
Untangling the Role of Tau in Huntington’s Disease Pathology
Abstract
There is increasing evidence for the presence of pathological forms of tau in tissues of both Huntington’s disease (HD) patients and animal models of this condition. While cumulative studies of the past decade have led to the proposition that this disorder could also be considered a tauopathy, the implications of tau in cellular toxicity and consequent behavioral impairments are largely unknown. In fact, recent animal work has challenged the contributory role of tau in HD pathogenesis/pathophysiology. This review presents the supporting and opposing arguments for the involvement of tau in HD, highlighting the discrepancies that have emerged. Reflecting on what is known in other tauopathies, the putative mechanisms through which tau could initiate and/or contribute to pathology are discussed, shedding light on the future research directions that could be considered to confirm, or rule out, the clinical relevance of tau in HD.
HISTORY
The classic views of Huntington’s disease (HD) pathology revolve around the role of the abnormal huntingtin (HTT) protein because the root cause of this neurodegenerative disorder is a genetic mutation within the huntingtin (HTT) gene which leads to an abnormal expansion of CAG repeats [1] characteristic of the mutant form of the protein, mutant HTT (mHTT). While this mutation is ubiquitously expressed in all cells of the body, neuronal populations, particularly those of the striatum and cortex, are more vulnerable to mHTT accumulation and subsequent dysfunction/death. The remarkable cell loss and brain atrophy that insures leads to a triad of motor, cognitive, and psychiatric symptoms that are increasingly debilitating to the patient and to this day, untreatable. While failure of recent clinical trials targeting mHTT suggest that reevaluation of this approach, both in terms of biology and methodology is needed, it may also suggest that other factors could be at play, warranting further investigation of alternative targets.
In the last decade, several groups have investigated the presence of a concomitant tau-related pathology in this disorder. Collectively, the data suggests that HD answers to several criteria of a secondary tauopathy [2, 3]. While the presence of abnormal tau has been repeatedly shown in brain tissues, the extent of its true contribution to the pathogenesis and pathophysiology of HD, however, remains unclear. The objective of this review is to untangle the role of tau in HD, by discussing the evidence accumulated over the last decades and by highlighting more recent findings, including unpublished data generated by our group. On one hand, we will describe the potential mechanisms by which tau may actively take part in HD pathology and evaluate the therapeutic relevance of tau targets. On the other hand, we will expose the possibility that tau abnormalities are only consequential events, weighing in on the findings that contradict or challenge the role of tau in HD. We hope that this review will clarify the reasons for the discrepancies reported in this field and bring forward arguments for pursuing tau research in HD.
TAU
Tau is a microtubule-associated protein which exists in six different isoforms in the healthy adult human brain. These isoforms are the result of alternative splicing of the tau gene MAPT (Microtubule Associated Protein Tau) located on chromosome 17. Differential splicing of exons 2, 3, and 10 gives rise to isoforms which vary by the number of inserts in the N-terminal region (0N, 1N, 2N) and the number of repeats in the microtubule-binding repeat domain (3R, 4R). In the healthy adult human brain, the ratio of 3R to 4R tau isoforms is 1:1. Under physiological conditions, tau exerts various functions notably in microtubule stabilization, axonal transport, synaptic plasticity and modulation of gene transcription [4–6]. While normal tau phosphorylation is essential to neuronal homeostasis, abnormally hyperphosphorylated tau may accumulate in the form of neurofibrillary tangles (NFTs) and/or neuropil threads [7], two entities that interfere with these fundamental functions. This has been traditionally associated with Alzheimer’s disease (AD); however, a number of human brain disorders have also been characterized by tau pathology. These include primary age-related tauopathy, progressive supranuclear palsy, corticobasal degeneration, frontotemporal dementia, Pick’s disease, and Parkinson’s disease, to name a few. Moreover longest human isoform of the tau protein possesses a total of 85 possible phosphorylation sites at various serine, threonine, and tyrosine residues [8]. Forty-five of these sites are associated with AD [9], while hyperphosphorylation of tau has been, thus far, detected at 5 distinct epitopes (Ser199, Ser202, Thr205, Ser396, and Ser404 residues) in HD [10–12].
SUPPORTING ARGUMENTS
Tau contributes to neuropathological and behavioral impairments in HD
The theory of a potential relationship between tau and HD emerged a few decades ago with the publication of a handful of clinical cases which also presented with AD-related neuropathological features [13, 14]. Postmortem histological examination indeed revealed the presence of both mHTT inclusions and NFTs in the brains of HD patients who suffered from severe dementia [11, 13–16]. These anecdotal observations were subsequently confirmed in a larger cohort of patients (n = 15) where the majority of HD patients (>80%) with advanced dementia additionally presented with AD-related neuropathological characteristics [17]. The co-occurrence of AD and HD is likely more common than previously thought and the presence of AD-related pathology may well be linked to cognitive deficits observed in HD [17].
In patients, the genetic variation in the tau gene MAPT has been associated with the manifestation of HD clinical phenotypes. As a matter of fact, HD patients with the H2 MAPT haplotype present a more rapid cognitive decline as compared to the H1 carriers [18], suggesting a link between tau and cognitive symptoms. While some studies have reported increases in total tau mRNA in humans [12] and in protein levels in mice and men [19], knock-out of the tau protein in the R6/1 transgenic mouse model of the disease improves motor functions as measured by the rotarod and open field tests [19]. In parallel, increases in 4R/3R tau mRNA as well as protein ratios have been observed in the striatum and cortex of R6/1 and HD94 mouse models of HD [19] and a similar increase in 4R mRNA and protein levels has been reported in both of these structures in postmortem HD patient samples [12, 18, 19]. Aberrant splicing of tau exon 2 was also identified in the putamen of HD patients, with an increase in 0N4R tau and a reduction in 1N3R tau isoform [12]. Splicing defects at exons 2, 3, and 10 of tau have additionally been noted in HD patients [12]. While splicing modifications of exon 10, such as an increase in 4R tau in cortex and striatum of HD patients, was described in more advanced stages (Grade 3 and beyond), splicing of other exons are detectable earlier in the disease course. A recent study illustrated that splicing alterations of exon 2 were restricted to the putamen, detected earlier in disease (Grade 2) and correlated with tau pathology and mHTT aggregation [20]. This suggests that regulation of exon 2 splicing could represent an initial phenomenon underlying region-specific neuropathology in HD.
Additional studies on postmortem HD brain samples have documented increases in phosphorylated tau (p-tau) protein levels at a number of epitopes (Ser199, Thr205, Ser396/Ser404) in the striatum of patients of late stage disease (Grades 3-4) [12, 20]. Hyperphosphorylated p-tau inclusions [18–20] as well as oligomeric forms of the protein [18] were also detected by immunohistochemistry in the striatum and cortex of HD brains. Similar observations collected in young-onset HD subjects suggest a correlation of tau pathology with disease progression rather than a phenomenon that was prompted by age [18]. To explore the implications of such findings, several research groups have also investigated and confirmed the presence of pathological tau in mouse models of HD, focusing on the phosphorylation state of the protein since murine tau has not been shown to aggregate. These studies demonstrated increased protein concentrations of p-tau (Ser202, Ser202/Thr205, Ser396/Ser404) in the cortex and striatum of R6/2 and KI140 mice [21, 22]. Hyperphosphorylated tau was also shown to be more prominent in the hippocampus, striatum and cortex as behavioral deficits became more apparent, and elevated p-tau was detected in the hippocampus of R6/2 mice prior to any measurable phenotypic changes [21]. Phosphoproteomic studies [23] also suggest that the dysregulation of tau phosphorylation is an early event in HD. Assessment of cortical samples of the R6/1 transgenic mouse model of HD revealed dysregulation of phosphorylation of more than 600 peptides in the beginning of the disease, prior to the appearance of motor impairments. Of these candidates, upregulation of the phosphorylation of tau at epitopes Thr181, Thr217, Ser356, and Ser396 was observed before motor onset in R6/1 mice. Hyperphosphorylation of tau at Ser396 was also measurable in the striatum and hippocampus of R6/1 mice before they exhibited behavioral signs of disease. Upregulations in phosphorylation, which occur prior to the onset of motor deficits/early stage of HD pathology, could cause imbalances of microtubule dynamics leading to axonal and cellular dysfunctions [23].
In line with these findings, administration of an anti-tau pS202 CP13 antibody to zQ175 mice, a knock-in model of HD, resulted in a decrease in hippocampal p-tau protein concentrations (Ser202), which was associated with behavioral improvements, notably cognitive and motor performances. Indeed, working and long-term memory of treated mice were ameliorated as measured by the spontaneous alternation in the Y-maze and by the habituation to the open field test. Motor impairments were also attenuated by antibody treatment as quantified by the increased distance travelled in the open field. Reduction of pSer202 tau further led to neuropathological changes specifically on proteinopathy with a decrease in HTT oligomer levels measured by dot blot assays and electrophoresis. These observations validate the link between tau phosphorylation and HTT conformation/toxicity, with notable consequences on behavioral outcomes [24]. Furthermore, intracerebral injections of human recombinant tau fibrils in the zQ175 progressive mouse model of HD seemingly precipitates the appearance of cognitive deficits and exacerbates anxiety-like behavior in treated mice. At the neuropathological level, treatment with tau fibrils significantly increases mHTT aggregate expression (measured by filter retardation assay) and number/size (evaluated by quantitative stereology analyses) in the HD-targeted brain regions (unpublished observations). These recent findings imply a strong impact of tau on the state of mHTT aggregation as well as HD-like features of a mouse model of the disease.
While there is accumulating and compelling evidence for the presence of tau pathology in HD, the protein has also been reported to behave, like several others, in a prion-like manner. Along these lines, NFTs and neuropil threads of p-tau (Ser202/Thr205, Thr231) were detected by immunohistochemistry in healthy grafted tissue delivered to the striatum of HD patients, where mHTT aggregates were also present. This finding suggests a possible propagation of tau pathology to the transplanted tissue, but also implies that tau may play an active role in the pathophysiology of HD [10]. Abnormalities associated to tau take different shapes in the HD brains. Whether it is an increase in p-tau, the accumulation of aggregated deposits or splicing defects leading to an imbalance of tau isoforms, pathological forms of the protein are, without a doubt, found in HD patients. Combined, these studies suggest that impairments in tau splicing and hyperphosphorylation occur early in the disease process, impacting both mHTT aggregation and behavioral phenotypes. The mechanisms by which tau and mHTT could directly or indirectly influence each other have only been superficially explored (see Fig. 1) but shedding light on this relationship will be critical to understand the true contribution of tau to HD pathology.
Fig. 1
From tau neuropathology to tau therapeutics. The upper panel illustrates the mechanisms of action potentially shared between tau and mHTT at the cellular level. The bottom panel represents the potential implications of tau on the neuropathology and symptomatology of HD and implying its clinic relevance as a biomarker and/or therapeutic target. Abbreviations: MAPT, microtubule associated protein tau; mHTT, mutant huntintin; NFTs, neurofibrillary tangles; p-tau, phosphorylated tau; SRSF6, serine/arginine-rich splicing factor-6; 3R, 3R isoform of tau; 4R, 4R isoform of tau; Δtau314, truncated tau314 protein.
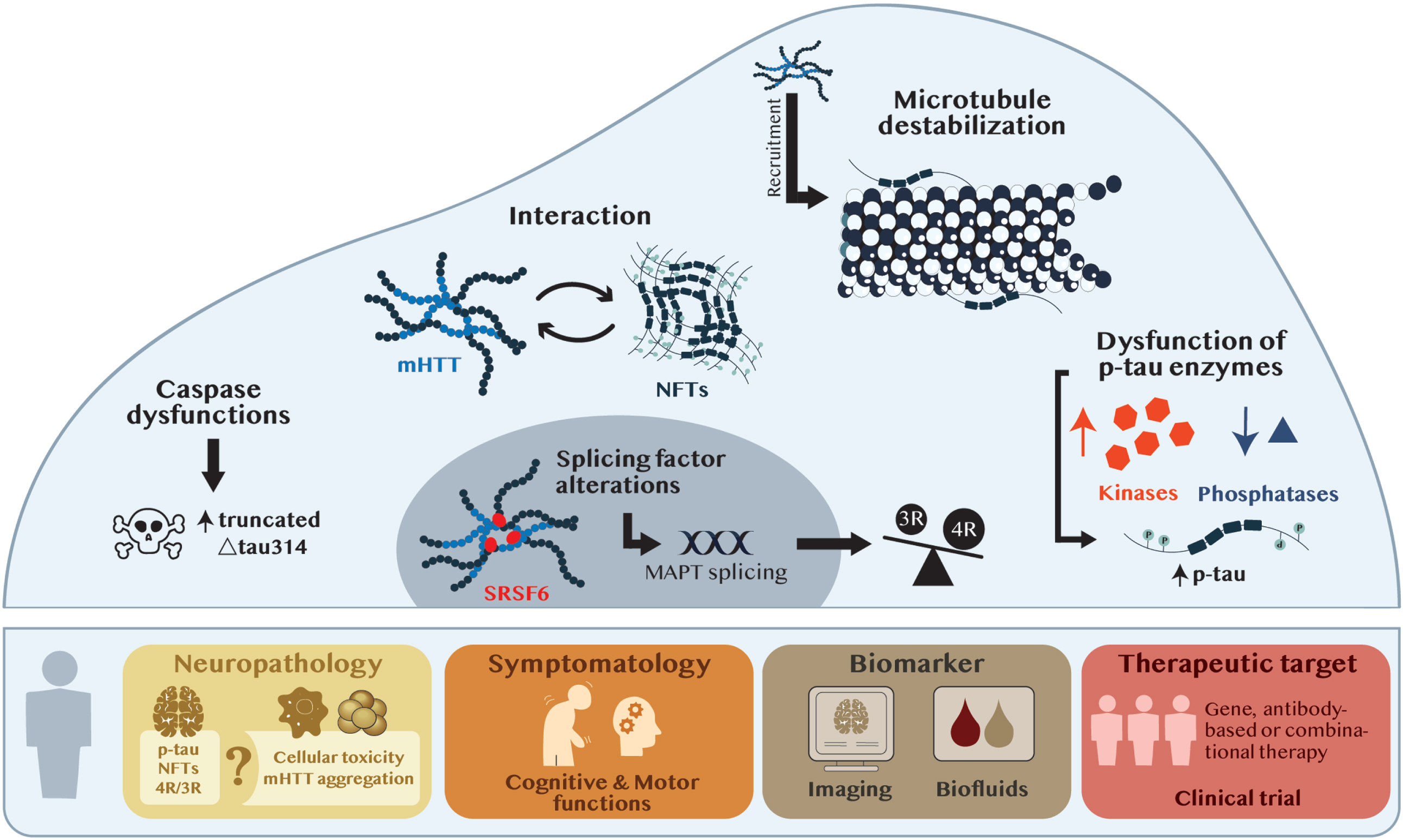
Tau and mHTT-induced pathology share common mechanisms of action
Tau phosphorylation is guided by dysregulations of kinases and phosphatases
Kinase dysregulations have been reported to play a role in HD pathology, notably GSK-3β, a kinase that phosphorylates both HTT and tau. Evidence supports the idea that hyperphosphorylated levels of tau could be mediated through compromised GSK-3β activity (see Fig. 1). In the hippocampus of postmortem HD samples, an increase in GSK-3β mRNA and protein levels was quantified by PCR and western blot in Grade 2 HD brains and coincided with elevated p-tau protein levels [25]. In mouse models of the disease, protein levels of phosphorylated GSK-3β at different epitopes was assessed as an indirect indicator of kinase activity. Increase in phosphorylated GSK-3β at Tyrosine 216 residue (p-GSK-3β-Tyr216) is believed to be associated with activated GSK-3β [26]. An augmentation in p-GSK-3β-Tyr216 levels was detected in the hippocampus of R6/2 mice before (3 weeks) and after (8 weeks) onset of disease [25]. Similar results were obtained in the hippocampus and cortex of the R6/1 mouse model [27] but no elevation of p-GSK-3β-Tyr216 was detected in the striatum [27]. The amount of the active form of GSK-3β was also increased in a hippocampal primary neuronal culture harvested from R6/2 brains [25]. Others have focused on the concentration of phosphorylated GSK-3β at the inhibitory Serine 9 residue (p-GSK-3β-Ser9), for which higher levels correlate with decreased GSK-3β activity [28, 29]. A reduction in p-GSK-3β-Ser9 protein concentration in the cortex and striatum of the N171-82Q and YAC128 mouse models was reported [30] implying, therefore, greater GSK-3β activity.
To further elucidate the role of this enzyme in HD, a few studies have investigated the effect of altering GSK-3β expression in cell and mouse models of the disease by genetic depletion or pharmacological inhibition. Knockdown of GSK-3β with a siRNA against GSK-3β (siRNAGSK - 3β) in primary hippocampal neurons from R6/2 mice resulted in a decrease in tau phosphorylation protein levels (Ser202/Thr205) and increased cell viability [25]. Daily intranasal treatment of R6/2 mice at 4 weeks of age with the L807mts GSK-3β inhibitor significantly diminished the number of striatal mHTT aggregates quantified by immunostaining, by enhancing autophagic clearance, and significantly ameliorating motor and coordination abilities in treated mice (clasping, ledge and Cat-Walk tests) [31]. These results indicate that increased levels of GSK-3β are primarily restricted to the hippocampus in HD human brains and that inhibition of this kinase in mouse models leads to beneficial outcomes both at the level of neuropathology and behavior. These studies combined suggest that the beneficial effect of GSK-3β inhibition on mHTT toxicity and behavioral phenotypes could be mediated by a decrease in tau phosphorylation and that, consequently, elevated p-tau through GSK-3β activity could contribute to both neuropathological and clinical features of HD.
A decrease in calcineurin mRNA expression using microarray analyses has also been outlined in the cerebral cortex and caudate nucleus of Grades 0–2 human HD brains [32], and protein levels decreased in the putamen, although only 3 cases (one juvenile onset and two Grade 4) were analyzed [33]. An in vitro study further demonstrated that pharmacological inhibition of this phosphatase with the Cyclosporine A or the CN585 inhibitors in the SHSY5Y neuronal human cell line led to a significant increase in tau phosphorylation at various residues (Ser 202, Ser202/Thr205, Ser396/Ser404) [21]. This suggests that calcineurin is responsible for the dephosphorylation of tau and that its inhibition induces tau hyperphosphorylation in vitro. Moreover, in the StHdhQ111/Q111 HD cell model, calcineurin protein concentrations were decreased when compared to the control StHdhQ7/Q7 cells [21]. A decrease in calcineurin mRNA [34] and protein [21, 22, 33] was also detected in the striatum, cortex and hippocampus of several mouse models of HD (R6/1, R6/2, KI140CAG, zQ175). One study reported that the marked increase in p-tau levels (Ser 202, Ser202/Thr205, Ser396/Ser404) in the R6/2 mice was accompanied by a significant reduction of calcineurin levels in the hippocampus, cortex and striatum of animals of either pre-phenotypic or full-blown phenotypes [21]. One other study showed that the decrease in calcineurin levels coincided with a decrease in activity in the striatum of R6/1 mice [33]. Taken together, these findings suggest that mHTT can induce calcineurin reduction, thereby promoting tau hyperphosphorylation early in the disease process, i.e., before the onset of behavioral impairments (see Fig. 1).
Tau splicing factors are altered in disease
Altered splicing in the HTT gene, regulated by SRSF6 (serine/arginine-rich splicing factor-6) through its binding to the CAG repeat in mHTT mRNA, has been linked to HD pathology via the production of a toxic N-terminal mHTT fragment [35, 36]. SRSF6 regulates the splicing of MAPT exon 10 [37, 38], also associated to HD pathology [19]. In human HD striatal sections, immunofluorescence revealed that SRSF6 accumulates within mHTT inclusions. Moreover, an increase in phosphorylated SRSF6 concentrations is measurable in homogenates from striatum of HD brains. Similar SRSF6 alterations (presence of SRSF6 in mHTT inclusion bodies and increase of SRSF6 phosphorylation levels) were observed in the R6/1 HD mouse model [19]. Together, these findings demonstrate that SRSF6 is sequestered in mHTT aggregates and is abnormally phosphorylated suggesting a deficiency of activity [39, 40]. This could explain for the tau exon 10 mis-splicing and, consequently, the 4R/3R isoform imbalance noted in HD (see Fig. 1).
Tau and mHTT influence microtubule health
Alterations in axonal transport, due to microtubule disruption, constitute another key feature of HD pathology. mHTT has been shown to affect axonal transport through alteration of microtubule functions [41]. The destabilization of the microtubule network is one of the primary toxic events taking place within neuronal cells in the pathology [42]. It has indeed been previously shown, by immunopurification assays, that mHTT can interact with β-tubulin and bind to microtubules provoking impairments in axonal transport [43]. It has also been demonstrated that tau recruits mHTT to this cellular compartment, further compromising stabilization [22]. In the phosphoproteomic analysis described above, prominent upregulation tau phosphorylation has been shown to occur in the cortex of the R6/1 mouse model of HD, prior to the onset of motor deficits. These findings indicate that both tau and mHTT, which bind microtubules, alter the dynamic properties of the cytoskeleton. These dysfunctions could occur through coordinated effects of the two proteins and provoke the hyperphosphorylation of tau. Microtubule disruption, in concert with tau hyperphosphorylation, could be an early event leading to exacerbation of cellular toxicity in HD [23].
Taken together, mHTT has been shown to 1) play a role in calcineurin dysregulation which triggers hyperphosphorylation of tau, 2) sequester the SFRF6 splicing factor resulting in an imbalance of tau isoforms and 3) to be recruited by tau where it binds to the microtubule network causing destabilization (refer to Fig. 1).
Tau toxic species are mediated by caspases
Several caspases, including caspases 2, 3, and 6 play a role in HD, notably by cleaving mHTT and promoting aggregation to exacerbate toxicity [44, 45]. In the caudate nucleus and prefrontal cortex of postmortem HD brains, an increase in caspase 2 protein concentrations has been associated with elevated levels of the truncated Δtau314 tau protein (detected by immunoprecipitation and western blot) [46]. Of note, in cellular and animal models of tauopathies, higher Δtau314 levels are coupled with impairments in synaptic functions and reported to accentuate memory problems [47], respectively. It has therefore been suggested that the severe cognitive deficits observed in HD patients could originate from an increase in caspase 2-mediated Δtau314 levels (see Fig. 1) within the prefrontal cortex [46].
Tau interacts with mHTT
The idea that mHTT and tau directly and closely interact is highly debated (see summary Table 1). Histological analyses of the cortex and striatum of HD postmortem samples have revealed that, although sparce, a co-localization of mHTT and p-tau (Ser202/Thr205) aggregates does exist [18, 48]. In another study also performed in HD brain samples, abnormal deposits of these two proteins were most frequently found in the same subset of cortical neurons [19], although no interaction between mHTT and tau per se was reported. The most convincing evidence of a direct interaction between the two proteins originates from an in vitro study demonstrating that mHTT and tau can physically interact and co-aggregate to form “ring-like” inclusions within the cytoskeleton (Biomolecular fluorescence complementation assay BiFC) [22].
Table 1
Supporting and opposing arguments for the role of tau in HD
 |
Discrepancies between studies concerning 1) kinase and phosphatase dysregulations; 2) tau and mHTT interactions; 3) the contribution of tau to HD-like symptomatology and 4) the presence of tau in biofluids. Abbreviations: BDNF, Brain-Derived Neurotrophic Factor; CSF, Cerebrospinal Fluid; GSK3B, Glycogen Synthase Kinase 3 Beta; HD, Huntington's Disease; mHTT, mutant huntintin; p-tau, phosphorylated tau; t-tau, total tau; ↑, increase; ↓ decrease; ✓, presence; ✗, absence.
OPPOSING ARGUMENTS
Does tau truly contribute to neuropathological and behavioral impairments in HD?
Age indubitably participates to the accumulation of cerebral tau deposits and almost all human brains are scattered with such abnormal protein elements after the age of 70 [49, 50]. Because signs of tau pathology have been primarily observed in late-stage disease, some have argued that protein phosphorylation and aggregation may be a consequence of HD-pathology and not a triggering event and/or stand-alone phenomenon. In favor of this position, augmentations in p-tau at various epitopes (Ser199, Thr205, Ser396/Ser404) have been quantified in human HD putamen samples of Grades 3 and 4 patients, but not Grade 2 [12]. While one report has demonstrated the presence of oligomeric tau [18] in postmortem striatal samples of all Grades, none have performed correlations with disease progression. While tau oligomers have been proposed to induce cellular toxicity and synaptic dysfunctions in mice [51], the pathological effects of tau aggregated NFTs remain unclear. Consequently, complementary studies focusing on the levels of misfolded soluble and oligomeric p-tau in both HD mouse models and human brains would be of critical importance to establish the true contribution of tau to the pathology. Aside from brain pathology, measurements of tau plasma levels in different mouse models of HD (zQ175, R6/2:Q200, R6/2:Q90) has unveiled significant increases but exclusively at more advanced stages of disease (12 months in zQ175, 12 weeks in R6/1:Q200 and 24 weeks in R6/2:Q90), whereas an increase in plasma neurofilament levels was quantifiable at early timepoints (beginning at 6 months in zQ175, 8 weeks in R6/2:Q200 and 4 weeks in R6/2:Q90) [52]. Based on these findings, it is clear that additional studies focusing on early-stage disease in both mouse models and human brains is critically needed to establish the involvement of tau in disease onset/progression.
The causal impact of tau on behavioral phenotypes also needs to be clarified (see Table 1). Although one study conducted in mice suggested that tau may be involved in motor aspects of the disease [19], correlation analysis failed to uncover a relationship between MAPT haplotype and motor function in HD patients [18]. Along these lines, reduction of tau expression in the R6/1 mouse model did not improve motor (rotarod and clasping tests), cognitive (Y-maze test) nor depression-like (Porsolt swim test) behaviors [53] in contrast to previous reports [19]. In R6/1 mice expressing the human tau P301L transgene, behavioral phenotypes characteristic of HD are unchanged suggesting that tau may not participate to the manifestation of HD-like symptoms [53]. The discrepancies between these studies [19, 53] could be partially explained by the different strains/genetic backgrounds of the selected mouse models, a factor which has been shown to influence behavioral outcomes in rodents [54] or by the difference in the paradigms used (rotarod and clasping). In this particular publication, authors themselves propose a few limitations to their work that we feel are important to reiterate. The transgenic R6/1 mouse model of HD progresses rapidly to a full-blown phenotype and presents with severe behavioral impairments which begin to manifest at approximately 3 months of age. In contrast, the P301L transgenic mouse model develops behavioral deficits at 5-6 months of age. The different disease course of the two mice may be one reason why the overall behavioral traits of the crossed R6/1 and P301L is not exacerbated. Additionally, animals were sacrificed at 5 months of age which is likely too early to measure the impact of tau on HD pathology. We would encourage to conduct a similar study on a milder and more progressive mouse model of HD, such as the knock-in zQ175 in which motor deficits appear at a similar timepoint, i.e., 5-6 months of age. This would be more appropriate to evaluate the precipitation and/or aggravation of behavioral impairments as a consequence of tau expression. Importantly, the neuropathological impact of tau depletion or overexpression on mHTT aggregation, mHTT and NFT interactions, p-tau and NFTs aggregates, for example, was not assessed by the authors. While interesting, the R6/1/P301L and MAPT–/–mouse studies may suffer from the use of suboptimal approach. For instance, the CBA/Bl6/129Sv background may not be characterized by increases in total tau or imbalances in tau isoforms and this is where postmortem analyses, absent from this study, would have been key to draw more definitive conclusions on the role of tau.
Do tau and mHTT-induced pathology truly share common mechanisms of action?
At a cellular level, the mechanisms by which tau may contribute to pathology also remain elusive and are therefore a subject of debate (refer to Table 1). Upregulation of kinases, such as GSK-3β, is one of the favored pathways by which tau undergoes hyperphosphorylation, but this idea, too, has been challenged in both humans and animals. While one study reported an increase in GSK-3β mRNA and protein levels in the hippocampus of postmortem HD brains [25], the opposite has been observed in other affected brain regions, notably the frontal cortex [55] and striatum in HD patients [27]. Similarly, levels of GSK-3β were measured in brain regions of HD mouse models and data implied significantly lower levels of GSK-3β protein concentrations in the striatum and cortex of R6/1 mice at 3.5 months of age [27]. Globally, the conflicting findings related to GSK-3β levels in human and mouse brains is most probably explained by the difference in the structures analyzed, where levels of GSK-3β were increased in the hippocampus but decreased in the striatum and cortex. Of note, in the few studies demonstrating an elevation of GSK-3β protein levels in mouse and human HD brains, the enzymatic potency was not investigated. Independent in vitro enzymatic assays on homogenates of human and mice brains revealed a significant decrease in GSK-3 activity in the striatum and cortex of human brains [27]. In R6/1 mice, a significant reduction in GSK-3 activity was only observed in the striatum of early-manifest (3.5 months) and late-stage animals (7.5 months) [27]. To further explore the role of GSK-3β function in HD, R6/1 mice were crossed with a transgenic mouse line with conditional GSK-3β overexpression [27]. R6/1 mice which had low levels of overexpression of GSK-3β also depicted attenuation of brain atrophy (cortical and hippocampal), improvement of motor performances (open field and rotarod tests) and amelioration of cognitive functions (fear conditioning test) when compared to the R6/1 mouse line alone [27]. To evaluate the activation state of this kinase, other groups have focused on the levels of N-terminal phosphorylated GSK-3β at the inhibitory serine-9 residue as an indirect indicator of GSK-3 activity. The combined in vitro and preclinical results demonstrated an increase in GSK-3β phosphorylation in serine-9 which suggests a lowering of GSK-3β activity (studies detailed in Table 1). Although it is clear that GSK-3β is altered in HD, dysregulations of this kinase seem to be region-specific. Based on the findings presented, upregulation of GSK-3β could be responsible for the hyperphosphorylation of tau in the HD hippocampus. However, the concomitant studies demonstrating a decrease in GSK-3β expression and function in the striatum and cortex indicate that other mechanisms could very well be at play.
Work on the implications of calcineurin phosphatase in HD, i.e. that decreased modulation of this enzyme is associated with increased p-tau, have also been challenged (see Table 1). Indeed, few studies have focused on the consequences of increased calcineurin activity in HD, and therefore the beneficial implications of its inhibition on pathology [56, 57]. In the HdhQ111/Q111 mouse model of the disease, while there was no change in calcineurin protein levels in various brain regions (substantia nigra, cortex, striatum) when compared to wild-type mice, a significant increase in the enzyme’s activity was observed in the cortex [56]. In primary cortical neurons from the HdhQ111/Q111 mouse model, genetic inhibition of calcineurin by RNA interference resulted in increased levels of phosphorylated HTT at serine 421 (p-HTT-Ser421) - which is also a substrate of this enzyme - and in the restoration of the microtubule-dependent BDNF (Brain-Derived Neurotrophic Factor) transport in vitro [56]. Pharmacological inhibition of this phosphatase using the FK506 drug in neuronal cells derived from a knock-in mouse model of HD led to a significant increase in p-HTT-Ser421 levels and this was accompanied by a decrease in neuronal death [57]. Although few in numbers, these studies suggest a neuroprotective effect of calcineurin inhibition on cell viability in vitro through an increase in HTT phosphorylation at the serine residue which has been shown to result in neuroprotective effects of striatal neurons [58]. However, the effects of calcineurin inhibition on behavioral improvements in HD mice has not been established and, to our knowledge, no increase in calcineurin levels has been detected in human HD postmortem brains. The relevance of calcineurin inhibition in HD therefore deserves further investigation.
The hypothesis of a physical interrelationship between tau and mHTT also remains controversial (some of the discrepancies found between studies are summarized in Table 1). While interaction between abnormal deposits of these two proteins has been observed in vitro, no co-localization between mHTT aggregates and p-tau (Ser202/Thr205, Ser396/Ser404) has been reported in either the R6/2 and KI140 mouse models of HD [21, 22] and similarly, co-immunoprecipitation experiments have failed to reveal a direct association between them, at least in mice [22]. In HD brains, one study reported that tau and mHTT did not co-precipitate and that each protein was found exclusively within its respective aggregate-type [19]. Although the idea of a dynamic interplay between these two proteins remains unclear, the effects of mHTT on tau phosphorylation and/or of tau on exacerbated mHTT aggregation are not necessarily mediated by direct interactions and the toxicity of one on the other could well take place via indirect pathways and intermediate steps.
THE CLINICAL RELEVANCE OF TAU
Can tau be a relevant biomarker?
Despite the monogenic nature of HD, clinical phenotypes present with prominent heterogeneity, in particular when it relates to age of onset and disease severity. The diagnosis is also preceded by a lengthy prodromal stage during which some patients may begin to experience subtle changes in cognition and behavior including unusual irritability, anxiety, or depression. Better understanding elements arising during the prodromal phase, such as specific biomarkers, and how they correlate with disease onset has become a major area of research as it offers the best hope to develop new treatments that could delay or even halt disease progression.
The quest for robust biomarkers may be accelerated by studying the data collected in the field of AD where tau pathology has been recognized and studied for decades. For example, we already know that increases in cerebrospinal fluid (CSF) total tau (t-tau) levels found in AD patients [59–61] are associated with cognitive decline [62]. Tau is additionally detectable in other biofluids and blood compartments such as plasma [63, 64], platelets [65], lymphocytes [66], saliva [67], and oral mucosa epithelium [68] which correlate with the degree of cognitive impairments. Similarly to AD, increases of CSF t-tau concentrations have recently been reported in HD patients and levels correlated with disease progression, cognitive decline and motor abnormalities [69]. Another study demonstrated that CSF t-tau is elevated in HD patients suffering from psychiatric symptoms when compared to patients without such impairments [70]. Increases in CSF levels of t-tau and p-tau in early manifest HD patients, when compared to pre-symptomatic carriers, have also been associated with apathy and cognitive disabilities and further correlated with brain damage measured by a reduction of cortical thickness and of the grey-matter volume [71].
While data on the potential to predict disease manifestation/severity is emerging, our group has begun to quantify levels of t-tau and p-tau in different blood compartments of HD gene carriers. An overall increase in t-tau is observed in manifest HD patients as compared to age- and sex-matched healthy controls in plasma, peripheral blood mononuclear cells (PBMCs) and platelets. Measures of t-tau in both plasma and platelets significantly correlate with disease stage with levels in platelets additionally correlating with cognitive ability. In PBMCs, increased t-tau levels are present at all stages of manifest while increased levels of tau phosphorylated at threonine 231 (p-tau231) is observed in the plasma at later stages (Stages 3–5). Together, these observations suggest that accumulation of tau in blood cells is an early event in HD and that t-tau levels in platelets are associated with changes in cognition (unpublished observations). These findings, although preliminary, suggest that tau pathology is a feature of HD which may contribute to disease presentation.
Biomarkers are not restricted to bodily fluids. Hence, a number of PET tracers have recently been developed and are currently being validated in order to visualize tau deposits in vivo [72]. A PET imaging study using the [18F]AV-1451 (T807) ligand highlighted that tau pathology in brains of Parkinson’s and Lewy body disease correlated with cognitive decline [73]. Imaging with T807 PET tracer, which binds to pathological tau aggregates, also showed a distinct tau deposition pattern associated with cognitive performances in AD patients [74]. More recently, tau pathology has begun to be investigated in HD brains using T807 which binds to tau aggregates with high affinity and good selectivity. This ligand allowed assessment of tau pathology in brains of three manifest HD patients, and in vivo imaging of tau pathology was restricted mainly to the striatum, and to a lesser extent in the cortex and cerebellum. The PET signal adopted different patterns between the three patients depending on the degree of cognitive and motor impairments as well as their disease burden scores. The subcortical and cortical signal was the weakest in the least cognitively impaired subject, whereas tau tracer uptake was the most prominent in the patient presenting the worse motor and disease burden scores [75]. T807 is currently in phase I for mapping cerebral tau in neurodegenerative disorders, including HD (https://clinicaltrials.gov/ct2/show/NCT04926259). The aim of this study is to compare the regional distribution and tau burden to the clinical symptomatology of patients with neurodegenerative disorders. CSF levels of t-tau and p-tau will further be measured and correlated with PET imaging results. Imaging techniques and the development of specific tau ligands will greatly help advance knowledge of the role of tau in HD.
Can tau be a relevant therapeutic target?
Given the monogenic nature of HD, gene therapies zooming in on the HTT gene with the aim to reduce the amount of mHTT in the brain have been at the forefront of recent clinical efforts. A number of these methodologies have therefore been developed to, selectively or non-selectively, dampen DNA or mRNA expression of the HD gene [76, 77]. In preclinical studies, non-allele specific silencing of HTT via RNA interference in the striatum of the N171-82Q mouse model of HD resulted in the reduction of wild-type and mutant HTT mRNA and protein levels, and improved motor performances in the rotarod test as well as longevity [78]. While encouraging in animal models, the application of these methods to the clinical realm has been met with significant challenges. In the last couple of years, various trials aiming to target the HTT gene have been terminated due to a lack of clinical improvements and/or appearance of adverse effects in treated patients [79]. Of these, the Generation-HD1 phase III trial, which used an intrathecal administration of a non-allele specific ASO targeting the HTT transcript, reported a significant decrease in CSF mHTT levels, but no improvement of clinical performances and, importantly, the manifestation of adverse effects. The Precision-HD1 and -HD2 phase Ib/IIa trials, which are two distinctive allele-selective ASOs administered intrathecally, have not met the primary outcome of safety and also reported undesirable side-effects, at least at the higher dose administered. Here, no significant reduction of CSF mHTT was even detected. Despite these setbacks, trials are being pursued to improve the safety and efficiency of these molecules, notably by rethinking the chemical structures of the drugs tested, identifying the appropriate therapeutical window in which to intervene as well as to establish patient population who will best respond to such treatments. Another type of approach targeting the intracellular mHTT protein makes use of therapeutic anti-HTT intrabodies. This strategy, by which the intrabody which recognizes the proline-rich domain of HTT (Happ1) is administered through intra-striatal adeno-associated virus delivery, has been tested in several HD mouse models (N171-82Q, BACHD, R6/2, YAC128). This resulted in the amelioration of motor abilities in the rotarod and narrow beam tests of all tested models [80] as well as cognitive functions in YAC128 mice (but not the BACHD) as observed in the object location memory task reflecting spatial learning [80]. This type of methodology has not reached clinical stages, however.
While we struggle to understand why targeting the main actor of HD pathology, mHTT, has not yielded the desired and expected benefits, other potential therapeutic targets must be considered. The wealth of evidence for the presence of pathological tau in HD, its contribution to neuropathological changes and correlation with some clinical features, certainly encourages to further research the validity of targeting tau as a therapy for HD. Several tau-oriented therapies have already been investigated for treatment of tauopathies, and particularly AD. These include strategies that address tau hyperphosphorylation with kinase inhibitors and phosphatase activators, the inhibition of tau aggregation, tau immunotherapies, stabilization of microtubules, and inhibition of the tau gene MAPT expression [81]. While these concepts are interesting from their own perspective, they have been at the center of extensive discussion in a review we have recently published [81]. Here, we will therefore focus exclusively on approaches that have been tested in the context of HD and that we judge may act via a tau-related mechanism.
Lithium, a GSK-3β inhibitor that has shown promising results in preclinical [82, 83] and clinical [84] studies conducted in AD, has also been tested in HD. Lithium reduces tau phosphorylation (Ser202, Ser396/Ser404) and tau aggregation in mouse models of AD [82, 83] and improves cognitive performances of AD patients [84]. Lithium treatment had a beneficial effect on motor performances of R6/2 mice [84] and a pilot study conducted in three HD cases suggested an amelioration of chorea and mood in patients [85]. The specific mechanism of Lithium and how this compound may impact p-tau levels in HD remains unclear but certainly worth exploring. Similarly, memantine is an approved treatment to alleviate psychological disturbances in AD and was shown to enhance the activity of the phosphatase PP2A. In HD, memantine improved motor performances of patients [87, 88] and accordingly, this molecule is currently tested in a phase II trial (MITIGATE-HD) (https://clinicaltrials.gov/ct2/show/NCT01458470). However, similarly to Lithium, the correlations between the beneficial outcomes of memantine and a potential reduction of tau pathological phosphorylated forms have not been investigated.
Therapeutic approaches targeting tau pathology have been designed to treat AD [89] building on a large body of evidence suggesting a strong connection between alterations of tau function and cognitive decline [90]. Based on the data that we have reviewed herein, HD pathology is also associated with pathological forms of tau and cognitive impairments, thus tau-targeting therapies may offer a completely new angle to treat HD-associated cognitive dysfunction.
PERSPECTIVE
Like in any fields, not all evidence brought forward for the role of tau in HD has been replicated or agreed upon. Despite some discrepancies, the overall landscape favorably supports the contribution of the protein to this pathology and the contrastive studies imply that supplementary studies are needed to validate its relevance to disease. Among some points of contingencies, a more careful selection of the experimental models and the stages of disease when to study this will be needed to confirm or refute the contribution of tau to HD pathology. Additional work on tau expression in the periphery, clinical correlations and in vivo PET imaging would be particularly valuable to enhance our understanding on tau’s role in HD. HD is a condition for which there are still no treatment, and the recent/ongoing data and arguments presented in this review enforce the importance of further investigating tau-related mechanisms underlying HD pathology and, if relevant, proposing tau as a potential therapeutical target.
ACKNOWLEDGMENTS
FC is a recipient of a Researcher Chair from the Fonds de Recherche du Québec en Santé (FRQS, 35059) providing salary support and operating funds, and receives funding from the Canadian Institutes of Health Research (CIHR, PJT-162164 and PJT-168865) to conduct her HD-related research. SS was supported by the Bourse de formation Desjardins pour la recherche et innovation from CHU de Québec.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes The Huntington’s Disease Collaborative Research Grou, Cell (1993) ;72: :971–83. https://doi.org/10.1016/0092-8674(93)90585-e. |
[2] | Gratuze M , Cisbani G , Cicchetti F , Planel E Is Huntington’s disease a tauopathy? Brain (2016) ;139: :1014–25. https://doi.org/10.1093/brain/aww021. |
[3] | Maxan A , Cicchetti F Tau: A common denominator and therapeutic target for neurodegenerative disorders, J Exp Neurosci (2380) ;12: :1179069518772380. https://doi.org/10.1177/1179069518772380. |
[4] | Harada A , Oguchi K , Okabe S , Kuno J , Terada S , Ohshima T , et al. Altered microtubule organization in small-calibre axons of mice lacking tau protein, Nature (1994) ;369: :488–91. https://doi.org/10.1038/369488a0. |
[5] | Sultan A , Nesslany F , Violet M , Bégard S , Loyens A , Talahari S , et al. Nuclear tau, a key player in neuronal DNA protection, J Biol Chem (2011) ;286: :4566–75. https://doi.org/10.1074/jbc.M110.199976. |
[6] | Guo T , Noble W , Hanger DP Roles of tau protein in health and disease, Acta Neuropathol (2017) ;133: :665–704. https://doi.org/10.1007/s00401-017-1707-9. |
[7] | Mudher A , Brion JP , Avila J , Medina M , Buée L EuroTau: Towing scientists to tau without tautology, Acta Neuropathol Commun (2017) ;5: :90. https://doi.org/10.1186/s40478-017-0491-z. |
[8] | Goedert M , Spillantini MG , Jakes R , Rutherford D , Crowther RA Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease, Neuron (1989) ;3: :519–26. https://doi.org/10.1016/0896-6273(89)90210-9. |
[9] | Morishima-Kawashima M , Hasegawa M , Takio K , Suzuki M , Yoshida H , Titani K , et al. Proline-directed and non-proline-directed phosphorylation of PHF-tau, J Biol Chem (1995) ;270: :823–9. https://doi.org/10.1074/jbc.270.2.823. |
[10] | Cisbani G , Maxan A , Kordower JH , Planel E , Freeman TB , Cicchetti F Presence of tau pathology within foetal neural allografts in patients with Huntington’s and Parkinson’s disease, Brain (2017) ;140: :2982–92. https://doi.org/10.1093/brain/awx255. |
[11] | Jellinger KA Alzheimer-type lesions in Huntington’s disease, J Neural Transm (1998) ;105: :787–99. https://doi.org/10.1007/s007020050095. |
[12] | St-Amour I , Turgeon A , Goupil C , Planel E , Hébert SS Co-occurrence of mixed proteinopathies in late-stage Huntington’s disease, Acta Neuropathol (2018) ;135: :249–65. https://doi.org/10.1007/s00401-017-1786-7. |
[13] | Reyes MG , Gibbons S Dementia of the Alzheimer’s type and Huntington’s disease, Neurology (1985) ;35: :273–7. https://doi.org/10.1212/wnl.35.2.273. |
[14] | Moss RJ , Mastri AR , Schut LJ The coexistence and differentiation of late onset Huntington’s disease and Alzheimer’s disease. A case report and review of the literature, J Am Geriatr Soc (1988) ;36: :237–41. https://doi.org/10.1111/j.1532-5415.1988.tb01807.x. |
[15] | McIntosh GC , Jameson HD , Markesbery WR Huntington disease associated with Alzheimer disease, Ann Neurol (1978) ;3: :545–8. https://doi.org/10.1002/ana.410030616. |
[16] | Myers RH , Sax DS , Schoenfeld M , Bird ED , Wolf PA , Vonsattel JP , et al. Late onset of Huntington’s disease, J Neurol Neurosurg Psychiatry (1985) ;48: :530–4. https://doi.org/10.1136/jnn48.6.530. |
[17] | Davis MY , Keene CD , Jayadev S , Bird T The co-occurrence of Alzheimer’s disease and Huntington’s disease: A neuropathological study of 15 elderly Huntington’s disease subjects, J Huntingtons Dis (2014) ;3: :209–17. https://doi.org/10.3233/JHD-140111. |
[18] | Vuono R , Winder-Rhodes S , de Silva R , Cisbani G , Drouin-Ouellet J , REGISTRY Investigators of the European Huntington’s Disease Network et al. The role of tau in the pathological process and clinical expression of Huntington’s disease, Brain (2015) ;138: :1907–18. https://doi.org/10.1093/brain/awv107. |
[19] | Fernández-Nogales M , Cabrera JR , Santos-Galindo M , Hoozemans JJM , Ferrer I , Rozemuller AJM , et al. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods, Nat Med (2014) ;20: :881–5. https://doi.org/10.1038/nm.3617. |
[20] | Petry S , Nateghi B , Keraudren R , Sergeant N , Planel E , Hébert SS , et al. Differential regulation of tau exon 2 and 10 isoforms in Huntington’s disease brain. Neuroscience. 2022. https://doi.org/10.1016/j.neuroscience.2022.07.014. |
[21] | Gratuze M , Noë A , Julien C , Cisbani G , Milot-Rousseau P , Oise Morin F , et al. Tau hyperphosphorylation and deregulation of calcineurin in mouse models of Huntington’s disease, Hum Mol Genet (2015) ;24: :86–99. https://doi.org/10.1093/hmg/ddu456. |
[22] | Blum D , Herrera F , Francelle L , Mendes T , Basquin M , Obriot H , et al. Mutant huntingtin alters tau phosphorylation and subcellular distribution, Hum Mol Genet (2015) ;24: :76–85. https://doi.org/10.1093/hmg/ddu421. |
[23] | Mees I , Tran H , Roberts A , Lago L , Li S , Roberts BR , et al. Quantitative phosphoproteomics reveals extensive protein phosphorylation dysregulation in the cerebral cortex of Huntington’s disease mice prior to onset of symptoms, Mol Neurobiol (2022) ;59: :2456–71. https://doi.org/10.1007/s12035-021-02698-y. |
[24] | Alpaugh M , Masnata M , de Rus Jacquet A , Lepinay E , Denis HL , Saint-Pierre M , et al. Passive immunization against phosphorylated tau improves features of Huntington’s disease pathology, Mol Ther (2022) ;30: :1500–22 . https://doi.org/10.1016/j.ymthe.2022.01.020. |
[25] | L’Episcopo F , Drouin-Ouellet J , Tirolo C , Pulvirenti A , Giugno R , Testa N , et al. GSK-3β-induced tau pathology drives hippocampal neuronal cell death in Huntington’s disease: Involvement of astrocyte-neuron interactions, Cell Death Dis (2016) ;7: :e2206. https://doi.org/10.1038/cddis.2016.104. |
[26] | Hughes K , Nikolakaki E , Plyte SE , Totty NF , Woodgett JR Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation, EMBO J (1993) ;12: :803–8. https://doi.org/10.1002/j.1460-2075.1993.tb05715.x. |
[27] | Fernández-Nogales M , Hernández F , Miguez A , Alberch J , Ginés S , Pérez-Navarro E , et al. Decreased glycogen synthase kinase-3 levels and activity contribute to Huntington’s disease, Hum Mol Genet (2015) ;24: :5040–52. https://doi.org/10.1093/hmg/ddv224. |
[28] | Medina M , Wandosell F Deconstructing GSK- The fine regulation of its activity, Int J Alzheimers Dis (2011) ;3: :4792–49. https://doi.org/10.4061/2011/479249. |
[29] | Saavedra A , García-Martínez JM , Xifró X , Giralt A , Torres-Peraza JF , Canals JM , et al. PH domain leucine-rich repeat protein phosphatase 1 contributes to maintain the activation of the PI3K/Akt pro-survival pathway in Huntington’s disease striatum, Cell Death Differ (2010) ;17: :324–35. https://doi.org/10.1038/cdd.2009.127. |
[30] | Chiu C-T , Liu G , Leeds P , Chuang D-M Combined treatment with the mood stabilizers lithium and valproate produces multiple beneficial effects in transgenic mouse models of Huntington’s disease, Neuropsychopharmacology (2011) ;36: :2406–21. https://doi.org/10.1038/n2011.128. |
[31] | Rippin I , Bonder K , Joseph S , Sarsor A , Vaks L , Eldar-Finkelman H Inhibition of GSK-3 ameliorates the pathogenesis of Huntington’s disease, Neurobiol Dis (2021) ;154: :1053–36. https://doi.org/10.1016/j.nbd.2021.105336. |
[32] | Hodges A , Strand AD , Aragaki AK , Kuhn A , Sengstag T , Hughes G , et al. Regional and cellular gene expression changes in human Huntington’s disease brain, Hum Mol Genet (2006) ;15: :965–77. https://doi.org/10.1093/hmg/ddl013. |
[33] | Xifró X , Giralt A , Saavedra A , García-Martínez JM , Díaz-Hernández M , Lucas JJ , et al. Reduced calcineurin protein levels and activity in exon-1 mouse models of Huntington’s disease: Role in excitotoxicity, Neurobiol Dis (2009) ;36: :461–9. https://doi.org/10.1016/j.nbd.2009.08.012. |
[34] | Luthi-Carter R , Strand A , Peters NL , Solano SM , Hollingsworth ZR , Menon AS , et al. Decreased expression of striatal signaling genes in a mouse model of Huntington’s disease, Hum Mol Genet (2000) ;9: :1259–71. https://doi.org/10.1093/hmg/9.9.1259. |
[35] | Sathasivam K , Neueder A , Gipson TA , Landles C , Benjamin AC , Bondulich MK , et al. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease, PNAS (2013) ;110: :2366–70. https://doi.org/10.1073/pnas.1221891110. |
[36] | Schilling J , Broemer M , Atanassov I , Duernberger Y , Vorberg I , Dieterich C , et al. Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington’s disease, J Mol Biol (2019) ;431: :1869–77. https://doi.org/10.1016/j.jmb.2019.01.034. |
[37] | Wang Y , Wang J , Gao L , Lafyatis R , Stamm S , Andreadis A Tau exons 2 and 10, which are misregulated in neurodegenerative diseases, are partly regulated by silencers which bind a SRp30c.SRp55 complex that either recruits or antagonizes htra2beta1, J Biol Chem (2005) ;280: :14230–9. https://doi.org/10.1074/jbc.M413846200. |
[38] | Corsi A , Bombieri C , Valenti MT , Romanelli MG Tau isoforms: Gaining insight into MAPT alternative splicing. Int J Mol Sci. 2022;23. https://doi.org/10.3390/ijms232315383. |
[39] | Yin X , Jin N , Gu J , Shi J , Zhou J , Gong C-X , et al. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) modulates serine/arginine-rich protein 55 (SRp55)-promoted tau exon 10 inclusion, J Biol Chem (2012) ;287: :30497–506. https://doi.org/10.1074/jbc.M112.355412. |
[40] | Fernández-Nogales M , Lucas JJ Altered levels and isoforms of tau and nuclear membrane invaginations in Huntington’s disease, Front Cell Neurosci (2020) ;13: :574. https://doi.org/10.3389/fncel.2019.00574. |
[41] | Szebenyi G , Morfini GA , Babcock A , Gould M , Selkoe K , Stenoien DL , et al. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport, Neuron (2003) ;40: :41–52. https://doi.org/10.1016/s0896-6273(03)00569-5. |
[42] | Trushina E , Heldebrant MP , Perez-Terzic CM , Bortolon R , Kovtun IV , Badger JD 2nd , et al. Microtubule destabilization and nuclear entry are sequential steps leading to toxicity in Huntington’s disease, PNAS (2003) ;100: :12171–6. https://doi.org/10.1073/pnas.2034961100 |
[43] | Hoffner G , Kahlem P , Djian P Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: Relevance to Huntington’s disease. J Cell Sci (2002) ;115: :941–8. https://doi.org/10.1242/jcs.115.5.941. |
[44] | Wellington CL , Singaraja R , Ellerby L , Savill J , Roy S , Leavitt B , et al. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells, J Biol Chem (1983) ;275: :1–8. https://doi.org/10.1074/jbc.M001475200. |
[45] | Carroll JB , Southwell AL , Graham RK , Lerch JP , Ehrnhoefer DE , Cao L-P , et al. Mice lacking caspase-2 are protected from behavioral changes, but not pathology, in the YAC128 model of Huntington disease, Mol Neurodegener (2011) ;6: :59. https://doi.org/10.1186/1750-1326-6-59. |
[46] | Liu P , Smith BR , Huang ES , Mahesh A , Vonsattel JPG , Petersen AJ , et al. A soluble truncated tau species related to cognitive dysfunction and caspase-2 is elevated in the brain of Huntington’s disease patients, Acta Neuropathol Commun (2019) ;7: :111. https://doi.org/10.1186/s40478-019-0764-9. |
[47] | Zhao X , Kotilinek LA , Smith B , Hlynialuk C , Zahs K , Ramsden M , et al. Caspase-2 cleavage of tau reversibly impairs memory, Nat Med (2016) ;22: :1268–76. https://doi.org/10.1038/nm.4199. |
[48] | Caparros-Lefebvre D , Kerdraon O , Devos D , Dhaenens CM , Blum D , Maurage CA , et al. Association of corticobasal degeneration and Huntington’s disease: Can tau aggregates protect Huntingtin toxicity? Mov Disord (2009) ;24: :1089–90. https://doi.org/10.1002/mds.22204. |
[49] | Nelson PT , Alafuzoff I , Bigio EH , Bouras C , Braak H , Cairns NJ , et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature, J Neuropathol Exp Neurol (2012) ;71: :362–81. https://doi.org/10.1097/NEN.0b013e31825018f7. |
[50] | Ziontz J , Bilgel M , Shafer AT , Moghekar A , Elkins W , Helphrey J , et al. Tau pathology in cognitively normal older adults, Alzheimers Dement (Amst) (2019) ;11: :637–45. https://doi.org/10.1016/j.dadm.2019.07.007. |
[51] | Lasagna-Reeves CA , Castillo-Carranza DL , Sengupta U , Clos AL , Jackson GR , Kayed R Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice, Mol Neurodegener (2011) ;6: :39. https://doi.org/10.1186/1750-1326-6-39. |
[52] | Bondulich MK , Byrne LM , Iqbal A , Nita IM , Cañibano-Pico M , Phillips JM , et al. D21 Analysis of blood and CSF biomarkers in mouse models of Huntington’s disease, J Neurol Neurosurg Psychiatry (2022) ;93: :A27 LP–A27. https://doi.org/10.1136/jnnp-2022-ehdn.77. |
[53] | Mees I , Li S , Beauchamp LC , Barnham KJ , Dutschmann M , Hannan AJ , et al. Loss-of-function and gain-of-function studies refute the hypothesis that tau protein is causally involved in the pathogenesis of Huntington’s disease, Hum Mol Genet.- (2022) ;31: :1997–2009. https://doi.org/10.1093/hmg/ddac001. |
[54] | Sultana R , Ogundele OM , Lee CC Contrasting characteristic behaviours among common laboratory mouse strains, R Soc Open Sci (2019) ;6: :190574. https://doi.org/10.1098/rsos.190574. |
[55] | Lim NKH , Hung LW , Pang TY , Mclean CA , Liddell JR , Hilton JB , et al. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain, Hum Mol Genet (2014) ;23: :4051–63. https://doi.org/10.1093/hmg/ddu119. |
[56] | Pineda JR , Pardo R , Zala D , Yu H , Humbert S , Saudou F Genetic and pharmacological inhibition of calcineurin corrects the BDNF transport defect in Huntington’s disease, Mol Brain (2009) ;2: :33. https://doi.org/10.1186/1756-6606-2-33. |
[57] | Pardo R , Colin E , Régulier E , Aebischer P , Déglon N , Humbert S , et al. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421, J Neurosci (2006) ;26: :1635–45. https://doi.org/10.1523/JNEUROSCI.3706-05.2006. |
[58] | Humbert S , Bryson EA , Cordelières FP , Connors NC , Datta SR , Finkbeiner S , et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt, Dev Cell (2002) ;2: :831–7. https://doi.org/10.1016/s1534-5807(02)00188-0. |
[59] | Vigo-Pelfrey C , Seubert P , Barbour R , Blomquist C , Lee M , Lee D , et al. Elevation of microtubule-associated protein tau in the cerebrospinal fluid of patients with Alzheimer’s disease, Neurology (1995) ;45: :788–93. https://doi.org/10.1212/wnl.45.4.788. |
[60] | Galasko D , Chang L , Motter R , Clark CM , Kaye J , Knopman D , et al. High cerebrospinal fluid tau and low amyloid beta42 levels in the clinical diagnosis of Alzheimer disease and relation to apolipoprotein E genotype, Arch Neurol (1998) ;55: :937–45. https://doi.org/10.1001/archneur.55.7.937. |
[61] | Andreasen N , Minthon L , Davidsson P , Vanmechelen E , Vanderstichele H , Winblad B , et al. Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice, Arch Neurol (2001) ;58: :373–9. https://doi.org/10.1001/archneur.58.3.373. |
[62] | Koychev I , Gunn RN , Firouzian A , Lawson J , Zamboni G , Ridha B , et al. PET tau and amyloid-β burden in mild Alzheimer’s disease: Divergent relationship with age, cognition, and cerebrospinal fluid biomarkers, J Alzheimers Dis (2017) ;60: :283–93. https://doi.org/10.3233/JAD-170129. |
[63] | Winston CN , Goetzl EJ , Akers JC , Carter BS , Rockenstein EM , Galasko D , et al. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile, Alzheimers Dement (Amst) (2016) ;3: :63–72. https://doi.org/10.1016/j.dadm.2016.04.001. |
[64] | Yang S-Y , Chiu M-J , Chen T-F , Horng H-E Detection of plasma biomarkers using immunomagnetic reduction: A promising method for the early diagnosis of Alzheimer’s disease, Neurol Ther (2017) ;6: :37–56. https://doi.org/10.1007/s40120-017-0075-7. |
[65] | Neumann K , Farías G , Slachevsky A , Perez P , Maccioni RB Human platelets tau: A potential peripheral marker for Alzheimer’s disease, J Alzheimers Dis (2011) ;25: :103–9. https://doi.org/10.3233/JAD-2011-101641. |
[66] | Kvetnoy IM , Hernandez-Yago J , Kvetnaia T V , Khavinson VK , Malinin V V , Yarilin AA , et al. Tau-protein expression in human blood lymphocytes: A promising marker and suitable sample for life-time diagnosis of Alzheimer’s disease, Neuro Endocrinol Lett (2000) ;21: , 313–8. |
[67] | Shi M , Sui Y-T , Peskind ER , Li G , Hwang H , Devic I , et al. Salivary tau species are potential biomarkers of Alzheimer’s disease, J Alzheimers Dis (2011) ;27: :299–305. https://doi.org/10.3233/JAD-2011-110731. |
[68] | Arredondo LF , Aranda-Romo S , Rodríguez-Leyva I , Chi-Ahumada E , Saikaly SK , Portales-Pérez DP , et al. Tau protein in oral mucosa and cognitive state: A cross-sectional study, Front Neurol (2017) ;8: :554. https://doi.org/10.3389/fneur.2017.00554. |
[69] | Rodrigues FB , Byrne L , McColgan P , Robertson N , Tabrizi SJ , Leavitt BR , et al. Cerebrospinal fluid total tau concentration predicts clinical phenotype in Huntington’s disease, J Neurochem (2016) ;139: :22–5. https://doi.org/10.1111/jnc.13719. |
[70] | Vinther-Jensen T , Börnsen L , Budtz-Jørgensen E , Ammitzbøll C , Larsen IU , Hjermind LE , et al. Selected CSF biomarkers indicate no evidence of early neuroinflammation in Huntington disease, Neurol Neuroimmunol Neuroinflammation (2016) ;3: :e287. https://doi.org/10.1212/NXI.0000000000000287. |
[71] | Martinez-Horta S , Perez-Perez J , Perez-Gonzalez R , Horta-Barba A , Sampedro F , Rivas-Asensio E , et al. Tau pathology contributes to specific patterns of structural brain damage and neuropsychological heterogeneity in Huntington’s disease [abstract]. Mov Disord. 2020;35. |
[72] | Sexton C , Snyder H , Beher D , Boxer AL , Brannelly P , Brion J-P , et al. Current directions in tau research: Highlights from Tau 2020, Alzheimers Dement (2022) ;18: :988–1007. https://doi.org/10.1002/alz.12452. |
[73] | Gomperts SN , Locascio JJ , Makaretz SJ , Schultz A , Caso C , Vasdev N , et al. Tau positron emission tomographic imaging in the Lewy body diseases, JAMA Neurol (2016) ;73: :1334–41. https://doi.org/10.1001/jamaneurol.2016.3338. |
[74] | Brier MR , Gordon B , Friedrichsen K , McCarthy J , Stern A , Christensen J , et al. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease, Sci Transl Med (2016) ;8: :338ra66. https://doi.org/10.1126/scitranslmed.aaf2362. |
[75] | Reetz K , Giehl K , Dogan I , Werner CJ , Hammes J , Schulz JB , et al. D26 Pathological tau signal in Huntington’s disease –an in vivo-AV-pet imaging report, J Neurol Neurosurg Psychiatry (2016) ;87: :A44 LP–A44. https://doi.org/10.1136/jnnp-2016-314597.125. |
[76] | Tabrizi SJ , Leavitt BR , Landwehrmeyer GB , Wild EJ , Saft C , Barker RA , et al. Targeting Huntingtin expression in patients with Huntington’s disease, N Engl J Med (2016) ;380: :2307–16. https://doi.org/10.1056/NEJMoa1900907. |
[77] | Fields E , Vaughan E , Tripu D , Lim I , Shrout K , Conway J , et al. Gene targeting techniques for Huntington’s disease, Ageing Res Rev (2021) ;70: :1013–85. https://doi.org/10.1016/j.arr.2021.101385. |
[78] | Boudreau RL , McBride JL , Martins I , Shen S , Xing Y , Carter BJ , et al. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice, Mol Ther (2009) ;17: :1053–63. https://doi.org/10.1038/mt.2009.17. |
[79] | Estevez-Fraga C , Rodrigues FB , Tabrizi SJ , Wild EJ Huntington’s disease clinical trials corner: April, J Huntingtons Dis (2022) ;11: :105–18. https://doi.org/10.3233/JHD-229002. |
[80] | Southwell AL , Ko J , Patterson PH Intrabody gene therapy ameliorates motor, cognitive, and neuropathological symptoms in multiple mouse models of Huntington’s disease, J Neurosci (2009) ;29: :13589–602. https://doi.org/10.1523/JNEUROSCI.4286-09.2009. |
[81] | Masnata M , Salem S , de Rus Jacquet A , Anwer M , Cicchetti F Targeting tau to treat clinical features of Huntington’s disease, Front Neurol (2020) ;11: :5807–32. https://doi.org/10.3389/fneur.2020.580732. |
[82] | Noble W , Planel E , Zehr C , Olm V , Meyerson J , Suleman F , et al. Inhibition of glycogen synthase kinase-3 by lithium correlates with reduced tauopathy and degeneration in vivo , PNAS (2009) ;102: :6990–5. https://doi.org/10.1073/pnas.0500466102. |
[83] | Serenó L , Coma M , Rodríguez M , Sánchez-Ferrer P , Sánchez MB , Gich I , et al. A novel GSK-3beta inhibitor reduces Alzheimer’s pathology and rescues neuronal loss in vivo , Neurobiol Dis (2009) ;35: :359–67. https://doi.org/10.1016/j.nbd.2009.05.025. |
[84] | Matsunaga S , Kishi T , Annas P , Basun H , Hampel H , Iwata N Lithium as a treatment for Alzheimer’s disease: A systematic review and meta-analysis, J Alzheimers Dis (2015) ;48: :403–10. https://doi.org/10.3233/JAD-150437. |
[85] | Wood NI , Morton AJ Chronic lithium chloride treatment has variable effects on motor behaviour and survival of mice transgenic for the Huntington’s disease mutation, Brain Res Bull (2003) ;61: :375–375. https://doi.org/10.1016/s0361-9230(03)00141-2. |
[86] | Danivas V , Moily NS , Thimmaiah R , Muralidharan K , Purushotham M , Muthane U , et al. Off label use of lithium in the treatment of Huntington’s disease: A case series, Indian J Psychiatry (2013) ;55: :81–3. https://doi.org/10.4103/0019-5545.105522. |
[87] | Beister A , Kraus P , Kuhn W , Dose M , Weindl A , Gerlach M The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. J Neural Transm Suppl. 2004:117-22. https://doi.org/10.1007/978-3-7091-0579-5_14. |
[88] | Cankurtaran ES , Ozalp E , Soygur H , Cakir A Clinical experience with risperidone and memantine in the treatment of Huntington’s disease, J Natl Med Assoc (2006) ;98: , 1353–5. |
[89] | Congdon EE , Sigurdsson EM Tau-targeting therapies for Alzheimer disease, Nat Rev Neurol (2018) ;14: :399–415. https://doi.org/10.1038/s41582-018-0013-z. |
[90] | Bejanin A , Schonhaut DR , La Joie R , Kramer JH , Baker SL , Sosa N , et al. Tau pathology and neurodegeneration contribute to cognitive impairment in Alzheimer’s disease, Brain.- (2017) ;140: :3286–300. https://doi.org/10.1093/brain/awx243. |
[91] | Wild EJ , Boggio R , Langbehn D , Robertson N , Haider S , Miller JRC , et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients, J Clin Invest (2015) ;125: :1979–86. https://doi.org/10.1172/JCI80743. |


