
Laminin α2 Deficiency-Related Muscular Dystrophy Mimicking Emery-Dreifuss and Collagen VI related Diseases
Abstract
Background: Laminin α2 deficient congenital muscular dystrophy, caused by mutations in the LAMA2 gene, is characterized by early muscle weakness associated with abnormal white matter signal on cerebral MRI.
Objective: To report on 4 patients with LAMA2 gene mutations whose original clinical features complicated the diagnosis strategy.
Methods: Clinical, electrophysiological, muscle imaging and histopathological data were retrospectively collected from all patients. DNA samples were analysed by next-generation sequencing or direct gene sequencing. Laminin α2 was analysed by western-blot and immunohistochemistry.
Results: The four patients achieved independent walking. All had proximal muscle weakness with scapular winging and prominent joint contractures without peripheral neuropathy. During follow-up, two patients suffered from refractory epilepsy associated with brain leukoencephalopathy in one, polymicrogyria and lissencephaly without white matter changes in the other. In two patients, the distribution of fatty infiltration resembles that of collagen-VI related myopathies. Dilated cardiomyopathy contstartabstractwith conduction defects, suggestive of Emery-Dreifuss myopathy, emerged in two of them within the 4th decade. Molecular diagnosis remained elusive for many years. Finally, targeted capture-DNA sequencing unveiled the involvement of the LAMA2 gene in two families, and led us to further identify LAMA2 mutations in the remaining family using Sanger sequencing.
Conclusions: This report extends the clinical and radiological features of partial Laminin α2 deficiency since patients showed atypical manifestations including dilated cardiomyopathy with conduction defects in 2, epilepsy in 2, one of whom also had sole cortical brain abnormalities. Importantly, clinical findings and muscle imaging initially pointed to collagen-VI related disorders and Emery-Dreifuss muscular dystrophy.
INTRODUCTION
Congenital muscular dystrophy (CMD) is a clinically and genetically heterogeneous group of neuromuscular disorders [1]. Laminin alpha 2-deficient CMD (MDC1A, OMIM #607855) is characterized by hypotonia at birth or within the first few months of life, contractures of large joints, elevated serum creatine kinase (CK) levels and abnormal white matter changes although cognitive abilities are normal [2– 4]. MDC1A, caused by a deficiency in the α2 chain of laminin-211 [5] previously named merosin [6], is the most represented form of CMD [7]. Typically, independent ambulation is not achieved in MDC1A with complete laminin-alpha 2 deficiency. Only a few reports have mentioned cardiac involvement associated with MDC1A (for reviews see [8, 9]). Atypical presentations such as adult onset limb-girdle muscular dystrophies with white matter abnormalities have been reported with partial laminin-alpha 2 deficiency [10, 11]. Numerous mutations of the LAMA2 gene encoding the α2 chain of laminin-211 have been identified, scattered along the 64 coding exons and leading to partial or complete protein deficiency [4].
Here we report four adult patients in whom the initial diagnosis suggested by clinical presentation and muscle biopsy findings was mainly consistent with either Bethlem myopathy (BM) or Emery-Dreifuss muscular dystrophy (EDMD). Peculiar features included dilated cardiomyopathy (two patients) and epilepsy (two patients) that occurred during follow-up. Ultimately, molecular diagnosis was achieved through next generation sequencing (NGS), revealing homozygous or compound heterozygous LAMA2 gene mutations. These findings extend the clinical spectrum of LAMA2 mutations to adults, clinically resembling Bethlem myopathy or Emery-Dreifuss muscular dystrophy and emphasize further the critical input of NGS in diagnosing atypical forms of myopathies.
MATERIAL AND METHODS
Clinical assessment
Four patients including two isolated cases (Patients #1 and #2) and two brothers (Patients #3 and #4) were included in this study (Fig. 1). Neurological assessment, CK levels, muscle imaging including whole-body muscle MRI for patients #1 and #2 and muscle CT-scan for patients #3 and #4, cardiac investigations including at least electrocardiogram (ECG) and echocardiography, pulmonary function tests, electroneuromyography and brain MRI or CT scan were retrospectively collected from medical files for each patient (Table 1). All biological samples (blood, skin and muscle biopsies) were obtained after informed consent from patients and their relatives.
Histopathology, immunohistochemistry
Muscle biopsy was performed in 3 out of the 4 patients. Biopsy specimens of deltoid muscle were analysed by light microscopy [12]. Immunohistochemical studies were carried out for all patients on 10μm cryosections for dystrophin, α-, β- and γ-sarcoglycans, dysferlin, α-dystroglycan, desmin, αB-crystallin, myotilin, myosin heavy chain, caveolin 3 and collagen VI. Laminin α2 chain immunostainings were performed after LAMA2 mutations identification and used 3 antibodies directed against the whole protein (NCL-merosin, Leica Novocastra), the N-terminal 300 KDa fragment (4H8-2, Abcam) or the C-terminal 80KDa fragment (MAB 1922, Chemicon International). Representative images were obtained with an Axioplan 2 microscope (Zeiss) equipped with a HBO 100 mercury lamp (Zeiss) and captured using Metaview software (Roper Scientific), using identical exposure time for each antibody.
Western blot analyses
Western blot analyses were performed on proteins extracted from muscle biopsy for dystrophin, α-, β- and γ-sarcoglycans, calpain, dysferlin, α-dystroglycan in all patients, and from lymphoblastoid cell lines (LCL) for emerin in patient #3 according to previous studies [13, 14]. After LAMA2 mutations identification, laminin α2 western blot was performed on muscle samples from patients #1, #2 and #3 as well as in 2 control subjects and a previously identified MDC1A patient with complete laminin α2 deficiency using the C-terminal 80KDa fragment antibody (MAB1922, Chemicon International).
Collagen VI immunolabelling on cultured fibroblasts
Skin biopsies were obtained from patients #1 and #2 and confluent fibroblasts cultures were subjected to two different protocols to detect collagen VI expression and secretion as previously described [15].
Genetics
DNA samples were extracted from blood using standard methods. Linkage studies were conducted using the Illumina Linkage-24 beads array platform, covering around 6000 SNPs and analysed using the Merlin software [16]. As one of the partners of the EU FP7-NMD-Chip consortium, we used the DNA capture array designed in this European project [17]. For patients #1 and #3 who developed cardiomyopathy during the course of their disease, 46 genes involved in cardiomyopathies were studied through a targeted DNA capture assay (TruSight Cardiomyopathy, Illumina). Validation and segregation of variants were subsequently performed using the Sanger sequencing method.
RESULTS
Clinical findings
The main clinical and muscle imaging findings in the 4 patients are reported in Table 1 and Fig. 1.
Patient #1 is of Turkish origin with no known parental consanguinity. He was first seen before he developed any cardiac disease. He was clinically diagnosed with Bethlem myopathy at 28 years of age, mainly due to keloid scars and joint contractures including Achilles tendons (lengthened at 15 and 25 years old) and finger flexors requiring surgical repair at 25. At 25 years of age, themuscle CT scan pattern was compatible with a collagen-VI related myopathy. Indeed, the rectus femoris had an area of central hypodensity. The fatty infiltration was localized in the peripheral region of the vastus lateralis. The semimembranosus, semitendinosus and biceps femoris, as well as adductor muscles were only mildly involved, whereas the gracilis was spared. In the legs, the fatty infiltration was localized between the gastrocnemius (lateralis and medialis) and the soleus. He then progressively developed dilated cardiomyopathy from his 30’s that suggested a diagnosis of EDMD.
Patient #2 is the eldest boy of consanguineous parents from Algeria. He achieved independent ambulation at 13 months, despite severe hypotonia during his first months of life. For a long time he was considered as suffering from an autosomal recessive form of limb girdle muscular dystrophy (LGMD). He experienced a femur fracture at 16 years which led to a worsening of lower limb muscle weakness. He was wheelchair dependent at 27 years although he could walk 5 meters at his last assessment at 32 years old. Muscle MRI, performed at 29 years of age, showed severe and diffuse fatty infiltration of the proximal muscles, lumbar extensors, and abdominal wall muscles. By contrast, the superficial layers of the vastus lateralis, the semimembranosus, the semitendinosus and the gracilis muscles were less affected. In the legs, the fatty infiltration surrounded the soleus muscle and was localized at the periphery of gastrocnemius medialis and lateralis, whereas the tibialis anterior, extensor and tibialis posterior muscles were almost preserved. Atypical features in the LGMD context included mixed partial and generalized epilepsy that he developed from 6 years of age which was successfully treated with carbamazepine until his 20 s. Afterwards, he developed severe pharmacoresistant partial-onset epilepsy with up to 30 partial seizures per day. Various antiepileptic drugs were tried that did not significantly change seizures frequency, except for Vigabatrin which was finally withdrawn because of visual field defects. Extensive brain imaging revealed bilateral occipital lissencephaly and polymicrogyria on T1 weighted sequence, without any white matter changes. Cardiac assessment was always normal during his follow-up. Occurrence of extensive joint contractures, follicular hyperkeratosis without keloids pointed to collagen VI-related myopathy as a possible diagnosis.
Patients #3 and #4 are brothers from non-consanguineous Portuguese parents. They had normal motor development and are still ambulatory at 50 and 54 years respectively. Due to extensive joint contractures, the presence of premature ventricular contractions and left ventricular dysfunction found in the youngest brother since 37 years of age, EDMD was suggested. Muscle CT scans, performed at 43 and 47 years of age, respectively, showed at the thigh level advanced hypodensity of the vastus lateralis and intermedius muscles, the semimembranosus, the semitendinosus, the biceps femoris and the adductors whereas the gracilis, sartorius, vastus medialis and rectus femoris muscles were spared. At the leg level, peroneus longus and extensor digitorum longus muscles were mildly affected in patient #4 while only the right peroneus longus muscle was mildly affected in patient #3. The other leg muscles including tibialis anterior, soleus, gastrocnemii and tibialis posterior were spared. In the upper limbs, deltoid and biceps brachialis muscles had a mild fatty infiltration. Atypical features included triceps brachii, quadriceps and gastrocnemius muscles hypertrophy and abnormal brain MRI that showed leukencephalopathyhy. In addition, patient #4 developed several episodes of loss of consciousness with eye rolling and postictal amnesia, unresponsiveness preceded by blurred vision, paraesthesia in the lower limbs and spreadingto the head. EEG was either normal or showed bi-occipital spike-waves predominating within the right side. Extensive neurological investigations including cerebrospinal fluid analysis, sensory and auditory evoked responses were normal while visual evoked responses showed delayed central and peripheral responses. Ophthalmologic examination, skin and rectal biopsies, arylsulfatase, beta-galactosidase, hexosaminidase and long chain fatty acid dosage were normal. The epilepsy was assumed to be post-traumatic and the patient was treated by phenobarbital, sodium valproate and finally by carbamazepine and diazepam.
CK levels were usually elevated in all patients ranging from 2.5 to 8 times normal levels. All electroneuromyographic studies, performed in adulthood, showed normal nerve conduction studies and myogenic pattern at needle electromyography.
Histological, immunohistological and biochemical findings
Muscle biopsies for patients #1 and #2 showed nonspecific dystrophic features including increased internal nuclei and connective tissue, leading to an initial suspicion of BM (Fig. 2A). Muscle biopsy from patient #3 revealed fibre size variation, moderate endomysial fibrosis, atrophic angulated myofibers and fiber type grouping suggesting a neuropathic pattern (data not shown). Routine immunohistochemical stainings, including α-dystroglycan, showed normal expression of all proteins analysed in the patients (data not shown). Muscle biopsy multiplex western blotting demonstrated that dystrophin, α- and γ-sarcoglycan, calpain 3, dysferlin and α-dystroglycan, were normally expressed, except in protein extracts from patient #3, which revealed mildly reduced amounts of α- and γ-sarcoglycan (data not shown). Western blot analysis on lymphoblastoid cells from patient #3 revealed normal emerin expression (data not shown). Collagen VI immunolabelling was normal in muscle cryosections and cultured dermal fibroblasts from patient #1 while reduced COLVI secretion was detected in fibroblasts from patient #2 (data not shown).
Genetic studies
According to the diagnostic pathway tree for each patient, several genes including LMNA (patients #1– 3), EMD (patients #1– 3), the 3 collagen VI encoding genes COL6A1-3 (patients #1 and #2), SGCA and SGCG (patient #3), FKRP (patients #1 and #3), FHL1 (patients #1 and #3), CAPN3 and DES (patient #1) and FSHD-1 (patient #2), were screened and ruled out. Importantly, no pathogenic mutation was found in patients #1 and #3 in the panel of 46 genes known to be associated with isolated or syndromic cardiomyopathies with or without skeletal muscle involvement. In parallel, linkage analysis was performed on patient #2 family and two regions presented a lod-score>1.8 (on chr2 and chr6).
In the context of the European NMD-Chip project, we turned to NGS technologies under the hypothesis of a recessive mode of inheritance. We identified a novel nonsense LAMA2 (NM_000426.3) mutation in patient #1 (c.4936G>T; p.Glu1646 *) and a previously reported one in patient #2 (c.2230C>T; p.Arg744 *) [18]. Both mutations are homozygous in the patients and lead to premature termination codons (PTC) in the laminin α2 chain (Table 1, Fig. 1). Mutations were confirmed by Sanger sequencing and screened in other family members. The father and the four non-affected siblings of patient #1 carried the c.4936G>T mutation in a heterozygous state, while the parents and the two non-affected siblings of patient #2 carried the c.2230C>T mutation in a heterozygous state. In light of these LAMA2 mutations and of the phenotypic similarities between patients #1 and #2 and the 2 brothers from the remaining family (patients #3 and #4), the 65 LAMA2 exons were directly sequenced in the latter with the Sanger method. Two compound heterozygous variants were identified (Table 1, Fig. 1): the first (c.1854_1861dup; p.Leu621Hisfs *7) had already been described as pathogenic [19] and the second (c.2461A>C; p.Thr821Pro) was previously submitted to the Leiden muscular dystrophy pages LAMA2 database (www.dmd.nl/). It has been predicted pathogenic by SIFT, Polyphen2 and UMD predictor tools [20]. The c.2461A>C was found at heterozygous state in the mother, whereas the c.1854_1861dup was transmitted by the father. The unaffected brother of the patients did not carry any of these mutations (Fig. 1).
Laminin α2 expression analyses
Immunohistochemical stainings of laminin α2 chain in patient #3, performed prior to any genetic analyses, was considered as normal (data not shown). In patients #1 and #2, laminin α2 chain stainings were performed post-mutation identification and were found to be abnormal (Fig. 2B), associated with an increased expression of laminin α5 (data not shown). In fact, patient #1 showed only slight irregularities and weakness of laminin α2 staining while patient #2 showed absence of laminin α2 staining when using the N-terminal 300 KDa and the C-terminal 80KDa fragment antibodies and a moderate reduction with the whole protein antibody (Fig. 2B). All these data were consistent with laminin α2 partial deficiency. Interestingly, western-blot studies in the 3 patients showed normal size but reduced laminin α2 chain amounts in muscle biopsy extracts particularly in patients #1 and #2 (Fig. 2C). For patient #3, laminin α2 amount appears to be reduced by half (Fig. 2C).
DISCUSSION
The clinical presentation of most patients carrying LAMA2 gene mutation is relatively similar with severe congenital hypotonia, delayed milestones with inability to achieve independent ambulation, extensive joint contractures, muscle weakness, normal mental development associated with white matter abnormalities revealed by brain MRI, and markedly raised CK levels [21, 22]. However, milder forms presenting as LGMD have been reported [10, 23]. Additional features have been rarely described [24] and may include subclinical cardiac involvement and neuronal migration defects associated with diffuse white matter changes leading to variable degrees of epilepsy and mental retardation [11, 23, 25, 26].
From the clinical point of view, our patients came to our attention at an adult age for diagnosis purpose after a long atypical disease course and the molecular diagnosis proved challenging. Over the years, the diagnostic hypothesis varied from BM (Patients #1 and #2) to EDMD (Patients #1, #3 and #4) and even to LGMD (Patients #2 and #4). However no mutation was found in the main candidate genes. The presence of extensive joint contractures combined with muscle imaging pattern resembling collagen VI related myopathy (Patient #1), follicular hyperkeratosis with non-conclusive muscle MRI pattern (Patient #2) led to misdiagnosis in these 2 patients. Although collagen VI secretion was reduced in culturedfibroblasts, COL6A1-3 genes screening remained normal in patients #1 and #2. We suspect this deficit in COLVI secretion to be non-specific and likely secondary to an as of yet uncharacterized altered homeostasis of the dermal fibroblasts from the patient. To our knowledge this pattern of fatty infiltration surrounding the muscle has not been yet reported in LAMA2 mutated patients. Therefore, LAMA2 gene analysis is warranted in patients with prominent joint contractures, especially those with secondary collagen VI abnormalities [27]. Patient #2 also presented with pharmacoresistant epilepsy and extensive bilateral occipital micropolygyria. Epilepsy and focal cortical dysplasia associated with cognitive deterioration have been reported in addition to the classical diffuse white matter abnormalities in some cases with total laminin α2 deficiency [25, 28, 29] and more recently in a 7 year-old patient with epilepsy, cognitive deterioration, extensive bilateral occipital polymicrogyria who lost ambulation [30]. These reports are in contrast with patient #2 who displayed only partial laminin α2 deficiency and is still able to walk a few steps within his 4th decade of life. Indeed patients showing a LGMD-like form of laminin α2-deficient CMD were already pointed out previously, indicating that partial laminin α2 deficiency should be kept in mind in the work-up diagnosis of adult forms of LGMD [10].
Cardiac involvement was rarely reported in MDC1A patients [31, 32]. The first comprehensive cardiac assessment in a series of MDC1A patients was reported by Spyrou et al. [33]. The authors noticed that the left ventricular ejection fraction in laminin α2-negative children was significantly lower (43% ± 11% ) compared with laminin α2-positive children (53% ±5% ). In a subsequent review, Jones et al. [24] identified a range of electrocardiogram and echocardiogram abnormalities in 7 out of 20 patients (35% ) with more than half being asymptomatic. In 2010, Germanyeh et al. [11] reported 5 patients with absent laminin α2 staining and cardiac abnormalities including mitral regurgitation, pulmonary hypertension, palpitations and wall motion hypokinesia on echocardiogram (Table 2). More recently, 3 patients (Table 2) have been described with cardiopathy: 2 with dilated cardiomyopathy, one of whom with ventricular arrhythmia [9] and one with impaired left ventricular contractility [34]. All of these reports combined with our patients (Patients #1 and #3) suggest that laminin α2 deficiency may indeed lead to an overt cardiomyopathy. To exclude any digenic phenomenon that would explain the presence of cardiac disease, we excluded other cardiomyopathy causative genes including those associated with cardiac and skeletal muscle diseases.
From the genetic point of view, three of the mutations reported here were previously reported with a clear deleterious effect on laminin α2 chain when isolated or associated with other mutation in a compound heterozygous state (Table 2). The 8 pb insertion c.1854_1861dup identified in patients #3 and #4 was identified in 3 patients with 2 showing classical MDC1A presentation [19]. The homozygous c.1854_1861dup mutation (p.Leu621Hisfs *7) or the compound heterozygous c.1854_1861dup and c.7750-1713_7899-2153del (p.Ala2584Hisfs *8) induce a frameshift resulting in the total absence of laminin α2 in muscle. The c.2230C>T (p.Arg744 *) homozygous mutation in patient #2 was previously reported in a brother and sister with similar presentation, although the sister was less severely affected [18]. The brother had a limb girdle muscular weakness starting at 15 years of age, severe contractures involving neck, elbows, ankles and knees; mild peripheral neuropathy, diffuse white matter abnormalities and normal cardiac evaluation at 39 years of age. Immunohistochemical and western blotting analyses disclosed partial laminin α2 chain deficiency. A smaller transcript arising from skipping of the exon containing the mutation and retaining the open reading frame was detected, thereby sustaining the presence of a partially functional laminin α2 chain. The other missense mutation identified in patients #3 and #4 (c.2461A>C, p.Thr821Pro) was already reported, associated with a missense variation (c.812C>T, p.Thr271Ile) at compound heterozygous state [34]. The patient had an exclusive central nervous system involvement showing among other things, refractory epilepsy associated with progressive cognitive regression since the age of 6, area of agyria in the occipital cortex on brain MRI, extensive white matter abnormalities with swelling and resulting widening of gyri. Surprisingly, there was normal muscular strength, tone and deep tendon reflexes, with the highest CK level being 1589 UI/L and no cardiac involvement. Laminin-α2 immunohistochemical studies revealed partial and irregular labelling [34]. The c.2461A>C, p.Thr821Pro was also reported in combination with an intronic mutation (c.5234 + 1G>A) in 2 patients: a Canadian one with classical MDC1A phenotype and cervical spine fusion, seizures and abnormal white matter (www.dmd.nl/) and in a Portuguese patient [34] (Table 2). In the latter, the phenotype was highly similar to the 2 brothers reported here withlimb-girdle weakness, rigid spine syndrome, brain white matter abnormalities and impaired left ventricular contractility, whereas the partial laminin α2 deficiency described in the third patient carrying the compound heterozygous c.1854_1861dup and missense mutation c.3832G>T, p.Gly1278Cys results from the production at the plasma membrane of a mutant form of laminin α2.
Beyond the classical MDC1A phenotype with absence of merosin expression [5], milder forms have been described in individuals with partial laminin α2 deficiency. In general, patients with absence of laminin α2 expression have an early presentation within the first months of life, typically do not acquire independent ambulation, usually require enteral feeding and ventilatory support [11]. By contrast, individuals with partial laminin α2 deficiency tend to have a milder form of CMD or may rarely have onset after infancy, delay in walking or proximal muscle weakness. They may show muscle pseudohypertrophy and/or rigid spine and are usually classified within the LGMD spectrum although they show characteristic white matter abnormalities [8, 10, 35– 39]. Our 4 patients may be included in the milder group since they all had preserved ambulation at advanced ages and none of them required respiratory support. All these features are in agreement with the partial laminin α2 deficiency of variable severity uncovered by immunolabelling and western blot, patient #2 displaying the most pronounced deficiency. Theoretically, the 2 homozygous mutations observed in patients #1 and #2 should lead to absence of laminin α2 and thus to a classical MDC1A phenotype. Since laminin α2 is still present, even at low levels, one may expect that a posttranscriptional event such as an aberrant splicing which restores the LAMA2 open reading frame as previously reported [19, 34]. For patients #3 and #4 the missense mutation in exon 18 may result in the translation of a protein carrying the mutated amino acid like previously described [34].
Taken together, these observations suggest that the diagnosis of laminin α2 deficient related diseases should be considered in adult patients presenting a myopathy with joint contractures and brain abnormalities (either white matter changes or structural abnormalities) or even cardiomyopathy. Since the cardiomyopathy may appear later in life, regular monitoring of cardiac function and electrocardiogram are required in these patients. Moreover, in atypical forms of LGMD without definite molecular diagnosis, brain MRI can offer a diagnostic clue either to suspect LAMA2 as a candidate gene or to contribute in the validation of LAMA2 mutations since NGS will be used more frequently for molecular genetic assessment of LGMDs.
In addition, all dystrophic muscle biopsies, regardless of clinical phenotype, should be studied with multiple antibodies against different regions of laminin α2 using immunohistochemistry or western blot to detect LAMA2 partial defects. This report also draws attention to the potential overlap between the 2 most common forms of CMD: MDC1A caused by the deficiency of laminin α2 chain and collagen VI-related muscular dystrophies.
CONFLICT OF INTEREST
Authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank patients and their family members for their cooperation. We also thank Corine Gartioux, Nathalie Deburgrave, Caroline Beugnet, Céline Ledeuil, Emmanuelle Lacène and Maud Beuvin for technical assistance for protein immunolabellings and western blot studies. Thanks to Delphine Bouteiller and Giovanni Stevanin for the help on the linkage study.
This work was financially supported by the Institut National de la Santéet de la Recherche Médicale; the UniversitéPierre et Marie Curie Paris 06, the Centre National de la Recherche Scientifique, the Association Française contre les Myopathies, Assistance Publique-Hôpitaux de Paris and the NMD-CHIP Consortium, a FP7 HEALTH project of the European Commission (Development of Targeted DNA Chips for High Throughput Diagnosis of NeuromuscularDisorders – Collaborative Project – FP7 Grant Agreement Number: HEALTH-F5-2008-223026; to GB, IN and VA).
REFERENCES
1 | Sparks S, Quijano-Roy S, Harper A, Rutkowski A, Gordon E, Hoffman EPCongenital muscular dystrophy overview2011 |
2 | Banker BQ(1994) The congenital muscular dystrophiesEngel AG, Franzini-Amstrong CMyology. 22nd ed12751289New YorkMcGraw-Hill |
3 | Tome FM(1999) The Peter Emil Becker Award lecture The saga of congenital muscular dystrophyNeuropediatrics30: 25565 |
4 | Bonnemann CG, Wang CH, Quijano-Roy S, Deconinck N, Bertini E, Ferreiro A(2014) Diagnostic approach to thecongenital muscular dystrophiesNeuromuscul Disord24: 4289311 |
5 | Tome FM, Evangelista T, Leclerc A, Sunada Y, Manole E, Estournet B(1994) Congenital muscular dystrophy withmerosin deficiencyComptes Rendus de l’Academie Des Sciences317: 4351357 |
6 | Aumailley M(2013) The laminin familyCell Adhesion & Migration7: 14855 |
7 | Norwood FL, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V(2009) Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic populationBrain132: Pt 1131753186 |
8 | Herrmann R, Straub V, Meyer K, Kahn T, Wagner M, Voit T(1996) Congenital muscular dystrophy with laminin alpha 2 chain deficiency: Identification of a new intermediate phenotype and correlation of clinical findings to muscle immunohistochemistryEuropean Journal of Pediatrics.155: 1196876 |
9 | Carboni N, Marrosu G, Porcu M, Mateddu A, Solla E, Cocco E(2011) Dilated cardiomyopathy with conduction defectsin a patient with partial merosin deficiency due to mutations in the laminin-alpha2-chain gene: A chanceassociation or a novel phenotype?Muscle & Nerve44: 5826828 |
10 | Gavassini BF, Carboni N, Nielsen JE, Danielsen ER, Thomsen C, Svenstrup K(2011) Clinical and molecularcharacterization of limb-girdle muscular dystrophy due to LAMA2 mutationsMuscle & Nerve44: 5703709 |
11 | Geranmayeh F, Clement E, Feng LH, Sewry C, Pagan J, Mein R(2010) Genotype-phenotype correlation in a largepopulation of muscular dystrophy patients with LAMA2 mutationsNeuromuscul Disord.20h: 4241250 |
12 | Dubowitz V. Muscle Biopsy: A pratical approach. Second edition. Eastbourne I, Tindal B, editors1985. |
13 | Manilal S, Recan D, Sewry CA, Hoeltzenbein M, Llense S, Leturcq F(1998) Mutations in Emery-Dreifuss musculardystrophy and their effects on emerin protein expressionHum Mol Genet7: 5855864 |
14 | Deburgrave N, Daoud F, Llense S, Barbot JC, Recan D, Peccate C(2007) Protein- and mRNA-basedphenotype-genotype correlations in DMD/BMD with pointmutationsand molecular basis for BMD with nonsense andframe-shift mutations in the DMD geneHuman Mutation.28: 2183195 |
15 | Deconinck N, Richard P, Allamand V, Behin A, Laforet P, Ferreiro ABethlem myopathy: Long-term follow-up identifies COL6 mutations predicting severe clinical evolutionJ Neurol Neurosurg Psychiatry2014 |
16 | Abecasis GR, Cherny SS, Cookson WO, Cardon LR(2002) Merlin–rapid analysis of dense genetic maps using sparse gene flow treesNat Genet30: 197101 |
17 | Nectoux J, de Cid R, Baulande S, Leturcq F, Urtizberea JA, Penisson-Besnier IDetection of TRIM32 deletions in LGMD patients analyzed by a combined strategy of CGH array and massively parallel sequencingEur J Hum Genet2014 |
18 | Di Blasi C, He Y, Morandi L, Cornelio F, Guicheney P, Mora M(2001) Mild muscular dystrophy due to a nonsense mutationin the LAMA2 gene resulting in exon skippingBrain124: Pt 4698704 |
19 | Oliveira J, Santos R, Soares-Silva I, Jorge P, Vieira E, Oliveira ME(2008) LAMA2 gene analysis in a cohort of 26 congenitalmuscular dystrophy patientsClin Genet.74: 6502512 |
20 | Frederic MY, Lalande M, Boileau C, Hamroun D, Claustres M, Beroud C(2009) UMD-predictor, a new prediction tool fornucleotide substitution pathogenicity – application to fourgenes: FBN1, FBN2, TGFBR1, and TGFBR2Human Mutation30: 6952959 |
21 | Fardeau M, Tomé FMS, Helbling-Leclerc A, Evangelista T, Ottolini A, Chevallay M(1996) Dystrophiemusculaire congénitale avec déficience en mérosine: Analyse clinique,histopathologique, immunocytochimique et génétiqueRev Neurol Paris152: 1119 |
22 | Quijano-Roy S, Sparks S, Rutkowski ALAMA2-related muscular Dystrophy2012 |
23 | Løkken N, Born AP, Duno M, Vissing JLAMA2-related myopathy; frequency among congenital and limb-girdlemuscular dystrophiesMuscle & Nerve2015 |
24 | Jones KJ, Morgan G, Johnston H, Tobias V, Ouvrier RA, Wilkinson I(2001) The expanding phenotype of lamininalpha2 chain (merosin) abnormalities: Case series and reviewJournal of Medical Genetics38: 10649657 |
25 | Pini A, Merlini L, Tomé FMS, Chevallay M, Gobbi G(1996) Merosin-negative congenital muscular dystrophy,occipital epilepsy with periodic spasms and focal cortical dysplasia. Report of three Italian cases in two familiesBrain and Development18: 316322 |
26 | Martinello F, Angelini C, Trevisan CP(1998) Congenital muscular dystrophy with partial merosin deficiency and lateonset epilepsyEuropean Neurology40: 13745 |
27 | Eymard B, Ferreiro A, Ben Yaou R, Stojkovic T(2013) Muscle diseaseswith prominent joint contractures: Main entities and diagnosticstrategyRev Neurol (Paris).169: 8-9546563 |
28 | Trevisan CP, Martinello F, Ferruzza E, Angelini C(1995) Divergence of central nervous system involvement in 2 Italiansisters with congenital muscular dystrophy, a clinical and neuroradiological follow-uEur Neurol35: 230235 |
29 | Philpot J, Cowan F, Pennock J, Sewry C, Dubowitz V, Bydder G(1999) Merosin-deficient congenital musculardystrophy: The spectrum of brain involvement on magnetic resonance imagingNeuromuscul Disord9: 8185 |
30 | Vigliano P, Dassi P, Di Blasi C, Mora M, Jarre L(2009) LAMA2 stop-codonmutation: Merosin-deficient congenital muscular dystrophy withoccipital polymicrogyria, epilepsy and psychomotor regressionEurJ Paediatr Neurol13: 17276 |
31 | Prelle A, Comi GP, Rigoletto C, Turconi C, Felisari G, Ciscato P(1997) An atypical case of partial merosindeficiency congenital muscular dystrophyJ Neurol244: 6391395 |
32 | Sewry CA, Philpot J, Sorokin LM, Wilson LA, Naom I, Goodwin F(1996) Diagnosis of merosin (laminin-2) deficientcongenital muscular dystrophy by skin biopsyThe Lancet347: 582584 |
33 | Spyrou N, Philpot J, Foale R, Camici PG, Muntoni F(1998) Evidence of left ventricular dysfunction in children withmerosin-deficient congenital muscular dystrophyAmerican Heart Journal136: 3474476 |
34 | Marques J, Duarte ST, Costa S, Jacinto S, Oliveira J, Oliveira ME(2014) Atypical phenotype in two patients withLAMA2 mutationsNeuromuscul Disord24: 5419424 |
35 | Hayashi YK, Ishihara T, Domen K, Hori H, Arahata K(1997) A benignallelic form of lamininα2 chain deficient musculardystrophyLancet349: 1147 |
36 | Cohn RD, Herrmann R, Sorokin L, Wewer UM, Voit T(1998) Laminin alpha 2 chain-deficient congenital muscular dystrophy:Variable expression in severe and mild casesNeurology51: 94101 |
37 | Naom I, D’Alessandro M, Sewry C, Philpot J, Manzur AY, Dubowitz V(1998) lamininα2-chain genemutations in two siblings presenting with limb-girdle muscular dystrophyNeuromusc Disord8: 495501 |
38 | He Y, Jones K, Vignier N, Morgan G, Chevallay M, Barois A, Estournet-Mathiaud B, Hori H, Mizuta T, Tomé FMS, North KN, Guiceheney PMild Congenital muscular dystrophy with primarypartial laminin a2 chain deficiency: Molecular studyNeurology2001 |
39 | Rajakulendran S, Parton M, Holton JL, Hanna MG(2011) Clinical and pathological heterogeneity in late-onset partialmerosin deficiencyMuscle & Nerve44: 4590593 |
Figures and Tables
Fig.1
Patients carrying LAMA2 mutations. Patient #1 presents distal and proximal joint contractures. The muscle CT scan showed a predominantly central hypodensity in the rectus femoris (yellow arrows), in the peripheral region of the vastus lateralis (asterisks) and the gastrocnemius medialis and lateralis muscles (red arrows). Patient #2 also presents distal and proximal joint contractures. Whole body MR showed diffuse fatty infiltration of pelvic. The superficial layers of the vastus lateralis, semimembranosus, semitendinosus and gracilis muscles were less affected. The fatty infiltration surrounded the soleus muscle and was localized at the periphery of gastrocnemius medialis and lateralis, wheras the tibialis anterior, the extensors and the tibialis posterior muscle were almost spared. Brain MRI disclosed a posterior polymicrogyria (blue arrows) and lissencephaly (white arrows). Patient #3 and #4 have mild ankle and elbow contractures. Muscle CT scan showed marked hypodensity of the vastus lateralis and intermedius, the semimembranosus, the semitendinosus, the biceps femoris and the adductor muscles, paraspinal muscles, mild involvement of right peroneus longus muscle in patient #3 and peroneus longus and extensor digitorum longus muscles in patient #4, whereas the gracilis, sartorius, vastus medialis, rectus femoris, gastrocnemii, soleus, tibialis anterior muscles were spared. In the upper limbs, deltoid and biceps brachialis muscles present a mild fatty infiltration. Brain MRI of patient #3 showed a symmetric leukoencephalopathy(green arrows). (Colours are visible in the online version of the article; http://dx.doi.org/10.3233/JAD-150093).
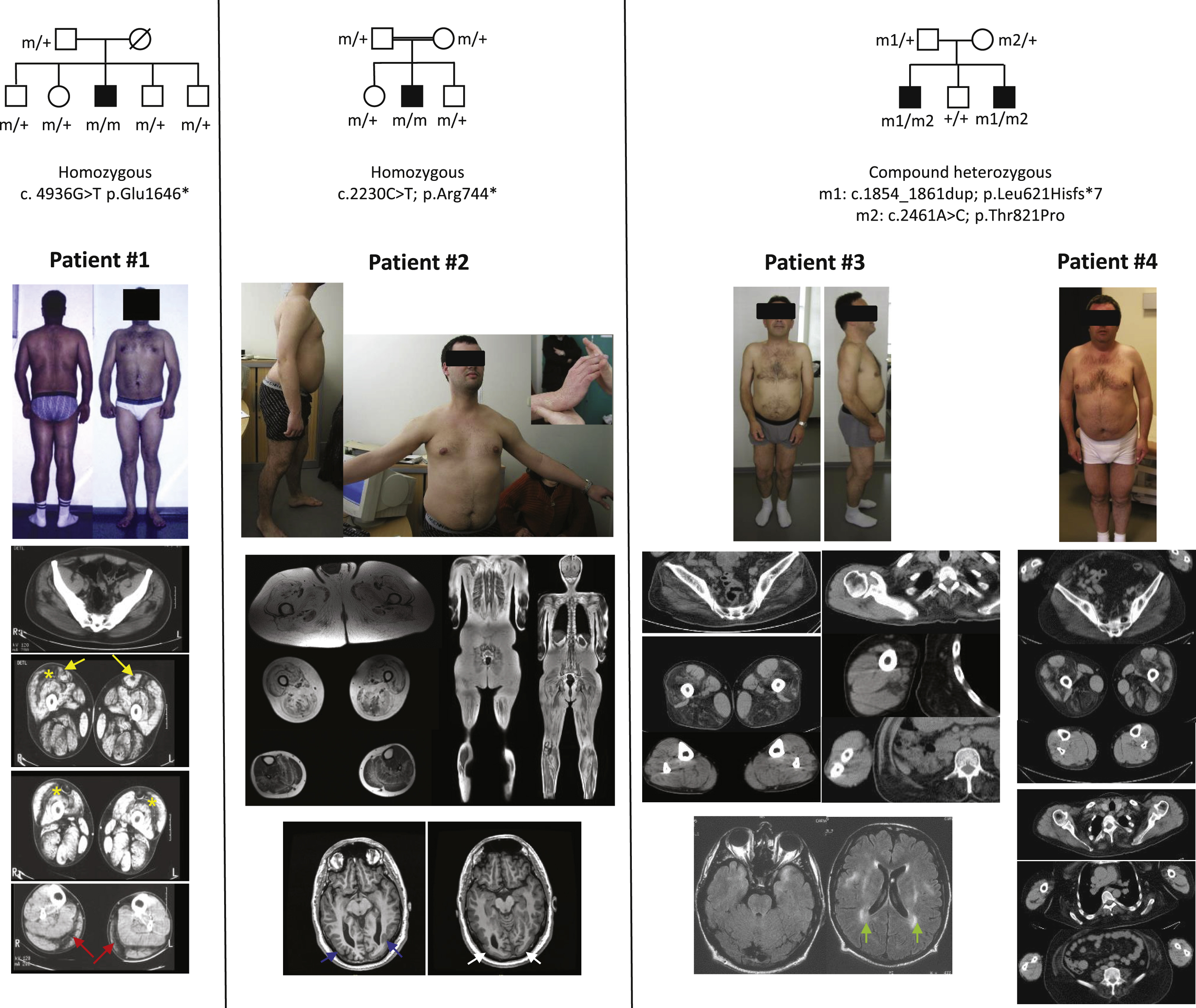
Fig.2
Muscle histology, immunolabelling with laminin α2 antibodies and western blot studies. Histology of muscle biopsies from patients #1 and #2 using Haematoxylin Eosin, modified Gomori trichrome and ATPase pH 9.4 are presented. Bar: 50μm. (B) Immunostaining on muscle cryosections from a control individual (CT), patient #1and patient #2. Three antibodies against different regions of the α2 chain of laminin were used: 4H8-2, NCL and MAB1922, Bar: 50μm. (C) Western blot analysis of muscle biopsies form patients #1– 3, using MAB1922 antibody with densitometry values below the blot.
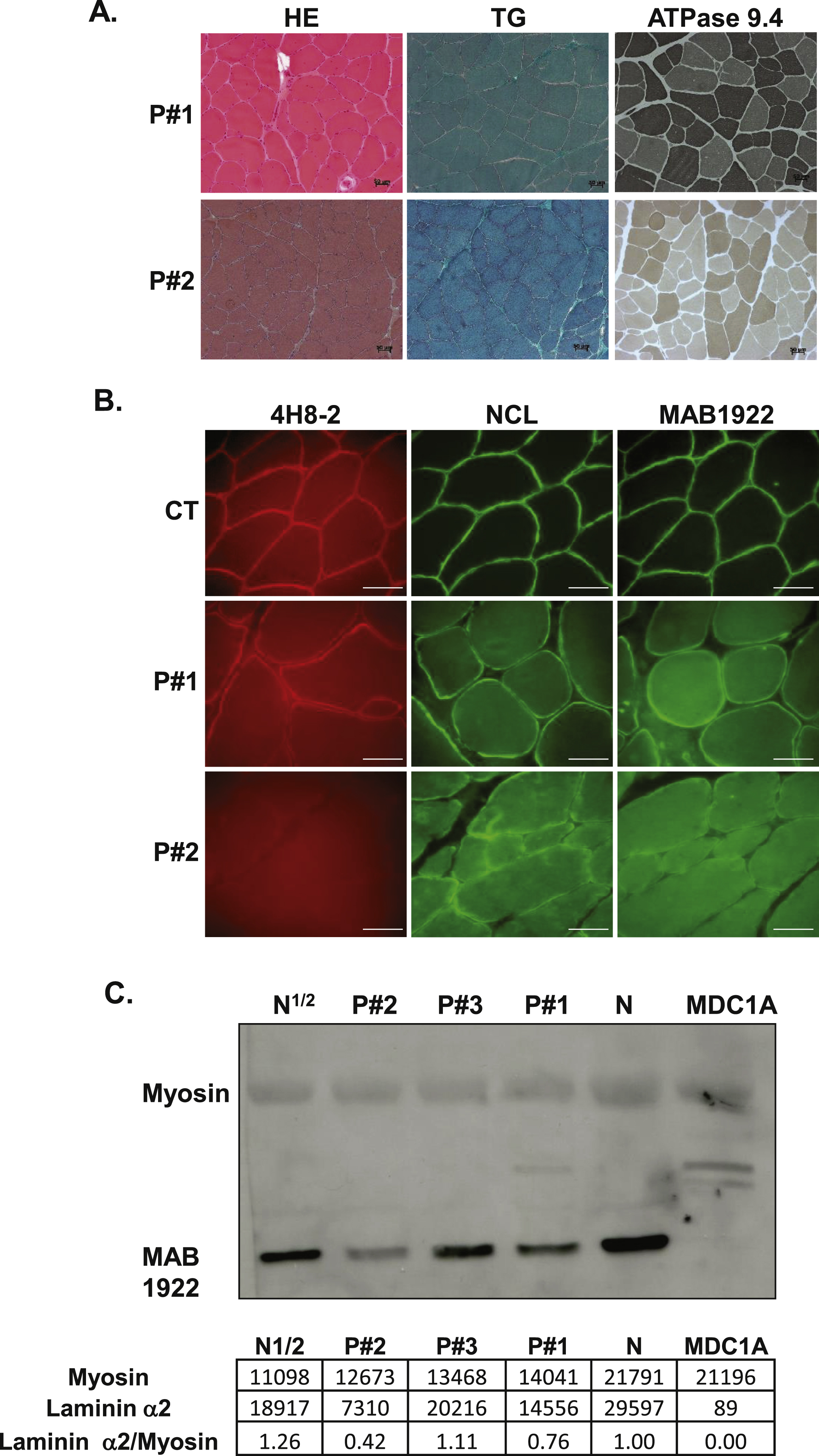
Table 1
Summary of the clinical features of the 4 patients
| Case # | Age at last assessment | First symptoms / age at onset | Maximal motor achievement / Walton scale at last assessment | CK level (X normal level) | Muscle weakness distribution | Joint Contractures | Fatty involvements on muscle imaging | Epilepsy (age at onset)/ Treatment | Brain imaging | Cardiac involvement / Last LVEF (% | Last measured FVC (%) | Other features | LAMA2 mutation (Status, acid changes) |
| 1 | 38 | Calves myalgia at running, tip-toe walking / 10y | WI at 1 / 6 | 8X | Prox (LL+UL), mild scapular winging | N, A, E, W, FF | GMa, GMe, GMm, all anterior and posterior compartment thigh muscles, deeper layers of GM, GL, VL, RF (central hypodensity) | No | Normal | DCM, sinusal rythm, VT, ICD at 35 y / 30 | 80 | Calf hypertrophy | Homozygous: c.4936G>T, p.Glu1646* |
| 2 | 32 | Severe hypotonia but achieve walking at 1 y / birth | WI at 1 / 3 | 2.5X | Prox (UL+LL), axial (flexors, extensors), scapular winging | N, A, H, E, W, FF | Periphery of LL, UL and axial muscles | Partial epilepsy (6 y) / Carbamazepine until 20 y then pharmacoresistant | Hippocampal polymicrogyria, complex bilateral cortical polymicrogyria and lissencephaly | No / normal | 78 | Follicular hyperkeratosis | Homozygous, c.2230C>T, p.Arg744* |
| 3 | 49 | Spine and elbows stiffness / 6y | WI at 1 / 3 | 4X | Prox (LL+UL), scapular winging | Sh, E, W, FF, K, A, RS | Del, BB, BH, GMa, GMe, GMm, PS, VL and hamstrings and paraspinal muscles, right PL | No | Brain MRI = Multiple and bilateral white matter hypersignals within frontal, parietal and temporal regions | DCM, sinusal rhythm, resuscitated sudden death due to VF / 35 | 52 | Quadriceps and calf hypertrophy | Compound heterozygous, c.1854 1861dup; p.Leu621Hisfs*7 / c.2461A>C, p.Thr821Pro |
| 4 | 47 | Delayed walking and frequent falls / 2.5y | WI at 2.5 / 3 | 3X | Prox (LL+UL), no scapular winging | Sh, E, FF, K, A, RS | Del, BB, BH, GMa, GMe, GMm, PS, VL, VI, all hamstrings muscles asymmetrically affected, paraspinal muscles, PL and EDL, sparing of VM, RF, SA, AL | Post-traumatic generalized epilepsy (18)/ successively treated by phenobarbital, sodium valproate and finally by carbamazepine and diazepam | Brain MRI = Multiple and bilateral white matter hypersignals in a patchy pattern within periventricular and infracortical regions involving also temporal lobes | No / 58 | – | VL hypertrophy | Compound heterozygous c.1854_1861dup; p.Leu621Hisfs *7 / c.2461A>C; p.Thr821Pro |
A: ankles, AL: adductor longus muscle, BH: biceps humeri muscle, CK: creatine kinase; DCM: dilated cardiomyopathy; E: elbows, ENMG: electroneuromyography; FF: finger flexors, FVC: forced vital capacity; GM: gastrocnemius medial head, GMa, GMe, GMm: gluteus maximus, medius and minimus muscles, GL: gastrocnemius lateral head, GR: gracilis muscle, H: hips, ICD: implantable cardioverter defibrillator; K: knees, LL: lower limbs; MP: myopathic pattern; MRI: magnetic resonance imaging; N: neck, NCV: nerve conduction velocities, PM: pacemaker; Prox (proximal), PS: psoas muscle, RF: rectus femoris muscle, RS: rigid spine, SA: sartorius muscle, Sh: shoulders, UL: upper limbs, VA: ventricular arrhythmia, VM: vastus medialis muscle, VI: vastus intermedius muscle, VL: vastus lateralis muscle, W: wrists, WI: walked independently, Y: year.
Table 2
Previous reports of patients with LAMA2 mutations either identified in our patients or with overt cardiac disease
| Ref. | Patient # | LAMA2 mutations / mRNA study | Laminin α2 staining | Overall phenotype | Cardiac involvement | Other features |
| [19] | 5 | Homozygous: c.1854_1861dup, p.Leu621Hisfs *7 / mRNA: frameshift | Absent | MDC1A | NI | No cognitive delay, no seizures. Brain MRI = white matter changes and no cortical abnormalities |
| [19] | 11 | c.1854_1861dup, p.Leu621Hisfs *7 / c.3832G>T, p.Gly1278Cys / mRNA: frameshift | Partial deficiency | Slowly progressive Spastic paraparesis | NI | Slight cognitive delay, no seizures. Brain MRI = white matter changes and no cortical abnormalities |
| [19] | 12 | c.1854_1861dup, p.Leu621Hisfs *7 / exon 56 deletion (c.7750-1713_7899-2153del) / mRNA: frameshift | Absent | MDC1A | NI | No cognitive delay, no seizures. Brain white matter changes and no cortical abnormalities |
| [18] | Brother and sister | Homozygous: c.2230C>T, p.Arg744 * / skipping of exon 15 | Partial deficiency | LGMD with contractures | Absent at 39 years | Mild peripheral neuropathy revealed by somatosensory brainstem evoked potentials testing, Brain MRI = diffuse white matter abnormalities in periventricular and subcortical areas |
| [34] | 1 | c.2461A>C, p.Thr821Pro / c.812C>T, p.Thr271Ile / No splicing defects in the vicinity of the mutations | Partial deficiency | Exclusive central nervous system involvement (cognitive impairment and refractory epilepsy) | Absent | Macrocephaly, refractory epilepsy with progressive cognitive regression, bilateral divergent strabismus, normal muscular strength, tone and deep tendon reflexes, highest CK level 1589 UI/L, area of agyria in the occipital cortex on brain MRI, extensive white matter abnormalities and swelling and widening of gyri. Normal motor function. |
| LDB | c.2461A>C; p.Thr821Pro / c.5234 + 1 G>A | NI | MDC1A | NI | Cervical spine fusion, seizures. Brain MRI = white matter abnormalities. | |
| [34] | 2 | c.2461A>C, p.Thr821Pro / c.5234 + 1 G>A, p.Val1765Serfs *21 | Partial deficiency | Rigid spine syndrome and limb-girdle weakness. | Impaired left ventricular contractility | Brain white matter abnormalities |
| [11] | 38 | c.4035T>G, p.Tyr1345 *, | Absent | MDC1A | Wall hypokinesia on echocardiogram | Ventilatory support, scoliosis, mild contractures, ophthalmoplegia, enteral feeding at 10y, typical MDC1A. White matter change on brain MRI, |
| homozygous | (at 13 years) | |||||
| [9, 18] | – | c.4405T>C, p.Cys1469Arg / | Partial deficiency | Myopathy | Dilated cardiomyopathy, ventricular arrhythmia | Mild wasting proximal leg muscles, calf hypertrophy, mild weakness all leg muscles; decreased knee reflexes, ankle reflexes brisk without other pyramidal signs; pes cavus, leukoencephalopathy |
| c.4645C>T, p.Arg1549 * | resembling Inclusion Body Myositis | |||||
| [11] | 44 | c.7881T>G, p.His2627Gln, | Absent | MDC1A | Wall hypokinesia on echocardiogram | Decreased lung capacity (<70% FVC), scoliosis, mild contractures, feeding problems |
| homozygous | (at 20 years) |
LDB: Leiden muscular dystrophy pages database, NI: not investigated.


