Follistatin Gene Therapy Improves Ambulation in Becker Muscular Dystrophy
Abstract
Follistatin is a ubiquitous secretory propeptide that functions as a potent inhibitor of the myostatin pathway, resulting in an increase in skeletal muscle mass. Its ability to interact with the pituitary activin-inhibin axis and suppress the secretion of follicle-stimulating hormone (FSH) called for caution in its clinical applicability. This limitation was circumvented by the use of one of the alternatively spliced follistatin variants, FS344, undergoing post-translational modification to FS315. This follistatin isoform is serum-based, and has a 10-fold lower affinity to activin compared to FS288. Preclinical studies of intramuscular delivery of the follistatin gene demonstrated safety and efficacy in enhancing muscle mass. We herein review the evidence supporting the utility of follistatin as a genetic enhancer to improve cellular performance. In addition, we shed light on the results of the first clinical gene transfer trial using the FS344 isoform of follistatin in subjects with Becker muscular dystrophy as well as the future directions for clinical gene therapy trials using follistatin.
Abbreviations
FS | follistatin |
FSH | follicle-stimulating hormone |
BMD | Becker muscular dystrophy |
AAV | adeno-associated virus |
INTRODUCTION
Monogenic diseases are the targets for most gene therapy offering the potential for gene replacement or gene restoration. Alternatively, the delivery of a surrogate gene has the potential for enhancement of cellular performance that bypasses the underlying gene defect. Follistatin gene therapy exemplifies this strategy. In this review, we provide a summary of follistatin as a potent inhibitor of the myostatin signaling pathway and the therapeutic applications in both inherited and acquired neuromuscular disorders. We also highlight the findings of the first human clinical trial delivering the follistatin gene in muscular dystrophy patients.
Myostatin signaling pathway and regulation of muscle growth
Myostatin is a muscle-specific secretory protein that negatively regulates muscle growth [1]. Initially designated growth differentiation factor-8 (GDF-8), myostatin is expressed early in embryonic myoblasts and under defined conditions at a later time in developing and adult skeletal muscles [2, 3]. Myostatin knockout mice (Mstn −/−) exhibit a doubling of muscle mass related to a combination of increased number of muscle fibers and increased muscle fiber size [1]. This has raised questions of a possible myostatin-activin A ligand for satellite cell signaling [3]. Studies by Lee et al. have shed light on this issue, demonstrating that myostatin-related muscle hypertrophy can occur in the absence of satellite cell function. Thus, the therapeutic potential for increasing muscle growth following myostatin inhibition can occur even in conditions where satellite cells are dormant or depleted, including muscular dystrophy and other conditions such as sarcopenia [4]. Myostatin down-regulates the expression of transcriptional regulators of muscle development such as MyoD and Pax-3, which results in suppression of myogenic cell differentiation and muscle fiber growth. Earlier studies of myostatin demonstrated a widespread improvement in individual muscle weights of myostatin null mice with a 2-3-fold increase compared to wild-type littermates [1]. While muscle gain in myostatin null mice was due to a combination of both muscle fiber hypertrophy and hyperplasia, the increase in muscle mass seen in the myostatin-mutated double-muscled cattle is predominantly from hyperplasia of muscle fibers rather than hypertrophy [5–8]. A report of a similar muscular phenotype in a young boy with a myostatin mutation highlighted the clinical significance of reduced myostatin levels in humans [9]. The effect of myostatin inhibition extends beyond its influence on muscle development. In addition to enhancement of muscle mass, myostatin deficiency leads to a reduction in fat deposition, suggesting an additional role in regulating adipogenesis [10, 11].
Myostatin is a member of the transforming growth factor-β (TGF- β) superfamily and is synthesized and secreted as a 376-amino acid precursor polypeptide formed by an N-terminal propeptide domain, a signal sequence for secretion, and a C-terminal domain [1] (Fig. 1). Myostatin activation is regulated through a proteolytic process that removes the signal peptide and cleaves the N-terminal releasing a latent myostatin dimer-propeptide complex that requires further modification to a myostatin dimer exerting its skeletal muscle effect through a signaling pathway initiated by the binding of the active C-terminus to a transmembrane activin receptor, ActRIIB. Activation of ActRIIB enhances the transphosphorylation of type I activin receptors, ActRIB (ALK4) and TGFbetaRI (ALK5), and a cascade of events culminating in the nucleus targeting the expression of myogenic regulatory genes [12–14]. Interference with myostatin’s binding to the ActRIIB receptor alters this pathway and the negative effect of myostatin on skeletal muscle. Follistatin is one of several myostatin-binding proteins capable of inactivating myostatin by preventing receptor binding and the effects on the subsequent signaling pathway [15].
Follistatin is a potent inhibitor of the myostatin pathway
Follistatin, a monomeric secretory protein coded for by the FST gene, is a ubiquitous glycoprotein expressed in nearly all tissues at variable concentrations [16]. It was first isolated from porcine ovarian follicular fluid in in vitro studies that identified its function as a suppressor of the release of follicle stimulating hormone (FSH) [17–21]. The mechanism by which follistatin inhibits FSH secretion was subsequently shown to be through the inhibition of activin [22]. Activin, a TGF-β ligand, enhances the synthesis and secretion of (FSH) through interaction with the same transmembrane receptors as myostatin [23]. Hence, the role follistatin plays in any tissue is fundamentally based in its ability to neutralize the effect of various TGF-β ligands that include myostatin and components of the activin-inhibin complex through competitiveinhibition of binding to the Act RIIB receptors [15, 22, 24, 25]. The monomeric follistatin protein is composed of two isoforms, FS-288 and FS-315, produced by alternative splicing of the approximately 6 kb FST gene precursor mRNA transcript [26] (Fig. 2). The FS315 isoform results from peptide cleavage of the FS344 variant, and the FS288 isoform is produced by cleavage of the FS317 variant [24, 26]. FS288 is the membrane-bound form of follistatin [27] and is a potent suppressor of FSH, while FS344 generates the serum circulating FS315 isoform that includes a C-terminal acidic region [24]. It is suggested that a third isoform FS303, abundantly isolated from porcine ovaries, is derived from proteolytic cleavage of this carboxyl terminal [27].
The intricate mechanism by which follistatin interferes with ligand binding to receptors in skeletal muscle tissue was studied in animal models in which follistatin was genetically manipulated. Follistatin-deficient mice have reduced muscle mass, skeletal defects, retarded growth and die within hours of birth [28]. Contrarily, transgenic mice expressing high levels of follistatin have a dramatic increase in muscle mass by 194–327% relative to controls that results from a net effect of an increase in fiber count as well as fiber diameter [29]. The effect of overexpressing follistatin is significantly greater than the increase in muscle size in the myostatin null animals [1]. This suggests that the mechanism by which follistatin enhances muscle growth is likely through regulating the action of several members of the TGF-β family and not exclusively through its ability to block the myostatin pathway. These findings favor follistatin as a gene therapy candidate in the treatment of muscular dystrophy with potential advantages over other myostatin-binding proteins.
Translational studies of myostatin inhibition
The dramatic enhancement of muscle mass demonstrated in pre-clinical studies employing a myostatin inhibition strategy in animal models favors a potential translational role for the treatment of muscle disease. The initial clinical study employed MYO-029, a myostatin-neutralizing antibody [30]. While it is true that the design of the study was randomized, double-blind and placebo-controlled, the attempt to include three different diseases, each caused by distinct pathogenic mechanisms, with all three groups underpowered for efficacy, permitted only an assessment of safety. Muscle strength assessment was a secondary outcome that showed no improvement. There were no treatment-emergent adverse events with the exception of a rash, with or without urticaria seen in slightly fewer than 10% of subjects. The study design called for dose escalation but the highest proportion of skin problems encountered in the high dose group limited enrollment. This in essence established a dose-limiting toxicity. Nevertheless, as an introductory effort to the clinic, the conclusion that systemic administration of myostatin inhibition to patients was safe, provided a template upon which to explore future clinical trials.
As a complementary approach to the targeted effort to inhibit myostatin using neutralizing antibodies, we designed a follistatin gene delivery study that would potentially enable a path to clinical translation. On the clinical side, Becker muscular dystrophy (as opposed to Duchenne) provided targeted and relatively focal muscle weakness and atrophy of the quadriceps muscle that limited ambulation and resulted in frequent falls. It was our intent to strengthen the quadriceps muscle by intramuscular injection of the follistatin gene. Proof of principle studies in preparation for the clinical trial were done in mdx mice receiving quadriceps and gastrocnemius muscle injections of adeno-associated virus serotype 1 (AAV1) encoding the follistatin isoform FS344 [31]. In these pre-clinical studies, the FS344 (FS315) isoform was favored because of a lower affinity for activin and a reduced potency for inhibiting FSH hormone secretion compared to FS317 (FS288). The study outcome supported this choice by lack of untoward effects on reproductive capacity in either male or female treated animals in the presence of significant muscle hypertrophy and improved muscle function. Of note in both non-human primate studies [32] and in our subsequent clinical trial [33] we found no effect of FS344 (FS315) gene therapy on any pituitary secreted hormone.
Of further interest, following gene transfer in the mdx mouse was the finding of muscle hypertrophy not only in the muscles receiving direct AAV1.FS344 injections (quadriceps and gastrocnemius), but also in adjacent non-injected lower limb muscles, paraspinal muscles and even extending to upper limb muscles. This suggested a remote effect related to circulating follistatin from secretion of injected muscles that were transduced by FS344. The high serum levels of follistatin supported this explanation. Histological assessment of the AAV1.FS344 treated muscle also showed favorable findings, given that endomysial connective tissue was reduced, an established hallmark of muscular dystrophy. The benefit of this treatment persisted for more than a year in treated mice. Similar long-term benefits from the FS344 transgene were seen in cynomolgus macaque non-human primates treated with the same cassette [32], with expression up to 15 months after gene transfer. Although an increase in muscle size and strength was also exhibited in the treated macaques, these findings were interpreted cautiously given that these animals did not have the muscle pathology seen in muscular dystrophy. The pre-clinical studies in both mice and non-human primates in the absence of toxicity enabled a Phase I/IIa clinical trial in patients with BMD (IND 14845) [33].
Phase I/IIa follistatin gene therapy trial in becker muscular dystrophy
In the planning of the first follistatin gene transfer clinical trial, factors taken into consideration in the selection of Becker muscular dystrophy (BMD) as the targeted patient population included the lack of treatment efficacy [34–37] compared to glucocorticoids in DMD [38, 39]. A potential advantage is the expression of a truncated dystrophin protein that partially protects the membrane and would potentially shield the muscle from any stress that results from increasing size related to follistatin therapy. The prominence of weakness of the quadriceps muscle (knee extensors) in BMD has already been mentioned above. Six ambulatory BMD patients with confirmed mutations in the dystrophin gene were enrolled in this dose-ascending gene therapy trial [33]. Patients in the low dose cohort (n = 3) received 3 × 1011 vg/kg/leg (total 6 × 1011 vg/kg/patient), while high dose injections were 6 × 1011 vg/kg/leg (1.2 × 1012 vg/kg/patient) for the remaining three subjects. Eligibility for the trial required knee extensor weakness ≥2 standard deviations below normal [40], ability to cooperate for testing, no evidence of cardiomyopathy, diabetes or organ system abnormalities of bone marrow, liver or kidney and finally willingness to practice contraception during the study. All patients were screened for human immunodeficiency virus infection and viral hepatitis. Prior to receiving AAV1.CMV.FS344 injections, all subjects were treated with prednisone for one month in an attempt to prevent an immune response to the AAV capsid as seen in other gene therapy trials [41–43]. T-cell responses towards the AAV1 capsid were monitored by IFN-γ ELISpot assays; all subjects had levels <50 spot forming cells/million peripheral blood mononuclear cells (PBMCs). In addition, serum neutralizing antibody titers to AAV1 were assessed by ELISA and were <1:50 at enrollment. Both assays were monitored throughout the trial and prednisone treatment was reduced according to immunological findings with a final taper by 60 days post-gene therapy for all subjects.
Baseline muscle biopsies as well as magnetic resonance imaging (MRI) studies were performed prior to gene therapy. The primary functional outcome measure was the distance walked on the 6-minute walk test (6MWT) assessed at predefined time points. Safety labs to assess for adverse effects of the gene therapy were drawn during scheduled follow up visits. This included a complete blood count, liver function tests, creatine kinase, amylase and serum sex hormones. Renal function was assessed using cystatin C because of reduced muscle mass that can falsely obscure renal toxicity [44]. A repeat muscle biopsy from the contralateral quadriceps muscle was obtained at 180 days post-treatment. Injections for both cohorts 1 and 2 were distributed in a similar manner, with four injections in three of the four muscles forming the quadriceps muscle (vastus lateralis, vastus medialis and rectus femoris). The procedure was very well tolerated by all patients and there were no immediate or late adverse effects directly related to gene transfer. Follow up data is available for up to 12 months on Cohort 1 (low dose) and 6 months for cohort 2 (high dose). In each cohort of three subjects, two patients improved in the distance walked measured by the 6MWT (Fig. 3). The two subjects who did not benefit from this treatment had both histological and radiological evidence of extensive fibrosis and advanced stage of disease that likely explains the lack of functional improvement (Fig. 4).
Clinical applicability of follistatin gene transfer in other muscle diseases
The excellent safety profile as well as the therapeutic benefit in the BMD patients paves the path for an extension of the trial to other muscle diseases. The potential role for follistatin in targeting inflammatory cells, promoting muscle regeneration and increasing muscle mass, offer a rationale for treating patients with sporadic inclusion body myositis (sIBM). This disease is the most common, yet most challenging, acquired muscle disorder seen in the adult population that is characterized by progressive degeneration and an inflammatory process that is refractory to immunosuppressive therapies. Patients with sIBM characteristically have weak quadriceps muscles, putting them at high risk for serious injury due to falls. In an attempt to reduce morbidity by improving quadriceps strength, a dose-escalation trial of AAV1.CMV.FS344 in patients with sIBM has been initiated. Ultrasound-guided intramuscular injections to the quadriceps muscle have been carefully planned based on areas of preserved skeletal muscle. The first three patients with sIBM have been injected with AAV1.CMV.FS344 at low-dose vector (2 × 1011 vg/kg) in a single limb as required by the FDA. No adverse events were encountered, and patients transiently improved the distance walked on the 6MWT (in spite of unilateral injections). Treatment has now extended to the high-dose sIBM group; bilateral intramuscular injections of 1.2 × 1012 vg/kg of vector have been delivered to three subjects and additional patients are being recruited. This trial is still in progress and the results are under evaluation.
Of particular relevance to myostatin inhibition, separate from follistatin, a randomized double-blind controlled proof-of-concept clinical study of the human monoclonal antibody against the activin receptor 2B (ACVR2B), bimagrumab (BYM338), has been tested in 11 subjects with sporadic inclusion body myositis (sIBM). After 8 weeks of treatment, an increase in muscle volume assessed by magnetic resonance imaging has been reported [45]. At 16 weeks after dosing, the 6-minute walk distance was significantly improved (+14.6% , p = 0.008). A phase III long-term safety, efficacy tolerability clinical trial is currently underway using this drug in patients with sIBM (NCT02250443).
In light of the promising results seen in BMD, a Phase I/IIa trial was recently launched aiming at treating a cohort of six boys with Duchenne muscular dystrophy (DMD) using AAV1.CMV.FS344 vector. A significantly higher dose of AAV will be used in the DMD follistatin gene therapy trial (2.4 × 1012 vg/kg) permitting a greater number of muscles to be targeted including glutei, quadriceps, and tibialis anterior muscles. Patients have been selected based on age, baseline 6MWT performance as well as preservation of skeletal muscle assessed by MRI at time of enrollment. All subjects will be followed for two years after gene transfer.
CONCLUSIONS
In summary, inhibition of the myostatin pathway remains a promising strategy in gene therapy for patients with muscular dystrophy. Follistatin is a safe and potent inhibitor of this pathway with strong potential for broad applicability that can be extended to other muscle diseases. Careful selection of targeted muscle groups at earlier stages of pathology may lead to the enhancing effects of follistatin on muscle mass and strength. The fundamental role of follistatin in augmenting myofiber growth supports its potential use in conjunction with other disease-specific gene replacement therapies [46]. It should also be noted that pre-clinical studies in Pompe mice show that follistatin could be used as an adjuvant therapy to alpha-glucosidase deficiency [47].
ACKNOWLEDGMENTS
The Parent Project Muscular Dystrophy supported the Clinical Trial. Staff for this trial and some of the materials and supplies were supplied by the Senator Paul D Wellstone Muscular Dystrophy Research Center, NICHD, NIH, Bethesda, MD #5U54HD066409-05. Jesse’s Journey supported some of the participating staff. The Myositis Association (TMA) helped bring this trial to the clinic by supporting the preclinicalstudies.
REFERENCES
1 | McPherron AC, Lawler AM, Lee SJ(1997) Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily memberNature387: 8390Epub 1997/05/01. |
2 | Gonzalez-Cadavid NF, Taylor WE, Yarasheski K, Sinha-Hikim I, Ma K, Ezzat S(1998) Organization of the human myostatin gene andexpression in healthy men and HIV-infected men with musclewastingProceedings of the National Academy of Sciences of theUnited States of America.95: 251493814943Epub 1998/12/09. |
3 | McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R(2003) Myostatin negatively regulates satellite cell activation and self-renewalThe Journal of Cell Biology162: 611351147Epub 2003/09/10. |
4 | Lee SJ, Huynh TV, Lee YS, Sebald SM, Wilcox-Adelman SA, Iwamori N(2353) Role of satellite cells versus myofibers in muscle hypertrophy induced by inhibition of the myostatin/activin signaling pathwayProceedings of the National Academy of Sciences of the United States of America109: 35E2353E2360Epub 2012/08/08. |
5 | Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J(1997) A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattleNature Genetics17: 17174Epub 1997/09/01. |
6 | Grobet L, Poncelet D, Royo LJ, Brouwers B, Pirottin D, Michaux C(1998) Molecular definition of an allelic series of mutations disrupting the myostatin function and causing double-muscling in cattleMammalian genome: Official journal of the International Mammalian Genome Society9: 3210213Epub 1998/03/21. |
7 | McPherron AC, Lee SJ(1997) Double muscling in cattle due to mutations in the myostatin geneProceedings of the National Academy of Sciences of the United States of America94: 231245712461Epub 1997/11/14. |
8 | Kambadur R, Sharma M, Smith TP, Bass JJ(1997) Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattleGenome Research7: 9910916Epub 1997/10/06. |
9 | Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W(2004) Myostatin mutation associated with gross muscle hypertrophy in a childThe New England Journal of Medicine350: 2626822688Epub 2004/06/25. |
10 | Lin J, Arnold HB, Della-Fera MA, Azain MJ, Hartzell DL, Baile CA(2002) Myostatin knockout in mice increases myogenesis and decreases adipogenesisBiochemical and Biophysical Research Communications291: 3701706Epub 2002/02/22. |
11 | Gangopadhyay SS (2013) Systemic administration of follistatin288 increases muscle mass and reduces fat accumulation in miceScientific Reports3: 2441Epub 2013/08/15. |
12 | Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L(2003) Myostatin signals through a transforming growthfactor beta-like signaling pathway to block adipogenesisMolecular and Cellular Biology23: 2072307242Epub 2003/10/01. |
13 | Langley B, Thomas M, Bishop A, Sharma M, Gilmour S, Kambadur R(2002) Myostatin inhibits myoblast differentiation by down-regulating MyoD expressionThe Journal of Biological Chemistry277: 514983149840Epub 2002/09/24. |
14 | Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN(2005) Regulation of muscle growth by multiple ligands signaling through activin type II receptorsProceedings of the National Academy of Sciences of the United States of America102: 501811718122Epub 2005/12/07. |
15 | Amthor H, Nicholas G, McKinnell I, Kemp CF, Sharma M, Kambadur R(2004) Follistatin complexes Myostatin and antagonises Myostatin-mediated inhibition of myogenesisDevelopmental Biology270: 11930Epub 2004/05/12. |
16 | Tortoriello DV, Sidis Y, Holtzman DA, Holmes WE, Schneyer AL(2001) Human follistatin-related protein: A structural homologue of follistatin with nuclear localizationEndocrinology142: 834263434Epub 2001/07/19. |
17 | Ling N, Ying SY, Ueno N, Esch F, Denoroy L, Guillemin R(1985) Isolation and partial characterization of a Mr 32,000 protein with inhibin activity from porcine follicular fluidProceedings of the National Academy of Sciences of the United States of America82: 2172177221Epub 1985/11/01. |
18 | Miyamoto K, Hasegawa Y, Fukuda M, Nomura M, Igarashi M, Kangawa K(1985) Isolation of porcine follicular fluid inhibin of 32K daltonsBiochemical and Biophysical Research Communications129: 2396403Epub 1985/06/14. |
19 | Rivier J, Spiess J, McClintock R, Vaughan J, Vale W(1985) Purification and partial characterization of inhibin from porcine follicular fluidBiochemical and Biophysical Research Communications133: 1120127Epub 1985/11/27. |
20 | Ueno N, Ling N, Ying SY, Esch F, Shimasaki S, Guillemin R(1987) Isolation and partial characterization of follistatin: A single-chain Mr 35,000 monomeric protein that inhibits the releaseof follicle-stimulating hormoneProceedings of the National Academy of Sciences of the United States of America84: 2382828286Epub 1987/12/01. |
21 | Robertson DM, Klein R, de Vos FL, McLachlan RI, Wettenhall RE, Hearn MT(1987) The isolation of polypeptides with FSH suppressing activity from bovine follicular fluid which arestructurally different to inhibinBiochemical and Biophysical Research Communications149: 2744749Epub 1987/12/16. |
22 | Thompson TB, Lerch TF, Cook RW, Woodruff TK, Jardetzky TS(2005) The structure of the follistatin: Activin complex revealsantagonism of both type I and type II receptor bindingDevelopmental Cell9: 4535543Epub 2005/10/04. |
23 | Pangas SA, Woodruff TK(2000) Activin signal transduction pathwaysTrends in endocrinology and metabolism: TEM11: 8309314Epub 2000/09/21. |
24 | Shimasaki S, Koga M, Esch F, Cooksey K, Mercado M, Koba A(1988) Primary structure of the human follistatin precursor and its genomic organizationProceedings of the National Academy of Sciences of the United States of America85: 1242184222Epub 1988/06/01. |
25 | Nakamura T, Takio K, Eto Y, Shibai H, Titani K, Sugino H(1990) Activin-binding protein from rat ovary is follistatinScience247: 4944836838Epub 1990/02/16. |
26 | Shimasaki S, Koga M, Esch F, Mercado M, Cooksey K, Koba A(1988) Porcine follistatin gene structure supports two forms of mature follistatin produced by alternative splicingBiochemical and Biophysical Research Communications152: 2717723Epub 1988/04/29. |
27 | Sugino K, Kurosawa N, Nakamura T, Takio K, Shimasaki S, Ling N(1993) Molecular heterogeneity of follistatin, an activin-binding proteinHigher affinity of the carboxyl-terminal truncated forms for heparan sulfate proteoglycans on the ovarian granulosa cell. The Journal of Biological Chemistry268: 211557915587Epub 1993/07/25. |
28 | Matzuk MM, Lu N, Vogel H, Sellheyer K, Roop DR, Bradley A(1995) Multiple defects and perinatal death in mice deficient in follistatinNature374: 6520360363Epub 1995/03/23. |
29 | Lee SJ, McPherron AC(2001) Regulation of myostatin activity and muscle growthProceedings of the National Academy of Sciences of the United States of America98: 1693069311Epub 2001/07/19. |
30 | Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM(2008) A Phase I/II trial of MYO-029 in adult subjects with muscular dystrophyAnn Neurol63: 543545 |
31 | Haidet AM, Rizo L, Handy C, Umapathi P, Eagle A, Shilling C(2008) Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitorsProceedings of the National Academy of Sciences of the United States of America105: 1143184322Epub 2008/03/13. |
32 | Kota J, Handy CR, Haidet AM, Montgomery CL, Eagle A, Rodino-Klapac LR(2009) Follistatin gene delivery enhances muscle growth and strength in nonhuman primatesScience Translational Medicine1: 66ra15Epub 2010/04/07. |
33 | Mendell JR, Sahenk Z, Malik V, Gomez AM, Flanigan KM, Lowes LP(2015) A phase 1/2a follistatin gene therapy trial for Becker muscular dystrophyMol Ther23: 192201 |
34 | Backman E, Henriksson KG(1995) Low-dose prednisolone treatment in Duchenne and Becker muscular dystrophyNeuromuscular disorders: NMD5: 3233241Epub 1995/05/01. |
35 | Cittadini A, Ines Comi L, Longobardi S, Rocco Petretta V, Casaburi C, Passamano L(2003) A preliminary randomized study of growth hormone administration in Becker and Duchenne muscular dystrophiesEuropean Heart Journal24: 7664672Epub 2003/03/27. |
36 | Witting N, Kruuse C, Nyhuus B, Prahm KP, Citirak G, Lundgaard SJ(2014) Effect of sildenafil on skeletal and cardiac muscle in Becker muscular dystrophyAnnals of Neurology76: 4550557Epub 2014/07/22. |
37 | Leung DG, Herzka DA, Thompson WR, He B, Bibat G, Tennekoon G(2014) Sildenafil does not improve cardiomyopathy inDuchenne/Becker muscular dystrophyAnnals of Neurology76: 4541549Epub 2014/07/22. |
38 | Mendell JR, Moxley RT, Griggs RC, Brooke MH, Fenichel GM, Miller JP(1989) Randomized, double-blind six-month trial of prednisone in Duchenne’s muscular dystrophyThe New England Journal of Medicine320: 2415921597Epub 1989/06/15. |
39 | Biggar WD, Gingras M, Fehlings DL, Harris VA, Steele CA(2001) Deflazacort treatment of Duchenne muscular dystrophyJ Pediatr138: 14550 |
40 | Tawil R, McDermott MP, Mendell JR, Kissel J, Griggs RC(1994) Facioscapulohumeral muscular dystrophy (FSHD): Design of natural history study and results of baseline testingFSH-DY GrouNeurology44: 3 Pt 1442446Epub 1994/03/01. |
41 | Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S(2010) Dystrophin immunity in Duchenne’s musculardystrophyThe New England Journal of Medicine363: 1514291437Epub 2010/10/12. |
42 | Mendell JR, Rodino-Klapac LR, Rosales-Quintero X, Kota J, Coley BD, Galloway G(2009) Limb-girdle muscular dystrophy type 2D gene therapy restores alpha-sarcoglycan and associated proteinsAnnals of Neurology66: 3290297Epub 2009/10/03. |
43 | Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC(2011) Adenovirus-associated virus vector-mediated genetransfer in hemophilia BThe New England Journal ofMedicine365: 2523572365Epub 2011/12/14. |
44 | Viollet L, Gailey S, Thornton DJ, Friedman NR, Flanigan KM, Mahan JD(2009) Utility of Cystatin C to Monitor Renal Function in Duchenne Muscular DystrophyMuscle Nerve40: 3438442 |
45 | Amato AA, Sivakumar K, Goyal N, David WS, Salajegheh M, Praestgaard J(2014) Treatment of sporadic inclusion body myositis with bimagrumabNeurology83: 2422392246Epub 2014/11/09. |
46 | Rodino-Klapac LR, Janssen PM, Shontz KM, Canan B, Montgomery CL, Griffin DMicro-dystrophin andfollistatin co-delivery restores muscle function in aged DMDmodelHuman Molecular Genetics2013Epub 2013/07/19. |
47 | Foley JW, Bercury SD, Finn P, Cheng SH, Scheule RK, Ziegler RJ(2010) Evaluation of systemic follistatin as an adjuvant to stimulate muscle repair and improve motor function in Pompe miceMolecular therapy: The journal of the American Society of Gene Therapy18: 915841591Epub 2010/06/17. |
Figures and Tables
Fig.1
The myostatin pathway and myostatin-binding proteins. Myostatin (M) is synthesized as a precursor protein. Activation is regulated through a proteolytic process in which the signal peptide (SP) is first removed, followed by a second cleavage that releases two fragments: an N-terminal propeptide domain of ∼28kD and the biologically active 12.5 kD C-terminal domain. The myostatin C-terminus circulates in the blood in a latent inactive state. The final activation requires cleavage at amino acid 76 to prevent the propeptide from binding to the C-terminus. Once activated, the myostatin dimer binds to the activin receptor type IIB (ActRIIB), which then enhances the transphosphorylation of type I activin receptors (ALK4 or ALK5). The intracellular path is through a series of Smads (Smad 2 and Smad 3) leading to the formation of the Smad complex (inclusive of Smad 4) that enters the nucleus to activate target gene transcription. Several proteins, including follistatin (FS), follistatin-related gene (FLRG) and growth and differentiation factor-associated serum protein-1 (GASP-1), have the ability to bind to myostatin leading to its inactivation and inhibition of the myostatin pathway.
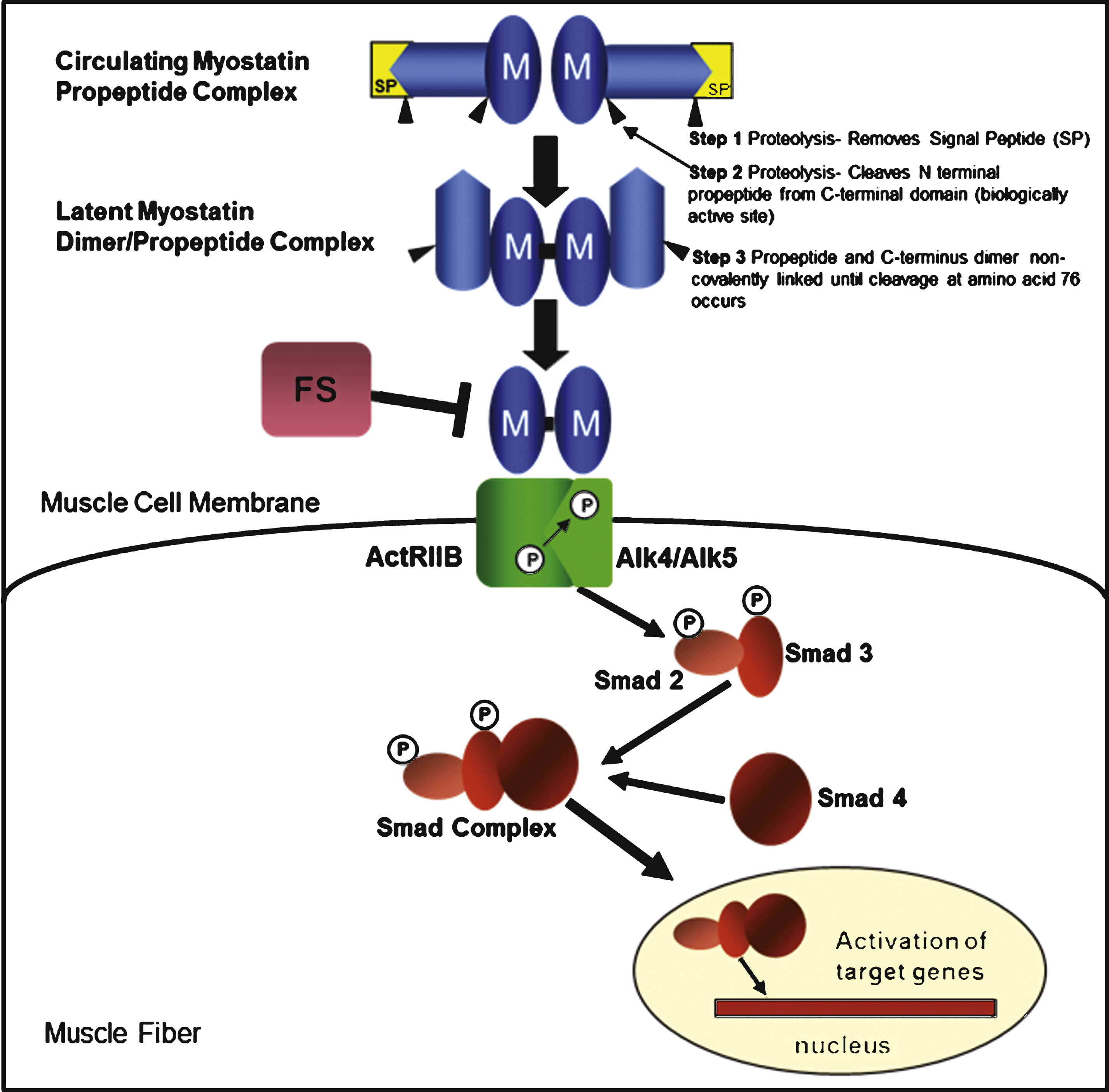
Fig.2
Alternative splicing of the follistatin gene produces two isoforms, FS317 and FS344. Alternative splicing occurs at the 3’ end of the gene between exon 5 and exon 6. Splicing out of intron 5 generates a stop codon immediately following the last amino acid of exon 5, and leads to the termination of the coding sequence for FS317. An alternative splice site results in the inclusion of exon 6 and generates FS344. After translation and prior to activation, follistatin undergoes further posttranslational modification by cleavage of the 29 amino acid signal peptide. This results in polypeptides FS315 (long-isoform from FS344) and FS288 (short-isoform from FS317).
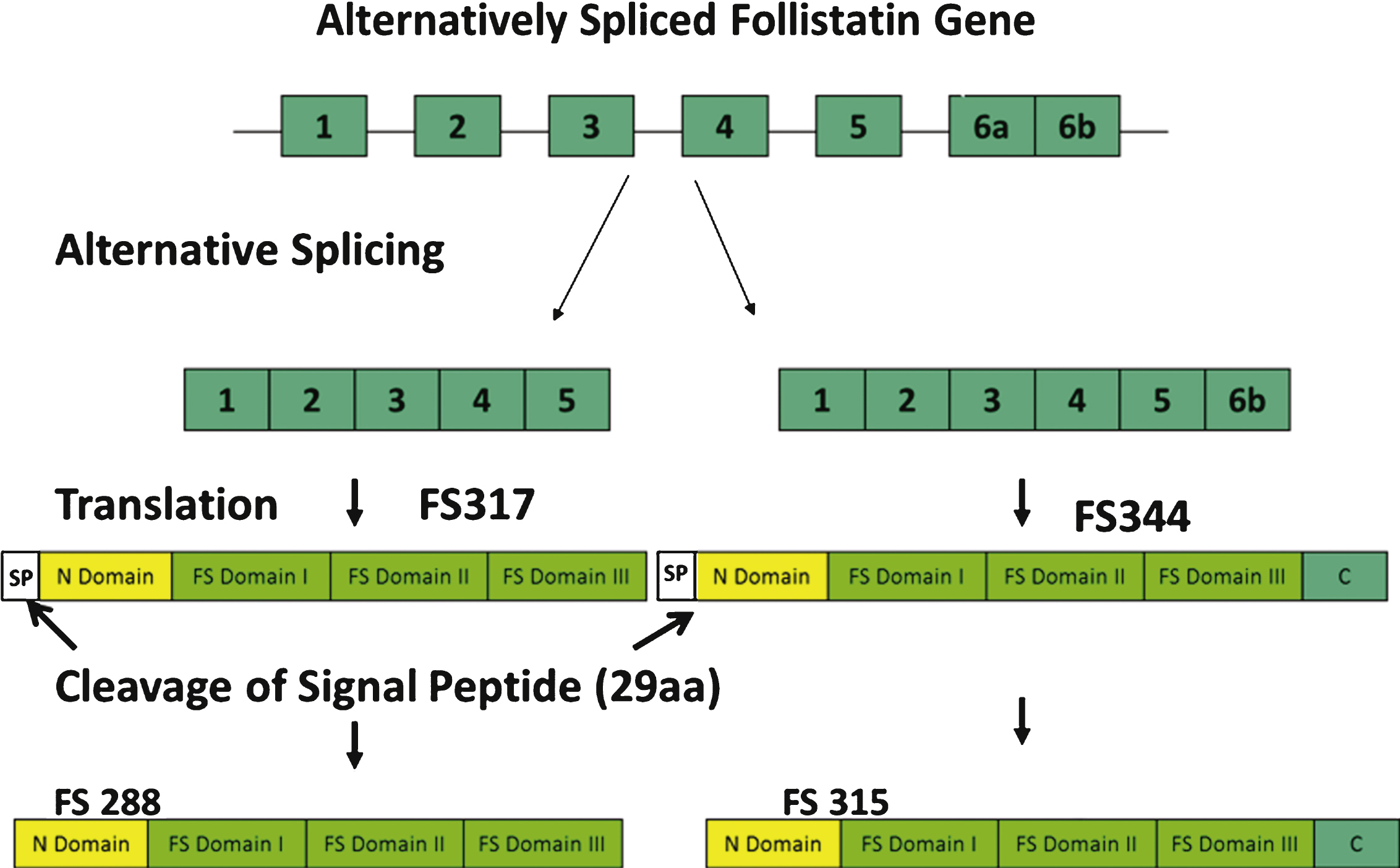
Fig.3
Six-minute walk test (6MWT) in follistatin-treated Becker muscular dystrophy (BMD) Subjects. This graph represents the average distance walked in meters (y axis) in all six subjects over a 180-day follow-up period (x axis). Despite two subjects who failed to improve on the 6MWT, we observed a statistically significant average improvement by 11.5% (p = 0.02) at six months post-gene therapy.
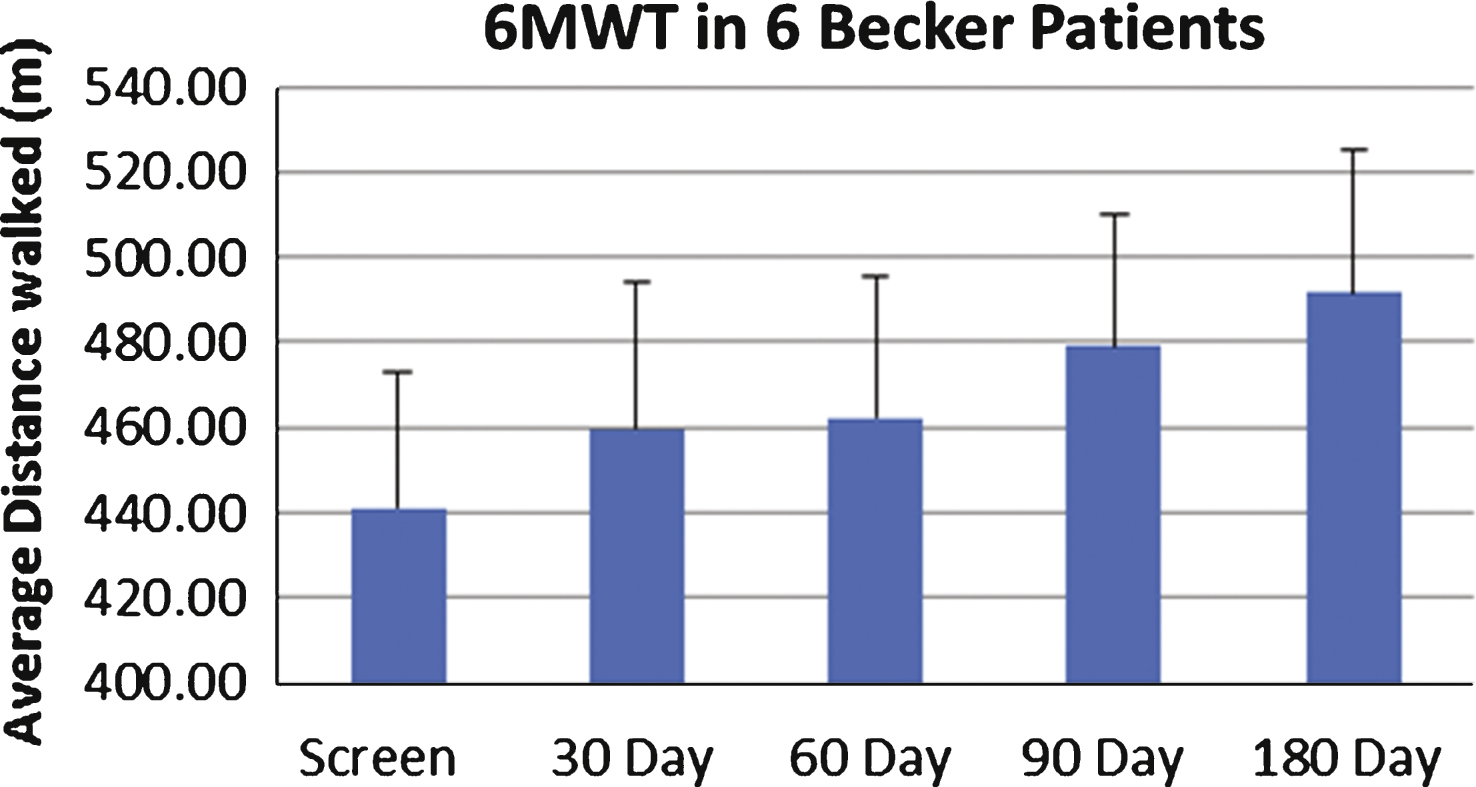
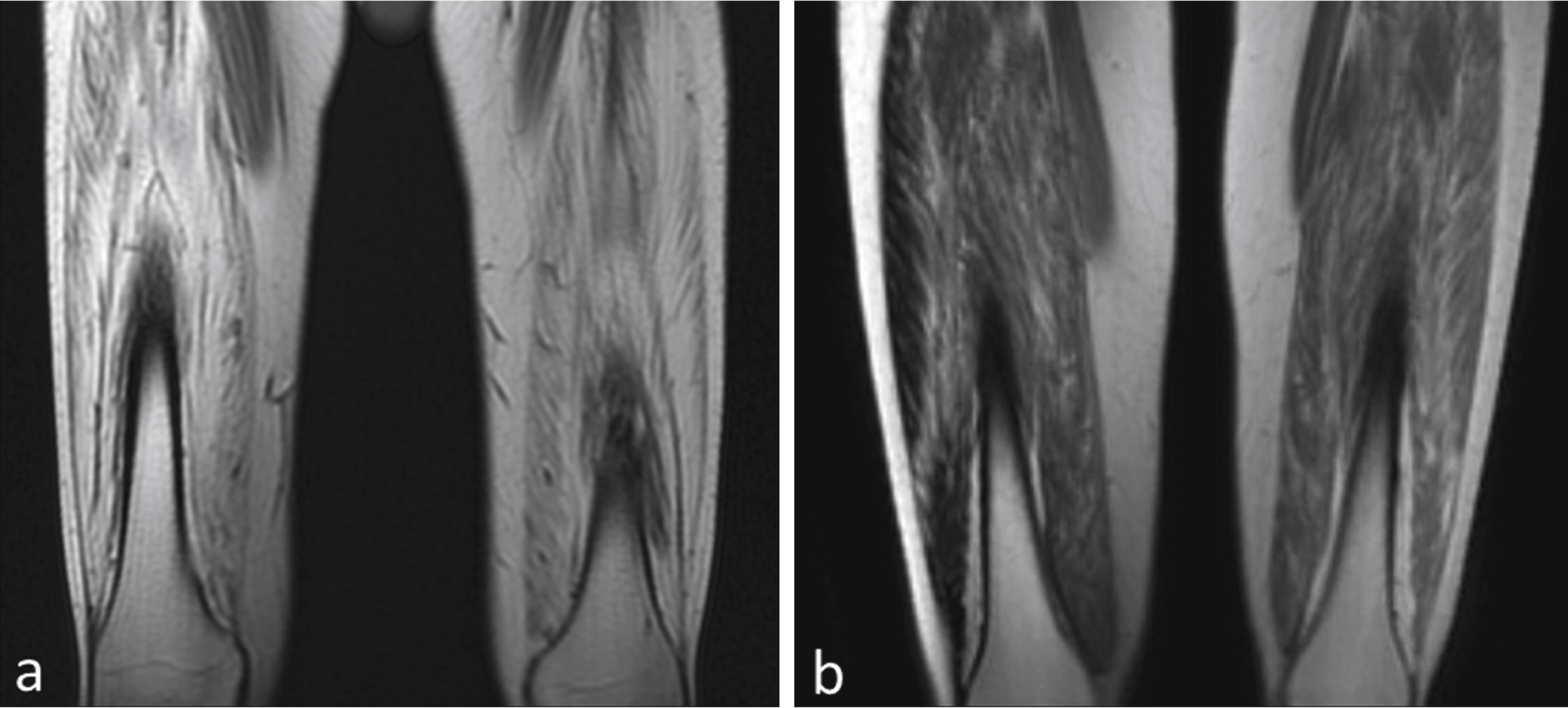
Fig.4
Degree of fibrosis correlates with treatment effect. (a) MRI of quadriceps muscles for the BMD patient with insignificant improvement (distance walked- 9 m on 6MWT) shows a much higher degree of skeletal muscle involvement compared to (b) the BMD subject who had most benefit (distance walked = 108 m on 6MWT).