through beta-oxidation. Patients with MADD are clinically a heterogeneous group with symptoms ranging from fatal metabolic decompensation with hypoketotic hypoglycemia, episodes of lethargy and encephalopathy to mild myopathy. The impact of MADD on skeletal muscle metabolism in vivo is unknown. We studied two women aged 45 and 20 years with MADD on riboflavin and L-carnitine treatment and 4 healthy controls (women aged 18-35 years). Peak oxidative capacity (VO2peak) was determined by an incremental exercise test on a cycle ergometer. All subjects performed a submaximal exercise test until exhaustion or <1 hour. Meanwhile, total fatty acid and carbohydrate oxidation rates (FAO and CHO) were studied with indirect calorimetry and palmitate rate of oxidation (PO) was measured using the stable isotope tracer, U-13C-palmitate. On a separate day, patients repeated the submaximal exercise test without the tracers, but were given intravenous glucose 10% as bolus of 2mL×kg-1 10 minutes before exercise and a continuous infusion of 4.7mL×kg-1 during exercise. We report preliminary results. Please see the table. The patients had lower VO2peak of 25.8 and 17.4 mL×min-1×kg-1 vs. 41.7±3.4 mL×min-1×kg-1 in the controls. The patients stopped after 58 and 50 minutes of submaximal exercise due to muscle pain and exertion. In the patients, FAO increased with exercise, but much less than in the controls. During exercise, CHO increased twice as much in the patients vs. the controls. The mean heart rates during exercise were higher in the patients than in the healthy controls, but dropped with glucose infusion. With the glucose infusion, CHO increased further while the FAO rather dropped than increased (table).
Our results show that MADD-patients have an impaired fat metabolism when challenged with prolonged exercise and that carbohydrate metabolism is upregulated to fuel the exercise. Riboflavin and L-carnitine treatment does not restore fat metabolism in these patients. However, we have demonstrated that a glucose infusion boosts the carbohydrate oxidation and eases the exercise reflected in a drop in heart rate.
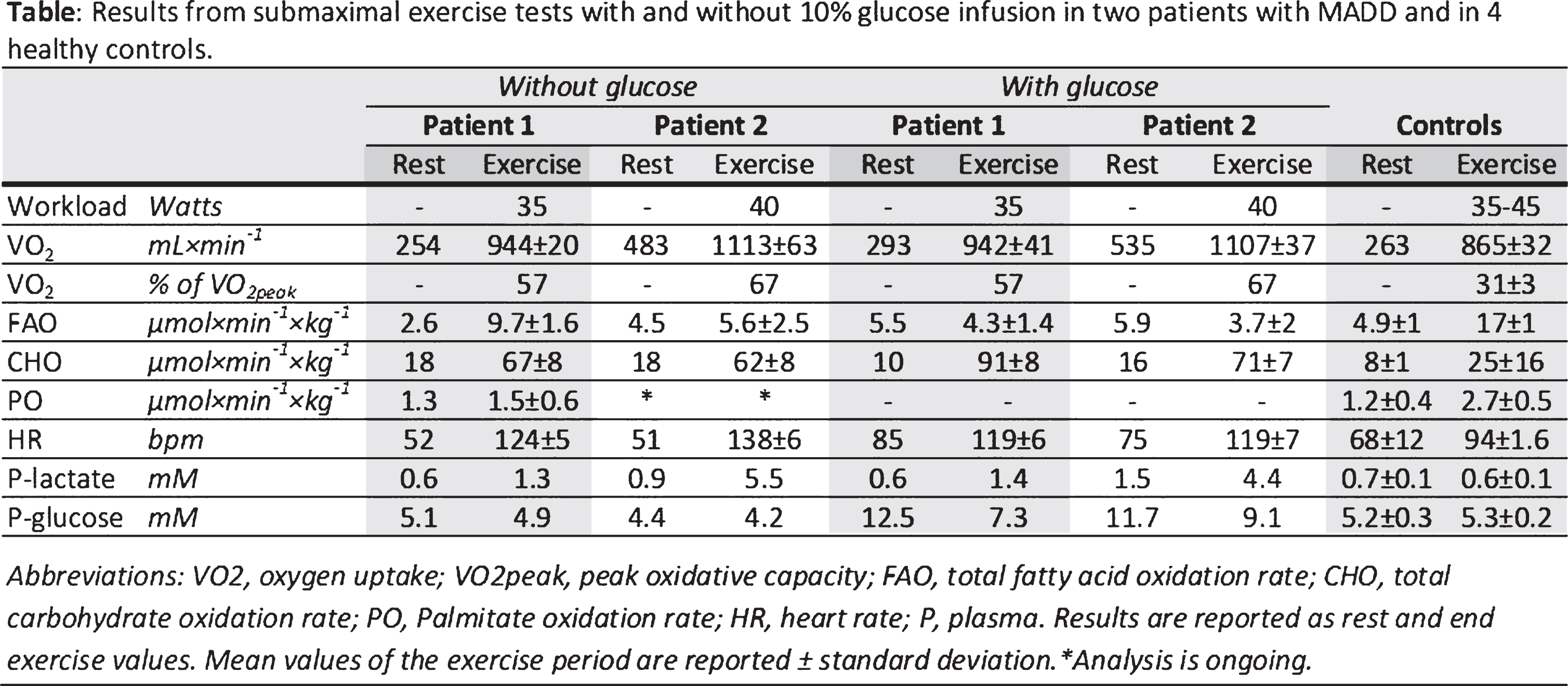
PS2Group1-060 / #358REGULATION OF LIPOPHAGY AND LIPOLYSIS IN LIPID STORAGE MYOPATHIES AND CPT DEFICIENCY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Corrado Angelini1, Elisabetta Tasca1, Anna C. Nascimbeni2
1Neuromuscular, Foundation San Camillo Hospital IRCCS, Venice, IT;
2Inserm, Hopital Necker, Paris, FR
Abstract: Aims. Triglycerides droplets are massively stored in muscle in Lipid Storage Myopathies (LSM). We studied in muscle regulators of lipophagy, the expression of the transcription factor-EB (TFEB) (a master regulator of lysosomal biogenesis), and markers of autophagy which are induced by starvation and exert a transcriptional control on lipid catabolism. Methods. We investigated the factors that regulate lipophagy in muscle biopsies from 6 patients with different types of LSM: 2 cases of riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency (MADD), 1 case of primary carnitine deficiency (CD), 2 cases of neutral lipid storage myopathy (NLSD-M), 1 case of carnitine-palmitoyl-transferase-II (CPT) deficiency. Results. Conventional morphology and electron microscopy documented the lipid accumulation and its dramatic resolution after treatment. Muscle immunofluorescence showed that while in MADD and NLSD-M there was a co-localized expression of TFEB and p62-SQSTM1 (marker of protein aggregates) in some atrophic fibers, in CD and CPT-II deficiency the reaction was almost normal. In regenerating fibers, TFEB localized in the cytoplasm (inactive form), whereas in atrophic fibers it localized in the nuclei (active form). Lipid-accumulated/atrophic fibers did not display p62-positive protein aggregates, indicating, together with the LC3-II (marker of autophagosomes) and p62-SQSTM1 analysis, that the autophagic flux is often preserved and lipophagy occurs. Conclusion. In atrophic and regenerating fibers of patients with NLSD-M we observed TFEB over-expression; in other conditions autophagy markers are increased, suggesting lipophagy active role on human lipid metabolism.
PS2Group1-061 / #319FATTY ACID OXIDATION DEFECTS PRESENTING AS PRIMARY MYOPATHY AND PROMINENT DROPPED HEAD SYNDROME
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Seena Vengalil1, Veeramani Preethish-Kumar2, Kiran Polavarapu2, Atchayaram Nalini1, Narayanappa Gayathri3, Rita Christopher4, Manjunath Mahadevappa1, Chandrajit Prasad5
1Neurology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
2Clinical Neurosciences, National Institute of Mental Health and Neurosciences, Bangalore, IN;
3Neuropathology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
4Neurochemistry, National Institute of Mental Health and Neurosciences, Bangalore, IN;
5Neuroimaging And Interventional Radiology, National Institute of Mental Health and Neurosciences, Bangalore, IN
Abstract: BACKGROUND: Inherited disorders of fatty acid oxidation can present with varied clinical manifestations ranging from asymptomatic or mild cases presenting in adulthood to severe multisystem involvement in neonatal period. Neurological manifestations can range from seizures, encephalopathy or Reye like syndrome to myopathy. Presentation as primary myopathy may be rare. Diagnosis needs a high index of suspicion and meticulous investigations to identify this potentially treatable disorder.Method : In this article, we describe a short series of twelve cases of metabolic myopathy due to fatty acid oxidation disorder confirmed by tandem mass spectrometry (TMS). These patients were seen in neuromuscular clinic of a tertiary hospital in South India between 2011 to 2016. Detailed description of clinical characteristics of each subtype as well as relevant investigations including muscle MRI, biopsy and TMS were done. RESULTS: Out of twelve cases, four had medium chain acyl CoA dehydrogenase deficiency (MCAD), four had Very long chain Acyl Co A dehydrogenase deficiency (VLCAD) , three had multiple acyl CoA dehydrogenase deficiency (MADD) and one had carnitine uptake defect. Mean age at onset was 12.75 , 10.5 and 19 years reaspectively for MCAD, VLCAD and MADD respectively with a mean delay between onset and diagnosis of 3.5, 4.7 and 11.3 years respectively. Single patient with carnitine uptake defect had onset at 29 years of age and was diagnosed one year later. Chronic muscular symptoms like exertional myalgia and mild proximal weakness of upper and lower limbs were the predominant symptoms. Other significant symptoms included weight loss and recurrent vomiting. We also found ptosis and bulbar weakness in a few of our patients which have not been previously described. All of them except one had severe neck extensor weakness presenting as head drop. Diagnosis was confirmed by tandem mass spectrometry which revealed characteristic findings. All patients are under regular follow up and have been doing well with carnitine, CoQ 10 and riboflavin supplements (for MADD). CONCLUSION: Fatty acid oxidation disorders presenting as primary myopathy are probably under diagnosed and should be kept in the differential diagnosis of patients presenting with acute or chronic muscular symptoms. Neck drop may be a prominent finding in these patients. TMS is a safe and less invasive investigation for diagnosis of these disorders and may help in avoiding invasive investigations like muscle biopsies. However diagnosis may be delayed as blood acyl carnitine may be normal at times of well being and hence high index of suspicion and repeated testing may be needed for accurate diagnosis and prompt treatment which will help in avoiding metabolic decompensations and mortality.
PS2Group1-062 / #316ENERGY DEFICIT IN THE MCARDLE MOUSE MODEL AFFECTS CALCIUM HOMEOSTASIS AND FORCE GENERATION
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Thomas Krag1, Tomas Pinos2, John Vissing1
1Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK;
2Vall d’Hebron Research Institute, Barcelona, ES
Abstract: McArdle disease is an inborn disorder caused by a lack of glycogen breakdown and due to a mutation in the muscle glycogen phosphorylase (myophosphorylase). We have recently reported that the mouse model of McArdle disease is significantly more affected by the disorder than the patients with McArdle disease are, with massive glycogen accumulation in muscle causing not only energy deficiency but also structural degeneration. This causes a significant decrease in contractile force as well as increased fatigue in glycolytic muscle. Using electron microscopy, we have found that in addition to the glycogen accumulations, glycolytic muscles have bloated sarcoplasmic reticulum (SR) and mitochondria as well as T-tubules in disarray. Using western blotting we found a significant increase in sarco/endoplasmic reticulum Ca2+-ATPase (SERCA2) as well as calsequestrin in tibialis anterior of the McArdle mouse suggesting that the bloated sarcoplasmic reticulum may be caused by an increase in sequestered calcium ions. The SERCA2-inhibitor phospholamban remained at wild-type level suggesting less overall inhibitory action. Bloated SR has also been found in patients with Pompe disease, suggesting energy deficit is the cause, whereby a larger part of the available pool of calcium-ions is withheld in SR due to the lower level of ATP. This is likely to have a direct effect on the level of force in muscle contractions and may explain why the severely affected McArdle mouse produces less muscle force. Hence, the McArdle mouse may be a model for studies of the link between energy deficit and lower contractile force.
PS2Group1-063 / #229ENZYME REPLACEMENT THERAPY IS BENEFICIAL AFTER 5 YEARS OF TREATMENT IN A LARGE GROUP OF ADULT POMPE PATIENTS
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Esther Kuperus1, Michelle E. Kruijshaar2, Stephan C.a. Wens2, Juna M. De Vries2, Marein M. Favejee2, Chris J.c. Van Der Meijden2, Dimitris Rizopoulos2, Esther Brusse2, Pieter A. Van Doorn2, Ans T. Van Der Ploeg2, Nadine A.m.e. Van Der Beek2
1Neurology, Faculty, Erasmus MC, Rotterdam, NL;
2Erasmus MC, Rotterdam, NL
Abstract: Background: Pompe disease is the first hereditary myopathy for which a registered treatment is available. Enzyme replacement therapy (ERT) has shown to be effective in adult patients, but data on the long-term effects in large groups of patients are lacking. We describe the effect of ERT in adult Pompe patients after a median follow-up of 5 years. Methods: In this nationwide, prospective study, muscle strength, muscle function and pulmonary function were measured at intervals of three to six months both prior to and after start of ERT. The treatment effect of ERT was defined as the difference between the outcomes at 5 years of treatment and the extrapolated natural course of disease. These differences were assessed using linear mixed-effects models for repeated-measures. Findings: One-hundred-two patients (median age 52.0 years) were included: 82 patients contributed both natural course and ERT data; 6 ERT data only; and 14 natural course data only. The median follow-up during the natural course was 1.1 years (range 7.9-0.1), and during ERT 5 years (range 0.2-7.3). In the first 2-3 years of ERT outcomes improved, after which they stabilized or declined. At 5 years, relative to the extrapolated natural course, treated patients had better muscle strength (manual muscle testing +6.6 percentage points (pp); hand-held dynamometry +9.6 pp; both p<0.001), pulmonary function (forced vital capacity in upright position +7.3 pp; in supine position +7.6 pp; maximal inspiratory pressure +20.8 pp; maximal expiratory pressure +17.3 pp; all p≤0.001), and self-reported activity levels (Rasch-built Pompe-specific Activity scale +10.8 pp, p=0.002). ERT did not improve muscle function tested with the Quick Motor Function Test (p=0.5). Gender, age and disease duration did not influence response to therapy for most outcome measures. Conclusion: We conclude that, at group level, long-term treatment with ERT positively affects muscle strength, pulmonary function and daily life activities. Although individual differences exist, both mildly affected and severely affected patients benefit from treatment.
PS2Group1-064 / #195SUGAR INFUSION IMPROVES EXERCISE CAPACITY IN PATIENTS WITH GLYCOGENIN-1 DEFICIENCY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Mads Stemmerik1, Pascal Laforêt2, Astrid Emilie Buch1, Karen L. Madsen1, John Vissing1
1Copenhagen Neuromuscular Center, 3342, Rigshospitalet, Copenhagen, DK;
2Centre De Référence Pathologie Neuromusculaire Paris Est, Hôpital Pitié-Salpêtrière, Paris, FR
Abstract: Glycogenin-1 Deficiency (GYG1) is an inborn error of glycogen synthesis. It is the most recently discovered muscle glycogenosis (Glycogen Storage Disease XV), and less than 15 patients have been identified worldwide. It has a variable clinical presentation, often slowly progressive with adult onset and hip-girdle muscle weakness. It is unknown if muscle glycogen breakdown is also affected in GYG1. We investigated fuel utilization during prolonged, moderate-intensity exercise, and the effect of intravenous glucose supplementation during this exercise. Three patients with genetically verified GYG1, and 6 healthy, sex-matched controls were included. Patients performed one hour of cycling exercise at 65% of their maximal oxidative capacity, while the controls cycled at workloads matching the patients. Stable isotope technique was used to measure fat and carbohydrate metabolism during exercise. Gas exchanges and heart rate (HR) were measured continuously during the exercise. The following day, patients repeated the exercise, this time receiving a bolus of 10% glucose of 2 ml*kg-1 prior to exercise and a constant infusion of 5 ml*min-1 during the exercise. All patients had reduced maximal oxidative (14.2, 33.9 and 36.5 ml*kg-1*min-1) and workload (60, 75 and 65 W) capacities compared to healthy controls (45.1 SD 6.4 ml*kg-1*min-1 and 270 SD 64 W). They were all able to complete 60 minutes of exercise. Patients had a lower carbohydrate oxidation (3.8 SD 2.5 mmol*min-1) and a similar fatty acid oxidation (15.7 SD 3.3 μmol*kg-1*min-1) compared to healthy controls (10.2 SD 1.6 mmol*min-1 and 16.1 SD 2.6 μmol*kg-1*min-1 respectively). With the glucose infusion, the total carbohydrate oxidation increased to 5.0 SD 2.5 mmol*min-1, and total fatty acid oxidation decreased to 9.0, SD 0.7 μmol*kg-1*min-1 in the patients during exercise. All calculations are based on indirect calorimetry. Stable isotope analyses are ongoing. Patients reported a subjective feeling of improved exercise tolerance, which was also reflected in a decrease in average HR of 12 bpm compared to the exercise test without the glucose infusion. The present findings demonstrate that although Glycogenin-1 Deficiency is primarily a defect in the glycogen build-up, glycogen breakdown is also affected as indicated by a lower oxidation of glucose during exercise and a beneficial effect of glucose supplementation. A likely explanation for this is an abnormal structure of glycogen that may impair breakdown. It needs to be determined if oral supplements of sucrose before exercise also will be helpful to patients.
PS2Group1-065 / #170EFFECT OF SENSORY INTEGRATION THERAPY ON FINE MOTOR FUNCTION IN MITOCHONDRIAL MYOPATHY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.8 Metabolic Myopathies / Mitochondrial Myopathies
Berkan Torpil, Semin Akel, Sedef Şahin
Occupational Therapy, Hacettepe University, Ankara, TR
Abstract: Backround: Mitochondrial Myopathies (MM) are a group of neuromuscular diseases caused by damage to the mitochondria-small, energy-producing structures that serve as the cells’ “power plants.” Purpose: The aim of the study is to examine the effect of sensory integration therapy on fine motor function. Material and Metods: Eleven-year-old boy with a diagnosis of MM referred to occupational therapy (OT) department for his motor function problems. As he represented movement problems that can be related to sensory problems during observation and clinic assessments, Dunn sensory profile was used to determine modulation and praxia problems for constructing treatment plan. Ayres Southern California Motor Performance and The Bruininks-Oseretsky Test of Motor Proficiency (BOTMP) were used to assess fine motor function. After the evaluation, OT program applied for 6 months, 2 days a week for 45 minutes. For modulation problems, appropriate sensory input elicitor activities were used. Sensory diet was given as home program. Results: According to Dunn Sensory Profile; there were problems in multisensory processing; proprioceptive, vestibular and auditory systems and sensory processing related to endurance/tone. After therapy BOTMP fine motor function score increased from 9 to 17. There were also significant positive changes in Ayres California Motor Performance score and sensory modulation which had an impact on fine motor skills; auditory, vestibular, multisensory, sensory processings related to endurance/tone. Moreover, frequency of falls also decreased after therapy. Conclusion: Sensory integration therapy improved fine motor functions and also decreased modulation and praxia problems in this child. Sensory problems of children with MM should be analysed in detail to tailor the best rehabilitation program and sensory integration therapy can be used to increase motor function.
PS2Group1-066 / #341PREVALENCE STUDY OF MUSCLE CHANNELOPATHIES IN ITALY
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.9 Muscle Channelopathies and Related Disorders
Lorenzo Maggi1, Mauro Lo Monaco2, Simona Portaro3, Giovanni Meola4, Jean Francois Desaphy5, Sabrina Lucchiari6, Serena Pagliarani6, Raffaele Dubbioso7, Pia Bernasconi8, Raffaella Brugnoni8, Paola Imbrici5, Giacomo Pietro Comi9, Renato Mantegazza8, Maria Trojano5, Adele D’Amico10, Lucio Santoro7, Elena Pegoraro11, Luisa Politano12, Tiziana Mongini13, Liliana Vercelli13, Gabriele Siciliano14, Giulia Ricci14, Diana Conte-Camerino5, Antonio Toscano3, Valeria Sansone15
1Neuromuscular Diseases And Neuroimmunology Unit, Foundation IRCCS Neurological Institute Carlo Besta, Milano, IT;
2Cattolica University, Roma, IT;
3University of Messina, Messina, IT;
4IRCCS Policlinico San Donato, University of Milan, Milano, IT;
5University of Bari, Bari, IT;
6University of Milan, Neurology Unit Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, IT;
7University of Naples Federico II, Napoli, IT;
8Foundation Neurological Institute Carlo Besta, Milano, IT;
9Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milano, IT;
10IRCCS Bambin Gesú Children’s Hospital, Roma, IT;
11University of Padua, Padova, IT;
12Second University of Naples, Napoli, IT;
13University of Turin, Torino, IT;
14University of Pisa, Pisa, IT;
15Centro Clinico NEMO, Milano, IT
Abstract: Background: Hereditary skeletal muscle channelopathies (SMC) are rare diseases whose prevalence in Italy is yet unclear. Aim of the study: To define the prevalence rate of SMC in Italy and the frequency and distribution of associated mutations. Methods: We reviewed clinical, laboratory, and genetic SMC data from 3 Italian neuromuscular laboratories. Results: Of 620 genetically confirmed SMC, 526 are non-dystrophic myotonias (73% CLCN1 and 27% SCN4A), 34 hyperkalemic periodic paralysis (SCN4A), 45 hypokalemic periodic paralysis (CACNA1S = 33; SCN4A = 12) and 15 Andersen-Tawil syndrome (KCJN2). All SCN4A (except an unreported 9-nucleotide deletion) and 71% of CLCN1 mutations are missense. Most CACNA1S and KCJN2 mutations are common mutations. Thirty-nine new mutations were detected (CLCN1 = 16; SCN4A:=20; CACNA1S = 2; KCNJ2 = 1). Conclusions: Frequency of specific SMC subtypes in Italy is similar to that in other countries. Our data confirm clinical and genetic heterogeneity with a limited number of mutations accounting for a large number of cases. This information will help for the search of a personalized therapy through functional and pharmacological studies of mutations.
PS2Group1-067 / #371A COHORT OF PEDIATRIC AGE PATIENTS WITH NON-DYSTROPHIC MYOTONIA
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.9 Muscle Channelopathies and Related Disorders
Joel Freitas1, Ana P. Sousa1, Teresa Coelho2, Manuela Santos3
1Neurophysiology, Centro Hospitalar do Porto, Porto, PT;
2Unidade Corino De Andrade E Serviço De Neurofisiologia, Hospital Santo António, Porto, PT;
3Neuromuscular - Pediatric Neurology, Centro Hospitalar do Porto, Porto, PT
Abstract: Introduction Non-dystrophic myotonias are a heterogeneous group of rare neuromuscular disorders caused by mutations in the skeletal muscle channels of chloride (CLCN1) and sodium (SCN4A). CLCN1 mutations can have dominant or recessive inheritance, whereas SCN4A have only dominant. Phenotypes can vary from mild myotonia to severe muscle stiffness, including congenital presentations. Objective Characterize a cohort of patients from a pediatric neuromuscular center, based on clinical, laboratory, neurophysiological and genetic features. Methods Retrospective review of consecutive patients with non-dystrophic myotonia[f1] [NJAGDF2] observed from 1995 until 2015. All clinical, ancillary, neurophysiologic and genetic information was compared between subgroups. Results Eight patients were identified, four with family history of myotonia. Six were male. Mean overall follow-up was 7,6 years. Three subgroups were identified: subgroup I - CLCN1 (4); subgroup II - SCN4A (3, from 2 families); subgroup III - unidentified mutations for CLCN1 and SCN4A (1, belonging to a family with hypokalemic periodic paralysis). In subgroup I the mean age of clinical onset was 3,5 years old (minimum 2 and maximum 6 years); 5,6 years (minimum 8 months, maximum 10 years) for subgroup II; 1,2 years for subgroup III. Congenital forms were observed in 2 patients, one from subgroup II and another from subgroup III. The mean age at first observation was 8,4 years for subgroup I; 8,6 years for subgroup II; 1,5 years for subgroup III. Gait disorder was the main clinical complaint on referral, in all patients. On first observation, myotonia was identified on the lower limbs in four patients, in one it was restricted only to toes, in the other four patients myotonia was present more generalized. Muscle hypertrophy was present in all patients, in some being focal, but through the follow-up other muscle groups were involved. Warm-upphenomenon was present in all patients with CLCN1 mutations (subgroup I), one patient with SCN4A mutation (subgroup II) and the patient from subgroup III. All patients had functional limitation throughout the follow-up, two patients with objective weakness. Both patients with congenital presentations (subgroup II and III) had speech difficulties, as well as swallowing limitations. All patients underwent different medication trials, but only in three patients there was a significant response to treatment; none tried mexiletine so far. Discussion Non-dystrophic myotonia represented a small percentage of patients in a pediatric neuromuscular center. The majority of patients had mutations in the chloride channel gene and one patient had no mutations identified. Both patients with congenital onset myotonia are clinically similar and negative for CLCN1 mutations. The presence of myotonia in early stages of live can be very subtle, which makes diagnosis a clinical challenge. [f1]observed [NJAGDF2]
PS2Group1-068 / #348THE ANTI-CONVULSANTS LACOSAMIDE, LAMOTRIGINE AND RUFINAMIDE REDUCE MYOTONIA IN ISOLATED HUMAN AND RAT SKELETAL MUSCLE
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.9 Muscle Channelopathies and Related Disorders
Thomas H. Pedersen, Martin Skov, Ole B. Nielsen
Department Of Biomedicine, Aarhus University, Aarhus, DK
Abstract: INTRODUCTION: In myotonia congenita loss of ClC-1 Cl--channel function results in skeletal muscle hyperexcitability and myotonia. Anti-myotonic treatment has typically targeted the voltage gated sodium channel in skeletal muscle (Nav1.4). This study explored whether three sodium channel modulating anti-epileptica (lacosamide, lamotrigine and rufinamide) can reduce myotonia in isolated rat and human muscles. METHODS: After dissection, muscles were rendered myotonic by ClC-1 inhibition by exposing the muscle to 9-AC, which inhibits ClC-1. The ability of lacosamide, lamotrigine or rufinamide to suppress myotonia was then assessed from sub-clinical to maximal clinical concentrations. Drug synergy was determined using isobole plots. RESULTS: All drugs were capable of abolishing myotonia in both rat and human muscles (Fig. 1). Lamotrigine and rufinamide completely suppressed myotonia at sub-maximal clinical concentrations while lacosamide had to be raised above the maximal clinical concentration to completely suppress myotonia. A synergistic effect of lamotrigine and rufinamide on myotonia was observed. CONCLUSION: These findings suggest lamotrigine and/or rufinamide could be used as anti-myotonic treatment in myotonia congenita.
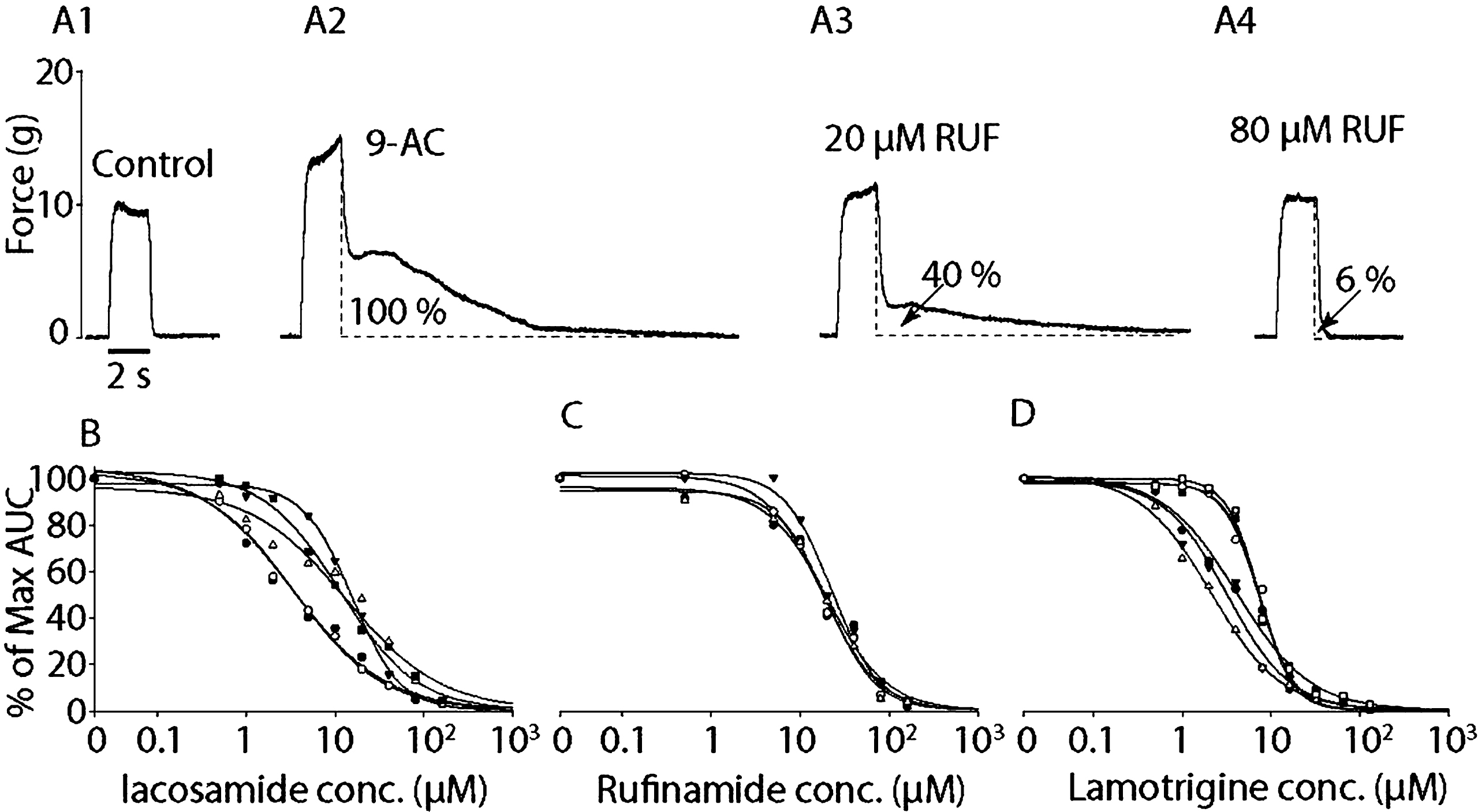
PS2Group1-069 / #299THYROTOXIC PERIODIC PARALYSIS: DOES MUSCLE MEMBRANE DYSFUNCTION UNDERLIE DISEASE PATHOGENESIS?
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.9 Muscle Channelopathies and Related Disorders
Nam-Hee Kim1, Joong-Yang Cho2, Kyung Seok Park3
1Neurology, Dongguk University Ilsan Hospital, Goyangsi, KR;
2Neurology, Inje university Ilsan hospital, Goyangsi, KR;
3Neurology, Seoul National University Bundang Hospital, Seongnamsi, KR
Abstract: Introduction: Thyrotoxic periodic paralysis (TPP) is a disease characterized by reversible episodes of paralysis concurrent with hyperthyroidism. TPP is clinically similar to hypokalemic periodic paralysis (HOPP), which is mediated by ion channel dysfunction. However, the specific mechanisms underlying TPP pathogenesis are largely unknown. We investigated whether or not TPP has a dysfunction at the level of muscle membrane as HOPP by measuring muscle fiber conduction velocity (MFCV). Methods: Thirteen TPP patients and 15 age-matched controls were included in the study. Clinical characterization and serial neurophysiological testing, including nerve conduction, prolonged exercise (PET) and MFCV tests were performed to assess electrophysiological function. Results: MFCV values were elevated until one year after an attack in TPP patients. In patients with positive PET results, MFCVs were significantly higher. The effects of hypokalemia or hyperthyroidism were non-significant. Discussion: Although clinical manifestations in TPP are similar to those observed in HOPP, TPP appears to feature a different pathogenic mechanism, in which MFCV values demonstrated an increase rather than a reduction. Further studies are needed to support these findings.
PS2Group1-070 / #253THE CULLIN 4A/B-DDB1-CEREBLON E3 UBIQUITIN LIGASE COMPLEX MEDIATES THE DEGRADATION OF CLC-1 CHANNELS RESULTING IN MYOTONIA CONGENITA
Topic: Group 1 – Muscle Diseases of Genetic Origin: Clinical Features, Pathophysiology, Therapy / 1.9 Muscle Channelopathies and Related Disorders
Ssu-Ju Fu, Yi-An Chen, Yi-Jheng Peng, Chih-Yung Tang
Department Of Physiology, College go Medicine, National Taiwan University, Taipei, TW
Abstract: Abstract: Mutations in human CLC-1 channels have been linked to the hereditary muscle disorder myotonia congenita. Human CLC-1 is encoded by the CLCN1 gene on chromosome 7, and more than 100 different mutations have been reported. A previous has characterized disease-associated CLC-1 A531V mutant protein may fail to pass the endoplasmic reticulum quality control system and display enhanced protein degradation and defective membrane trafficking. Currently the molecular basis of protein degradation for CLC-1 channels is virtually unknown. Here we aim to identify the E3 ubiquitin ligase of CLC-1 channels. Method: cDNAs of WT and mutant CLC-1 transfected in to HEK293T cells for whole-cell patch and biochemistry analyses. Recombinant lentivirus was generated by co-transfecting HEK293T cells with the packaging plasmid pCMV-Δ R8.91, the envelope plasmid pMD.G, and shRNA expressing constructs. Skeletal muscle fibers were isolated from adult Wistar rats. Result: This CLC-1 mutant displays dramatically enhanced proteasomal protein degradation, thereby manifesting a diminished whole-cell current density and a reduction in the total protein level. Pharmacological and biochemical inhibition of CUL4A/B notably reduced CLC-1 protein turn-over and ubiquitination. Co-immunoprecipitation analyses further revealed that CLC-1 co-existed in the same protein complex with CUL4A/B, as well as the previously identified CUL4A/B-associated binding partners DDB1 and CRBN. Disruption of DDB1 or CRBN expression also resulted in enhanced CLC-1 protein level. Taken together, we propose that the CUL4A/B-DDB1-CRBN E3 ligase complex catalyses the ubiqutination and regulates the degradation of CLC-1 channels. Conclusion: Suppression of ubiquitination effectively enhanced the protein level as well as the functional expression of the A531V mutant may shed light on the therapeutic potential of cullin inhibitor in correcting disease-related protein expression defects in CLC-1 channels.
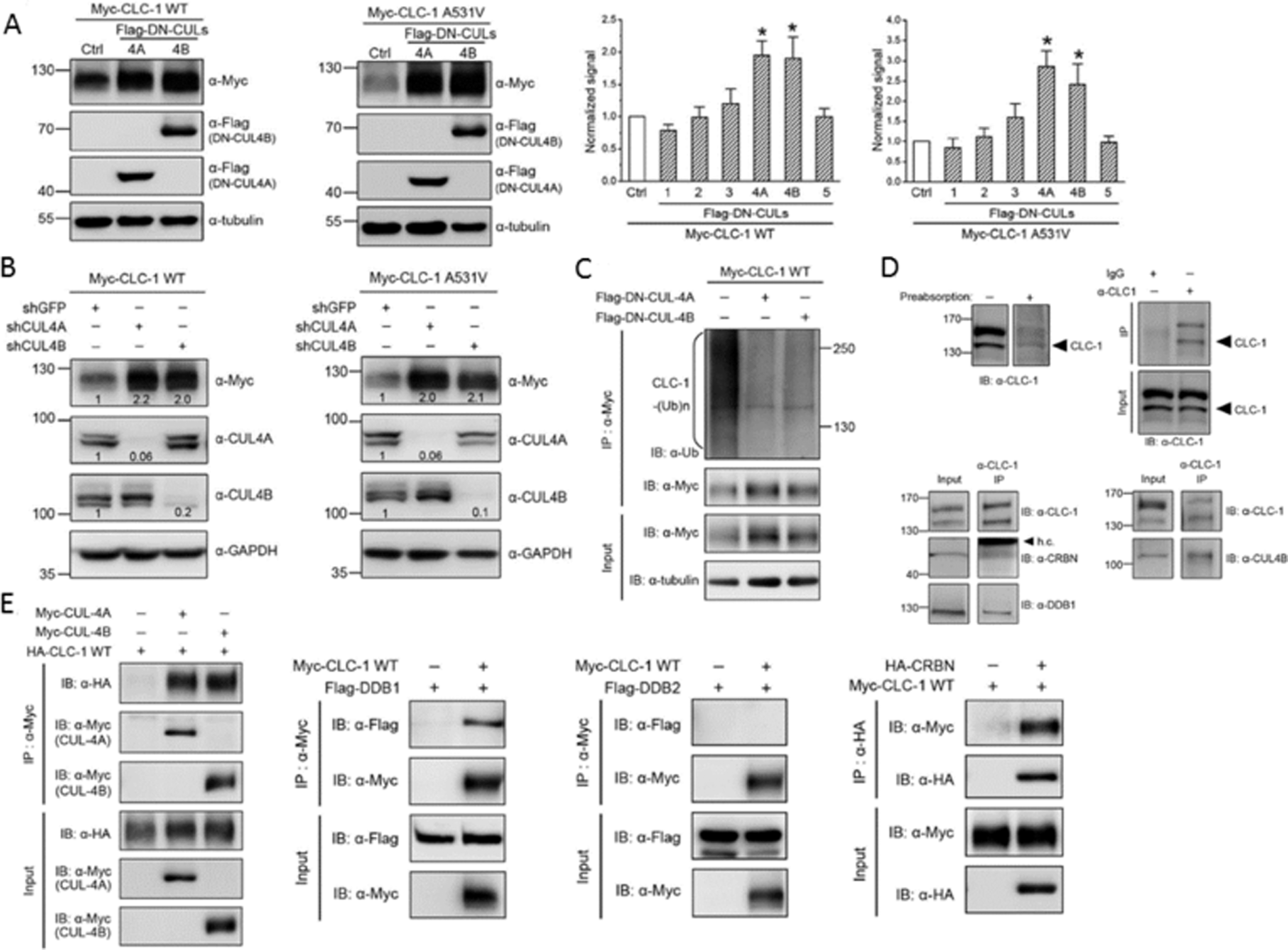
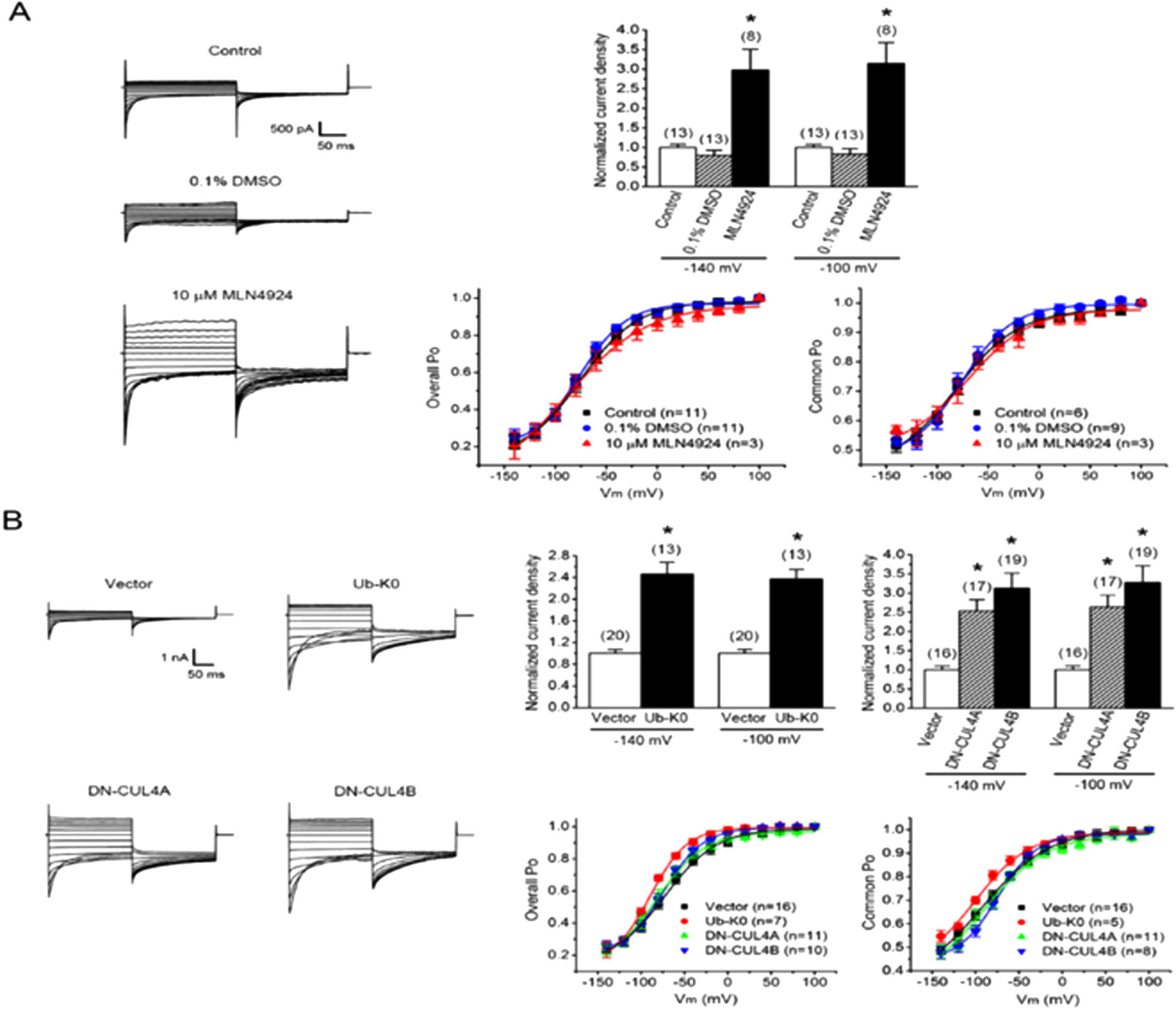
PS2Group1-071 / #494ELECTROMYOGRAPHY AND THE RISK OF PNEUMOTHORAX
Topic: Group 6 – Novel Diagnostic Methods in Neuromuscular Diseases / 6.4 Electrodiagnosis
Charles Kassardjian1, Cullen M. O’Gorman2, Eric J. Sorenson3
1Division Of Neurology, St. Michael’s Hospital, University of Toronto, Toronto, ON, CA;
2Neurology, Griffith University, Brisbane, QLD, AU;
3Neurology, Mayo Clinic, Rochester, MN, US
Abstract: Introduction: Pneumothorax is a feared complication of electromyography (EMG). Data on the frequency, severity and risk factors for pneumothorax after needle EMG are lacking. Objective: To determine the frequency of iatrogenic pneumothorax after needle EMG examination, and to determine the timing, risk factors and morbidities associated with iatrogenic pneumothorax caused by EMG. Methods: A retrospective chart review of cases diagnosed with pneumothorax in temporal association with an EMG examination was performed. Clinical, electrophysiological and radiological data were reviewed to determine if the EMG was causative of the pneumothorax. Results: Out of 64,490 EMG studies, 7 cases were identified with a probable or definite association between the EMG and pneumothorax. Mean age was 39.6 years; there were 4 males and 3 females, and 2 patients were obese. Pneumothoraces were symptomatic and presented with 24 hours of EMG in all cases. Pneumothorax occurred after needle of the diaphragm and serratus anterior in one case each. In 5 cases, multiple high-risk muscles were sampled. Muscles with the highest frequency of pneumothorax were serratus anterior (0.445%), followed by diaphragm (0.149%) and trapezius (0.117%). Summary/Conclusion The frequency of symptomatic iatrogenic pneumothorax after needle EMG of selected high-risk muscles appears low, with the serratus anterior and diaphragm possibly carrying a higher risk. Despite the apparent safety of EMG, needle examination of chest-wall muscles should be performed by experienced examiners with attention to meticulous technique. NOTE: The content of this abstract was recently published in Muscle & Nerve (2016 Apr;53(4):518-21), PMID: 26333600.
PS2Group3-001 / #487CLINICAL AND EPIDEMIOLOGICAL FEATURE OF MYASTHENIA GRAVIS IN CHILEAN POPULATION
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Gabriel Cea1, David Martinez1, Radrigo Salinas1, Andres Stuardo2
1Neurology, University of Chile, Santiago, CL;
2Neurology, Hospital Salvador, Santiago, CL
Abstract: Introduction: Myasthenia gravis is an autoimmune condition of the neuromuscular junction and systematic descriptions of its epidemiology are scarce. Data on myasthenia graves prevalence and incidence vary widely. Method: We performed a survey that included demographic and clinical questions to more than four hundreds patients from all regions of Chile, with the help of the Chilean Myasthenia Gravis Corporation. We also, carried out a prevalence study of myasthenia gravis using the method of capture and recapture and the hospital prescription register of pyridostigmine. Results: Prevalence in adult population was estimated to be 8.36 × 100000 in the area covered by our hospital. Four hundred and five patients responded the survey and were interviewed personally or over the phone in relation to sensitive clinical questions. Two hundred and seventy-nine were women and one hundred and twenty-six were men (ratio 2,2). When the analysis included patients older than 40 years there was 97 women and 73 men (ratio 1.3) and 33 women against 34 men (ratio 0.97) in older than 60 years old. The median age of onset of symptoms was 35.0 years (range 1 to 89 years) and patients older than 60 years of age were 16.5% of the entire survey group. The onset was ocular in 188 patients (46.4%), oculobulbar in 47 patients (11.6%), bulbar 36 patients (8.9%), limbs 47 patients (11.6%) and generalized in 87 patients (21.4 %). In fifty-three patients the diagnosis of thymoma was made (13,3%), 22 patients were 40 years old or younger. Two hundreds and twelve patients (52,4%) were admitted to hospital for thymectomy or medical treatment. One hundred and forty one patients underwent thymectomy, 52 for thymoma and in 88 patients for non-thymomatous myasthenia gravis (25%). Associated autoimmune diseases were reported in 59 patients, such as hypothyroidism in 27, rheumatoid arthritis in 11, thrombocytopenia in 3 and systemic lupus erythematous in 2. At least one of these diseases was reported in 134 members (33%) of the families of the patients. Seventy-eight patients had to change work due to the disease and 68 needed some kind of help to carry out their daily activities. Conclusions: The prevalence of myasthenia gravis in Chile is within the range of prevalence described worldwide. We do not see in our series the inversion in the women to men ratio observed in other countries in older patients. The proportion of patients with an onset of myasthenic symptoms older that 60 years showed an increase compared to historical reports. The presence of thymoma in this age group is low (4.5%).
PS2Group3-002 / #236EXOME SEQUENCING IDENTIFIES TARGETS IN OPHTHALMOPLEGIC MYASTHENIA GRAVIS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Melissa Nel1, Mahjoubeh Jalali Sefid Dashti2, Junaid Gamieldien2, Jeannine M. Heckmann3
1University of Cape Town, Cape Town, ZA;
2Sanbi, University of Western Cape, Cape Town, ZA;
3Medicine (neurology Division), University of Cape Town, Cape Town, ZA
Abstract: Background: While extraocular muscles (EOMs) are affected early in Myasthenia Gravis (MG), we identified a distinct ophthalmoplegic complication (OP-MG) in patients with African-genetic ancestry that remains treatment-resistant. This phenotype most commonly affects acetylcholine receptor-antibody positive cases with juvenile-onset MG. A previous candidate approach identified two African-specific gene expression traits linked to OP-MG, decay accelerating factor and transforming growth factor beta-1. We speculate that OP-MG may result from a complex genetic network activated in EOMs by autoimmune MG, in these and as yet unknown targets. Objective: To use the unbiased approach of whole exome sequencing in a highly selected cohort of juvenile-onset OP-MG vs control MG individuals to discover unknown OP-MG susceptibility variants. Method: Next-generation sequencing (Agilent v5, Otogenetics, USA) of the coding and untranslated regions (UTRs) was performed on genomic DNA from 15 African ancestry individuals at 50x coverage (11 OP-MG vs 4 control-MG). Read alignment and variant calling were carried out. Filtering strategies were employed to further prioritize a set of genes for Sanger sequencing validation in OP-MG (n=22) and control=MG (n=44) groups. Results: 356 variants were predicted to impact function; 60% were in the 3’UTR and 25% were novel i.e. not present in the 1000 Genome African/ExAC/NHLBI-ESP databases. Two filtering strategies were then applied: unbiased prioritization according to statistical strength of association between OP-MG/MG-controls (p<0.05), and functional filtering related to biological relevance for MG. Unbiased filtering showed seven likely deleterious variants associating with OP-MG in the discovery set (P<0.05); the two variants showing the strongest association are implicated in myogenesis or membrane protection against complement. Motif analysis of the variant locations predicted the loss of miRNA binding sites in the 3’UTR of DDX17 and the loss of a transcription factor binding site in the 5’UTR of ST8SIA1. Verification by Sanger sequencing is underway. To explore the biological relevance in our gene hit list, we searched existing public transcriptomic and miRNA data showing ≥2-fold altered regulation in either human/mouse EOM vs limb muscle under basal or MG conditions. A 3’UTR T>C variant in interleukin 6-receptor (rs113444481) was identified for MG relevance by 2 different public datasets. Motif analysis indicated potential loss of two miRNA binding sites and the loss of a RNA-protein binding site. Sanger validation of the rare (1% in 1000 Genome African database) variant and its association with OP-MG is being carried out (21% OP-MG vs 8% control-MG; p = 0.043). Discussion: Exome sequencing in a small well-characterized discovery sample has identified several potentially pathogenic variants that associate with OP-MG. Preliminary results suggest novel gene variants which cross-talk with known MG-associated pathways.
PS2Group3-004 / #476SFEMG FINDINGS IN OCULAR COMPLICATIONS OF COSMETIC BOTOX INJECTIONS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Daniela A. Navarrete1, Carlos S. Navarrete2, Raul H. Muñoz1, Vera Bril3, Mireya E. Balart4
1Medicine, Universidad Mayor, Santiago, CL;
2Neurology, Universidad de los Andes, Santiago, CL;
3Toronto General Hospital, Toronto, CA;
4Neurology, Clinica Davila, Santiago, CL
Abstract: Background: Jitter abnormalities have been described after therapeutic use of Botox injections for dystonia, not only in the treated muscles but in other muscles distant from the injection site. However, after cosmetic Botox, only minor side effects have been described and the SFEMG results are not as clear. Introduction: We report 5 patients treated with small doses of Botox (less than 45 IU) for cosmetic reasons. These cases were referred to our laboratory for possible ocular myasthenia between 2012 and 2014. All patients had a history of Botox injections for wrinkles in the forehead and lateral orbicular areas. The patients complained of unilateral ptosis and diplopia but no other symptoms. They had been free of Botox injections for more than 6 months before their clinic appointment. Methods: We performed SFEMG with a Cadwell Wave machine using concentric disposable needles having 0.02 mm2 lead off surface. We collected 20 MCD measurements with the mean jitter ranging from 80 to 120 microseconds in all cases. All had increased fiber density and neuromuscular blocking was minimal to absent. All patients had negative testing for anti-acetylcholine receptor antibodies. No other antibodies were measured. Patients were followed for a year after the last Botox injections and all improved clinically. Conclusions: 1.- Contrary to what is mentioned in the literature, some patients treated with cosmetic Botox for wrinkles can have undesirable ocular symptoms persisting for more than 6 months after treatment and leading to the suspicion of ocular myasthenia. 2.-The lack of neuromuscular blocking and increase in fiber density observed consistently in these patients’ results differ from findings in ocular MG, and should be validated in a larger series of patients.
PS2Group3-005 / #460A PHASE II TRIAL TO ASSESS THE EFFICACY, SAFETY AND FEASIBILITY OF 20% SUBCUTANEOUS IMMUNOGLOBULIN IN PATIENTS WITH MYASTHENIA GRAVIS EXACERBATION - SAFETY AND FEASIBILITY
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Zaeem A. Siddiqi1, Derrick Blackmore1, Ashley Mallon2
1Neurology, University of Alberta Hospital, Edmonton, AB, CA;
2Neurology, University of Alberta Hospital, Edmonton, CA
Abstract: OBJECTIVE: An interim analysis to assess the safety and feasibility of 20% Subcutaneous Immunoglobulin (SCIg; (Hizentra) in patients with myasthenia gravis (MG) exacerbation. BACKGROUND: The University of Alberta MG program is the primary site of an ongoing investigator initiated, multicenter clinical trial to assess the feasibility, safety and efficacy of 20% SCIg in MG exacerbation. METHODS: A dose of 2gm/kg of SCI is infused in a flexible regimen over 4 weeks. Patient reported adverse effects, infusion site reactions, incidence of hemolysis, serum Ig levels, and patient compliance and satisfaction are monitored during the study period. RESULTS: Twelve patients have completed the study with a total of 148 infusions at four abdominal sites (total sites = 604). All patients were able to infuse the full-prescribed dose. Mean total dose was 180.8gms (range: 104 to 204gms), mean volume 904ml per patient (range 520–1020 ml). All patients needed only a single training session. Pump rather than manual infusions were associated with improved compliance. There was no incidence of hemolysis or any other serious adverse event. Minor infusion site reactions (mild redness, and itching) and a mild flu like syndrome and headache were reported with some infusions. None caused discontinuation of infusions or any significant effect on routine activities. Two patients intolerant to IVIg were able infuse the full SCIg dose. Serum IgG levels gradually increased by about 200% at study end. Patient reported survey scores based on various measures indicate reasonable satisfaction with SCIg infusions (mean score: 4.9/7; range: 4-7).CONCLUSIONS: Preliminary results indicate that despite its large volume SCIg appears to be well tolerated at standard dose for neuromuscular diseases i.e. 2 gm/kg, when infused over 4 weeks. Infusion site reactions as well as systemic side effects appear to be mild and rare. Further studies are needed to confirm these preliminary results.
PS2Group3-006 / #459A PHASE II TRIAL TO ASSESS THE EFFICACY, SAFETY AND FEASIBILITY OF 20% SUBCUTANEOUS IMMUNOGLOBULIN IN PATIENTS WITH MYASTHENIA GRAVIS EXACERBATION - INTERIM ANALYSIS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Zaeem A. Siddiqi1, Derrick Blackmore2, Ashley Mallon3, Cecile Phan1
1Neurology, University of Alberta Hospital, Edmonton, AB, CA;
2University of Alberta Hospital, Edmonton, CA;
3Neurology, University of Alberta Hospital, Edmonton, CA
Abstract: OBJECTIVE: A preliminary analysis of efficacy of 20% Subcutaneous immunoglobulin (SCIg) Hizentra@) in an ongoing study on patients with worsening myasthenia gravis (MG). BACKGROUND: Recent studies in immune-mediated neuromuscular diseases indicate that SCIg may be an effective alternative to intravenous immunoglobulin (IVIG). Patients on chronic IVIg therapy can switch to SCIg without any loss of efficacy and report improved quality of life due to flexibility to their treatment schedule with SCIg. No prior studies have explored the efficacy of SCIg in MG. METHODS: This is an ongoing investigator-initiated, open label, multicenter, pilot study to assess the efficacy of SCIg in patients with moderately severe MG (MGFA Class II & III). Patients received 2gm/kg of SCIg in a flexible dosing regimen over four weeks. The primary outcome measure for efficacy was change in Quantitative MG (QMG) Scores from baseline to six weeks. Secondary measures included change in MG Composite (MGC), Manual Muscle Testing (MMT) and MG activities of daily living (MG ADL) scores. RESULTS: To date of 20 eligible patients 16 were enrolled. One patient was unable to infuse SCIG due to hand weakness and three withdrew consent. 12 patients (6 males & 6 females; mean age 58 years; 9 acetylcholine receptor antibody & 1 Anti-MuSK antibody positive, 2 seronegative,) completed the study. Most patients showed improvement in QMG scores (mean reduction 5.75 points; p<.05) and in secondary outcome measures (mean reduction MGC 11.25; mean MMT 12.4; mean MG ADL 3.75) from baseline. In general, peak improvement occurred during the third week of treatment. Transient worsening of the MG occurred in two patients initially but neither had to be withdrawn. CONCLUSIONS: Our interim analysis suggests that SCIg may be effective in patients with moderate MG exacerbation. This is an ongoing study and further data will be needed to corroborate these preliminary findings.
PS2Group3-007 / #458SUBCUTANEOUS IMMUNOGLOBULIN IN MYASTHENIA GRAVIS: TRIAL DESIGN AND PROGRESS UPDATE
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Zaeem A. Siddiqi1, Derrick Blackmore2, Ashley Mallon1
1Neurology, University of Alberta Hospital, Edmonton, AB, CA;
2Neurology, University of Alberta Hospital, Edmonton, CA
Abstract: OBJECTIVE: To present the clinical trial design of a phase II investigator initiated multi-center open label study to assess the feasibility, safety and efficacy of 20% SCIg (Hizentra) in patients with moderately severe myasthenia gravis (MG). BACKGROUND: Intravenous immunoglobulin (IVIg) is an effective therapy for patients with worsening MG. Recently, the subcutaneous form of immunoglobulin (20% SCIG; Hizentra) has become available and has been used in neuromuscular conditions amenable to IVIg therapy. SCIg appears to be associated with fewer adverse effects, comparable efficacy and the added advantage of flexibility due to self infusion by the patients. The use of SCIg has not been systematically studied in patients with MG. METHODS: This is a two-year multicenter open label, single phase, investigator-initiated phase II clinical trial in patients with moderately severe MG (MGFA Grade II and III). Enrolled subjects are trained to self-infuse 20% SCIg at 2gm/kg over 3 to 4 weeks in a dose escalating manner. Automated pumps allow accurate and convenient administration of the study drug concurrently at at four infusion sites. The dosing regimen is flexible allowing two to four infusions per week. The primary efficacy outcome measure is the change in the QMG Score for Disease Severity from baseline to day 42 (6 weeks). In addition, scores on MG Composite, MG-ADL, and SF36 scales, infusion related adverse effects, laboratory monitoring for safety and patient satisfaction with SCIg treatment will be assessed. The trial was approved by Health Canada and the local institution ethics board. RESULTS/CONCLUSION: The trial started in January 2015 and data collection is complete for sixteen of thirty expected patients. One more Canadian sties is being initiated. We anticipate providing an update on the overall progress of this ongoing clinical trial. Support by CSL Behring Investigator Initiated Research Grant
PS2Group3-008 / #446DISTAL MYASTHENIA GRAVIS: AN UNUSUAL PRESENTATION WITH SLOWLY PROGRESSIVE DISTAL WEAKNESS AND ATROPHY
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Renata A. Andrade1, Alexandre M.S. Januario2, João E. Magalhaes3
1Neuropsychiatry, Federal University of Pernambuco, Recife, BR;
2Federal University of Pernambuco, Recife, BR;
3State University of Pernambuco, Recife, BR
Abstract: A 47-year-old female patient were refered to a neurological center complaining of right hand weakness and atrophy beginning 18 years ago. Two years later, started with weakness also in the left hand, with very slow progression in both sides. In the last two years, she begans to suffer frequent falls . She denied diplopia, ptosis, cramps, twitching or muscle fatigability. She brought to medical appointment an previous diagnosis investigation with normal values of muscle enzymes and several electromyography revealed an myopathic pattern with low amplitude and short duration motor unit potentials in all limbs, with spontaneous activity. She had also undergone muscle biopsy performed on right forearm, which revealed just neurogenic atrophy, with no myophatic specific finds or inflammation. At clinical evaluation we found an predominant involvement of finger extension with milder involvement of finger flexion, wrist flexion and extension, and normal strength in lower limbs. No bulbar, ocular weakness or evidence of fatigability were noticed. General clinical exam was unremarkable. After the referral to our center was performed a new electromyography with 3Hz repetitive nerve stimulation that showed normal nerve conduction studies, myopathic motor unit potentials at weakness muscle and important decrement ,above 30% in the trigeminal nerve- masseter and 15% at radial nerve- anconeous muscle. Serum acetylcholine receptor antibody assay performed subsequently was positive. The assay for muscle- specific tyrosine kinase antibodies was negative. Myasthenia gravis characteristically involves ocular; bulbar, and proximal limb muscles, with typical fatigability of symptoms. An initial presentation with distal weakness and atrophy is extremely unusual, and although muscle atrophy may occur in about 10% of patients with myasthenia gravis, severe atrophy is rare. Here we describe a patient with very slow progression of weakness and atrophy in hands, during 20 years, when she starts to complain of troubles in walking. During her medical follow-up was diagnosed as a chronic myopathy based on the clinical picture and complementary exams, especially the pattern found in electromyography. On this case the diagnosis of myasthenia gravis was made with the high titers of acetylcoline receptor antibody and characteristic decrement obtained on repetitive stimulation test. Although rare, cases of patients with myasthenia gravis with distal weakness and neurogenic atrophy have been reported in the literature, and despite the myopathic pattern found, repetitive nerve stimulation was not performed in previous evaluations of this patient. It’s already known that electromyography finds in myopathic disorders overlap with those of neuromuscular juction diseases; short duration, small-amplitude motor unit action pontential can be seen in neuromuscular junction disorders, and even spontaneous activity such as fibrillation potentials have been described in this kind of patients. With this case we conclude that neurophysiological testes for a neuromuscular juction disorder should be performed in patients who will undergo a electromyography with a hypothesis of myopathy.
PS2Group3-009 / #448LATE ONSET MYASTHENIA GRAVIS (LOMG): WHEN DOES IT START? Â
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Nestor D. Genco, Mariela Bettini, Maria Ines Araoz, Felipe Peralta Calderon, Edgardo Cristiano, Hernan Gonorazky, Marcelo F. Rugiero
Neurology, hospital italiano de buenos aires, buenos aires, AR
Abstract: Introduction Late onset Myasthenia Gravis (LOMG) is a group of MG patients with probably distinctive clinical and immunogenetic characteristics different from early onset Myasthenia Gravis (EOMG). The onset of LOMG is not well defined and it varies according different authors. Objective: The aim of this study is determine the age of cut off for the LOMG group of patients. Methods Retrospective analysis of medical records of patients with MG, with at least 2 years of follow up. A multivariate analysis was performed with clinical and demographic variables proposed to establish LOMG, with age of cut off at 40,50 and 65 years old. Results 215 MG patients were included. 3 categories were obtained: Group 1: age at onset <40 years old (EOMG)(N= 47), Group 2: age at onset between 40 and 65 years old (Intermediate Onset) (N=63) and Group 3: age at onset > 65 years old (LOMG)(N=105). Female predominance was higher in group 1 (p<0.001). Ocular MG was more frequent in group 3 (p 0.01). Group 2 and 3 had higher rate of thymoma (p<0.001). Remission was more frequent in Group 3 (p <0.001). The rest of the analyzed variables did not show statistical differences. CONCLUSION In our population of MG patients, 2 ages of cutt off were obtained according to demographic characteristics, thymic pathology, remission rate and ocular form. This allow us to recocnize 3 groups: Early Onset (<40 y)(EOMG), Intermediate onset (between 40 and 65 y) and Late Onset (>65 y)
PS2Group3-010 / #429ROLE OF THYMECTOMY IN PREVENTING MYASTHENIC CRISIS IN GENERALIZED MYASTHENIA GRAVIS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Gillella K.R. Pavan1, A.K. Meena2, R.V. Kumar3, S.A. Jabeen1, K.M. Rukmini1, Rupam Borgohain1
1Neurology, Nizam’s Institute of Medical Sciences, hyderabad, IN;
2Neurology, Nizam’s Institute of Medical Sciences, Hyderabad, IN;
3Cardio-thoracic Surgery, Nizam’s Institute of Medical Sciences, hyderabad, IN
Abstract: Background: Myasthenic crisis is the most serious life-threatening event in generalized myasthenia gravis (GMG) patients. The objective of this study was to assess the role of thymectomy in preventing myasthenic crisis in GMG. Methods: Clinical records from 274 myasthenic patients diagnosed and treated in our neurology clinic during 2000 to 2015 were reviewed. Forty four patients were excluded because of unconfirmed diagnosis, ocular form of Myasthenia gravis, contraindication to surgery, and loss to follow-up. The Osserman classification was used to assess the initial severity of the disease. Frequency and severity of the attacks were compared between thymectomy and non-thymectomy groups of patients with GMG with appropriate statistical tests according to the nature of variables. Multivariate logistic regression analysis was used to assess the predictors of myasthenic crisis in the group of patients with thymoma and non-thymoma, with or without thymectomy. Results: 108 patients were in thymectomy group and the other 122 patients were only on medical therapy. These two groups had no significant differences with respect to age at onset, gender, Osserman score in baseline and follow up period. 93 patients (40.4% of all 230 patients) had reported 96 attacks of myasthenic crisis. 28 patients of 108 (25.9%) were in thymectomy group and 65(53.2%) were in the other group. There was significant difference between the two groups in number of patients with crisis (p < 0.001). In addition, these attacks were more severe in group of non-thymectomized patients as the duration of ICU admission was longer and they needed longer ventilatory support during their attacks. Regression model showed thymectomy and lower age at onset as two predictors of decrement in myasthenic crisis rate in non-thymomatous GMG patients. Conclusions: Thymectomy seems to have a preventive role in decreasing the rate and severity of myasthenic crisis in GMG.
PS2Group3-011 / #384OCULAR VERSUS Ã, GENERALIZED MYASTHENIA GRAVIS: 17 YEARS’ EXPERIENCE OF A TURKISH NEUROMUSCULAR CLINIC
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Belgin Mutluay1, Aysun Soysal1, Fikret Aysal2, Ayhan Koksal3, Mesude Ozerden3, Ayten C. Dirican1, Dilek Atakli1, Sevim Baybas1
1Neurology, Bakirkoy Research and Training Hospital for Psychiatry,Neurology and Neurosurgery, Istanbul, TR;
2Neurology, Medipol University, Istanbul, TR;
3Bakirkoy Research and Training Hospital for Psychiatry, Neurology and Neurosurgery, Istanbul, TR
Abstract: BACKGROUND: According to clinical presentation of myasthenia gravis (MG), demographics as well as specific features are influenced by genetic and geographical characteristics OBJECTIVE: The aim of this retrospective study was to determine characteristics of myasthenia gravis patients followed up at our neuromuscular patient outpatient clinic in Istanbul. METHOD: Patients with a preliminary diagnose of MG attending our neuromuscular clinic during 1997 to 2015, were evaluated. Patients complaining of eyelid drop, double vision or both were investigated for clinical characteristics, laboratory and electrophysiological findings, and participated in the study when they were followed up for at least one year. All patients underwent repetitive nerve stimulation test and were tested for acetylcholine receptor antibody. When necessary, ice pack and pyridostigmine tests and single fiber EMG was performed for diagnostic support. RESULTS: Conversion to GMG was seen in 63% of all patients, with a mean time 13.6±24.72 months. Gender, age, age at onset revealed no significant correlation for ocular (OMG) and generalized myasthenia (GMG) groups (p= 0.47; 0.67; 0.7; respectively). Onset with diplopia, ptosis or both together did not differ in groups. Acetylcholine receptor antibody positivity was detected in 83% OMG and 67% GMG patients. Thymoma was seen in four OMG and six GMG patients. Immunosuppressant treatment was markedly preferred in GMG patients in contrast to OMG (p<0.000). DISCUSSION: Acetylcholine receptor antibody positivity and electrophysiological investigations are the main diagnostic tools for MG. Although life expectancy has been changed in the last decades, prognosis for ocular onset MG is still a debate. In order to determine factors implicated in the conversion to GMG, gender, age or ethnic factors have been shown to influence. GMG was reported to be prominent in young female and OMG was seen in ages over 50 years. However, no age or sex difference was found in our study. Besides, conversion to GMG was shorter in our patients in contrast to former data. CONCLUSION: Although a neuromuscular junction disease, myasthenia characteristics differ according to ethnicity and/or environmental changes. Rapid transformation to generalized MG may suggest early introduction of immunosuppressant in conjunction to corticosteroid treatment for OMG patients.
PS2Group3-012 / #329MYCOPHENOLATE MOTETILN MYASTHENIA. EXPERIENCES AN ACADEMIC NEUROMUSCULAR CLINIC.
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Ratna K. Bhavaraju-Sanka, Mithila M. Fadia, Alejandro Tobon, Carlayne Jackson
Neurology, UT Health Science Center at San Antonio, San Antonio, TX, US
Abstract: A retrospective review was performed on all MG patients seen in the neuromuscular clinic since 2009 who were started on MMF. Data that was collected included: demographics, antibody status, Myasthenia gravis activities of daily living (MGADL) score, and the use of prednisone or other agents to treat MG at start of MMF and at 12 months. A total of 67 patients had received MMF and 37 patients were included in the study. The other patients were excluded due to lack of enough information at the time of initiation of therapy. 26 of the 37 patients (70%) were scetylcholien receptor (Achr) antibody positive. 22 of the 37 were females (59%). 21/37 (56%) showed improvement in MGADL at 12 months with an average improvement of 3.5 points. 8/37 patients were able to reduce the daily dose of prednisone by almost 19 mg. Seven patients were initially on intravenous immunoglobulin but were able to be weaned off of the infusions. Two patients who were getting plasmapharesis were able to stop the exchanges. Two patients continue to need rituximab despite MMF. One patient was moved from MMF to cyclosporine and one to azathioprine due to lack of efficacy. Our experience shows that MMF is effective in certain subsets of patients with MG. It is well tolerated and relatively inexpensive. More studies are needed to identify the specific subset of population that would benefit from this medication. In addition, more consensus guidelines are needed from experts to assist with insurance coverage for this potentially useful medication.
PS2Group3-013 / #374BULBAR MYASTHENIA PRESENTING AS UNILATERAL PALATAL PALSY
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Veena Vasi1, Andrew Thompson1, Keith Trimble2, Sandya Tirupathi3
1General Paediatrics, Royal Belfast Hospital for Sick Children, Belfast, GB;
2Department Of Otolaryngology/head & Neck Surgery, Royal Belfast Hospital for Sick Children, Belfast, GB;
3Paediatric Neurology, Royal Belfast Hospital For Sick Children, Belfast, GB
Abstract: Backgound: Velopharyngeal insufficiency is an uncommon problem in children that typically manifests as hypernasal speech, dysphagia and nasal regurgitation. The causes of velopharyngeal insufficiency can be divided into structural, functional and dynamic impairment (congenital/acquired neurological deficit) . Myasthenia can present with only bulbar symptoms and some cases can be termed as isolated VPI. Objective: To raise awareness of the subtle presentation of bulbar myasthenia and suggest appropriate investigations in similar cases. Method: To report a case of Velopharyngeal insufficiency in a 7 year old boy who was diagnosed with antibody positive bulbar myasthenia gravis and to do a literature review of similar cases. Case: A 7 year old boy presented to general paediatrics with acute onset of slurred speech and swallowing difficulties ongoing for few weeks which may have worsened with tiredness. There was no infective focus, trauma, previous surgeries or any weakness. There were no abnormalities on systemic examination. Neurology was consulted. A detailed examination revealed dysarthria and a deviated uvula to the right leading to suspicion of left palatal palsy. A further detailed assessment was carried out by the ENT team and speech and language therapy. Flexible laryngoscopy revealed pooling of secretions in the hypopharynx and left palatal paralysis. Videofluoroscopy revealed difficulty in swallowing and poor pharyngeal contractions with increased risk of choking and aspiration. Perpetual speech evaluation revealed hypernasal speech with mild dysphagia. Routine bloods, biochemistry, ASOT and CRP were normal. Virology and bacteriology were negative to infections. CT and MRI Brain was carried out which were normal and showed no significant abnormality along the course of the vagus nerve to explain palatal paralysis. However, a small incidental intracystic lesion in the right thyroid lobe was found. Tensilon test was inconclusive. Anti-acetylcholine receptor antibodies were sent. The child was initially treated with a short course of steroids for isolated palatal palsy. Results: Tensilon test was inconclusive as patient was not cooperative and there was no clear improvement in speech. Anti-acetylcholine receptor antibodies were sent. They were reported to be high on two occasions at 0.48 and 0.56 (<0.45 nmol/l) confirming the diagnosis of bulbar myasthenia. The patient’s symptoms responded well to treatment with pyridostigmine. Conclusion: Literature search reveals similar cases being labelled as functional disorders or palatal insufficiency in speech therapy and cleft palate clinics without a more specific diagnosis. In spite of the fact that myasthenia gravis is usually mentioned in the differential diagnosis of velopharyngeal incompetence, it is rarely ruled out by specific studies. In the absence of a definitive aetiology in which a structural defect or functional abnormality does not explain a patient’s Velopharyngeal insufficiency, a neurological factor must be considered even if fatigability is questionable. The clinical picture of Myasthenia gravis may be subtle and limited to bulbar features and the diagnosis may remain elusive to physicians at the outset.
PS2Group3-014 / #334PREPURTAL MYASTHENIA_ IS THERE A POSTINFECTIOUS ENTITY?
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Sandya Tirupathi1, Clare Loughran2
1Paediatric Neurology, Royal Belfast Hospital for Sick Children, Belfast, GB;
2Department Of Neurology, Royal belfast Hospital for sSck Children, Belfast, GB
Abstract: Objective: Myasthenia gravis(MG) is an acquired autoimmune disorder of neuromuscular junction. Prepubertal children have a higher prevalence of ocular symptoms(OMG), bulbar symptoms, lower frequency of acetylcholine receptor antibodies(AChR), and higher probability of achieving remission. We aimed to define presenting features and outcomes in our cohort. Methods: We reviewed our prepubertal children with MG diagnosis. Results: Five children, Three males(M) and two females(F). The age of onset was 3(F1), 2.5(M1), 1.5(F2), and 10(M2) and 6 (M3) years. All patients were tensilon test positive. They had electrophysiology tests including the repetitive nerve stimulation(RNS) tests. All patients also had antibodies sent against AChR and Muscle specific Kinase(MuSK). Neuroimaging was negative in three who presented with acute ptosis and also in the fourth who presented with swallowing difficulties and a slurred speech. Three children(F1,M1,F2) presented acutely following viral infections which included Chicken pox, diarrhoeal illness and herpetic stomatitis respectively. F1 presented with ptosis and bilateral divergent squint and was initially diagnosed with internuclear opthalmoplegia. Months later was diagnosed with OMG following positive AChR antibodies. Ptosis has resolved with pyridostigmine but the opthalmoplegia partially persists. F2 and M1 presented with ptosis and generalised weakness(M1). RNS showed a decrement in both. They are antibody negative. The symptoms resolved with pyridostigmine in both. M2 presented over a few weeks with difficulty in swallow and slurred speech. He is antibody (AChR antibody) positive and has a refractory course with life threatening crises necessitating thymectomy, plasma exchange, steroids and rituximab. M3 presented with slurred speech and swallowing difficulties. There was no previous infection. Palatal insufficiency was noted with an isolated left palatal palsy picture. His antibodies (AChR antibodies) were positive. Symptom improvement was seen with initial immune therapy and further improvement after pyridostigmine therapy. Discussion: Could the relatively higher remission rates and lower levels of acetylcholine receptor antibodies seen in prepubertal MG be related to a postinfectious autoimmune phenomenom? Or is there a genetic predisposition which is triggered by infection? A high index of suspicion is necessary to make an early diagnosis.
PS2Group3-015 / #333ANTIGEN-SPECIFIC DEPLETION OF PLASMA CELLS IN MYASTHENIA GRAVIS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Andreas Pelz1, Qingyu Cheng2, Adriano Taddeo3, Andreas Radbruch2, Andreas Meisel4, Falk Hiepe5, Siegfried Kohler4
1Department Of Experimental Neurology, Charité University Medicine Berlin, Berlin, DE;
2German Rheumatism Research Center Berlin, Berlin, DE;
3Department Of Plastic And Hand Surgery, Inselspital, Universitätsspital Bern, Bern, CH;
4Department Of Neurology, Charité University Medicine Berlin, Berlin, DE;
5Department Of Rheumatology And Clinical Immunology, Charité University Medicine Berlin, Berlin, DE
Abstract: Myasthenia gravis (MG) is an autoimmune disease characterized by the presence of autoantibodies directed against components of the neuromuscular junction, predominantly (>80%) against the acetylcholine receptor (AChR). Clinically, patients suffer from muscle weakness and receive treatment with acetylcholinesterase inhibitors and immunosuppressive drugs. However, these fail to target the long-lived autoantibody-producing plasma cells (PCs), which reside as post-mitotic cells in survival niches in the bone marrow and are fueling disease pathogenesis. Their high protein turnover renders PCs vulnerable against proteasome inhibitors, such as bortezomib, but this treatment results in the unselective depletion of all PCs and leads to the loss of antibody-mediated protective immunity. Therefore, a specific treatment against pathogenic PCs in myasthenia gravis and other antibody-mediated autoimmune diseases is still lacking. We have developed a method to specifically deplete autoimmune PCs while sparing the protective PC population and are establishing this method in the mouse model of myasthenia gravis (EAMG). Our approach uses an affinity matrix, consisting of an antibody fragment against the PC marker Syndecan-1 (CD138), coupled to the AChR autoantigen. While all PCs capture the matrix via CD138, only autoreactive PCs secrete high concentrations of anti-AChR antibodies and label themselves with antibodies against the antigen of the affinity-matrix, i.e. AChR. Hence, only anti-AChR secreting PCs carry antibodies with exposed Fc domains on their surface, which render them vulnerable for complement dependent lysis, antibody dependent cellular cytotoxicity and/or phagocytosis, while the protective PCs remain unharmed. In first proof-of-concept experiments in vitro, we were able to selectively deplete anti-AChR PCs from spleen or bone marrow cultures of EAMG-mice, while unrelated PCs were not changed. We are currently translating this technique to treat EAMG mice in vivo and to see whether we can effectively deplete autoreactive PCs and ameliorate disease symptoms. In parallel, we are already working with samples from MG patients in order to enable a fast translation to humans.
PS2Group3-016 / #325CLINICAL FOLLOW-UP OF THE PREGNANCY IN MYASTHENIA GRAVIS PATIENTS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Renata D. Ducci, Paulo J. Lorenzoni, Claudia S.k. Kay, Lineu C. Werneck, Rosana H. Scola
Neuromuscular Service, Hospital de Clínicas - Universidade Federal do Paraná, Curitiba, BR
Abstract: OBJECTIVE: To analyze the outcome and impact of pregnancy in women with myasthenia gravis (MG). METHODS: Obstetric and clinical data were retrospectively analyzed before, during and after pregnancy. Predictors of outcome were studied. RESULTS: We included 35 pregnancies from 21 MG patients. In the course of MG symptoms in 30 pregnancies with live births, 50% deteriorated (mainly in the second trimester, p=0.028), 30% improved and 20% remained unchanged. The deterioration group had more frequent abnormal repetitive nerve stimulation (RNS) (p=0.028) and lower scores in myasthenia gravis composite (MGC) (p=0.045) before pregnancy. The improvement group was associated with higher MGC scores (p=0.012) before pregnancy. The no-change group was associated with longer duration of MG (p=0.026) and normal RNS (p=0.008) before pregnancy. In the second pregnancy, the course of MG was different from the previous in 65.3%. Obstetric complications were reported in 20 pregnancies, the most common was preterm premature rupture of membranes (PPROM) (25.8%) and the most severe were abortion (11.4%) and fetal death (2.9%). Abortions were statistically associated with azathioprine use (p=0.045). Most of the patients had caesarean section (66.7%). Spinal anaesthesia was performed in 73.3%. Transient neonatal myasthenia gravis occurred in 12.9% of live-born babies, no predictors were found. CONCLUSIONS: The severity and duration of MG, RNS and treatment influence MG and pregnancy. Pregnant MG patients have increasing rates of PPROM and caesarean delivery. Our data suggests that duration of MG, MGC and RNS before pregnancy could be useful to help to predict the course of MG during pregnancy.
PS2Group3-017 / #297CHOLINERGIC TRANSMISSION OF OUTER HAIR CELL IMPAIRED IN MYASTHENIA GRAVIS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Nam-Hee Kim1, Joong-Yang Cho2, Kyung Seok Park3
1Neurology, Dongguk University Ilsan Hospital, Goyangsi, KR;
2Neurology, Inje university Ilsan hospital, Goyangsi, KR;
3Neurology, Seoul National University Bundang Hospital, Seongnamsi, KR
Abstract: Introduction: Nicotinic acetylcholine receptors (nAChRs) are located on outer hair cell (OHC) which is source of otoacoustic emission (OAE), and can be inhibited by alpha-bungarotoxin like muscular nAChR in myasthenia gravis (MG). The classical techniques such as repetitive stimulation, autoantibody, and response to ACh esterase inhibitor (AChEI) are sometimes not helpful for diagnosis of MG. The purpose of this study is to evaluate OAE for the possible role in diagnosis of MG. Methods: We performed transient evoked otoacoustic emissions (TEOAEs) and distortion product otoacoustic emissions (DPOAEs) on 30 ears of 15 MG with normal hearing, and on 20 ears of 10 controls. Results: Mean age was not different. TEOAE were significantly lower in MG (3.41 dB SPL) than in controls (8.69 dB SPL) (p < 0.01). DPOAE were lower in MG at higher frequencies between 2,026 and 4,053 Hz (p < 0.01). TEOAE and DPOAE were significantly lower in repetitive stimulation positive group and in AChR antibody positive group. TEOAE and DPOAE were correlated with titers of antibody. Conclusions: The decrease of OAE in MG is probably related to the reduced cholinergic transmission at OHC level. This study supports the role of ACh in the efferent function of OHC, as well as the impaired AChRs on OHC in MG. Furthermore, more reduced OAE in repetitive stimulation positive or AChR antibody positive groups with the correlation between OAE and antibody titer suggests impaired OHC function by AChR antibodies in MG. Consequently, measuring TEOAEs and DPOAEs may be useful in the diagnosis of MG.
PS2Group3-018 / #252RITUXIMAB FOR THE TREATMENT OF PEDIATRIC AUTOIMMUNE NEUROMUSCULAR DISORDERS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Cam-Tu Emilie Nguyen1, Elie Haddad2, Guy D’Anjou2, Jean Mathieu3, Michel Vanasse2
1Pediatrics (pediatric Neurology), London Health Sciences Centre, London, ON, CA;
2CHU Sainte-Justine, Montreal, QC, CA;
3Université de Sherbrooke, GRIMN and Jonquière Neuromuscular Clinic, Jonquiere, QC, CA
Abstract: Background: Myasthenia gravis (MG) and chronic inflammatory demyelinating polyneuropathy (CIDP) are chronic autoimmune neuromuscular disorders which can affect both adult and children. Occasionally, steroids, intravenous immunoglobulins and plasma exchange fail to control disease activity or cause serious side effects. There is currently very little data on the use of newer steroid-sparing immunomodulatory agents, such as Rituximab, in children. Objective: To describe our single-center experience with the use of Rituximab (RTX) in the treatment of pediatric patients with MG or CIDP, including the long-term clinical follow-up of these patients. Methods: Retrospective chart review of all pediatric patients with MG or CIDP who received RTX at our institution since 2008. Clinical presentation, age at diagnosis, diagnostic investigations, serological profile, medications and hospitalizations before and after RTX treatment, time from diagnosis to RTX treatment, pre- and post-treatment modified Rankin scale (for all patients), pre-treatment Myasthenia Gravis Foundation of America (MGFA) class (for patients with MG), post-treatment MGFA postintervention status (for patients with MG), adverse effects of treatment and length of follow-up were recorded. Results: We identified four pediatric patients with MG and two with CIDP who received RTX. The mean age at diagnosis was 8 years (range: 2-14) and the average time between diagnosis and RTX initiation was 27 months (range: 8-43). The typical RTX dose given was 375 mg/m2/dose every week for 4 weeks or 750 mg/m2/dose every week for 2 weeks, with a maximum of 1000 mg/dose. The average length of follow-up was 37 months (range: 14-68). At the time of their last follow-up, four out of six patients (66.7%) had shown improvement compared to their baseline clinical status, as measured on the modified Rankin scale. Two patients, both of whom had MG, were clinically unchanged. However, no patient achieved complete remission and all six patients were still on at least one immunomodulatory treatment. Three out of the four patients who showed improvement were able to significantly decrease their other immunomodulatory medications. No adverse effect of RTX was noted. Discussion: In four out of six patients in our cohort, RTX therapy resulted in improvement of their clinical condition, but did not lead to complete remission. These four patients achieved sustained clinical improvement after a single course of RTX, whereas the two other patients had no significant change in their clinical status after several courses of RTX. Conclusion: A single course of RTX can result in significant and sustained clinical improvement for some pediatric patients with MG or CIDP, but larger studies are required to determine which patients would benefit most.
PS2Group3-019 / #269EPIDEMIOLOGICAL AND CLINICAL CHARACTERISTICS OF ELDERLY MG
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Jiwon Yang1, Byung-Nam Yoon2, Seol-Hee Baek3, Jung-Joon Sung4
1Department Of Neurology, Gachon University, Gil Medical Center, Incheon, KR;
2Neurology, InHa University Hospital, Incheon, KR;
3Department Of Neurology, Seoul National University Hospital, Seoul, KR;
4Neurology, Seoul National University Hospital, Seoul, KR
Abstract: Background Myasthenia gravis (MG) is the most common neuromuscular junction disorder associated with autoimmune mechanism. It has been known to be prevalent in young adult and female, however, the increase is more found in the elderly patients at age over 50 years now than before. We aimed to identify the epidemiological and clinical characteristics of elderly MG. Methods We reviewed medical records of who admitted to the Gachon Medical Center for management of myasthenia gravis between Jan 2009 and Dec 2013. Total 46 patients were included in this study. We analyzed the clinical characteristics according to the age of onset >50 and ≤50 years: type of onset (ocular or generalized), sex, thymic pathology, interval time to generalize, initial acetylcholine-receptor (AchR) autoantibody titer, associated autoimmune disease, clinical course and other clinical features. Results Of the 46 patients, 28 (61%) were young-MG (age of onset ≤50 years) and 18 (39%) were elderly-MG (age of onset >50 years). The mean age of onset was 37.7±10.5 years in the young-MG and 63.7±12.2 years in the elderly-MG. The proportion of female (50.0% vs. 83.3%, p = 0.030), ocular-type onset (60.7% vs. 88.9%, p = 0.049) and its mean concentration of AchR autoantibody (2.6 vs. 6.3 nmol/L, p = 0.016), normal thymus (32.1% vs. 61.1%, p = 0.072) was significantly larger in the elderly-MG. The interval time to generalize from ocular-type onset (p = 0.915), the number of immunosuppressant agent (p = 0.157), and associated thyroid disease (p = 1.00) was not significantly different. Conclusion Unlike other previous study for describing the elderly-MG, our results showed that non-thymomatous MG was common among older patients with female predominance. The mean concentration of autoantibodies to acetylcholine receptor of ocular-type onset was higher in the elderly-MG than the young. It needs to be studied with larger populations to determine whether the non-thymic immune-mediated mechanism is present in the elderly-MG.
PS2Group3-020 / #239IMPACT OF REFRACTORY MYASTHENIA GRAVIS ON EMPLOYMENT
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Audra N. Boscoe1, Gary R. Cutter2, Haichang Xin2
1Health Outcomes Research, Alexion Pharmaceuticals, Lexington, MA, US;
2Univ. of Alabama-Birmingham, Birmingham, AL, US
Abstract: Background: Myasthenia gravis (MG) is a debilitating disease that can substantially interfere with patients’ activities of daily living, particularly among the subset of patients whose disease is refractory to conventional therapies. The impact of refractory MG on employment has not been heretofore evaluated. Objective: The objective of this analysis was to compare the employment status of patients with refractory MG to that of patients with non-refractory MG. Methods: This retrospective analysis of the enrollment data from the Myasthenia Gravis Foundation of America’s MG Patient Registry included U.S. residents aged ≥18 years who reported having been diagnosed with MG by their doctor ≥3 years prior to completing the enrollment survey. Respondents were classified as refractory if they had 1) past and/or current use of ≥3 of the following immunosuppressant therapies: prednisone, azathioprine, mycophenolate, mycophenolic acid, cyclosporine, tacrolimus, or methotrexate), OR 2) past and/or current use of ≥1 immunosuppressant therapy (listed above) AND use of ≥1 therapy typically reserved for disease resistant to conventional therapies: IVIg, cyclophosphamide, rituximab, or ≥4 rounds of plasmapheresis in the past year. Respondents who did not meet the criteria for refractory MG were classified as non-refractory. Antibody status was not factored into the analysis. Outcome measures included current employment status (full-time, part-time, not employed), status among those who were not employed (e.g. homemaker, student, disabled, etc.), whether or not their MG required them to cut back on work hours over the previous 6 months (among patients employed part- or full-time), and missed work days over the previous 6 months. Results: A total of 589 patients met inclusion criteria, 176 of whom (29.9%) were classified as refractory and 413 of whom (70.1%) were classified as non-refractory. Refractory patients were younger than non-refractory patients (mean age 52.3 years vs 56.0 years, respectively; p=0.005) and were more likely to be female (72.7% vs 64.2% for non-refractory patients; p=0.043). Refractory patients were significantly less likely to be employed full-time than non-refractory patients (21.0% vs 35.0%; p=0.001), and significantly more likely to be non-employed (68.8% vs 55.6%; p=0.003). Among the non-employed, refractory patients were significantly more likely than non-refractory patients to report being disabled (59.5% vs 34.1%, p<0.001). Non-employed, non-refractory patients were significantly more likely to be retired for reasons not related to their MG (39.3% vs 14.0% for non-employed, refractory patients, p<0.001), a difference that is likely attributable to the older age of the non-refractory group. Non-employed, refractory patients were more likely to report being retired due to MG (24.8% vs 19.2% for non-employed, non-refractory patients), but this difference did not reach statistical significance (p=0.118). Among patients who reported being predominantly employed full-time or part-time over the previous six months, there was no difference between groups with respect to reduced work hours due to MG or missed work days. Conclusions: Refractory MG patients are more likely to be of working age than non-refractory patients, yet many are non-employed, and a strikingly high percentage report being disabled. Having MG that is refractory to conventional therapies has a significant negative impact on employment.
PS2Group3-021 / #238IMPACT OF REFRACTORY MYASTHENIA GRAVIS ON QUALITY OF LIFE
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Audra N. Boscoe1, Gary R. Cutter2, Haichang Xin2
1Health Outcomes Research, Alexion Pharmaceuticals, Lexington, MA, US;
2Univ. of Alabama-Birmingham, Birmingham, AL, US
Abstract: Background: Myasthenia gravis (MG) is a debilitating disease that can substantially interfere with patients’ activities of daily living and negatively impact their quality of life (QOL). We hypothesized that the QOL of patients whose disease is refractory to conventional therapies is worse than that of patients with non-refractory MG. Objective: The objective of this analysis was to compare the QOL of patients with refractory MG to patients with non-refractory MG. Methods: This retrospective analysis of the enrollment data from the Myasthenia Gravis Foundation of America’s MG Patient Registry included U.S. residents aged ≥18 years who reported having been diagnosed with MG by their doctor ≥3 years prior to completing the enrollment survey. Respondents were classified as refractory if they had 1) past and/or current use of ≥3 of the following immunosuppressant therapies: prednisone, azathioprine, mycophenolate, mycophenolic acid, cyclosporine, tacrolimus, or methotrexate), OR 2) past and/or current use of ≥1 immunosuppressant therapy (listed above) AND use of ≥1 therapy typically reserved for disease resistant to conventional therapies: IVIg, cyclophosphamide, rituximab, or ≥4 rounds of plasmapheresis in the past year. Respondents who did not meet the criteria for refractory MG were classified as non-refractory. Antibody status was not taken into account because it was patient-reported. Although many respondents were able to report a value from their physicians, it was from unknown labs and frequently missing. Quality of life was assessed using the MG-QOL15, a validated questionnaire consisting of 15 questions with responses to each question scored from 0 (not at all) to 4 (quite a bit) and possible cumulative scores ranging from 0 to 60, with higher scores representing worse QOL. Results: A total of 589 patients met inclusion criteria, 176 of whom (29.9%) were classified as refractory and 413 of whom (70.1%) were classified as non-refractory. Refractory patients were younger than non-refractory patients (mean age 52.3 years vs 56.0 years, respectively; p=0.005) and were more likely to be female (72.7% vs 64.2% for non-refractory patients; p=0.043). Total MG-QOL15 scores were significantly higher (worse QOL) for refractory patients (24.2 vs 18.4 for non-refractory patients; p<0.001). Refractory patients had significantly higher (worse) scores than non-refractory patients on 11 of the 15 questions. Specifically, patients with refractory MG reported significantly greater difficulties with eating, personal grooming, speaking, and walking, more trouble meeting the needs of their family, greater limitations in their social activities, less ability to enjoy hobbies and fun activities, more trouble getting around public places, a more negative impact of MG on their occupational skills and job status, greater need to make plans around their MG, and more trouble with depression than non-refractory patients (p<0.05 for all). Refractory patients reported feeling more frustrated than non-refractory patients, although this did not achieve statistical significance (p=0.056). No significant differences were found between groups with respect to MG’s impact on vision or driving, or patients’ feeling overwhelmed by their MG. Conclusions: Patients with refractory MG have significantly worse quality of life than patients with non-refractory MG.
PS2Group3-022 / #146CLINICAL AND ECONOMIC BURDEN OF REFRACTORY GENERALIZED MYASTHENIA GRAVIS IN THE UNITED STATES
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Nicole M. Engel-Nitz1, Audra N. Boscoe2, Ryan Wolbeck1, Jonathan Johnson1, Nicholas Silvestri3
1Optum, Eden Prairie, MN, US;
2Health Outcomes Research, Alexion Pharmaceuticals, Lexington, MA, US;
3Neurology, SUNY at Buffalo Jacobs School of Medicine and Biomedical Sciences, Buffalo, NY, US
Abstract: Objective: To assess the burden of refractory generalized myasthenia gravis (MG) in terms of clinical burden, healthcare resource utilization, and costs by comparing patients with refractory versus non-refractory generalized MG and versus controls without MG. Methods:This retrospective analysis of a national administrative claims database included individuals aged ≥18 years with at least 24 months of continuous health plan enrollment between January 1, 2000, and December 31, 2014. Generalized MG patients had ≥2 medical claims with a primary ICD-9-CM diagnosis code for MG (358.0x) with neurology listed as the provider specialty. Refractory MG was defined as: current or past treatment with either 1) ≥3 immunosuppressant therapies (azathioprine, cyclosporine, mycophenolate, methotrexate, or oral corticosteroids), 2) ≥1 immunosuppressant therapy and ≥1 therapy typically reserved for disease resistant to conventional therapies (cyclophosphamide or rituximab), or 3) maintenance treatment with plasmapheresis. MG patients not identified as refractory constituted the non-refractory cohort. Controls consisted of age- and gender-matched individuals with no diagnosis codes for MG. A refractory date was identified as the first date at which a patient qualified as refractory based on the above criteria. For non-refractory and control patients, index dates were randomly selected to match the distribution of refractory dates in the refractory patients. Myasthenic crises, exacerbations, healthcare resource use, and costs were analyzed for the one-year period following the refractory/index date. Results: A total of 403 refractory patients, 3,811 non-refractory patients, and 403 non-MG controls were included in the analysis. Refractory patients were younger than non-refractory patients (56.5 years vs 59.7 years, p<0.001) and more likely to be female (57.6% vs 52.2%, p=0.04). Refractory patients were significantly more likely than non-refractory and control patients to be hospitalized during the one-year follow-up (mean 1.0 admissions for refractory patients vs 0.4 admissions for non-refractory patients and 0.2 for controls; p<0.001 for both). Refractory patients also had significantly longer lengths of hospital stay (mean 10.7 vs 3.7 days [non-refractory] and 1.7 days [controls], respectively; p<0.001 for both). Refractory patients also had significantly more emergency department and ambulatory visits than non-refractory patients and controls. Healthcare costs for refractory patients were more than 4 times higher than costs for non-refractory patients ($109,004 vs. $24,196, respectively, p<0.001) and almost 10 times higher than costs for controls ($11,582; p<0.001). Compared with non-refractory patients, more than three times as many refractory patients had a myasthenic crisis (21.3% vs. 6.1%, p<0.001) during the one-year follow-up, and 71.2% of refractory patients had an exacerbation compared to 32.4% of non-refractory patients (p<0.001). Conclusions: Refractory MG patients differ from non-refractory patients in ways beyond the higher number and types of therapies they have received. Healthcare resource utilization and costs are significantly higher for refractory MG patients than for non-refractory patients and non-MG controls. Although the literature suggests the majority of myasthenic crises tend to occur within two years of disease onset, the high rates of crises seen in our study suggest that refractory patients are at ongoing risk of crises and may even experience them at a higher rate than non-refractory patients.
PS2Group3-023 / #237MYASTHENIA GRAVIS INDUCED BY A MEK INHIBITOR: FIRST CASE REPORTED
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Majed Majed Alabdali1, Vera Bril2, Catherine Maurice3
1Neurology, Toronto General Hospital, Toronto, AB, CA;
2Toronto General Hospital, Toronto, CA;
3Neurology, Princess Margaret Hospital, Toronto, ON, CA
Abstract: Low-grade serous ovarian cancer, representing 10% of all ovarian cancer diagnoses, is renown for its poor response rate to chemotherapy, despite its indolent course. The MILO study is an international, randomized phase III trial comparing binimetinib, a targeted agent, to standard chemotherapy in patients diagnosed with low-grade serous ovarian cancer. This oral drug is designed to down-regulate cancer cell proliferation and survival via the inhibition of MEK-1 and MEK-2, involved in the RAS/RAF/MEK/ERK signal cascade. We present the case of a 77 year-old lady started on binimetinib in November 2014, in a clinical trial context. She developed head drop correlating with the administration of this drug. The clinical presentation, evolution of the symptoms and electromyography/single fibre results confirmed the diagnosis of myasthenia gravis (MG). Anti-acetylcholine receptor (AChR) antibodies were not detected. The patient improved after withdrawal of the MEK inhibitor, and the institution of Mestinon and Prednisone. MEK inhibitors have been reported to cause myopathy, but this is the first description of drug-induced MG arising from this treatment. MG is a rare autoimmune disease with an estimated prevalence of 10-20 per 100,000. The neuromuscular junction is attacked by autoantibodies directed against the nicotinic AChR, producing symptoms of fatigable muscle weakness. Certain drugs have been reported to induce or exacerbate MG including penicillamine, chloroquine and quinidine. MEK inhibitor treatment can be added to this list. In an era where targeted therapies may revolutionize the treatment of certain cancers and will be used more frequently, it is crucial to recognize their potential impact on the nervous system to understand, prevent and treat adequately their adverse events.
PS2Group3-024 / #225INCIDENCE AND PREVALENCE OF MYASTHENIA GRAVIS IN KOREA: A POPULATION-BASED STUDY USING THE NATIONAL HEALTH INSURANCE CLAIMS DATABASE
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Su-Yeon Park1, Jee-Eun Kim2, Jin Yong Lee3, Nam Gu Lim4, Yoon-Ho Hong5
1Department Of Neurology, Korea cancer center hospital, Seoul, KR;
2Department Of Neurology, Seoul Medical Center, Seoul, KR;
3Public Health Medical Service, Boramae Medical Center, Seoul, KR;
4Department Of Medical Administration, Daejeon Health Sciences College, Daejeon, KR;
5Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, KR
Abstract: Background and Purpose There have been a few national population-based epidemiological studies of myasthenia gravis (MG) with wide variation of incidence and prevalence rates worldwide. Herein we report the first nationwide population-based epidemiological study of MG in Korea. Methods We attempted to estimate the incidence and prevalence rates of MG using the Korean National Health Insurance claims database for 2010 to 2013. Cases with MG were defined as those having claim records with a principal diagnosis of MG and the prescription of acetylcholinesterase inhibitors or immunosuppressive agents including corticosteroids and azathioprine within 2 years after the diagnosis. The year 2010 was set as a washout period, such that patients were defined as incident cases if their first records of MG were observed in 2011. Results: In 2011 there were 1,236 incident cases, and the standardized incidence rate was 2.44 per 100,000 person-years. The standardized prevalence rates were 9.67 and 10.66 per 100,000 persons in 2010 and 2011, respectively. The incidence and prevalence rates peaked in the elderly population aged 60 to 69 years for both sexes. Conclusions This is one of the largest national population-based epidemiological studies of MG, and it has confirmed the high incidence and prevalence rates of MG in the elderly population of South Korea.
PS2Group3-025 / #123CLINICAL CHARACTERISTICS OF MUSK-MG IN KOREA: COMPARISON WITH DOUBLE SERONEGATIVE MG
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Kee Hong Park1, Jung-Joon Sung2, Suk-Won Ahn3, Byung-Nam Yoon4, Ji-Eun Kim5, Seol-Hee Baek6, So Hyun Ahn2, Yoon-Ho Hong7
1Neurology, Gyeongsang National University Hospital, Jinju, KR;
2Neurology, Seoul National University Hospital, Seoul, KR;
3Neurology, Chung-Ang University Hospital, Seoul, KR;
4Neurology, InHa University Hospital, Incheon, KR;
5Neurology, Seoul Medical Center, Seoul, KR;
6Department Of Neurology, Seoul National University Hospital, Seoul, KR;
7Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, KR
Abstract: Background & objective: Myasthenia gravis (MG) is an autoimmune disease caused by auto-antibodies against components of muscle membrane at the neuromuscular junction. Anti-acetylcholine receptor (AChR) antibody positive patients accounts for about 85% of generalized MG patients, while the seropositivity rate of MuSK antibody varies widely depending on the region and ethnicity. MuSK-MG is known to have different clinical manifestation and treatment response from AChR-MG. Here, we investigated the seropositivity rate of MuSK antibody and clinical characteristics of MuSK MG patients in Korea. Methods: Serum samples of 82 patients (75 MG, 2 LEMS, 4 motor neuron disease and 1 orbital pseudotumor) from 15 hospitals were collected and anti-MuSK antibody was assayed by commercial ELISA kit. Clinical characteristics of AChR-negative generalized MG patients were analyzed, comparing MuSK-positive and negative groups. Results: Among the MG patients, 60 patients were negative to AChR antibodies (8 ocular, and 52 generalized MG), and 15 were positive. Frequency of anti-MuSK antibody positivity was 37% (n=21) in AChR-seronegative generalized MG group, and none was positive to both antibodies. Noted was female predominance (71.4% vs. 41.9%), rare incidence of thymoma (0% vs. 6.5%), less frequent use of acetylcholinesterase inhibitors (28.6% vs. 67.7%), and more frequent crisis (33.3% vs 16.1%) in MuSK-positive compared to double-seronegative patients. There was no difference regarding onset age, bulbar predominance, symptom severity at onset, current symptom severity measured using MG composite score, and the rate of remission between groups. Conclusions: MuSK-MG accounts for 37% of AChR-seronegative MG patients in Korea. Demographic and clinical features of MuSK-MG in Korea are similar to those previously reported in Western countries. Future studies are warranted for serological diagnosis and further characterizing of double seronegative generalized MG.
PS2Group3-026 / #191LATE-ONSET MYASTHENIA GRAVIS: COMPARISON WITH EARLY-ONSET AND VERY LATE-ONSET MYASTHENIA GRAVIS
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Eun Bin Cho1, Ju-Hong Min2, Sujin Lee3, Cindy W. Yoon4, Hye-Jin Cho2, Jin Myoung Seok2, Hye Lim Lee2, Byoung Joon Kim2
1Neurology, Gyeongsang National University Changwon Hospital, Changwon, KR;
2Samsung Medical Center, Seoul, KR;
3International St. Mary’s Hospital, Incheon, KR;
4Inha University School of Medicine, Incheon, KR
Abstract: Purpose: To identify the clinical characteristics of patients with myasthenia gravis (MG) according to age at onset. Methods: We retrospectively recruited 227 non-thymomatous MG patients with adult onset who had been followed up for more than one year. The patients were classified based on the age of symptom onset as “early-onset MG” (EOMG, <50 years; N=135), “late-onset MG” (LOMG, 50–64 years; N=53), and “very late-onset MG” (VLOMG, ≥65 years; N=39). Clinical features and serological findings were compared between these groups.Results: LOMG patients showed more frequent ocular MG (55%) and less frequent thymic hyperplasia (9%) compared to EOMG patients (31% and 38%; p=0.006 and p<0.001, respectively), and no female preponderance compared to VLOMG patients (female, 49% vs.77%; p=0.014). However, there were no significant differences between VLOMG and EOMG patients, except less frequent thymic hyperplasia (p<0.001) in VLOMG patients. When analyzing female patients only, less frequent secondary generalization (10%) were additionally found in LOMG patients, compared to EOMG (47%, p= 0.008) and VLOMG (59%, p=0.004) patients. Anti-acetylcholine receptor antibody (HR, 5.48; 95% CI, 1.73–17.37; p=0.004) was independently associated with secondary generalization in female EOMG patients. Conclusion: Our study suggests that LOMG patients, especially female, were characterized by frequent ocular MG and less frequent secondary generalization, distinguished from EOMG and VLOMG patients. Further large epidemiologic studies in Korea are needed to determine the characteristics of MG patients according to the age at onset and gender.
PS2Group3-027 / #148DISEASES OF NEUROMUSCULAR JUNCTION, CLINICAL FEATURES, PATHOPHYSIOLOGY, THERAPY
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Varleen -. Kaur
-, -, patiala, IN
Abstract: TITLE:-Diseases of neuromuscular junction,clinical features,and pathophysiology. BACKROUND:-Myasthenia gravis(MG) is an autoimmune neuromuscular disease that leads to fluctuating muscle weakness and fatigue.In most of the cases muscle weakness is caused by circulating antibodies that block acetylcholine receptors at post synaptic neuromuscular junction,inhibhiting the excitatory efffects of the neurotransmitter acetylcholine on nicotinic receptors.Consequently 85% of individuals with Myasthenia gravis have variable deficits,such as fatigue and generalised weakness, ocular symptoms like ptosis and diplopia.Majority of patients have generalised weakness, in axial ,bulbar and limb muscles resulting in reduced balance,functional ability,strength and fitness resulting in deteoration of quality of life.Women mainly develop Myasthenia gravis at the age of 20-30 years, whereas Men tend to develop at the age of 50-70 years.The pathophysiology behind is the variation in certain genes.The antibody in myasthenia gravis attacks the normal human protein targeting protein called muscle-specific kinase.Human leukocyte antigen have been associated with myasthenia gravis receptibity. Objective:-To maintain balance,functional ability,strength and to improve quality of life. Method:-Six individuals with myasthenia gravis were included in a Balance Exercise(BE) training program for 5 weeks(4 times per week). Results:-Majority of cases showed sustained improvement without any noticeable adverse effect. Discussion:-Overall,balance exercise appears to be promising intervention to improve quality of life.But future reserach must involve large sample size with longer intervention period. Conclusion:-Balance exercise(BE) proves to be effective in improving balance,coordination,strength and functional ability in individuals.
PS2Group3-028 / #271FETAL AND ADULT ACETYLCHOLINE RECEPTOR FUNCTION IN MYASTHENIC SYNDROMES
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.2 Myasthenic Syndromes
Hakan Cetin, David Beeson, Richard Webster, Angela Vincent
Nuffield Department Of Clinical Neurosciences, University of Oxford, Oxford, GB
Abstract: Background: Myasthenic syndromes are characterised by muscle weakness and increased fatigability due to an impairment of neuromuscular transmission. Autoimmune or genetic factors compromise the function of muscle nicotinic acetylcholine receptors (AChR), which have two isoforms composed of five subunits in muscle (fetal AChR with gamma and adult AChR with epsilon subunits). The full significance of the developmental switch from fetal to adult AChR at late gestation remains elusive. Understanding the functional properties of both isoforms will provide greater insight into disease mechanisms. Methods: AChR whole-cell currents were measured by performing patch-clamping experiments on different cell lines. The rhabdomyosarcoma cell line TE671 expresses fetal AChR endogenously. CN21 cells derived from TE671 cells and have stably been transfected with the human epsilon-subunit, so that adult AChR predominate. HEK cells were transiently transfected with fetal or adult AChR subunits. Using a fast perfusion system, we performed application protocols consisting of two acetylcholine pulses: a desensitising pulse of 3s duration was followed by a 50ms test pulse. The interval between both pulses increased with each sweep until full recovery from desensitisation. Results: In both cell lines, desensitisation rates were similar between fetal and adult AChR (current decay time constant of 188 ± 22ms vs. 212 ± 29ms in HEK cells and 118 ± 7ms vs. 105 ± 8ms in TE671 and CN21 cells, respectively). Current decay was significantly faster in the muscle cell line compared to HEK cells. Interestingly, recovery from desensitisation was faster in adult receptors compared to fetal AChR (recovery time constant of 680 ± 106ms compared to 1363 ± 199ms in HEK cells (p=0.0044) and 864 ± 70ms compared to 2008 ± 296ms in the muscle cell line, p=0.0006). Recovery rates tended to be slower in the muscle cell line, but the difference failed to reach significance. Interpretation: Our findings provide important clues for the significance of the developmental switch from fetal to adult AChR around the time of birth. The expression of adult AChR with faster recovery rates from desensitisation could be a necessary adjustment of the neuromuscular junction to changing motor neuron firing patterns, the frequency of which increase around the time of birth. Adult AChR could thus facilitate the robust and reproducible high-frequent neuromuscular transmission in the adult. Our findings are relevant to patients with genetic or autoimmune myasthenic syndromes causing loss of adult AChR and where fetal AChR expression is sustained in mature muscle. Funding: This study was funded by the Austrian Science Fund (FWF) and Nuffield Department of Clinical Neurosciences.
PS2Group3-029 / #178MILD LIMB GIRDLE CONGENITAL MYASTHENIC SYNDROME CAUSED BY HOMOZYGOUS C.686-2A>G MUTATION IN GFPT-1
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.3 Congenital Myasthenia
Hacer Durmus1, Serdar Ceylaner2, Yesim Parman1, Feza Deymeer1, Piraye Oflazer-Serdaroglu1
1Neurology, Istanbul Faculty of Medicine, Istanbul, TR;
2Molecular Biology And Genetic, Intergen, Ankara, TR
Abstract: Glutamine-fructose-6-phosphate transaminase 1 (GFPT1) is the initial and rate-limiting enzyme in the biosynthesis of N-acetylglucosamine, an essential substrate for O- and N-glycosylation. A long muscle-specific isoform is encoded by GFPTL1 which is identical to GFAT1 except for a 54 bp insert at nucleotide position 686 of the coding sequence, resulting in a 16 or 18 amino acid insert at position 229 of the protein. Mutations that disrupt the function of this isoform are very rare. Mutation c.686-2A.G eliminated the first 4 nucleotides of the muscle-specific exon and resulted in 56 missense amino acids and a stop codon. Only one patient with very severe phenotype, carrying compound heterozygous mutation c.686-A>G and a missense mutation was published before. It was assumed that mutation c.686-A>G was responsible for severe disease phenotype. Here, we present two siblings from a consanguineous family with homozygous c.686-A>G mutation in GFPT1 and mild phenotype. Both patients presented with delayed motor milestones, difficulty with climbing stairs and easy fatigability at the age 13 and 8 years. Last neurologic examination of the older brother at the age 15 years revealed weakness in neck flexors and dominantly in proximal upper and lower extremities without any cranial involvement. Neurologic examination of the other sibling at age 10 revealed neck flexor and mild proximal weakness with independent ambulation. Both patients partially responded to pyridostigmine and salbutamol treatments. Creatine kinase level was mildly elevated in both, nerve conduction studies were normal, and needle electromyography showed mild myopathic pattern. Repetitive nerve stimulation (RNS) study at 3 Hz, showed over 10% decremental response. Muscle biopsy of the first sibling revealed myopathic changes, type 1 predominance and autophagic vacuoles; internal nuclei were abundant. GFPT1-myasthenia is heterogeneous, mutations that even hamper the function of muscle isoform GFPTL1 can cause a mild phenotype.
PS2Group3-030 / #290CONGENITAL MYASTHENIC SYNDROMES: REPORT ON 8 CASES FROM INDIA
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.3 Congenital Myasthenia
Atchayaram Nalini1, Veeramani Preethish-Kumar2, Kiran Polavarapu2, Seena Vengalil1, Xin-Ming Shen3, Andrew G. Engel3
1Neurology, National Institute of Mental Health and Neurosciences, Bangalore, IN;
2Clinical Neurosciences, National Institute of Mental Health and Neurosciences, Bangalore, IN;
3Neurology, Mayo Clinic, Minnesota, US
Abstract: Background: Congenital myasthenic syndromes (CMS) comprise a diverse group of inherited neuromuscular transmission disorders. Objective: To describe the clinical, electrophysiological and genetic pattern in 8 cases of CMS. Methods: The genomic DNA was isolated from peripheral blood and sequencing analysis was performed at the Mayo clinic, USA. Results: Mutations were identified in 8 patients. All eight were sporadic cases with onset in infancy or early childhood. Repetitive nerve stimulation revealed significant decrement and the Neostigmine test was positive in all. Patients 1 to 6 have homozygous mutations in the AChR epsilon subunit (CHRNE) predicted to cause endplate AChR deficiency. Patient 1 has a novel c.905C>T mutation (p.Pro302Arg) in the M1 and M2 linker and Patient 2 carries c.712C>T (p.Arg218Trp) mutation in the pre-M1 region of the extracellular domain. Patients 3 and 4 have frame shifting mutations: Patient 3 has five base pair (bp) duplication (c.183_187dup) in the extracellular domain; Patient 4 carries a twenty four bp deletion (c.1216_1219+20del24) in exon10 in the long cytoplasmic loop. Patients 5 and 6 were homozygous for (c.1267del G), a 1-bp deletion in exon 12 that has been previously reported in Roma gypsies and Indian/Pakistani patients. Patient 7 carries two heterozygous mutations in RAPSYN, the common N88K mutation and a c.272G>T (p.R91L) previously reported once. Patient 8 is homozygous for the missense mutation in c. 652G>A (p.Asp218Asn) in exon 5 of DOK7. This is a novel mutation that is likely to disturb normal splicing. Patients with mutations in CHRNE mutation cases had early onset ptosis with severe restriction of extra ocular movements and fatigable limb weakness. Patients with RAPSN and DOK7 mutations had early onset fatigable limb weakness and developed ptosis and diplopia later. Patients with CHRNE and RAPSYN showed sustained improvement with pyridostigmine. The patient with DOK7 mutation first showed mild improvement when treated with 30 mg pyridostigmine per day but then his weakness worsened. Salbutamol therapy has just been initiated. Conclusion: CMS are genetically mediated and potentially treatable disorders. Many patients have identifiable mutations but some are yet to be described. However, this clinical entity is commonly underdiagnosed and mislabelled. As response drug therapy varies depending on the type of mutation. Genetic analysis is essential for choice of appropriate therapy, for detection of novel mutations in known CMS, and for identifying novel CMS.
PS2Group3-031 / #497PERSISTENT DIFFUSE DEEP T WAVE INVERSION: AN ECG MANIFESTATION OF MYASTHENIA GRAVIS IN CRISISÂ
Topic: Group 3 – Diseases of Neuromuscular Junction: Clinical Features, Pathophysiology, Therapy / 3.1 Myasthenia Gravis
Jose Eduardo D.L. Duya, Michael Joseph Agbayani
Medicine, University of the Philippines Philippine General Hospital, Manila, PH
Abstract: Introduction: Myasthenia gravis(MG) is an autoimmune disorder directed against acetylcholine receptors. Despite the absence of these receptors in cardiomyocytes, ECG changes, tachyarrhythmias, myocarditis, and sudden death have been documented. We report 2 cases of MG presenting with deep diffuse persistent T wave inversions as a marker for possible MG related cardiac disease. Case 1: A 68 year old female, diagnosed with MG, post thymectomy for malignant thymoma in 2013, was admitted for progressive weakness, pleuritic chest pain and cough. ECG showed sinus rhythm, low voltage complexes on limb leads, poor R wave progression, prolonged QT interval, diffuse T wave inversion on all leads. Troponin I level was borderline elevated however, monitoring of troponin was negative. Echocardiography revealed concentric left ventricular hypertrophy with good contractility. ECG monitoring showed deepening diffuse symmetric T wave inversion. Due to the low CAD risk, this was interpreted as non-ischemic and was attributed to MG’s autoimmunity. Antibiotics, pyridostigmine, prednisone and plasmapheresis were given. She remained stable through out the course. Repeat ECG a month showed normalization of T wave inversion. Case 2: A 29 year old female was admitted for MG crisis. ECG revealed sinus tachycardia, with upright T waves. On Day 8, patient developed sepsis induced hypotension and repeat ECG showed 2mm ST elevation on V2-V3 with 3 mm T wave inversion on lateral leads. Serial ECG showed deepening of T wave inversion on V2-V6 (deepest 7 mm). The cardiac enzymes, echocardiogram and electrolytes were normal. With medical management, the patient was discharged improved. Conclusion: The dynamic ECG changes were attributed to possible immunologic myocarditis, which can present with deep T wave inversions. This case report highlights that clinicians should be aware that MG can present with this ECG feature, albeit seemingly alarming, usually follows a benign course and resolves with the resolution of MG crisis.
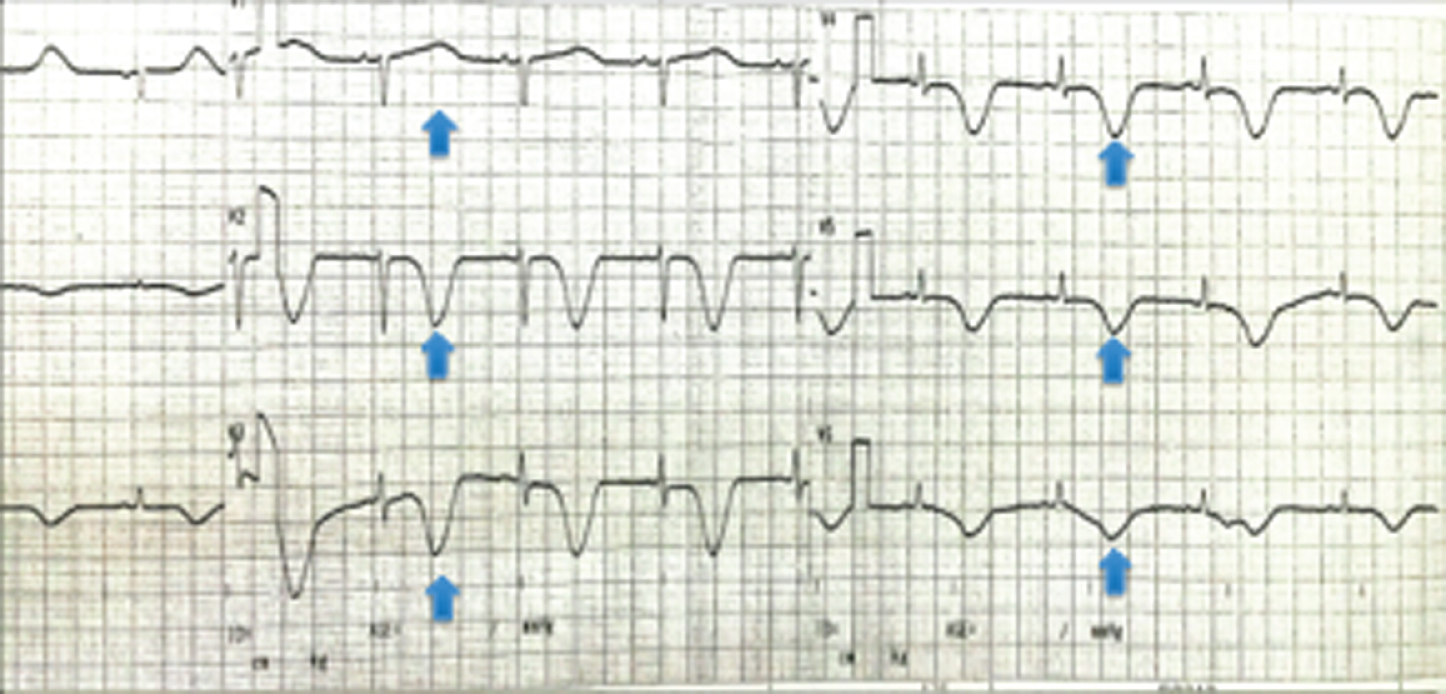
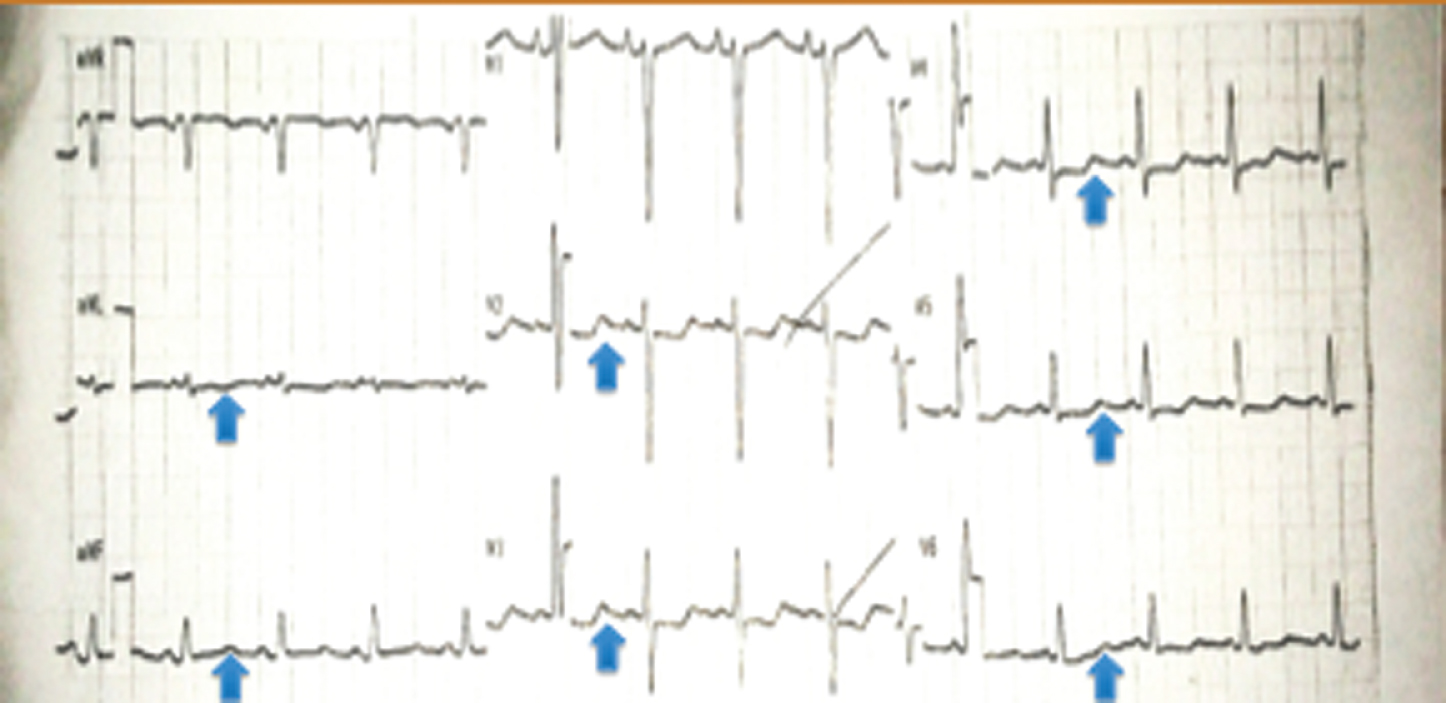
PS2Group5-001 / #337PRESERVATION OF MOTOR NEURON EXCITABILITY DURING THE CUTANEOUS SILENT PERIOD IN AMYOTROPHIC LATERAL SCLEROSIS
Topic: Group 5 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy / 5.1 Biology, Genetics
1Neurology, Kangdong Sacred Heart Hospital, Seoul, KR;
2Ewha Womans University Medical Center, Seoul, KR
Abstract: Objective: Electrical stimulation of a cutaneous nerve can transiently suppress electromyographic (EMG) activity in a voluntarily contracting muscle. This period of electrical silence, known as the cutaneous silent period (CSP), is usually evoked in upper limb muscles by stimulating cutaneous nerves in the digits. To determine motor neuron excitability in the patients with ALS, we examined F-waves in the abductor pollicis brevis (APB) muscle generated in response to antidromic stimuli during the CSP. Methods: Five patients with ALS (5 males; 43–68 years old) and 13 healthy subjects (6 males and 7 females; 34–70 years old) were studied. Result: The latencies of the onset and endpoint of the CSP in the normal subjects were 84.3 ± 10.9 and 122.4 ± 9.7 ms (mean ± 1 SD), respectively. These latencies were within the normal limits determined in earlier studies. In the patients with ALS, the onset was 103.9 ±14.2 ms and the endpoint was 137.6 ±22.1 ms, showing that the CSP was delayed. F-waves in the APB muscle were recorded with and without conditioned cutaneous nerve stimulation to produce CSPs in the patients with ALS. Of the 5 patients, F-waves were seen in four in the control condition. No F-waves were seen in one patient with marked APB muscle atrophy. Three out of the 4 patients generated F-waves in the test condition. The latencies of the F-waves in the test and control conditions were similar. Conclusion: Although additional data is needed to fully understand the CSP, the preservation of F-waves during the CSP appears to support the hypothesis that the motor neurons remain excitable in ALS patients.
PS2Group5-002 / #154PARKINSON DISEASE IS NOT ASSOCIATED WITH C9ORF72 REPEAT EXPANSIONS AND C19ORF12 MUTATIONS IN IRANIAN PATIENTS
Topic: Group 5 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy / 5.1 Biology, Genetics
Afagh Alavi1, Elahe Elahi2, Maryam Malakouti Nejad3, Gholamali Shahidi4
1Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran., Tehran, IR;
2School Of Biology, College Of Science, University of Tehran, Tehran, IR;
3Department Of Biotechnology, College Of Science, University of Tehran, Tehran, IR;
4Department Of Neurology, Hazrat Rasool Hospital, Iran University of Medical Sciences, Tehran, IR
Abstract: Expansion of the hexanucleotide repeat (HR) in intron 1 of the C9orf72 gene is the most common cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) in European countries. C9orf72 expansions have also been observed in some Parkinson’s disease (PD), Alzheimer disease (AD) and essential tremor patients and some other disorders. The mechanism by which the mutation causes disease is unknown. During a recent screening of the gene among 80 Iranian ALS patients, we observed the expansion mutation in C9orf72in a single family with three individuals affected with ALS, PD, or FTD. The HR was observed in all of three patients. Mutations in the C19orf12 are associated with a type of neurodegeneration with brain iron accumulation (NBIA); as mitochondrial membrane protein-associated neurodegeneration (MPAN) and have been linked to cases of hereditary spastic paraplegia type 43 (HSP-43), pallid-pyramidal syndrome (PPS), one Parkinson’s disease and two unrelated amyotrophic lateral sclerosis. Thus, the several diseases may cause by mutations in C19orf12gene. MPAN and PPS are associated with Parkinsonism and lewy body formation in substantia nigra. It suggests there are overlaps between pathogenesis of many neurodegenerative disorders. Identification of the new PD causing genes is very important and may provide new pathways and insights into disease pathogenesis and crucial for therapeutic development in future. On the other hand, Iranian population is heterogeneous and two of the PD genes, FBXO7 and SYNJ1 have been identified in Iranian patients. So, it seems the Iranian PD patients are appropriate for identifying of the other disease causing genes. Together, these findings prompted screening of C9orf72 expansions and C19orf12 in a cohort of 186 Iranian PD patients using a repeat primed (RP) and normal polymerase chain reaction (PCR) protocols, respectively. No pathogenic expansion in C9orf72 and disease causing mutation in C19orf12 were found in our cases. We conclude that abnormal C9orf72 HR and C19orf12 variants are not a major contributor of PD in Iranian patients.
PS2Group5-003 / #180MULTI-MODALITY, CERVICAL SPINAL CORD MRI IN ALS: A VALIDATION STUDY
Topic: Group 5 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy / 5.2 Biomarkers in MND
Barry J. Bedell1, Thanh Nguyen2, Vincent Auclair2, Julien Cohen-Adad3, Maxime Descoteaux4, Felix C. Morency5, Angela Genge6, Jim Paskavitz7, Guillaume Gilbert8, Donald G. Mclaren2, Alex P. Zijdenbos2
1Cns, Biospective Inc. & McGill University, Montreal, QC, CA;
2Biospective Inc., Montreal, QC, CA;
3Ecole Polytechnique & Functional Neuroimaging Unit, CRIUGM, Unviersite de Montreal, Montreal, QC, CA;
4Imeka Solutions and Unversite de Sherbrooke, Sherbrooke, QC, CA;
5Imeka Solutions, Sherbrooke, QC, CA;
6Montreal Neurological Institute, McGill University, Montreal, QC, CA;
7Biogen Inc., Cambridge, MA, US;
8Philips Healthcare Canada, Montreal, QC, CA
Abstract: Introduction: The development of disease-modifying therapeutic agents for amyotrophic lateral sclerosis (ALS) creates the need for objective, quantitative outcome measures that are sensitive to change in disease progression. Magnetic Resonance Imaging (MRI) is currently widely used in clinical trials of other neurological diseases, including Alzheimer’s disease and multiple sclerosis. Previous single-center studies indicate that multi-modality MRI may be sensitive to change in ALS (Cohen-Adad, 2013; El Mendili, 2014). In order to serve as a useful biomarker in multi-center clinical trials, the image acquisition and processing/analysis methods must be reliable and robust to differences in MRI scanners across sites. To this end, we have optimized an acquisition protocol and fully-automated processing pipeline for structural MRI, magnetization transfer contrast (MTC), and diffusion tensor imaging (DTI), and collected data from healthy volunteers and ALS subjects to assess the intra- and inter-site reliability. Materials & Methods: Healthy volunteer subjects (n=7; age = 33 ± 3 years) underwent MRI scanning on 3T Philips Ingenia and 3T Siemens TIM Trio systems located at two different sites to assess inter-site reliability. Each subject was scanned twice on the same day at each site to also assess intra-site reliability. ALS subjects (n=5; age = 51 ± 10 years) were scanned twice on the same day on the 3T Siemens scanner to compare intra-site reliability to that of the healthy volunteers. For each subject, 3D T2-weighted fast spin-echo (0.9×0.9×0.9 mm), 3D magnetization transfer (MT)-weighted (0.6×0.6×5.0 mm), and single-shot, reduced field-of-view DTI (1.0×1.0×5.0 mm) scans were obtained from the cervical spinal cord. Images were processed using a modified version of the Spinal Cord Toolbox (SCT) (Fonov, 2014; https://sourceforge.net/projects/spinalcordtoolbox), which was integrated into Biospective’s PIANO™ software. The output measures included: (1) spinal cord cross-sectional area (CSA), cerebrospinal fluid (CSF)-normalized MT signal (MT-CSF), and DTI measures. The intra- and inter-site test-retest reliability was assessed using the within-subject coefficient-of-variation. Results: CSA showed mean cord intra-site reliabilities of <1% in both healthy volunteers and ALS subjects, and inter-site reliability of <1% in healthy volunteers. MT-CSF showed intra-site reliabilities of <1.5% in healthy volunteers and ALS subjects, and inter-site reliability of <3.5% in healthy volunteers. Fractional anisotropy (FA) showed the best test-retest reliability amongst the DTI parameters with intra-site reliability of <2.5% for healthy and ALS subjects. Discussion: The primary objective of this validation study was to assess the intra- and inter-site test-retest variability of promising spinal cord MRI measures for use in multi-center clinical trials of ALS and other neurological diseases. The combination of a thoroughly-tested image acquisition protocol and fully-automated image processing software allowed us to obtain a high degree of reliability. Based on the improved understanding of methodological variability in the various modalities gained from this study, it should be possible to optimize clinical trial designs for the assessment of novel therapeutics. References: Cohen-Adad, J., et al., Amytroph. Lateral Scler. Frontotemporal Degener., 14: 30-38, 2013. El Mendili, M.-M., et al., PLoS ONE, 9(4): e95516, 2014. Fonov, V., et al., NeuroImage, 15: 817-827, 2014.
PS2Group5-004 / #20IMMUNOHISTOCHEMICAL STUDIES OF VALOCIN-CONTAINING PROTEIN IN THE SKIN OF PATIENTS WITH AMYOTROPHIC LATERAL SCLEROSIS
Topic: Group 5 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy / 5.2 Biomarkers in MND
Seiitsu Ono
Neurology, Teikyo University Chiba Medical Center, Ichihara, JP
Abstract: Background: So far studies of the skin of amyotrophic lateral sclerosis (ALS) have shown unique pathological and biochemical abnormalities in collagen, elastic fibers, and the ground substance. The lack of bedsore formation even in the terminal stages in ALS patients is considered characteristic. The valosin-containing protein (VCP) is one of the most evolutionarily conserved proteins that is ubiquitous and abundant in cells accounting for more than 1% of total cellular proteins. VCP was implicated in the pathogenesis of neurodegenerative diseases. Specifically VCP was found in the pathologic lesions in Alzheimer’s disease, Parkinson’s disease, ALS, and polyglutamine repeat diseases. It is unknown, however, whether VCP-positive (VCP+) structures are present in ALS skin. Objective: To make immunohistochemical studies of VCP in the skin of ALS patients. Methods: Skin biopsy specimens were taken from the left biceps from 20 sporadic ALS patients (61.0±9.4 years) and 20 control subjects with other neurologic disorders (62.3±9.8 years). Routine formalin-fixed paraffin-embedded 6 μm sections were immunostained according to standard techniques. VCP+ cells were labeled in each section in the epidermis. The percentages of VCP+ cells in the epidermis were calculated by dividing the number of VCP+ cells by the total cell count in the same region. A densitometroic analysis was performed using image analysis system. Total VCP content was obtained by delimiting the positive areas for immunolabeling on each VCP+ cell, determining their range of gray levels, and marking the areas to be analyzed. After that, they were measured and evaluated by an image analysis program. The integrated optical density was parameter tested and the amount of VCP for each patient was represented by the total of the different measurements taken. Results: Numerous VCP+ cells were observed in the epidermis in ALS patients, which became more marked as ALS progressed, and a small number of cells were seen in controls. VCP immunoreactivity of VCP+ cells was markedly positive in the epidermis and moderately positive in some dermal blood vessels and glands in ALS patients. These findings became more conspicuous as ALS progressed. On the other hand, VCP+ cells of the epidermis, dermal blood vessels and glands in control subjects showed a weak positive reaction even after repeated antigen-retrieval trials. The proportion of VCP+ cells in the epidermis in ALS patients (64.0±8.8%) was significantly higher (p<0.001) than in controls (16.6±16.0%). There was a significant positive relationship (r=0.59, p<0.01) between the proportion and duration of illness in ALS patients. The optical density of VCP+ cells in the epidermis in ALS patients (17.7±4.0) is markedly stronger (p<0.001) than in controls (12.4±4.2). There was a significant positive relation (r=0.61, p<0.01) between the immunoreactivity and duration of illness in ALS patients. Conclusion: The data suggest that changes of VCP in ALS skin are likely to be related to the disease process and that metabolic alterations of VCP may take place in the skin of patients with ALS.
PS2Group5-005 / #441LOWER MOTOR NEURONE DISEASE WITH BICLONAL IGM-PARAPROTEINAEMIA
Topic: Group 5 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy / 5.3 Epidemiology, Clinic, Treatment
Jochen Schaefer1, Heike Kostka2, Lisa Klingelhoefer2, Karsten Conrad2
1Neurology, Uniklinikum C.G.Carus Dresden, Dresden, DE;
2Uniklinikum C.G.Carus, Dresden, DE
Abstract: A 59-year old man developed proximal weakness, muscle atrophy and impairment of fine motor function of his right arm; one year later weakness of his left shoulder and hand ensued. There were no sensory deficits, thus amyotrophic lateral sclerosis was diagnosed. On examination, there was symmetric upper limb weakness and atrophy with proximal accentuation and general areflexia. Mild weakness of bilateral hip flexion was also detected. Upper motor neuron signs were not present. Nerve conduction studies revealed demyelination of both ulnar nerves without conduction block, EMG showed active denervation in the arms, legs and paravertebral muscles. Nerve ultrasound showed swelling of cervical nerve roots, median and ulnar nerve. Electrophoresis demonstrated biclonal IgM-gammopathy with high titres of anti-GM1 (1: 100,000) and anti-GD1a (1: 1000,000). We treated the patient with two courses of IVIG, which led to a reduction of the antibody titres, but no relevant clinical improvement. Therefore the patient has been receiving rituximab since with no further deterioration, but neither improvement, of his upper limb weakness. Clinically, the current case resembles brachial amyotrophic diplegia (flail arm syndrome). Differential diagnosis mainly comprises paraproteinaemic lower motor neuron syndrome (LMNS) and dysimmune (multifocal) motor neuropathy (MMN). The latter is supported by the presence of anti-GM1-IgM in very high titres and the finding of enlarged nerve roots on ultrasound, whilst symmetrical weakness, lack of conduction block and in particular the demonstration of paravertebral denervation rather suggest generalized LMNS. Also, the lack of response to IVIG favours LMNS rather than MMN, because more than 90% of MMN improve on IVIG. A puzzling aspect is the enlargement of cervical nerve roots and proximal parts of the peripheral nerves which indicates an inflammatory etiology rather than a degenerative process like generalized LMNS. Previous studies (Strigl-Pill, 2006; Simon, 2013) had indicated a poor response to IVIG for patients with symmetrical and proximal localization of motor axonal neuropathy, elevated CK and generalized EMG changes, independent of the presence of a conduction block. These prognostic parameters are reflected in our patient’s lack of response to IVIG despite a drop in anti-GM1 antibody titres after the first dose of IVIG. Nevertheless, Rituximab treatment seems to have stabilized, but not improved, the course of the disease.
PS2Group5-007 / #140FUNCTIONAL OUTCOMES IN HEREDITARY SPASTIC PARAPLEGIA: A PROSPECTIVE COHORT STUDY
Topic: Group 5 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy / 5.3 Epidemiology, Clinic, Treatment
Nicolas Chrestian1, Nicolas Dupré2, Ziv Gan-Or3, Jean Denis Brisson2, Anna Szuto4, Shiyi Chen4, Kym Boycott5, Jodi Warman-Chardon5, Sohnee Ahmed4, Peter Ray4, Oksana Suchowersky6, Guy Rouleau3, Grace Yoon7
1Département De Neuropédiatrie, Centre Hospitalier Université Laval, Quebec city, QC, CA;
2Hôpital de l,Enfant Jésus, Z, QC, CA;
3Montreal Neurological Institute, McGill, B, QC, CA;
4Sickkids, X, ON, CA;
5CHEO, L, ON, CA;
6University hospital of alberta, Edmonton, CA;
7Sickkids, X, CA
Abstract: Objective: To describe the clinical, genetic, and epidemiological features of HSP (Hereditary Spatic Paraplegia) in Canada, and to identify which clinical, radiologic and genetic factors determine functional outcome for patients with HSP. Background: Many subtypes of HSP have been reported worldwide, but prevalence, clinical subtypes, genetic characterization, and functional impacts of this group of disease remain poorly assessed in Canada. These informations are crucial in the diagnosis and management of affected patients. Methods: We conducted a multicenter prospective observational study of patients who met the clinical criteria for diagnosis of HSP in the provinces of Alberta, Ontario and Quebec. Standardized clinical evaluations were carried out at all study sites. The characteristics of the participants were analyzed using descriptive statistics. The main outcome measure was the spastic paraplegia rating scale (SPRS). We also used the SPATAX-EUROSPA disability stage score to assess disability. Results: A total of 534 patients were identified with HSP across the country and 158 patients had a confirmed genetic diagnosis. Mutations were identified in 15 different genes; the most common were SPAST (SPG4, 46%), ATL1 (SPG3A, 20%), KIAA1840 (SPG11, 8%), PGN (SPG7, 6%), KIAA0196 (SPG8, 5%) and PLP1 (SPG 2, 3%). Diagnosis of SPG4 and SPG7 were associated with older age at symptom onset. SPG4 and SPG3A were less associated with learning disabilities compared to other subtypes of HSP and SPG11 was strongly associated with progressive cognitive deficits. SPG3A was associated with better functional outcome compared to other HSP subtypes. The strongest predictor for significant disability was an abnormal brain MRI (p=0.04). Conclusion: The most important predictors of disability in our patients with HSP were SPG11 mutations and abnormal brain MRI. Accurate molecular characterization of well-phenotyped cohorts and international collaboration will be essential to establish the natural history of these rare degenerative disorders.
PS2Group5-008 / #310GENE THERAPY FOR SPINAL MUSCULAR ATROPHY TYPE 1 SHOWS POTENTIAL TO IMPROVE SURVIVAL AND MOTOR FUNCTIONAL OUTCOMES
Topic: Group 5 – Motor Neuron Diseases: Clinical Features, Pathophysiology, Therapy / 5.4 Spinal Muscular Atrophy / Neuronopathies
Samiah Al-Zaidy1, Richard Shell2, Doug M. Sproule3, William D. Arnold1, Louise Rodino-Klapac4, John T. Kissel1, Thomas W. Prior5, Carlos Miranda4, Linda Lowes6, Lindsay Alfano6, Katherine Berry6, Christopher Petek6, Kathleen Church6, Lyndsey Braun4, Jessica Cardenas3, Sarah Corcoran6, Kathrin Meyer4, Shibi Likhite4, Arthur H. Burghes7, Kevin D. Foust8, Brian K. Kaspar4, Jerry R. Mendell4
1Neurology, Nationwide Children’s Hospital, Columbus, OH, US;
2Pulmonary Medicine, Nationwide Children’s Hospital, Columbus, OH, US;
3Medical Affairs, AveXis, Inc, Bannockburn, IL, US;
4Center For Gene Therapy, Nationwide Children’s Hospital, Columbus, OH, US;
5Molecular Pathology, The Ohio State University, Columbus, OH, US;
6Nationwide Children’s Hospital, Columbus, OH, US;
7Molecular Genetics, The Ohio State University, Columbus, OH, US;
8Neuroscience, The Ohio State University, Columbus, OH, US
Abstract: Spinal muscular atrophy (SMA) is the most common genetic cause of infant death with mutations of survival motor neuron 1 gene affecting 1 in 10,000 live births. SMA manifests as symmetrical weakness of proximal extremity, axial, intercostal and bulbar muscles. Longitudinal studies in SMA type I with 2 SMN2 copies show progressive weakness with a need for continuous non-invasive ventilation or death by a median age of 10.5m. The change over time in CHOP INTEND (CI) score is -1.27 points/year for all SMA type I infants. In this ongoing gene therapy trial, the criteria for enrollment for the first 10 patients was as follows: onset of symptoms before 6mos, homozygous loss of the SMN1 gene, 2 copies of SMN2, and no c.859G>C exon 7 mutation. Two cohorts received intravenous AVXS-101(scAAV9.CB.SMN), Group 1 (n=3) 6.7x1013 vg/kg and Group 2 (n=7) 2.0x1014 vg/kg. The secondary endpoint for this safety study is death or need for >16 hours/day of noninvasive ventilation for at least 2 weeks. The minimal therapeutic dose cohort-1 had a mean age of 6.3±0.75m and a CI of 16.33±10.50 at baseline. Currently, this cohort is age 23.6±1.95m with a CI of 22.3±13.65. The therapeutic dose cohort-2 was stratified by age at gene-transfer. Group 2A (Pts 6 and 10) were treated at the earliest time points in the study 1.9m and 0.9m, respectively with CI of 47 and 50 at baseline. Currently, this group is age 12.2m and 5.8m; and has a normal CI of 64. Group 2B (Pts 4, 5, 7, 9) had a mean age of 4.6±0.86m with a CI of 29.25±3.69 at baseline. Currently, this group is age 14.5±3.44m with a CI of 52±2.45. Group 2C (Pt 8) was 7.9m with a CI of 12 at baseline. Currently, this patient is age 15.4m with a CI of 11. Electrophysiology studies showed increased compound motor action potentials associated with early treatment. It appears that an older age at enrollment (i.e. Pt 8 at 7.9m) with low CI results in stabilization with the therapeutic dose. Treatment-related serious adverse events in this trial were limited to transient liver enzyme elevations without clinical manifestations and correlated with high level IFN-g ELISpot assays to AAV9 capsid. This transaminasemia responded well to short-term prednisolone treatment. In summary, gene therapy for SMA type 1 appears to be neuroprotective allowing motor development to continue. At a higher CI score of 47-50 with an enrollment age of 1-2m, group 2A normalized to a score of 64. At a mid-level CI score with enrollment at age 4.6±0.86m, improvement for group 2B appears to be equivalent to the SMA Type 2 phenotype. This trial provides evidence that gene therapy may provide patient benefit when compared to natural history. Natural history would predict that CI would decrease by 1.27 points/year and event-free survival of 75% of SMA Type 1 patients at 13.6m, whereas 6 of 10 patients receiving gene therapy have passed the 13.6 month milestone. These results support the need for further studies of this promising therapeutic approach.
PS2Group7-001 / #440BIOENERGETIC IMPAIRMENT IN CONGENITAL MUSCULAR DYSTROPHY TYPE 1A AND LEIGH SYNDROME
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.1 Muscle Homeostasis / Muscle Regeneration
Cibely C. Fontes-Oliveira1, Maarten Steinz1, Peter Schneiderat2, Madeleine Durbeej1
1Experimental Medical Science - Unit Of Muscle Biology, Lund University, Lund, SE;
22friedrich-baur-institute, Department Of Neurology, Ludwig-Maximilians-University of Munich, Munich, DE
Abstract: Skeletal muscle is a tissue that requires high amounts of energy in order to perform its function. Consequently, disturbances in mitochondrial function are linked to pathological conditions that affect skeletal muscle. Congenital muscular dystrophy type 1A (MDC1A) is a severe form of muscular dystrophy caused by mutations in the gene encoding laminin α2 chain, an extracellular matrix protein. Leigh syndrome is a neurometabolic disease caused by mutations in genes related to mitochondrial function. Mutations in SURF1, one protein related to assembly and function of complex IV, also called cytochrome c oxidase or COX, is the most common cause of COX-deficient Leigh syndrome. In both diseases skeletal muscle tissue is severely affected and the common characteristics are muscle weakness which leads hypotonia and respiratory problems. Considering that mitochondria have a key role in bioenergetics and cell signaling, we have characterized the metabolic pattern in MDC1A and Leigh Syndrome. Skeletal muscle cells from MDC1A and Leigh patients and control subjects were utilized in this study. We performed expression analysis of several genes related to apoptosis, proteolysis and energy production. Mitochondrial and glycolytic functions were monitored in real-time. A dysregulated expression of genes related to mitochondrial biogenesis and glucose metabolism, muscle atrophy and apoptosis were observed in human MDC1A and Leigh myotubes compared to control cells. Using the Seahorse technology we observed impaired mitochondrial function and a compensatory upregulation of the glycolytic function in muscle cells from MDC1A and Leigh patients. We have for the first time demonstrated disturbances in bioenergetic state of muscle cells from MDC1A and Leigh Syndrome. Thus, metabolism can be a pharmacological target for these diseases.
PS2Group7-002 / #165TRANSFORMING GROWTH FACTOR-Î2 INHIBITS ADIPOGENESIS IN REGENERATING GLYCEROL-INJURED MUSCLE
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.1 Muscle Homeostasis / Muscle Regeneration
Mohamed A. Mahdy, Katsuhiko Warita, Yoshinao Hosaka
Anatomy, Tottori University, Tottori, JP
Abstract: Fibrosis and adipogenesis are characteristic features of several muscle diseases such as muscle dystrophies, inflammatory myopathies and sarcopenia, they negatively affect muscle function. Chemically-induced injuries provide good models to study the mechanism of muscular dystrophies and consequently to develop new therapies for treatment. Recently we have reported that glycerol injury induced muscle regeneration with adipogenesis and progressive deposition of intramuscular connective tissue in normal mice. However, the effect of transforming growth factor-β (TGF-β) on skeletal muscle regeneration and adipogenesis is unclear. Therefore, the aim of the present study was to investigate the effect of TGF-β on muscle regeneration and adipogenesis following glycerol injury. Mice were divided into three groups. The early treatment group was injected with TGF-β combined with glycerol. The late treatment group was injected with TGF-β at day 4 after glycerol injury. The control group was injected with glycerol only. Injections were performed into tibialis anterior muscles of adult mice. Muscle samples were collected at day 7 after glycerol injury. Early TGF-β treatment inhibited adipogenesis significantly while late treatment decreased adipogenesis. Moreover, muscle regeneration was impaired in TGF-β treated muscles compared to the glycerol-injured muscles. Furthermore, TGF-β reduced macrophages infiltration resulting in significantly larger necrotic area compared to glycerol-injured muscle. On other hand, TGF-β injection reduced mRNA expression of both myogenic and adipogenic factors compared to the glycerol-injured muscle. In conclusion, the inhibitory effect of TGF-β is much higher during early stages of muscle regeneration and adipogenesis.
PS2Group7-003 / #201ROLE OF INTERLEUKIN-6 ON UPREGULATION OF MYOSIN HEAVY CHAIN TYPE IIB MESSENGER RNA IN MOUSE MYOCYTES
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.2 Muscle Structure / Muscle Development / Muscle Growth
Junko Yamaji1, Yoshiaki Mori2, Reiko Hiroshima2, Ayako Miyazaki3
1Nutrition Science, Kansai University of Welfare Sciences, Kashiwara, JP;
2Rehabilitation sciences, Kansai University of Welfare Sciences, Kashiwara, JP;
3Clinical Laboratory Medicine, Hyogo College of Medicine, Nishinomiya, JP
Abstract: Backgroud Interleukin-6 (IL-6) is a cytokine from immune cells during infection or after trauma and is also known as a myokine, a cytokine from muscle, produced in skeletal muscle during muscle contraction (1). Our recent study indicated that IL-6- and/or calcineurin-mediated mechanism enhance expression of myosin heavy chain class I (MyHC I), IL-6 and heat shock protein (HSP) 70 mRNAs in C2C12 cells, murine skeletal muscle myoblast cell line, and the myotube differentiation (2, 3). In the present study, we examined the effects of IL-6 and calcineurin activators including La3+ (2, 3), chlorogenic acid (4), which is a plant polyphenol compound presented in coffee beans, potatoes and many other plants, or unsaturated fatty acids (5) including oleic acid and linoleic acid on expression levels of myosin heavy chain class IIb(MyHC IIb) isoform and IL-6 mRNAs in C2C12 cells. Methods C2C12 cells were maintained until semi-confluence in D-MEM containing 15%FBS and then differentiation from C2C12 myoblast cells to myotubes were initiated by medium exchange to D-MEM containing 2%FBS. C2C12 cells were incubated in D-MEM containing 2%FBS with or without agents, which are IL-6 and anti-IL-6 antibody as a IL-6 receptor antagonist, or chlorogenic acid, oleic acid and linoleic acid as calcineurin activators, and cyclosporine A as a calcineurin inhibitor, at the beginning of differentiation. After 24-hr incubation, the medium supplemented with agents was removed from cell culture dish and cells were maintained in differentiation medium for 3 days. The mRNA expression levels were measured by quantitative RT-PCR method using Taqman probes. Quantitative RT-PCR method was performed by a LightCycler Nano system (Roche Applied Science). MyHC mRNA expression level expressed as the cycle time (Ct) values normalized by the Ct of GAPDH as house keeping gene expression level. Results and Discussion First, we demonstrated the effect of IL-6 on MyHC IIb mRNA expression level in C2C12 cells. The MyHC IIb mRNA level was significantly upregulated by medium supplemented with IL-6. Second, we examined the effect of calcineurin activators, its inhibitor, or IL-6 antagonist. This mRNA expression level was increased by chlorogenic acid or by unsaturated fatty acids such as oleic acid and linoleic acid, but not by La3+. The MyHC IIb mRNA expression was decreased by anti-IL-6 receptor antibody or by cyclosporine A with calcineurin activator. Third, we examined the effect of calcineurin activators on IL-6 mRNA level. Then, the expression level of IL-6 mRNA were significantly upregulated by chlorogenic acid, oleic acid and linoleic acid. These results suggested that production of IL-6 induced by calcineurin activation might enhance MyHC IIbmRNA in C2C12 cells. COI:No References 1. Pedersen, B. K. & Febbraio, M. A. Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol. Rev. 88, 1379–1406, 2008. 2. Mori, Y. et al. Neuromuscular Disorders, 24 (9-10), 877-8, 2014 3. Yamaji, J. et al. Neuromuscular Disorders 24 (9-10), 804, 2014. 4. Tong, L. et al. IUBMB Life. 59 (6), 402-7, 2007. 5. Kessen, U. et al. J Biol Chem 31, 274 (53), 37821-6, 1999.
PS2Group7-004 / #241UPREGULATION OF MYOSIN HEAVY CHAIN TYPE I AND INTERLEUKIN-6 MESSENGER RNA LEVELS BY EXOGENOUS APPLICATION OF CALCINEURIN ACTIVATORS IN C2C12 SKELETAL MYOCYTES
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.2 Muscle Structure / Muscle Development / Muscle Growth
Yoshiaki Mori1, Junko Yamaji2, Reiko Hiroshima1, Ayako Miyazaki3
1Rehabilitation Sciences, Kansai University of Welfare Sciences, Kashiwara, JP;
2Nutritionsciences, Kansai University of Welfare Sciences, Kashiwara, JP;
3Clinical Laboratory Medicine, Hyogo College of Medicine, Nishinomiya, JP
Abstract: Introduction: Calcineurin is a protein phosphatase known as protein phosphatase 3 and calcium-dependent serine-threonine phosphatase. Calcineurin activates nuclear factor of activated T cell (NFAT) by dephosphorylating it, and the activated NFAT is translocated into the nucleus, where it upregulates the expression of interleukin-6 (IL-6) mRNA [1]. Our previous study using differentiated C2C12 cells indicated that myosin heavy chain type I (MyHC I) mRNA expression were significantly increased by the application of La3+ to the culture medium. The upregulation of the mRNA levels by the La3+ were abolished by the co-administration of cyclosporine A [2]. Thus, the effects of La3 on the mRNA levels are considered as a result of calcineurin activation. In the present study, we examined the effects of other calcineurin activators such as chlorogenic acid or unsaturated fatty acids on expression levels of MyHC I and IL-6 mRNA in C2C12 cells. Methods: C2C12 cells were induced to differentiate to myotubes by medium exchange to D-MEM containing 2%FBS. The cells were incubated in D-MEM containing 2%FBS with calcineurin activators, with or without cyclosporine A at the beginning of differentiation and removed after 24hr, and were maintained in differentiation medium for 3 days. MHC I, HSP 70, and IL-6 mRNA expression level in C2C12 cells were measured by the real-time PCR method. Results: First, we confirmed the effects of La3+, which was known to activate calcineurin Ca2+-independent manner [3], on MyHC I and IL-6 mRNA levels in C2C12 cells. The MyHC I and IL-6 mRNA levels were significantly increased by the application of La3+, but were decreased by the application of cyclosporine A with or without La3+, indicating that the effects of La3+ on these mRNA levels was mediated by the calcineurin activation. Second, we examined the effect of other calcineurin activators [4, 5] on MyHC I and IL-6 mRNA expression. The MyHC I mRNA level was significantly increased by the application of chlorogenic acid which is the ester of caffeic acid and (-)-quinic acid, but was decreased by the application of cyclosporine A with or without chlorogenic acid. The MyHC I mRNA level was significantly increased by the application of unsaturated fatty acid, oleic acid or linoleic acid. IL-6 mRNA level was also significantly increased by chlorogenic acid, oleic acid and linoleic acid. Conclusion These results indicate that the application of calcineurin activators upregulates MyHC I and IL-6 mRNA in a Ca2+-independent manner. Further experiments need to be done to clarify the effectiveness of exogenous calcineurin activators on the amount of skeletal muscle mass. References: 1. Allen DL, Uyenishi JJ, Cleary AS, Mehan RS, Lindsay SF, Reed JM. Am J Physiol Regul Integr Comp Physiol, 298(1):R198-210, 2010. 2. Mori Y, Yamaji J, Hiroshima R, Nakano T, Miyazaki A, Watanabe M. Neuromuscul Disord, 24(9-10):877-8, 2014 3. Hu J, Yang X, Wang K. J Biol Inorg Chem, 10(6):704-11, 2005. 4. Tong L, Song Y, Jia Z, Zhang W, Wei Q. IUBMB Life, 59(6):402-7, 2007. 5. Kessen U, Schaloske R, Aichem A, Mutzel R. J Biol Chem, 31;274(53):37821-6, 1999.
PS2Group7-005 / #254CYTOPLASMIC NOTCH AND MEMBRANAL BETA-CATENIN LINK CELL FATE CHOICE TO EMT DURING MYOGENESIS.
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.2 Muscle Structure / Muscle Development / Muscle Growth
Christophe Marcelle
Neuromyogene Institute, Inmg, University Lyon1, Lyon, FR
Abstract: How cells in the embryo coordinate epithelial plasticity with cell fate decision in a fast changing cellular environment is largely unknown. In chick embryos, skeletal muscle formation is initiated by migrating neural crest cells that, in passing, trigger myogenesis in selected epithelial somite progenitor cells, which rapidly translocate into the nascent muscle to differentiate. Here, we uncovered at the heart of this response a signalling module encompassing NOTCH, GSK-3b, SNAI1 and WNT. This module transduces the activation of NOTCH from neural crest cells into i) an inhibition of GSK-3b activity by non-transcriptional NOTCH signalling; ii) a SNAI1-induced epithelial to mesenchymal transition leading to iii) the recruitment of membranal b-catenin to trigger myogenesis independently of WNT ligand. Our results intimately associate the initiation of myogenesis to a change in cell adhesion and may reveal a general principle for coupling cell fate changes to EMT in many developmental and pathological processes.
PS2Group7-006 / #268FUNCTIONS OF THE SIL1-BIP CHAPERONE SYSTEM IN MAINTAINING MUSCLE FIBER INTEGRITY
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.3 Muscle Atrophy / Degeneration
Andreas Roos1, Laxmikanth Kollipara2, Stephan Buchkremer3, José Andrés González Coraspe3, Joachim Weis3, René Zahedi2
1John Walton Muscular Dystrophy Research Centre, Institute of Genetic Medicine, Newcastle University, Newcastle, GB;
2Bioanalytics, Protein Dynamics, Leibniz-Institut für Analytische Wissenschaften -ISAS- e.V., Dortmund, DE;
3University Hospital Rwth Aachen, Institute of Neuropathology, Aachen, DE
Abstract: The SIL1-BiP machinery represents a major chaperone-system of the Sarcoendoplasmic Reticulum (SR/ ER) and is involved in protein processing, modulation of cellular stress responses as well as of Ca2+ homeostasis. BiP functions in an ATP-dependent manner and thus depends on the co-chaperone function of SIL1 linking ATP to BiP. Prompted by the fact that the SR stress response plays a significant role in diverse muscular disorders, we hypothesized that the SIL1-BiP chaperone-system is essential in skeletal muscle cells to deal with stress and that disturbances in chaperone cycling lead to clinically relevant cellular perturbations. In order to address this question, we examined level of both proteins in stressed cells and diseased muscle. Apart from that, functional relevance of this chaperone-system was addressed by comprehensive investigation of SIL1/Sil1-deficient skeletal muscle: using immunoblotting and immunohistochemistry, we studied the level of SIL1 and BiP in stressed muscle cells and tissue as well as in de-innervated mouse muscles and in muscles derived from a mouse model of caveolinopathy. In addition, muscle biopsies of sporadic inclusion body myositis (sIBM) patients were investigated. In order to elucidate the functional role of this chaperone-system, we moreover examined muscle biopsies of Marinesco-Sjögren syndrome (MSS) patients with SIL1mutation and woozy mice lacking functional SIL1 using electron microscopy, immunoblotting, immunohistochemistry as well as label-free and iTRAQ-based proteome profiling. The investigation of SIL1 and BiP in stressed muscle cells and tissue as well as in muscle biopsy specimen of different (neuro)muscular disorders revealed increased level of both proteins. These findings underline the importance of the BiP chaperone system in muscle fiber integrity and the need to further elucidate its particular functions in this context. Doing so, ultrastructural examinations of SIL1-deficient muscle revealed marked dilatation of the SR, peculiar alterations of the myonuclear envelope with irregular proliferation of the nuclear lamina and buildup of autophagic vacuoles and severe mitochondrial changes. Morphological alterations could be correlated with the findings of our unbiased proteomic profiling as well as with the results of our targeted approaches focusing on protein degradation and thereby identifying substrates of the impaired SIL1-BiP machinery. To conclude, our comprehensive studies highlighted involvement of the SIL1/Sil1-BiP machinery in the etiopathology of (neuro)muscular disorders and moreover demonstrate the need of proper function of this machinery in muscle fiber maintenance especially with regard to defects in various aspects of BiP functions.
PS2Group7-007 / #387ANTI-INFLAMMATORY EFFECT OF ORAL-FORMULATED TACROLIMUS IN EXPERIMENTAL AUTOIMMUNE ENCEPHALOMYELITIS (EAE) MICE
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.6 Immune Mechanisms in Neuromuscular Diseases
Suk-Won Ahn1, Myung-Jin Kim2, Dae-Woong Kang3, Chang-Seop Kim4, Jung-Joon Sung5, Yoon-Ho Hong6
1Neurology, Chung-Ang University Hospital, seoul, KR;
2Chung-Ang University Hospiatl, seoul, KR;
3Chung-Ang University Hospital, Seoul, KR;
4Chung-Ang-University Hospital, seoul, KR;
5Department Of Neurology, Seoul National University Hospital, Seoul, KR;
6Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Center, Seoul, KR
Abstract: Introduction: It is well established that multiple sclerosis (MS) is a T-lymphocyte mediated autoimmune disease characterized by the mechanisms hypothesized to underlie CNS inflammation include T-cell autoimmunization, environmental factor, altered genetic influence, impaired vitamin D deficiency and combined autoimmune disease. However, the underlying mechanisms and complete treatment for MS remain uncertain. Although the many DMTs are presumed to be significantly effective compared to randomized control groups, the preventive efficacy for MS relapse is limited, and DMTs may induce serious adverse effects. Therefore, we tested the oral-formulated Tacrolimus (FK506; macrolide lactone immunosuppressant) for the MS using EAE mice model. Method: Total 64 mice with approximately 80 days were randomly divided into 3 experimental groups: an EAE mice group without treatment, an EAE mice group with 5mg/kg Tacrolimus treatment and an EAE mice group with 10mg/kg Tacrolimus treatment. For 14 days before sacrificing EAE mice, the original oral form of Tacrolimus was administrated 5 mg/kg or 10mg/kg.The control group with EAE mice was used for comparative studies. Clinical score, spinal cord statining and western blot were evaluated. Results: After immunization, EAE scores of each group are constantly increased as time goes by and comparing with each group shows significant difference in all days except 9 days. The more group taking large dose of Tacrolimus gets a lower score. Group of control EAE mice shows myelination in spine and inflammation in perivascular area. Result of taking Tacrolimus groups’ staining present decreased myelination and inflammation, and also group of 10mg/kg Tacrolimus EAE mice has rare myelination and inflammation than group of 5mg/kg Tacrolimus EAE mic. After 14 days of immunization, control EAE mice group and Tacrolimus EAE group are compared histological immunization using Western blot. Tacrolimus EAE mice group has weak band line of all immunized biomarker. Discussion: Our results revealed that tacrolimus plays a therapeutic role by inhibiting the activity of autoimmunization in MS pathogenesis. Furthermore, the therapeutic effect of tacrolimus may lead to an inactivation of the CD4 T cell pathway and decrement of inflammatory cells. In conclusion, the present study suggests that tacrolimus administration could be a promising neuroprotective strategy for treating MS.
PS2Group7-008 / #265MUSCLE ADAPTATIONS AND MOTOR DYSFUNCTION IN PERIPHERAL NEURODEGENERATIVE DISEASES MODELS
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.9 Others
Ole B. Nielsen1, Jeppe Bayley2, Martin Broch-Lips2, Anders Riisager2, Thomas H. Pedersen2
1Department Of Biomedicine, Aarhus University, Aarhus, DK;
2Aarhus University, Aarhus, DK
Abstract: Introduction and aim: Muscle atrophy in association with diseases involving progressive degeneration of peripheral nerves is often paralleled by failure of neuromuscular transmission and a transition of the affected muscles towards a slow-twitch and more fatigue resistant phenotype (Midrio. Eur J Appl Physiol 98: 1-21, 2006). Theoretically, transmission failure could include axonal damage of motor nerves causing compromised action potential propagation, reduced safety factor of neuromuscular transmission, and decreased excitability of muscle fibers. For most peripheral neurodegenerative diseases the specific involvement of these steps in the failure of neuromuscular signal transmission is, however, largely unknown. Likewise, the mechanisms leading to prolonged relaxation time following contractions remains elusive. Generally, muscle relaxation depends on re-uptake of calcium ions from the cytoplasm to the sarcoplasmic reticulum, which in turn depends on the transport activity of the sarcoplasmic calcium ATPases (SERCA) (Eshima et al. 56: 381-389, 2014). Based on this, we here examined the motor dysfunction and its mechanisms in mouse models of type II diabetes (the db/db mouse) and amyotrophic lateral sclerosis (the wobbler mouse). Methods: Experiments were conducted in accordance with institutional, national, and EU guidelines (Directive 2010/63/EU) for the care and use of laboratory animals, and were approved by Aarhus University. Contractile and electrical function was measured in nerve-muscle preparations of soleus and extensor digitorum longus muscles isolated from 6-week old wobbler and 8-week old db/db mice using various protocols for electrical stimulation. The preparations were incubated at optimal length for isometric contractions in standard bicarbonate Krebs-Ringer solution at 30 degrees Celsius during experiments. SERCA protein was quantified from the difference in calcium dependent and non-specific phosphorylation in crude muscle homogenates. Results: Compared to controls, the muscle-nerve preparations of both wobbler and db/db mice displayed muscle atrophy, reduced axonal excitability, and force deficit when stimulated via the motor nerve. In addition, muscles from wobbler mice exhibited reduced excitability of the muscle fibers and a reduced safety factor for neuro-muscular signal transmission as judged from a tolerance test to tubocurarine. In both disease models, muscle relaxation after contractions was slowed causing a left-shift in the force-frequency relation. Fiber type specific examinations of wobbler mice showed that this adaptation rather was related to ~40 % reduction SERCA content in all fiber types than to changes in myosin fiber type composition of the muscles. Conclusions: Force deficit in wobbler and db/db muscles was caused by atrophy and failure of the neuromuscular signal transmission that was related to axonal dysfunction of motor nerves and reduced muscle fiber excitability. The slowed muscle relaxation generally observed in peripheral neurodegenerative diseases was present in both animal models and could to a large extent be explained by decreased SERCA pump content, causing slower clearance of cytosolic calcium at end of contractions. Together, the results argue that muscle fiber adaptations contribute substantially to the motor dysfunction observed in degenerative diseases of motor nerves.
PS2Group7-009 / #245NORMAL MITOCHONDRIAL TRANSPLANTATION MAY BE USEFUL FOR THE TREATMENT OF MITOCHONDRIA-ASSOCIATED NEUROMUSCULAR DISEASES: EVIDENCES IN VITRO
Topic: Group 7 – Basic Sciences in Neuromuscular Diseases / 7.9 Others
Xianpeng Jiang1, Robert L. Elliott2, Jonathan F. Head2
1Research Division, Sallie A. Burdine Breast Foundation, Baton Rouge, LA, US;
2Sallie A. Burdine Breast Foundation, Baton Rouge, LA, US
Abstract: Accumulating evidences suggest that mitochondrial dysfunction or mitochondrial DNA deletion or mutation causes many neuromuscular diseases or symptoms such as amyotrophic lateral sclerosis, Leigh syndrome, etc. We recently published that transplantation of normal mitochondria into human breast cancer cells reversed cancer glycolytic metabolism. In the present study, we hypothesize that introduction of exogenous normal mitochondria into mitochondria-depleted neural cells might improve mitochondrial function and metabolism of neural cells. Mitochondrial DNA of mouse NSC-34 motor neuron is depleted by long-term culture in medium containing 400 ng/ml ethidium bromide. Mitochondria of parent NSC-34 are transferred into mitochondrial DNA-depleted NSC-34 cells (Rho0) by co-culture. Polymerase chain reaction (PCR) and real time PCR are used to examine mitochondrial DNA and mRNA expression. After 100 days culture with ethidium bromide, PCR showed no amplification of mitochondrially encoded cytochrome c oxidase I (MT-CO1) and mitochondrially encoded NADH dehydrogenase 1 (MT-ND1), but amplification of nuclear gene β-actin (ACTB). One day post-culture of NSC-34 (Rho0) with the isolated NSC-34 mitochondria, mitochondrial MT-CO1and MT-ND1 DNA genes are amplified. Moreover, MT-CO1 and MT-ND1 are still amplified at 3 and 6 days post-transplantation of mitochondria. Real time PCR showed that NSC34 (Rho) cells have very low levels of mRNA expression of mitochondrial genes MT-CO1 and MT-ND1 (approximately 10% of parent NSC-34 cells), but regained mRNA expression to normal levels of NSC-34 at 3 days post-mitochondrial transplantation. In addition, mRNA expression of hexokinase (HK2), lactate dehydrogenase A (LDHA) and glucose transporter 1(SLC2A1) are upregulated in NSC34 (Rho0) cells, but suppressed by the transplantation of NSC 34 mitochondria. These results suggest that the isolated NSC-34 mitochondria can easily enter into the mitochondrial DNA-depleted NSC 34 cells (Rho0) and effectively restore mitochondrial DNA depletion and gene expression. Mitochondrial organelle transplantation might be a potential therapy for mitochondria-associated neuromuscular diseases.
Author index
Aartsma-Rus, Annemieke PS1Group8-011 / #285, S110
Aban, Inmaculada B. PL 2.2 / #25 S4
Abraham, Alon PS1Group4-030 / #186, S86
Acevedo, Lorena PS2Group1-046 / #475, S165
Aciksoz, Semra PS1Group4-035 / #372, S89
Ackerl, Michael PS1Group4-006 / #349, S64
Adam, Berit PS2Group1-050 / #185, S167
Adams, David PL 2.7 / #29, S8
PS1Group4-010 / #219, S68
WS 3.4.1 / #63, S30
Agbayani, Michael Joseph PS2Group3-031 / #497, S203
Ahmed, Ausma PS1Group6-011 / #298, S97
Ahmed, Sohnee PS2Group5-007 / #140, S208
Ahn, Jin-Young PS1Group4-012 / #207, S71
Ahn, So Hyun PS1Group4-007 / #335, S65
PS1Group4-009 / #281, S67
PS2Group3-025 / #123, S200
Ahn, Suk-Won PS1Group4-032 / #462, S88
PS1Group4-036 / #164, S89
PS2Group3-025 / #123, S200
PS2Group7-007 / #387, S213
Aizawa, Tetushi PS1Group6-007 / #143, S94
Ajroud-Driss, Senda PS1Group8-005 / #163, S106
Akel, Semin PS2Group1-065 / #170, S179
Alabdali, Majed Majed PS1Group4-030 / #186, S86
PS2Group3-023 / #237, S199
Alavi, Afagh PS2Group5-002 / #154, S205
Albulaihe, Hana PS1Group4-030 / #186, S86
Al-Bustani, Najwa PS2Group1-021 / #381, S147
Alfano, Lindsay N. PS1Group2-009 / #362, S60
PS1Group8-004 / #353, S105
PS2Group1-006 / #369, S136
Alfano, Lindsay PS2Group5-008 / #310, S208
Alifano, Marco PS2Group1-009 / #283, S138
Aljaafari, Danah PS1Group4-030 / #186, S86
Allen, Jeffrey A. PS1Group4-002 / #449, S62
PS1Group8-005 / #163, S106
Almeida-Becerril, Tomas PS2Group1-008 / #377, S138
Alsulaiman, Abdulla PS1Group4-030 / #186, S86
Al-Zaidy, Samiah PS1Group2-009 / #362, S60
PS2Group5-008 / #310, S208
Amato, Anthony A. WS 4.2.1 / #68, S33
WS 7.3.2 / #99, S47
Amato, Anthony PS1Group2-004 / #393, S57
Amburgey, Kimberly WS 4.5.2 / #470, S37
Andersen, Grete PS2Group1-051 / #443, S168
Andersen, Linda K. PS1Group4-024 / #273, S80
Andrade, Renata A. PS2Group3-008 / #446, S189
Angelini, Corrado PS2Group1-031 / #347, S155
PS2Group1-060 / #358, S176
Antoine, Jean Christophe PS1Group4-010 / #219, S68
PS1Group8-013 / #227, S112
Appourchaux, Philippe PS1Group8-034 / #233, S127
PS1Group8-035 / #231, S129
Araoz, Maria Ines PS1Group4-019 / #434, S76
PS2Group3-009 / #448, S189
Arimura, Kimiyoshi PS1Group8-032 / #331, S126
Arnold, William D. PS2Group5-008 / #310, S208
Asakura, Masanori PS2Group1-020 / #224, S146
Atakli, Dilek PS2Group3-011 / #384, S191
Atalaia, Antonio PS2Group1-048 / #137, S166
Atassi, Nazem PL 2.1 / #24, S4
Atilano-Miguel, Salvador PS2Group1-008 / #377, S138
Attie, Kenneth M. PS2Group1-032 / #266, S156
Auclair, Vincent PS2Group5-003 / #180, S205
Avaria, María De Los Ángeles PS2Group1-026 / #478, S151
Ayoglu, Burcu PS1Group8-011 / #285, S110
Aysal, Fikret PS2Group3-011 / #384, S191
Azulay, Jean-Philippe PS1Group8-013 / #227, S112
Baek, Seol-Hee PS1Group4-007 / #335, S65
PS1Group4-009 / #281, S67
PS2Group3-019 / #269, S196
PS2Group3-025 / #123, S200
Bai, Johnny-Wei PS1Group8-015 / #301, S114
Balart, Mireya E. PS2Group3-004 / #476, S186
Ball, Laura J. PS1Group8-016 / #415, S115
Baranello, Giovanni PS2Group1-018 / #282, S144
Barnes, Aileen M. PS2Group1-042 / #295, S162
Barnett, Carolon PS1Group4-030 / #186, S86
Barnett-Tapia, Carolina PS1Group8-003 / #433, S104
PS1Group8-027 / #395, S123
Barohn, Richard J. PL 2.3 / #416, S5
PL 2.4 / #26, S6
WS 3.1.1 / #57, S28
WS 3.1.2 / #58, S29
WS 8.2.1 / #106, S50
WS 8.2.2 / #107, S51
Barohn, Richard LB 1.1 / #378, S15
PS2Group1-058 / #356, S173
Barresi, Rita PS2Group1-035 / #168, S158
Barrot, Claire-Cécile PS1Group6-017 / #274, S102
Bartoli, Marc PS2Group1-026 / #478, S151
Basak, Tulay PS1Group8-017 / #357, S115
Basran, Raveen WS 7.2.2 / #97, S46
Battini, Roberta PS2Group1-018 / #282, S144
Baudoin, Christian PS1Group6-015 / #263, S100
Baybas, Sevim PS2Group3-011 / #384, S191
Bayley, Jeppe PS2Group7-008 / #265, S214
Bazinet, Richard PS1Group4-037 / #171, S90
WS 5.3.2 / #79, S39
Bedell, Barry J. PS2Group5-003 / #180, S205
Bednarik, Josef PS1Group8-036 / #196, S130
Beek, Nadine A.m.e. Van Der PS2Group1-063 / #229, S178
Beeson, David PS2Group3-028 / #271, S201
Behin, Antony PS1Group6-015 / #263, S100
Bellevue, Ashley PS2Group1-032 / #266, S156
Bellgard, Matthew PS1Group8-021 / #439, S118
Bellis, Michela De PS2Group1-010 / #354, S139
Beltran, Sergi PS1Group8-021 / #439, S118
Benatar, Michael LB 1.1 / #378, S15
PL 2.3 / #416, S5
PL 4.1 / #33, S10
Berardinelli, Angela PS2Group1-018 / #282, S144
Berger, Melvin PS1Group8-005 / #163, S106
Berianu, Florentina PS1Group2-008 / #157, S59
Bernasconi, Pia PS2Group1-066 / #341, S180
Berner, Todd PS1Group2-003 / #405, S56
Bernert, Günther PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Béroud, Christophe PS1Group8-021 / #439, S118
Berry, Katherine M. PS1Group2-009 / #362, S60
PS1Group8-004 / #353, S105
PS2Group1-006 / #369, S136
Berry, Katherine PS2Group5-008 / #310, S208
Bertini, Enrico PS2Group1-018 / #282, S144
PS2Group1-043 / #270, S163
Besson, Gérard PS1Group8-013 / #227, S112
Bettini, Mariela PS1Group4-019 / #434, S76
PS2Group3-009 / #448, S189
Bettinson, Karen PS2Group1-030 / #355, S154
PS2Group1-033 / #292, S157
Betto, Romeo PS2Group1-029 / #363, S154
Bettolo, Chiara Marini PS2Group1-035 / #168, S158
Bevilacqua, Jorge A. PS2Group1-026 / #478, S151
PS2Group1-046 / #475, S165
Bevilacqua, Jorge PS1Group2-001 / #409, S55
Bhavaraju-Sanka, Ratna K. PS2Group3-012 / #329, S191
Bhunnoo, Anila PS2Group1-001 / #436, S132
Bianchini, Elisa PS2Group1-029 / #363, S154
Biglari, Habibeh Nejad PS1Group4-004 / #404, S63
Bishop, Courtney PS1Group6-003 / #422, S92
Blackmore, Derrick PS1Group8-019 / #464, S117
PS2Group3-005 / #460, S187
PS2Group3-006 / #459, S187
PS2Group3-007 / #458, S188
Bladen, Catherine L. PS2Group1-033 / #292, S157
Blain, Alison PS1Group8-011 / #285, S110
Blanchette, Chris M. PS1Group4-002 / #449, S62
Blavier, André PS1Group8-021 / #439, S118
Bleecker, Jan De PL 2.3 / #416, S5
Boes, Brian L. PS2Group1-032 / #266, S156
Boland, Anne PS1Group4-017 / #491, S75
Bolduc, Véronique PS2Group1-021 / #381, S147
Bonne, Gisèle PS1Group4-017 / #491, S75
Bonnemann, Carsten G. PS2Group1-041 / #414, S161
PS2Group1-042 / #295, S162
Bonnemann, Carsten PL 2.5 / #27, S7
WS 1.1.1 / #36, S19
WS 5.1.1 / #468, S38
Borgohain, Rupam PS2Group3-010 / #429, S190
Boscoe, Audra N. PS2Group3-020 / #239, S196
PS2Group3-021 / #238, S197
PS2Group3-022 / #146, S198
Bouchard, Jean-Pierre PS2Group1-021 / #381, S147
Bouhour, Francoise PS1Group6-015 / #263, S100
Boulet, Genevieve PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Boulis, Nicholas M. PL 2.1 / #24, S4
Boycott, Kym PS2Group5-007 / #140, S208
Bradbury, Margaret PS1Group6-014 / #235, S99
Brais, Bernard PS2Group1-021 / #381, S147
PS2Group1-045 / #492, S164
Brajkovic, Leposava PS2Group1-049 / #413, S167
Brannagan, Thomas H., Iii PS1Group8-005 / #163, S106
Braun, Lyndsey PS2Group5-008 / #310, S208
Breiner, Ari PS1Group4-030 / #186, S86
PS1Group6-012 / #248, S97
Brent, Michael H. PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Brevard, Julie PS1Group2-004 / #393, S57
Briand, Audrey PS1Group6-015 / #263, S100
Bril, Vera PS1Group4-001 / #461, S61
PS1Group4-030 / #186, S86
PS1Group6-009 / #293, S95
PS1Group6-010 / #300, S96
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
PS1Group8-003 / #433, S104
PS1Group8-015 / #301, S114
PS1Group8-027 / #395, S123
PS2Group3-004 / #476, S186
PS2Group3-023 / #237, S199
WS 5.3.2 / #79, S39
WS 5.5.1 / #82, S40
WS 6.3.2 / #89, S43
PS1Group4-037 / #171, S90
Brisson, Jean Denis PS2Group5-007 / #140, S208
Broch-Lips, Martin PS2Group7-008 / #265, S214
Brožková, Dana Šafka PS1Group4-020 / #343, S76
PS1Group6-013 / #294, S98
Brudno, Michael PS1Group8-021 / #439, S118
Brugnoni, Raffaella PS2Group1-066 / #341, S180
Brunner-Krainz, Michaela PS2Group1-057 / #417, S173
Bruno, Claudio PS2Group1-018 / #282, S144
Brusse, Esther PS2Group1-063 / #229, S178
Buch, Astrid Emilie PS1Group8-010 / #286, S109
PS2Group1-059 / #276, S175
PS2Group1-064 / #195, S179
Buchkremer, Stephan PS2Group7-006 / #268, S213
Buhour, Francoise PS1Group8-013 / #227, S112
Burghes, Arthur H. PS2Group5-008 / #310, S208
Burlot, Ludovic PS1Group8-034 / #233, S127
Burns, Anthony WS 2.1.2 / #47, S23
WS 2.1.3 / #48, S23
Burns, Ted M. PL 2.3 / #416, S5
Burns, Ted PS1Group8-005 / #163, S106
WS 5.5.2 / #83, S41
Burnyte, Birute PS1Group6-016 / #318, S101
Bushby, Kate PS2Group1-030 / #355, S154
PS2Group1-033 / #292, S157
PS2Group1-035 / #168, S158
Buttle, Sarah PS1Group8-038 / #364, S131
Buyse, Gunnar PS1Group8-001 / #426, S103
PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
PS1Group8-008 / #338, S108
Byrne, Barry PS2Group1-058 / #356, S173
Calderon, Felipe Peralta PS2Group3-009 / #448, S189
Cammish, Phillip PS2Group1-023 / #345, S149
Campbell, Craig PS1Group2-010 / #305, S61
PS1Group4-008 / #308, S66
PS1Group8-030 / #317, S124
PS1Group8-031 / #315, S125
PS2Group1-005 / #400, S135
WS 8.5.2 / #160, S52
Campion, Giles PS2Group1-007 / #385, S137
PS2Group1-011 / #340, S140
Cañada, Andrés PS1Group8-021 / #439, S118
Capogrosso, Roberta F. PS2Group1-010 / #354, S139
Cardas, Ruxanda PS1Group4-017 / #491, S75
Cardenas, Jessica PS2Group5-008 / #310, S208
Cardinez, Nancy PS1Group6-011 / #298, S97
Cardoso, Marcio PS1Group4-018 / #389, S75
PS2Group1-022 / #194, S148
Carotti, Marcello PS2Group1-029 / #363, S154
Carvalho, Mauricio PS1Group2-006 / #324, S58
Casar, Juan C. PS2Group1-024 / #326, S149
Cassereau, Julien PS1Group4-010 / #219, S68
Castiglioni, Claudia PS2Group1-026 / #478, S151
PS2Group1-046 / #475, S165
Catteruccia, Michela PS2Group1-018 / #282, S144
PS2Group1-043 / #270, S163
Caviedes, Pablo PS2Group1-026 / #478, S151
Cea, Gabriel PS2Group3-001 / #487, S185
Cerino, Mathieu PS2Group1-026 / #478, S151
Cetin, Hakan PS2Group3-028 / #271, S201
Ceylaner, Serdar PS2Group3-029 / #178, S202
Chaiton, Abraham PS1Group6-002 / #179, S91
Chandrasekaran, Krish PS1Group4-029 / #445, S86
Chao, Hua Chuan PS1Group4-015 / #200, S72
PS1Group4-025 / #259, S80
Chapon, Francoise PS1Group6-015 / #263, S100
Chastonay, Sabine De PS2Group1-041 / #414, S161
Chaves, Marcelo PS1Group4-019 / #434, S76
Chavez, Juan PS1Group6-005 / #302, S93
Chen, Chen PS1Group4-029 / #445, S86
Chen, Jinghan J. PS2Group1-019 / #242, S145
Chen, Shiyi PS2Group5-007 / #140, S208
Chen, Yi-An PS2Group1-070 / #253, S182
Cheng, Qingyu PS2Group3-015 / #333, S193
Cherney, David Z. PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Chettiath, Tobin PS1Group2-003 / #405, S56
Chmelikova, Magda PS1Group8-036 / #196, S130
Cho, Eun Bin PS2Group3-026 / #191, S200
Cho, Hye-Jin PS2Group3-026 / #191, S200
Cho, Joong-Yang PS2Group1-068 / #348, S181
PS2Group3-017 / #297, S194
Choi, Jae-Phil PS1Group4-012 / #207, S71
Choi, Kyomin PS1Group4-007 / #335, S65
PS1Group4-009 / #281, S67
Choi, Seok-Jin PS1Group4-007 / #335, S65
PS1Group4-009 / #281, S67
Chou, Cheng Ta PS1Group4-015 / #200, S72
Chrestian, Nicolas PS2Group5-007 / #140, S208
Christopher, Rita PS2Group1-061 / #319, S177
Church, Kathleen PS1Group2-009 / #362, S60
PS2Group5-008 / #310, S208
Cimbalistiene, Loreta PS1Group6-016 / #318, S101
Cisse, Ousmane Alfa PS1Group8-034 / #233, S127
Ciszewski, Lucasz PS2Group1-055 / #120, S171
Cocanougher, Benjamin T. PS2Group1-041 / #414, S161
Coelho, Teresa PS1Group4-018 / #389, S75
PS1Group4-021 / #193, S77
PS2Group1-022 / #194, S148
PS2Group1-067 / #371, S180
Coffey, Christopher LB 1.1 / #378, S15
Cohen-Adad, Julien PS2Group5-003 / #180, S205
WS 2.1.3 / #48, S23
WS 2.1.4 / #49, S24
Cohn, Ronald D. PL 3.3 / #32, S9
PS2Group1-042 / #295, S162
Collares, Daniel PS1Group2-006 / #324, S58
Colson, Serge PS2Group1-053 / #260, S170
Comi, Giacomo Pietro PS2Group1-018 / #282, S144
PS2Group1-066 / #341, S180
Conner, Ed PS2Group1-023 / #345, S149
Conrad, Karsten PS2Group5-005 / #441, S207
Conte, Talita C. PS2Group1-021 / #381, S147
Conte-Camerino, Diana PS2Group1-066 / #341, S180
Conwit, Robin LB 1.1 / #378, S15
Cook, Albert A. PS1Group8-005 / #163, S106
Cooper, Anneli PS2Group1-050 / #185, S167
Copenheaver, Deborah PS1Group6-014 / #235, S99
Coppard, Nick PS1Group8-001 / #426, S103
Coraspe, José Andrés González PS2Group1-034 / #264, S157
PS2Group7-006 / #268, S213
Corcia, Philippe PS1Group4-010 / #219, S68
Corcoran, Sarah PS2Group5-008 / #310, S208
Cornblath, David PS1Group4-001 / #461, S61
Costa, Elisa PS2Group1-013 / #314, S141
Courrier, Sebastien PS2Group1-026 / #478, S151
Couto, Jillian M. PS2Group1-050 / #185, S167
Couttet, F. PS1Group8-001 / #426, S103
Covaleski, Anna Paula P.M. PS1Group4-034 / #383, S88
Cozzoli, Anna PS2Group1-010 / #354, S139
Creange, Alain PS1Group8-013 / #227, S112
Cripe, Linda H. PS1Group8-004 / #353, S105
Cristiano, Edgardo PS1Group4-019 / #434, S76
PS2Group3-009 / #448, S189
Cristofari, Gael PS1Group6-015 / #263, S100
Cruz, Oriana D.R. PS2Group1-008 / #377, S138
Cudkowicz, Merit LB 1.1 / #378, S15
PL 2.1 / #24, S4
Cuisset, Jean-Marie PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Culm-Merdek, Kerry PS2Group1-058 / #356, S173
Cumming, Sarah PS2Group1-050 / #185, S167
Cutter, Gary R. PL 2.3 / #416, S5
PS2Group3-020 / #239, S196
PS2Group3-021 / #238, S197
PL 2.2 / #25, S4Dabilgou, Anselme PS2Group1-001 / #436, S132
Dahlqvist, Julia PS2Group1-051 / #443, S168
D’Amico, Adele PS2Group1-018 / #282, S144
PS2Group1-043 / #270, S163
PS2Group1-066 / #341, S180
Damme, Philip Van PS2Group1-058 / #356, S173
D’Angelo, Maria Grazia PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
PS2Group1-018 / #282, S144
D’Anjou, Guy PS2Group3-018 / #252, S195
Dashti, Mahjoubeh Jalali
Sefid PS2Group3-002 / #236, S185
Dastgir, Jahannaz PS2Group1-042 / #295, S162
Davis, Aileen PS1Group8-038 / #364, S131
WS 5.5.1 / #82, S40
Decker, Amy PS1Group6-014 / #235, S99
Decostre, Valérie PS2Group1-011 / #340, S140
Decoupade, Catherine PS1Group8-034 / #233, S127
PS1Group8-035 / #231, S129
Deikin, Alexey PS2Group1-014 / #304, S142
Delanoe, Catherine PS1Group4-017 / #491, S75
Deleuze, Jean-Francois PS1Group4-017 / #491, S75
Delmont, Emilen PS1Group8-014 / #226, S112
Denvir, Martin PS2Group1-050 / #185, S167
Derouault, Paco PS1Group6-017 / #274, S102
Desaphy, Jean Francois PS2Group1-066 / #341, S180
Descoteaux, Maxime PS2Group5-003 / #180, S205
Desnuelle, Claude PS1Group4-010 / #219, S68
PS1Group6-015 / #263, S100
PS1Group8-013 / #227, S112
PS1Group8-014 / #226, S112
PS2Group1-053 / #260, S170
PS2Group1-058 / #356, S173
Deymeer, Feza PS2Group3-029 / #178, S202
Díaz, Jorge PS2Group1-026 / #478, S151
PS2Group1-046 / #475, S165
Diaz-Manera, Jordi PS1Group6-004 / #419, S93
Dicaire, Marie-Josée PS2Group1-021 / #381, S147
Dickson, George PS2Group1-055 / #120, S171
Dilworth, Jeff PS1Group8-029 / #365, S123
Dimachkie, Mazen LB 1.1 / #378, S15
PL 2.6 / #28, S7
WS 7.3.1 / #98, S46
Dimitrieva, Tatiana PS2Group1-014 / #304, S142
Dinonno, Wendy PS2Group1-042 / #295, S162
Dirican, Ayten C. PS2Group3-011 / #384, S191
Dixon, Jeremy PS1Group8-030 / #317, S124
Dodig, Dubravka PS1Group6-006 / #328, S94
Doix, Aude C. PS2Group1-053 / #260, S170
Domingos, Joana Pisco PS1Group8-011 / #285, S110
Donisa-Dreghici, R. PS1Group8-001 / #426, S103
Donkervoort, Sandra PS2Group1-041 / #414, S161
PS2Group1-042 / #295, S162
Donoghue, C. PS2Group1-006 / #369, S136
Doorn, Pieter A. Van PS2Group1-063 / #229, S178
Doria, Valentina PS2Group1-043 / #270, S163
Dowling, James J. PL 1.3 / #23, S3
WS 4.5.1 / #75, S37
Dowling, James WS 4.5.2 / #470, S37
Drave, Alassane PS2Group1-001 / #436, S132
Duan, Dongsheng PL 3.1 / #30, S8
Dubbioso, Raffaele PS2Group1-066 / #341, S180
Dubé, Marie-Pierre PS2Group1-021 / #381, S147
Duboc, Denis PS2Group1-009 / #283, S138
Ducci, Renata D. PS1Group2-005 / #391, S58
PS1Group2-006 / #324, S58
PS2Group3-016 / #325, S194
Duchemin-Pelletier, Eve PS2Group1-027 / #401, S152
Duda, P. PS2Group1-006 / #369, S136
Dumonceaux, Julie PS2Group1-055 / #120, S171
Duncan, Alexis PS2Group1-050 / #185, S167
Duno, Morten PS2Group1-044 / #213, S163
Duong, Tina PS2Group1-011 / #340, S140
Dupré, Nicolas PS2Group5-007 / #140, S208
Durand-Zaleski, Isabelle PS1Group8-014 / #226, S112
Durbeej, Madeleine PS2Group7-001 / #440, S209
Durmus, Hacer PS2Group3-029 / #178, S202
Durn, Billie PS1Group4-001 / #461, S61
Duya, Jose Eduardo D.L. PS2Group3-031 / #497, S203
Dvorchik, Igor PS1Group2-009 / #362, S60
Dworzak, J. PS2Group1-006 / #369, S136
Dyck, P James B. PS1Group4-016 / #187, S74
WS 4.2.2 / #69, S33
Eagle, Michelle PS2Group1-011 / #340, S140
PS2Group1-030 / #355, S154
Elahi, Elahe PS2Group5-002 / #154, S205
Eldelekli, Devrim PS1Group8-015 / #301, S114
Elfring, Gary PS2Group1-005 / #400, S135
Elliott, Robert L. PS2Group7-009 / #245, S215
Encisa, Clarissa PS1Group8-038 / #364, S131
Engel, Andrew G. PS2Group3-030 / #290, S203
Engel-Nitz, Nicole M. PS2Group3-022 / #146, S198
England, John D. LB 1.2 / #500, S15
PS1Group2-002 / #406, S55
WS 5.4.1 / #80, S40
Ernst, Beat PS1Group4-013 / #206, S71
Evangelista, Teresinha PS2Group1-035 / #168, S158
Eymard, Bruno PS1Group6-015 / #263, S100
Fadia, Mithila M. PS2Group3-012 / #329, S191
Fadic, Ricardo PS1Group4-003 / #442, S63
PS2Group1-024 / #326, S149
Fajkusova, Lenka PS1Group8-036 / #196, S130
Falappa, Cesar M. PS1Group6-009 / #293, S95
Fanon, Laurie PS2Group1-009 / #283, S138
Farooqi, Mohammed A.M. PS1Group6-009 / #293, S95
PS1Group6-010 / #300, S96
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
PS1Group8-015 / #301, S114
Farrugia, Maria Elena PS2Group1-037 / #149, S159
PS2Group1-050 / #185, S167
Fattori, Fabiana PS2Group1-043 / #270, S163
Favejee, Marein M. PS2Group1-063 / #229, S178
Fecchio, Chiara PS2Group1-029 / #363, S154
Fehlings, Michael G. WS 2.1.1 / #46, S22
Fehlings, Michael WS 2.1.3 / #48, S23
Feldman, Eva L. PL 2.1 / #24, S4
Feng, Jia PS2Group1-054 / #192, S170
Ferlini, Alessandra PS2Group1-043 / #270, S163
Fernandes, J Americo PS1Group4-016 / #187, S74
Fernandes, Mathieu PS2Group1-027 / #401, S152
Fernández, Jose Maria PS1Group8-021 / #439, S118
Ferreira, Sara A. PS1Group8-009 / #432, S109
Findlay, Iain PS2Group1-050 / #185, S167
Finkel, Richard S. PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Flanigan, Kevin PS1Group2-009 / #362, S60
PS1Group8-004 / #353, S105
WS 1.4.2 / #43, S21
WS 3.3.2 / #62, S30
WS 8.5.1 / #112, S52
Flynn, Lauren PS2Group1-041 / #414, S161
Foley, A. R. PS2Group1-042 / #295, S162
PS2Group1-041 / #414, S161
Fonseca, Isabel PS1Group4-021 / #193, S77
Fontes-Oliveira, Cibely C. PS2Group7-001 / #440, S209
Fournier-Mehouas,
Manuella PS2Group1-053 / #260, S170
Foust, Kevin D. PS2Group5-008 / #310, S208
Freitas, Joel PS2Group1-067 / #371, S180
Frikke-Schmidt, Ruth PS1Group8-010 / #286, S109
Fu, Ssu-Ju PS2Group1-070 / #253, S182
Fuentealba, Mario PS1Group2-001 / #409, S55
PS1Group8-025 / #412, S121
Fuerst, Thomas PS1Group6-005 / #302, S93
Fujimura, Harutoshi PS2Group1-020 / #224, S146
Gagnon, Cynthia PS1Group8-030 / #317, S124
Gallano, Pia PS2Group1-046 / #475, S165
Gamieldien, Junaid PS2Group3-002 / #236, S185
Gan-Or, Ziv PS2Group5-007 / #140, S208
Garcia, Jeremy PS2Group1-053 / #260, S170
Gargaun, Elena PS1Group4-017 / #491, S75
Gartner, Michael R. PS2Group1-032 / #266, S156
Gauthier-Darnis, Marc PS1Group8-013 / #227, S112
PS1Group8-014 / #226, S112
Gayathri, Narayanappa PS2Group1-025 / #202, S150
PS2Group1-052 / #367, S169
PS2Group1-061 / #319, S177
Gejman, Roger PS2Group1-024 / #326, S149
Gelmont, David PS1Group2-002 / #406, S55
Geloven, Nan Van PS1Group4-001 / #461, S61
Genco, Nestor D. PS2Group3-009 / #448, S189
Genestet, Steve PS1Group4-010 / #219, S68
Genge, Angela PS2Group5-003 / #180, S205
Gibertini, Sara PS2Group1-031 / #347, S155
Gidaro, Teresa PS1Group4-017 / #491, S75
Gilbert, Guillaume PS2Group5-003 / #180, S205
Girdea, Marta PS1Group8-021 / #439, S118
Giustino, Arcangela PS2Group1-010 / #354, S139
Glass, Jonathan D. PL 2.1 / #24, S4
Glasser, Chad E. PS2Group1-032 / #266, S156
Goemans, Nathalie M. PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
PS1Group8-008 / #338, S108
PS2Group1-006 / #369, S136
PS2Group1-007 / #385, S137
PS2Group1-011 / #340, S140
PS2Group1-017 / #287, S144
Goker-Alpan, Ozlem PS2Group1-058 / #356, S174
Goldstein, Jonathan LB 1.1 / #378, S15
Gomes-Da-Silva, Monica M. PS1Group2-006 / #324, S58
Gonorazky, Hernan PS2Group3-009 / #448, S189
Gonzalez-Hormazabal,
Patricio PS2Group1-026 / #478, S151
González-Quereda, Lidia PS2Group1-046 / #475, S165
Goobie, Sharan PS1Group2-010 / #305, S61
Gorni, Ksenjia PS2Group1-018 / #282, S144
Gorokhova, Svetlana PS2Group1-026 / #478, S151
Gorson, Kenneth C. PS1Group8-005 / #163, S106
Goutman, Stephen A. PL 2.1 / #24, S4
Govoni, Alessandra PS2Group1-018 / #282, S144
Goyal, Namita PS2Group1-056 / #479, S172
Graham, Eileen PS2Group1-035 / #168, S158
Granlund, Lindsey PS1Group2-004 / #393, S57
Grecmalová, Dagmar PS1Group4-020 / #343, S76
Gregory, Helen PS2Group1-050 / #185, S167
Grisold, Anna WS 6.4.1 / #279, S43
Grisold, Wolfgang PS1Group4-006 / #349, S64
PS2Group1-057 / #417, S173
WS 6.4.1 / #279, S43
Groot, Imelda De PS2Group1-011 / #340, S140
Gruis, Kirsten PS1Group2-004 / #393, S57
Grynpas, Marc PS2Group1-019 / #242, S145
Gualandi, Francesca PS2Group1-043 / #270, S163
Guignat, Laurence PS2Group1-009 / #283, S138
Guptil, Jeffrey T. PS1Group4-002 / #449, S62
Gut, Ivo Glynne PS1Group8-021 / #439, S118
Haberlová, Jana PS1Group4-020 / #343, S76
Haddad, Elie PS2Group3-018 / #252, S195
Haenggi, Pascal PS1Group4-013 / #206, S71
Hafler, David LB 1.1 / #378, S15
Haller, Ronni WS 8.1.1 / #104, S50
Halpern, Elise M. PS1Group6-009 / #293, S95
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
PS1Group8-015 / #301, S114
Hamilton, Mark J. PS2Group1-050 / #185, S167
Hammans, Simon PS2Group1-048 / #137, S166
Hankin, Monty PS2Group1-032 / #266, S156
Harris, Elizabeth PS2Group1-033 / #292, S157
Hartung, Hans-Peter PS1Group4-001 / #461, S61
Hashiguchi, Akihiro PS1Group4-022 / #280, S78
PS1Group8-026 / #255, S122
Hathazi, Denisa PS2Group1-034 / #264, S157
Hauwe, Marleen Van Den PS1Group8-008 / #338, S108
PS2Group1-007 / #385, S137
PS2Group1-011 / #340, S140
Hayashi, Yukiko K. PS2Group1-004 / #407, S134
Head, Jonathan F. PS2Group7-009 / #245, S215
Heckmann, Jeannine M. PS2Group3-002 / #236, S185
Hegde, Madhuri R. PS1Group6-018 / #234, S102
Heidari, Morteza PS2Group1-002 / #471, S133
Heletea, Melissa PS1Group8-031 / #315, S125
Heo, Jae-Hyeok PS1Group4-012 / #207, S71
Hepple, Russell PS2Group1-021 / #381, S147
Herrendorff, Ruben PS1Group4-013 / #206, S71
Hershberger, Ray E. PS2Group1-028 / #366, S153
Hiepe, Falk PS2Group3-015 / #333, S193
Higuchi, Yujiro PS1Group4-022 / #280, S78
Hilsden, Heather PS2Group1-033 / #292, S157
Hilton-Jones, David PS2Group1-048 / #137, S166
Hiroshima, Reiko PS2Group7-003 / #201, S210
PS2Group7-004 / #241, S211
Hodge, Megan PS2Group1-054 / #192, S170
Hoey, Lynda PS1Group8-031 / #315, S125
Hogan, Mark J. PS1Group2-009 / #362, S60
Hong, Yoon-Ho PS1Group4-007 / #335, S65
PS1Group4-009 / #281, S67
PS1Group4-032 / #462, S88
PS1Group4-036 / #164, S89
PS2Group3-024 / #225, S199
PS2Group3-025 / #123, S200
PS2Group7-007 / #387, S213
Horobin, Joanna PS1Group2-004 / #393, S57
Hosaka, Yoshinao PS2Group7-002 / #165, S210
Hoskens, Jasmine PS1Group8-008 / #338, S108
Howard, James F. PL 2.3 / #416, S5
Hsiao, Cheng-Tsung PS1Group4-026 / #215, S82
Hu, Ying PS2Group1-042 / #295, S162
Hudson, Judith PS2Group1-025 / #202, S150
PS2Group1-035 / #168, S158
Huemer, Martina PS2Group1-057 / #417, S173
Hufschmitt, Anne PS1Group4-010 / #219, S68
Hughes, Richard PS1Group4-011 / #208, S69
PS1Group4-014 / #203, S72
Huh, So-Young PS1Group8-024 / #427, S120
Huq, Safawa LB 1.1 / #378, S15
Iannaccone, Susan WS 1.5.2 / #45, S22
WS 7.4.1 / #100, S47
Idiaquez, Juan PS1Group4-003 / #442, S63
Iglseder, Stephan PS2Group1-057 / #417, S173
Ilic, Vera PS2Group1-049 / #413, S167
Illa, Isabel PS1Group4-011 / #208, S69
WS 7.1.1 / #94, S45
Imbrici, Paola PS2Group1-066 / #341, S180
Inashkina, Inna PS1Group6-016 / #318, S101
Ishii, Akiko PS1Group6-007 / #143, S94
Ishii, Kazuhiro PS1Group6-007 / #143, S94
Iwata, Yuko PS2Group1-020 / #224, S146
Izenberg, Aaron WS 5.2.1 / #129, S38
Jabeen, S.A. PS2Group3-010 / #429, S190
Jackson, Carlayne PS2Group3-012 / #329, S191
Jackson, Sandra PS2Group1-047 / #181, S166
Jacob, Saiju PL 2.3 / #416, S5
Jain, Mina PS2Group1-041 / #414, S161
Jakobsen, Johannes PS1Group2-002 / #406, S55
James, Meredith PS2Group1-030 / #355, S154
PS2Group1-033 / #292, S157
Janiczek, Robert PS1Group6-003 / #422, S92
Januario, Alexandre M.S. PS2Group3-008 / #446, S189
Japp, Alan PS2Group1-050 / #185, S167
Jara, Lilian PS2Group1-026 / #478, S151
Jeanpierre, Marc PS1Group6-015 / #263, S100
Jiang, Xianpeng PS2Group7-009 / #245, S215
Jing Wang, Jing PL 2.3 / #416, S5
Johe, Karl PL 2.1 / #24, S4
Johnson, Jonathan PS2Group3-022 / #146, S198
Johnsson, Diana PS1Group8-011 / #285, S110
Johnston, Megan PS1Group8-029 / #365, S123
PS1Group8-030 / #317, S124
Jones, Kelvin PS1Group8-030 / #317, S124
Kabore, Jean PS2Group1-001 / #436, S132
Kafaie, Jafar PS1Group4-031 / #327, S87
Kafal, Ayman R. PS1Group8-002 / #463, S104
Kalina, Zdenek PS1Group4-020 / #343, S76
Kaliyaperumal, Rajaram PS1Group8-021 / #439, S118
Kalsi-Ryan, Sukhvinder WS 2.1.3 / #48, S23
Kaminski, Henry J. PL 2.2 / #25, S4
Kane, Marie-Louise PS1Group4-005 / #386, S64
Kang, Dae-Woong PS1Group4-032 / #462, S88
PS2Group7-007 / #387, S213
Kang, Sa-Yoon PS1Group8-024 / #427, S120
Kapral, Moira WS 5.5.1 / #82, S40
Karim, Rabail PS1Group8-023 / #437, S120
Kaspar, Brian K. PS1Group2-009 / #362, S60
PS2Group5-008 / #310, S208
Kassardjian, Charles PS1Group8-030 / #317, S124
PS2Group1-071 / #494, S184
Katz, Jon WS 4.1.2 / #67, S33
Katz, Sherri PS1Group8-031 / #315, S125
Katzberg, Hans PS1Group4-030 / #186, S86
PS1Group8-003 / #433, S104
PS1Group8-029 / #365, S123
WS 5.2.1 / #250, S38
WS 6.2.1 / #251, S41
Kaur, Varleen PS2Group3-027 / #148, S201
Kawai, Mitsuru PS1Group8-012 / #267, S111
PS2Group1-003 / #428, S133
Kay, Claudia S.k. PS1Group2-005 / #391, S58
PS1Group2-006 / #324, S58
PS2Group3-016 / #325, S194
Kaye, E. M. PS2Group1-006 / #369, S136
Keenan, Hillary A. PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Kelly, Dylan PS1Group6-009 / #293, S95
Khan, Aneal PS1Group8-030 / #317, S124
Kim, Bongje PS1Group4-007 / #335, S65
Kim, Byoung Joon PS2Group3-026 / #191, S200
Kim, Chang-Seop PS1Group4-032 / #462, S88
PS2Group7-007 / #387, S213
Kim, Dae-Seong PS1Group8-024 / #427, S120
Kim, Hye-Ok PS1Group4-012 / #207, S71
Kim, Jee-Eun PS1Group4-012 / #207, S71
PS2Group3-024 / #225, S199
Kim, Ji-Eun PS1Group4-036 / #164, S89
PS2Group3-025 / #123, S200
Kim, Jong-Kuk PS1Group8-024 / #427, S120
Kim, Jun-Soon PS1Group4-007 / #335, S65
PS1Group4-009 / #281, S67
Kim, Min-Ky PS1Group4-012 / #207, S71
Kim, Minsoo PS1Group4-031 / #327, S87
Kim, Myung-Jin PS1Group4-032 / #462, S88
PS2Group7-007 / #387, S213
Kim, Nam-Hee PS2Group1-068 / #348, S181
PS2Group3-017 / #297, S194
Kim, Raymond PS1Group8-027 / #395, S123
Kim, Sun-Young PS1Group8-024 / #427, S120
Kim, Wookyung PS2Group5-001 / #337, S204
Kimonis, Virginia PS2Group1-056 / #479, S172
Kimura, En PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Kissel, John T. PL 2.3 / #416, S5
PL 4.3 / #480, S10
PS1Group8-005 / #163, S106
PS2Group1-028 / #366, S153
PS2Group5-008 / #310, S208
WS 7.4.2 / #481, S48
Kissel, John LB 1.1 / #378, S15
Kleinsteuber, Karin PS2Group1-026 / #478, S151
Klingelhoefer, Lisa PS2Group5-005 / #441, S207
Klingels, Katrijn PS1Group8-008 / #338, S108
PS2Group1-011 / #340, S140
Knak, Kirsten L. PS1Group4-024 / #273, S80
Koga, Yasutoshi PS1Group6-007 / #143, S94
Kohler, Siegfried PS2Group3-015 / #333, S193
Koksal, Ayhan PS2Group3-011 / #384, S191
Kollipara, Laxmikanth PS2Group7-006 / #268, S213
Kollmer, Jennifer WS 2.2.1 / #50, S25
Komaki, Hirohumi PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Komarova, Marina I. PS2Group1-016 / #307, S143
Kongkham, Paul PS1Group8-027 / #395, S123
Korngut, Lawrence PS1Group8-029 / #365, S123
PS1Group8-030 / #317, S124
PS1Group8-031 / #315, S125
WS 1.4.1 / #42, S21
Kostka, Heike PS2Group5-005 / #441, S207
Krag, Thomas PS2Group1-062 / #316, S177
Krahn, Martin PS2Group1-026 / #478, S151
Kroger, Hans PS2Group1-005 / #400, S135
Kroksmark, Anna-Karin PS2Group1-007 / #385, S137
Kruijshaar, Michelle E. PS2Group1-063 / #229, S178
Krůtová, Marcela PS1Group4-020 / #343, S76
PS1Group6-013 / #294, S98
Kulkarni, Abhaya WS 5.5.1 / #82, S40
Kumar, R.V. PS2Group3-010 / #429, S190
Kuperus, Esther PS2Group1-063 / #229, S178
Kyelem, Julie M. A. PS2Group1-001 / #436, S132
Kyung Seok Park PS2Group3-017 / #297, S194
Lace, Baiba PS1Group6-016 / #318, S101
Lacour, Arnaud PS1Group4-010 / #219, S68
Ladha, Shafeeq PS2Group1-058 / #356, S173
Laforêt, Pascal PS1Group6-015 / #263, S100
PS2Group1-058 / #356, S173
PS2Group1-059 / #276, S175
PS2Group1-064 / #195, S179
Lagha, Nadira PS1Group6-015 / #263, S100
Lagler, Florian PS2Group1-057 / #417, S173
Lahaut, Pauline PS2Group1-053 / #260, S170
Laing, Nigel PS2Group1-021 / #381, S147
Lair, Séverine PS1Group8-021 / #439, S118
Lamont, Phillipa PS2Group1-021 / #381, S147
Lassuthova, Petra PS1Group4-020 / #343, S76
PS1Group6-013 / #294, S98
Lauria, Giuseppe WS 3.4.2 / #493, S31
Laurie, Steven PS1Group8-021 / #439, S118
Lawo, John-Philip PS1Group4-001 / #461, S61
Lawson, Margaret L. PS1Group8-031 / #315, S125
Leach, Meganne E. PS2Group1-042 / #295, S162
Lee, Elaine PS1Group6-018 / #234, S102
Lee, Hye Lim PS2Group3-026 / #191, S200
Lee, Jin Yong PS2Group3-024 / #225, S199
Lee, Sujin PS2Group3-026 / #191, S200
Lee, Yi-Chung PS1Group4-015 / #200, S72
PS1Group4-026 / #215, S82
PS1Group4-027 / #142, S84
PS1Group4-028 / #141, S85
Leger, Jean Marc PS1Group4-011 / #208, S69
PS1Group4-014 / #203, S72
WS 4.4.2 / #244, S36
Leikin, Sergey PS2Group1-042 / #295, S162
Leinonen, Mika PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Lemoing, Anne Gaelle PS1Group4-017 / #491, S75
Levy, Nicolas PS2Group1-026 / #478, S151
Lewis, Evan PS1Group4-037 / #171, S90
WS 5.3.2 / #79, S39
Lewis, Richard A. PS1Group4-001 / #461, S61
PS1Group8-005 / #163, S106
Lewis, S PS1Group2-009 / #362, S60
PS2Group1-006 / #369, S136
Lia, Anne-Sophie PS1Group6-017 / #274, S102
Liao, Yi-Chu PS1Group4-026 / #215, S82
PS1Group4-027 / #142, S84
PS1Group4-028 / #141, S85
Likhite, Shibi PS2Group5-008 / #310, S208
Lim, Nam Gu PS2Group3-024 / #225, S199
Lin, Chun-Keng PS1Group6-008 / #228, S95
Lin, Kon Ping PS1Group4-015 / #200, S72
PS1Group4-025 / #259, S80
PS1Group4-026 / #215, S82
PS1Group4-028 / #141, S85
Lin, Zhengning PS1Group6-003 / #422, S92
PS2Group1-007 / #385, S137
Lindsay, Sally PS1Group8-033 / #258, S127
Lindy, Amanda PS1Group6-014 / #235, S99
Lins, Otávio G. PS1Group4-034 / #383, S88
Liu, An-Bang PS1Group6-008 / #228, S95
Liu, Yo-Tsen PS1Group4-027 / #142, S84
PS1Group4-028 / #141, S85
Lochmüller, Hanns PS1Group8-011 / #285, S110
PS1Group8-021 / #439, S118
PS2Group1-023 / #345, S149
PS2Group1-025 / #202, S150
PS2Group1-034 / #264, S157
PS2Group1-048 / #137, S166
Longman, Cheryl PS2Group1-037 / #149, S159
PS2Group1-050 / #185, S167
Lorenzoni, Paulo J. PS1Group2-005 / #391, S58
PS1Group2-006 / #324, S58
PS2Group3-016 / #325, S194
Löscher, Wolfgang PS2Group1-057 / #417, S173
Loughran, Clare PS2Group3-014 / #334, S193
Lovblom, Leif E. PS1Group6-009 / #293, S95
PS1Group6-010 / #300, S96
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
PS1Group8-015 / #301, S114
Lovblom, Leif PS1Group4-030 / #186, S86
Lovshin, Julie A. PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Lowes, Linda P. PS1Group2-009 / #362, S60
PS1Group8-004 / #353, S105
PS2Group1-006 / #369, S136
Lowes, Linda PS2Group5-008 / #310, S208
WS 8.4.1 / #110, S52
Luca, Annamaria De PS2Group1-010 / #354, S139
Lucchiari, Sabrina PS2Group1-066 / #341, S180
Ludovic, Burlot PS1Group8-035 / #231, S129
Luo, Xiaohui PS2Group1-005 / #400, S135
Lysy, Zoe PS1Group6-012 / #248, S97
Lytvyn, Yuliya PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Mabaya, Gracia PS1Group8-030 / #317, S124
PS1Group8-031 / #315, S125
MacArthur, Daniel PL 1.1 / #21, S3
Macdonald, Christopher PS1Group8-029 / #365, S123
Maddison, Paul PS2Group1-048 / #137, S166
Madsen, Karen L. PS2Group1-059 / #276, S175
PS2Group1-064 / #195, S179
Magalhães, Francisco B. PS1Group2-006 / #324, S58
Magalhaes, João E. PS2Group3-008 / #446, S189
Maggi, Lorenzo PS2Group1-031 / #347, S155
PS2Group1-066 / #341, S180
Magy, Laurent PS1Group8-013 / #227, S112
Mah, Jean K. PS1Group8-029 / #365, S123
PS1Group8-031 / #315, S125
PS2Group1-054 / #192, S170
Mahadevan, Anita PS2Group1-052 / #367, S169
Mahadevappa, Manjunath PS2Group1-061 / #319, S177
Mahdy, Mohamed A. PS2Group7-002 / #165, S210
Mahenderin, Tania PS1Group8-033 / #258, S127
Main, Marion PS2Group1-011 / #340, S140
Majewski, Jacek PS2Group1-045 / #492, S164
Makareeva, Elena PS2Group1-042 / #295, S162
Mallon, Ashley PS2Group3-005 / #460, S187
PS2Group3-006 / #459, S187
PS2Group3-007 / #458, S188
Malyszczak, Witold PS1Group4-011 / #208, S69
PS1Group4-014 / #203, S72
Mammen, Andrew WS 7.1.2 / #95, S45
Mantegazza, Renato PL 2.3 / #416, S5
PS2Group1-031 / #347, S155
PS2Group1-066 / #341, S180
Mantuano, Paola PS2Group1-010 / #354, S139
Marcel, Sebastien PS1Group8-013 / #227, S112
Marcelle, Christophe PS2Group7-005 / #254, S212
Marini, Joan C. PS2Group1-042 / #295, S162
Marini-Bettolo, Chiara PS2Group1-048 / #137, S166
Mariot, Virginie PS2Group1-055 / #120, S171
Marková1, Simona PS1Group6-013 / #294, S98
Marques, Wilson PS1Group4-034 / #383, S88
Marsollier, Anne-Charlotte PS2Group1-055 / #120, S171
Marti, Pilar PS1Group6-004 / #419, S93
Martin, Dana PL 3.1 / #498, S9
Martinez, David PS2Group3-001 / #487, S185
Masson, Cécile PS1Group4-017 / #491, S75
Masson, Gwendal Le PS1Group8-013 / #227, S112
Mathieu, Jean PS2Group1-021 / #381, S147
PS2Group3-018 / #252, S195
Mathieu, Yves PS2Group1-026 / #478, S151
Matsui, Misa PS2Group1-020 / #224, S146
Matsumura, Tsuyoshi PS2Group1-020 / #224, S146
Matsuura, Eiji PS1Group4-022 / #280, S78
PS1Group8-026 / #255, S122
Maurice, Catherine PS1Group8-027 / #395, S123
PS2Group3-023 / #237, S199
Mayer, Oscar H. PS1Group8-001 / #426, S103
PS1Group8-007 / #342, S107
Mayhew, Anna PS2Group1-011 / #340, S140
PS2Group1-030 / #355, S154
PS2Group1-033 / #292, S157
Mazanec, Radim PS1Group6-013 / #294, S98
Mazzone, Elena S. PS2Group1-011 / #340, S140
Mcadam, Laura PS1Group8-030 / #317, S124
PS1Group8-033 / #258, S127
PS1Group8-038 / #364, S131
Mccolly, Markus PS1Group2-009 / #362, S60
Mccomb, Rodney PS1Group2-007 / #189, S59
Mcconville, John PS1Group4-005 / #386, S64
Mccuaig, Jeanna PS1Group8-027 / #395, S123
Mcdonald, Craig M. PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
PS2Group1-005 / #400, S135
Mcdonald, Craig WS 8.4.2 / #111, S52
Mcghie, Josephine PS2Group1-050 / #185, S167
Mckeown, Anne PS2Group1-050 / #185, S167
Mclaren, Donald G. PS2Group5-003 / #180, S205
Mcmillan, Hugh PS1Group4-008 / #308, S66
PS1Group8-031 / #315, S125
Mcwilliam, Catherine PS2Group1-050 / #185, S167
Medne, Livija WS 4.5.2 / #469, S37
Meena, A.K. PS2Group3-010 / #429, S190
Mehdizadeh, Mahshid PS2Group1-002 / #471, S133
Meier, Thomas PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Meijden, Chris J.c. Van Der PS2Group1-063 / #229, S178
Meisel, Andreas PS2Group3-015 / #333, S193
Menager, Pauline PS2Group1-027 / #401, S152
Mendell, J. R. PS2Group1-006 / #369, S136
PS1Group2-009 / #362, S60
PS1Group8-004 / #353, S105
PS2Group5-008 / #310, S208
Mengel, Eugen PS2Group1-058 / #356, S173
Meola, Giovanni PS2Group1-066 / #341, S180
Mercier, Chrystelle PS1Group4-010 / #219, S68
Mercuri, Eugenio PS2Group1-005 / #400, S135
PS2Group1-006 / #369, S136
PS2Group1-011 / #340, S140
PS2Group1-018 / #282, S144
Merillou, Stéphane PS1Group6-017 / #274, S102
Merkies, Ingemar S.j. PS1Group4-001 / #461, S61
Merkies, Ingemar WS 4.4.1 / #72, S35
Merkies, S.J. Ingemar PS1Group4-011 / #208, S69
PS1Group4-014 / #203, S72
Messina, Sonia PS2Group1-011 / #340, S140
PS2Group1-018 / #282, S144
Meyer, Kathrin PS2Group5-008 / #310, S208
Michalski, Catherine PS1Group8-034 / #233, S127
PS1Group8-035 / #231, S129
Michaud, Joris PS2Group1-027 / #401, S152
Micule, Ieva PS1Group6-016 / #318, S101
Mielke, Orell PS1Group4-001 / #461, S61
Miller, Natalie F. PS1Group2-009 / #362, S60
PS1Group8-004 / #353, S105
Millet, Anne-Laure Bedat PS1Group4-010 / #219, S68
Min, Ju-Hong PS2Group3-026 / #191, S200
Miranda, Carlos PS2Group5-008 / #310, S208
Miranda, Natalia PS2Group1-026 / #478, S151
Mitchell, Jane PS1Group8-030 / #317, S124
PS2Group1-019 / #242, S145
Mitsuhashi, Satomi PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Miyake, Zenshi PS1Group6-007 / #143, S94
Miyazaki, Ayako PS2Group7-003 / #201, S210
PS2Group7-004 / #241, S211
Moggio, Maurizio PS2Group1-018 / #282, S144
PS2Group1-031 / #347, S155
Mojbafan, Marzieh PS2Group1-038 / #139, S160
Momma, Kazunari PS1Group8-012 / #267, S111
Monaco, Mauro Lo PS2Group1-066 / #341, S180
Monckton, Darren G. PS2Group1-050 / #185, S167
Mongini, Tiziana PS2Group1-018 / #282, S144
PS2Group1-066 / #341, S180
Moore, Ursula PS2Group1-030 / #355, S154
PS2Group1-033 / #292, S157
Moore-Clingenpeel, Melissa PS1Group2-009 / #362, S60
Mora, Marina PS2Group1-031 / #347, S155
Morales, Ana PS2Group1-028 / #366, S153
Morandi, Lucia PS2Group1-031 / #347, S155
Moreau, Thibault PS1Group4-010 / #219, S68
Morency, Felix C. PS2Group5-003 / #180, S205
Mori, Yoshiaki PS2Group7-003 / #201, S210
PS2Group7-004 / #241, S211
Moriyama, Tetsuya PS1Group6-007 / #143, S94
Mori-Yoshimura, Madoka PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Morovvati, Saeid PS2Group1-040 / #485, S161
Morovvati, Yashar PS2Group1-040 / #485, S161
Moser, Herman PS2Group1-057 / #417, S173
Mota, Vanessa V.D.L. PS1Group4-034 / #383, S88
Moulinas, Rémi PS1Group6-017 / #274, S102
Moxley, III, Richard T. WS 3.2.2 / #60, S30
Mozaffar, Tahseen PS1Group2-004 / #393, S57
PS2Group1-056 / #479, S172
Muelas, Nuria PS1Group6-004 / #419, S93
Muñoz, Raul H. PS2Group3-004 / #476, S186
Muntoni, Francesco PS1Group8-011 / #285, S110
PS2Group1-005 / #400, S135
Muppidi, Srikanth PL 2.3 / #416, S5
Murai, Hiroyuki PL 2.3 / #416, S5
Murata, Miho PS2Group1-004 / #407, S134
Mutluay, Belgin PS2Group3-011 / #384, S191
Myllyharju, Johanna PS2Group1-042 / #295, S162
Nagaraju, Kanneboyina PS1Group2-004 / #393, S57
Nakamagoe, Kiyotaka PS1Group6-007 / #143, S94
Nakamura, Harumasa PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Nakamura, Tomonori PS1Group4-022 / #280, S78
Nalini, Atchayaram PS2Group1-012 / #322, S141
PS2Group1-025 / #202, S150
PS2Group1-052 / #367, S169
PS2Group1-061 / #319, S177
PS2Group3-030 / #290, S203
Nallamilli, Babi R. PS1Group6-018 / #234, S102
Nam, Tai-Seung PS1Group8-024 / #427, S120
Napon, Christian PS2Group1-001 / #436, S132
Naraghi, Ali WS 2.2.2 / #51, S26
Narayanan, Unni G. PS1Group8-038 / #364, S131
Nascimbeni, Anna C. PS2Group1-060 / #358, S176
Nascimento, Osvaldo J. LB 1.3 / #499, S15
Naudina, Maruta S. PS1Group6-016 / #318, S101
Navarrete, Carlos S. PS2Group3-004 / #476, S186
Navarrete, Daniela A. PS2Group3-004 / #476, S186
Nectoux, Juliette PS1Group4-017 / #491, S75
Nejad, Maryam Malakouti PS2Group5-002 / #154, S205
Nel, Melissa PS2Group3-002 / #236, S185
Network, Dmd Italian PS2Group1-006 / #369, S136
Neupauerová, Jana PS1Group4-020 / #343, S76
PS1Group6-013 / #294, S98
Newbould, Rexford PS1Group6-003 / #422, S92
Ney, John P. PS1Group8-005 / #163, S106
Ng, Eduardo PS1Group6-009 / #293, S95
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
Ngo, Mylan PS1Group6-009 / #293, S95
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
Nguyen, Cam-Tu Emilie PS1Group2-010 / #305, S61
PS1Group4-008 / #308, S66
PS2Group3-018 / #252, S195
Nguyen, Thanh PS2Group5-003 / #180, S205
Niceta, Marcello PS2Group1-043 / #270, S163
Nielsen, Ole B. PS2Group7-008 / #265, S214
Nigro, Vincenzo PS2Group1-031 / #347, S155
Nigumann, Pilvi PS1Group6-015 / #263, S100
Nikolenko, Nikoletta PS2Group1-048 / #137, S166
Niks, Erik PS1Group8-011 / #285, S110
Nilsson, Peter PS1Group8-011 / #285, S110
Nishino, Ichizo PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Nobile-Orazio, Eduardo PS1Group4-011 / #208, S69
PS1Group4-014 / #203, S72
Noel, Martin PS1Group2-003 / #405, S56
Nohara, Seitaro PS1Group6-007 / #143, S94
Nonaka, Ikuya PS1Group8-012 / #267, S111
Noohi, Forough PS2Group1-045 / #492, S164
Noone, Josh M. PS1Group4-002 / #449, S62
Norwood, Fiona PS2Group1-035 / #168, S158
Nowak, Richard LB 1.1 / #378, S15
PL 2.3 / #416, S5
Nozuma, Satoshi PS1Group8-026 / #255, S122
O’Brien, Fanny PL 2.3 / #416, S5
O’Connor, Kevin LB 1.1 / #378, S15
O’Ferrall, Erin K. PS2Group1-021 / #381, S147
Oflazer-Serdaroglu, Piraye PS2Group3-029 / #178, S202
Ogata, Katsuhisa PS1Group8-012 / #267, S111
O’Gorman, Cullen M. PS2Group1-071 / #494, S184
Oliveira, Jorge PS2Group1-022 / #194, S148
Ong, Tuyen PS2Group1-005 / #400, S135
Ono, Seiitsu PS2Group5-004 / #20, S206
Orszag, Andrej PS1Group6-009 / #293, S95
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
Oskoui, Maryam PS1Group8-030 / #317, S124
Østergaard, Sofie T. PS1Group8-010 / #286, S109
PS2Group1-036 / #174, S159
Ostrovski, Ilia PS1Group6-009 / #293, S95
PS1Group6-011 / #298, S97
Ouaja, Rabye PS1Group4-010 / #219, S68
PS1Group4-011 / #208, S69
PS1Group4-014 / #203, S72
PS1Group8-013 / #227, S112
PS1Group8-014 / #226, S112
PS1Group8-034 / #233, S127
PS1Group8-035 / #231, S129
Özdemir, Coşkun PS1Group8-018 / #361, S116
Ozerden, Mesude PS2Group3-011 / #384, S191
Padua, Luca PS1Group4-011 / #208, S69
Pagliarani, Serena PS2Group1-066 / #341, S180
Panasisi, Barbara PS2Group1-018 / #282, S144
Pane, Marika PS2Group1-018 / #282, S144
Pang, K PS1Group4-005 / #386, S64
Paolantonacci, Philippe PS1Group8-034 / #233, S127
PS1Group8-035 / #231, S129
Papakonstantinou, Anastasios PS1Group8-021 / #439, S118
Paquet, Jean-Michel PS1Group8-013 / #227, S112
Park, Jin-Sung PS1Group8-024 / #427, S120
Park, Kee Duk PS2Group5-001 / #337, S204
Park, Kee Hong PS2Group3-025 / #123, S200
Park, Ki-Jong PS1Group8-024 / #427, S120
Park, Kyung Seok PS2Group1-068 / #348, S181
Park, Sang-Soon PS1Group4-012 / #207, S71
Park, Su-Yeon PS1Group4-012 / #207, S71
PS2Group3-024 / #225, S199
Park, Tai-Hwan PS1Group4-012 / #207, S71
Parman, Yesim PS2Group3-029 / #178, S202
Parmova, Olesja PS1Group8-036 / #196, S130
Parojcic, Aleksandra PS2Group1-049 / #413, S167
Pasanisi, Barbara PS2Group1-031 / #347, S155
Paschall, Justin PS1Group8-021 / #439, S118
Paskavitz, Jim PS2Group5-003 / #180, S205
Pasnoor, Mamatha PS1Group8-005 / #163, S106
WS 8.3.2 / #109, S52
Patil, Parag G. PL 2.1 / #24, S4
Pattee, Gary L. PS1Group8-016 / #415, S115
Paul, Marine PS2Group1-009 / #283, S138
Paul, Narinder PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Pavan, Gillella K.R. PS2Group3-010 / #429, S190
Pearsall, R. S. PS2Group1-032 / #266, S156
Pearson, Brenda LB 1.1 / #378, S15
Pedersen, Karen B.H. PS1Group8-010 / #286, S109
PS2Group1-051 / #443, S168
Pedersen, Thomas H. PS2Group7-008 / #265, S214
Pegoraro, Elena PS1Group6-015 / #263, S100
PS2Group1-018 / #282, S144
PS2Group1-030 / #355, S154
PS2Group1-066 / #341, S180
Peltz, Stuart W. PS2Group1-005 / #400, S135
Pelz, Andreas PS2Group3-015 / #333, S193
Pena, Loren D.M. PS2Group1-058 / #356, S173
Peng, Yi-Jheng PS2Group1-070 / #253, S182
Pereon, Yann PS1Group4-010 / #219, S68
Peric, Stojan PS2Group1-049 / #413, S167
Perkins, Bruce A. PS1Group6-009 / #293, S95
PS1Group6-010 / #300, S96
PS1Group6-011 / #298, S97
PS1Group6-012 / #248, S97
PS1Group8-015 / #301, S114
WS 5.3.1 / #78, S38
Perkins, Bruce PS1Group4-030 / #186, S86
PS1Group4-037 / #171, S90
WS 5.3.2 / #79, S39
Pesovic, Jovan PS2Group1-049 / #413, S167
Pestronk, Alan PS2Group1-058 / #356, S173
Petek, Christopher PS2Group5-008 / #310, S208
Peterman, Robert PS1Group2-003 / #405, S56
Peterson, E PS2Group1-006 / #369, S136
Petiot, Philippe PS1Group6-015 / #263, S100
Petrini, Stefania PS2Group1-043 / #270, S163
Petrucelli, Leonard PL 4.2 / #34, S10
Petty, Richard K. PS2Group1-037 / #149, S159
PS2Group1-050 / #185, S167
Pfister, Hélène PS1Group4-013 / #206, S71
Phan, Cecile PS2Group3-006 / #459, S187
Philips, Margaret PS2Group1-048 / #137, S166
Piccione, Jr, Ezequiel PS1Group4-016 / #187, S74
Pini, Antonella PS2Group1-018 / #282, S144
Pinos, Tomas PS2Group1-062 / #316, S177
Pires, Melo PS2Group1-013 / #314, S141
PS2Group1-022 / #194, S148
Piscia, Davide PS1Group8-021 / #439, S118
Ploeg, Ans T. Van Der PS2Group1-063 / #229, S178
Plourde, Annie PS1Group8-030 / #317, S124
Pogoryelova, Oksana PS2Group1-023 / #345, S149
PS2Group1-025 / #202, S150
Polavarapu, Kiran PS2Group1-012 / #322, S141
PS2Group1-025 / #202, S150
PS2Group1-052 / #367, S169
PS2Group1-061 / #319, S177
PS2Group3-030 / #290, S203
Politano, Luisa PS2Group1-018 / #282, S144
PS2Group1-066 / #341, S180
Pollard, John PS1Group4-023 / #204, S79
Polydefkis, Michael James WS 2.4.2 / #55, S28
Pons, Carlos Casasnovas PL 2.3 / #416, S5
Popplewell, Linda PS2Group1-055 / #120, S171
Portaro, Simona PS2Group1-066 / #341, S180
Pouget, Jean PS1Group4-010 / #219, S68
Poulin, Chantal PS1Group4-008 / #308, S66
Pour, Yalda Nili PS2Group1-038 / #139, S160
Poydenot, Pauline PS2Group1-027 / #401, S152
Prasad, Chandrajit PS2Group1-025 / #202, S150
PS2Group1-061 / #319, S177
Preethish-Kumar, Veeramani PS2Group1-012 / #322, S141
PS2Group1-025 / #202, S150
PS2Group1-052 / #367, S169
PS2Group1-061 / #319, S177
PS2Group3-030 / #290, S203
Preisler, Nicolai PS2Group1-059 / #276, S175
Previtali, Stefano Carlo PS2Group1-018 / #282, S144
Price, April PS1Group8-031 / #315, S125
Prior, Thomas W. PS2Group5-008 / #310, S208
Probyn, Linda WS 6.5.2 / #93, S45
Propp, Roni PS1Group8-038 / #364, S131
Protasio, Joan PS1Group8-021 / #439, S118
Provost, Sylvie PS2Group1-021 / #381, S147
Puget, Sophie PS1Group4-011 / #208, S69
PS1Group4-014 / #203, S72
PS1Group8-013 / #227, S112
PS1Group8-014 / #226, S112
PS1Group8-035 / #231, S129
Puppo, Francesca PS2Group1-026 / #478, S151
Purushottam, Meera PS2Group1-012 / #322, S141
Quasthoff, Stefan PS2Group1-057 / #417, S173
Querol, Luis WS 7.1.1 / #94, S45
Rabbat, Christopher J. PS1Group2-003 / #405, S56
Radbruch, Andreas PS2Group3-015 / #333, S193
Radhakrishnan, Dhenuka PS1Group8-031 / #315, S125
Rahman, Md Mizanur PS1Group8-037 / #496, S131
Rahman, Monika PS2Group1-050 / #185, S167
Rao, Supriya PS2Group1-023 / #345, S149
Rapôso, Catarina PS1Group8-009 / #432, S109
Ravenscroft, Gina PS2Group1-021 / #381, S147
Ray, Peter PS2Group5-007 / #140, S208
Reeve, Russell PS1Group2-002 / #406, S55
Reichmann, Heinz PS2Group1-047 / #181, S166
Reilly, Mary M. WS 4.5.1 / #74, S36
WS 7.2.1 / #96, S46
Reshetov, Denis A. PS2Group1-014 / #304, S142
PS2Group1-016 / #307, S143
Reza, Mojgan PS1Group8-011 / #285, S110
Ricci, Federica PS2Group1-018 / #282, S144
Ricci, Giulia PS2Group1-066 / #341, S180
Ricotti, Valeria PS2Group1-011 / #340, S140
Riebling, Peter PS2Group1-005 / #400, S135
Riisager, Anders PS2Group7-008 / #265, S214
Riley, Patty PS1Group8-005 / #163, S106
Rivera, Mario PS2Group1-046 / #475, S165
Rizopoulos, Dimitris PS2Group1-063 / #229, S178
Robb, Yvonne PS2Group1-050 / #185, S167
Roberts, Mark PS2Group1-048 / #137, S166
Rocha, Thalita PS1Group8-009 / #432, S109
Rodino-Klapac, Louise PS1Group2-009 / #362, S60
PS2Group1-006 / #369, S136
PS2Group5-008 / #310, S208
Rodrigues, Paula PS1Group2-005 / #391, S58
Rodríguez, María José PS2Group1-046 / #475, S165
Rodriguez-Cruz, Maricela PS2Group1-008 / #377, S138
Rogaeva, Ekaterina WS 6.1.1 / #84, S41
Rogers, Mark PS2Group1-048 / #137, S166
Roggenbuck, Jennifer PS2Group1-028 / #366, S153
Roos, Andreas PS1Group8-021 / #439, S118
PS2Group1-034 / #264, S157
Roos, Andreas PS2Group7-006 / #268, S213
Roos, Marco PS1Group8-021 / #439, S118
Rouhart, Francois PS1Group8-013 / #227, S112
Rouleau, Guy PS2Group5-007 / #140, S208
Rudnicki, Stacy WS 1.2.1 / #38, S19
WS 1.2.2 / #39, S20
Rufibach, Laura PS1Group6-018 / #234, S102
Ruggieri, Alessandra PS2Group1-031 / #347, S155
Rugiero, Marcelo F. PS1Group4-019 / #434, S76
PS2Group3-009 / #448, S189
Rukmini, K.M. PS2Group3-010 / #429, S190
Rummey, Christian PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Runken, Micheal C. PS1Group4-002 / #449, S62
Russell, James W. PS1Group4-029 / #445, S86
WS 6.3.1 / #88, S42
Sacchetto, Roberta PS2Group1-029 / #363, S154
Sacconi, Sabrina PS1Group6-015 / #263, S100
PS2Group1-053 / #260, S170
Saez, Maria S. PS1Group4-019 / #434, S76
Sagi, Avinash R. PS1Group4-029 / #445, S86
Sahenk, Z PS1Group2-009 / #362, S60
PS2Group1-006 / #369, S136
Şahin, Sedef PS2Group1-065 / #170, S179
Saito, Toshio PS2Group1-020 / #224, S146
Sakoda, Saburo PS2Group1-020 / #224, S146
Salbach, Nancy M. PS1Group8-038 / #364, S131
Salgado, David PS1Group8-021 / #439, S118
Salinas, Radrigo PS2Group3-001 / #487, S185
Salo, Antti M. PS2Group1-042 / #295, S162
Salviati, Leonardo PS1Group6-015 / #263, S100
Sanarica, Francesca PS2Group1-010 / #354, S139
Sandona, Dorianna PS2Group1-029 / #363, S154
Sansone, Valeria PS2Group1-066 / #341, S180
Santoro, Lucio PS2Group1-066 / #341, S180
Santos, Manuela PS2Group1-013 / #314, S141
PS2Group1-022 / #194, S148
PS2Group1-067 / #371, S180
Santos, Rosário PS2Group1-022 / #194, S148
Saperstein, David WS 2.4.1 / #54, S27
Saporta, Mario WS 4.3.2 / #71, S35
Savarese, Marco PS2Group1-031 / #347, S155
Savic-Pavicevic, Dusanka PS2Group1-049 / #413, S167
Sayers, Matthew PS1Group4-005 / #386, S64
Sayın, Yakup PS1Group8-018 / #361, S116
Scarr, Daniel PS1Group6-009 / #293, S95
PS1Group6-010 / #300, S96
PS1Group6-011 / #298, S97
Schaefer, David PS1Group8-005 / #163, S106
Schaefer, Jochen PS2Group1-047 / #181, S166
PS2Group5-005 / #441, S207
Schara, Ulrike PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Schneiderat, Peter PS2Group7-001 / #440, S209
Scholtes, Cheryl PS1Group8-031 / #315, S125
Schoser, Benedikt PS2Group1-058 / #356, S173
Scola, Rosana H. PS1Group2-005 / #391, S58
PS1Group2-006 / #324, S58
PS2Group3-016 / #325, S194
Seeman, Pavel PS1Group4-020 / #343, S76
PS1Group6-013 / #294, S98
Seferian, Andreea M. PS1Group4-017 / #491, S75
Sekar, Deepha PS2Group1-012 / #322, S141
Sendra, Isabel Illa PL 2.3 / #416, S5
Seok, Jin Myoung PS2Group3-026 / #191, S200
Sernadela, Pedro PS1Group8-021 / #439, S118
Servais, Laurent PS1Group4-017 / #491, S75
PS2Group1-011 / #340, S140
Seze, Jerome De PS1Group4-010 / #219, S68
Sguarezi, Jetro G.D. PS1Group8-009 / #432, S109
Shafi, Raheel PS2Group1-058 / #356, S173
Shahidi, Gholamali PS2Group5-002 / #154, S205
Shambaugh, Claudia PS2Group1-056 / #479, S172
Shell, Richard PS2Group5-008 / #310, S208
Shen, Xin-Ming PS2Group3-030 / #290, S203
Shephard, Allyson PS1Group8-031 / #315, S125
Sherman, Alexander WS 4.1.1 / #66, S32
Sherman, Matthew L. PS2Group1-032 / #266, S156
Shin, Jin-Hong PS1Group8-024 / #427, S120
Shoesmith, Christen WS 1.2.1 / #38, S19
WS 1.2.2 / #39, S20
Shontz, K. PS2Group1-006 / #369, S136
Shook, Steven WS 6.5.1 / #92, S44
Short, Gerard PS2Group1-058 / #356, S173
Shy, Michael WS 2.3.1 / #52, S26
Siciliano, Gabriele PS2Group1-066 / #341, S180
Siddiqi, Zaeem A. PS1Group8-019 / #464, S117
PS2Group3-005 / #460, S187
PS2Group3-006 / #459, S187
PS2Group3-007 / #458, S188
Signorovitch, James PS2Group1-017 / #287, S144
Silvestri, Nicholas PS2Group3-022 / #146, S198
WS 6.2.2 / #156, S42
Simschitz, Phillip PS2Group1-057 / #417, S173
Skrinar, Alison PS2Group1-023 / #345, S149
Smith, A. Gordon WS 8.3.1 / #108, S51
Sobue, G. PS1Group4-001 / #461, S61
Sole, Guilhem PS1Group4-010 / #219, S68
PS1Group8-013 / #227, S112
PS1Group8-014 / #226, S112
Soloway, Aimee PS1Group8-019 / #464, S117
Sommer, Claudia PS1Group2-002 / #406, S55
Song, Jinlin PS2Group1-017 / #287, S144
Song, Sook-Hee PS1Group4-012 / #207, S71
Soomro, Aleena F. PS1Group8-022 / #438, S120
Soomro, Bashir PS1Group8-023 / #437, S120
Soong, Bing-Wen PS1Group4-027 / #142, S84
Sorenson, Eric J. PS2Group1-071 / #494, S184
Sorroche, Patricia PS1Group4-019 / #434, S76
Šoukalová, Jana PS1Group4-020 / #343, S76
Sousa, Ana P. PS1Group4-018 / #389, S75
PS2Group1-013 / #314, S141
PS2Group1-067 / #371, S180
Sousa, Sandra PS1Group4-021 / #193, S77
PS2Group1-022 / #194, S148
Soysal, Aysun PS2Group3-011 / #384, S191
Spagnolo, Paolo PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Spiegel, Robert PS2Group1-005 / #400, S135
Spies, Judy PS1Group4-023 / #204, S79
Spitali, Pietro PS1Group8-011 / #285, S110
Spring, Penny PS1Group4-023 / #204, S79
Sproule, Doug M. PS2Group5-008 / #310, S208
Srour, Myriam PS2Group1-021 / #381, S147
Statland, Jeffrey M. WS 7.5.2 / #103, S49
Stavusis, Janis PS1Group6-016 / #318, S101
Steck, Andreas PS1Group4-013 / #206, S71
Steinz, Maarten PS2Group7-001 / #440, S209
Stemmerik, Mads PS2Group1-059 / #276, S175
PS2Group1-064 / #195, S179
Stewart, William PS2Group1-037 / #149, S159
Stojanovic,
Vidosava M. Rakocevic PS2Group1-049 / #413, S167
Stojkovic, Tania PS1Group6-015 / #263, S100
Stoltz, Randall PS1Group6-005 / #302, S93
Straathof, Chiara S. PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
Strandberg, Kristin PS1Group8-011 / #285, S110
Straub, Volker PS1Group8-021 / #439, S118
PS2Group1-035 / #168, S158
PS2Group1-037 / #149, S159
PS2Group1-048 / #137, S166
PS2Group1-058 / #356, S173
WS 1.1.2 / #466, S19
WS 5.1.1 / #467, S37
Strautmanis, Jurgis PS1Group6-016 / #318, S101
Stuardo, Andres PS2Group3-001 / #487, S185
Stulnig, Thomas PS2Group1-057 / #417, S173
Sturtz, Franck PS1Group6-017 / #274, S102
Suchowersky, Oksana PS2Group5-007 / #140, S208
Suchy, Sharon PS1Group6-014 / #235, S99
Sun, Jade PS2Group1-032 / #266, S156
Sung, Jung-Joon PS1Group4-007 / #335, S65
PS1Group4-009 / #281, S67
PS1Group4-032 / #462, S88
PS1Group4-036 / #164, S89
PS2Group3-019 / #269, S196
PS2Group3-025 / #123, S200
PS2Group7-007 / #387, S213
Suzuki, Mikiya PS1Group8-012 / #267, S111
Swallow, Elyse PS2Group1-017 / #287, S144
Szigyarto, Cristina Al-Khalili PS1Group8-011 / #285, S110
Szuto, Anna PS2Group5-007 / #140, S208
’T Hoen, Peter A.C. PS1Group8-021 / #439, S118
Taddeo, Adriano PS2Group3-015 / #333, S193
Taipa, Ricardo PS2Group1-013 / #314, S141
PS2Group1-022 / #194, S148
Taivassalo, Tanja PS2Group1-021 / #381, S147
Takahashi, Toshiaki PS1Group8-012 / #267, S111
Takashima, Hiroshi PS1Group4-022 / #280, S78
PS1Group8-026 / #255, S122
PS1Group8-032 / #331, S126
Takeda, Shin’Ichi PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Takeuchi, Fumi PS2Group1-003 / #428, S133
PS2Group1-004 / #407, S134
Tamaoka, Akira PS1Group6-007 / #143, S94
Tamura, Takuhisa PS1Group8-012 / #267, S111
Tanaka, Yuzo PS1Group8-012 / #267, S111
Tanant, Véronique PS2Group1-053 / #260, S170
Tang, Chih-Yung PS2Group1-070 / #253, S183
Tapia, Carolina Barnett WS 5.5.1 / #82, S40
Tarnopolsky, Mark PS1Group6-006 / #328, S94
WS 1.3.1 / #482, S20
Tartaglia, Marco PS2Group1-043 / #270, S163
Tartaglia, Maria Carmela WS 6.1.2 / #85, S41
Tasca, Elisabetta PS2Group1-060 / #358, S176
Tasca, Giorgio PS2Group1-043 / #270, S163
Tashiro, Yuichi PS1Group8-026 / #255, S122
Tawil, Rabi WS 7.5.1 / #102, S49
Tenenbaum, Florence PS2Group1-009 / #283, S138
Terada, Makoto PS1Group6-007 / #143, S94
Tétreault, Martine PS2Group1-045 / #492, S164
PS2Group1-021 / #381, S147
Thaisetthawatkul, Pariwat PS1Group2-007 / #189, S59
PS1Group4-016 / #187, S74
PS1Group4-038 / #188, S91
Thomassen, Jesper Q. PS1Group8-010 / #286, S109
Thomé, Rodolfo PS1Group8-009 / #432, S109
Thompson, Andrew PS2Group3-013 / #374, S192
Thompson, Mark PS1Group8-021 / #439, S118
Thompson, Rachel PS1Group8-021 / #439, S118
Thomsen, Carsten PS2Group1-051 / #443, S168
Thornton, Charles PL 3.2 / #31, S9
WS 3.2.1 / #59, S29
Thurberg, Beth L. PS2Group1-058 / #356, S173
Tirupathi, Sandya PS1Group4-005 / #386, S64
PS2Group3-013 / #374, S192
PS2Group3-014 / #334, S193
Tobon, Alejandro PS2Group3-012 / #329, S191
Tomidokoro, Yasushi PS1Group6-007 / #143, S94
Tonekaboni, Seyed Hasan PS2Group1-038 / #139, S160
PS1Group4-004 / #404, S63
Torpil, Berkan PS2Group1-065 / #170, S179
Toscano, Antonio PS2Group1-066 / #341, S180
Touzaka, Naoki PS1Group6-007 / #143, S94
Trangulao, Alejandra PS2Group1-026 / #478, S151
PS2Group1-046 / #475, S165
Trimble, Keith PS2Group3-013 / #374, S192
Trivedi, Jaya PS2Group1-058 / #356, S173
WS 5.4.2 / #81, S40
Trojano, Maria PS2Group1-066 / #341, S180
Trucco, Federica PS2Group1-018 / #282, S144
Trujillo, Oscar PS1Group4-003 / #442, S63
PS2Group1-024 / #326, S149
Truong, Laetitia PS1Group4-016 / #187, S74
Tsai, Pei-Chien PS1Group4-026 / #215, S82
PS1Group4-027 / #142, S84
PS1Group4-028 / #141, S85
Tsuji, Hiroshi PS1Group6-007 / #143, S94
Tulinius, Már PS2Group1-007 / #385, S137
Turner, Chris PS2Group1-048 / #137, S166
Uhlén, Mathias PS1Group8-011 / #285, S110
Urtizberea, J. A. PS2Group1-026 / #478, S151
Utkus, Algirdas PS1Group6-016 / #318, S101
Utsugisawa, Jr., Kimiaki PL 2.3 / #416, S5
Vajsar, Jiri PS1Group4-008 / #308, S66
PS2Group1-015 / #309, S142
Valdrez, Katia PS1Group4-018 / #389, S75
PS1Group4-021 / #193, S77
Valencia, Alfonso PS1Group8-021 / #439, S118
Van Schaik, Ivo N. PS1Group4-001 / #461, S61
Vanasse, Michel PS1Group4-008 / #308, S66
PS2Group3-018 / #252, S195
Vasavada, Ruhi PS2Group1-041 / #414, S161
Vasi, Veena PS2Group3-013 / #374, S192
Vengalil, Seena PS2Group1-012 / #322, S141
PS2Group1-025 / #202, S150
PS2Group1-052 / #367, S169
PS2Group1-061 / #319, S177
PS2Group3-030 / #290, S203
Vercelli, Liliana PS2Group1-066 / #341, S180
Verdegem, Lise Van PS1Group8-008 / #338, S108
Verinaud, Liana PS1Group8-009 / #432, S109
Vial, Christophe PS1Group4-010 / #219, S68
PS1Group6-015 / #263, S100
Vidal, Brian PS2Group1-032 / #266, S156
Vidaud, Michel PS1Group6-015 / #263, S100
Vieira, Emília PS2Group1-022 / #194, S148
Vilchez, Juan J. PS1Group6-004 / #419, S93
Vincent, Angela PS2Group3-028 / #271, S201
Vissing, Christoffer PS2Group1-051 / #443, S168
Vissing, John PL 2.3 / #416, S5
PS1Group4-024 / #273, S80
PS1Group8-010 / #286, S109
PS2Group1-044 / #213, S163
PS2Group1-051 / #443, S168
PS2Group1-058 / #356, S173
PS2Group1-059 / #276, S175
PS2Group1-062 / #316, S177
PS2Group1-064 / #195, S179
WS 1.3.2 / #41, S21
WS 8.1.2 / #105, S50
Vita, Giuseppe PS2Group1-018 / #282, S144
Vlodavets, Dmitry V. PS2Group1-014 / #304, S142
PS2Group1-016 / #307, S143
Vohanka, Stan PS1Group8-036 / #196, S130
Voit, Thomas PS1Group8-006 / #346, S107
PS1Group8-007 / #342, S107
PS2Group1-055 / #120, S171
Vries, Juna M. De PS2Group1-063 / #229, S178
Vroom, Elizabeth PS2Group1-011 / #340, S140
Vu, Tuan PL 2.3 / #416, S5
Wahbi, Karim PS2Group1-009 / #283, S138
Waite, Melissa PS2Group1-041 / #414, S161
Walker, Francis WS 2.5.1 / #56, S28
WS 3.5.1 / #65, S32
Walton, Timothy PS1Group8-005 / #163, S106
Wang, Jichuan PS1Group8-016 / #415, S115
Wang, Min-Xia PS1Group4-023 / #204, S79
Wang, Susanne PS1Group6-003 / #422, S92
PS2Group1-007 / #385, S137
Wanschitz, Julia PS2Group1-057 / #417, S173
Ward, Susan J. PS2Group1-017 / #287, S144
Warita, Katsuhiko PS2Group7-002 / #165, S210
Warman-Chardon, Jodi PS2Group5-007 / #140, S208
Watanabe, Masahiko PS1Group6-007 / #143, S94
Watanabe, Osamu PS1Group8-026 / #255, S122
PS1Group8-032 / #331, S126
Webster, Richard PS2Group3-028 / #271, S201
Weir, Shannon PS1Group8-038 / #364, S131
Weis, Joachim PS2Group1-034 / #264, S157
PS2Group7-006 / #268, S213
Weisman, Alanna PS1Group6-010 / #300, S96
PS1Group8-015 / #301, S114
Wencel, Marie PS2Group1-056 / #479, S172
Wens, Stephan C.a. PS2Group1-063 / #229, S178
Wentworth, B. PS2Group1-006 / #369, S136
Werlauff, Ulla PS2Group1-011 / #340, S140
PS2Group1-044 / #213, S163
Werneck, Lineu C. PS1Group2-005 / #391, S58
PS1Group2-006 / #324, S58
PS2Group3-016 / #325, S194
Westergard, Shelley PS1Group8-027 / #395, S123
Wilcox, Alison PS2Group1-050 / #185, S167
Windish, Hillarie P. PS1Group6-018 / #234, S102
PS2Group1-033 / #292, S157
Wittenbach, Jason PS2Group1-041 / #414, S161
Witting, Nanna PS1Group8-010 / #286, S109
PS2Group1-044 / #213, S163
Wolbeck, Ryan PS2Group3-022 / #146, S198
Wolever, Thomas PS1Group4-037 / #171, S90
WS 5.3.2 / #79, S39
Wolfe, Gil I. PL 2.2 / #25, S4
WS 1.5.1 / #44, S22
Wolff, Jodi PS1Group8-001 / #426, S103
Wong-Baeza, Carlos PS2Group1-008 / #377, S138
Wood, Libby PS2Group1-048 / #137, S166
Wu, Hsien-Tsai PS1Group6-008 / #228, S95
Wu, Tong PS1Group6-009 / #293, S95
PS1Group6-011 / #298, S97
Xin, Haichang PS2Group3-020 / #239, S196
PS2Group3-021 / #238, S197
Yamaguchi, Tetsuto PS1Group6-007 / #143, S94
Yamaji, Junko PS2Group7-003 / #201, S210
PS2Group7-004 / #241, S211
Yamamoto, Fumiko PS1Group6-007 / #143, S94
Yanagiha, Kumi PS1Group6-007 / #143, S94
Yang, Jiwon PS2Group3-019 / #269, S196
Yatabe, Kana PS1Group8-012 / #267, S111
Yatsuga, Shuichi PS1Group6-007 / #143, S94
Yonemoto, Naohiro PS2Group1-004 / #407, S134
Yoon, Byung-Nam PS1Group4-036 / #164, S89
PS2Group3-019 / #269, S196
PS2Group3-025 / #123, S200
Yoon, Cindy W. PS2Group3-026 / #191, S200
Yoon, Grace PS2Group5-007 / #140, S208
WS 3.3.1 / #153, S30
Yoon, Sung-Hee S. PS2Group1-019 / #242, S145
Yoshimura, Akiko PS1Group4-022 / #280, S78
Young, Peter PS2Group1-058 / #356, S173
Yu, Hui Jing PS1Group6-005 / #302, S93
Yuan, Junhui PS1Group4-022 / #280, S78
Yun, Pomi PS2Group1-041 / #414, S161
Zacherle, Emily PS1Group4-002 / #449, S62
Zadeh, Abdolazim Nejati PS2Group1-038 / #139, S160
Zadeh, Galareh PS1Group8-027 / #395, S123
WS 6.4.2 / #91, S44
Zahedi, René PS2Group1-034 / #264, S157
PS2Group7-006 / #268, S213
Zamani, Gholamreza PS2Group1-002 / #471, S133
Zamorano, Ivonne PS2Group1-046 / #475, S165
Zeinali, Sirous PS2Group1-038 / #139, S160
Zhang, Xiaosha PS2Group1-032 / #266, S156
Zijdenbos, Alex P. PS2Group5-003 / #180, S205
Zotova, Eugeniia PS2Group1-014 / #304, S142
Zou, Yaqun PS2Group1-042 / #295, S162
Züchner, Stephan PL 1.2 / #22, S3
WS 4.3.1 / #70, S34

