
Genome Editing Gene Therapy for Duchenne Muscular Dystrophy
Abstract
Duchenne muscular dystrophy (DMD) is a severe genetic disorder caused by loss of function of the dystrophin gene on the X chromosome. Gene augmentation of dystrophin is challenging due to the large size of the dystrophin cDNA. Emerging genome editing technologies, such as TALEN and CRISPR-Cas9 systems, open a new erain the restoration of functional dystrophin and are a hallmark of bona fide gene therapy. In this review, we summarize current genome editing approaches, properties of target cell types for ex vivo gene therapy, and perspectives of in vivo gene therapy including genome editing in human zygotes. Although technical challenges, such as efficacy, accuracy, and delivery of the genome editing components, remain to be further improved, yet genome editing technologies offer a new avenue for the gene therapy of DMD.
INTRODUCTION
Duchenne muscular dystrophy (DMD)
Duchenne muscular dystrophy (DMD) is one of the most common and severe forms of genetic muscle disorders and is described by the progressive degeneration of proximal muscular tissue with the characteristic of calf muscle enlargement. Muscle weakness becomes obvious around 3 to 5 years of age, resulting in compromised running, climbing, or standing (known as Gowers’ sign). Patients at infant stage show a massive elevation of creatine kinase (CK) or creatine phosphokinase (CPK) levels (>10,000 IU/L in comparison to normal levels of <200 IU/L) in the blood, which is an indication of muscle or heart damage, since CK is a muscle specific kinase mediating the production of energy storage material phosphocreatine in muscle. Between ages 7 and 12, DMD patients gradually start to rely on wheel chairs due to difficulties in walking and a waddling gait. By their teenage years, they may require assistance in activities that involve the use of arms and legs. With time, muscle weakness affects not only proximal muscle tissues, but also cardiac and respiratory function. The development of artificial ventilators in the 1980s have helped improve life expectancy, however, cardiac or respiratory defects remain the main cause of mortality in DMD patients, with a life expectancy of around the age of 30.
Dystrophin gene
The genetic cause of DMD was first identified by Dr. Louis M. Kunkel’s group in 1986 [1], and the identified cDNA was named “Dystrophin” [2]. The dystrophin gene spans about 2.2 Mbp on the X chromosome and consists of 79 exons for the Dp427 m isoform, whichis mainly expressed in skeletal muscle cells. Dystrophin protein has an actin-binding domain on the N’-terminus and a dystrogly can complex-binding domain on the C’-terminus, and the two are connected by a rod domain which consists of spectrin-like repeats (Fig. 1). The function of dystrophin protein is to physically anchor the cellular skeletal actin fibers and sarcolemma membrane in muscle fibers [3]. Muscle cells without functional dystrophin become vulnerable during physical muscle movements, resulting in spontaneous cellular death which induces inflammation and fibrosis [4].
DMD mutation patterns
Due to the large size of the dystrophin gene, mutation patterns found in DMD patients largely vary. Approximately 60–70% of DMD patients harbor a large deletion of one or more exons, with many occurring with in exons 44 to 54 [5–9].
Notably, a milder form of DMD, Becker muscular dystrophy (BMD), is also caused by mutations in the same dystrophin gene. Complete loss of functional dystrophin protein due to out-of-frame mutations is classified as DMD, while partial loss of functionality (such as smaller protein size) or decreased level of dystrophin protein expression due to in-frame deletions, insertions, or small amino acid substitutions are classified as BMD.
BMD patients suffer from weakened muscles and mobility in their old age, although the disease phenotype is highly heterogeneous. Depending on the type of genetic mutation, functionality of the resulting dystrophin protein ranges from barely functional (similar to DMD) to almost normal [3, 10]. Therefore, the symptoms of BMD patients vary, and some patients have nearly normal life spans.
Considering the function of dystrophin as a structural protein that connects cytoskeletal actin fibers and the sarco lemma membrane, minor amino acid substitutions or truncation at the rod domain is not critical for its function. This is a unique and important property when it comes to designing strategies on how to restore the function of dystrophinprotein for treating DMD.
Antisense oligonucleotide-mediated exon skipping
The deletion mutations found in roughly two thirds of exons are not in multiples of three base pairs, thus resulting shifts of one or two base pairs in the protein reading frame. Notably, if an exon with the sizeof (3n+1) bp is deleted, further translational skipping of another adjacentor neighboring exon with the size of(3n+2) bp would restore the out-of-frame mutation. This approach is known as exon skipping. Although exon skipping causes some amino acid deletion, in general truncations in the rod domain are tolerated, as seen in BMD patients.
To induce exon skipping, several methods have been investigated, including the use of anti sense oligonucleotides to bind to a targeted exon. Several different types of anti sense oligonucleotides have been tested and demonstrated as effective and functional in various animal models [11–16]. Encouragingly, anti sense oligonucleotide analogues are now being investigated in clinical trials, with some showing promising results [17–20]. However, due to the transient nature of antisense oligonucleotide analogues, DMD patients are required to be repeatedly treated throughout their lifetime (Fig. 2).
Gene augmentation therapy
The concept of gene therapy, which is to treat or cure congenital disorders, emerged through the development of viral vectors in the early 1970s. Since then, the field of gene therapy has significantly expanded with the concept of supplying a healthy gene to replace the function of the mutated gene [21–24]. So far, several genedelivery vectors based on viruses have been developed, such as retroviral vector [25–27], lentiviral vector [28–30], herpes simplex virus (HSV)-based vector [31,32], adenoviral vector (HDAdv) [33,34], adeno-retroviral hybrid vector [35], and adeno-associated virus (AAV) vector [36–40]. Each vector has its own advantages and disadvantages, as listed in Table 1.
In the case of gene therapy for DMD, there are three major challenges: the large size of dystrophin cDNA (14 kbp for the Dp427 m isoform); the transduction of muscle tissues that are surrounded by layers of connective tissues; and the abundance of affected tissue, which consists of 30∼40% of body weight. In general, oversized viral vectors have significantly lower production efficiency, therefore leading to poor transduction efficiency. HSV vectors [31,32] and HDADv vectors [33, 34] have been demonstrated to deliver full-length dystrophin cDNA. However, other vectors require the dystrophin cDNA to be cut down in size by trimming the rod domain (as seen in mild BMD patients), resulting in mini-dystrophin (6.2 kb) or micro-dystrophin genes (3.6 to 4.2 kb) [41]. As an alter native,the utrophin gene (official gene symbol: UTRN), which shares both structural and functional similarities with the dystrophin gene, was shown to functionally replace defective dystrophin [42, 43].
Apart from viral vectors, non-viral vectors have also been investigated for DMD gene therapy. Sleeping Beauty is an engineered DNA transposon originally derived from Tc1/mariner transposons in fish [44] and is found to have high chromosomal integration ability of a foreign gene in human cells. Dr. Rita Perlingeiro’s group demonstrated that Sleeping Beauty transposon vector can transduce the micro-utrophin gene, which is a truncated form of the utrophin gene [45]. To overcome the packaging size limit of a gene transfer vector, HAC (human artificial chromosome) vectors have been investigated to deliver a 2.4 Mb human dystrophin genomic region that includes regulatory elements [46]. Despite the promise of delivering the full dystrophin genomic region, the current delivery efficiency of HAC vectors is low, thus requiring anextensive selection process for HAC vector transduction.
Genome editing technologies for DMD
Due to difficulties in delivering a functional dystrophin gene into myogenic cells, alternative approaches were anticipated. In this regard, the development of engineered nucleases opened a new era of gene therapy for DMD. Artificial nucleases, which are engineered to cut genomic DNA once at the designated target sequence, can induce DNA repair pathways, such as non-homologous end joining (NHEJ) and homologous directed repair (HDR). The NHEJ pathway results in the generation of small deletions or sometimes insertions, whereas the HDR pathway uses a repair template called the donor DNA.
Meganucleases for DMD
The first demonstration of genome editing technology for the dystrophin gene was reported in 2010 by Dr. Jacques Tremblay’s group in collaboration with the biotechnology company Cellectis [47]. They used meganucleases, which are homing end on ucleases that contain a large DNA recognition domain to target 12- to 40-bp DNA sequences. For example, I-SceI mega nuclease derived from the mitochondrial DNA of Saccharomyces cerevisiae binds and cutsan 18-bp recognition sequence (5′-TAGGGATAACAGGGTAAT-3′). This recognition sequence does not exist in the human genome, therefore, it can be used to introduce a specific DNA cleavage only at the target sequence without attacking human endogenous sequences. Similarly I-CreI, a mega nuclease derived from Chlamydomonas reinhardtii, recognizes a 22-bp DNA sequence (5′-CAAAACGTCGTACGACGTTTTG-3′). Cellectis has engineered I-CreI mega nuclease variants to recognize the human RAG1 gene (I-CreI-RAG1, 5′-TGTTCTCAGGTACCTCAGCCAG-3′) [48]. To demonstrate a proof-of-principle, Dr. Tremblay’s group first inserted a mega nuclease target site (either I-SceI or I-CreI-RAG1 target site) into the middle of the dog dystrophin gene with a frame-shift mutation. They then checked whether over expression of the corresponding mega nuclease (I-SceI or I-CreI-RAG1) could induce a small deletion or insertion at the target site to alter the protein reading frame. Indeed, restored expression of the dog dystrophin protein was detected in 293FT cells, human myoblasts, and mouse muscle fibers. This report was the first demonstration of engineered nucleases restoring out-of-frame mutations of the dystrophin gene solely by the initiation of DNA cleavage. However, due to the lack of a natural targeting site for mega nucleases in the human dystrophin gene and because custom engineering of mega nucleases require laborious screening processes, custom engineering nucleases that can be targeted to an endogenous dystrophin sequence are desired.
ZFN mediated indels for DMD
Zinc finger domain is one of the most abundant DNA binding domains in mammals, and most of itspossible combinations of DNA binding motifs have been identified. Custom conjugation of several zinc finger domain swith a nuclease domain allows scientists to target specific sequences of interest. To target one target site, typically three or four zing finger domains are conjugated with FokI nuclease domain to form zinc finger nuclease (ZFN). FokI domain cleaves DNA only when it is dimerized, so a pair of zinc finger nucleases (ZFNs) is used to cleavage the target sequence and also to increase specificity.
Dr. Jacques Tremblay’s group investigated the utility of ZFNs that target endo genous the human dystrophin gene together with several mega nucleases and found that ZFNs can induce small deletions (typically 1–20 bp in size) or small insertions (typically 1–5 bp in size) at the target site of the human dystrophin gene [49]. These results demonstrate that ZFNs are able to restore the reading frame of endo genous dystrophin simply by introducing a small deletion or insertion (indel).
ZFN-mediated knock-in for DMD
Site-specific DNA cleavage not only resulted in NHEJ mediated indels, but also enhances the efficiency of HDR. In human DMD myoblasts carrying a deletion of exons 45–52, Dr. George Dickson’s group demonstrated that mega nucleases targeting intron 44 of the dystrophin gene facilitated the insertion of cDNA corresponding to the exon 45–52 region in a bulk cell population [50]. This is a promising gene therapy approach for patients who carry deletions of multiple exons. Unfortunately, restored dystrophin protein expression was not detected, presumably due to the low incidence of cDNA knock-in events or the insertion of an artificial exon altering the expression pattern of dystrophin protein. Further enhancement of knock-in events or selection of a collected cell pool is required.
While ZFN is a powerful tool to edit a target site of interest, it does have drawbacks. Firstly, construction of an active ZFN pair is challenging, as simple assembly of the corresponding zinc finger domain does not guarantee full DNA binding activity due to cross interactions between adjacent zinc finger domains (known as context dependency). Secondly, even though zinc finger domains that recognize all possible combinations of trinucleotide motifs (43 = 64) have been identified, there are some biases or preferences to certain sequences, such as “GNN”. Therefore, designable target sequences are limited (Fig. 3).
TALEN for DMD
To this end, a more flexible DNA targeting domain is preferred. TALE (Transcriptional Activator-Like Effector) domain, which was originally derived from the plant pathogenic bacteria Xanthomonas, has a unique structure for its DNA binding motif. TALE domain consists of a tandem repeat of 34 a mino acid residues, also known as repeat variable di-residue (RVD), with variations at the 12th and 13th amino acids. Each RVD recognizes a single nucleotide sequence, and there is less context dependency between adjacent RVDs. Therefore, the conjugation of TALE domain with FokI nuclease, which is known as TALEN, provides a much more flexible DNA binding platform for targeting a specific DNA sequence of interest (Fig. 3).
Thanks to its greater flexibility in target sites, the TALEN system has high probability at targeting exonic regions, which are typically less than 200 bp in size. By targeting the protein-coding region in exons, the dystrophin coding frame can be modulated when the correct size of a deletion or insertion is introduced. Deletion is solely mediated by programmable nucleases via the NHEJ repair pathway and since a donor template is not required, the gene delivery process can be simplified. This NHEJ-mediated approach is unique in its targeting the rod domain region of the dystrophin gene, as small truncations or amino acid changes can be tolerated or have only minor effects on dystrophin protein function, as seen in BMD patients. Dr. Charles Gersbach’s group utilized the TALEN system to target exon 51 in fibroblasts or an immortalized myoblast cell line, which was originally derived from a DMD patient with deletion of exons 48–50 [51]. Successful introduction of a small deletion (i.e. 5 bp) in exon 51 resulted in a frame shift of the protein reading frame and restored dystrophin protein expression in differentiated myoblasts. We tested a similar approach in induced pluripotent stem (iPS) cells derived from a DMD patient who lacks exon 44 [52]. By targeting exon 45 with TALEN, we successfully introduced a small deletion or insertion (i.e. 1 bp insertion), which restored the proper protein reading frame to express dystrophin protein after myogenic differentiation. However, the frame shift approach is highly stochastic, as the size of the indelby programmable nucleases cannot be controlled. Therefore, out of all the indelclones analyzed, only one-third of cells showed a proper protein reading frame.
To enhance the likelihood of reading frame correction, we also tested the possibility of skipping exon 45 by targeting a splicing acceptor site. As consensus sequences of splicing acceptor sites mainly consist of a 2-bp “AG” sequence, we initially thought the disruption of a splicing acceptor site should be more efficient than the frame shifting approach. However, it was difficult to design TALENs or other programmable nuclease swith high specificity, due to the low complexity of the polypyrimidine tract (which is a “C and T” rich region) around the splicing acceptor site. Nevertheless, by targeting around 10 bp downstream from the splicing acceptor site, we managed to show that some NHEJ-mediated deletions can reach the splicing acceptor site and succeeded in skipping exon 45 to restore the proper reading frame of dystrophin protein [52]. A similar approach is also reported in skipping exon 51 by using ZFNs [53].
For NHEJ-mediated deletion approaches, the control or prediction of the deletion pattern is important. Interestingly, deep sequencing analysis of deletion patterns identified apredominant deletion pattern (up to 30–40% of the deletion events) harboring microhomology sequences (typically 3–5 bp in size) on both sides of the cleavage site [52, 54]. By using web tools to predict microhomology-mediated end joining patterns [52, 55], we sought to enhance the success rate of the frame shift approach.
Thus, by taking advantage of iPS cells and their propensity to self-renew indefinitely, we examined a knock-in approach of missing exon 44 by conjugating antibiotic selection followed by subsequent removal of the selection cassette by the Cre-loxP system. We conducted TALEN-mediated targeting experiments and observed high knock-in events amongst antibiotic resistant clones. Successful expression of full-length dystrophin protein was detected by immunostaining and western blotting upon differentiation into myoblasts. Two rounds of sub cloning is demanding and time consuming, but homologous recombination (HR)-mediated targeted knock-in of a missing exon might be a feasible approach in DMD patient-derived iPS cells.
CRISPR-Cas9 for DMD
The TALEN system is versatile enough to target almost any DNA sequence, however, construction of TALEN is laborious, especially for newcomers to genome editing experiments. To construct one TALEN, a library of RVD domains is required to conjugate 14 to 20 RVD domains into an expression vector [56–59]. The assembly process normally takes two to three steps of cloning cycles (i.e. two to three weeks) depending on the assembly systems.
Mean while, bacterial adaptive immunity systems [60], namely CRISPR (clustered regularly interspaced short palindromic repeats) and Cas (CRISPR associated) proteins [61], have been identified as a DNA cleavage machinery against invasive DNA elements, such as bacteriophages [62–64]. Soon after this finding, Drs. Jennifer Dundna and Emmanuelle Charpentier’s groups recognized that the combination of Cas9 protein and two small RNAs, crRNA and tracrRNA, can induce a DNA double strand break at a site complementary to the crRNA sequence [65]. Furthermore, crRNA and tracrRNA can be conjugated into a single guide RNA (sgRNA) by a tetranucleotide linker loop to simplify the system. This finding has had significant impact on the genome editing field, as only the construction of ansgRNA (∼100 bp in size) is required to determine the target sequence, and the Cas9 protein component is common for any target sequences. Construction of sgRNA is simple and straight forward, as target sequences can be synthesized and inserted into an expression vector driven by a PolIII promoter.
One major limitation of the CRISPR system as a genome editing tool is the requirement of a “proto spacer adjacent motif (PAM)” sequence for the target site, as this sequence is essential for the initial step of the target sequence recognition [66]. For example, the most widely used CRISPR-Cas9 system derived from Streptococcus pyogenes requires a “NGG” PAM sequence at the 3’-end of the target sequence, while Staphylococcus aureus derived Cas9 requires a “NNGRRT” PAM sequence [67]. This feature somehow reduces the target ability of CRISPR-sgRNA at given sequences, especially when compared with the TALEN system, however, the CRISPR-Cas9 system is still flexible enough to target roughly every 10∼20 bp of DNA sequence (Fig. 3).
Strikingly, it is reported that DNA cleavage activity with the Streptococcus pyogenes-derived CRISPR-Cas9 system is greater than that with the TALEN system in human embryonic stem (ES) cells and iPS cells [68]. In this regard, we tested the CRISPR-Cas9 system to correct dystrophin mutations in patient-derived iPS cells, finding the resulting cleavage activity and HR-mediated knock-in efficiency was comparable with those using the TALEN system [52]. Considering the ease of construction, the CRISPR system is significantly easier to use by non-cloning specialists (Table 2).
Another concern about the CRISPR system is its targeting specificity. There are a number of reports on off-target mutagenesis even with several base pair mismatches, especially in immortalized cancer cell lines [69–73]. On the other hand, several other groups reported high specificity and minimum off-target mutation load by whole genome sequencing in human pluripotent stem cells [74, 75]. We tested the risk of off-target mutagenesis by karyotyping, CNV analysis, and exome sequencing, and found minimum incidents of undesired mutagenesis in DMD-iPS cells. Therefore, at least in human pluripotent stem cells, the CRISPR system can be regarded as a precise recognition machinery. Further study is required to shed light on whether immortalized cell lines have abnormal DNA repair pathways similar to cancer, or whether pluripotent stem cells equip a particular DNA repair mechanism to maintain genomic integrity, such as Zscan4 [76] or Filia genes [77].
Strategies to correct DMD mutations must considerer the type of mutation in the patient, as genome editing approach is highly custom tailored. Therefore, pathogenic mutation patterns must be carefully examined before deciding on the appropriate therapeutic strategy. To cover as many mutation types as possible, Dr. Gersbach’s group took advantage of the CRISPR-Cas9 system for its ability to target multiple exons simultaneously [78]. It is difficult to target all the mutations known to cause DMD, but this approach greatly expands the range of treatable DMD patients. Targeting other types of mutations, especially large bulk deletions or duplication of multiple exons, is a future challenge.
Ex vivo gene therapy
Considering the stochastic nature of the deletion patterns as well as the risk of off-target mutagenesis by custom nuclease treatment, it is advantageous to predetermine the edited DNA sequence in isolated cells from patients in advance and then, only after proving the gene correction was successful, transplant the cells back to the patient. This “ex vivo” gene therapy approach is particularly useful for evaluating off-target mutagenesis. It has been shown that myoblast cells can be isolated from patient’s muscle biopsy and immortalized by transduction of a transforming factor, such as TERT (telomerase). Such immortalized myoblasts can be expanded unlimitedly and can form fused myofibers once transplanted back to muscle tissue [79, 80]. Such cells serve as ideal targets for genome editing, however, the risk of introducing transforming factors must be carefully examined.
As another cell source to be expanded in vitro are the patient’sown fibro blast cells, which can be isolated and immortalized by transforming genes. Such fibro blast cells can be differentiated into myogenic cells by forced expression of the MyoD (also known as MYOD1) gene, which is a major transcriptional factor of myogenic development [81]. Again, oncogenic risk of the cells due to the introduction of transforming factors must be examined carefully.
In comparison to most somatic cells, except for neoplastic cells, ES cells have unlimited self-renewal capacity and grow rapidly while maintaining normal karyotyping. In addition, ES cells remain pluripotent for the differentiation into different cell types even after massive expansion. Establishment of ES cells requires fertilized embryos, however, the invention of iPS cell technology now allows us to establish cells almost identical to ES cells from the patient’s own somatic cells by the transient expression of transcriptional and transforming factors [82, 83]. So far, hundreds of patient-derived iPS cell lines have been established, including DMD patient-derived iPS cells, which were first reported in 2008 [84].
Due to its unlimited self-renewal capacity and differentiation ability into myoblasts, iPS cell holds great promise as a cell source for ex vivo gene therapy. Dr. Oshimura’s group utilized HAC technology to deliver the full-length human dystrophin gene into DMD-iPS cells to restore dystrophin expression [85]. Viral vector-mediated gene delivery into iPS cells is another option [45], however, strong transcriptional silencing activity inpluripotent stem cells may hamper the stable expression of therapeutic transgenes [86]. Therefore, genome editing approaches have advantages over gene augmentation approaches when targeting ES or iPS cells.
To materialize iPS cell-mediated ex vivo gene therapy, efficient differentiation into the proper cell type after genomic correction is essential (Fig. 4). Differentiation of patient-derived iPS cells into myoblasts is one promising approach, as differentiated myoblasts have the ability to fuse and form myofibers both in vitro and in vivo [87, 88]. Considering that human adult muscle can regenerate damaged muscle tissue from muscle stem cells called satellite cells, another potential approach would be the differentiation of patient-derived iPS cells into muscle stem cells or myogenic progenitor cells [89–91]. It is yet to be determined which cell type is best for engraftment and long-term therapeutic effect.
Genome editing in zygotes
Apart from ex vivo gene therapy approaches, in vivo gene therapy approaches, where genome editing nucleases are directly injected into in vivo tissues, might be possible. Compared with ex vivo gene therapy, which requires the preparation of both the genome editing nuclease and appropriate target cells, in vivo gene therapy is a simpler procedure, as it only requires the preparation of the genome editing nuclease for therapeutic application.
Muscle is one of the most abundant tissues in the human body, and DMD affects the entire muscle mass, therefore, correction of the systematic phenotype is a great challenge. Due to the lack of effective DNA delivery methods into adult tissues, one might consider introducing gene therapy for DMD at the early developmental stage. Dr. Eric Olson’s group reported “prevention” of the muscular dystrophy phenotype in mdx mice by direct injection of CRISPR-Cas9 mRNA, sgRNA and single-strandedoligodeoxynucleotide (ssODN) as a donor template into mouse zygotes [92]. Since totipotent zygotes can give rise to the entire population of cells in our body, gene correction in zygotes is an effective way to correct systemic cells in theory. However, it also brings the ethical debate about germline editing, since these edits will be transmitted to the offspring regardless of the outcome. Current genome editing technology cannot offer 100% correction efficiency and often suffers from several limitations, including mosaicism, random indel patterns, low HR efficiency and off-target mutagenesis. Agroup at Sun Yat-sen University tested the cleavage activity of CRISPR-Cas9 system in human tripronuclear zygotes at the HBB (=β-globin) gene locus [93] and found a surprisingly high off-target mutagenesis rate (up to 7 embryos out of 11 embryos investigated), which deviates from the observations made by many reports of mouse zygotes and human pluripotent stem cell lines, but is consistent with those of cancer cell lines instead. It is not clear from the experimental settings described why this high off-target mutagenesis rate was observed. Possibilities include impaired DNA repair pathways due to the nature of the tripronuclear zygotes, possible problems with the target DNA sequence, contamination of inaccurate sgRNA products during in vitro transcription, or something else. Regardless the conclusions of the report, genome editing procedures are still immature for application into human embryos.
Will germ line editing be necessary to treat DMD patients? To correct the dystrophin mutation, it is first necessary to diagnose where the genetic mutation is located. Recent deep sequencing technologies require only small amounts of DNA template for the identification of genomic mutations from biopsies of pre-implantation embryos. However, the inheritance of DMD follows an X-linked recessive fashion, therefore all female embryos and half of male embryos are not affected by the mutations. This begs the question, will it be necessary to select the affected male embryos and correct the mutated dystrophin gene by genome editing despite the various technical risks described above? Ethically, it would be more appropriate to select the non-affected population of embryos than genetically manipulate the affected one. Of course, the selection of embryos brings up another ethical issue, which is related to the rights of early human embryos. Considering the severity, incurability and lethality of DMD, pre-implantation diagnosis could be justified, and most parents will probably choose to give up on the embryo if DMD is detected. Further discussion will be necessary among scientists, patients, and broadersociety.
Genome editing in vivo
One of the most exciting approaches for DMD gene therapy is in vivo treatment of patient’s muscle tissues by genome editing nucleases. It has been investigated that some serotypes of AAV vector (i.e. AAV serotype 9) can effectively deliver a gene of interest into a wide variety of tissues, including skeletal muscles and cardiac tissues [94, 95].
For conventional gene augmentation therapy, consistent and high-level expression of a therapeutic transgene is essential. However, for genome editing technologies, a simple “hit-and-away” strategy is feasible, as transient expression of the genome editing nuclease is sufficient to induce a stable alteration of the genomic DNA sequence.
Cas9 protein from Streptococcus pyogenes is relatively large, and its cDNA size is about 4.2 kb. Considering the packaging limit of AAV vector, which is typically 4 to 5 kb including the promoter and termination signal, the use of smaller Cas9 is desirable for efficient in vivo delivery by this vector. Dr. Feng Zhang’s group screened several orthogonal Cas9 sequences and found that Cas9 from Staphylococcus aureus has comparable genome editing activity with S. pyogenes Cas9, but significantly smaller cDNA size (around 3.2 kb). The group demonstrated efficient delivery of S. aureus Cas9 into the mouse liver by tail vein injection of AAV2 ITR vector pseudo-typed with AAV8 capsid [67]. Delivery of S. aureus Cas9 into muscle tissues to correct the dystrophin mutation in mice and/or larger animal models of DMD is the next important step towards DMD genome editing gene therapy. Important to note, targeting muscle satellite cells is an attractive approach by Cas9, but AAV6 vector has poor transduction efficiency of muscle satellite cells compared with proliferating myoblasts [96]. Optimizing the proper gene delivery would significantly progress gene targeting in vivo muscle tissues.
Future challenges
The development of easy and efficient genome editing tools, such as TALENs and CRISPR-Cas9 systems, have opened a new era for DMD gene therapy. The collection of disease mutations in cultured cell or mouse models is the beginning of a long journey for clinical applications. Ex vivo gene therapy approaches require determination of the most applicable cell type based on feasibility of the genome editing, engraftment ability after transplantation, immunogenicity, and functional integration into the myofibers. The application of gene correction into satellite cells might be an ultimate hallmark for ex vivo gene therapy, but cultivation, maintenance and transplantation of such satellite cells are still challenging. In vivo gene therapy approaches may be less demanding in terms of technical requirements, however, current genome editing and gene delivery techniques do not offer sufficiently accurate and precise therapeutic application. Immune responses against the corrected dystrophin protein must also be take into consideration [97]. Overcoming these technical limitations would open the door for safe and effective gene therapy for DMD.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
ACKNOWLEDGMENTS
The author would like to thank Hongmei Lisa Li for her technical contributions, and Drs. Mandy Siu Yu Lung and Peter Karagiannis for their critical reading of the manuscript. A. H. is supported in part by JSPS KAKENHI grant (15H05581) and AMED Research Center Network for Realization of Regenerative Medicine grants.
REFERENCES
1 | Monaco AP, Neve RL, Colletti-Feener C, Bertelson CJ, Kurnit DM, Kunkel LM (1986) Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene Nature 323: 6089 646 650 |
2 | Hoffman EP, Brown RH, Kunkel LM (1987) Dystrophin: The protein product of the Duchenne muscular dystrophy locus Cell 51: 6 919 928 |
3 | Blake DJ, Weir A, Newey SE, Davies KE (2002) Function and genetics of dystrophin and dystrophin-related proteins in muscle Physiol Rev 82: 2 291 329 |
4 | Fairclough RJ, Wood MJ, Davies KE (2013) Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches Nat Rev Genet 14: 6 373 378 |
5 | Sironi M, Pozzoli U, Cagliani R, Giorda R, Comi GP, Bardoni A (2003) Relevance of sequence and structure elements for deletion events in the dystrophin gene major hot-spot Hum Genet 112: 3 272 288 |
6 | Aartsma-Rus A, van Deutekom JCT, Fokkema IF, van Ommen G-JB, Dunnen den JT (2006) Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that confirm the reading-frame rule Muscle Nerve 34: 2 135 144 |
7 | Tuffery-Giraud S, Béroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L (2009) Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: A model of nationwide knowledgebase Hum Mutat 30: 6 934 945 |
8 | Flanigan KM, Dunn DM, Niederhausern von A, Soltanzadeh P, Gappmaier E, Howard MT (2009) Mutational spectrum of DMD mutations in dystrophinopathy patients: Application of modern diagnostic techniques to a large cohort Hum Mutat 30: 12 1657 1666 |
9 | Nakamura H, Kimura E, Mori-Yoshimura M, Komaki H, Matsuda Y, Goto K (2013) Characteristics of Japanese Duchenne and Becker muscular dystrophy patients in a novel Japanese national registry of muscular dystrophy (Remudy) Orphanet J Rare Dis 8: 60 |
10 | Nicolas A, Raguénès-Nicol C, Ben Yaou R, Hir SA-L, Chéron A, Vié V (2015) Becker muscular dystrophy severity is linked to the structure of dystrophin Hum Mol Genet 24: 5 1267 1279 |
11 | Yin H, Lu Q, Wood M (2008) Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice Mol Ther 16: 1 38 45 |
12 | Aartsma-Rus A, Fokkema I, Verschuuren J, Ginjaar I, van Deutekom J, van Ommen GJ (2009) Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations Hum Mutat 30: 3 293 299 |
13 | Heemskerk HA, de Winter CL, de Kimpe SJ, van Kuik-Romeijn P, Heuvelmans N, Platenburg GJ (2009) In vivo comparison of 2′-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping J Gene Med 11: 3 257 266 |
14 | Yin H, Betts C, Saleh AF, Ivanova GD, Lee H, Seow Y (2010) Optimization of peptide nucleic acid antisense oligonucleotides for local and systemic dystrophin splice correction in the mdx mouse Mol Ther 18: 4 819 827 |
15 | Aoki Y, Yokota T, Nagata T, Nakamura A, Tanihata J, Saito T (2012) Bodywide skipping of exons 45-55 in dystrophic mdx52 mice by systemic antisense delivery Proceedings of the National Academy of Sciences 109: 34 13763 13768 |
16 | Aoki Y, Yokota T, Wood MJA (2013) Development of Multiexon Skipping Antisense Oligonucleotide Therapy for Duchenne Muscular Dystrophy BioMed Research International 2013: 6176 1 8 |
17 | van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M (2007) Local dystrophin restoration with antisense oligonucleotide PRO051 N Engl J Med 357: 26 2677 2686 |
18 | Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C (2009) Local restoration of dystrophin expression with the morpholino oligomer AVI-in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study The Lancet Neurology 8: 10 918 928 |
19 | Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K (2011) Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study Lancet 378: 9791 595 605 |
20 | Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N (2011) Systemic administration of PRO051 in Duchenne’s muscular dystrophy N Engl J Med 364: 16 1513 1522 |
21 | Chamberlain JS (2002) Gene therapy of muscular dystrophy Hum Mol Genet 11: 20 2355 2362 |
22 | Pichavant C, Aartsma-Rus A, Clemens PR, Davies KE, Dickson G, Takeda S (2011) Current status of pharmaceutical and genetic therapeutic approaches to treat DMD Mol Ther 19: 5 830 840 |
23 | Foster H, Popplewell L, Dickson G (2012) Genetic Therapeutic Approaches for Duchenne Muscular Dystrophy Hum Gene Ther 23: 7 676 687 |
24 | Verhaart IEC, Aartsma-Rus A (2012) Gene therapy for Duchenne muscular dystrophy Curr Opin Neurol 25: 5 588 596 |
25 | Dunckley MG, Wells DJ, Walsh FS, Dickson G (1993) Direct retroviral-mediated transfer of a dystrophin minigene into mdx mouse muscle in vivo Hum Mol Genet 2: 6 717 723 |
26 | Fassati A, Wells DJ, Sgro Serpente PA, Walsh FS, Brown SC, Strong PN (1997) Genetic correction of dystrophin deficiency and skeletal muscle remodeling in adult MDX mouse via transplantation of retroviral producer cells J Clin Invest 100: 3 620 628 |
27 | Fassati A, Wells DJ, Walsh FS, Dickson G (1996) Transplantation of retroviral producer cells for in vivo gene transfer into mouse skeletal muscle Hum Gene Ther 7: 5 595 602 |
28 | MacKenzie TC, Kobinger GP, Kootstra NA, Radu A, Sena-Esteves M, Bouchard S (2002) Efficient transduction of liver and muscle after in utero injection of lentiviral vectors with different pseudotypes Mol Ther 6: 3 349 358 |
29 | Li S, Kimura E, Fall BM, Reyes M, Angello JC, Welikson R (2005) Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin Gene Ther 12: 14 1099 1108 |
30 | Kimura E, Li S, Gregorevic P, Fall BM, Chamberlain JS (2010) Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression Mol Ther 18: 1 206 213 |
31 | Huard J, Krisky D, Oligino T, Marconi P, Day CS, Watkins SC (1997) Gene transfer to muscle using herpes simplex virus-based vectors Neuromuscul Disord 7: 5 299 313 |
32 | Akkaraju GR, Huard J, Hoffman EP, Goins WF, Pruchnic R, Watkins SC (1999) Herpes simplex virus vector-mediated dystrophin gene transfer and expression in MDX mouse skeletal muscle J Gene Med 1: 4 280 289 |
33 | DelloRusso C, Scott JM, Hartigan-O’Connor D, Salvatori G, Barjot C, Robinson AS (2002) Functional correction of adult mdx mouse muscle using gutted adenoviral vectors expressing full-length dystrophin Proc Natl Acad Sci USA 99: 20 12979 12984 |
34 | Kawano R, Ishizaki M, Maeda Y, Uchida Y, Kimura E, Uchino M (2008) Transduction of full-length dystrophin to multiple skeletal muscles improves motor performance and life span in utrophin/dystrophin double knockout mice Mol Ther 16: 5 825 831 |
35 | Roberts ML, Wells DJ, Graham IR, Fabb SA, Hill VJ, Duisit G (2002) Stable micro-dystrophin gene transfer using an integrating adeno-retroviral hybrid vector ameliorates the dystrophic pathology in mdx mouse muscle Hum Mol Genet 11: 15 1719 1730 |
36 | Yuasa K, Miyagoe Y, Yamamoto K, Nabeshima Y, Dickson G, Takeda S (1998) Effective restoration of dystrophin-associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs FEBS Lett 425: 2 329 336 |
37 | Wang B, Li J, Xiao X (2000) Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model Proc Natl Acad Sci USA 97: 25 13714 13719 |
38 | Fabb SA, Wells DJ, Serpente P, Dickson G (2002) Adeno-associated virus vector gene transfer and sarcolemmal expression of a 144 kDa micro-dystrophin effectively restores the dystrophin-associated protein complex and inhibits myofibre degeneration in nude/mdx mice Hum Mol Genet 11: 7 733 741 |
39 | Gregorevic P, Blankinship MJ, Allen JM, Crawford RW, Meuse L, Miller DG (2004) Systemic delivery of genes to striated muscles using adeno-associated viral vectors Nat Med 10: 8 828 834 |
40 | Gregorevic P, Allen JM, Minami E, Blankinship MJ, Haraguchi M, Meuse L (2006) rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice Nat Med 12: 7 787 789 |
41 | van Deutekom JCT, van Ommen G-JB (2003) Advances in Duchenne muscular dystrophy gene therapy Nat Rev Genet 4: 10 774 783 |
42 | Tinsley JM, Potter AC, Phelps SR, Fisher R, Trickett JI, Davies KE (1996) Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene Nature 384: 6607 349 353 |
43 | Tinsley J, Deconinck N, Fisher R, Kahn D, Phelps S, Gillis JM (1998) Expression of full-length utrophin prevents muscular dystrophy in mdx mice Nat Med 4: 12 1441 1444 |
44 | Ivics Z, Hackett PB, Plasterk RH, Izsvak Z (1997) Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells Cell 91: 4 501 510 |
45 | Filareto A, Parker S, Darabi R, Borges L, Iacovino M, Schaaf T (2013) An ex vivo gene therapy approach to treat muscular dystrophy using inducible pluripotent stem cells Nat Commun 4: 1549 |
46 | Hoshiya H, Kazuki Y, Abe S, Takiguchi M, Kajitani N, Watanabe Y (2009) A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4MB entire human dystrophin gene Mol Ther 17: 2 309 317 |
47 | Chapdelaine P, Pichavant C, Rousseau J, Pâques F, Tremblay JP (2010) Meganucleases can restore the reading frame of a mutated dystrophin Gene Ther 17: 7 846 858 |
48 | Grizot S, Smith J, Daboussi F, Prieto J, Redondo P, Merino N (2009) Efficient targeting of a SCID gene by an engineered single-chain homing endonuclease Nucleic Acids Res 37: 16 5405 5419 |
49 | Rousseau J, Chapdelaine P, Boisvert S, Almeida LP, Corbeil J, Montpetit A (2011) Endonucleases: Tools to correct the dystrophin gene J Gene Med 13: 10 522 537 |
50 | Popplewell L, Koo T, Leclerc X, Duclert A, Mamchaoui K, Gouble A (2013) Gene correction of a duchenne muscular dystrophy mutation by meganuclease-enhanced exon knock-in Hum Gene Ther 24: 7 692 701 |
51 | Ousterout DG, Perez-Pinera P, Thakore PI, Kabadi AM, Brown MT, Qin X (2013) Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients Mol Ther 21: 9 1718 1726 |
52 | Li HL, Fujimoto N, Sasakawa N, Shirai S, Ohkame T, Sakuma T (2015) Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9 Stem Cell Reports 4: 1 143 154 |
53 | Ousterout DG, Kabadi AM, Thakore PI, Perez-Pinera P, Brown MT, Majoros WH (2015) Correction of dystrophin expression in cells from duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases Mol Ther 23: 3 523 532 |
54 | Koike-Yusa H, Li Y, Tan E-P, Velasco-Herrera MDC, Yusa K (2014) Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library Nat Biotechnol 32: 3 267 273 |
55 | Bae S, Kweon J, Kim HS, Kim J-S (2014) Microhomology-based choice of Cas9 nuclease target sites Nat Methods 11: 7 705 706 |
56 | Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C (2011) Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting Nucleic Acids Res 39: 12 e82 |
57 | Sander JD, Cade L, Khayter C, Reyon D, Peterson RT, Joung JK (2011) Targeted gene disruption in somatic zebrafish cells using engineered TALENs Nat Biotechnol 29: 8 697 698 |
58 | Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK (2012) FLASH assembly of TALENs for high-throughput genome editing Nat Biotechnol 30: 5 460 465 |
59 | Sakuma T, Ochiai H, Kaneko T, Mashimo T, Tokumasu D, Sakane Y (2013) Repeating pattern of non-RVD variations in DNA-binding modules enhances TALEN activity Sci Rep 3: 3379 |
60 | Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S (2007) CRISPR provides acquired resistance against viruses in prokaryotes Science 315: 5819 1709 1712 |
61 | Jansen R, Embden JDAV, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes Mol Microbiol 43: 6 1565 1575 |
62 | Sorek R, Kunin V, Hugenholtz P (2008) CRISPR–a widespread system that provides acquired resistance against phages in bacteria and archaea Nat Rev Microbiol 6: 3 181 186 |
63 | Marraffini LA, Sontheimer EJ (2008) CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA Science 322: 5909 1843 1845 |
64 | Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA Nature 468: 7320 67 71 |
65 | Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity Science 337: 6096 816 821 |
66 | Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9 Nature 507: 7490 62 67 |
67 | Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ (2015) In vivo genome editing using Staphylococcus aureus Cas9 Nature 520: 7546 186 191 |
68 | Ding Q, Regan SN, Xia Y, Oostrom LA, Cowan CA, Musunuru K (2013) Enhanced efficiency of human pluripotent stem cell genome editing through replacing TALENs with CRISPRs Cell Stem Cell 12: 4 393 394 |
69 | Cradick TJ, Fine EJ, Antico CJ, Bao G (2013) CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity Nucleic Acids Res 41: 20 9584 9592 |
70 | Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V (2013) DNA targeting specificity of RNA-guided Cas9 nucleases Nat Biotechnol 31: 9 827 832 |
71 | Pattanayak V, Ramirez CL, Joung JK, Liu DR (2011) Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection Nat Methods 8: 9 765 770 |
72 | Lin Y, Cradick TJ, Brown MT, Deshmukh H, Ranjan P, Sarode N (2014) CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences Nucleic Acids Res 42: 11 7473 7485 |
73 | Cho S-W, Kim S, Kim Y, Kweon J, Kim HS, Bae S (2014) Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases Genome Res 24: 1 132 141 |
74 | Smith C, Gore A, Yan W, Abalde-Atristain L, Li Z, He C (2014) Whole-Genome Sequencing Analysis Reveals High Specificity of CRISPR/Cas9 and TALEN-Based Genome Editing in Human iPSCs Cell Stem Cell 15: 1 12 13 |
75 | Veres A, Gosis BS, Ding Q, Collins R, Ragavendran A, Brand H (2014) Low Incidence of Off-Target Mutations in Individual CRISPR-Cas9 and TALEN Targeted Human Stem Cell Clones Detected by Whole-Genome Sequencing Cell Stem Cell 15: 1 27 30 |
76 | Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee S-L (2010) Zscan4 regulates telomere elongation and genomic stability in ES cells Nature 464: 7290 858 863 |
77 | Zhao B, Zhang W-D, Duan Y-L, Lu Y-Q, Cun Y-X, Li C-H (2015) Filia Is an ESC-Specific Regulator of DNA Damage Response and Safeguards Genomic Stability Cell Stem Cell 16: 6 684 698 |
78 | Ousterout DG, Kabadi AM, Thakore PI, Majoros WH, Reddy TE, Gersbach CA (2015) Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy Nat Commun 6: 6244 |
79 | Skuk D, Goulet M, Roy B, Chapdelaine P, Bouchard J-P, Roy R (2006) Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells J Neuropathol Exp Neurol 65: 4 371 386 |
80 | Mamchaoui K, Trollet C, Bigot A, Negroni E, Chaouch S, Wolff A (2011) Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders Skelet Muscle 1: 34 |
81 | Chaouch S, Mouly V, Goyenvalle A, Vulin A, Mamchaoui K, Negroni E (2009) Immortalized skin fibroblasts expressing conditional MyoD as a renewable and reliable source of converted human muscle cells to assess therapeutic strategies for muscular dystrophies: Validation of an exon-skipping approach to restore dystrophin in Duchenne muscular dystrophy cells Hum Gene Ther 20: 7 784 790 |
82 | Takahashi K, Yamanaka S (2006) Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors Cell 126: 4 663 676 |
83 | Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors Cell 131: 5 861 872 |
84 | Park I-H, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A (2008) Disease-specific induced pluripotent stem cells Cell 134: 5 877 886 |
85 | Kazuki Y, Hiratsuka M, Takiguchi M, Osaki M, Kajitani N, Hoshiya H (2010) Complete genetic correction of ips cells from Duchenne muscular dystrophy Mol Ther 18: 2 386 393 |
86 | Hotta A, Ellis J (2008) Retroviral vector silencing during iPS cell induction: An epigenetic beacon that signals distinct pluripotent states J Cell Biochem 105: 4 940 948 |
87 | Goudenege S, Lebel C, Huot NB, Dufour C, Fujii I, Gekas J (2012) Myoblasts derived from normal hESCs and dystrophic hiPSCs efficiently fuse with existing muscle fibers following transplantation Mol Ther 20: 11 2153 2167 |
88 | Tanaka A, Woltjen K, Miyake K, Hotta A, Ikeya M, Yamamoto T (2013) Efficient and reproducible myogenic differentiation from human iPS cells: Prospects for modeling Miyoshi Myopathy in vitro PLoS ONE 8: 4 e61540 |
89 | Darabi R, Arpke RW, Irion S, Dimos JT, Grskovic M, Kyba M (2012) Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice Cell Stem Cell 10: 5 610 619 |
90 | Xu C, Tabebordbar M, Iovino S, Ciarlo C, Liu J, Castiglioni A (2013) A zebrafish embryo culture system defines factors that promote vertebrate myogenesis across species Cell 155: 4 909 921 |
91 | Shelton M, Metz J, Liu J, Carpenedo RL, Demers S-P, Stanford WL (2014) Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells Stem Cell Reports 3: 3 516 529 |
92 | Long C, McAnally JR, Shelton JM, Mireault AA, Bassel-Duby R, Olson EN (2014) Prevention of muscular dystrophy in mice by CRISPR/Cas9-mediated editing of germline DNA Science 345: 6201 1184 1188 |
93 | Liang P, Xu Y, Zhang X, Ding C, Huang R, Zhang Z (2015) CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes Protein Cell 6: 5 363 372 |
94 | Inagaki K, Fuess S, Storm TA, Gibson GA, Mctiernan CF, Kay MA (2006) Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8 Mol Ther 14: 1 45 53 |
95 | Zincarelli C, Soltys S, Rengo G, Rabinowitz JE (2008) Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection Mol Ther 16: 6 1073 1080 |
96 | Arnett AL, Konieczny P, Ramos JN, Hall J, Odom G, Yablonka-Reuveni Z (2014) Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells Mol Ther Methods Clin Dev 1 |
97 | Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S (2010) Dystrophin immunity in Duchenne’s muscular dystrophy N Engl J Med 363: 15 1429 1437 |
Figures and Tables
Fig.1
Structure of the dystrophin gene and protein. (a) Human dystrophin gene is located on chromosome Xp 21.2, spans 2.22 Mb in size, and consists of 79 exons for the muscle isoform Dm 427 m. DMD patients suffer from functional loss of dystrophin protein, mainly caused by premature truncation due to a large deletion around exons 40 to 54. (b) Dystrophin protein translated from the Dm427 m isoform consists of 3685 amino acids and is 427 kDa. The protein can be divided into three functional domains: an actin-binding domain, rod domain, and cysteine (Cys)-rich C’-terminal domain. The actin-binding domain at the N’-terminal is important for binding to cytoskeletal actin fibers, and the Cys-rich C’-terminal domain is critical for the binding to the dystroglycan complex at the sarcolemma membrane. The center rod domain consists of several spectrin-like repeats, which form a three-helix bundle-like structure.
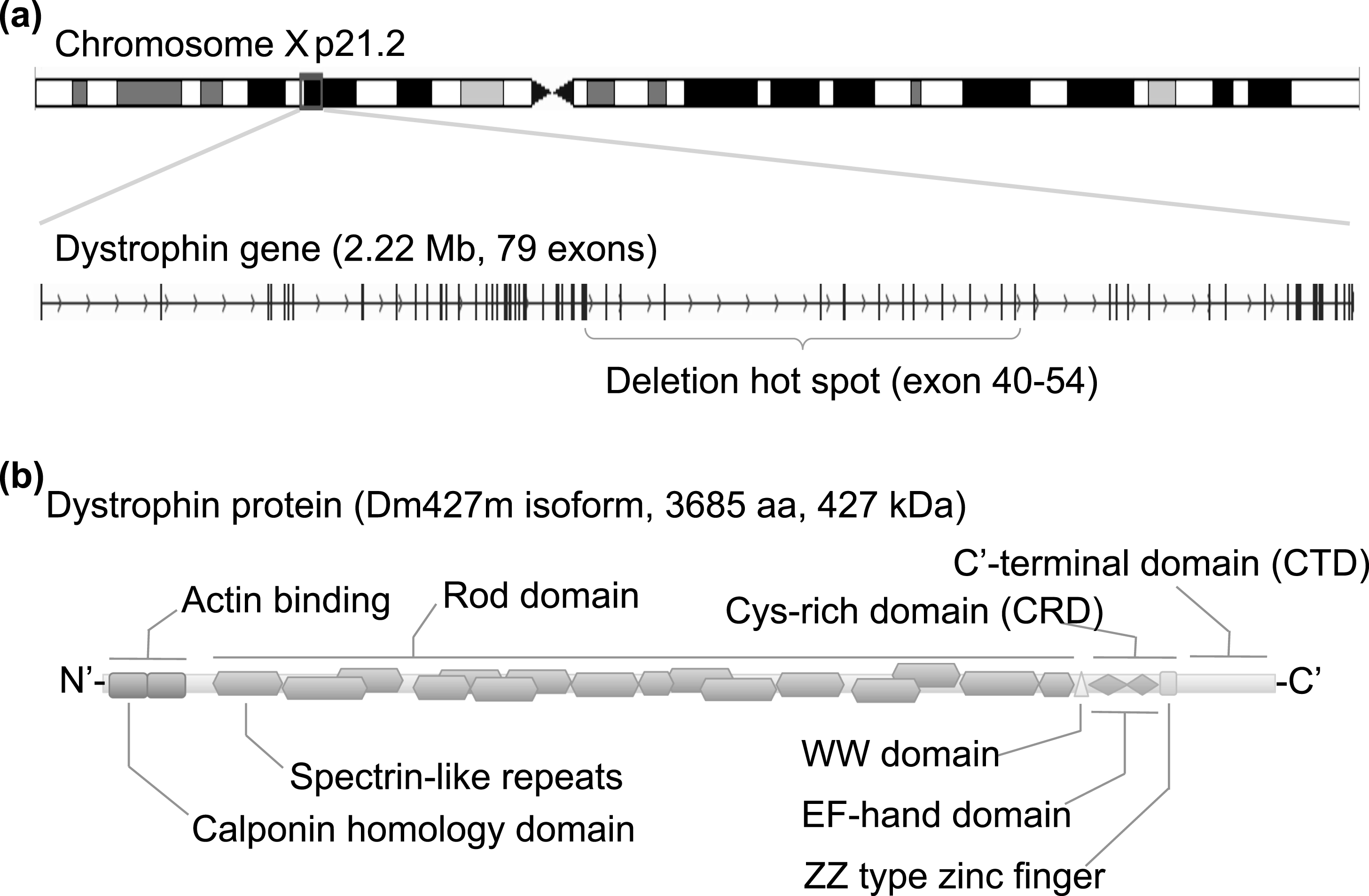
Fig.2
Three therapeutic strategies target distinct biological processes. Gene augmentation approaches deliver functional cDNA, such as micro-dystrophin or micro-utrophin cDNA, to compensate the function of dystrophin protein. Retroviral vectors or DNA transposonvectors stably integrate the therapeutic cDNA into chromosomes randomly, whereas HDAv, AAV, HSV, or HAC vectors remain within the nucleus without integration. Due to the large size of the dystrophin or utrophin cDNA, transduction efficiency is one of the biggest obstacles. Genome editing approaches modify the mutated gene specifically, but off-target mutagenesis is a concern. The delivery of programmable nucleases is unexplored in the context of muscle tissue. Exon skipping uses antisense oligonucleotides to modulate the splicing patterns of a particular exon. Systematic delivery is feasible for antisense oligonucleotides, but the effect is transient. Risk of off-target effects or posttranslational suppression of the target gene should also be considered.
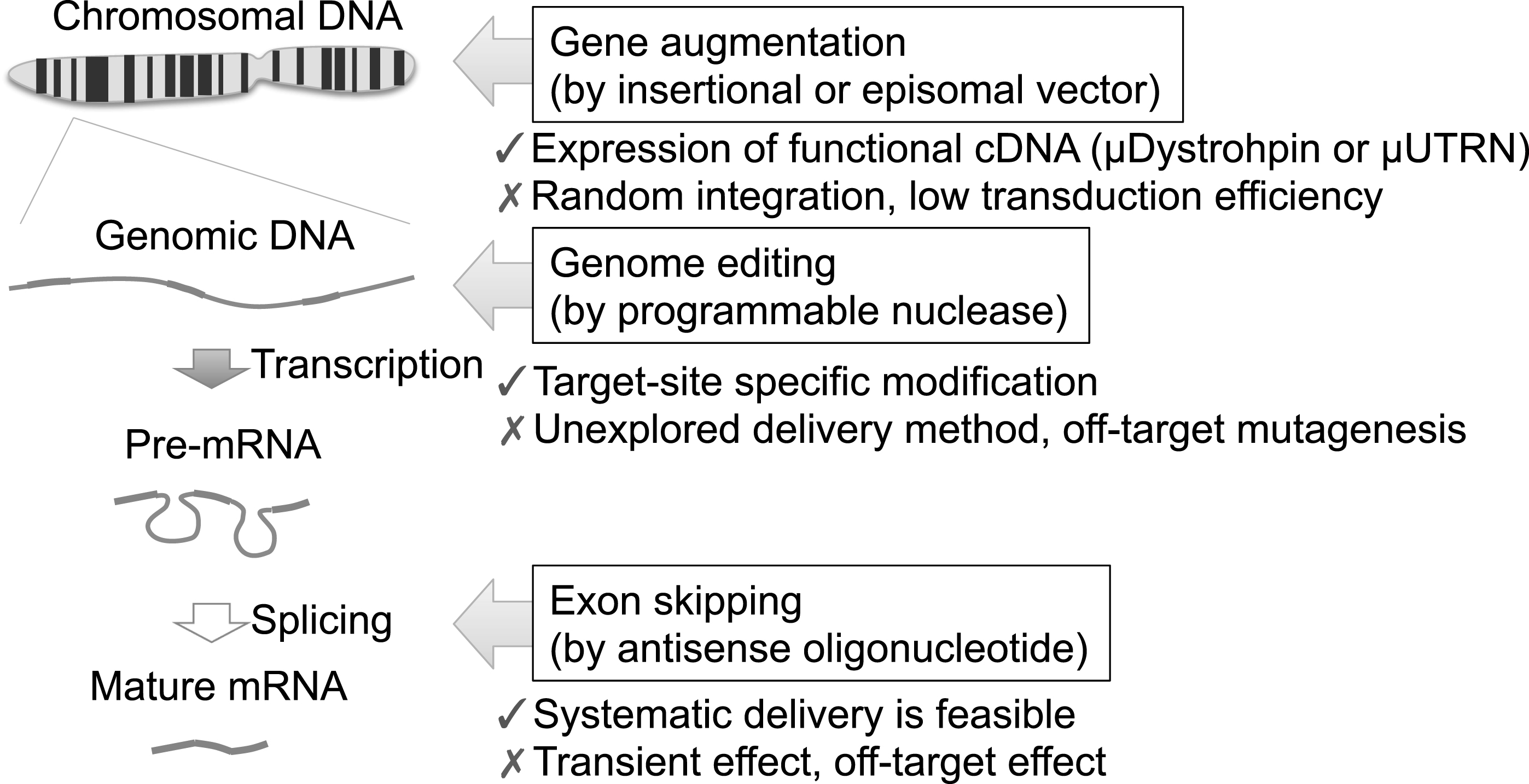
Fig.3
Targetability of programmable nucleases. Programmable nucleases, such as ZFNs, TALENs, and CRISPR-Cas9 systems, have preferences or requirements for their target DNA sequences. The targeting of a particular site of interest can be limited for some nucleases. For example, ZFNs have a sequence preference for “5′-NNMNNMNNC [5 to 7bp spacer] GNNKNNKNN-3′” (where M = C or A, K = G or T, N = A or C or G or T), although this preference depends on the assembly method of the zinc finger domains. TALENs offer much more flexible targetability, as the size of the RVD domain (i.e. typically 15 to 20 repeats) and spacer length (i.e. 14 to 19 bp) can be variable. One major requirement, however, is a “T” 1 bp before the RVD domain. Therefore, the consensus sequence is “5′-TN (×15 ∼20) [14∼19 bp spacer] N (×15 ∼20) A-3′”. CRISPR-Cas9 requires a protospacer adjacent motif (PAM) to recognize its target sequence; e.g. “5′-NGG-3′” for Cas9 derived from Streptococcus pyogenes, and “5′-NNGRRT-3′” for Cas9 derived from Staphylococcus aureus. Each targetable sequence is plotted at around exon 45 of the dystrophin gene. Note that “targetable sequence” only accounts for the patterns matched with the above consensus motifs and does not take other factors, such as the epigenetic status (i.e. DNA CpG methylation), specificity in the human genome, or risk of off-target cleavages, into consideration. Sequence specificity or sequence uniqueness is one of the most important factors for choosing a proper nuclease, as nucleases targeting repeat sequences have high risk for off-target mutagenesis and cellular toxicity. In this regard, we have developed a database to visualize the uniqueness of DNA sequences by stacking a set of unique k-mer sequences (where k = 10 to 16), as indicated in the red histogram on top.
![Targetability of programmable nucleases. Programmable nucleases, such as ZFNs, TALENs, and CRISPR-Cas9 systems, have preferences or requirements for their target DNA sequences. The targeting of a particular site of interest can be limited for some nucleases. For example, ZFNs have a sequence preference for “5′-NNMNNMNNC [5 to 7bp spacer] GNNKNNKNN-3′” (where M = C or A, K = G or T, N = A or C or G or T), although this preference depends on the assembly method of the zinc finger domains. TALENs offer much more flexible targetability, as the size of the RVD domain (i.e. typically 15 to 20 repeats) and spacer length (i.e. 14 to 19 bp) can be variable. One major requirement, however, is a “T” 1 bp before the RVD domain. Therefore, the consensus sequence is “5′-TN (×15 ∼20) [14∼19 bp spacer] N (×15 ∼20) A-3′”. CRISPR-Cas9 requires a protospacer adjacent motif (PAM) to recognize its target sequence; e.g. “5′-NGG-3′” for Cas9 derived from Streptococcus pyogenes, and “5′-NNGRRT-3′” for Cas9 derived from Staphylococcus aureus. Each targetable sequence is plotted at around exon 45 of the dystrophin gene. Note that “targetable sequence” only accounts for the patterns matched with the above consensus motifs and does not take other factors, such as the epigenetic status (i.e. DNA CpG methylation), specificity in the human genome, or risk of off-target cleavages, into consideration. Sequence specificity or sequence uniqueness is one of the most important factors for choosing a proper nuclease, as nucleases targeting repeat sequences have high risk for off-target mutagenesis and cellular toxicity. In this regard, we have developed a database to visualize the uniqueness of DNA sequences by stacking a set of unique k-mer sequences (where k = 10 to 16), as indicated in the red histogram on top.](https://content.iospress.com:443/media/jnd/2015/2-4/jnd-2-4-jnd150116/jnd-2-4-jnd150116-g003.jpg)
Fig.4
Ex vivo gene therapy approaches using iPS cells. A scheme for iPS cell-mediated ex vivo gene therapy approaches for DMD. Skin fibroblasts or monocytes from peripheral blood are reprogrammed to iPS cells by transient expression of the Yamanaka factors. The dystrophin mutation can then be repaired using genome engineering technologies. Such corrected iPS cells can be further differentiated into myoblasts to form myofibers. Either myoblasts or myofibers can be transplanted to patients, but only for transient recovery, as myoblasts or myofibers will eventually die after cellular turnover. An ideal approach would be to differentiate iPS cells into satellite cells, which are muscle stem cells, to gain long-term self-renewal and regeneration capacity in the myofibers. Currently, ex vivo expansion of primary satellite and genome editing is challenging, but progress here could circumvent the use of iPS cells.
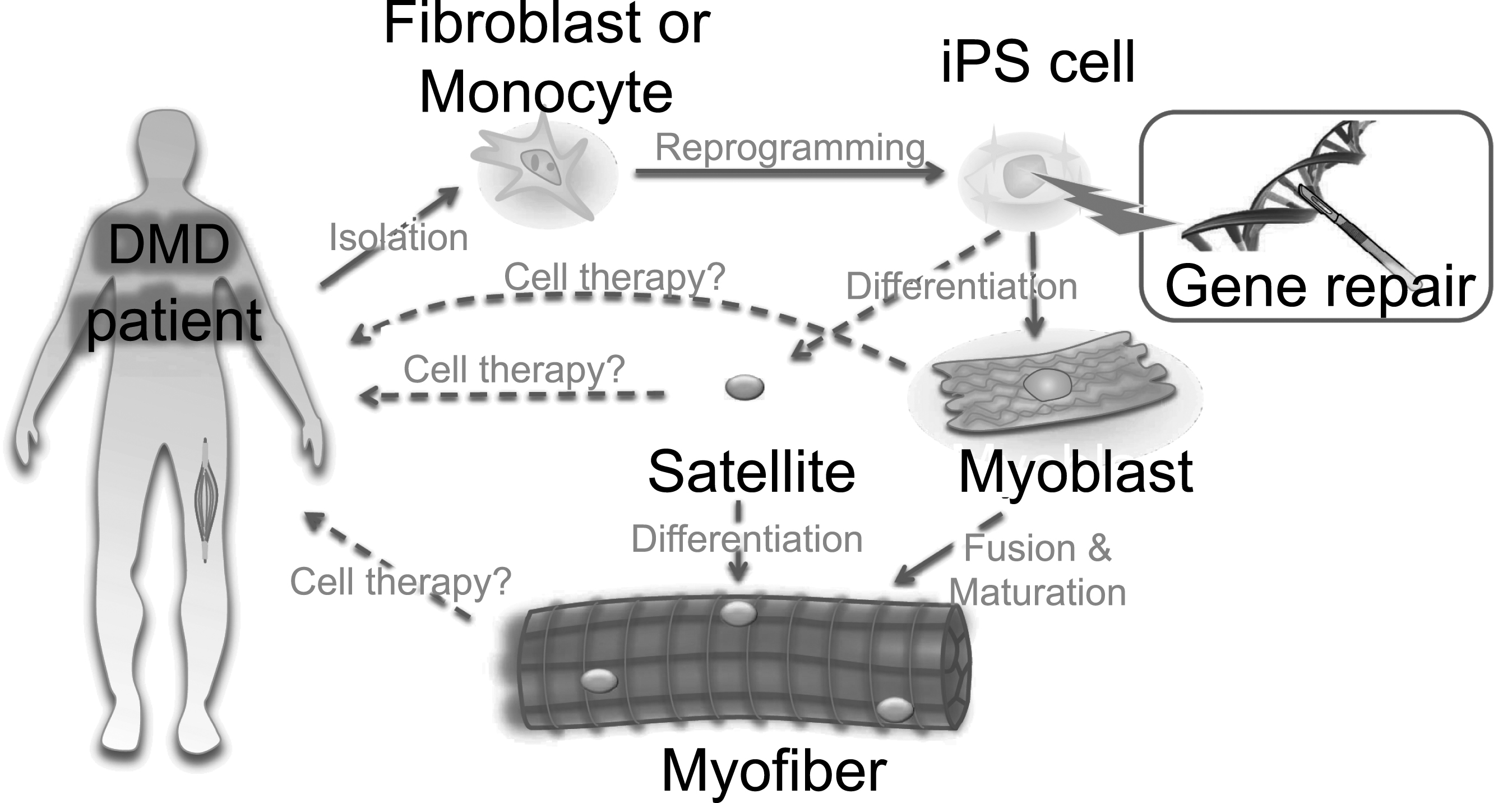
Table 1
Advantages and disadvantages of commonly used viral vectors for DMD gene therapy
| Vector | Packaging size limit | Genomic integration (stable expression) | Infection into non-dividing cells | Major risk/concern in gene therapy application |
| Retrovirus | ∼8 kb | Yes | No | Leukemia |
| Lentivirus | ∼8 kb | Yes | Yes | Leukemia, recombination with HIV |
| Herpes simplex virus | ∼150 kb | No | Yes | Cytotoxicity and immunogenicity |
| Adenovirus (helper-dependent) | ∼30 kb | No | Yes | Immunogenicity against viral capsid |
| Adeno-associated virus (AAV) | ∼4 kb | No | Yes | Inhibited by neutralizing antibody |
Table 2
Advantages and disadvantages of genome editing technologies
| Programmable nuclease | DNA recognition motif | Construction procedures | Sequence targetability |
| Meganuclease | Homing endonuclease LAGLIDADG motifs | Very complicated | Low |
| ZFNs | Zinc finger domains | Complicated | Relatively low |
| TALENs | TAL effector RVD domain | Relatively complicated | Very high |
| CRISPR-Cas9 | crRNA (DNA-RNA heteroduplex) | Easy | Relatively high |


