
Transthyretin-Related Familial Amyloid Polyneuropathy (TTR-FAP): A Single-Center Experience in Sicily, an Italian Endemic Area
Abstract
Background: Familial amyloid polyneuropathy related to transthyretin gene (TTR-FAP) is a life-threatening disease transmitted as an autosomal dominant trait. Val30Met mutation accounts for the majority of the patients with large endemic foci especially in Portugal, Sweden and Japan. However, more than one hundred other mutations have been described worldwide. A great phenotypic variability among patients with late- and early-onset has been reported.
Objective: To present a detailed report of TTR-FAP patients diagnosed in our tertiary neuromuscular center, in a 20-year period.
Methods: Clinical informations were gathered through the database of our center.
Results: The study involved 76 individuals carrying a TTR-FAP mutation. Three phenotypes were identified, each corresponding to a different TTR variant, homogeneous within and heterogeneous between each other: i) Glu89Gln mutation, characterised by 5th – 6th decade onset, neuropathy as presenting symptoms, early heart dysfunction, cardiomyopathy as major cause of mortality followed by dysautonomia and cachexia; ii) Phe64Leu mutation, marked by familiarity reported in one-half of cases, late onset, severe peripheral neuropathy, moderate dysautonomia and mild cardiomyopathy, death for wasting syndrome; iii) Thr49Ala mutation, distinguished by onset in the 5th decade, autonomic disturbances as inaugural symptoms which may remain isolated for many years, moderate polyneuropathy, cachexia as major cause of mortality followed by cardiomyopathy.
Conclusions: This survey highlighted a prevalence of 8.8/1,000,000 in Sicily Island. Good knowledge of the natural history of the disease according to different TTR mutations allow clinicians to optimise multiprofessional care for patients and to offer carriers a personalized follow-up to reveal first signs of the disease.
INTRODUCTION
Familial amyloid polyneuropathy (FAP) associated with mutations in the transthyretin (TTR) gene is the most common form of genetic amyloidosis. It is a progressive devastating disease transmitted as an autosomal dominant trait, with fatal outcome occurring within ten years after onset. It accounts several thousand cases worldwide, with Val30Met mutation identified in most patients with endemic foci in Portugal, Sweden and Japan. However, more than one hundred other mutations have been described, with varying geographic distributions, and organ involvement [1].
In peripheral nerves, amyloid accumulates around endoneurial capillaries. Damage of unmyelinated fibers occurs early. As the disease progresses, density of small and then larger myelinated fibers is reduced, and blood vessels are frequently invaded and destroyed by amyloid [1]. The harmful result of endoneurial amyloid deposits in ganglia and nerves occurs through a mechanical effect and also through a toxic effect, which could be also triggered by Schwann cells [2]. Suggested pathogenic mechanisms include endoneurial oedema, nerve ischemia, oxidative stress, inflammation, apoptosis, each of which could become therapeutic targets [3–6].
The most typical presentation of TTR-FAP is a progressive length-dependent sensory-motor polyneuropathy, which usually begins with loss of thermal and pain sensation in the feet and slowly ascends up the limbs. Another type of clinical appearance starts with focal deficits resulting from local deposits of amyloid [1]. Both are associated with variable autonomic disturbances and extra-neurological manifestations, especially a cardiomyopathy. Some patients with an early-onset presentation deteriorate quickly because of autonomic dysfunction and rapid progression of the sensory-motor deficit. Conversely, in many patients with a late onset (6th to 8th decade), the polyneuropathy progresses slowly, often with a cardiac involvement but with less autonomic dysfunction. Diagnosis is based on family history, neurographic evidence of a prevalent axonal polyneuropathy, identification of amyloid deposits in the tissues, and detection of TTR mutation. The diagnosis can be challenging in sporadic cases and when clinical manifestations are not typical [7, 8]. Diagnostic pitfalls include inadequate attention of neurologists to autonomic symptoms, decreased nerve conduction velocity and increased protein content in the cerebrospinal fluid leading to a wrong diagnosis of chronic inflammatory demyelinating polyneuropathy (CIDP), no detection of amyloid at biopsy, coincident diabetes mellitus or monoclonal gammopathy [9, 10].
Treatment often requires a multidisciplinary approach for symptomatic management of orthostatic hypotension, cardiac failure, gastrointestinal disorders, malnutrition, neuropathic pain. Liver transplantation (LT) provides a specific therapy by allowing for the suppression of the main source of mutant TTR [11]. However, its effectiveness is higher in Val30Met versus non-Val30Met patients, is influenced by nutritional status, age, severity of neuropathy and cardiac involvement, and is associated to surgery risks and unending use of immunosuppressants. Tafamidis meglumine (Vyndaqel, Pfizer), a selective TTR kinetic stabilizer that inhibits the amyloid cascade, has been approved by European Medicines Agency in 2011, and it seems promising [12–16]. Very recently, antisense technology and interfering RNA therapeutics have been developed, respectively by Isis Pharmaceutical Inc. and Alnylam Pharmaceuticals Inc. Both treatments displayed potent and dose-dependent suppression of mutant and normal TTR levels in humans and randomized, double-blind, placebo controlled phase 3 studies are ongoing [17].
In the common Val30Met mutation, a considerable phenotypic variation with late- and early-onset has been reported in endemic areas, suggesting that unknown genetic or environmental factors are important in the clinical expression [18, 19]. Some other mutations are thought to be associated with particular phenotypes, although a great variability among patients carrying the same mutation has also been described [20]. In Northern Italy, Val30Met is carried by roughly one fourth of the patients with non-endemic distribution [20]. In South Italy, according to our experience in a tertiary neuromuscular center, Val30Met mutation is absent, and only three mutations are found with an endemic distribution. The three cohorts are quite homogeneous but different from each other in age of onset, phenotype, severity, diagnostic difficulty and management. We report here their clinical and laboratory characteristics and phenotype-to-genotype correlations.
MATERIALS AND METHODS
A retrospective, observational study was performed involving 76 individuals (36 M; 40 F) carrying a TTR mutation, all of Sicilian origin, and followed longitudinally at Neuromuscular Center of the AOU Policlinico Hospital, Messina, Italy in a 20-year period (1995–2015). Our Center is the only neuromuscular center in a wide area including Sicily and the near Calabria Region. The study included living symptomatic patients (n. 34), deceased patients (n. 23) and asymptomatic carriers (n. 19). The latter were relatives of patients, alive or dead, who had asked for a genetic testing because of 50% risk for TTR-FAP and had received a positive DNA result [21]. Two symptomatic patients carrying a TTR mutation (Glu89Gln n. 1 and Phe64Leu n. 1) with associated other cause of peripheral neuropathy (diabetes mellitus and multiple myeloma, respectively) were not included in this survey but were accounted for prevalence calculation.
We characterized the family history, clinical manifestations, course, laboratory findings, and treatments. Demographic details and geographical distribution of affected families including place of residence and place of origin were also investigated. The prevalence of TTR-FAP (the number of patients per 1,000,000 persons) was estimated by correction of the number of patients with the data availability at December 31st, 2014 and population in Sicily region at January 1st, 2014 according to National Institute of Statistics. The total population in Sicily on that date was 5,094,937.
All individuals were questioned in a similar manner about symptoms indicative of motor (distal weakness), sensory (paraesthesias, numbness, distal burning, pain) and autonomic (impotence, diarrhoea, persistent constipation, symptoms of gastric and bladder paresis, orthostatic hypotension, syncope, abnormal sweating) dysfunction. Compound Autonomic Dysfunction Test (CADT) was also used to evaluate the main symptoms of autonomic dysfunction [22]. Periodically, they also had neurological examination, routine urine and blood analysis, electromyography and electroneurography [23]. The Charcot-Marie-Tooth neuropathy score (CMTNS), which is a reliable and valid composite score comprising symptoms, signs, and neurophysiological tests of sensory and motor functions, was used [24].
A battery of cardiovascular autonomic tests was performed. The four tests on predominantly parasympathetic function were: beat to beat variability during quiet breathing and deep breathing; heart rate response during the Valsalva manoeuvre and in the standing position; the two tests of predominantly sympathetic function were blood pressure responses to standing and sustained handgrip [25, 26]. Based on the results of the above mentioned tests, an ‘autonomic score’ was given to each subject in the range 0–12 (0 for normal, 1 for borderline and 2 for abnormal test). We considered the presence of a definite autonomic dysfunction if the autonomic score was ≥4, and a borderline status when the score was in the range of 2–3 [27, 28].
Cardiological evaluation included electrocardiography (ECG), 2D-echocardiography with Doppler examination, cardiac magnetic resonance imaging (MRI) and Technetium-99m-diphosphonate (99mTc-DPD) scintigraphy [29–32]. New York Heart Association (NYHA) classification was used to assess cardiac functional capacity. Left inter-ventricular septum (IVS) thickening due to amyloidosis was defined as end-diastolic thickness of the left ventricular walls (anterior septal thickness or posterior wall thickness) greater than 12 mm (in the absence of any other cause of ventricular hypertrophy). 99mTc-DPD scintigraphy image analysis was performed by experienced analysts; two sets of images were obtained (5 min. and 3 hours after radiotracer injection). Visual scoring of cardiac retention was: score 0, absent cardiac uptake and normal bone uptake; score 1, mild cardiac uptake, lower than bone uptake; score 2, moderate cardiac uptake associated with reduced bone uptake; or score 3, strong cardiac uptake with mild/absent bone uptake. Semiquantitative analysis of whole-body retention (WBR), heart retention (HR), and heart/whole body (H/WB) ratio were evaluated from region-of-interest (ROI) drawings in the standard manner. In brief, rectangular ROIs were drawn over the heart, and irregular ROIs were drawn over the kidneys and bladder on anterior images. These ROIs were copied and mirrored on posterior images, and geometric means of the 2 projections were calculated for each ROI. All ROIs were corrected for background counts. Total counts in the images were considered as whole-body counts. Early whole-body counts were used to represent the injected activity. WBR was evaluated by comparing counts in the late images (corrected for decay and scan speed, and subtracting the activity in the urinary tract and bladder) with the counts in the early whole-body images. HR was evaluated by comparing decay-corrected counts of the heart in late images with counts in early whole-body images. The H/WB ratio was calculated by dividing counts in the heart by counts in late whole-body images [33].
Almost all subjects have been also enrolled in THAOS – the Transthyretin Amyloidosis Outcomes Survey – which is a worldwide, longitudinal, observational survey designed to characterize differences in geographically dispersed patient populations [34]. The protocol was approved by our Local Ethics Committee.
Variables are presented as mean ± standard deviation. Fisher’s exact test was used for categorical data. Continuous variables were analyzed using Student’s t-test or one-way analysis of variance (ANOVA) followed by Tukey-Kramer post hoc test. A level of significance of p < 0.05 was considered.
RESULTS
Glu89Gln mutation
Forty subjects carried a heterozygous Glu89Gln mutation. 38/40 (95%) referred a positive family history. They belonged to 8 apparently unrelated families, all located in the South-East of Sicily (Syracuse and Catania provinces) and of Sicilian descent (Fig. 1). 17 were males (42.5%) and 23 were females (57.5%). 12 subjects are asymptomatic carriers (age range: 21–47 years), 20 living symptomatic patients (age range: 39–63 years) and 8 deceased (Table 1).
The asymptomatic carriers had a follow-up of 4.5 ± 3.7 years (range: 1–10) since the time of genetic test result. When investigated with neurographic, autonomic and cardiological tests, all had normal results at last follow-up.
Age of onset was 49 ± 7.9 years (range: 37–66; n. 28). Male patients had a mean age of onset of 47.2 ± 5.2 years, whereas females 50.4 ± 9.3 (nosignificant difference). Presenting symptoms were distal paraesthesia (n. 14), carpal tunnel syndrome (CTS) (n. 7 monolateral and n. 1 bilateral), gait and balance disorder (n. 3), orthostatic hypotension (n. 2), stipsis/diarrhoea (n. 1). Age at diagnosis in 8 probands was 56.3 ± 5.3 years (range: 46–63) versus age at onset of 50.5 ± 7.1 years (range: 38–60). Therefore, the interval between onset and diagnosis was 5.8 ± 3.8 years (range: 1–10). Previous wrong diagnosis were lumbar radiculopathy, lumbar stenosis, spastic colitis.
Living patients had a follow-up ranging from 0.5 to 10 years. At last follow-up, autonomic involvement (orthostatic hypotension, diarrhea/stypsis, erectile dysfunction, urinary incontinence, xerostomia) paralleled sensory and motor dysfunction in 12/20 cases; 4 patiens had only sensory disturbances, 1 sensory-motor dysfunction, and 3 had yet only CTS.
Heart involvement with an increased IVS thickness ranging 15 to 24 mm (NYHA class II to IV) was found in 10/20 symptomatic patients. Its severity was proportional to both sensory-motor peripheral neuropathy and dysautonomia. One patient had a borderline IVS thickness of 14 mm (NYHA class II), one 13 mm (NYHA class I) and one 12 mm (NYHA class I). The former had mild muscle weakness at upper limbs, mild pinprick at upper and lower limbs, stypsis and xeroftalmia; the second had only mild pinprick at upper limbs; the latter had CTS.
Whereas most of the symptomatic patients referred as first symptoms those indicative of a peripheral neuropathy, among non-symptomatic subjects who had received a positive DNA results and were followed with neurographic, autonomic and cardiological tests once every one (two) year(s), the heart resulted to be firstly affected, even though without symptoms. Among ECG, echocardiography, cardiac MRI and 99mTc-DPD scintigraphy, the latter resulted the most sensitive technique [35]. A representative case is shown in Fig. 2. She was the daughter of an affected patient, and had the genetic diagnosis at 46 years when she was still without symptoms and signs.
Age at death was 63.4 ± 5.1 years (range: 58–71) with a disease duration of 7.6 ± 3.7 years (range: 3–13). The major cause of mortality resulted heart failure and sudden death most likely due to arrhythmias, followed by dysautonomia and cachexia, and the last, complications of bedridden condition.
Phe64Leu mutation
Twenty-eight subjects carried the Phe64Leu mutation (all were heterozygous). Some of these patients have been already described [36]. 15/28 (54%) referred a positive family history. They belonged to 19 apparently unrelated families, most of them located in the North of Sicily (Messina and Palermo provinces) and of Sicilian descent (Fig. 1). 16 were males (57.1%) and 12 were females (42.9%). 6 subjects are asymptomatic carriers (age range: 32–63 years), 10 living symptomatic patients (age range: 58–80 years) and 12 deceased. The asymptomatic carriers had a follow-up of 5.3 ± 3.7 years (range: 2–10) since the time of genetic test result (Table 1).
Age of onset was 64.1 ± 7.4 years (range: 44–75; n. 22). Male patients had a mean age of onset of 64.9 ± 7.2 years, whereas females 62.2 ± 8.5 (not significant). Presenting symptoms were distal paraesthesia (n. 10; 2/10 had also diarrhoea), CTS (n. 10 monolateral), gait and balance disorder with orthostatic hypotension or diarrhoea and weight loss (n. 2). Age at diagnosis in 19 probands was 72.4 ± 4.8 years (range: 64–78) versus age at onset of 66.3 ± 4.9 years (range: 54–76). Therefore, the interval between onset and diagnosis was 6.1 ± 3.7 years (range: 1–11). Previous wrong diagnosis included compressive radiculopathy, lumbar stenosis, CIDP.
While autonomic disturbances were not present in most patients when seen the first time, orthostatic hypotension, diarrhea/stypsis, erectile dysfunction, urinary incontinence, xerostomia occurred in all of them approximately within 4 years from the onset.
At last follow-up, heart involvement with an increased IVS thickness ranging 12 to 16.5 mm (NYHA class I to II) was found only in 5/10 symptomatic patients.
Age at death was 77.6 ± 3.8 years (range: 69–82) often in a severe tetraparetic status with a disease duration of 11.3 ± 5.1 years (range: 3–20). The main cause of death was wasting syndrome, followed by dysautonomia and then heart failure.
Thr49Ala mutation
Eight subjects carried the Thr49Ala mutation (all heterozygous). 7/8 (88%) referred a positive family history. They belonged to 4 apparently unrelated families, located in the South Center of Sicily (Agrigento province) and of Sicilian descent (Fig. 1). 3 were males (37.5%) and 5 were females (62.5%). One 35-year-old female is an asymptomatic carrier, 4 subjects are living symptomatic patients (age range: 41–58 years) and 3 deceased (Table 1).
Age of onset was 43.7 ± 7.7 years (range: 33–55; n. 7). Male patients had a mean age of onset of44.7 ± 11 years, whereas females 43 ± 6 (not significant). Presenting symptoms were autonomic disturbances (syncope, orthostatic hypotension, stypsis, weight loss, impotence) in 4 patients, distal paraesthesia in 2, and CTS in one. Age at diagnosis in 4 probands was 46.5 ± 6.9 years (range: 41–56) versus age at onset of 42 ± 4.7 years (range: 37–46). The interval between onset and diagnosis was 4.5 ± 3.9 years (range: 1–10). Previous wrong diagnosis was vasovagal syncope in an already reported 46-year-old man [37]. He had recurrent episodes of syncope for 4 years as an overt and isolated symptom. Later, he experienced paresthesia in the hands and impotence, and clinical and neurophysiological signs of axonal polyneuropathy and mixed parasympathetic and sympathetic dysfunction were detected.
Autonomic disturbances paralleled sensory/motor dysfunction in 5 patients, and were more severe in 2. At last follow-up, heart involvement with an increased IVS thickness ranging 13 to 20 mm (NYHA class I to III) was found in all four symptomatic patients with a long disease duration.
Age of death was 55 ± 6.6 years (range: 49–62) with a disease duration of 10.7 ± 2.3 years (range: 8–12). The first cause of death was dysautonomia and cachexia due to diarrhoea and malnutrition syndrome, followed by cardiomyopathy.
Statistics among different mutations
Table 1 summarizes clinical features according to mutation in our cohort and statistics. Age of onset resulted significantly higher in Phe64Leu than in both Glu89Gln (Δ: 15.1 years; p < 0.001) and Thr49Ala mutation (Δ: 20.4 years; p < 0.001). Moreover, Thr49Ala had an earlier onset of 5.3 years than Glu89Gln (p < 0.05). Fig. 3 shows estimated penetrance curve according to mutation. Similarly, diagnosis in probands occurred later in Phe64Leu than in both Glu89Gln (Δ: 16.1 years; p < 0.001) and Thr49Ala mutation (Δ: 25.9 years; p < 0.001). In Thr49Ala, age at diagnosis was lower than in Glu89Gln (Δ: 9.8 years; p < 0.05). Interval between onset of symptoms and TTR-FAP diagnosis was similar in the three mutations.
Life expectancy was not different among patients carrying the three mutations, with a high range from 3 to 20 years. Consequentially, age at death in the different mutations corresponded to differences in age at onset. It was significantly higher in Phe64Leu than in both Glu89Gln (Δ: 14.2 years; p < 0.001) and Thr49Ala mutation (Δ: 22.6 years; p < 0.001). Moreover, in Thr49Ala mutation, death occurred earlier than in Glu89Gln (Δ: 8.4 years; p < 0.05).
Carpal tunnel syndrome
When considering all the patients together, CTS alone was recorded as inaugural symptom in 19/57 patients (33%). 4/19 still have no other complaints for a mean period of 5.6 years. In 15/19 CTS remained the only symptom for a period ranging from 1 to 12 years (mean, 4.6 years), before another clinical manifestation occurred. The latter was: distal paraesthesias in 6, muscle weakness in 3, walking difficulty, orthostatic hypotension and constipation in 2 each. There was no significant association between type of second symptom or length of time interval and any of the three mutations.
Nerve biopsy
A nerve biopsy was performed in sixteen sporadic or doubtful cases, all limited to the period 1995–2005. It showed a severe fibre loss in all patients. Deposits of Congo red-positive amyloid were found in 13/16.
Treatment
23 patients had symptomatic treatment for neuropathic pain with gabapentin or pregabalin. 15 patients received midodrine or fludrocortisone acetate for orthostatic hypotension. Symptoms of irritable bowel syndrome were alleviated in 36. 6 patients had LT and 2/6 died. In none of them, LT modified the course of the disease. In the last two years, 17 patients started treatment with tafamidis meglumine. They have been enrolled in an Italian multicenter observational study of 61 patients, most of them with non-Met30 mutation and in all disease stages, followed by a homogenous protocol for 24 months. Preliminary results have shown: stabilization of nutritional status; 30–35% of responders’ rate independently from disease severity; few and minor adverse events. However, all neurological and cardiological outcome measures significantly worsened [16].
Prevalence
On 31st December 2014, 36 symptomatic patients with TTR-FAP were living in Sicily region. The prevalence of the disease was 7.1/1,000,000. We asked two major reference centers for TTR-FAP, one in North Italy, Pavia, and one in Center Italy, Rome, to provide us the number of living patients of Sicilian origin and with Sicilian residency, who were followed by them. Individuals who had moved from Sicily to another Italian city for study or work reasons were excluded. Other nine living symptomatic patients, 8 with Glu89Gln mutation and 1 with Phe64Leu mutation, were identified, leading to a total prevalence rate of 8.8/1,000,000.
DISCUSSION
Although TTR-FAP is a seemingly monogenic disease, literature highlights the considerable phenotypic heterogeneity in patients with either the hereditary or sporadic form, in endemic and non-endemic areas. Val30Met is the most studied mutation, in which similarities as well differences in age at onset, system/organ involvement and complications have been reported in diverse or even the same geographical areas [18, 38–40]. Mean age of onset ranges from 32–35 years in endemic areas of Brasil, Portugal and Japan to 56.7 years in Sweden [41]. An even later age of onset (61 years) has been reported in sporadic cases in a series from a non-endemic area [7, 42]. In the same mutation, gender analysis reported a later age of onset in women in Portugal and Brasil, whereas no difference in Sweden, Cyprus and Majorca [41]. Possible factors contributing to the differences among populations, but also within populations, include associate polymorphisms, mitochondrial function, environmental or external causes as diet influencing amyloid deposition, and are now under investigation [19, 43]. On the contrary, very few reports examined other mutations in details, but often they described small numbers of cases, or miscellanea of mutations all together [20, 44]. Differences in natural history among mutations and within mutations may have a number of important consequences in planning measures to overcome diagnostic difficulty and therapeutic management. With the promise of new disease-modifying gene/RNA therapies on the horizon, it has become increasingly important to have a good knowledge of the natural history of the disease, according to different mutations.
Our findings, which are based on a relatively large series of patients followed in the only tertiary neuromuscular center in Sicily, provide additional informations on three non-Val30Met mutations, so far described in some isolated patients, most of which of Sicilian origin [44–48]. The present study is the first epidemiological survey of TTR-FAP in an Italian area. We estimated its prevalence in Sicily Region to be 8.8/1,000,000. It is lower than the prevalence of 151, 104 and 3.72/100,000 in endemic areas as North Portugal, North Sweden and Cyprus, respectively [39, 49, 50], but higher than that of 0.87–1.1/1,000,000 in an even endemic area as Japan (having Nagano prefecture the highest prevalence of 11–15.5) [19] and that of approx. 3-4/1,000,000 in France [51] (David Adams, personal communication). We can assume that the prevalence is underestimated because of possible late onset, isolated cases and diagnostic pitfalls. A major challenge is to create a national registry to know the distribution of the disease in all Italian territory and to plan adequate multidisciplinary care forthe patients.
This survey reported phenotype-to-genotype correlations in 76 patients belonging to 31 Sicilian families, carrying three different TTR mutations (Glu89Gln, Phe64Leu, Thr49Ala) geographically distributed in three major areas of the island. They could be inherited from three common ancestors, and haplotype analyses should be performed to confirm this hypothesis. It is summarized that there are three phenotypes of FAP in Sicily, each corresponding to a different TTR variant, which are homogeneous within and heterogeneous between each other: i) Glu89Gln mutation, characterised by onset in the 5th – 6th decade, prevalent distal paraesthesias/CTS as presenting symptoms, early heart dysfunction but with fatigue, palpitation, dyspnea appearing later, heart failure and sudden death as major cause of mortality followed by dysautonomia and cachexia; ii) Phe64Leu mutation, marked by familiarity reported in one-half of cases, late onset from 5th to 8th decade, prevailing distal paraesthesias/CTS at onset, organ involvement with severe peripheral neuropathy, moderate dysautonomia and mild cardiomyopathy, death from 7th to 9th decade for wasting syndrome followed by dysautonomia; iii) Thr49Ala mutation, distinguished by an earlier onset in the 5th decade, autonomic disturbances as inaugural symptoms which may remain isolated for many years, moderate polyneuropathy, dysautonomia and cachexia as major causes of mortality followed by cardiomyopathy. In contrast, comparison among the three mutations revealed no sex predominance, no sex difference in age of onset, same interval between onset and diagnosis from 1 to 11 years, same life expectancy from 3 to 20 years. However, duration of the disease appeared shorter in Glu89Gln patients (7.6 years), most likely due to the cardiomyopathy which represents the first cause of death. Although the disease profile associated with mutations with an exclusively/predominantly cardiac involvement has been little defined, patients with non-endemic Val30Met or with non-Val30Met mutations display a more frequent and severe heart phenotype [10, 20, 52]. The only large prospectively followed cohort of patients with one of the so-called “cardiac” mutations, Thr60Ala, showed in sixty patients that: i) clinical presentation was cardiac in 42% but 96% of the patients had echocardiographic evidence of amyloidosis at diagnosis; ii) the median age of onset of symptoms was 63 years; iii) prognosis was poor, reflecting frequency and severity of cardiac involvement [53]. Our experience on asymptomatic carriers of Glu89Gln mutation followed with periodic check-ups suggests that heart involvement occurs in their forties and before that of nervous system. Unfortunately, the absence of neurological symptoms and indolent course of cardiomyopathy may cause the patient to seek medical attention far along. The present survey is one of the most numerous on non-Val30Met patients reporting phenotype-to-genotype correlations. Studies regarding epidemiological data from different countries are very important worldwide and should be encouraged.
CTS alone was the first symptom/sign in one third of our cohort, occurring bilaterally only in one patient. CTS remained the only manifestation for a period of 1–12 years. The occurrence of a short interval supports, as postulated by some authors, that the electrophysiological abnormality at the distal portion of the median nerve may be the consequence of polyneuropathy rather than an entrapment injury [54]. On the other hand, occurrence of a long interval between CTS signs and appearance of other complaints could suggest a coincidental presence of an idiopathic CTS, because of the high CTS prevalence of 7.8% in working populations [55]. However, life expectancy in our cohort should be misleadingly lengthened when considering CTS as presenting symptom. This is not the case since the mean values of 7.6 – 11.3 years in our study are in accordance with the known mean duration of the disease of 10 years [10].
In conclusion, TTR-FAP is a progressive and fatal disease that is increasingly diagnosed worldwide. This analysis of data in Sicily Region highlighted a prevalence of 8.8/1,000,000, absence of the common Val30Met mutation, and presence of only three TTR variants (Glu89Gln, Phe64Leu, Thr49Ala) with homogeneous within and heterogeneous clinical characteristics between each other. Neurologists must be aware of diagnostic pitfalls of TTR-FAP and gene sequencing should be done in all suspected cases. Genetic testing should be also encouraged in relatives of diagnosed cases when they are able to understand its medical, social and psychological consequences. Good knowledge of the natural history of the disease according to different TTR mutations allow clinicians to optimise multiprofessional care for patients and to offer carriers a personalized follow-up to reveal first signs of the disease.
DISCLOSURES
G.V. is the local PI of clinical trials in TTR-FAP supported by Pfizer, Alnylam, Isis. A.M., M.R., G.D.B., C.S., and L.G. are involved in the same trials.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the patients and their families. The authors thank Gian Maria Fabrizi (Verona) for molecular analysis, Mario Sabatelli, Marco Luigetti and Marco Di Girolamo (Rome) and Giampaolo Merlini and Laura Obici (Pavia) for referring Sicilian patients followed at their centers.
REFERENCES
1 | Planté-Bordeneuve V, Said G(2011) Familial amyloid polyneuropathyLancet Neurol10: 1210861097 |
2 | Murakami T, Sango K, Watabe K, Niimi N, Takaku S, Li Z, Yamamura KI, Sunada Y(2015) Schwann cells contribute to neurodegeneration intransthyretin amyloidosisJ Neurochem10.1111/jnc.13068Feb 17. [Epub ahead of print] |
3 | Said G(2003) Familial amyloid polyneuropathy: Mechanisms leading tonerve degenerationAmyloid10 Suppl 1: 712 |
4 | Mazzeo A, Aguennouz M, Messina C, Vita G(2004) Immunolocalization andactivation of transcription factor nuclear factor kappa B indysimmune neuropathies and familial amyloidotic polyneuropathyArch Neurol61: 710971102 |
5 | Said G, Planté-Bordeneuve V(2009) Familial amyloid polyneuropathy:A clinico-pathologic studyJ Neurol Sci284: 1-2149154 |
6 | Gonçalves NP, Vieira P, Saraiva MJ(2014) Interleukin-1 signalingpathway as a therapeutic target in transthyretin amyloidosisAmyloid21: 3175184 |
7 | Planté-Bordeneuve V, Ferreira A, Lalu T, Zaros C, Lacroix C, Adams D, Said G(2007) Diagnostic pitfalls in sporadic transthyretinfamilial amyloid polyneuropathy (TTR-FAP)Neurology69: 7693698 |
8 | Cappellari M, Cavallaro T, Ferrarini M, Cabrini I, Taioli F, Ferrari S, Merlini G, Obici L, Briani C, Fabrizi GM(2011) Variablepresentations of TTR-related familial amyloid polyneuropathy inseventeen patientsJ Peripher Nerv Syst16: 2119129 |
9 | Dohrn MF, Röcken C, De Bleecker JL, Martin JJ, Vorgerd M, Van den Bergh PY, Ferbert A, Hinderhofer K, Schröder JM, Weis J, Schulz JB, Claeys KG(2013) Diagnostichallmarks and pitfalls in late-onset progressivetransthyretin-related amyloid-neuropathyJ Neurol260: 1230933108 |
10 | Ando Y, Coelho T, Berk JL, Cruz MW, Ericzon BG, Ikeda S, Lewis WD, Obici L, Planté-Bordeneuve V, Rapezzi C, Said G, Salvi F(2013) Guideline of transthyretin-related hereditary amyloidosis forcliniciansOrphanet J Rare Dis8: 31 |
11 | Herlenius G, Wilczek HE, Larsson M, Ericzon BG(2004) Ten years ofinternational experience with liver transplantation for familialamyloidotic polyneuropathy: Results from the Familial AmyloidoticPolyneuropathy World Transplant RegistryTransplantation77: 16471 |
12 | Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V, Lozeron P, Suhr OB, Campistol JM, Conceição IM, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Wilson A, Grogan DR(2012) Tafamidis for transthyretin familial amyloid polyneuropathy: Arandomized, controlled trialNeurology79: 8785792 |
13 | Coelho T, Maia LF, da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, Conceiçao I, Schmidt HH, Trigo P, Kelly JW, Labaudinière R, Chan J, Packman J, Grogan DR(2013) Long-termeffects of tafamidis for the treatment of transthyretin familialamyloid polyneuropathyJ Neurol260: 1128022814 |
14 | Lozeron P, Théaudin M, Mincheva Z, Ducot B, Lacroix C, Adams D(2013) Effect on disability and safety of Tafamidis in late onset ofMet30 transthyretin familial amyloid polyneuropathyEur J Neurol20: 1215391545 |
15 | Merlini G, Planté-Bordeneuve V, Judge DP, Schmidt H, Obici L, Perlini S, Packman J, Tripp T, Grogan DR(2013) Effects of tafamidis ontransthyretin stabilization and clinical outcomes in patients withnon-Val30Met transthyretin amyloidosisJ Cardiovasc Transl Res6: 610111020 |
16 | Cortese A, Russo M, Obici L, Cavallaro T, Fabrizi GM, Manganelli F, Santoro L, Luigetti M, Sabatelli M, Schenone A, Grandis M, Mauro A, Pradotto LG, Mazzeo A, Gentile L, Piscosquito G, Calabrese D, Vita G, Merlini G, Pareyson D(2015) Anobservational study on tafamidis for transthyretin-relatedfamilial amyloid polyneuropathy in ItalyJ Peripher Nerv Syst20: S1S8S9(abstract) |
17 | Obici L, Merlini G(2014) An overview of drugs currently underinvestigation for the treatment of transthyretin-relatedhereditary amyloidosisExpert Opin Investig Drugs23: 912391251 |
18 | Conceição I, De Carvalho M(2007) Clinical variability in type Ifamilial amyloid polyneuropathy (Val30Met): Comparison betweenlate- and early-onset cases in PortugalMuscle Nerve35: 1116118 |
19 | Kato-Motozaki Y, Ono K, Shima K, Morinaga A, Machiya T, Nozaki I, Shibata-Hamaguchi A, Furukawa Y, Yanase D, Ishida C, Sakajiri K, Yamada M(2008) Epidemiology of familial amyloid polyneuropathy inJapan: Identification of a novel endemic focusJ Neurol Sci270: 1-2133140 |
20 | Rapezzi C, Quarta CC, Obici L, Perfetto F, Longhi S, Salvi F, Biagini E, Lorenzini M, Grigioni F, Leone O, Cappelli F, Palladini G, Rimessi P, Ferlini A, Arpesella G, Pinna AD, Merlini G, Perlini S(2013) Disease profile and differential diagnosis of hereditarytransthyretin-related amyloidosis with exclusively cardiacphenotype: An Italian perspectiveEur Heart J34: 7520528 |
21 | Graceffa A, Russo M, Vita GL, Toscano A, Dattola R, Messina C, Vita G, Mazzeo A(2009) Psychosocial impact of presymptomatic genetictesting for transthyretin amyloidotic polyneuropathyNeuromusculDisord19: 14448 |
22 | Denier C, Ducot B, Husson H, Lozeron P, Adams D, Meyer L, Said G, Planté-Bordeneuve V(2007) A brief compound test for assessment ofautonomic and sensory-motor dysfunction in familial amyloidpolyneuropathyJ Neurol254: 1216841688 |
23 | Kimura J2001Electrodiagnosis in Diseases of Nerve and Muscle:Principles and Practice3rd edNew YorkOxford UniversityPress |
24 | Shy ME, Blake J, Krajewski K, Fuerst DR, Laura M, Hahn AF, Li J, Lewis RA, Reilly M(2005) Reliability and validity of the CMT neuropathyscore as a measure of disabilityNeurology64: 712091214 |
25 | Vita G, Princi P, Calabro R, Toscano A, Manna L, Messina C(1986) Cardiovascular reflex tests. Assessment of age-adjusted normalrangeJ Neurol Sci75: 3263274 |
26 | Di Leo R, Musumeci O, de Gregorio C, Recupero A, Grimaldi P, Messina C, Coglitore S, Vita G, Toscano A(2007) Evidence ofcardiovascular autonomic impairment in mitochondrial disordersJ Neurol254: 1114981503 |
27 | Vita G, Princi P, Messina C(1991) Multivariate analysis ofcardiovascular reflexes applied to the diagnosis of autonomicneuropathyJ Neurol238: 5251255 |
28 | Di Leo R, Rodolico C, De Gregorio C, Recupero A, Coglitore S, Annesi G, Toscano A, Messina C, Vita G(2004) Cardiovascular autonomiccontrol in myotonic dystrophy type A correlative study withclinical and genetic dataNeuromuscul Disord14: 2136141 |
29 | Di Bella G, Minutoli F, Mazzeo A, Vita G, Oreto G, Carerj S, Anfuso C, Russo M, Gaeta M(2010) MRI of cardiac involvement intransthyretin familial amyloid polyneuropathyAJR Am JRoentgenol195: 6W394W399 |
30 | Di Bella G, Minutoli F, Pingitore A, Zito C, Mazzeo A, Aquaro GD, Di Leo R, Recupero A, Stancanelli C, Baldari S, Vita G, Carerj S(2011) Endocardial and epicardial deformations in cardiac amyloidosis andhypertrophic cardiomyopathyCirc J75: 512001208 |
31 | Di Bella G, Pizzino F, Minutoli F, Zito C, Donato R, Dattilo G, Oreto G, Baldari S, Vita G, Khandheria BK, Carerj S(2014) The mosaic ofthe cardiac amyloidosis diagnosis: Role of imaging in subtypes andstages of the diseaseEur Heart J Cardiovasc Imaging15: 1213071315 |
32 | Di Bella G, Minutoli F, Madaffari A, Mazzeo A, Russo M, Donato R, Zito C, Aquaro GD, Piccione MC, Pedri S, Vita G, Pingitore A, Carerj S(2014) Left atrial function in cardiac amyloidosisJCardiovasc Med (Hagerstown)Sep 12. [Epub ahead of print] |
33 | Rapezzi C, Quarta CC, Guidalotti PL, Pettinato C, Fanti S, Leone O, Ferlini A, Longhi S, Lorenzini M, Reggiani LB, Gagliardi C, Gallo P, Villani C, Salvi F(2011) Role of (99m)Tc-DPD scintigraphy indiagnosis and prognosis of hereditary transthyretin-relatedcardiac amyloidosisJACC Cardiovasc Imaging4: 6659670 |
34 | Coelho T, Maurer MS, Suhr OB(2013) THAOS - The TransthyretinAmyloidosis Outcomes Survey: Initial report on clinicalmanifestations in patients with hereditary and wild-typetransthyretin amyloidosisCurr Med Res Opin29: 16376 |
35 | Minutoli F, Di Bella G, Sindoni A, Vita G, Baldari S(2013) Effectiveness of skeletal scintigraphy in transthyretin-relatedamyloidosisInt J Cardiol168: 549884989 |
36 | Russo M, Mazzeo A, Stancanelli C, Di Leo R, Gentile L, Di Bella G, Minutoli F, Baldari S, Vita G(2012) Transthyretin-related familialamyloidotic polyneuropathy: Description of a cohort of patientswith Leu64 mutation and late onsetJ Peripher Nerv Syst17: 4385390 |
37 | Vita G, Mazzeo A, Di Leo R, Ferlini A(2005) Recurrent syncope aspersistently isolated feature of transthyretin amyloidoticpolyneuropathyNeuromuscul Disord15: 3259261 |
38 | Koike H, Misu K, Ikeda S, Ando Y, Nakazato M, Ando E, Yamamoto M, Hattori N, Sobue G(2002) Type I (transthyretin Met30) familial amyloidpolyneuropathy in Japan: Early- vs late-onset formArch Neurol59: 1117711776 |
39 | Dardiotis E, Koutsou P, Papanicolaou EZ, Vonta I, Kladi A, Vassilopoulos D, Hadjigeorgiou G, Christodoulou K, Kyriakides T(2009) Epidemiological, clinical and genetic study of familialamyloidotic polyneuropathy in CyprusAmyloid16: 13237 |
40 | Hellman U, Suhr O(2012) Regional differences and similarities of FAP inSwedenAmyloid19 Suppl 1: 5354 |
41 | Reinés JB, Vera TR, Martín MU, Serra HA, Campins MM, Millán JM, Lezaun CG, Cruz MR(2014) Epidemiology oftransthyretin-associated familial amyloid polyneuropathy in theMajorcan area: Son Llàtzer Hospital descriptive studyOrphanet J Rare Dis9: 29 |
42 | Plante-Bordeneuve V(2014) Update in the diagnosis and management oftransthyretin familial amyloid polyneuropathyJ Neurol261: 612271233 |
43 | Bonaïti B, Olsson M, Hellman U, Suhr O, Bonaïti-Pellié C, Planté-Bordeneuve V(2010) TTR familialamyloid polyneuropathy: Does a mitochondrial polymorphismentirely explain the parent-of-origin difference in penetrance?Eur J Hum Genet18: 8948952 |
44 | Luigetti M, Conte A, Del Grande A, Bisogni G, Madia F, Lo Monaco M, Laurenti L, Obici L, Merlini G, Sabatelli M(2013) TTR-relatedamyloid neuropathy: Clinical, electrophysiological andpathological findings in 15 unrelated patientsNeurol Sci34: 710571063 |
45 | Salvi F, Ferlini A, Plasmati R, Rubboli G, Michelucci R, Forti A, Saraiva MJM, Costa PP, Altland K, Tassinari CA(1990) Familialamyloidotic polyneuropathy in ItalyArquivos Med3: 1924 |
46 | Ii S, Minnerath S, Ii K, Dyck PJ, Sommer SS(1991) Two-tiered DNA-baseddiagnosis of transthyretin amyloidosis reveals two novel pointmutationsNeurology41: 6893898 |
47 | Almeida MR, Ferlini A, Forabosco A, Gawinowicz M, Costa PP, Salvi F, Plasmati R, Tassinari CA, Altland K, Saraiva MJ(1992) Twotransthyretin variants (TTR Ala-49 and TTR Gln-89) in two Siciliankindreds with hereditary amyloidosisHum Mutat1: 3211215 |
48 | Ferlini A, Salvi F, Uncini A, El-Chami J, Winter P, Altland K, Repetto M, Littardi M, Campoleoni A, Vezzoni P, Patrosso MC(1996) Homozygosity and heterozygosity for the transthyretin leu64mutation: Clinical, biochemical and molecular findingsClinGenet49: 1014 |
49 | Sousa A, Andersson R, Drugge U, Holmgren G, Sandgren O(1993) Familialamyloidotic polyneuropathy in Sweden: Geographical distribution,age of onset, and prevalenceHum Hered43: 5288294 |
50 | Sousa A, Coelho T, Barros J, Sequeiros J(1995) Genetic epidemiology offamilial amyloidotic polyneuropathy (FAP)-type I in Póvoa doVarzim and Vila do Conde (north of Portugal)Am J Med Genet60: 6512521 |
51 | Adams D, Lozeron P, Theaudin M, Mincheva Z, Cauquil C, Adam C, Signate A, Vial C, Maisonobe T, Delmont E, Franques J, Vallat JM, Sole G, Pereon Y, Lacour A, Echaniz-Laguna A, Misrahi M, Lacroix C(2012) French Network for FAP. Regional difference and similarity offamilial amyloidosis with polyneuropathy in FranceAmyloid19 Suppl 1: 6164 |
52 | Rapezzi C, Longhi S, Milandri A, Lorenzini M, Gagliardi C, Gallelli I, Leone O, Quarta CC(2012) Cardiac involvement inhereditary-transthyretin related amyloidosisAmyloid19 Suppl 1: 1621 |
53 | Sattianayagam PT, Hahn AF, Whelan CJ, Gibbs SD, Pinney JH, Stangou AJ, Rowczenio D, Pflugfelder PW, Fox Z, Lachmann HJ, Wechalekar AD, Hawkins PN, Gillmore JD(2012) Cardiac phenotype andclinical outcome of familial amyloid polyneuropathy associatedwith transthyretin alanine 60 variantEur Heart J33: 911201127 |
54 | Koike H, Morozumi S, Kawagashira Y, Iijima M, Yamamoto M, Hattori N, Tanaka F, Nakamura T, Hirayama M, Ando Y, Ikeda S, Sobue G(2009) Thesignificance of carpal tunnel syndrome in transthyretin Val30Metfamilial amyloid polyneuropathyAmyloid16: 3142148 |
55 | Dale AM, Harris-Adamson C, Rempel D, Gerr F, Hegmann K, Silverstein B, Burt S, Garg A, Kapellusch J, Merlino L, Thiese MS, Eisen EA, Evanoff B(2013) Prevalence and incidence of carpal tunnelsyndrome in US working populations: Pooled analysis of sixprospective studiesScand J Work Environ Health39: 5495505 |
Figures and Tables
Fig.1
Geographical distribution of TTR-FAP patients families in Sicily according to different mutations.
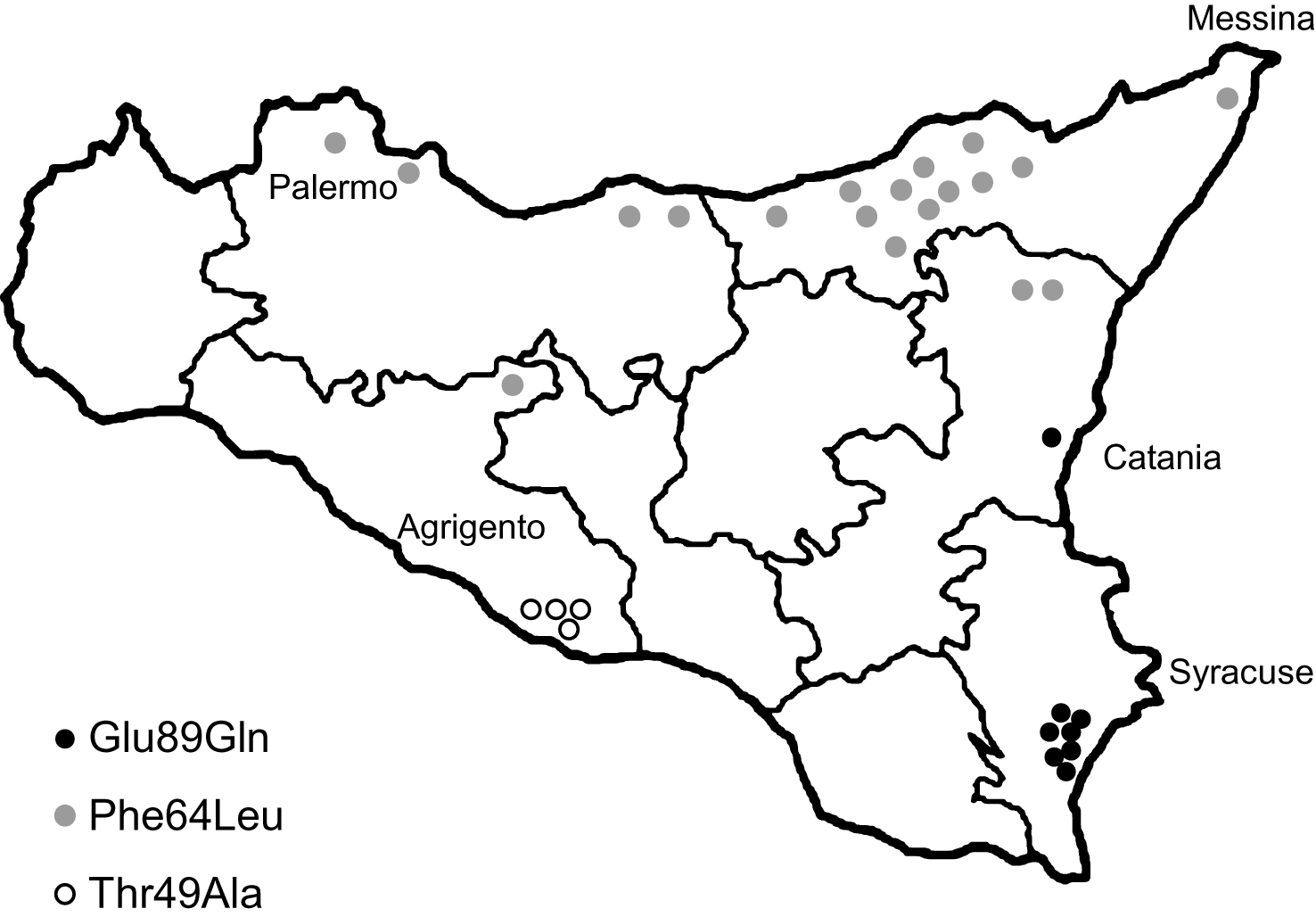
Fig.2
Clinical findings in an asymptomatic Glu89Gln carrier, showing higher sensitivity of 99mTc-DPD scintigraphy in detecting heart involvement. She was followed every two years with neurographic, autonomic and cardiological tests. Asterisk indicates a pathological result. At first control in 2006, at 46 years of age, Charcot-Marie-Tooth neuropathy score (CMTNS), compound autonomic dysfunction test (CADT), inter-ventricular septum thickness (IVST) and ejection fraction (EF) were normal; cardiac MRI and 99mTc-DPD scintigraphy were also normal; the latter showed no significant cardiac uptake (score 0) and normal indexes of semiquantitative analysis (HR, WBR, H/WB ratio; see Materials and Methods). In 2008, CMTNS, CADT, IVST, EF and cardiac MRI were still normal. On the contrary, 99mTc-DPD scan showed a mild cardiac uptake (score 1) confirmed by some pathological semiquantitative indexes. After two years, in 2010, there was a mild peripheral and autonomic nerve involvement at CMTNS and CADT, IVST was increased, cardiac MRI showed a minimal signal hyperintensity (arrow), and scintigraphy worsened to score 2.
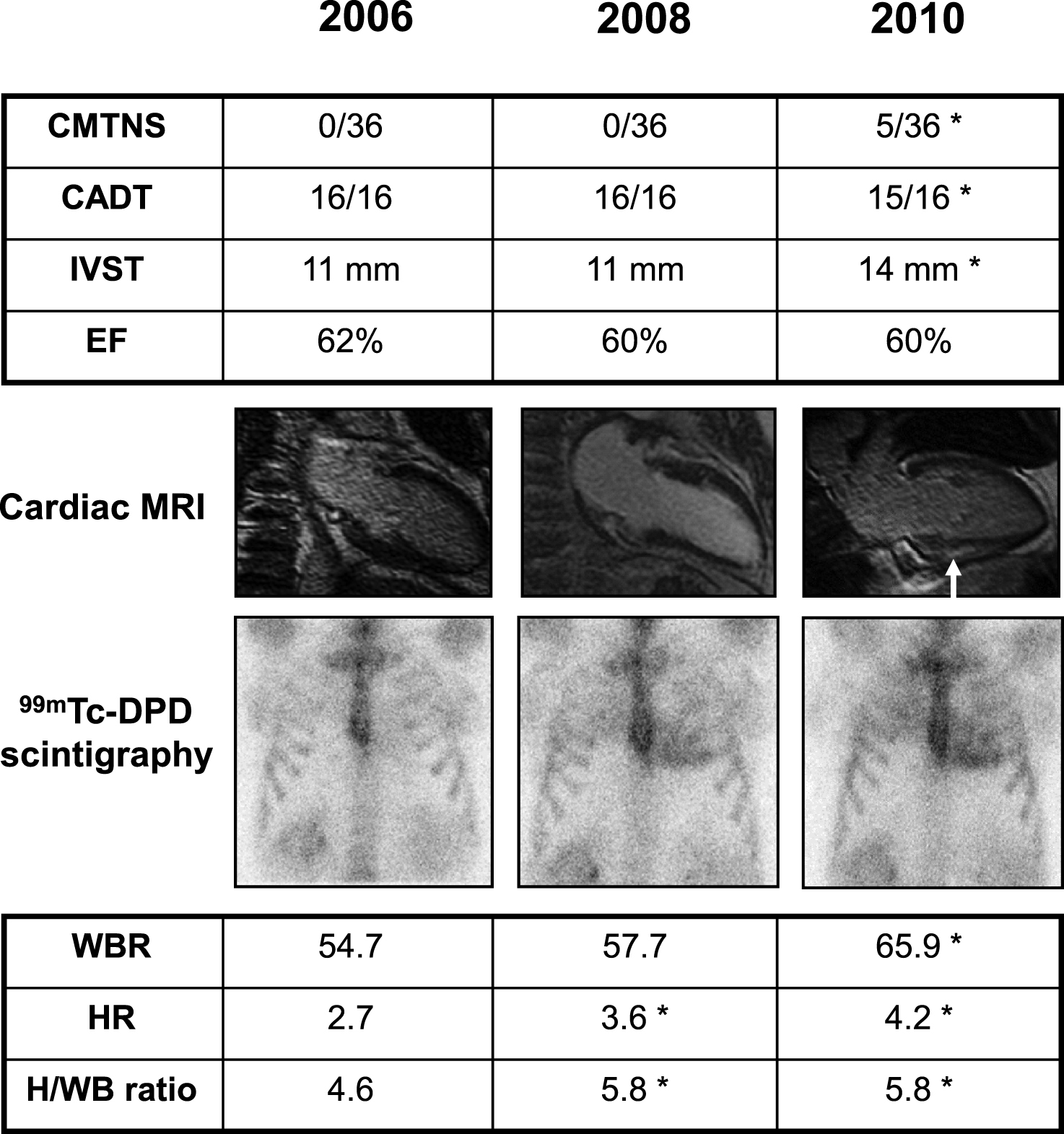
Fig.3
Estimated penetrance curve according to mutation.
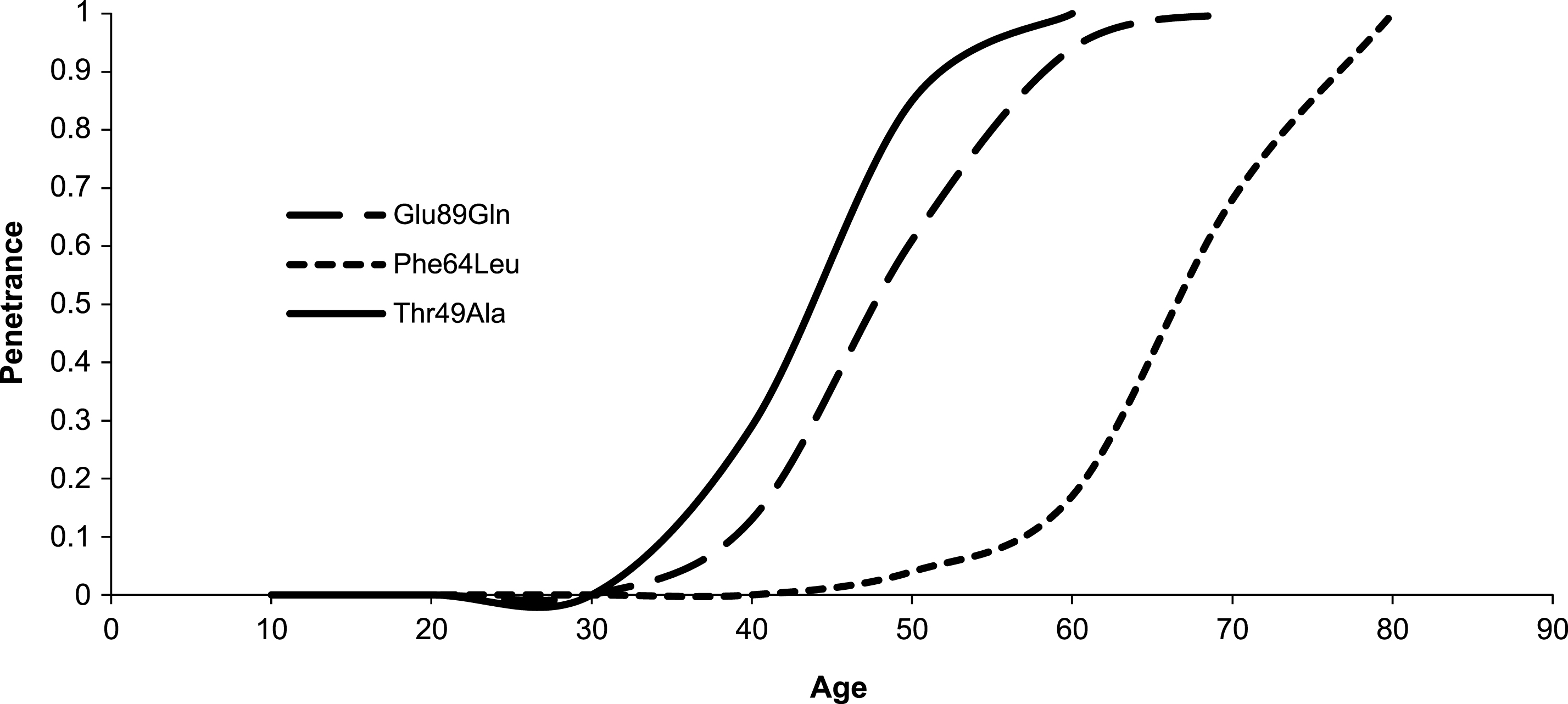
Table 1
Clinical characteristics of 76 TTR-FAP patients according to mutation in Sicily region
| Glu89Gln | Phe64Leu | Thr49Ala | p value * | |
| Patients (n.) | 40 | 28 | 8 | |
| asymptomatic carrier | 12 | 6 | 1 | |
| living symptomatic | 20 | 10 | 4 | |
| deceased | 8 | 12 | 3 | |
| Familial cases | 38/40 (95%) | 15/28 (54%) | 7/8 (88%) | |
| Ratio M/F | 17/23 | 16/12 | 3/5 | n.s. |
| Age of onset (years) | 49 ± 7.9 (37–66) | 64.1 ± 7.4 (44–75) | 43.7 ± 7.7 (33–55) | 0.0001 |
| Prevalent symptoms at onset | distal paraesthesias | distal paraesthesias | autonomic disturbances | |
| Age at diagnosis in probands (years) | 56.3 ± 5.3 (46–63) | 72.4 ± 4.8 (64–78) | 46.5 ± 6.9 (41–56) | 0.0001 |
| Interval between onset and diagnosis (years) | 5.8 ± 3.8 (1–10) | 6.1 ± 3.7 (1–11) | 4.5 ± 3.9 (1–10) | n.s. |
| Involvement | ||||
| peripheral neuropathy | moderate | severe | moderate | |
| dysautonomia | moderate | moderate | severe | |
| heart dysfunction | severe | mild | moderate | |
| Age at death (years) | 63.4 ± 5.1 (58–71) | 77.6 ± 3.8 (69–82) | 55 ± 6.6 (49–62) | 0.0001 |
| Life expectancy (years) | 7.6 ± 3.7 (3–13) | 11.3 ± 5.1 (3–20) | 10.7 ± 2.3 (8–12) | n.s. |
| Prevalent cause of death | cardiomyopathy | cachexia, dysautonomia | dysautonomia, cachexia |
*by ANOVA. Mean ± standard deviation (range).


