Myotonic Dystrophy Type 2: An Update on Clinical Aspects, Genetic and Pathomolecular Mechanism
Abstract
Myotonic dystrophy (DM) is the most common adult muscular dystrophy, characterized by autosomal dominant progressive myopathy, myotonia and multiorgan involvement. To date two distinct forms caused by similar mutations have been identified. Myotonic dystrophy type 1 (DM1, Steinert’s disease) is caused by a (CTG)n expansion in DMPK, while myotonic dystrophy type 2 (DM2) is caused by a (CCTG)n expansion in CNBP. Despite clinical and genetic similarities, DM1 and DM2 are distinct disorders. The pathogenesis of DM is explained by a common RNA gain-of-function mechanism in which the CUG and CCUG repeats alter cellular function, including alternative splicing of various genes. However additional pathogenic mechanism like changes in gene expression, modifier genes, protein translation and micro-RNA metabolism may also contribute to disease pathology and to clarify the phenotypic differences between these two types of myotonic dystrophies.
This review is an update on the latest findings specific to DM2, including explanations for the differences in clinical manifestations and pathophysiology between the two forms of myotonic dystrophies.
Introduction
Myotonic dystrophies (DMs) represent a group of dominantly inherited, multisystemic diseases that share the core features of myotonia, muscle weakness, muscular dystrophy, early-onset cataracts (younger than 50 years), cardiac conduction defects and endocrine disorders [1]. Clinicians considered myotonic dystrophy to be a single disease until 1909 when Steinert and colleagues first clearly described the “classic” form of myotonic dystrophy which was called Steinert’s disease (OMIM 160900) [1]. The gene defect responsible for myotonic dystrophy of Steinert was discovered in 1992 and found to be caused by expansion of a CTG repeat in the 3’ untranslated region of myotonic dystrophy protein kinase gene (DMPK; OMIM 605377), a gene located on chromosome 19q13.3 encoding a protein kinase [2–4]. After the discovery of this gene defect, DNA testing identified a group of patients previously diagnosed as having myotonic dystrophy with dominantly inherited myotonia, proximal greater than distal weakness, and cataracts but lacking the gene defect responsible for myotonic dystrophy of Steinert [5–8]. In Europe, the disease was termed proximal myotonic myopathy(PROMM, OMIM 602668) [5, 6] or proximal myotonic dystrophy (PDM) [8] while in the United States was termed myotonic dystrophy with no CTG repeat expansion or myotonic dystrophy type 2 (DM2) [7]. Later studies demonstrated that many of the families identified as having DM2, PROMM or PDM had the same disease, a disorder that results from an unstable tetranucleotide CCTG repeat expansion in intron 1 of the nucleic acid-binding protein (CNBP) gene (previously known as zinc finger 9 gene, ZNF9; OMIM 116955) on chromosome 3q21 [9, 10]. The existence of two types of myotonic dystrophy has created a need to develop a diagnostic classification. To address this need, the International Myotonic Dystrophy Consortium developed a new nomenclature and guidelines for DNA testing [11]. Myotonic dystrophy of Steinert, the classic form of myotonic dystrophy that results from an unstable trinucleotide repeat expansion on chromosome 19, is now termed myotonic dystrophy type 1 (DM1). Patients with the clinical picture of myotonic dystrophy type 2/proximal myotonic myopathy, who have positive DNA testing for the unstable tetranucleotide repeat expansion on chromosome 3, are now classified as having myotonic dystrophy type 2 (DM2) [7, 12, 13].
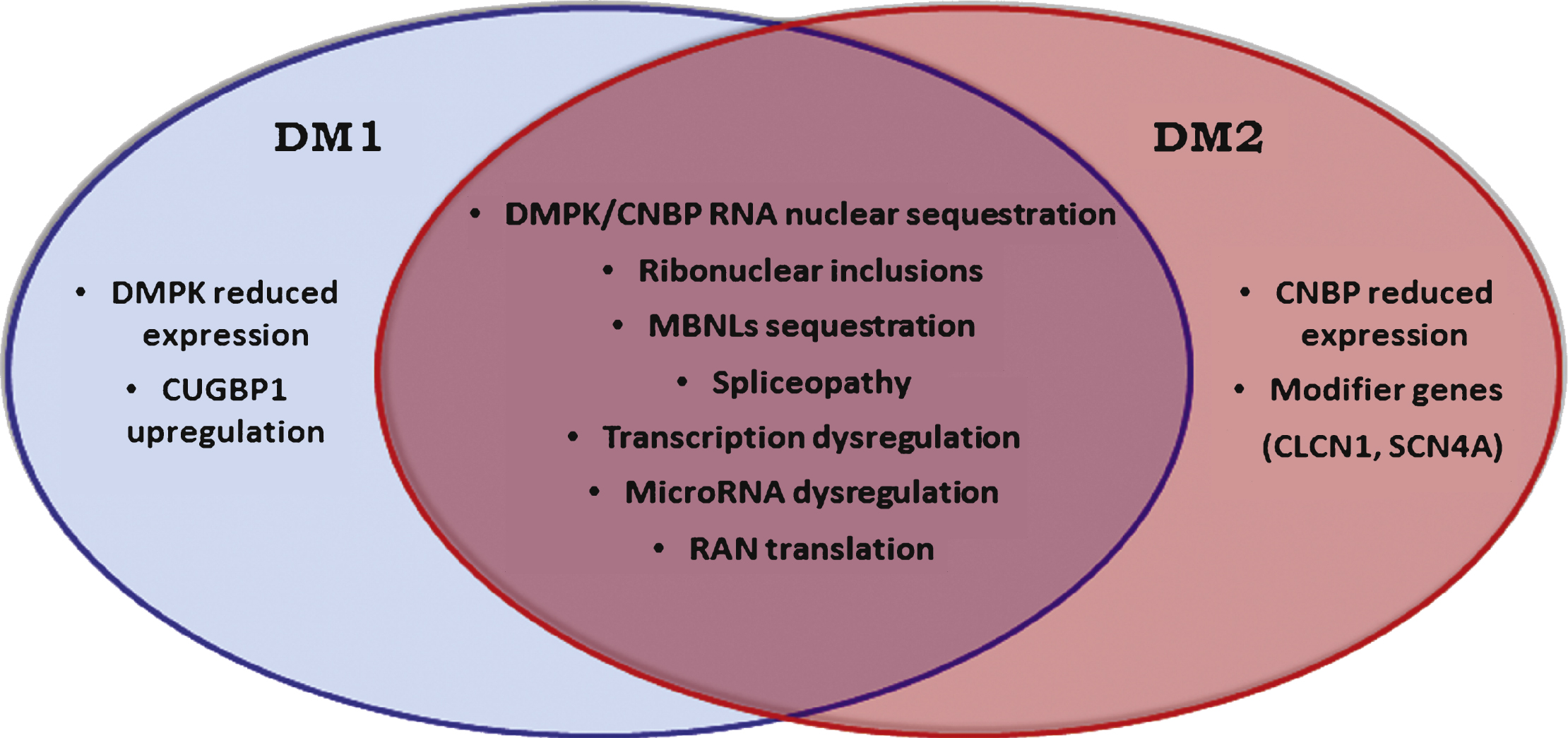
Although DM1 and DM2 have similar symptoms, there are also a number of dissimilar features making them clearly separate diseases (Fig. 1). In this review, we summarize the latest findings specific to DM2, including explanations for the differences in clinical manifestations and pathophysiology between the two forms of myotonic dystrophies.
MYOTONIC DYSTROPHY TYPE 2
Clinical features
DM2/PROMM typically appears in adult life and has variable manifestations, such as early-onset cataracts (younger than 50 years), varying grip myotonia, thigh muscle stiffness, and muscle pain, as well as weakness [5–7, 12–15]. Symptoms of DM2 usually begin in the second to sixth decade (median age 48 years) and patients as well as their care providers ascribe them to overuse of muscles, “pinched nerves,” “sciatica,” arthritis, fibromyalgia or statin use [16]. Younger patients may complain of stiffness or weakness when running up steps. Early in the presentation of DM2 there is only mild weakness of hip extension, thigh flexion, and finger flexion. Extensive pain is frequently reported in DM2 and includes abdominal, musculoskeletal, and exercise-related pain. The pain tends to come and go without obvious cause and usually fluctuates in intensity and distribution over the limbs. It may share similar features with fibromyalgia and a prior diagnosis of fibromyalgia is relatively common [17]. For many patients the first symptom is grip myotonia. However, in others the myotonia is not apparent and the presentation resembles an indolent form of limbgirdle dystrophy. Direct percussion of forearm extensor and thenar muscles is the most sensitive clinical test for myotonia in DM2 but it may be absent in several patients. Myotonia of grip is sometimes prominent and often has a jerky quality that seems to differ from that in DM1 and the nondystrophicmyotonia. Myotonia is often less apparent in DM2 compared with patients with DM1. It is more difficult to elicit myotonia on standard EMG testing in DM2 compared to DM1 except for proximal muscles such as the tensor fascia lata and vastus lateralis muscles. In cases of late-onset DM2, myotonia may only appear on electromyographic testing after examination of several muscles [13]. Facial weakness is mild in DM2 as is muscle wasting in the face and limbs. Weakness of neck flexors is frequent. Trouble arising from a squat is common, especially as the disease progresses. Calf muscle hypertrophy occasionally is prominent [5, 7, 15].
Cataracts develop before 50 years of age and appear as iridescent, posterior capsular opacities on slit-lamp in patients with DM2. The cataracts in DM2 have an appearance identical to that observed in DM1.
Cardiac problems appear to be less severe and frequent in patients with DM2 than in patients with DM1 [18–22]. Clinically significant cardiac features in DM2 include arrhythmias, atrioventricular conduction defects, and even overt dilated cardiomyopathy [23]. A high prevalence of atrial fibrillation and left ventricular dysfunction have been reported in DM2 patients [24]. Recently, Sansone et al. [22] performed an observational study in a relatively large cohort of DM2 patients (n = 104) on the frequency, severity and progression of cardiac involvement in this disease. The obtained results demonstrate that the frequency and severity of cardiac involvement are reduced in DM2 compared to DM1 and that progression is slower and less severe. However, a careful cardiac evaluation is recommended in DM2 patients to identify subjects at risk for potential major cardiac arrhythmias [22]. The overall risk of heart disease in DM2 patients was very close to that of DM1 patients. Indeed, sudden death, pacemaker implantation, and severe cardiac arrhythmias have been described in small numbers of patients [20–22, 24].
Contrary to DM1, in DM2 no ventilatory insufficiency has been reported. This preservation of pulmonary function lessens the tendency for right heart strain that occurs with pulmonary failure in DM1.
Central nervous system involvement represents one of the major differences between DM1 and DM2. Although retarded DM2 individuals have been reported, these occurrences may be either accidental or an infrequent disease consequence [13, 14]. The type of cognitive impairment that occurs in DM2 is similar to but less severe than that of DM1. Cognitive abnormalities and a reduction in cerebral blood flow in the frontal and temporal poles occur in patients with DM2 and occasionally there are alterations in the whitematter of the brain [25]. In a study where DM2 patients has been compared to age-matched DM1 patients, a specific type of “avoidant” personality and a significant impairment in frontal lobe function (especially limited ability to perform executive functions) have been observed in both groups of patients although these abnormalities were milder in DM2 patients [26]. Similar observations have been reported in a more recent study performed in a larger cohort of DM2 patients who showed significant dysexecutive syndrome and certain impairment of episodic verbal memory that are reflective of frontal (especially frontostriatal) and temporal lobe dysfunction [27]. At histopathological level, the analysis of post mortem DM2 brain samples showed the occurrence of a tau pathology similar to that observed in DM1 patients characterized by the presence of neurofibrillary tangles and Marinesco bodies in brain tissue [28, 29]. These intraneuronal aggregates are found in brain areas of patients affected by numerous different neurodegenerative disorders.
Other manifestations, such as hypogonadism, glucose intolerance, excessive sweating and dysphagia may also occur and worsen over time [7, 12, 13, 21, 22, 25, 26, 30–32]. DM2 affects the function of the testes [12]. Primary hypogonadism is occasionally presents in DM2 while appear to be more common in DM1 [7]. Similarly, the frequency of insulin resistance and glucose intolerance seems lower in DM2 than in DM1 [30, 31]. Pregnancy and menses may also exacerbate muscle pain, myotonia and muscle cramps in DM2 [32]. Premature labor and preterm deliveries appear to be more frequent compared to normal in DM2 [33]. Recently Passeri et al. [34] studied vitamin D, parathyroid function, calcium, and phosphate in male DM1 and DM2 patients and reported that hyperparathyroidism occurred in almost one fifth of patients with DM without any significant differences between DM1 and DM2 patients. Hyperparathyroidism in DM was secondary to vitamin D deficiency. Indeed the level of vitamin D (25-hydroxyvitamin D) was significantly lower in all myotonic dystrophy patients compared to healthy subjects with no difference between DM1 and DM2 [34].
Muscle histopathology
The histological features of skeletal muscle biopsy in DM1 and DM2 are very similar, and sufficiently characteristic that a diagnosis of DM can be suggested based on muscle biopsy alone [1, 13, 35]. In both diseases, affected muscles show a high number of central nuclei and a markedly increased variation in fiberdiameter that commonly ranges from less than 10μm to greater than 100μm. Basophilic regenerating fibers, splitting fibers, fibrosis and adipose deposition occur in both diseases to a variable degree depending on the extent of muscle involvement. However the comparison of muscle biopsy findings in classic DM1 with those in DM2 has indicated that specific features are present in DM2 muscle biopsy helping the diagnosis of DM2. Severely atrophic fibers with pyknotic nuclear clumps similar in appearance to the severely atrophic fibers in neurogenic atrophy are frequently found in DM2 biopsy also before the occurrence of muscle weakness. A predominant type 2 fiber atrophy in contrast to the type 1 atrophy observed in DM1, has been described in DM2 [35–38]. Moreover, in DM2 muscle biopsy central nucleation selectively affects type 2 fibers and the atrophic nuclear clumps express fast myosin isoform (type 2 fiber) indicating that DM2 is predominantly a disease of type 2 myofibers [37]. Recently, Cardani et al. [39] have studied the progression of the muscular involvement in relation to the evolution of skeletal muscle histopathology and biomolecular findings to better prognosticate patients with DM2. Data confirm that disease progression in DM2 is slow since histological and biomolecular alterations observed in skeletal muscle are minimal even after a 10-year interval. However, muscle morphological alterations evolve more rapidly over time than the molecular changes thus indicating that muscle biopsy is a sensitive tool to assess disease progression at muscular level [39].
Genetic
DM2 results from an unstable tetranucleotide repeat expansion, CCTG, in intron 1 of the CNBP gene on chromosome 3q21 [9, 10]. The (CCTG)n repeat is a part of the complex repetitive motif (TG)n(TCTG)n(CCTG)n. In contrast to the DM1 associated (CTG)n repeat, the DM2 associated (CCTG)n repeat tract is generally interrupted in healthy range alleles by one or more GCTG, TCTG or ACTG motifs, while it is typically uninterrupted in the expanded alleles [10, 40, 41]. The size of the CCTG repeat is below 30 repeats in normal individuals and up to about 11,000 repeats in DM2 patients [10, 13]. The smallest pathogenic size reported vary between 55–75 CCTG determined by Southern blot [10, 41]. The cause for the unstable expansion is unknown, however several hypothesis have been proposed such as Alu-mediated repeats development or unequal crossing over [40, 42–44]. The size of the CCTG repeat appears to increase over time in the same individual, and, like DM1, it is a dynamic gene defect thus the threshold size of the disease-causing mutation remains to be determined [13]. The size of CCTG repeat expansion in leukocyte DNA in DM2 seems to relate in large part to the age of the patient and not necessarily to the severity of symptoms or manifestations. Somatic instability, present in both DM1 and DM2, gives rise to intra-tissue, inter-tissue, and cell-type variability and somatic mosaicism over a patient’s lifetime [10, 40, 45, 46]. In DM2 the mutation usually contracts in the next generation being shorter in the children [13]. This may explain some distinct features of DM2 such as the missing of a congenital form, the lack of genetic anticipation and the later onset [47].
Contrary to DM1, at present in DM2 there is no clear evidence of the existence of a congenital form or of anticipation [48, 49]. Anticipation, i.e. progressively earlier and more severe manifestation of a disease in a family, has been well-established in DM1 [1]. In this disease, the biologic basis for anticipation is the tendency of the CTG trinucleotide repeat expansion to increase in length in successive generations, which is correlated with earlier onset and increased disease severity in the offspring. On the contrary, in DM2 anticipation has been described only with clinical criteria in few families [13, 48, 49], but no longer CCTG repeat expansions in patients with earlier age at onset has been observed.
Clinical features in DM2 are much more variable than in adult onset DM1 [8, 50]. The core features of the disease, muscle weakness, myotonia and cataracts, may be absent, and signs such as myotonia may even vary over time both when clinically assessed and when recorded by electromyography (EMG) [47]. Moreover, no correlation of the size of the repeat expansion with the clinical outcome has been shown in DM2 [13].This variability in phenotype may be explained by the existence of molecular modifiers or of additional mechanisms. Myotonia is generally mild and inconsistent in DM2 and it has been correlated with the disruption of the alternative splicing of the muscle chloride channel CLCN1 gene, encoding for a skeletal muscle chloride channel. However recent studies indicate that, unlike DM1, co-segregation of heterozygous recessive CLCN1 mutations in DM2 patients is more frequent than in healthy subjects and modifies the DM2 phenotype [51, 52]. CLCN1 gene maps to chromosome 7q35 and when mutated causes myotonia congenita (recessive Becker disease OMIM no. 255700; dominant Thomsen disease OMIM no. 160800) [53]. DM2 patients with co-segregating recessive CLCN1 mutations showed more severe muscle stiffness and more severe clinical and EMG myotonia than those having exclusively the CCUG expansion. Nevertheless, recently it has been observed that CLCN1 is not the only gene that alters the myotonic phenotype in DM2 patients. Bugiardini et al. [54] have described a DM2 patient with severe and early onset myotonia without mutation in CLCN1 gene. However the screening of SCN4A gene revealed a novel mutation c.215C>T (p.Pro72Leu) localized in the cytoplasmic N-terminus. This novel missense mutation P72L is remarkable because it represents the first mutation described in the cytoplasmic N terminus of Nav1.4 [54]. SCN4A codes for Nav1.4 a voltage gate sodium channel expressed in skeletal muscle and is another gene implicated in myotonic disorders (Myotonia, Potassium-aggravated OMIM 608390) [55]. The study of the biophysical alteration of P72L by whole-cell voltage clamping in a heterologous expression system showed the Nav1.4 mutant gain of function effect suggesting that the mutation described may act as a modulating factor increasing the severity of myotonia [54]. Investigation of other DM2 patients presenting atypical phenotype with no mutation on CLCN1 gene, leaded Meola and collaborators to identify a DM2 patient who presented an early severe myotonia since he was 12 years old and mexiletine treatment resulted ineffective in reducing myotonia. Genetic analysis of SCN4A gene showed a G2717C base exchange in exon 14 predicting an S906T substitution. This variant is considered a benign polymorphism however electrophysiological studies revealed that it affects the fast and slow gating processes [56]. Thus it is possible that the additive effect of the DM2 mutations and the S906T polymorphism may have created the atypical severe phenotype observed in this patient (Meola et al., unpublished data). These observations suggest that CLCN1 or SCN4A mutations may contribute to exaggerate the DM2 phenotype in these patients who could be more easily identified and diagnosed than DM2 patients without the modifier gene. Consequently, due to less apparent symptoms or with absent or minimal myotonic discharged, a large number of DM2 patients may remain undiagnosed even in clinical centers with considerable experience with DM2 [57].
There are few epidemiologic studies of DM2. The exact prevalence of DM2 is not known. DM1 affects at least 1 in 8,000 people worldwide but the prevalence of the two types of myotonic dystrophy varies among different geographic and ethnic populations. In most populations DM1 appears to be more common than DM2, however recent studies suggest that DM2 may be as common as DM1 among people in Germany, Poland and Finland [47, 58].
To date, DM2 mutations have been identified predominantly in European Caucasians and most patients are of northern and eastern European descent [40, 59]. Single kindred of Afghan [49, 59] and Japanese [60] origin have been identified. In the United States, clinical experience suggests that DM2 is roughly 5-fold less common than DM1. Haplotype analysis indicates that the European DM2 mutations originate from a single founder, between approximately 4,000–11,000 years ago [40].
Pathophysiology
Although genetically distinct, DM1 and DM2 share a common pathogenic mechanism (Fig. 2). Experimental evidence supports an RNA gain-of-function mechanism in which expanded CUG/CCUG-containing transcripts accumulate in the cell nuclei as foci which are responsible for the pathologic features common to both disorders. The mutant RNAs form imperfect double-stranded structure leading to deregulation of several RNA binding factors, including the muscleblind-like proteins (MBNLs) and RNA-binding protein 1 (CUGBP1) which are antagonist regulators of alternative splicing [61–65]. The functional loss of MBNL1, due to its sequestration in mutant RNA foci, or the upregulation of CUGBP1 result in abnormal expression of embryonic isoforms in adult tissues. The alteration of pre-mRNA processing strengthens the hypothesis of a spliceopathy which leads to an expression of isoforms inadequate for a particular tissue or developmental stage [66]. For example, among the symptoms of DM2, myotonia and insulin resistance are correlated with the disruption of the muscle chloride channel ClC-1 and the insulin receptor (IR) alternative splicing, respectively [30, 31, 67, 68].
An open question in the field of DM is to clarify the pathomechanism underlying the phenotypic differences between DM1 and DM2. Clinical signs in DM1 and DM2 are similar, but there are some distinguishing features. This suggests that other cellular and molecular pathways are involved besides the shared toxic-RNA gain of function hypothesized. Disease-specific manifestations may result from differences in spatial and temporal expression patterns of DMPK and CNBP genes. Similarly, changes in the expression of neighbouring genes may define disease-specific manifestations. While it is clear that MBNL1 is depleted from nucleoplasm through recruitment into ribonuclear inclusions both in DM1 and DM2 even when clinical symptoms and muscle alterations are very mild, CUGBP1 overexpression has been clearly demonstrated in DM1 but not in DM2 muscle biopsies [69–73]. Thus, the role of CUGBP1 in DM2 is particularly intriguing with contradictory results being reported [39, 69, 71, 74, 75]. CUGBP1 may have a role in the pathogenesis of splicing abnormalities because it has been demonstrated that is overexpressed in DM1 myoblasts, skeletal muscle and heart tissues [30, 71, 72] due to PKC-mediated hyperphosphorylation and subsequent protein stabilisation and upregulation [73]. However, in a recent work on the expression of CUGBP1 in human skeletal muscle from DM1 and DM2 patients, Cardani et al. [69] demonstrate that this protein is overexpressed in muscle biopsies from patients affected by the adult classical form of DM1 but not in muscle from DM2 patients suggesting that sequestration of MBNL1 evidently has a central role in splicing misregulation in both types of DM while in DM1 CUGBP1 overexpression might be an additional pathogenic mechanism notshared by DM2.
Another possible explanation for the clinical differences between the two DM types is the reduction of DMPK or CNBP protein levels in DM1 and DM2 respectively [3, 69, 76–79]. The transcripts from the mutant DMPK allele are retained in the nucleus and therefore are not efficiently translated leading to a partial (around 50% ) reduction of DMPK protein [77, 79]. However, while Dmpk knockout young mice do not develop a multisystemic phenotype mimicking myotonic dystrophy [80–82], reduction of CNBP levels is sufficient to produce multiorgan symptoms resembling those of DM as observed in heterozygous Cnbp+/− knockout mice [83]. Some studies also suggest that CCTG expansions cause reduction of CNBP protein in DM2 patients, but there are conflicting data on this point [39, 69, 75, 76, 78]. Reduction of CNBP expression has been reported in skeletal muscle tissue and cells from DM2 patients compared with those from non-DM2 subjects, including patients with DM1, thus explaining some of the phenotypic disparities between the two types of DM [69, 75, 78]. Although these effects may contribute to pathogenesis at some level, they do not appear to be the major determinants of disease.
Spliceopathy is a fundamental molecular feature of myotonic dystrophies shared by both DM1 and DM2, and a recent global analysis of alternative splicing in DM muscle biopsy specimens demonstrate a similar pattern of altered gene expression in DM2 as in DM1 [84]. More recently, Perfetti et al. [85] validates many known DM2-affected splice events and further expands its number identifying new aberrant splicing events in DM2 skeletal muscle. Moreover, this study also demonstrated that the affected genes are involved in numerous pathways and networks important for muscle physio-pathology, suggesting that the identified variants may contribute to DM2 pathogenesis [85]. In DM2 patients, symptoms such as muscle weakness and myotonia undergo progressive worsening with increasing age, however, this aggravation is not accompanied by a worsening of alternative splicing of several genes [39].
Recent data demonstrate that MBNL1-containing foci in DM2 cells also colocalize with snRNPs and hnRNPs, splicing factors involved in the early phases of transcript processing [86]. Moreover, in a study in situ by immunoelectron microscopy on muscle biopsies from DM and healthy subjects, an accumulation of splicing and cleavage factors in myonuclei of both DM1 and DM2 patients has been demonstrated suggesting an impairment of post-transcriptional pre-mRNA pathways which could lead to the multiple pathological dysfunctions observed in dystrophic patients [87].
In addition to splicing defects, deregulation of miRNA may be an important marker or additional mechanism of pathophysiology in myotonic dystrophies. Indeed, it has been demonstrated that miRNA expression and intracellular distribution is deregulated in many human diseases [88–93] and the highly regulated pathways of miRNA has been demonstrated to be altered in skeletal muscle in both DM1 and DM2 [90–92]. However, the miRNA profiling appears to be different in these two diseases potentially contributing to the differences observable between these two DM types. A subset of 11 miRNAs that are specifically deregulated in skeletal muscle of DM2 patients has been identified, however, only miR-193b-3p, miR-208a and miR-381 were similarly modulated in DM1 patients [92].
Recently a novel molecular mechanism that may contribute to the pathogenesis of several microsatellite expansion disorders, including myotonic dystrophies, has been described by Zu and collaborators [94]. RNA transcripts containing expanded CAG or CUG repeats can be translated in absence of a starting ATG and this non–canonical translation, called Repeat Associated Non-ATG translation (RAN-translation) occurs across expanded repeats in all reading frames to produce potentially toxic homopolymeric proteins [94, 95]. In DM1 RAN translation results in the accumulation of polyglutamine expansion proteins in previously established DM1 mouse models and human tissue [94]. Antibodies developed specifically against DM1 polyGln proteins, detect polyGln nuclear aggregates in DM1 mouse tissues and DM1 patient cardiac myocytes, leukocytes, and myoblasts not detectable in control tissues. RAN-translation products appear to be toxic to cells and may contribute to DM1 pathology. More recently RAN translation has been found to occur across intronic DM2 CCUG transcripts producing a tetra-repeat expansion protein with a repeating Leu-Pro-Ala-Cys (LPAC) motif. Moreover an LPAC antibody shows strong immunostaining in human DM2 autopsy brain but not in controls. Immunostaining has been observed in neurons, astrocytes and glia in frontal cortex, hippocampus and basal ganglia. These data suggest that RAN translation may be common to both DM1 and DM2 and that RAN proteins may be responsible for some of the CNS features of DM [96].
Another open question in the field of DM is the cause for the muscle weakness and wasting. Patients with DM2, in contrast to patients with classic DM1, usually have only mild muscle wasting. However, there is an uncommon, adult-onset variant of DM2, termed proximal myotonic dystrophy [8] that causes severe wasting of proximal arm and thigh muscles as the illness progresses. Recently, muscle weakness has been associated with bridging integrator 1 (BIN1) missplicing both in in DM1 and DM2 [97]. BIN1 is a lipid-binding protein that is involved in the biogenesis of the T tubule network in muscle and in the regulation of the excitation–contraction coupling. Moreover, to date there are no definitive explanation for the histopathological alterations observed in DM skeletal muscle which include fiber atrophy-hypertrophy, increased number of central nuclei, and presence of fibers with nuclear clumps. A possible explanation is the combined effects of misregulated splicing of several genes involved in calcium regulation and EC coupling, such as RyR1, SERCA and CaV1.1, which may contribute to the muscle degeneration in DM [98–100]. In a recent study Vihola and collaborators [101] have investigated the molecular basis of muscle weakness and wasting and the differences in muscle histopathology between DM1 and DM2. They identified differences in muscle-gene expression and splicing between DM1 and DM2 patients. In particular, the aberrant splicing isoform of TNNT3 is twice as frequent in DM2 compared to DM1. Moreover, in DM1 and DM2 a different protein expression pattern has been found in the highly atrophic fibers [101].
The presence of very atrophic fibers in DM skeletal muscle has been attributed to defects in myoblast fusion or differentiation and premature senescence. However, in vitro primary myoblasts obtained from muscle of adult DM patients do not show evident morphological abnormalities and are capable of normally differentiating [74, 102, 103]. Conversely, recent data demonstrated that DM2 myoblasts are characterized by senescence related features mainly consisting in the early appearance of cytological alterations and impairment of the pre-mRNA maturation pathways [104]. Moreover, Renna et al. [105] reported that DM1 and DM2 myoblasts are characterized by a premature proliferative growth arrest compared to healthy myoblasts through a mechanism similar to senescence since both DM1 and DM2 cells expressed biomarkers usually observed in senescent cells. These data suggest that the in vivo regenerative capacity of satellite cells in DM1 and DM2 muscle might be constitutively impaired [105]. Furthermore, contrary to DM1, the p16 pathway is not responsible for the premature growth arrest observed in DM2 myoblasts which stop dividing with telomeres shorter than controls suggesting that CCTG expansion might interfere with the telomere homeostasis in DM2 cells [105–108]. Hence, it appears that CTG and CCTG expansions trigger in vitro a mechanism of myoblast premature senescence through two different pathways, which could explain the different histological alterations observed between DM1 and DM2 skeletal muscle as for example the selective type 2 fibre atrophy present in DM2 muscle.
Diagnosis
The wide clinical spectrum phenotype makes the DM2 clinical diagnosis very difficult. Moreover, contrary to DM1, conventional PCR and Southern blot analysis are not adequate for a definitive molecular diagnosis in DM2 due to the extremely large size and somatic instability of the expansion mutation [10, 40]. The copy number of DM2 CCTG is below 30 in phenotypically normal individuals and up 11.000 in patients [109]. A complex genotyping diagnostic procedure is commonly used consisting of a three step molecular protocol [13, 47]. A conventional PCR assay across the mutation locus using probes binding to mutation flanking sequences, called short-range PCR, can be used for mutation exclusion. In all DM2 patients, a single PCR product representing the normal allele can be identified because the DNA polymerase fail to amplify the mutant allele due to length and stable secondary structure. All individuals showing two alleles for the marker are excluded from having the DM2 mutation, however, identical allele size on two normal alleles occurs in 12% of the population. All patients appearing to have one allele need further molecular analysis to determine whether or not they carry a DM2 expansion. Because of the incomplete sensitivity of Southern analysis, a long-range PCR method amplifying the CCTG repeat by PCR and probing the resultant product with an internal probe to assure specificity can be used [13, 110]. This protocol is time-consuming and requires careful evaluation of results, due to somatic length mosaicism that can give very weak and smeared signals, thus limiting the sensibility of the technique with the possibility of false negative results. Several alternative and highly sensitive methods have been developed for DM2 mutation verification including a tetraplet-primed PCR [111]. A modified Southern method using field–inversion electrophoresis (FIGE) is particularly efficient in determining the mutation length [40]. However, these methods are still too long and complicated to be part of routine laboratory diagnostics. Recently, a new genetic test “Myotonic Dystrophy type 2 kit-FL”, based on the combination of Long-PCR and Southern Blot Analysis, has been developed and validated to identify the DM2 disease. The advantage of this assay is that all reagents are pre-packaged and ready to use. The analytical results, evaluated on a total of 106 DNA samples, in terms of sensitivity, specificity and accuracy were very high (Meola et al, unpublished data). Nevertheless, ribonuclear foci and splicing changes are present before any histological abnormality manifestations [31, 52, 112]. This could be important for an early diagnosis before the spectrum of clinical signs of muscle disease appears. So a more practical tool to obtain a definitive DM2 diagnosis in few hours is represented by in situ hybridization (ISH) which is a method that allows the direct visualization of the mutant RNA on muscle biopsy [113, 114]. By using specific probes for CCUG expansions, it permits a differential diagnosis between DM2 and DM1. Therefore it may be a simple approach for DM2 diagnosis, which can be performed in a rapid and sensitive manner in any pathology laboratory. ISH with CAGG probe should be considered as a routine laboratory procedure to confirm or refute the clinical suspicion of DM2. It should also be applied routinely to screen patients with myotonic disorders [113, 114]. Since ISH technique has a limited sensitivity towards short pathogenic repeats, the diagnostic methods based on genomic DNA have the first priority and the muscle biopsy is performed only in special cases as a complimentary tool for a definitive biomolecular diagnosis of DM2 [115]. Moreover, since MBNL1 is sequestered by mutant RNA foci, it is possible to visualize the nuclear accumulation of MBNL1 by immunofluorescence on muscle sections. However, although MBNL1 represents a histopathological marker both of DM1 and DM2, it does not allow to distinguish between these two pathologies [116].
Management
No treatments are currently available that fundamentally alter the course of myotonic dystrophies. The management of DM is based on genetic counselling, preserving function and independence, preventing cardiopulmonary complications and providing symptomatic treatment of myotonia, hypersomnolence, and pain. In general the management of DM2 is similar to that of DM1, but there is less need for supportive care, such as bracing, scooters, or wheelchairs. Cataracts require monitoring, and serial monitoring with an electrocardiogram is necessary to check for covert dysrhythmia. Disturbances in cardiac rhythm are less frequent in DM2, but abnormalities do occur [13, 19–21]. Hypogonadism and insulin resistance need monitoring as in DM1. Myotonia tends to be less marked and less troublesome in DM2, but in specific circumstances antimyotonia therapy is helpful, especially if muscle stiffness is frequent and persistent or if pain is prominent. Cognitive difficulties also occur in DM2 as in DM1 but become manifest in adult life and appear to be associated with decreased cerebral blood flow to frontal and anterior temporal lobes [25, 117] and decreased brain volume [118, 119]. The changes are less severe than in DM1. Their etiology is unknown but may relate to the toxic effect of intranuclear accumulations of abnormally expanded RNA. Management of these brain symptoms is similar to that for DM1.
A frequent and difficult problem in DM2 is the peculiar muscle pain described earlier [16, 17]. The exact mechanism underlying the pain is unknown, and there is no well-established, effective treatment. Carbamazepine or mexiletine along with nonsteroidal anti-inflammatory medications or tylenol ameliorate this pain in some patients.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work was supported by CMN – Centro per lo Studio delle Malattie Neuromuscolari and FMM-Fondazione malattie miotoniche.
REFERENCES
1 | Harper PS(2001) Myotonic Dystrophy3rd editionSaundersLondon |
2 | Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hutnter K, Stanton VP, Thirion JP, Hudson T(1992) Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3’ end of a transcript encoding a protein kinase family memberCell68: 799808 |
3 | Fu YH, Pizzuti A, Fenwick RGJr, King J, Rajnarayan S, Dunne PW, Dubel J, Nasser GA, Ashizawa T, de Jong P(1992) An unstable triplet repeat in a gene related to myotonic muscular dystrophyScience255: 12561258 |
4 | Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barceló J, O’Hoy K(1992) Myotonic dystrophy mutation: An unstable CTG repeat in the 3’ untranslated region of the geneScience255: 12531255 |
5 | Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Otto M, Heine R, Moxley RT3rd(1994) Proximal myotonic myopathy: A new dominant disorder with myotonia, muscle weakness, and cataractsNeurology44: 14481452 |
6 | Meola G, Sansone V, Radice S, Skradski S, Ptacek L(1996) A family withan unusual myotonic and myopathicphenotype and no CTG expansion (proximal myotonic myopathysyndrome): A challenge for future molecular studiesNeuromusculDisord6: 143150 |
7 | Thornton CA, Griggs RC, Moxley RT3rd(1994) Myotonic dystrophy with no trinucleotide repeat expansionAnn Neurol35: 269272 |
8 | Udd B, Krahe R, Wallgren-Pettersson C, Falck B, Kalimo H(1997) Proximal myotonic dysrtrophy: A family with autosomal dominant muscular dystrophy, cataracts, hearing loss and hypogonadism: Heterogeneity of proximal myotonic syndromes?Neuromuscul Disord4: 217288 |
9 | Ranum LP, Rasmussen PF, Benzow KA, Koob MD, Day JW(1998) Genetic mapping of a second myotonic dystrophy locusNature Gen19: 196198 |
10 | Liquori CL, Ricker K, Moseley ML, Jacobsen JFKW, Naylor SL, Day JW, Ranum LP(2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9Science29: 38643867 |
11 | Ashizawa T, Baiget M(2000) New nomenclature and DNA testing guidelines for myotonic dystrophy type 1 (DM1). The International Myotonic Dystrophy Consortium (IDMC)Neurology54: 12181221 |
12 | Day JW, Roelofs R, Leroy B, Pech I, Benzow K, Ranum LP(1999) Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2)Neuromuscul Disord9: 1927 |
13 | Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress WC, Koch MC, Beilman GJ, Harrison AR, Dalton JC, Ranum LP(2003) Myotonic dystrophy type Molecular, diagnostic and clinical spectrumNeurology60: 657664 |
14 | Ricker K, Koch MC, Lehmann-Horn F, Pongratz D, Speich N, Reiners K, Schneider C, Moxley RT3rd(1995) Proximal myotonic myopathy. Clinical features of a multisystem disorder similar to myotonic dystrophyArch Neurol52: 2531 |
15 | Meola G, Cardani R(1852) Myotonic dystrophies: An update on clinical aspects, genetic, pathology, and molecular pathomechanismsBiochim Biophys Acta594606 |
16 | George A, Schneider-Gold C, Zier S, Reiners K, Sommer C(2004) Musculoskeletal pain in patients with myotonic dystrophy type 2Arch Neurol61: 19381942 |
17 | Auvinen S, Suominen T, Hannonen P, Bachinski LL, Krahe R, Udd B(2008) Myotonic dystrophy type 2 found in two of sixty-three persons diagnosed as having fibromyalgiaArthritis Rheum58: 36273631 |
18 | Meola G, Sansone V, Marinou K, Cotelli M, Moxley RT3rd, Thornton CA, De Ambroggi L(2002) Proximal myotonic myopathy: A syndrome with a favourable prognosis?J Neuro Sc19: 389396 |
19 | Flachenecker P, Schneider C, Cursiefen S, Ricker K, Toyka KV, Reiners K(2003) Assessment of cardiovascular autonomic function in myotonic dystrophy type 2 (DM2/PROMM)Neuromuscul Disord13: 289293 |
20 | Moxley RTIII, Meola G, Udd B, Ricker K(2002) Report of the 84th ENMC Workshop: PROMM (Proximal Myotonic Myopathy) and Other Myotonic Dystrophy-Like Syndromes: 2nd Worksho13-15th October, Loosdrecht, The NetherlandsNeuromuscul Disord12: 306317 |
21 | Schoser BG, Ricker K, Schneider-Gold C, Hengstenberg C, Durre J, Bultmann B, Kress W, Day JW, Ranum LP(2004) Sudden cardiac death in myotonic dystrophy type 2Neurology63: 24022404 |
22 | Sansone VA, Brigonzi E, Schoser B, Villani S, Gaeta M, De Ambroggi G, Bandera F, De Ambroggi L, Meola G(2013) The frequency and severity of cardiac involvement in myotonic dystrophy type 2 (DM2): Long-term outcomesInt J Cardiol168: 11471153 |
23 | Lee TM, Maurer MS, Karbassi I, Braastad C, Batish SD, Chung WK(2012) Severe dilated cardiomyopathy in a patient with myotonic dystrophy type 2 and homozygous repeat expansion in ZNF9Congest Heart Fail18: 183186 |
24 | Wahbi K, Meune C, Bécane HM, Laforêt P, Bassez G, Lazarus A, Radvanyi-Hoffman H, Eymard B, Duboc D(2009) Left ventricular dysfunction and cardiac arrhythmias are frequent in type 2 myotonic dystrophy: A case control studyNeuromuscul Disord19: 468472 |
25 | Meola G, Sansone V, Perani D, Colleluori A, Cappa S, Cotelli M Fazio F, Thornton CA, Moxley RT(1999) Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathyNeurology5: 10421050 |
26 | Meola G, Sansone V, Perani D, Scarone S, Cappa S, Dragoni C, Cattaneo E, Cotelli M, Gobbo C, Fazio F, Siciliano G, Mancuso M, Vitelli E, Zhang S, Krahe R, Moxley RT(2003) Executive dysfunction and avoidant personality trait in myotonic dystrophy type 1 (DM1) and in proximal myotonic myopathy (DM2/PROMM)Neuromuscular Disord13: 813821 |
27 | Peric S, Mandic-Stojmenovic G, Stefanova E, Savic-Pavicevic D, Pesovic J, Ilic V, Dobricic V, Basta I, Lavrnic D, Rakocevic-Stojanovic V(2015) Frontostriatal dysexecutive syndrome: A core cognitive feature of myotonic dystrophy type 2J Neurol262: 142148 |
28 | Sergeant N, Sablonnie‘re B, Schraen-Maschke S, Ghestem A, Maurage CA, Wattez A, Vermersch P, Delacourte A(2001) Dysregulation of human brain microtubuleassociated tau mRNA maturation in myotonic dystrophy type 1Hum Mol Genet10: 21432155 |
29 | Maurage CA, Udd B, Ruchoux MM, Vermersch P, Kalimo H, Krahe R, Delacourte A, Sergeant N(2005) Similar brain tau pathology in DM2/PROMM and DM1/Steinert diseaseNeurology65: 16361638 |
30 | Savkur RS, Philips AV, Cooper TA(2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophyNature Genet29: 4047 |
31 | Savkur RS, Philips AV, Cooper TA, Dalton JC, Moseley ML, Ranum LP, Day JW(2004) Insulin receptor splicing alteration in myotonic dystrophy type 2Am J Hum Genet74: 13091313 |
32 | Newman B, Meola G, O’Donovan DG, Schapira AHV, Kingston H(1999) Proximal myotonic myopathy (PROMM) presenting as myotonia during pregnancyNeuromuscul Disord9: 144149 |
33 | Rudnik-Schoneborn S, Schneider-Gold C, Raabe U, Kress W, Zerres K, Schoser BG(2006) Outcome and effect of pregnancy in myotonic dystrophy type 2Neurology66: 579580 |
34 | Passeri E, Bugiardini E, Sansone VA, Valaperta R, Costa E, Ambrosi B, Meola G, Corbetta S(2013) Vitamin D, parathyroid hormone and muscle impairment in myotonic dystrophiesJ Neurol Sci331: 132135 |
35 | Schoser BG, Schneider-Gold C, Kress W, Goebel HH, Reilich P, Koch MC, Pongratz DE, Toyka KV, Lochmüller H, Ricker K(2004) Muscle pathology in 57 patients with myotonic dystrophy type 2Muscle Nerve29: 275281 |
36 | Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A, Mancinelli E, Rouche A, Hogrel JY, Laforêt P, Maisonobe T, Pellissier JF, Krahe R, Eymard B, Udd B(2003) Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2Neurology60: 18541857 |
37 | Bassez G, Chapoy E, Bastuji-Garin S, Radvanyi-Hoffman H, Authier FJ, Pellissier JF, Eymard B, Gherardi RK(2008) Type 2 myotonic dystrophy can be predicted by the combination of type 2 muscle fiber central nucleation and scattered atrophyJ Neuropathol Exp Neurol67: 319325 |
38 | Pisani V, Panico MB, Terracciano C, Bonifazi E, Meola G, Novelli G, Bernardi G, Angelini C, Massa R(2008) Preferential central nucleation of type 2 myofibers is an invariable feature of myotonic dystrophy type 2Muscle Nerve38: 14051411 |
39 | Cardani R, Giagnacovo M, Rossi G, Renna LV, Bugiardini E, Pizzamiglio C, Botta A, Meola G(2014) Progression of muscle histopathology but not of spliceopathy in myotonic dystrophy type 2Neuromuscul Disord24: 10421053 |
40 | Bachinski LL, Udd B, Meola G, Sansone V, Bassez G, Eymard B, Thornton CA, Moxley RT, Harper PS, Rogers MT, Jurkat-Rott K, Lehmann-Horn F, Wieser T, Gamez J, Navarro C, Bottani A, Kohler A, Shriver MD, Sallinen R, Wessman M, Zhang S, Wright FA, Krahe R(2003) Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: A single shared haplotype indicates an ancestral founder effectAm J Hum Genet73: 835848 |
41 | Bachinski LL, Czernuszewicz T, Ramagli LS, Suominen T, Shriver MD, Udd B, Siciliano MJ, Krahe R(2009) Premutation allele pool in myotonic dystrophy type 2Neurology72: 490497 |
42 | Dere R, Wells RD(2006) DM2 CCTG*CAGG repeats are crossover hotspots that are more prone to expansions than the DM1 CTG*CAG repeats in Escherichia coliJ Mol Biol360: 2136 |
43 | Dere R, Napierala M, Ranum LP, Wells RD(2004) Hairpin structure-forming propensity of the (CCTG.CAGG) tetranucleotide repeats contributes to the genetic instability associated with myotonic dystrophy type 2J Biol Chem279: 4171541726 |
44 | Kurosaki T, Ueda S, Ishida T, Abe K, Ohno K, Matsuura T(2012) The unstable CCTG repeat responsible for myotonic dystrophy type 2 originates from an AluSx element insertion into an early primate genomePLoS One7: e38379 |
45 | Wong LJ, Ashizawa T, Monckton DG, Caskey CT, Richards CS(1995) Somatic heterogeneity of the CTG repeat in myotonic dystrophy is age and size dependentAm J Hum Genet56: 114122 |
46 | Monckton DG, Wong LI, Ashizawa T, Caskey CT(1995) Somatic mosaicism, germline expansions, germline reversions and intergenerational reductions in myotonic dystrophy males: Small pool PCR analysesHum Mol Genet4: 18 |
47 | Udd B, Meola G, Krahe R, Thornton C, Ranum L, Day J, Bassez G, Ricker K(2003) Report of the 115th ENMC workshop: Myotonic dystrophies3rd workshop, 14-16 February Naarden, the Netherlands. Neuromuscul Disord13: 589596 |
48 | Schneider C, Ziegler A, Ricker K, Grimm T, Kress W, Reimers CD, Meinck H, Reiners K, Toyka KV(2000) Proximal myotonic myopathy: Evidence for anticipation in families with linkage to chromosome 3qNeurology3: 383388 |
49 | Schoser BG, Kress W, Walter MC, Halliger-Keller B, Lochmuller H, Ricker K(2004) Homozygosity for CCTG mutation in myotonic dystrophy type 2Brain127: 18681877 |
50 | Udd B, Meola G, Krahe R, Thornton C, Ranum LP, Bassez G, Kress W, Schoser B, Moxley R(2006) 140th ENMC International Workshop: Myotonic Dystrophy DM2/PROMM and other myotonic dystrophies with guidelines on managementNeuromuscul Disord16: 403413 |
51 | Suominen T, Schoser B, Raheem O, Auvinen S, Walter M, Krahe R, Lochmuller H, Kress W, Udd B(2008) High frequency of co-segregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and GermanyJ Neurol255: 17311736 |
52 | Cardani R, Giagnacovo M, Botta A, Rinaldi F, Morgante A, Udd B, Raheem O, Penttila S, Suominen T, Renna LV, Sansone V, Bugiardini E, Novelli G, Meola G(2012) Co-segregation of DM2 with a recessive CLCN1 mutation in juvenile onset of myotonic dystrophy type 2J Neurol259: 20902099 |
53 | Zhang J, Bendahhou S, Sanguinetti MC, Ptácek LJ(2000) Functional consequences of chloride channel gene (CLCN1) mutations causing myotonia congenitalNeurology54: 937942 |
54 | Bugiardini E, Rivolta I, Binda A, Soriano Caminero A, Cirillo F, Cinti A, Giovannoni R, Botta A, Cardani R, Wicklund MP, Meola G(2015) SCN4A mutation as modifying factor of Myotonic Dystrophy Type 2 phenotypeNeuromuscul Disord25: 301307 |
55 | Catterall WA, Goldin AL, Waxman SG(2005) International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channelsPharmacol Rev57: 397409 |
56 | Kuzmenkin A, Jurkat-Rott K, Lehmann-Horn F, Mitrovic N(2003) Impaired slow inactivation due to a polymorphism and substitutions of Ser-906 in the II-III loop of the human Nav1.4 channelPflugers Arch447: 7177 |
57 | Young NP, Daube JR, Sorenson EJ, Milone M(2010) Absent, unrecognized, and minimal myotonic discharges in myotonic dystrophy type 2Muscle Nerve41: 758762 |
58 | Suominen T, Bachinski LL, Auvinen S, Hackman P, Baggerly KA, Angelini C, Peltonen L, Krahe R, Udd B(2011) Population frequency of myotonic dystrophy: Higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in FinlandEur J Hum Genet19: 776782 |
59 | Liquori CL, Ikeda Y, Weatherspoon M, Ricker K, Schoser BG, Dalton JC, Day JW, Ranum LP(2003) Myotonic dystrophy type Human founder haplotype and evolutionary conservation of the repeat tractAm J Hum Genet73: 849862 |
60 | Saito T, Amakusa Y, Kimura T, Yahara O, Aizawa H, Ikeda Y, Day JW, Ranum LP, Ohno K, Matsuura T(2008) Myotonic dystrophy type 2 in Japan: Ancestral origin distinct from Caucasian familiesNeurogenetics9: 6163 |
61 | Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS(1996) Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophyNucl Acids Res24: 44074414 |
62 | Philips AV, Timchenko LT, Cooper TA(1998) Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophyScience280: 737741 |
63 | Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS(2000) Recruitment of human muscleblind proteins to (CUG) (n) expansions associated with myotonic dystrophyEMBO J19: 44394448 |
64 | Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS(2003) A muscleblind knockout model for myotonic dystrophyScience302: 19781980 |
65 | Ho TH, Charlet-B N, Poulos MG, Singh G, Swanson MS, Cooper TA(2004) Muscleblind proteins regulate alternative splicingEMBO J23: 31033112 |
66 | Osborne J, Thornton CA(2006) RNA-dominant diseasesHum Mol Genet15: R162R169 |
67 | Charlet-B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA(2002) Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicingMol Cell10: 4553 |
68 | Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA(2002) Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophyMol Cell10: 3544 |
69 | Cardani R, Bugiardini E, Renna LV, Rossi G, Colombo G, Valaperta R, Novelli G, Botta A, Meola G(2013) Overexpression of CUGBP1 in skeletal muscle from adult classic myotonic dystrophy type 1 but not from myotonic dystrophy type 2PlosOne8: e83777 |
70 | Jiang H, Mankodi A, Swanson MS, Moxley RT, Thornton CA(2004) Myotonic dystrophy type 1 is associated with nuclear foci of mutant RNA, sequestration of muscleblind proteins and deregulated alternative splicing in neuronsHum Mol Genet13: 30793088 |
71 | Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA(2006) Failure of MBNL1-dependent post-natal splicing transitions in myotonic dystrophyHum Mol Genet15: 20872097 |
72 | Mankodi A, Lin X, Blaxall BC, Swanson MS, Thornton CA(2005) Nuclear RNA foci in the heart in myotonic dystrophyCirc Res97: 11521155 |
73 | Kuyumcu-Martinez NM, Wang GS, Cooper TA(2007) Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylationMolCell28: 6878 |
74 | Salisbury E, Schoser B, Schneider-Gold C, Wang GL, Huichalaf C, Jin B, Sirito M, Sarkar P, Krahe R, Timchenko NA, Timchenko LT(2009) Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patientsAm J Pathol175: 748762 |
75 | Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, Schoser B, Puymirat J(2009) Absence of a differentiation defect in muscle satellite cells from DM2 patientsNeurobiol Dis36: 181190 |
76 | Huichalaf C, Schoser B, Schneider-Gold C, Jin B, Sarkar P, Timchenko L(2009) Reduction of the rate of protein translation in patients with myotonic dystrophy 2J Neurosci29: 90429049 |
77 | Maeda M, Taft CS, Bush EW, Holder E, Bailey WM, Neville H, Perryman MB, Bies RD(1995) Identification, tissue-specific expression, and subcellular localization of the 80- and 71-kDa forms of myotonic dystrophy kinase proteinJ Biol Chem270: 2024620249 |
78 | Raheem O, Olufemi SE, Bachinski LL, Vihola A, Sirito M, Holmlund-Hampf J, Haapasalo H, Li YP, Udd B, Krahe R(2010) Mutant (CCTG)n expansion causes abnormal expression of Zinc finger protein 9 (ZNF9) in myotonic dystrophy type 2Am J Pathol177: 30253036 |
79 | Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE(1997) Expansion of a CUG trinucleotiderepeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcriptsProc Natl Acad Sci U S A94: 73887393 |
80 | Jansen G, Groenen PJ, Bachner D, Jap PH, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ, Molenaar PC, Nederhoff MG, van Echteld CJ, Dekker M, Berns A, Hameister H, Wieringa B(1996) Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in miceNat Genet13: 316324 |
81 | Reddy S, Smith DB, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D, Balice-Gordon RJ, Housman D(1996) Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathyNat Genet13: 325335 |
82 | Berul CI, Maguire CT, Gehrmann J, Reddy S(2000) Progressive atrioventricular conduction block in a mouse myotonic dystrophy modelJ Interv Card Electrophysiol4: 351358 |
83 | Chen W, Wang Y, Abe Y, Cheney L, Udd B, Li YP(2007) Haploinsuffciency for Znf9 in Znf9/ mice is associated with multiorgan abnormalities resembling myotonic dystrophyJ Mol Biol368: 817 |
84 | Nakamori M, Sobczak K, Puwanant A, Welle S, Eichinger K, Pandya S, Dekdebrun J, Heatwole CR, McDermott MP, Chen T, Cline M, Tawil R, Osborne RJ, Wheeler TM, Swanson MS, Moxley RT3rd, Thornton CA(2013) Splicing biomarkers of disease severity in myotonic dystrophyAnn Neurol74: 862872 |
85 | Perfetti A, Greco S, Fasanaro P, Bugiardini E, Cardani R, Manteiga JM, Riba M, Cittaro D, Stupka E, Meola G, Martelli F(2014) Genome wide identification of aberrant alternative splicing events in myotonic dystrophy type 2PLoS One9: e93983 |
86 | Perdoni F, Malatesta M, Cardani R, Giagnacovo M, Mancinelli E, Meola G, Pellicciari C(2009) RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type An immunocytochemical studyEur J Histochem53: 151158 |
87 | Malatesta M, Giagnacovo M, Cardani R, Meola G, Pellicciari C(2011) RNA processing is altered in skeletal muscle nuclei of patients affected by myotonic dystrophyHistochem Cell Biol135: 419425 |
88 | Eisenberg I, Alexander MS, Kunkel LM(2009) miRNAS in normal anddiseased skeletal muscleJ Cell Mol Med13: 211 |
89 | Greco S, De Simone M, Colussi C, Zaccagnini G, Fasanaro P, Pescatori M, Cardani R, Perbellini R, Isaia E, Sale P, Meola G, Capogrossi MC, Gaetano C, Martelli F(2009) Common micro-RNA signature in skeletal muscle damage and regeneration induced by Duchenne muscular dystrophy and acute ischemiaFASEB J23: 33353346 |
90 | Gambardella S, Rinaldi F, Lepore SM, Vihola A, Loro E, Angelini C, Vergani L, Novelli G, Botta A(2010) Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patientsJ Transl Med8: 4854 |
91 | Perbellini R, Greco S, Sarra-Ferraris G, Cardani R, Capogrossi MC, Meola G, Martelli F(2011) Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1Neuromuscul Disord21: 8188 |
92 | Greco S, Perfetti A, Fasanaro P, Cardani R, Capogrossi MC, Meola G, Martelli F(2012) Deregulated microRNAs in myotonic dystrophy type 2, PLoS One7e39732 |
93 | Perfetti A, Greco S, Bugiardini E, Cardani R, Gaia P, Gaetano C, Meola G, Martelli F(2014) Plasma microRNAs as biomarkers for myotonic dystrophy type 1Neuromuscul Disord24: 509515 |
94 | Zu T, Gibbensa B, Dotya NS, Gomes-Pereira M, Huguetd A, Stone MD, Margolis J, Petersong M, Markowski TW, Ingram MAC, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LPW(2011) Non-ATG–initiated translation directed by microsatelliteexpansionsProc Natl Acad Sci U S A108: 260265 |
95 | Pearson CE(2011) Repeat associated non-ATG translation initiation: OneDNA, two transcripts, seven reading frames, potentially ninetoxic entities!PLoS Genet7: e1002018 |
96 | Zu T, Cleary J, Liu Y, Reid T, Banez-Coronel M, Xia G, Ashizawa T, Yachnis A, Ranum LPW(2013) RAN proteins from intronic CCTG expansions in DM2 patient brainsIDMC9, 16-19 October San Sebastian, Spain. Abstract |
97 | Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N, Kokunai Y, Tsuburaya R, de la Grange P, Dembele D, Francois V, Precigout G, Boulade-Ladame C, Hummel MC, Lopez de Munain A, Sergeant N, Laquerrière A, Thibault C, Deryckere F, Auboeuf D, Garcia L, Zimmermann P, Udd B, Schoser B, Takahashi MP, Nishino I, Bassez G, Laporte J, Furling D, Charlet-Berguerand N(2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophyNat Med17: 720725 |
98 | Kimura T, Nakamori M, Lueck JD, Pouliquin P, Aoike F, Fujimura H, Dirksen RT, Takahashi MP, Dulhunty AF, Sakoda S(2005) Altered mRNA splicing of the skeletal muscle ryanodine receptor and sarcoplasmic/endoplasmic reticulum Ca2+-ATPase in myotonic dystrophy type 1Hum Mol Genet14: 21892200 |
99 | Hino S, Kondo S, Sekiya H, Saito A, Kanemoto S, Murakami T, Chihara K, Aoki Y, Nakamori M, Takahashi MP, Imaizumi K(2007) Molecular mechanisms responsible for aberrant splicing of SERCA1 in myotonic dystrophy type 1Hum Mol Genet16: 28342843 |
100 | Tang ZZ, Yarotskyy V, Wei L, Sobczak K, Nakamori M, Eichinger K, Moxley RT, Dirksen RT, Thornton CA(2012) Muscle weakness in myotonic dystrophy associated with misregulated splicing and altered gating of Ca(V)1.1 calcium channelHum Mol Genet21: 13121324 |
101 | Vihola A, Bachinski LL, Sirito M, Olufemi SE, Hajibashi S, Baggerly KA, Raheem O, Haapasalo H, Suominen T, Holmlund-Hampf J, Paetau A, Cardani R, Meola G, Kalimo H, Edström L, Krahe R, Udd B(2010) Differences in aberrant expression and splicing of sarcomeric proteins in the myotonic dystrophies DM1 and DM2Acta Neuropathol119: 465479 |
102 | Cardani R, Baldassa S, Botta A, Rinaldi F, Novelli G, Mancinelli E(2009) Ribonuclear inclusions and MBNL1 nuclear sequestration do not affect myoblast differentiation but alter gene splicing in myotonic dystrophy type 2Neuromuscul Disor19: 335343 |
103 | Loro E, Rinaldi F, Malena A, Masiero E, Novelli G, Angelini C, Vergani L, Novelli G, Botta A(2010) Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cellsCell Death Differ17: 13151324 |
104 | Malatesta M, Giagnacovo M, Renna LV, Cardani R, Meola G, Pellicciari C(2011) Cultured myoblasts from patients affected by myotonic dystrophy type 2 exhibit senescence-related features: Ultrastructural evidenceEur J Histochem55: e26 |
105 | Renna LV, Cardani R, Botta A, Rossi G, Fossati B, Costa E, Meola G(2014) Premature senescence in primary muscle cultures of myotonic dystrophy type 2 is not associated with p16 inductionEur J Histochem58: 2444 |
106 | Furling D, Coiffier L, Mouly V, Barbet JP, Lacau St Guily J, Taneja K, Gourdon G, Junien C, Butler-Browne GS(2001) Defective satellite cells in congenital myotonic dystrophyHum Mol Genet10: 20792087 |
107 | Thornell LE, Lindstöm M, Renault V, Klein A, Mouly V, Ansved T, Butler-Browne G, Furling D(2009) Satellite cell dysfunction contributes to the progressive muscle atrophy in myotonic dystrophy type 1Neuropathol Appl Neurobiol35: 603613 |
108 | Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Brown G, Mouly V, Furling D(2009) Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cellsAm J Pathol174: 14351442 |
109 | Day JW, Ranum LP(2005) Genetics and molecular pathogenesis of the myotonic dystrophiesCurr Neurol Neurosci Re5: 5559 |
110 | Bonifazi E, Vallo L, Giardina E, Botta A, Novelli G(2004) A long PCR-based molecular protocol for detecting normal and expanded ZNF9 alleles in myotonic dystrophy type 2Diagn Mol Pathol13: 164166 |
111 | Catalli C, Morgante A, Iraci R, Rinaldi F, Botta A, Novelli G(2010) Validation of sensitivity and specificity of tetraplet primed PCR (TP-PCR) in the molecular diagnosis of myotonic dystrophy type 2J Mol Diagn12: 601606 |
112 | Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA(2001) Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2Hum Mol Genet10: 21652170 |
113 | Cardani R, Mancinelli E, Sansone V, Rotondo G, Meola G(2004) Biomolecular identification of (CCTG)n mutation in myotonic dystrophy type 2 (DM2) by FISH on muscle biopsyEur J Histochem48: 437442 |
114 | Sallinen R, Vihola A, Bachinski LL, Huoponen K, Haapasalo H, Hackman P, Zhang S, Sirito M, Kalimo H, Meola G, Horelli-Kuitunen N, Wessman M, Krahe R, Udd B(2004) New methods for molecular diagnosis and demonstration of the (CCTG)n mutation in myotonic dystrophy type 2 (DM2)Neuromuscul Disord14: 274283 |
115 | Meola G, Bugiardini E, Cardani R(2012) Muscle biopsyJ Neurol259: 601610 |
116 | Cardani R, Mancinelli E, Rotondo G, Sansone V, Meola G(2006) Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2Eur J Histochem50: 177182 |
117 | Meola G, Sansone V(2007) Cerebral involvement in myotonic dystrophiesMuscle Nerve36: 294306 |
118 | Akiguchi I, Nakano S, Shiino A, Kimura R, Inubushi T, Handa J, Nakamura M, Tanaka M, Oka N, Kimura J(1999) Brain proton magnetic resonance spectroscopy and brain atrophy in myotonic dystrophyArch Neurol56: 325330 |
119 | Chang L, Ernst T, Osborn D, Seltzer W, Leonido-Yee M, Poland RE(1998) Proton spectroscopy in myotonic dystrophy: Correlations with CTGrepeatsArch Neurol55: 305311 |
Figures and Tables
Fig.1
Myotonic dystrophies are multisystemic diseases with a core pattern of clinical presentation which also presents a number of dissimilar features making them clearly separate diseases.
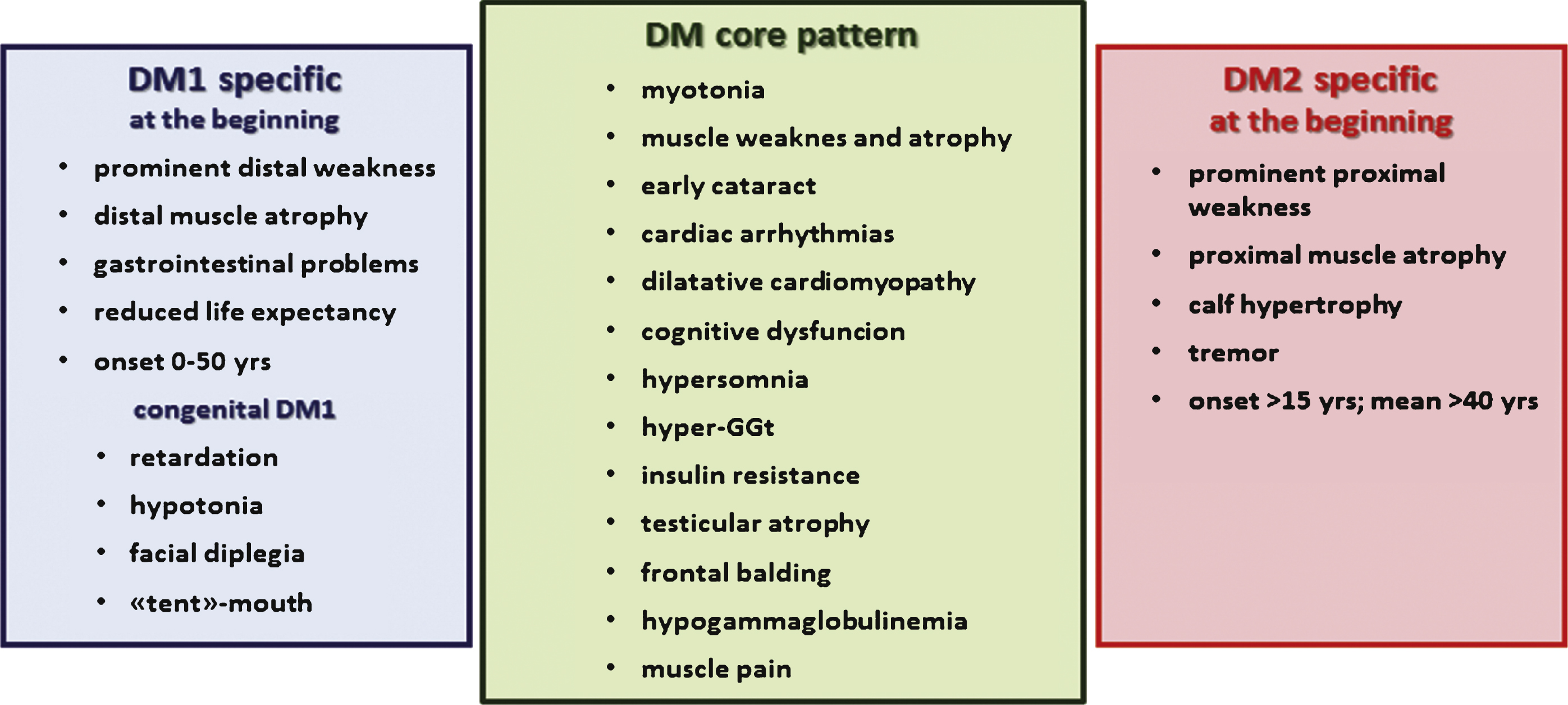
Fig.2
Common and specific postulated pathological mechanisms underlying myotonic dystrophy type 1 and type 2.