
Towards Standardizing Nomenclature in Huntington’s Disease Research
Abstract
The field of Huntington’s disease research covers many different scientific disciplines, from molecular biology all the way through to clinical practice, and as our understanding of the disease has progressed over the decades, a great deal of different terminology has accrued. The field is also renowned for its collaborative spirit and use of standardized reagents, assays, datasets, models, and clinical measures, so the use of standardized terms is especially important. We have set out to determine, through a consensus exercise involving basic and clinical scientists working in the field, the most appropriate language to use across disciplines. Nominally, this article will serve as the style guide for the Journal of Huntington’s Disease (JHD), the only journal devoted exclusively to HD, and we lay out the preferred and standardized terminology and nomenclature for use in JHD publications. However, we hope that this article will also serve as a useful resource to the HD research community at large and that these recommended naming conventions will be adopted widely.
INTRODUCTION
Huntington’s disease (HD, MIM# 143100) is an autosomal-dominant neurodegenerative condition caused by a CAG-repeat expansion in the first exon of the gene huntingtin (HTT), and this expansion lengthens a polyglutamine segment in the encoded huntingtin protein (HTT). Symptoms involve impairment in motor, cognitive, and behavioral/psychiatric domains, generally appearing during midlife with slowly progressive decline over the course of two decades. There is a wealth of scientific and clinical literature related to the disorder, but no disease-modifying treatment has been approved for HD as yet.
The primary authors of this position paper recognized a need for consistency of nomenclature in preclinical and clinical HD research. We assembled working groups to survey the current use of terms in the following fields: clinical genetics, genes and proteins, animal and cell models, and neuropathology. These working groups met over a series of conference calls, contributed writing and each of the members reviewed the recommendations across all four fields, culminating in this consensus paper. Specific HD-related terms are ranked as follows:
Preferred usage – Authors are advised to use this form of the term.
Acceptable alternative usage – Authors may use this form if done consistently in text.
Non-preferred usage – JHD will require justification by authors and approval of editors.
Not acceptable – Term is misleading or inappropriate and will not be accepted in JHD.
OVERVIEW OF SECTIONS
1. Clinical and Genetic Terms
2. Gene and Protein Nomenclature
3. Preclinical Animal and Cellular Models
4. Neuroanatomy and Neuropathology
5. Glossary of JHD preferred terms
6. References
1. CLINICAL AND GENETIC TERMS
There are good arguments to be made for the use of both “Huntington disease” and “Huntington’s disease”. That George Huntington did not himself have the disease, but rather first published a description of its clinical features, is the basis for using “Huntington disease”, but much like Parkinson’s disease and Alzheimer’s disease, the more commonly accepted term and the term generally used by the patient community is “Huntington’s disease”. For publications in JHD, “Huntington’s disease” is preferred, but we recognize that both forms are acceptable as long as authors are consistent. A common grammatical error to be avoided is capitalization of “disease”, which is not correct in text, despite the standard abbreviation “HD”.
The human huntingtin gene’s Human Genome Organization (HUGO) Nomenclature Committee-approved symbol is HTT and the mutant gene, containing the expanded CAG repeat, is referred to as the mutant huntingtin gene (mHTT). Previous historical designations of the gene such as “IT-15” or “HD gene” are not acceptable usage in JHD. The protein product of an HTT allele that has an expanded, disease-associated CAG repeat is called “mutant huntingtin protein” (standard abbreviation, mHTT). This is the preferred term in JHD, rather than other terms such as “variant huntingtin protein” which are not acceptable (although required in some other journals such as JAMA). Acceptable alternatives include “expanded huntingtin protein”, “expanded repeat huntingtin protein” or “expanded polyglutamine huntingtin protein”, although these are not the standard or preferred terms in JHD.
The preferred JHD term for the protein produced from an HTT allele that is not associated with a disease phenotype (CAG-repeat length less than 36) is huntingtin (HTT). The terms “normal” and “non-polyglutamine expanded huntingtin” are not acceptable for JHD.
HD is most commonly an adult-onset disorder with an average age of symptom onset in the mid-forties. Although the HTT CAG repeat is highly polymorphic in the population with a continuum of repeat sizes, the relationship between repeat length and development of clinical signs of HD is often predictable (Table 1). HD is caused by a single HTT CAG-repeat expanded allele, such that repeats of greater than 35 are associated with a risk of developing HD during that individual’s lifetime.
Table 1
Classification of HTT CAG-repeat lengths (adapted from [1])
| CAG-repeat count | Allele classification | Disease status |
| <27 | Wild-type or non-expanded | Will not develop disease phenotype |
| 27–35 | Intermediate | Will not develop disease phenotype |
| 36–39 | Reduced penetrance | May or may not develop disease phenotype |
| 40+ | Full penetrance | Will develop disease phenotype |
The preferred term for an individual known to have such an allele is “person with HD” (PwHD). The term “huntingtin gene expansion carrier” (HGEC) is non-preferred but can be used because of its current use in clinical practice (3). The commonly used term “Huntington disease gene expansion carrier (HDGEC)” is not acceptable, as it does not use the preferred gene name. HD with clinical onset before the age of 20 is usually associated with large CAG-repeat expansions in HTT of greater than ∼50. The preferred term for this is “juvenile-onset Huntington’s disease” (JoHD). The term “juvenile HD” is not acceptable, as this is less precise and can lead to confusion in individuals who had clinical onset before age 20 but are currently much older. The older term “Westphal variant” is non-preferred, but can be used to describe the common clinical phenotype seen in juvenile-onset HD. A form of chorea-predominant HD with a slowly progressive phenotype is often seen with clinical onset after the age of 70 and the term “late-onset HD” is acceptable for this form of HD, but the term “senile chorea” is not acceptable and should no longer be used to refer to HD.
For individuals who have CAG-repeat expansions of less than 36, the preferred term is “non-huntingtin gene expansion carrier (non-HGEC)” but “non-Huntington’s disease gene expansion carrier (non-HDGEC)” is not acceptable. The protocol of the study being reported should provide a definition of the individuals who were used as controls. Importantly, control participants in HD studies are not necessarily “healthy” because they may have other diseases. A subset of these individuals who have HTT CAG repeats of 27–35 should be referred to as “carriers of an intermediate allele”. The great majority of these individuals have no known risk of developing HD during their lifetime, but they do have a low risk of passing on an expanded (HD-associated) allele to their offspring, typically through the paternal lineage, due to intergenerational instability of the CAG repeat. In some contexts, these alleles can also be referred to as “pre-mutation alleles”. Although this is an acceptable alternative usage, most notably when discussing the development of HD in an offspring who has inherited a fully penetrant repeat from a parent with an intermediate CAG repeat, the preferred term for individuals with CAG-repeat expansions of 36–39 is “carriers of reduced penetrance alleles”. Alternatively, authors can use the acceptable alternative usage “incomplete penetrance alleles” since many individuals with repeat sizes in this range do not live long enough to manifest overt symptoms or obtain a clinical diagnosis of HD. These terms are outlined in Table 1.
Although some cognitive and psychiatric deficits may precede the onset of overt motor symptoms by many years, the current formal criteria for clinical diagnosis of HD are based on the determination by an appropriate health care professional that a person with a CAG-expanded HTT allele has developed the unequivocal presence of a constellation of otherwise unexplained extrapyramidal movement deficits such as chorea, dystonia, bradykinesia, or rigidity (Fig. 1). This is a clinical judgement by an experienced clinician and can be captured through the diagnostic confidence score (1–4), where a score of 4 represents >99% confidence of motor symptom onset [2]. Thus, it is assumed that “HD clinical diagnosis” is based on the onset of motor signs, unless otherwise defined in the manuscript. Other terms, such as “psychiatric onset or diagnosis” and “cognitive diagnosis” should be avoided unless specifically defined in the manuscript. Similarly, variations of “genetic diagnosis of HD” are non-preferred and should be avoided in JHD manuscripts.
Fig. 1
The HD-Integrated Staging System (HD-ISS): Cumulative staging framework and landmarks. Graphical representation of the temporal sequence of Stage progression and the associated landmark assessments that define Stage entry. (Note: time not to scale). From accompanying editorial: “Refining the Language of Huntington’s Disease Progression with the Huntington disease integrated staging system (HD-ISS)”.
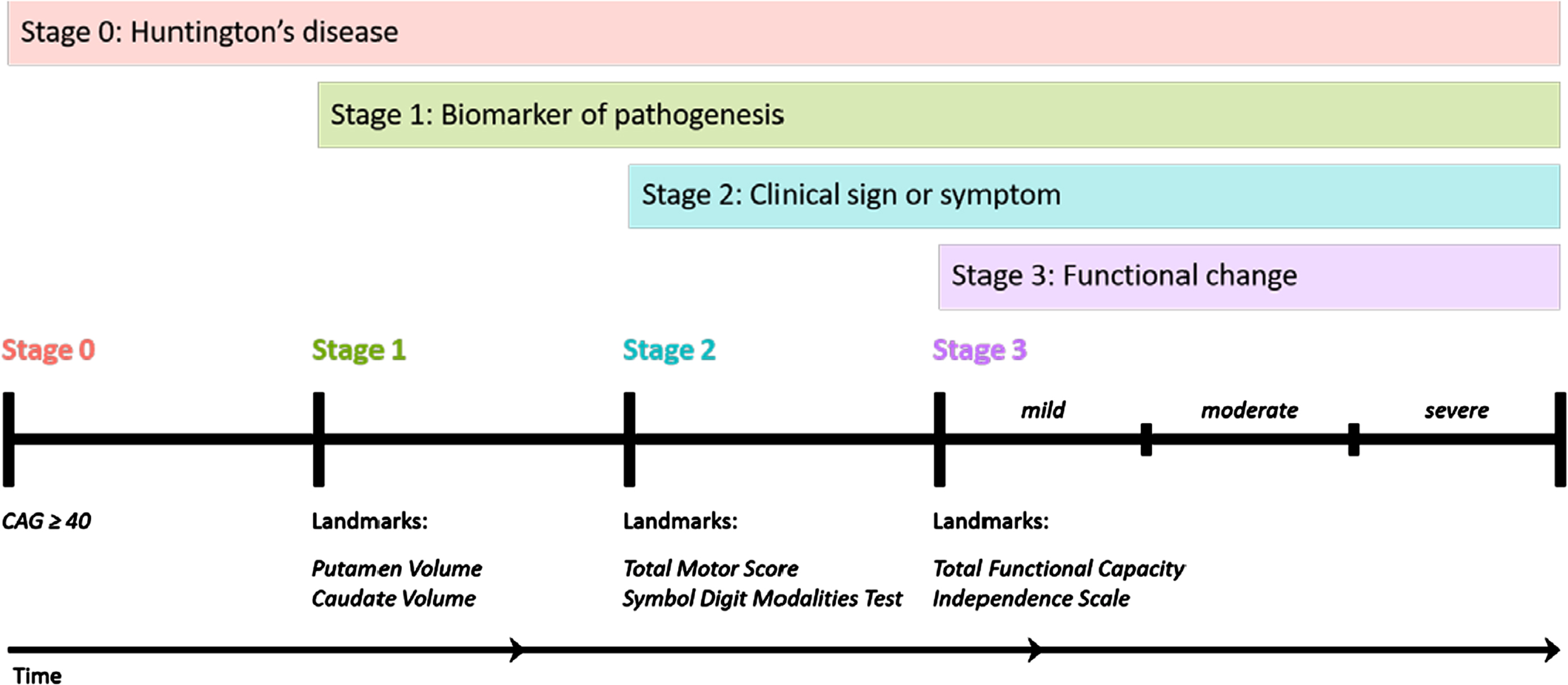
The period following motor diagnosis is considered “manifest HD” and preceding motor diagnosis “premanifest HD”. Premanifest HD can be divided into presymptomatic and prodromal phases, depending on whether any signs or symptoms consistent with HD are present. There are no precise consensus definitions of the terms presymptomatic and prodromal phases, therefore these terms should be avoided unless specifically defined in the manuscript.
The preferred term for individuals who have a CAG-expanded HTT allele at any disease stage is “persons with HD (PwHD)”. Manifest HD, the period after clinical motor diagnosis, can be divided into several stages based on functional scales but must be clearly defined for use in JHD manuscripts. Definitions such as “Shoulson-Fahn Stages (I–V)”, “early, moderate, advanced stage HD” that are based on specific scores in the Unified Huntington’s Disease Rating Scale, (UHDRS; [2] (e.g., the Total Functional Capacity (TFC) scale), are acceptable.
PwHD can have an onset of symptoms at any age and, when taking into account large groups of PwHD, the age of clinical motor diagnosis is inversely correlated with the length of the HTT CAG-repeat expanded allele. In addition, the phenomenon of anticipation (i.e., the tendency toward younger age at symptom onset in subsequent generations) is due to the propensity of the CAG-repeat expansion to further expand on intergenerational transmission. This tends to occur with paternal transmission of expanded HTT CAG repeats (the “sex-of-parent effect”).
The HD-Integrated Staging System (HD-ISS) (Fig. 1) is an evidence-based staging system that is based on a biologic definition of HD [3]. In the HD-ISS, each disease case is defined as the presence of a CAG expansion in HTT exon 1 of either (i) 40 or more CAG repeats, OR (ii) 36 or more CAG repeats and the presence of a disease-specific biomarker or clinical syndrome. The HD-ISS encompasses the entire disease course and indexes progression based on specific landmark assessments that determine stage entry (Fig. 1). Stage 0 starts at birth, and people with HD enter HD-ISS Stage 1 when they surpass a threshold for a biomarker of pathogenesis (caudate or putamen volume loss as determined by MRI). Stage 2 entry is based on the presence of specific clinical signs or symptoms (Total Motor Score (TMS) or Symbol Digit Modalities Test (SDMT)), and Stage 3 is marked by functional changes based on a threshold for the Total Functional Capacity and the Independence Scale.
The former terminology referring to the clinical phases can be mapped onto the HD-ISS. For example, clinical motor diagnosis occurs most frequently by the end of HD-ISS Stage 2. Still the main qualitative dichotomy among HD-ISS stages is presymptomatic (Stages 0 and 1) versus symptomatic (Stages 2 and 3). The former terminology has been widely used over many decades, so we expect a relatively long transition period as publications slowly adopt HD-ISS language. We anticipate that the HD-ISS will eventually become the standard research framework for cohort stratification and for inclusion and exclusion criteria in new observational studies and clinical trials, and increased use of the HD-ISS, in turn, will influence the commonly accepted terminology (see accompanying editorial in this issue). We therefore recommend using terminology consistent with the HD-ISS but acknowledge that other terms may still be acceptable if specifically defined in the manuscript.
Prognostic scores and clinical combination variables
CAG-Age Product (CAP)
The discovery that CAG length is a strong predictor of symptom onset and progression, particularly the emergence of clinically diagnosable motor symptoms, led to the development of prognostic scores. These scores make use of two variables—age and CAG length—either to predict landmark events over the course of the disease, or to provide a time referential (often the x-axis in a graph) to depict disease progression across age adjusted for CAG-repeat length. The interaction of age and CAG-repeat length are incorporated, which motivates the term CAG-age product (CAP).
There are various scoring systems in use because the statistical models to calculate them were developed from different perspectives, and they all used different datasets for validation. Ideally, only one scoring system should be used to enhance research comparability. To this end, Warren et al. [4] presented a general formula for CAP:
The L and K parameters define three of the major CAP scores, with L = 35.5 and K = 1 used for the Penney et al. version [5] (sometimes referred to as the disease burden score), L = 36.66 and K = 1 defining the version based on PREDICT-HD [6] and L = 30 and K = 6.49 defining CAP100, so-named because a value of 100 is associated with clinical motor diagnosis in Enroll-HD [4]. CAP100 has the advantage of the anchor value, and is the score we recommend for general use.
Expected age of clinical motor diagnosis
An alternative approach for the risk of HD progression has been proposed by Langbehn et al. [7, 8] based on survival analysis modeling. It too incorporates age and CAG length. For a given CAG length, the probability of not experiencing clinical motor diagnosis by a given age is estimated by
Perhaps the most commonly cited statistic related to the above formula is the expected age of clinical motor diagnosis onset (from birth) for a given CAG length,
referred to as the expected age of clinical motor diagnosis.
The underlying data for this model included a maximum CAG length of 56, and these formulae may be inaccurate for extreme CAG lengths associated with juvenile onset HD.
PIN score
Prognostic value is enhanced when clinical variables are included along with age and CAG length. This motivated the prognostic index for HD (PIHD) and the prognostic index normed (PIN) [9]. Both measures incorporate the UHDRS Total Motor Score (TMS) and Symbol Digit Modalities Test (SDMT), along with a version of CAP (with L = 34, K = 1):
PIN has been thoroughly examined by Langbehn et al. [10, 11] and is recommended for general use when a researcher wants to consider clinical information along with age and CAG length.
Combined/composite UHDRS
The combined (or composite) UHDRS (cUHDRS) was proposed as a more sensitive outcome measure than using a single clinical variable. The cUHDRS has been used as the primary endpoint in some recent pivotal trials [12]. The outcome combines scaled versions of the TMS, SDMT, Stroop word reading test (SWR), and Total Functional Capacity (TFC):
Smaller cUHDRS scores indicate greater decline (greater progression).
HD-CAB
Another combination outcome measure is the HD Cognitive Assessment Battery (HD-CAB) [13]. The HD-CAB combines six cognitive measures, Hopkins Verbal Learning Test (HVLT-R), Trail Making Test Part A & B (TMT A & B), Paced Tapping Test (PTAP), SDMT (see above), Emotion Recognition (EMO), and One Touch Stockings of Cambridge (OTS). The tests are combined by first computing a Z-score for each (Z = (score – mean)/SD), and then computing the mean of the Z-scores. Similar to the cUHDRS, smaller values indicate greater cognitive decline.
2. GENE AND PROTEIN NOMENCLATURE
The variable length of the polyglutamine domain in human HTT makes it difficult to specify a standard naming convention for the downstream amino acid residues towards the carboxy terminus. For example, serine 421 is a highly conserved phosphorylation site in HTT and has been shown to protect against the toxicity of the expanded polyglutamine tract in mHTT [14]. But with the variable length of the amino-terminal polyglutamine region in both human wtHTT and mHTT, serine 421 is not necessarily the 421st amino acid residue in both HTT proteins, or indeed in other HTT variants.
The convention adopted by many groups is to utilize a fixed polyglutamine length no matter what the actual length, thereby creating consistency across all human HTT variants (Table 2). There are three predominant conventions used in the HD field: the use of a fixed polyglutamine length of 0 [15], of 23 [16, 17], and of 25 for CAGCAA repeats used in cell lines. For the purposes of publication in JHD, we recommend that a fixed polyglutamine length of 23 be used as the convention for human HTT (which has been used for the serine 421 example). To avoid any confusion, authors must always define the convention upfront and provide the sequence (including any tags on the proteins) as a supplement or provide a reference if previously published.
Table 2
Species gene and protein nomenclature. Huntingtin gene and protein symbol nomenclature in different animal species with associated polyglutamine length and HTT length [19, 22] from NCBI: XP_028704080.1, (XP_045247979.1, NP_001254674.1, NP_001136110.1, NP_999129.1, NP_077333, XP_030126485.3, XP_041443615.1, XP_031751173.1, NP509663.3; UniProt XP_645159.1.
| Species | Huntingtin gene symbol | Huntingtin protein symbol | Polyglutamine repeat length | HTT residue length |
| Human/higher mammals | ||||
| Human (Homo sapiens) | HTT | HTT | Q23 | 3144 |
| Human, HD (Homo sapiens) | N/A | mHTT | Q>35 | 3144 |
| Human, HD (Homo sapiens), Exon 1 | N/A | mHTT1a | Q≥42 | ≥107 |
| Rhesus Macaque (Macaca mulatta) | HTT | HTT | Q11 | 3132 |
| Crab-eating macaque (cynomolgus, Macaca fascicularis) | HTT | HTT | Q12 | 3155 |
| Marmoset (Callithrix jacchus) | HTT | HTT | Q9 | 3131 |
| Sheep (Ovis aires) | HTT | HTT | Q10 | 3127 |
| Miniature pig (Sus scrofa domesticus) | HTT | HTT | Q18 | 3139 |
| Dog (Canis familiaris) | HTT | HTT | Q10 | 3130 |
| Rodents | ||||
| Mouse (Mus musculus) | Htt | HTT | Q7 | 3119 |
| Rat (Rattus norvegicus) | Htt | HTT | Q8 | 3120 |
| Birds | ||||
| Song bird (zebra finch, Taeniopygia guttata) | htt | HTT | Q4 | 3095 |
| Amphibians and fish | ||||
| Frog (Xenopus laevis; Xenopus tropicalis) | htt | HTT | Q4; Q4 | 3130; 3123 |
| Zebrafish (Danio rerio) | htt | Htt | Q4 | 3121 |
| Invertebrates | ||||
| Fruit Fly (Drosophila melanogaster) | htt | HTT | Q0 | 3758 |
| Roundworm (Caenorhabditis elegans) | htt | HTT | Q0 | 2022 |
| Amoeba (Dictyostelium discoideum) | htt | HTT | Q0 | 3095 |
| Unicellular microorganisms | ||||
| Bacteria (most genera) | htt | Htt | N/A | N/A |
| Budding yeast (Saccharomyces cerevisae) | HTT | Htt | N/A | N/A |
Furthermore, the animal species used in preclinical research have different endogenous wildtype pure or interrupted CAG repeats and polyglutamine lengths in their huntingtin orthologs and proteins (Table 2). For example, in several but not all mouse strains, murine HTT contains a polyglutamine repeat of 7, thus in the example above, serine 421 would be serine 399 using a fixed polyglutamine length domain of 7. For the purposes of publication in JHD, we recommend that a fixed polyglutamine length of 7 be used as the convention for all murine HTT with the supporting sequence as a supplement.
It should be noted that the Human GenomeVariation Society has created guidelines for the no-menclature of proteins with variable domains (https://varnomen.hgvs.org/recommendations/protein/variant/repeated/). They suggest identifying the position of the first residue of the repeat and then indicate the length of the repeat. For example, “p.Gln18[23]” is the abbreviation for a repeated amino acid sequence with the first glutamine residue located at position 18 and is present in 23 copies (HD glutamine-repeat based on HTT reference sequence (GenBank NP_002102.4)). They also provide an example of estimated repeat domain sizes with “p.(Gln18)[(70_80)]” indicating that the predicted glutamine amino acid repeat, starting at position 18, has an estimated size of 70 to 80 repeats and everything downstream stays with the numbering of the reference sequence. The Human Genome Variation Society does not seem to have an explicit convention for the residue nomenclature beyond a variable domain to keep the residue numbering consistent across multiple variable domain protein variants, but the downstream residues should be in the context of the numbering of the reference sequence. Here, we recommend that a fixed polyglutamine length of 23 be used as the convention for the human HTT reference sequence no matter what the repeat length is, thereby providing a consistent number of critical residues after the variable domain towards the C-terminus (e.g., serine 421).
Another variable domain of HTT to consider is the proline-rich domain (PRD) within exon 1 that displays sequence heterogeneity. The uninterrupted proline repeat most often is 7 or 10 residues in length in human HTT, but many other variants (longer and shorter) are also observed. For JHD publications, the PRD of the DNA construct or protein must be defined upfront and either the sequence provided as a supplement or referenced if previously published.
The first exon of HTT, when expressed on its own, encodes the HTT exon1 protein, which has been used for multiple studies and HD models including the highly utilized R6/1 and R6/2 mouse models [18]. The HTT exon 1 protein is designated as including 1–90 amino acids using the fixed Q23 polyglutamine convention. Furthermore, recent findings have shown that alternative processing of the HTT pre-mRNA by the activation of cryptic polyA sites in intron 1 can generate small single-exon transcripts called HTT1a in humans and Htt1a in mice [19, 20]. The HTT1a and Htt1a transcripts encode the HTT exon 1 protein (1–90) terminating in a proline residue, the correct nomenclature for which is HTT1a. Additional splice variants are encoded within exon 49/50 and need to be defined when relevant to the manuscript.
Another challenge in designating a repeat-region nomenclature convention is the recent finding that it is the number of pure CAGs in HTT that influences the timing of the emergence of clinical symptoms and that numbering requires consideration of the CAA interruptions within the CAG repeat [21]. Recent considerations by the Clinical Genetics Working Group and GeM-HD consortium have recommended that pure CAG repeats be designated as (CAG)n, where n is the number of repeat units. Thus, the most common human HTT sequence, with its single CAA interruption, would be (CAG)nCAACAG. We note that both CAG and CAA translate into a glutamine residue, so this variance does not affect the HTT sequence. Including the downstream PRD would yield (CAG)nCAACAGCCGCCA(CCG)n, and other sequence variations in these segments can be denoted in a similar manner.
3. PRECLINICAL ANIMAL AND CELLULAR MODELS
Animal models
There are a number of common names as well as formal/standardized nomenclature for preclinical animal models of HD (e.g., “JAX mouse nomenclature” from Jackson Labs, https://www.jax.org/jax-mice-and-services/solutions-by-therapeutic-area/neurobiology/huntingtons-disease-mouse-model-resource). For JHD, standardized nomenclature should be included in the materials and methods. The nature of the control animals should also be defined as siblings, wildtype animals, or other categories. Within the text, a common/accepted name can be used with standardized nomenclature cited in the methods section. If animals are obtained from, or are available at, Jackson Labs (JAX), then their standardized nomenclature followed by the common name is used with a link to the JAX website. CAG-repeat length should be defined if there are different variants of the mouse line. For example, R6/2, YAC128 and BACHD mice would be cited as follows:
• B6CBA-Tg(HDexon1)62Gpb/1J standardized nomenclature=R6/2 common name with CAG repeat of 160+/- 5 (https://www.jax.org/strain/002810)
• FVB-Tg(YAC128)53Hay/J standardized nome-nclature=YAC128 common name (https://www.jax.org/strain/004938)
• FVB/N-Tg(HTT*97Q)IXwy/J standardized no-menclature=BACHD common name (https://www.jax.org/strain/008197)
If an animal is obtained from an alternative repository, the name of the repository and the same information as for JAX models should be included. If the animal model was obtained from a research lab, then the methods should include species and strain background, transgene or method of knock-in, pure CAG versus mixed CAG/CAA codon repeats, or a reference describing the model. Finally, metadata for the experiment should be included in materials and methods, including age of animals, genotype and CAG-repeat measurements from tail snips (reported with mean and standard deviation for in-house breeding). If animals are provided by a repository, JHD manuscripts can include metadata from the repository. Potential exceptions include BACHD (original mixed repeats) or YAC128 that are stable in germline.
Cellular models
For cell models, authors should include full descriptions of the source (e.g., American Type Culture Collection (ATCC) or research lab with reference), the gene introduced into the system (HTT sequence, epitope tags, pure CAG versus mixed CAG/CAA codon repeats, promoter used, transient versus stable lines) as well as quality-control metrics (e.g., karyotype, source of cell line). For HD donor-derived cell lines (e.g., iPSCs), nomenclature should be clearly defined in the materials and methods, and include the source of the lines, for instance through the NINDS repository (https://nindsgenetics.org/). Recommended nomenclature is to include the repository ID number in the materials and methods (e.g., NDS00091) or to provide a reference source for the line and to provide CAG-repeat sizing as repeat lengths can be unstable over time. Common naming within the text should include CAG/polyglutamine repeat length such as CAG53 (or 55Q if allele has penultimate CAACAG codons). For controls, materials and methods should include whether these are family controls, non-family controls, or gene-corrected lines.
4. NEUROANATOMY and NEUROPATHOLOGY
The anatomical names used to reference brain regions should be appropriate to the species. The abbreviations for these brain regions should be those recommended here, in atlases, or in common use. Non-standard abbreviations should be avoided. If multiple terms or abbreviations are available, literature source(s) should be cited for the terminology used. When many non-standard abbreviations are used in the text they should be listed and defined at the beginning of the JHD manuscript. It is helpful to cite the stereotaxic atlas used for brain region terminology, injections, or recordings. For figures, use inserts of sectioned brain to indicate the location of high magnification images, injection sites, or dissected tissue.
Authentication of HD in human postmortem brain
The research use of postmortem tissue from a PwHD must meet genetic and neuropathological criteria, which include in order of preference: 1) PCR analysis of HTT CAG-repeat length should show 36 or more, and the brain region of determination should be specified, and if the cortex then the specific region; 2) western blot or ELISA-based assays for detection of mHTT; and 3) the presence of inclusions by immunohistochemistry.
A grading system describing the extent of atrophy and cellular changes in the postmortem neostriatum is a widely accepted standard [23]. The Vonsattel criteria are based on shape changes in the neostriatum and use a grading system from 0–4, with grades 1–4 denoting increasing degrees of atrophy, and “0” denoting no pathology.
The basal ganglia and some of its subcortical connections in human and nonhuman primate and mouse brain
The nuclei that comprise the basal ganglia are grouped differently depending on whether a neuroanatomical/neuropathological or phylogenetic classification is adopted. For JHD submissions, the former is most commonly used and therefore is preferred. Table 3 summarizes the classifications, terms, nuclei, and abbreviations for regions of the basal ganglia in human and nonhuman primate with the neuroanatomical/neuropathological classification shown at the top and the phylogenetic classification shown at the bottom. Also included in Table 3 are some of the major interconnections of basal ganglia nuclei. Table 5 reviews the terms, brain regions and abbreviations for the basal ganglia in mouse.
Table 3
The basal ganglia and some connecting subcortical structures in human and non-human primates
| Classification | Term | Consists of these regions: | Abbreviations |
| Neuroanatomical/neuropathological | Basal ganglia | Caudate nucleus Putamen Globus pallidus Amygdala Substantia nigra Subthalamic nucleus Pedunculopontine nucleus | Cd Put GP SN STN PPN |
| Corpus striatum | Caudate nucleus, putamen, nucleus accumbens and globus pallidus | ||
| Lenticular nuclei | Putamen and globus pallidus | ||
| Cortex Brodmann terminology function (motor, sensory, visual, etc.) | Projections to caudate nucleus and putamen | ||
| Substantia nigra, pars compacta | Projections to caudate nucleus and putamen | SNpc | |
| Parafascicular nucleus | Projections to caudate nucleus and putamen | Pf | |
| Medial dorsal nucleus | Projections to caudate nucleus and putamen | MD | |
| Substantia nigra, pars reticulata | Inputs from caudate nucleus and putamen | SNpr | |
| Phylogenetic | Neostriatum | Caudate nucleus Putamen Nucleus accumbens | Cd Put NAc or NAcc |
| Paleostriatum | Internal globus pallidus External globus pallidus | iGP or GPi eGP or GPe |
References for Table 3: [24–29]. Allen Atlas for Human Brain Online open access [30] https://help.brain-map.org/display/humanbrain/Allen+Human+Brain+Atlas.
Table 4
Neuron types in human and nonhuman primate neostriatum
| Neuron type | Abbreviation | Neurotransmitters/peptides | References |
| Medium spiny neuron | MSN | GABA, enkephalin, substance P, dynorphine, calbindin-D28 | [32, 33] |
| Medium aspiny neuron | Somatostatin, parvalbumin, nitric oxide synthase, calretinin | [34] | |
| Large aspiny neuron | Acetylcholine | [35] | |
| Neuron types 1-V | 1-V | [33] | |
| Unique interneuron | Tac3+ | [31] |
Table 5
The basal ganglia and some connecting subcortical structures in mouse
| Term | Consists of these regions: | abbreviation |
| Striatal complex | Dorsal striatum, ventral striatum | |
| Dorsal striatum | Caudate-putamen (or caudoputamen), Globus pallidus | CP, CPu GP |
| Globus pallidus | Dorsal pallidum and ventral pallidum or lateral globus pallidus and medial globus pallidus (or entopeduncular nucleus) Globus pallidus external part Globus pallidus internal part | lGP mGP GPe entopeduncular nucleu |
| Ventral striatum | Nucleus accumbens Olfactory tubercle Ventromedial parts of caudate putamen | NAc OT CP |
| Substantia nigra | Substantia nigra compacta, sends afferents to CP Substantia nigra reticulata, receives output from CP | SNc SNr |
| Subthalamic nucleus | Sends afferents to GP | STN |
| Parafascicular nucleus | Sends afferents to CP | Pf |
| Ventromedial nucleus | Sends afferents to CP | VM |
| Ventral anterior nucleus | Receives output from GP | VA |
| Ventral lateral nucleus | Receives output from GP | VL |
Neuron types in human and nonhuman primate neostriatum
The terms for neuron types are based on morphology and/or neurotransmitter/neuropeptide content. Single nucleus RNA sequencing has defined a unique type of interneuron in the primate neostriatum [31]. Table 4 classifies neuron types and provides some pertinent references for human and nonhuman primates.
Neuron types in mouse and rat caudate putamen
The basis for classification of neurons in the rodent caudate putamen may include morphology, electrophysiology, location, pathways, neurotransmitter/peptide content, output, or receptor content. Table 6 summarizes these categories and includes some common abbreviations.
Table 6
Neuron types in mouse and rat caudate putamen
| Classification | Name | Abbreviation |
| Morphology | Medium spiny neurons Medium aspiny neurons Large aspiny neurons Types I–V in rat | MSN |
| Electrophysiology | Fast spiking interneurons; Low-threshold Ca2+ spiking interneurons; Spontaneously active cholinergic interneurons; Neuropeptide Y | FS LTS Spontaneously active ChIs NPY-PLTS NPY-neurogliaform |
| Location/pathway | Striosomes: patch and/or matrix; Direct or indirect pathway | |
| Neurotransmitter or peptide content | Gamma amino butyric acid Acetylcholine Parvalbumin Somatostatin Nitric oxide synthase Calbindin D28k or calcium binding protein-D28k Calretinin Tyrosine hydroxylase Caldec Methionine enkephalin Leucine enkephalin Substance P | GABA ACh PV Som NOS CaBP-D28k CR TH RasGRP2 Met-ENK Leu-ENK SP |
| Output | Spiny projection neuron | SPN |
| Receptor content | Metabotropic glutamate receptors N-methyl-D-aspartic acid (NMDA) receptor subtypes alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid Dopamine receptor D1 Dopamine receptor D2 Adenosine receptors Nicotinic acetylcholine receptors | mGluR1 NMDAR2A, or NMDAR2B AMPAR D1R, DrD1 D2, DrD2 A2a nAChr |
| Gene clusters based on single-cell RNA-seq | Subgroups of MSNs and non-neuronal cells enriched with different transcripts |
Localization and accumulation of mutant huntingtin in human and mouse
Aggregates represent a continuum of species that reflect disruption of protein folding and homeostasis. Various terms have been used to describe the presence, subcortical location, and type of accumulation of mutant huntingtin or fragments of mutant huntingtin in cells and in brain. These terms include inclusions, fibrils and oligomers, aggregates, and aggresomes. Table 7 provides a list of terms, the context in which they have been used, and relevant publications. These terms, as defined, should be used for manuscripts in JHD.
Table 7
Terms used for the localization and accumulation of mutant huntingtin protein in human and mouse tissues and cells
| Term | Meaning | References |
| Inclusions Also: inclusion bodies (IB), nuclear inclusions (NI), perinuclear inclusions, cytoplasmic inclusions, neuropil inclusions, axonal inclusions | Visible by light microscopy. Description often relates to subcellular location. | [49, 50] 7,900 refs in PubMed |
| Oligomers, fibrils and protofibrils | Smaller assemblies not visible by light microscopy that may be resolved by EM. | [51] |
| Aggregates | A collective term that includes all assemblies of mHTT. | [52–57]14,000 refs in PubMed |
| Aggresomes | Inclusions at a perinuclear location at the microtubule organizing center surrounded by a vimentin cage. | [58] 1,000 refs in PubMed |
| By antibody: EM48, 4C9, 1C2 (anti-TBP), MW8, S830, PHP1 and PHP2, p62 | Antibodies with known or unknown epitopes that vary in the amount, composition and location of accumulated mHTT | [52, 59–64] |
5. GLOSSARY OF JHD PREFERRED ABBREVIATIONS
cUHDRS | composite Unified Huntington’s Disease Rating Scale |
HD | Huntington’s disease |
HD-CAB | HD Cognitive Assessment Battery |
HD-ISS | HD Integrated Staging System |
HGEC | huntingtin gene expansion carrier |
Non-HGEC | non-huntingtin gene expansion carrier |
mHTT | mutant HTT |
wtHTT | wild type HTT |
PBA | Problem Behaviors Assessment |
PCA | Principal Component Analysis |
PwHD | persons with HD |
SDMT | Symbol Digit Modalities Test |
SWR | Stroop Word Reading Test |
TFC | Total Functional Capacity |
TMS | Total Motor Score |
UHDRS | Unified Huntington’s Disease Rating Scale |
ACKNOWLEDGMENTS
We thank the HD community for their continual support and inspiration.
FUNDING
The authors have no funding to report.
REFERENCES
[1] | Walker FO . Huntington’s disease. Lancet. (2007) ;369: (9557):218–28. |
[2] | Unified Huntington’s Disease Rating Scale: Reliability and consistency. Huntington Study Group. Mov Disord. (1996) ;11: (2):136–42. |
[3] | Tabrizi SJ , Schobel S , Gantman EC , Mansbach A , Borowsky B , Konstantinova P , et al. A biological classification of Huntington’s disease: The Integrated Staging System. Lancet Neurol. (2022) ;21: (7):632–44. |
[4] | Warner JH , Long JD , Mills JA , Langbehn DR , Ware J , Mohan A , et al. Standardizing the CAP Score in Huntington’s Disease by Predicting Age-at-Onset. J Huntingtons Dis. (2022) ;11: (2):153–71. |
[5] | Penney JB Jr , Vonsattel JP, MacDonald ME, Gusella JF, Myers RH. CAG repeat number governs the development rate of pathology in Huntington’s disease. Ann Neurol. (1997) ;41: (5):689–92. |
[6] | Zhang Y , Long JD , Mills JA , Warner JH , Lu W , Paulsen JS , et al. Indexing disease progression at study entry with individuals at-risk for Huntington disease. Am J Med Genet B Neuropsychiatr Genet. (2011) ;156B: (7):751–63. |
[7] | Langbehn DR , Brinkman RR , Falush D , Paulsen JS , Hayden MR , International Huntington’s Disease Collaborative G. A new model for prediction of the age of onset and penetrance for Huntington’s disease based on CAG length. Clin Genet. (2004) ;65: (4):267–77. |
[8] | Langbehn DR , Hayden MR , Paulsen JS , and the P-HDIotHSG. CAG-repeat length and the age of onset in Huntington disease (HD): A review and validation study of statistical approaches. Am J Med Genet B Neuropsychiatr Genet. (2010) ;153B: (2):397–408. |
[9] | Long JD , Langbehn DR , Tabrizi SJ , Landwehrmeyer BG , Paulsen JS , Warner J , et al. Validation of a prognostic index for Huntington’s disease. Mov Disord. (2017) ;32: (2):256–63. |
[10] | Langbehn DR , Fine EM , Meier A , Hersch S . Utility of the Huntington’s Disease Prognostic Index Score for a Perimanifest Clinical Trial. Mov Disord. (2022) ;37: (5):1040–6. |
[11] | Langbehn DR , Hersch S . Clinical Outcomes and Selection Criteria for Prodromal Huntington’s Disease Trials. Mov Disord. (2020) ;35: (12):2193–200. |
[12] | McColgan P , Thobhani A , Boak L , Schobel SA , Nicotra A , Palermo G , et al. Tominersen in Adults with Manifest Huntington’s Disease. N Engl J Med. (2023) ;389: (23):2203–5. |
[13] | Stout JC , Queller S , Baker KN , Cowlishaw S , Sampaio C , Fitzer-Attas C , et al. HD-CAB: A cognitive assessment battery for clinical trials in Huntington’s disease 1,2,3. Mov Disord. (2014) ;29: (10):1281–8. |
[14] | Kratter IH , Zahed H , Lau A , Tsvetkov AS , Daub AC , Weiberth KF , et al. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J Clin Invest. (2016) ;126: (9):3585–97. |
[15] | Weiss A , Grueninger S , Abramowski D , Giorgio FP , Lopatin MM , Rosas HD , et al. Microtiter plate quantification of mutant and wild-type huntingtin normalized to cell count. Anal Biochem. (2011) ;410: (2):304–6. |
[16] | Macdonald D , Tessari MA , Boogaard I , Smith M , Pulli K , Szynol A , et al. Quantification assays for total and polyglutamine-expanded huntingtin proteins. PLoS One. (2014) ;9: (5):e96854. |
[17] | Weiss A , Abramowski D , Bibel M , Bodner R , Chopra V , DiFiglia M , et al. Single-step detection of mutant huntingtin in animal and human tissues: A bioassay for Huntington’s disease. Anal Biochem. (2009) ;395: (1):8–15. |
[18] | Mangiarini L , Sathasivam K , Mahal A , Mott R , Seller M , Bates GP . Instability of highly expanded CAG repeats in mice transgenic for the Huntington’s disease mutation. Nat Genet. (1997) ;15: (2):197–200. |
[19] | Neueder A , Landles C , Ghosh R , Howland D , Myers RH , Faull RLM , et al. The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci Rep. (2017) ;7: (1):1307. |
[20] | Sathasivam K , Neueder A , Gipson TA , Landles C , Benjamin AC , Bondulich MK , et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc Natl Acad Sci U S A. (2013) ;110: (6):2366–70. |
[21] | Genetic Modifiers of Huntington’s Disease Consortium. Electronic address ghmhe, Genetic Modifiers of Huntington’s Disease C. CAG Repeat Not Polyglutamine Length Determines Timing of Huntington’s Disease Onset. Cell. (2019) ;178: (4):887–900 e14. |
[22] | Tartari M , Gissi C , Lo Sardo V , Zuccato C , Picardi E , Pesole G , et al. Phylogenetic comparison of huntingtin homologues reveals the appearance of a primitive polyQ in sea urchin. Mol Biol Evol. (2008) ;25: (2):330–8. |
[23] | Vonsattel JP , Myers RH , Stevens TJ , Ferrante RJ , Bird ED , Richardson EP Jr . Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. (1985) ;44: (6):559–77. |
[24] | Mai JK , Assheuer J , Paxinos G . Human Brain Atlas: Academic Press; (1997) . |
[25] | Mai JK , Paxinos G , Voss T . Atlas of the Human Brain: Academic Press; (2007) . |
[26] | Carpenter MB . Human Neuroanatomy. 8th Edition ed: Williams and Wilkins; (1983) . |
[27] | Nauta WJ , Smith GP , Faull RL , Domesick VB . Efferent connections and nigral afferents of the nucleus accumbens septi in the rat. Neuroscience. (1978) ;3: (4-5):385–401. |
[28] | Brodmann K . Localisation in the Cerebral Cortex (1909) . |
[29] | Annese J . In Retrospect: Brodmann’s brain map. Nature. (2009) ;461: :884. |
[30] | Allen Human Brain Atlas 2019 [Available from: https://help.brain-map.org/display/humanbrain/Allen+Human+Brain+Atlas |
[31] | Krienen FM , Goldman M , Zhang Q , R CHDR , Florio M , Machold R , et al. Innovations present in the primate interneuron repertoire. Nature. (2020) ;586: (7828):262–9. |
[32] | DiFiglia M , Pasik P , Pasik T . A Golgi study of neuronal types in the neostriatum of monkeys. Brain Res. (1976) ;114: (2):245–56. |
[33] | Braak H , Braak E . Neuronal types in the striatum of man. Cell Tissue Res. (1982) ;227: (2):319–42. |
[34] | Wu Y , Parent A . Striatal interneurons expressing calretinin, parvalbumin or NADPH-diaphorase: A comparative study in the rat, monkey and human. Brain Res. (2000) ;863: (1-2):182–91. |
[35] | Geula C , Tokuno H , Hersh L , Mesulam MM . Human striatal cholinergic neurons in development, aging and Alzheimer’s disease. Brain Res. (1990) ;508: (2):310–2. |
[36] | Hunnicutt BJ , Jongbloets BC , Birdsong WT , Gertz KJ , Zhong H , Mao T . A comprehensive excitatory input map of the striatum reveals novel functional organization. Elife. (2016) ;5. |
[37] | Paxinos G , Franklin KBJ . The Mouse Brain in Stereotaxic Coordinates. 2nd ed. San Diego, Ca: Academic Press; (2001) . |
[38] | Ullmann JF , Watson C , Janke AL , Kurniawan ND , Paxinos G , Reutens DC . An MRI atlas of the mouse basal ganglia. Brain Struct Funct. (2014) ;219: (4):1343–53. |
[39] | Reiner A , Deng YP . Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci Ther. (2018) ;24: (4):250–80. |
[40] | Foster NN , Barry J , Korobkova L , Garcia L , Gao L , Becerra M , et al. The mouse cortico-basal ganglia-thalamic network. Nature. (2021) ;598: (7879):188–94. |
[41] | Gokce O , Stanley GM , Treutlein B , Neff NF , Camp JG , Malenka RC , et al. Cellular Taxonomy of the Mouse Striatum as Revealed by Single-Cell RNA-Seq. Cell Rep. (2016) ;16: (4):1126–37. |
[42] | Marin O , Anderson SA , Rubenstein JL . Origin and molecular specification of striatal interneurons. J Neurosci. (2000) ;20: (16):6063–76. |
[43] | Tepper JM , Tecuapetla F , Koos T , Ibanez-Sandoval O . Heterogeneity and diversity of striatal GABAergic interneurons. Front Neuroanat. (2010) ;4: :150. |
[44] | Alegre-Cortes J , Saez M , Montanari R , Reig R . Medium spiny neurons activity reveals the discrete segregation of mouse dorsal striatum. Elife. (2021) ;10. |
[45] | Friedman A , Hueske E , Drammis SM , Toro Arana SE , Nelson ED , Carter CW , et al. Striosomes Mediate Value-Based Learning Vulnerable in Age and a Huntington’s Disease Model. Cell. (2020) ;183: (4):918–34 e49. |
[46] | Crittenden JR , Graybiel AM . Basal Ganglia disorders associated with imbalances in the striatal striosome and matrix compartments. Front Neuroanat. (2011) ;5: :59. |
[47] | Chang HT , Wilson CJ , Kitai ST . A Golgi study of rat neostriatal neurons: Light microscopic analysis. J Comp Neurol. (1982) ;208: (2):107–26. |
[48] | Ibanez-Sandoval O , Tecuapetla F , Unal B , Shah F , Koos T , Tepper JM . A novel functionally distinct subtype of striatal neuropeptide Y interneuron. J Neurosci. (2011) ;31: (46):16757–69. |
[49] | Davies SW , Turmaine M , Cozens BA , DiFiglia M , Sharp AH , Ross CA , et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. (1997) ;90: (3):537–48. |
[50] | DiFiglia M , Sapp E , Chase KO , Davies SW , Bates GP , Vonsattel JP , et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. (1997) ;277: (5334):1990–3. |
[51] | Sathasivam K , Lane A , Legleiter J , Warley A , Woodman B , Finkbeiner S , et al. Identical oligomeric and fibrillar structures captured from the brains of R6/2 and knock-in mouse models of Huntington’s disease. Hum Mol Genet. (2010) ;19: (1):65–78. |
[52] | Gutekunst CA , Li SH , Yi H , Mulroy JS , Kuemmerle S , Jones R , et al. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J Neurosci. (1999) ;19: (7):2522–34. |
[53] | Scherzinger E , Lurz R , Turmaine M , Mangiarini L , Hollenbach B , Hasenbank R , et al. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo . Cell. (1997) ;90: (3):549–58. |
[54] | Hazeki N , Tsukamoto T , Yazawa I , Koyama M , Hattori S , Someki I , et al. Ultrastructure of nuclear aggregates formed by expressing an expanded polyglutamine. Biochem Biophys Res Commun. (2002) ;294: (2):429–40. |
[55] | Thakur AK , Wetzel R . Mutational analysis of the structural organization of polyglutamine aggregates. Proc Natl Acad Sci U S A. (2002) ;99: (26):17014–9. |
[56] | Tanaka M , Machida Y , Nishikawa Y , Akagi T , Hashikawa T , Fujisawa T , et al. Expansion of polyglutamine induces the formation of quasi-aggregate in the early stage of protein fibrillization. J Biol Chem. (2003) ;278: (36):34717–24. |
[57] | Bennett EJ , Bence NF , Jayakumar R , Kopito RR . Global impairment of the ubiquitin-proteasome system by nuclear or cytoplasmic protein aggregates precedes inclusion body formation. Mol Cell. (2005) ;17: (3):351–65. |
[58] | Lu M , Banetta L , Young LJ , Smith EJ , Bates GP , Zaccone A , et al. Live-cell super-resolution microscopy reveals a primary role for diffusion in polyglutamine-driven aggresome assembly. J Biol Chem. (2019) ;294: (1):257–68. |
[59] | Li H , Li SH , Cheng AL , Mangiarini L , Bates GP , Li XJ . Ultrastructural localization and progressive formation of neuropil aggregates in Huntington’s disease transgenic mice. Hum Mol Genet. (1999) ;8: (7):1227–36. |
[60] | Ko J , Isas JM , Sabbaugh A , Yoo JH , Pandey NK , Chongtham A , et al. Identification of distinct conformations associated with monomers and fibril assemblies of mutant huntingtin. Hum Mol Genet. (2018) ;27: (13):2330–43. |
[61] | Bayram-Weston Z , Jones L , Dunnett SB , Brooks SP . Comparison of mHTT Antibodies in Huntington’s Disease Mouse Models Reveal Specific Binding Profiles and Steady-State Ubiquitin Levels with Disease Development. PLoS One. (2016) ;11: (5):e0155834. |
[62] | Legleiter J , Lotz GP , Miller J , Ko J , Ng C , Williams GL , et al. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J Biol Chem. (2009) ;284: (32):21647–58. |
[63] | Landles C , Sathasivam K , Weiss A , Woodman B , Moffitt H , Finkbeiner S , et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J Biol Chem. (2010) ;285: (12):8808–23. |
[64] | Bjorkoy G , Lamark T , Brech A , Outzen H , Perander M , Overvatn A , et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. (2005) ;171: (4):603–14. |
[65] | Huntington’s Disease Sheep Collaborative Research G, Reid SJ , Patassini S , Handley RR , Rudiger SR , McLaughlan CJ , et al. Further molecular characterisation of the OVT73 transgenic sheep model of Huntington’s disease identifies cortical aggregates. J Huntingtons Dis. (2013) ;2: (3):279–95. |
[66] | Orlowski D , Glud AN , Palomero-Gallagher N , Sorensen JCH , Bjarkam CR . Online histological atlas of the Gottingen minipig brain. Heliyon. (2019) ;5: (3):e01363. |
[67] | Baxa M , Hruska-Plochan M , Juhas S , Vodicka P , Pavlok A , Juhasova J , et al. A transgenic minipig model of Huntington’s Disease. J Huntingtons Dis. (2013) ;2: (1):47–68. |


