
Mutant Huntingtin Protein Interaction Map Implicates Dysregulation of Multiple Cellular Pathways in Neurodegeneration of Huntington’s Disease
Abstract
Background:
Huntington’s disease (HD) is a genetic neurodegenerative disease caused by trinucleotide repeat (CAG) expansions in the human HTT gene encoding the huntingtin protein (Htt) with an expanded polyglutamine tract.
Objective:
HD models from yeast to transgenic mice have investigated proteins interacting with mutant Htt that may initiate molecular pathways of cell death. There is a paucity of datasets of published Htt protein interactions that include the criteria of 1) defining fragments or full-length Htt forms, 2) indicating the number of poly-glutamines of the mutant and wild-type Htt forms, and 3) evaluating native Htt interaction complexes. This research evaluated such interactor data to gain understanding of Htt dysregulation of cellular pathways.
Methods:
Htt interacting proteins were compiled from the literature that meet our criteria and were subjected to network analysis via clustering, gene ontology, and KEGG pathways using rigorous statistical methods.
Results:
The compiled data of Htt interactors found that both mutant and wild-type Htt interact with more than 2,971 proteins. Application of a community detection algorithm to all known Htt interactors identified significant signal transduction, membrane trafficking, chromatin, and mitochondrial clusters, among others. Binomial analyses of a subset of reported protein interactor information determined that chromatin organization, signal transduction and endocytosis were diminished, while mitochondria, translation and membrane trafficking had enriched overall edge effects.
Conclusion:
The data support the hypothesis that mutant Htt disrupts multiple cellular processes causing toxicity. This dataset is an open resource to aid researchers in formulating hypotheses of HD mechanisms of pathogenesis.
INTRODUCTION
Huntington’s disease (HD) is a progressive neurodegenerative disease caused by CAG triplet repeat expansion mutations in exon 1 of the HTT gene that is autosomal dominant [1–6]. Neurodegeneration in the brain, and especially in striatum, causes a characteristic involuntary movement disorder, chorea, but patients also suffer from deficits in cognitive and psychiatric functions, as well as weight loss and other symptoms resulting from neurodegeneration in brain [1–8]. HD is fatal, and there is a need for drugs that prevent or delay neurodegeneration [9]. HD is a spectrum disorder whereby the CAG repeat length mutation varies among patients. HD develops in adulthood in patients with 40–50 CAG repeats, and longer expansions of more than 60 CAG repeats causes progressively younger age-of-onset and faster HD disease progression [10, 11]. Individuals with expansions between 36 and 39 may or may not develop HD [1–6, 11, 12]. The normal CAG range in humans is 15–21, with a median of 18 [13, 14].
The HTT gene mutation in HD patients was identified in 1993 [1] and has been intensely studied. Yet, gaps remain in our understanding of how the HTT gene product, the mutant huntingtin (Htt) protein, causes neuronal cell toxicity and death leading to characteristic neurodegeneration, disease symptoms, and fatality [1–6, 11, 15]. Htt is a large protein [1, 16] of approximately 3,044 amino acids with varying polyglutamine (polyQ(n)) lengths encoded by the spectrum of CAG repeats. The expanded polyQ(n) peptide sequence domain follows the first 17 amino acids at the amino terminus of Htt. An ongoing challenge in the field has been to determine the cellular functions of the Htt protein and how the expanded polyQ(n) leads to dysfunction in cells to cause repeat-dependent neurodegeneration in HD. The functions of wild-type (wt) Htt are also not fully understood. Htt is processed into multiple protein fragments that are localized to different cellular compartments and may participate in different cellular pathways [17–19].
To address the question of how mutant Htt initiates the HD disease process, researchers have sought to identify proteins that interact with Htt that may potentially reveal the molecular pathways affected by aberrant protein interactions with mutant Htt. Reports over the last 25 years have identified numerous proteins that interact with mutant Htt and wild-type (wt) Htt, involving molecular, cellular, and animal HD model systems using a variety of protein technologies to identify Htt interactors. These extensive data can provide valuable insight into potential cellular functions of the Htt protein. However, a gap in the field is that a complete dataset of all of these potential Htt protein interactors that defines the Htt lengths and polyglutamine expansion lengths of native Htt protein complexes has not yet been fully curated and analyzed. Therefore, the goals of this study were to 1) create a database of all of these Htt protein interactors reported in the literature that meet the criteria of indicating the Htt fragments lengths or full-length Htt, number of glutamines within the Htt form, and analysis of native Htt protein complexes, 2) conduct molecular network analyses to reveal potential cellular pathways of Htt protein interactors that are enriched with expanded poly(Q)n, and 3) provide this dataset and analysis to the public. Results show that both mutant and wt Htt interact with a large number of proteins (2,740 for control wt Htt, and 2,631 for mutant Htt in HD); further, a majority of these protein interactors are shared by wt and mutant Htt forms. Mutant and wt Htt protein interactors cluster into twelve gene ontology (GO) biological functions; several clusters of interactors are significantly enriched or diminished with expanded mutant Htt poly(Q)n which include protein translation, signal transduction, membrane trafficking, and chromatin organization. Assessment of cluster components by KEGG pathway analysis revealed significant inclusion of Htt interactors in functional systems of mitochondria, RNA splicing, and protein modification by the ubiquitin-proteasome pathway. These findings support the hypothesis that Htt has multiple functions in the cell that can potentially be disrupted with CAG expansion mutation of the HTT gene. This new publicly available dataset of wt and mutant Htt protein interactors can enhance future experimental investigation of the functional consequences of Htt protein interactions in the HD disease process.
METHODS
Acquisition of primary research publications
PubMed was searched for primary research articles that identified Htt interacting proteins by searching for “huntingtin protein interaction” combined with curation of individual articles for direct identification of proteins interacting with Htt protein forms. Articles were excluded if (i) the study did not evaluate a protein-protein interaction, (ii) the report was a review article and did not report original data, (iii) the article was not about HD or Htt but the article came up in the search, (iv) the article showed that a protein did not interact with Htt, (v) the study used only short synthetic motifs (such as only polyQ) but not a protein segment containing Htt primary sequences. After curation, 193 articles were compiled that investigated proteins interacting with Htt; these articles are provided as references [20–212] (in alphabetical order within the set of citations).
2.2Dataset recording
Information from the 193 articles [20–212], meeting the above criteria for studies of Htt interacting proteins, were compiled for analysis with respect to (i) the number of polyQ(n) residues in the Htt protein form used in study, (ii) the species of Htt (human, rodent, or other), (iii) whether the Htt protein utilized was full-length or a fragment, (iv) the known Htt interactor protein used as “bait” in the study if not Htt, (iv) the species of the interactor protein(s) identified, (v) the assays and techniques used to identify the protein interactor, (vi) information about the identified protein interactor with respect to its full name, official gene symbol, and accession number, (vii) how the polyQ(n) expansion affects the interaction between Htt and the interactor protein, if known, ‘+’ indicates more abundant levels interacting with mutant Htt than wt Htt, and ‘-‘ indicates less abundant levels interacting with mutant Htt than wt Htt, and ‘=’ indicates similar levels, and if blank was not determined), (viii) the age of the model organism used, (ix) article reference information, (x) the protein function assigned based on the authors’ conclusions from the study, or if not indicated (e.g., was from a large dataset) the protein function was based on general cell biological knowledge (verified by genecards [213] or uniprot [214], (xi) if the article tested functional consequences of the interaction between Htt and the protein interactor in a cellular or animal model system and the authors’ conclusion, and finally (xii) if the study tested the interaction in human HD and/or control donor cells or tissue, and the functional consequence of the interaction. This Htt protein interaction data is compiled in Supplementary Table 1 (tab for Master Table).
Clustering analysis of Htt interactors
Among reported Htt protein interactors, we sought to identify groups of proteins that shared functional categories, known as “clusters,” of different molecular and cellular pathways. To accomplish this, Htt protein interactors were integrated with the STRING human database [215] to gain information about known connections among interactors, and facilitate analysis of highly connected gene sets, representing the protein sets of this study, to determine the likelihood of an interaction (“edge”) between proteins in the dataset. STRINGdb retrieved information from these types of open source data with respect to (i) genomic/protein context predictions, (ii) high-throughput lab experiments, (iii) gene/protein co-expression, (iv) automated text mining, and (v) previous knowledge in databases. STRING edge confidence assessment selected only edges with high confidence values (>700). An Htt + STRING subnetwork was built from these edges. To allow for reliable clustering, genes were included in the subnetwork if they are in the Htt dataset and have at least 2 neighbors in the background STRING interactome. Next, we quantified structure in Htt + STRING subnetwork by applying Louvain modularity maximization [216] clustering algorithm to break up nodes into highly connected sets related to protein function. These clusters were annotated for biological function by functional enrichment with gene ontology (GO) terms [217, 218] and KEGG [219] pathways, using ToppGene [220]. Clusters were annotated with the most significantly enriched term/pathway. Htt protein interactors in each cluster is provided in Supplementary Table 1.
Select enriched KEGG pathways were examined further. Interactor proteins in significant KEGG pathways were mapped onto KEGG pathway diagrams [219]. HTT interactor proteins were color coded in these diagrams for the effect of the mutant poly(q) expansion on the interaction of red = more abundant, yellow = no reported effect, blue = less abundant, and purple = mixed reports of both more and less abundant interactions.
Data provided in approximately half of the articles tested whether the mutant Htt polyQ(n) expansion in the HD model resulted in higher or lower abundance levels of a particular protein interacting with mutant compared to wt Htt. These data were included in the Htt + STRING dataset as “edge effects.” The significance of these edge effects was evaluated within each cluster by a binomial test of the number of proteins with higher abundance interactions (n +) and lower abundance (n –). N = n + + n –, with a null hypothesis of 68% positive edges (the rate of positive edges in the full dataset). Significance is defined as p < 0.05 with Benjamini-Hochberg multiple test correction.
RESULTS
Strategy for data acquisition of Htt interacting proteins and bioinformatic evaluation
Data for Htt interacting proteins was compiled from the literature [20–212] obtained by search of PubMed for bioinformatic analyses (Fig. 1). Htt interactors were organized into a dataset that indicates the conditions of identifying interactors with respect to Htt fragment length and species, polyglutamine (polyQ(n)) length of Htt, native Htt protein complexes, model system and its age, assay type for identifying interactors, interacting proteins identified, increased or decreased interaction with mutant Htt, and references (Supplementary Table 1). The Htt interactor dataset was evaluated by bioinformatics tools of STRINGdb, gene ontology, ToppGene, and KEGG pathway mapping with statistical scoring. This unique study compiled and curated articles reporting Htt protein interactors in the literature that meet our criteria that the interactor information includes (1) the lengths of Htt fragments or full-length Htt fragment used in the protein interaction studies, (2) the number of polyglutamines in the mutant and wild-type forms of Htt, and (3) native Htt protein interaction complexes were evaluated without general cross-linking agents, since such cross-linking can include non-specific associated proteins.
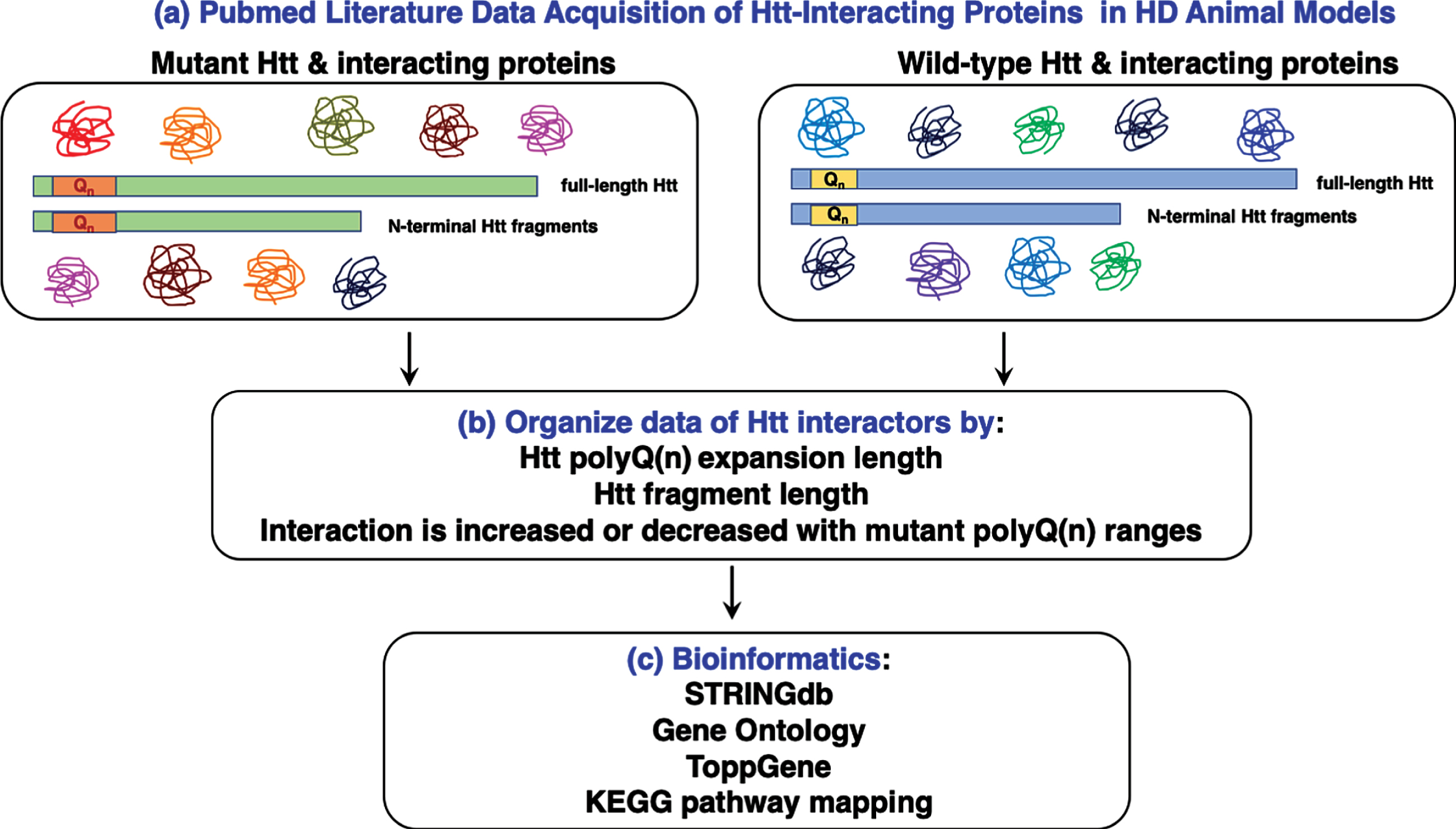
Fig. 1
Work-flow for bioinformatics analysis of huntingtin (Htt) interacting proteins reported in the literature. Literature searches for proteins interacting with mutant Htt and wt Htt were conducted with the PubMed resource (a). Information about Htt interactors was organized by experimental parameters of the reported studies, including the polyQ(n) expansion length, Htt fragment length, and whether the interaction is increased or decreased with mutant polyQ(n) Htt fragments (b). Compiled Htt interacting proteins (Supplementary Table 1) were subjected to bioinformatics analysis assessing statistically significant clusters and KEGG pathways using tools that included STRINGdb, gene ontology, and ToppGene (c).
Compilation of Htt interacting proteins of mutant Htt and wild-type (wt) Htt
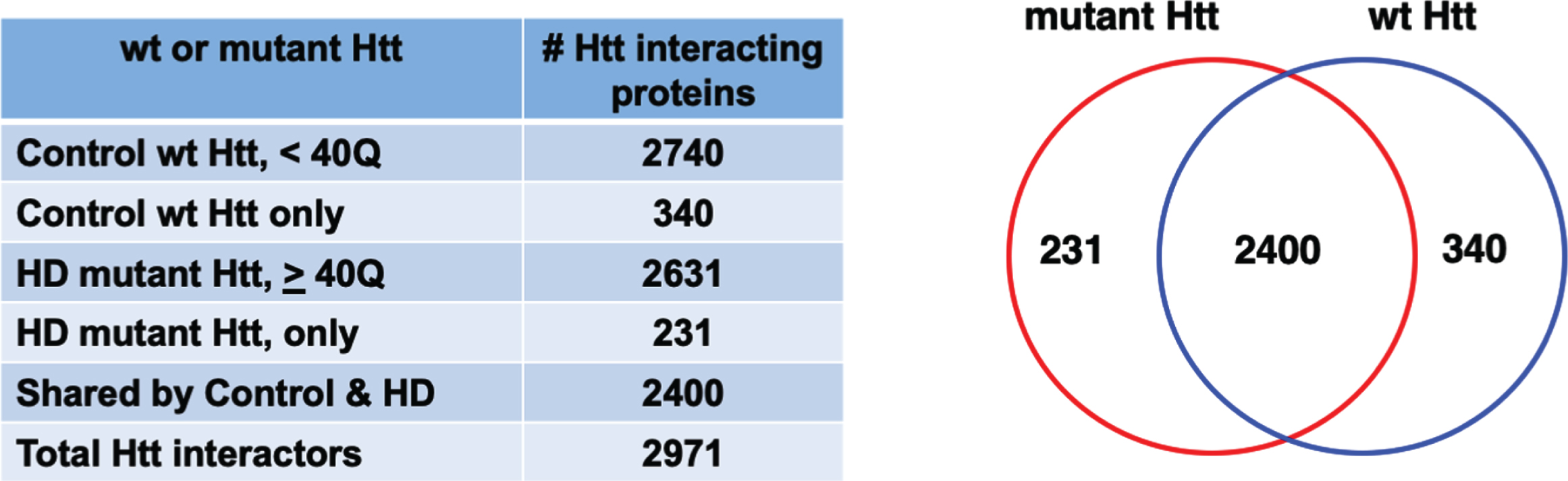
The compiled data of Htt protein interactors in HD animal models shows that a large number of proteins interact with both wild-type (wt) Htt and mutant Htt (Fig. 2). A PubMed search and curation of articles identified 193 articles that investigated non-human model systems from yeast to mammalian systems (Supplementary Table 1, Master Table). In total, 9,821 protein interactions were reported, with many protein interactors identified in multiple studies. When replicate proteins were removed, a total of 2,971 distinct Htt interacting protein identifications were found. Proteins interacting with wt Htt of < 40Q numbered 2,740, and proteins interacting with mutant Htt of ≥40 Q numbered 2,631. A major portion of the interactors were shared by wt Htt and mutant Htt, numbering 2,400 proteins. Notably, 231 interacting proteins were unique to mutant Htt which included cellular functions of transport, localization, organelles, and catalytic activities based on gene ontology (GO) analysis (Supplementary Figure 1). Furthermore, 340 interactors were unique to wt Htt which included general cellular functions of localization, vesicles, intracellular, cytosol, and enzyme binding (Supplementary Figure 1). Most prior studies have not reported data that indicated whether a protein is a primary interactor of Htt or a tertiary complex member, with the exception of proteins proposed to enzymatically modify Htt such as calmodulin (gene: CALM, [207], N-terminal acetyltransferase (gene: NAA10) [20], Tumor Necrosis Factor 6-E3 ubiquitin ligase (gene: TRAF6) [212].
Fig. 2
Protein counts for interactors of wt and mutant Htt. The numbers of distinct proteins found to interact with control wt Htt and HD mutant Htt are shown by a Venn diagram. Proteins were found that only interacted with mutant Htt or only with control wt Htt. Many proteins interacted with both wt and mutant Htt.
Parameters for identification of mutant Htt protein interactors
All studies utilized cellular, animal and in vitro non-human HD model systems to identify potential Htt interactors [20–212]. Some studies used human HD tissue or cells in follow-up experiments to confirm interactions. Studies used fragments of Htt or full-length Htt, the majority of which were cloned human or mouse amino-terminal Htt containing the polyQ(n) region (Supplementary Table 1, Master Table). Within the non-pathological human poly(Q)n range of < 40Q, the studies identified 2,740 protein interactions. Within the adult HD range of ≥40Q and < 60Q, 469 Htt protein interactors were identified (Supplementary Table 1, Table 1). In the juvenile range of ≥60Q to ≤120Q, 681 proteins were identified. In the especially long expansion range of > 120Q, 1,835 proteins were identified (Supplementary Table 1). Analysis of the distribution of Htt interacting Figure 2).
Table 1
Clusters of Htt Interactors: increased or decreased Htt interactions indicated by positive and negative edges
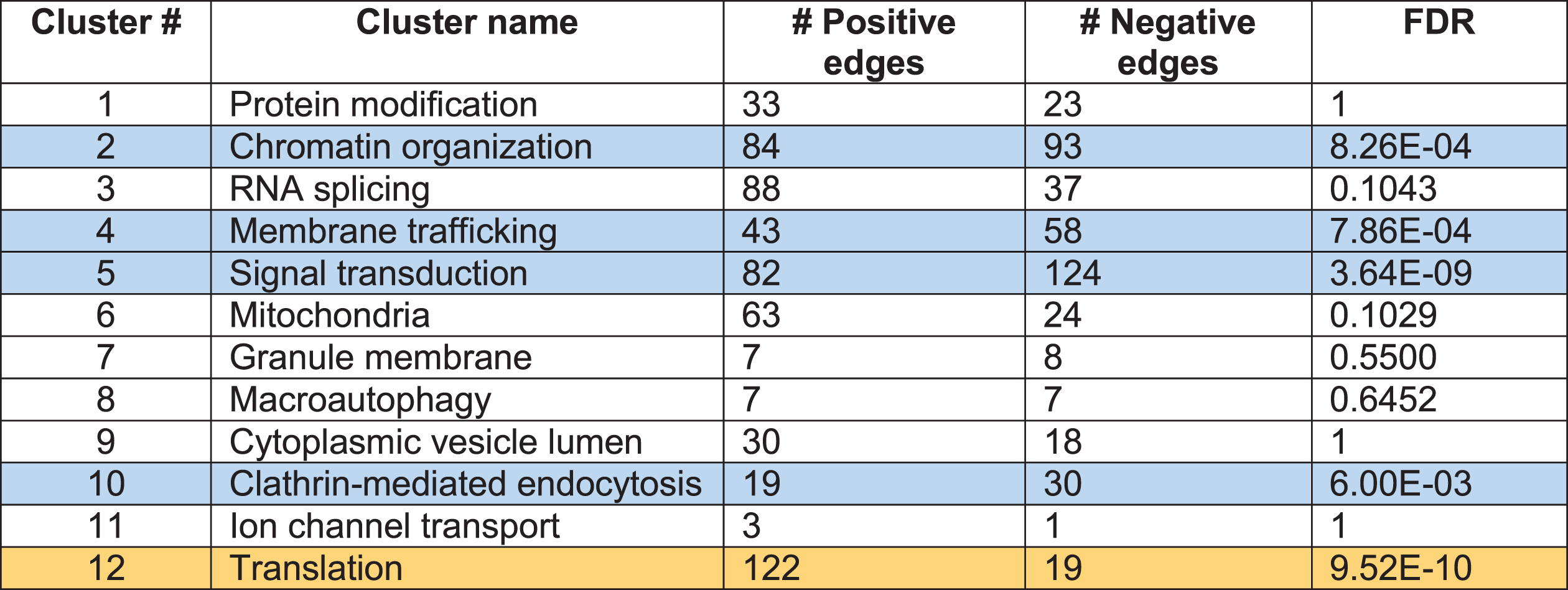 |
FDR (false discovery rate) values are corrected for Benjamini-Hochberg. Positive edge (yellow): proteins interact more abundantly with mutant Htt. Negative edge (blue): proteins interact less abundantly with mutant Htt.
While there is a clear inverse correlation in human HD age-at onset and polyQ(n) length that spans decades [11], for practical experimental designs, researchers used cellular models (age of < 1 week), or animal models with ages of 1 day (Drosophila larvae) to 24 months (transgenic and knockin mice) (Supplementary Table 1, Master Table). Animal model studies showed neurodegenerative and behavioral phenotypes with expression of mutant Htt, e.g., retinal degeneration in developing Drosophila, motor impairment in C. elegans, and motor and behavioral dysfunction and HD-like neuropathology in transgenic mice.
A variety of approaches were used to test Htt protein interactions using either hypothesis-driven or unbiased identification experimental designs. Hypothesis-driven studies tested approximately 10 or fewer potential Htt protein interactors, while unbiased approaches using Htt protein complex purification and mass spectrometry sequencing identified as many as approximately 1,000 proteins [16, 52, 87, 97, 140, 152]. Yeast 2-hybrid studies identified up to approximately 150 proteins [74, 155]. Approximately half of all potential Htt protein interactors were identified in studies that utilized strategies to determine whether proteins interacted at higher or lower levels with mutant compared to wt Htt (Supplementary Table 1, Master Table). Such results showed whether a protein interactor was present at higher or lower abundance in mutant Htt complexes, e.g., by a darker or lighter band on western blot, or by detection of higher or lower levels of a protein by mass spectrometry. These data were used in our study to determine significant enrichment of gene ontology (GO) clusters in mutant Htt compared to wt Htt.
Clustering analysis correlates Htt protein interactors with diverse cellular functional roles that may be enriched or diminished in HD-like phenotypes
We hypothesized that Htt protein interactors could be statistically sorted into functional categories to reveal information about the types of molecular pathways in the cell that are dysfunctional in HD, ultimately leading to cell toxicity and death. Accordingly, the list of Htt protein interactors was integrated with the STRING human database [215] (Search Tool for the Retrieval of Interacting Genes/Proteins) to gain knowledge of potential interactions (“edges”) between proteins. To allow for reliable clustering, genes were included in the subnetwork if they are in the Htt protein interactor dataset and have at least two neighbors in the background STRING interactome.
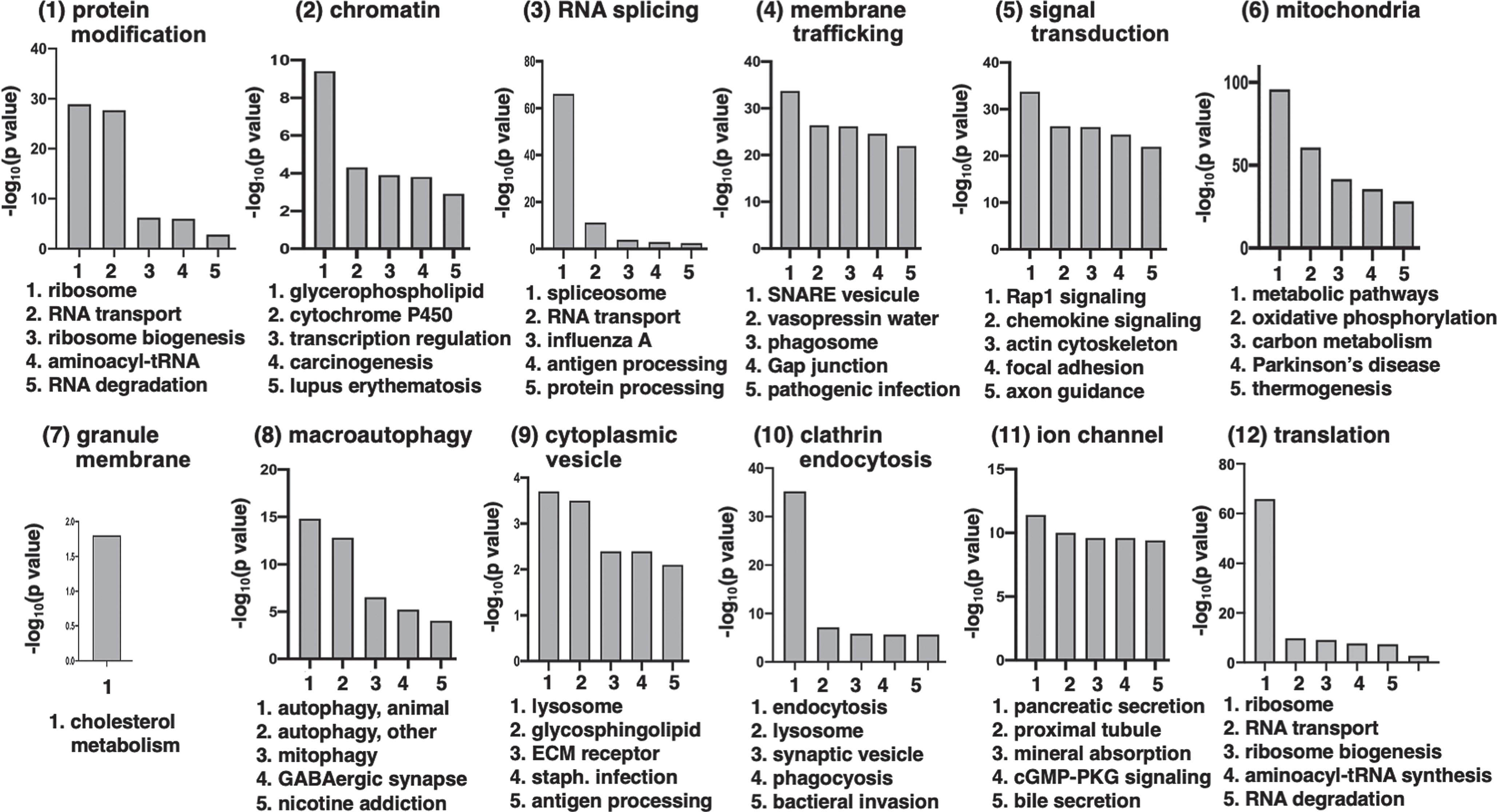
We then quantified structure in Htt + STRING subnetwork by applying Louvain modularity maximization clustering algorithm [216] to break up nodes into highly connected groups. Louvain modularity maximization is an optimized algorithm that runs on a scale of –1 to 1 to determine the density of connections of possible clusters as the algorithm runs. Modularity is designated as the variable Q, the density of connections inside the community compared to links between communities. The resulting clusters are shown in Fig. 3 for mutant Htt and wt Htt. The twelve clusters indicated functions of 1) protein modification, 2) chromatin organization, 3) RNA splicing, 4) membrane trafficking, 5) signal transduction, 6) mitochondria, 7) granule membrane, 8) macroautophagy, 9) cytoplasmic vesicle, 10) clathrin-mediated endocytosis, 11) ion channel transport, and 12) translation. Several of these pathways have been investigated in the field for HD cell death mechanisms including mitochondria, macroautophagy, protein modification, and protein translation which significantly clustered in our analysis.
Fig. 3
Clustering analysis of Htt interactors by gene ontology (GO). Htt interactors were analyzed for shared functional categories, “clusters,” in different cellular molecular pathways. The cluster networks are illustrated in color-coded format (see key in figure) for interactions that were lost by mutant Htt (mHtt), weaker with mHtt, stronger with mHtt, gained interaction with mHtt, or represented a neighbor.
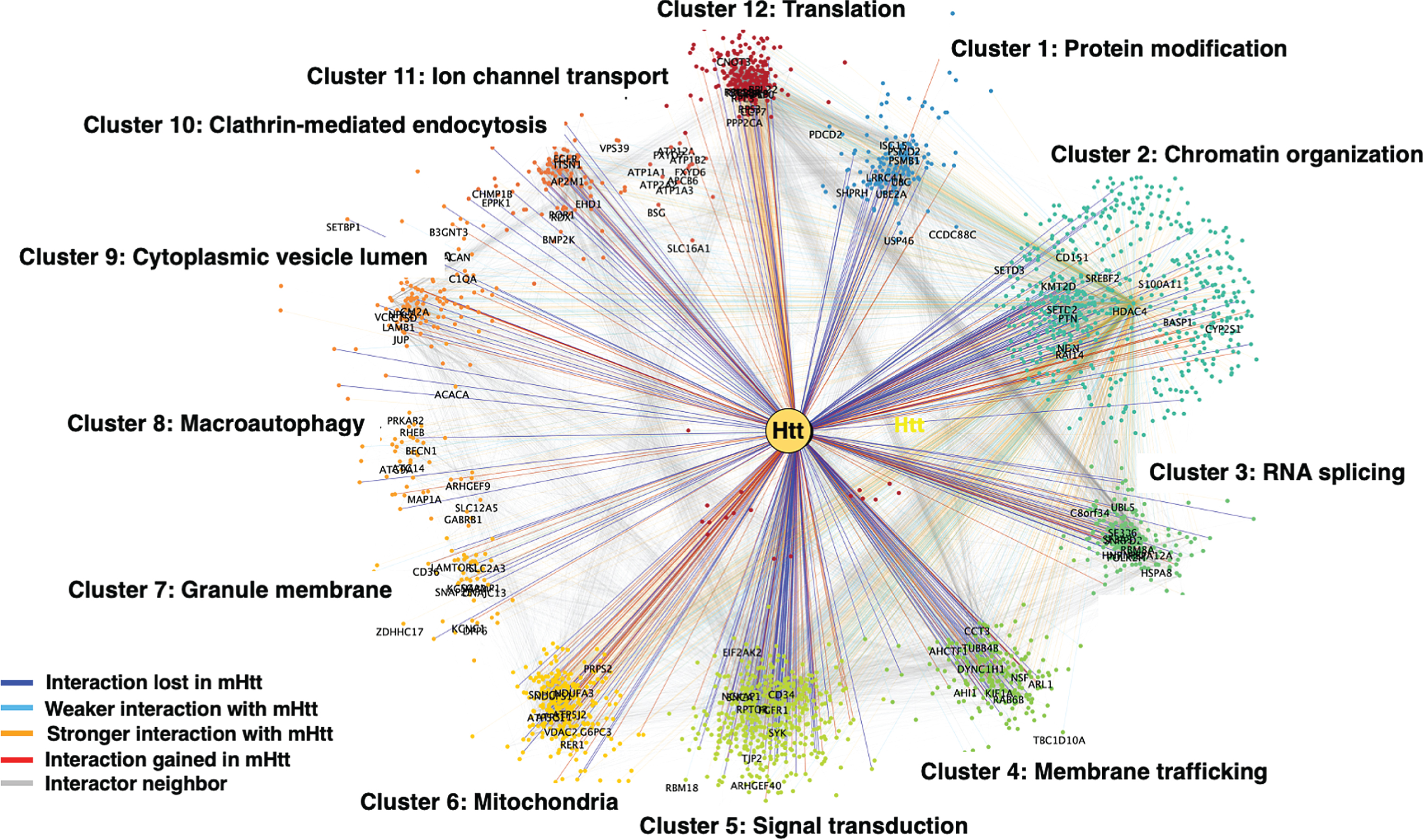
In the studies that reported differing levels of a protein interacting with mutant Htt compared to wt Htt, we conducted a statistical test for proteins within each cluster in the mutant Htt interactor dataset to determine whether an interactor protein interacted with mutant Htt at significantly greater or lower abundance levels, that is, if a protein was reported to interact with higher or lower abundance with mutant Htt, or the interaction was lost or gained compared to wt Htt protein interactors. We analyzed these data with a binomial test (N = n + + n –) for each cluster to determine the probability of observing n + higher abundance or n – lower abundance interactions in the mutant Htt interactor dataset out of the total interactions, N in each cluster. Results (Table 1) showed that the function of translation was significantly enriched in mutant Htt interactions, while signal transduction, chromatin organization, membrane trafficking, and clathrin-mediated endocytosis clusters were significantly diminished.
Clustered Htt interacting protein identifications achieved by multi-disciplinary techniques
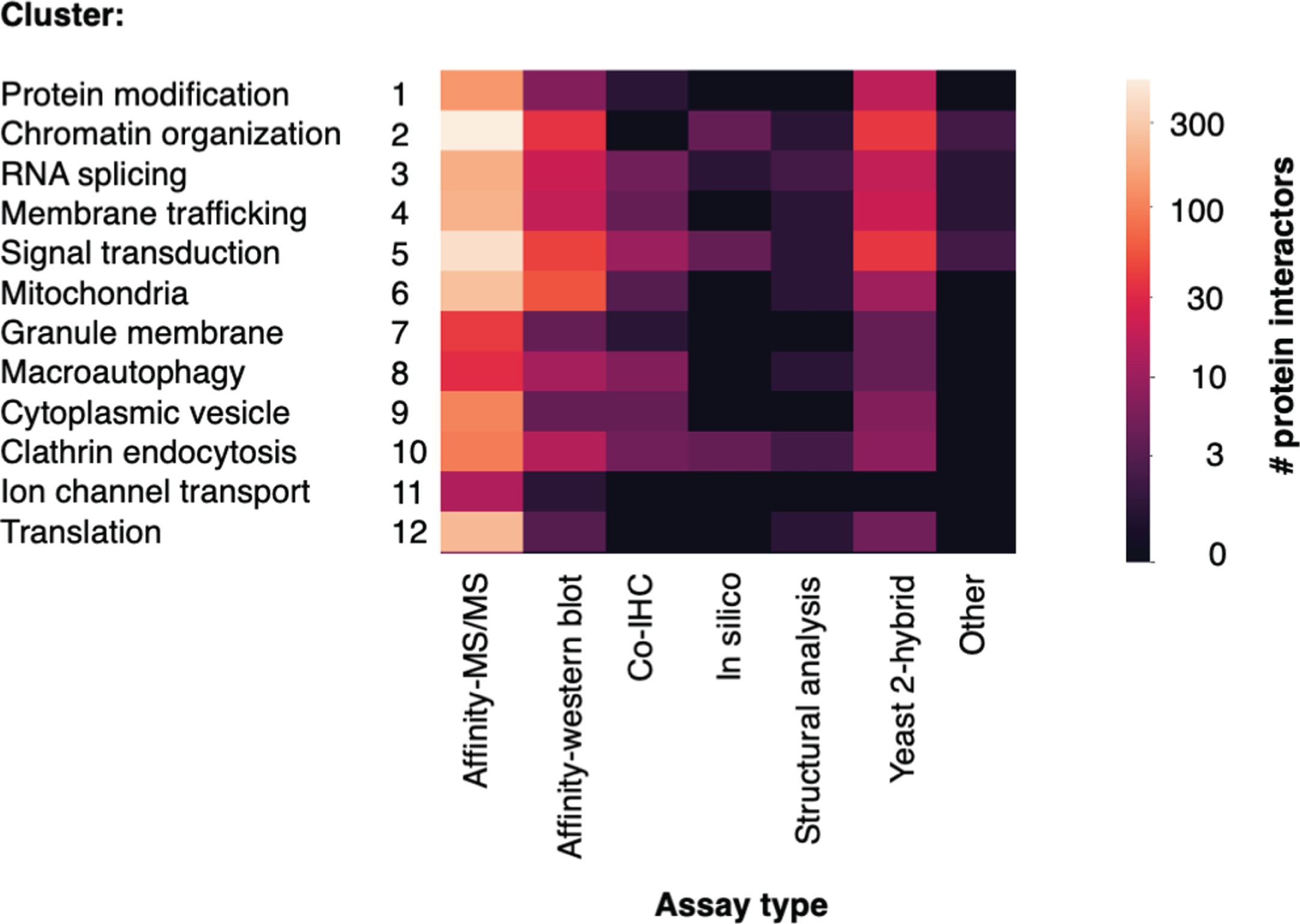
Various assay methods and techniques were used to identify Htt interactor proteins. The number of interactor proteins in each cluster identified by assay type are displayed in a heatmap (Fig. 4). Larger numbers of proteins were identified by high-throughput approaches, particularly methods that purified Htt complexes from tissue or cell lysate, followed by identification of interactors by mass spectrometry (Affinity-MS/MS) and yeast 2-hybrid. Other assays identified interactors by hypothesis-driven approaches; numerous articles utilized immuno-affinity purification of Htt complexes, followed by western blot to detect a proposed interactor. Additional studies used co-immunohistochemistry to detect physical proximity between Htt and an interactor protein. Fewer articles utilized in silico or structural analysis approaches, or other assay types. Higher numbers of interactors were identified by affinity-MS/MS, yeast 2-hybrid, and affinity-western blot assays in the chromatin organization, signal transduction, mitochondria, and translation clusters.
Fig. 4
Number of Htt interactors identified by assay type for each cluster. The identification of Htt interactors within each cluster by the type of experimental assay is illustrated. The number of interactors determined according to assay types are color-coded (see key).
Htt poly(Q)n repeat lengths used for identification of clusters of Htt interacting proteins
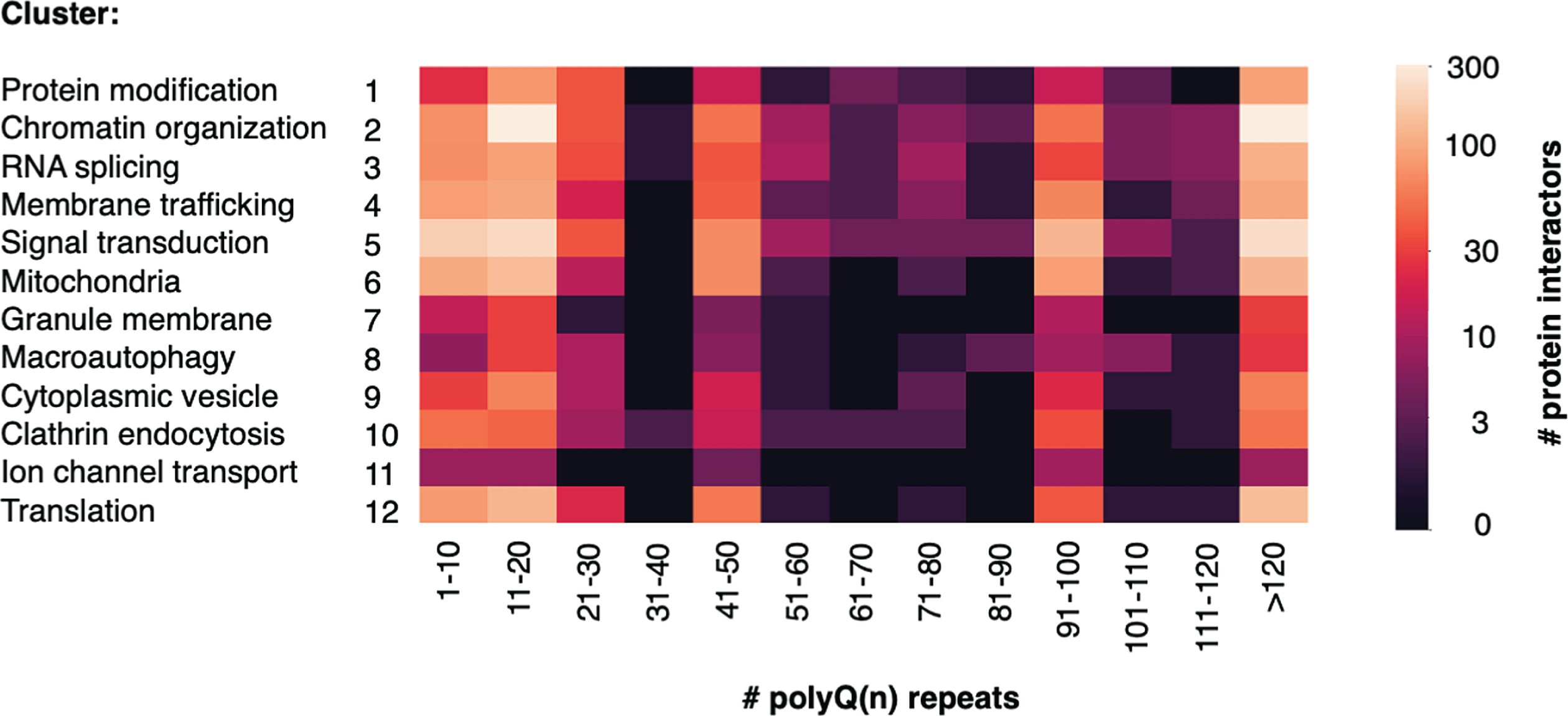
We next queried the data to determine how many proteins in each cluster were identified by varying polyQ(n) repeat length, assessed in lengths of increasing 10Q (Fig. 5). The largest numbers of proteins were identified in the wt range of < 30Q, and in the very high juvenile range of > 90Q. Notably, fewer interactor proteins were identified with polyQ(n) Htt lengths found in human adult HD of 40–60Q and juvenile HD of 60–120Q, likely due to fewer studies using Htt with these ranges of polyQ(n). HD model systems, particularly genetic mouse models, frequently used especially high polyQ(n) repeat lengths to induce HD pathology and phenotypes within the lifespan of a mouse of less than two years [52, 97, 167]. It will be of interest in future studies to identify Htt interactors within the human Htt polyQ(n) range in HD because proteins that interact with very high polyQ(n) mutant Htt may or may not interact with mutant Htt present in human HD brain.
Fig. 5
Number of Htt interactors identified by Htt polyQ(n) length within each cluster. The number of polyQ(n) expansions in Htt used for identification of Htt interactors within each cluster category is indicated. Low to high numbers of interacting proteins identified are color-coded (see key).
It was also found that the majority of studies used N-terminal fragments of mutant Htt and wt Htt containing the polyQ(n) domain for determination of Htt protein interactors (Supplementary Figure 2). Models using N-terminal fragments of 171 amino acids in length were prevalent; the 171 residue fragment corresponds to exons 1–3 [1, 221].
Cellular pathways within mutant Htt interactor clusters reveal potential molecular toxicity mechanisms
The most significant gene ontology (GO) pathways within each cluster were identified by ToppGene [220] with statistical evaluation. All significant pathways for each cluster are provided in Supplementary Table 1 (Master Table, tabs for clusters 1–12). Kyoto Encyclopedia of Genes and Genomes (KEGG) curates proteins in well-defined cellular pathways [219]. The significant GO terms for each cluster were filtered for KEGG pathways. A summary of the top KEGG pathways for each cluster is provided in Supplementary Table 2. The most significant KEGG pathways for the clusters of translation, signal transduction, chromatin organization, and membrane trafficking are described with respect to Htt interactor components below.
Protein translation
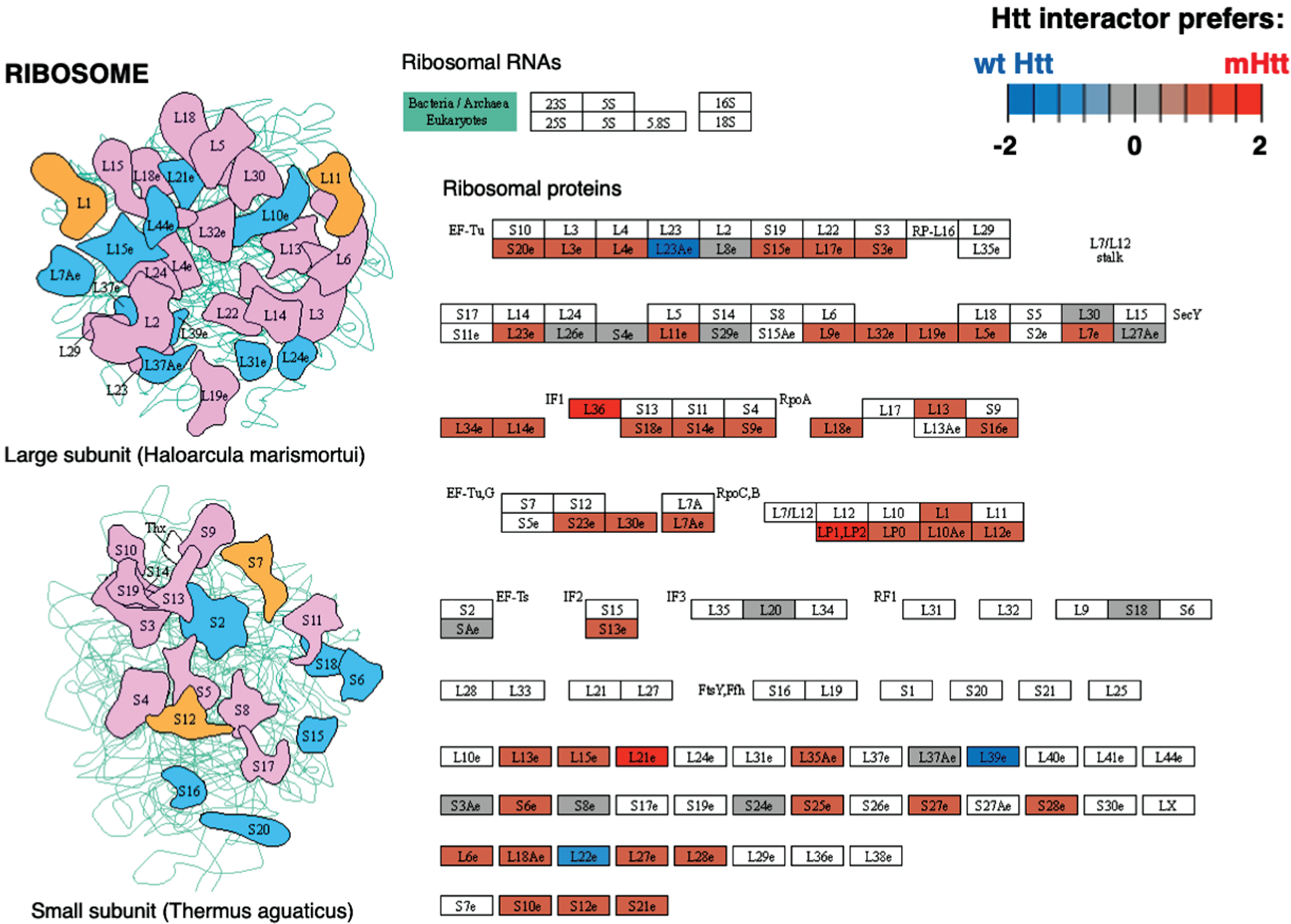
Cluster 12 for translation was identified by binomial analysis as the cluster with the most number of positive edges (Table 1). Ribosome was the most significant pathway within the translation cluster (Supplementary Table 2), meaning that protein interactors in this cluster overall had more abundant interactions with mutant compared to wt Htt (Fig. 6). Elongation factors within both the large and small subunits and aminoacyl tRNA transferases interact at higher levels with mutant compared to wt Htt. These results are consistent with a study showing that mutant Htt impedes ribosomal translocation during translation elongation and suppresses protein synthesis [222]. These data together indicate the ribosome as potentially impacted through mutant Htt protein interactions.
Fig. 6
Ribosome KEGG pathway of the translation cluster. The Ribosome pathway represented the most significant pathway of the translation cluster (Supplementary Table 2). Preferred interactions of mutant Htt (mHtt) or wt Htt with pathway components are illustrated by red to blue color, respectively (see color key). Proteins that were more abundant interactors of mutant Htt or gained mutant Htt interactors are shown in red shades, while blue shades indicate proteins with less abundant interactors with mutant Htt or the interaction is lost; gray shades indicate similar levels of more or less abundant interactions.
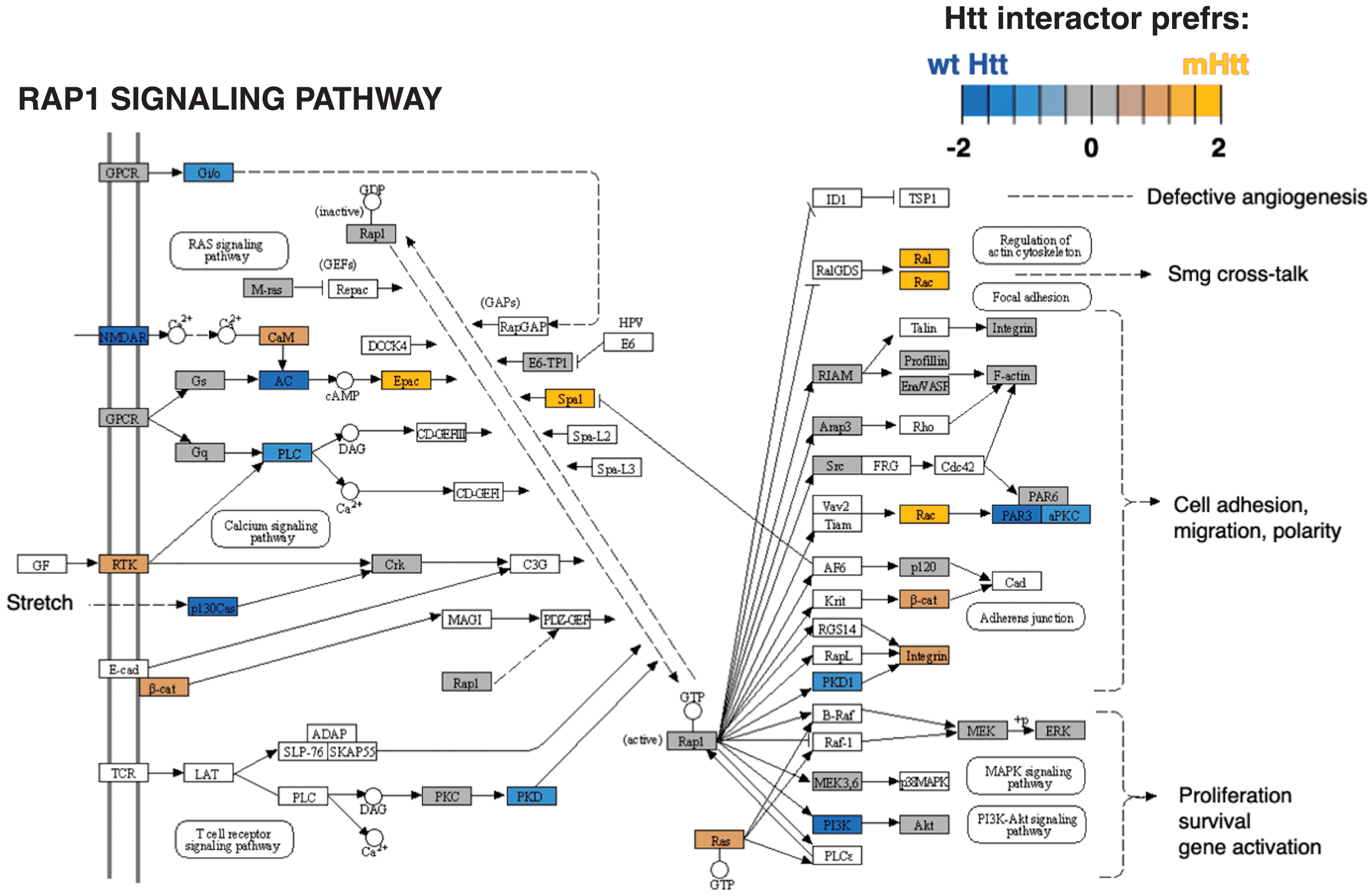
Signal transduction
Cluster 5 for signal transduction had significantly more negative edges as determined by binomial analysis (Table 1), indicating that protein interactors in this cluster overall had less abundant interactions with mutant Htt compared to wt Htt. The most significant KEGG pathways for cluster 5 (Supplementary Table 2) was the RAP1 signaling pathway (Fig. 7). The RAP1 signaling pathway has not been described as a pathway of cell toxicity and death in HD. Future investigations of this pathway mechanism may reveal new disease mechanisms and potential therapeutic targets.
Fig. 7
Rap1 signaling KEGG pathway of the signal transduction cluster. The Rap1 signaling pathway represented the top significant pathway of the signal transduction cluster (Supplementary Table 2). Preferred interactions of mutant Htt (mHtt) or wt Htt with pathway components are illustrated by the red to blue color color key.
Chromatin organization
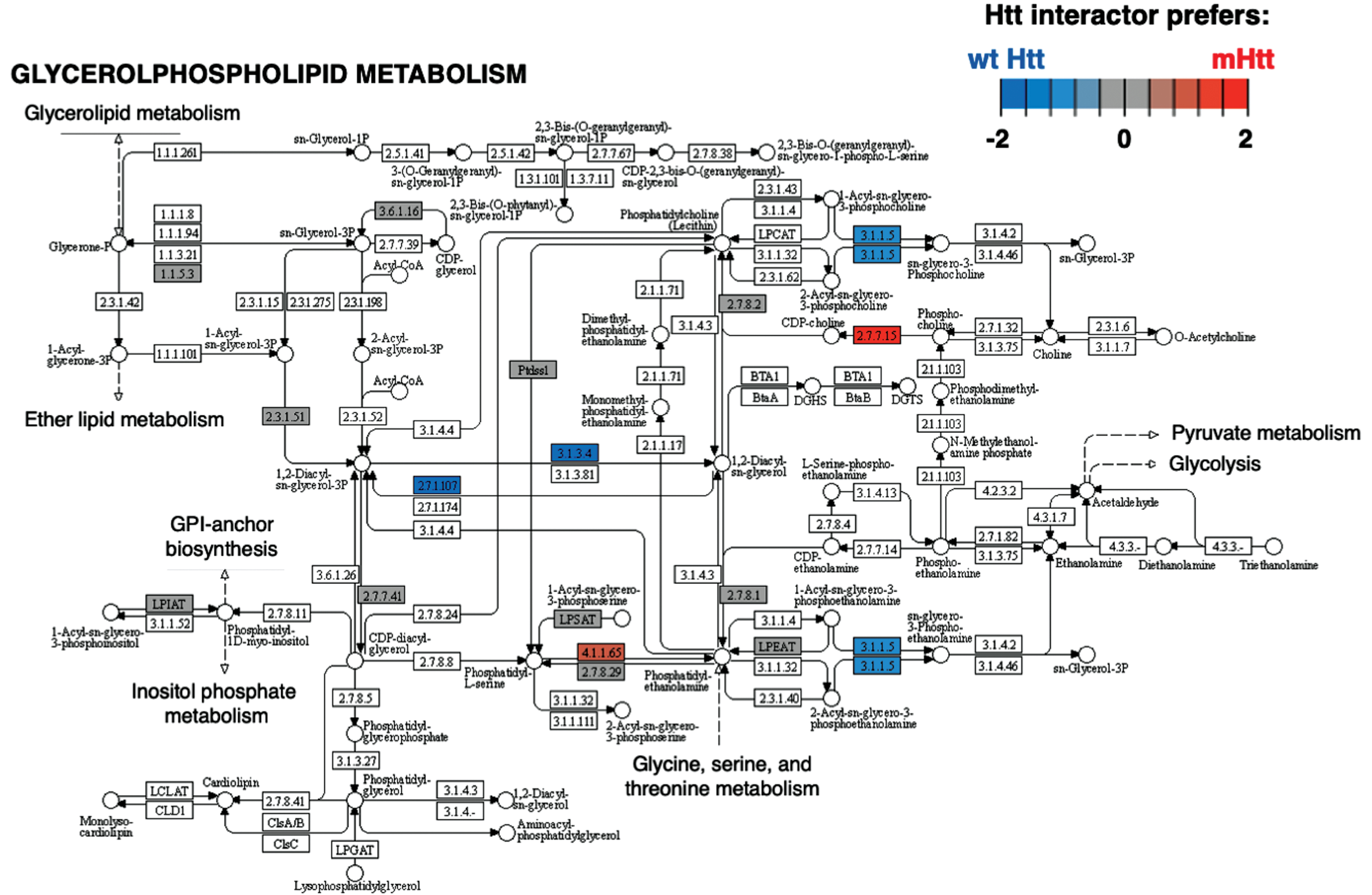
Cluster 2 for chromatin organization had significantly more negative edges according to binomial analysis (Table 1). The most significant KEGG pathways within this cluster (Supplementary Table 2) is glycerophospholipid metabolism (Fig. 8). Mutant Htt has aberrant interactions with glycerophospolipids, and biosynthetic and metabolic processes are disrupted [223]. Further investigations into Htt interacting proteins in glycerophospholipid pathways could hold promise for better understanding of mutant Htt associated membrane disruption.
Fig. 8
Glycerophospholipid metabolism KEGG pathway of the chromatin cluster. The Glycerophospholipid metabolism pathway of the chromatin organization cluster was found to be most significant (Supplementary Table 2). Preferred interactions of mutant Htt (mHtt) or wt Htt with pathway components are illustrated by red to blue color key.
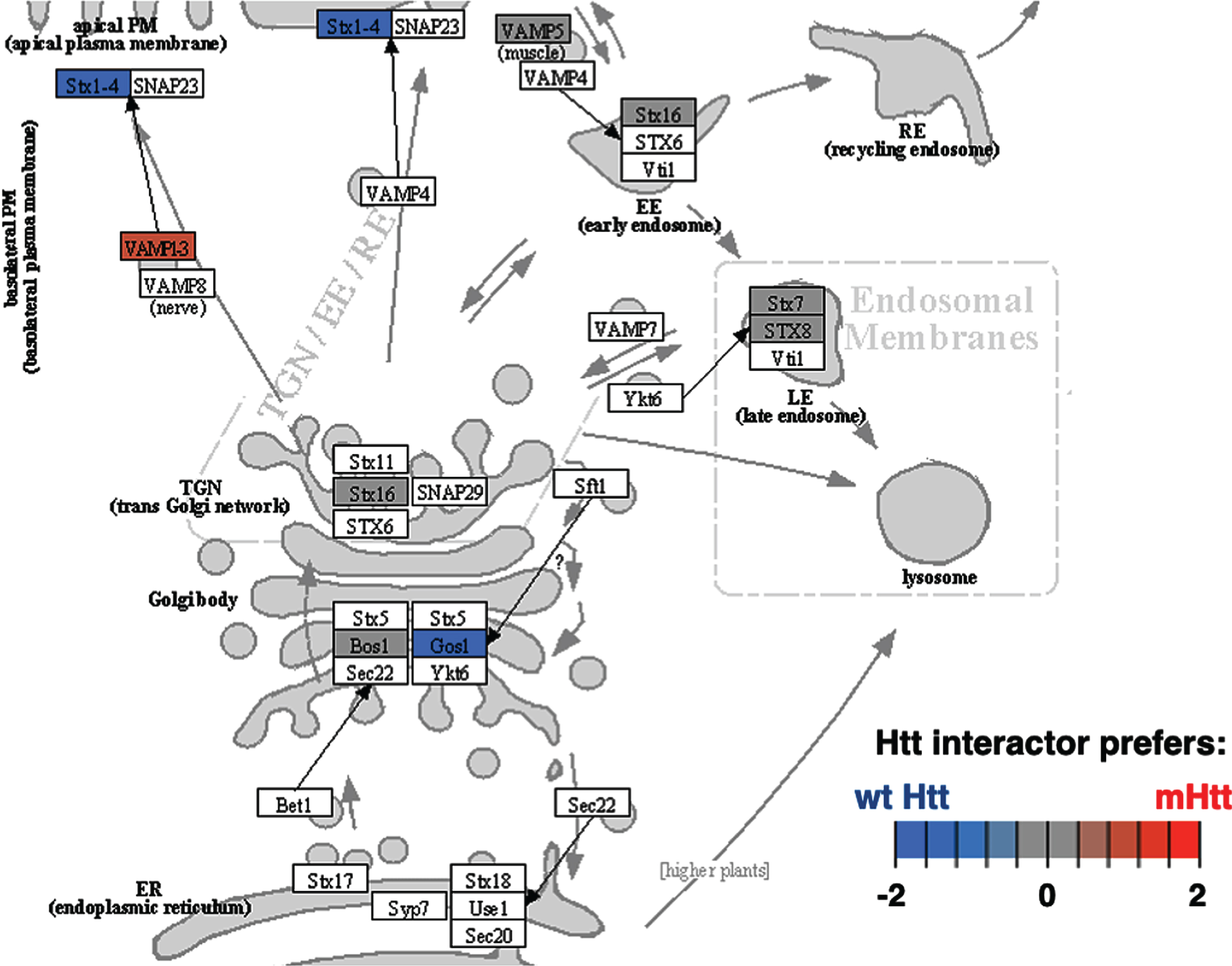
Membrane trafficking
Cluster 4 for membrane trafficking also had significantly more positive edges (Table 1). The most significant KEGG pathway of cluster 3 was SNARE interactions in vesicular transport (Fig. 9 and Supplementary Table 2). Disruption of SNARE proteins in astrocytes by mutant Htt cause impaired gliotransmission associated with behavioral disruptions in HD model mice [224]. Evaluation of Htt protein interactors associated with SNARE vesicular transport may reveal key mechanisms underlying psychiatric and behavioral changes in HD.
Fig. 9
SNARE interactions in vesicular transport KEGG pathway of the membrane trafficking cluster. The SNARE interactions for vesicular transport of the membrane trafficking cluster was highly significant with respect to interactions with Htt (Supplementary Table 2). Preferred interactions of mutant Htt (mHtt) or wt Htt with pathway components are illustrated by red to blue color key.
Significant KEGG pathways of cluster functions of Htt protein interactors
Mitochondria
Among KEGG pathways assessed, oxidative phosphorylation of mitochondria represents a highly significant cellular system of Htt interacting proteins (Fig. 10 and Supplementary Table 2). Among 71 components of the oxidative phosphorylation KEGG pathway, 70 of these proteins were found as Htt interactors. Mutant Htt forms displayed preferences for interacting with components of oxidative phosphorylation mechanisms of mitochondria. Htt interactors of the mitochondria cluster were also represented by KEGG pathways of metabolism, carbon metabolism, and thermogenesis with oxidative phosphorylation as the top pathways in this cluster (Fig. 11 and Supplementary Table 2). Mitochondrial dysfunctions that compromise energy production are consistent with weight loss that occurs in HD [11].
Fig. 10
Oxidative phosphorylation KEGG pathway of the mitochondrial cluster. The oxidative phosphorylation pathway of mitochondria was of high significance for interactions with Htt (Supplementary Table 2). Preferred interactions of mutant Htt (mHtt) or wt Htt with pathway components are illustrated by red to blue color key.
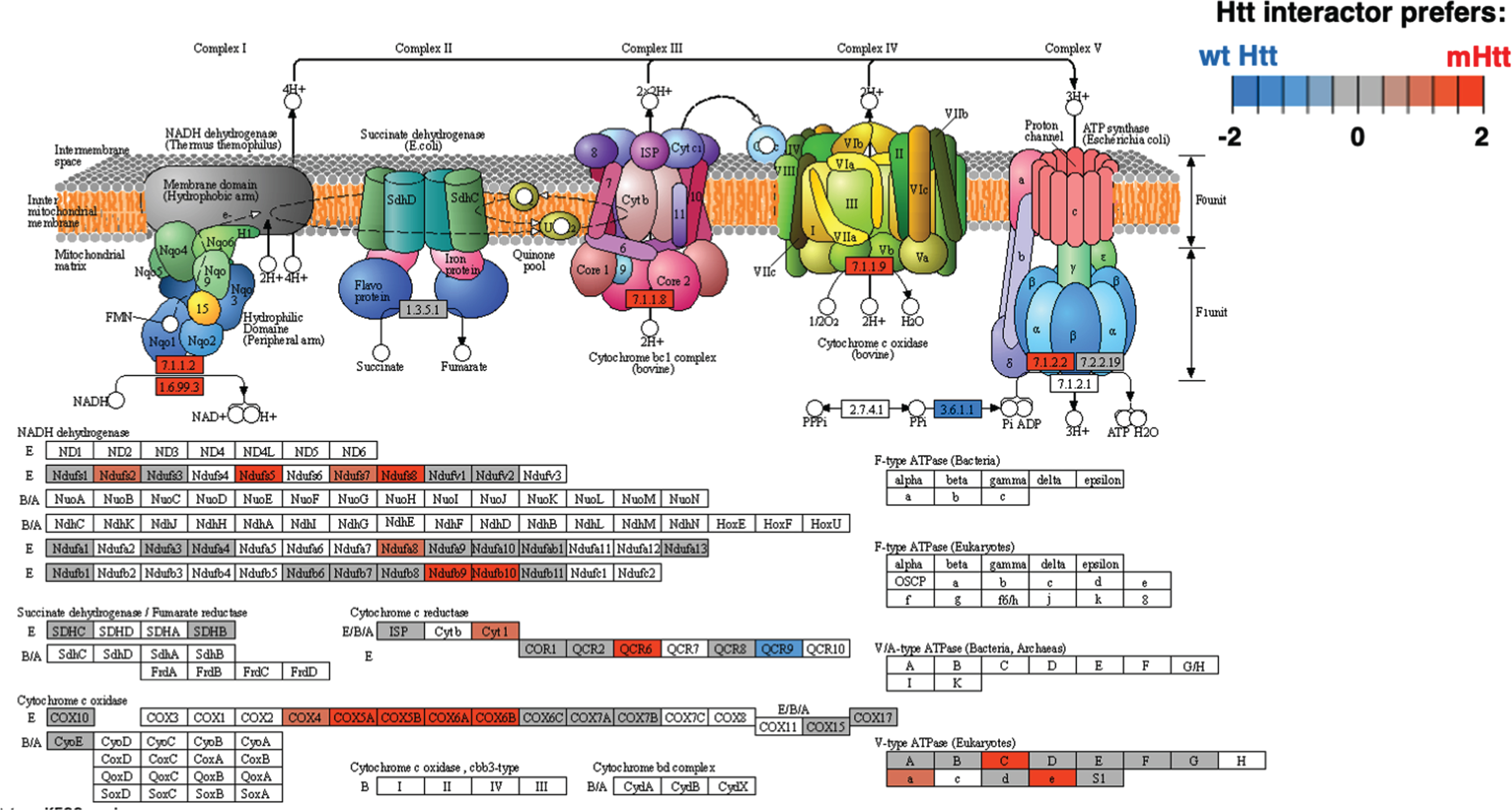
Fig. 11
Top significant KEGG pathways within each cluster of Htt interactor functions. The top five most significant KEGG pathways for each of the 12 clusters of Htt interactor functions are illustrated. The 12 clusters are shown in panels 1–12, respectively, with graphs of the most significant KEGG pathways by -log10(p value). These significant p values are well below the significance level of p < 0.05, which corresponds to -log10(p value) of greater than 1.3.
RNA splicing and protein modification
The RNA splicing cluster (#3) of Htt interactors includes significant KEGG pathways of the spliceosome and RNA transport (Fig. 11 and Supplementary Table 2). Dysregulated RNA splicing occurs in human HD brain and in HD models and variant splicing of human HTT RNA has been observed [225, 226].
The protein modification cluster of Htt interactors (cluster 1 shown in Fig. 11) contains significant KEGG pathways for ubiquitin-mediated proteolysis and the proteasome (Supplementary Table 2). Dysregulation of the ubiquitin-proteasome system in HD participates in the clearance of unwanted proteins, resulting in altered protein homeostasis [227, 228].
Htt interactor dataset available to the public
This dataset of Htt interactor proteins has provided a comprehensive dataset of published reports of wt and mutant Htt protein interactors in non-human HD model systems (provided in Supplementary Table 1) with information on length of Htt fragment or full-length Htt, length of poly-glutamine within Htt, and use of native Htt protein complexes. This Htt interactor data set is publicly available at the website shown at the end of the discussion. A variety of non-human model systems and assays were used to determine potential Htt protein interactors, and the systems used did not affect the associated functions of the proteins identified. Bioinformatics analyses reveal that significant modularity clusters in functional classes of protein translation, signal transduction, chromatin organization, mitochondria, and others that may be enriched or diminished with polyQ(n) expansion in mutant Htt. Access to these data will allow researchers in the field to formulate hypotheses and plan studies to investigate the key roles of Htt protein in HD cellular mechanisms of toxicity and cell death.
DISCUSSION
This study achieved collation and statistical bioinformatics analysis of published Htt protein interactors identified in non-human HD model systems that meet the criteria that the studies included 1) defining the lengths of Htt fragments or full-length Htt fragment used in the interaction studies, 2) indicating the number of poly-glutamines in the mutant and wild-type forms of Htt, and 3) assessment of native Htt protein interaction complexes without cross-linking agents, which avoids general cross-linking that may include non-specific associated proteins. A high number of proteins that potentially complex with wt Htt were identified as 2,740 interactors, and with mutant Htt that were identified as 2,631 interactors (Fig. 2). The majority of interactors were shared, 2400 proteins, by the wt Htt and mutant Htt groups. Htt is a large protein of 3044 amino acids whose N-terminal region contains the polyQ(n) domain encoded by the CAG repeat expansion of the human HTT gene. The exact function of Htt is under much investigation, but the data compiled here indicate diverse roles in cellular pathways. In the human brain, Htt is processed into multiple endogenous fragments [17] and each may have distinct cellular processes, although localization to specific cellular compartments and organelles is not known. The presence of the mutant polyQ(n) does not substantially affect the molecular weights of fragments outside of the mutation, although some fragments are present at higher levels in the human brain and are different in motor cortices compared to striatum [17]. Amino terminal regions of mutant Htt, containing the expanded polyQ(n) have been identified as diffuse and aggregated forms of nuclear, cytoplasmic and synaptic distribution [229–233]. Whether the expanded polyQ(n) fragment in particular initiates toxic sequalae or in addition to other endogenous fragments is an area of ongoing investigation.
These data compiled in this study provide a valuable resource to the field for formulating hypothesis related to HD pathogenesis and molecular mechanisms. There is currently a great need for effective drugs to slow or stop the neurodegeneration in HD. Therapeutic targeting of Htt interactor proteins are a potential strategy for drug development.
We note that recent articles recently reported data sets of Htt protein interactors but they did not meet our criteria for including information on Htt fragment length, number of polyglutamine residues in the expansion, and assessment of native Htt complexes [234–236] Articles by Aaronson et al. (2021) [234] and Haenig et al. (2020) [235] did not define Htt fragment lengths or number of Htt polyglutamines for all Htt interactors compiled. Another article by Sap et al. (2021) [236], used a general cross-linking agent formaldehyde to isolate Htt complexes, but formaldehyde is a general cross-linking agent that can include non-specific associated proteins. Therefore, information from these three articles were not included in this study.
Binomial analysis of edge effects found that Htt interacting proteins involved in translation were significantly enhanced, while signal transduction, chromatin organization, membrane trafficking, and clathrin-mediated endocytosis functions were significantly diminished in mutant Htt compared to wt. These results suggest diverse roles of Htt in different cell compartments. Among these clusters, significant KEGG pathways of the ribosome, Rap 1 signaling, glycerophospholipid metabolism, and SNARE interactions in vesicular transport were indicated. Among these pathways, glycerophospholipid synthesis and metabolism dysfunction was found to be dysregulated in HD genetic mouse models [237].
All studies utilized animal or cellular model systems to identify interactors. HD human brains have not been used as a starting material to identify interactors, likely due to the limited availability of such tissue. While some studies utilized Htt with polyQ(n) expansions in the adult human HD range (40Q - 50Q) and juvenile HD range (60Q - 100Q), the majority utilized especially high expansions (>100Q). There is a particular deficit in the reduced penetrance range, 30Q - 40Q. A small subset of articles reported Htt protein-protein interactions in human brain (Supplementary Table 1, Master Table) in follow-up studies subsequent to interactor identification in an HD model. Because some Htt interactors may interact at artificially high polyQ(n) ranges but not in the human HD range (40Q - 100Q), future studies should aim to use the ‘human’ range of polyQ(n).
Disruption or enhancement of key mutant Htt protein interactions is a logical approach to identify and test potential therapeutic targets in experimental systems for reduction of mutant Htt mediated toxicity and phenotypes. HD genetic mouse models expressing polyQ(n) expanded human Htt have HD-like neuropathology, motor dysfunction and cognitive behavioral phenotypes [238, 239]. Some articles in the compiled dataset tested the consequence of mutant Htt interactors on mouse model HD-like phenotypes (Supplementary Table 1, Master Table). All of these protein interactors were chosen by the authors based on hypothesis driven approaches and demonstrate improvement or exacerbation of HD-like pathology as a consequence of mutant Htt protein interactions. Results of these studies illustrate that aberrant mutant Htt protein interactions cause HD-like phenotypes in mouse genetic models.
The reported Htt interactors in animal model systems can be fruitful for understanding possible mechanisms of mutant Htt in the human disease process. We, therefore, propose the strategy that this list of candidate Htt interactors from animal model systems may be assessed in human HD brain tissues using stringent and rigor in the experimental design to elucidate the different clusters/pathways that may be present in human HD brain. Such future studies will be of value to understand mutant Htt protein interaction mechanisms in the human HD brain.
In summary, this study provides the Htt interacting protein dataset resource to the field that may enhance formulation of hypotheses and designing experiments to evaluate Htt initiation of neurodegenerative cellular pathways via Htt protein interactors. The key unknown mechanistic question of HD pathogenesis is why neurons become toxic and die because of the expanded polyQ(n) mutation. Our clustering and pathway analyses suggest protein interactions in significant GO biological processes for pharmacological or genetic (e.g., siRNA) intervention in model systems. While drugs are available for HD patients that may manage symptoms, there is a great need for development of drugs to delay or prevent fatal neurodegeneration and emaciation. Investigation of the key pathways related to HD-like phenotypes determined by statistical analysis in this study may prove to be effective therapeutic targets.
ACKNOWLEDGMENTS
This research was supported by grants from the Huntington’s Disease Society of America (HDSA) awarded to S. Podvin, C. Mosier, and V. Hook. This project was partially supported by the National Institutes of Health, grant UL1TR001442 of CTSA. The content is solely the responsibility of the authors and does not necessary represent the official view of the NIH. Review by our Institutional Review Board has indicated that for this project use of unidentified human tissue samples is exempt,
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
This compilation of Htt interacting proteins is provided to the public via the location of https://www.ndexbio.org/viewer/networks/8acef3e9-ccdc-11e8-aaa6-0ac135e8bacf
The data is also provided in Supplementary Tables 1 and 2.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JHD-220538.
REFERENCES
[1] | The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. (1999) ;72: :971–83. doi: 10.1016/0092-8674(93)90585. |
[2] | Vonsattel JP , DiFiglia M , Huntington disease, J Neuropathol Exp Neurol (1998) ;57: :369–84. |
[3] | Petersén A , Mani K , Brundin P , Recent advances on the pathogenesis of Huntington’s disease, Exp Neurol (1999) ;157: :1–18. doi: 10.1006/exnr.1998.7006. |
[4] | Ross CA , Tabrizi SJ , Huntington’s disease: From molecular pathogenesis to clinical treatment, Lancet Neurol (2011) ;10: :83–98. doi: 10.1016/S1474-4422(10)70245-3. |
[5] | Zuccato C , Valenza M , Cattaneo E , Molecular mechanisms and potential therapeutical targets in Huntington’s disease, Physiol Rev (2010) ;90: :905–81. doi: 10.1152/physrev.00041.2009. |
[6] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al., Huntington disease, Nat Rev Dis Primers (2015) ;1: :15005. doi: 10.1038/nrd2015.5. |
[7] | Waldvogel HJ , Kim EH , Tippett LJ , Vonsattel JP , Faull RL , The neuropathology of Huntington’s disease, Curr Top Behav Neurosci (2015) ;22: :33–80. doi: 10.1007/7854_2014_354. |
[8] | Hamilton JM , Wolfson T , Peavy GM , Jacobson MW , Corey-Bloom J ; Huntington Study GrouRate and correlates of weight change in Huntington’s disease, J Neurol Neurosurg Psychiatry (2004) ;75: (2):209–12. doi: 10.1136/jnn2003.017822. |
[9] | Kumar A , Kumar Singh S , Kumar V , Kumar D , Agarwal S , Rana MK , Huntington’s disease: An update of therapeutic strategies, Gene (2015) ;556: (2):1091–7. doi: 10.1016/j.gene.2014.11.022. |
[10] | Snell RG , MacMillan JC , Cheadle JP , Fenton I , Lazarou LP , Davies P , et al., Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease, Nat Genet (1993) ;4: (4):393–7. doi: 10.1038/ng0893-393. |
[11] | Podvin S , Reardon HT , Yin K , Mosier C , Hook V , Multiple clinical features of Huntington’s disease correlate with mutant HTT gene CAG repeat lengths and neurodegeneration, J Neurol (2019) ;266: (3):551–64. doi: 10.1007/s00415-018-8940-6. |
[12] | Ha AD , Jankovic J , Exploring the correlates of intermediate CAG repeats in Huntington disease, Postgrad Med (2011) ;123: (5):116–21. doi: 10.3810/pgm.2011.09.2466. |
[13] | Kremer B , Goldberg P , Andrew SE , Theilmann J , Telenius H , Zeisler J , et al., A worldwide study of the Huntington’s disease mutation, The sensitivity and specificity of measuring CAG repeats. N Engl J Med (1994) ;330: (20):1401–6. doi: 10.1056/NEJM199405193302001. |
[14] | Mühlau M , Winkelmann J , Rujescu D , Giegling I , Koutsouleris N , Gaser C ,et al., Variation within the Huntington’s disease gene influences normal brain structure, PLoS One. (2012) ;7: (1):e29809. doi: 10.1371/journal.pone.0029809. |
[15] | Saudou F , Humbert S , The biology of huntingtin, Neuron (2016) ;89: (5):910–26. doi: 10.1016/j.neuron.2016.02.003. |
[16] | Zhuang Y , Ma F , Li-Ling J , Xu X , Li Y , Comparative analysis of amino acid usage and protein length distribution between alternatively and non-alternatively spliced genes across six eukaryotic genomes, Mol Biol Evol (2003) ;20: (12):1978–85. doi: 10.1093/molbev/msg203. |
[17] | Mende-Mueller LM , Toneff T , Hwang SR , Chesselet MF , Hook VY , Tissue-specific proteolysis of Huntingtin (htt) in human brain: Evidence of enhanced levels of N- and C-terminal htt fragments in Huntington’s disease striatum J Neurosci (2001) ;21: (6):1830–7. doi: 10.1523/JNEUROSCI.21-06-01830.2001. |
[18] | Schilling G , Klevytska A , Tebbenkamp AT , Juenemann K , Cooper J , Gonzales V , et al., Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models, J Neuropathol Exp Neurol (2007) ;66: (4):313–20. doi: 10.1097/nen.0b013e318040b2c8. |
[19] | Landles C , Sathasivam K , Weiss A , Woodman B , Moffitt H , Finkbeiner S , et al., Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease, J Biol Chem (2010) ;285: (12):8808–23. doi: 10.1074/jbc.M109.075028. |
[20] | Arnesen T , Starheim KK , Van Damme P , Evjenth R , Dinh H , Betts MJ , et al., The chaperone-like protein HYPK acts together with NatA in cotranslational N-terminal acetylation and prevention of Huntingtin aggregation, Mol Cell Biol (2010) ;30: (8):1898–909. doi: 10.1128/MCB.01199-09. |
[21] | Ashkenazi A , Bento CF , Ricketts T , Vicinanza M , Siddiqi F , Pavel M , et al., Polyglutamine tracts regulate beclin 1-dependent autophagy. Nature. (2017) ;545: (7652):108–11. doi: 10.1038/nature22078. |
[22] | Atwal RS , Xia J , Pinchev D , Taylor J , Epand RM , Truant R , Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity, Hum Mol Genet (2007) ;16: (21):2600–15. doi: 10.1093/hmg/ddm217. |
[23] | Bailleul PA , Newnam GP , Steenbergen JN , Chernoff YO , Genetic study of interactions between the cytoskeletal assembly protein sla1 and prion-forming domain of the release factor Sup35 (eRF3) in Saccharomyces cerevisiae, Genetics (1999) ;153: (1):81–94. doi: 10.1093/genetics/153.1.81. |
[24] | Baksi S , Basu S , Mukhopadhyay D , Mutant huntingtin replaces Gab1 and interacts with C-terminal SH3 domain of growth factor receptor binding protein 2 (Grb2), Neurosci Res (2014) ;87: :77–83. doi: 10.1016/j.neures.2014.06.009. |
[25] | Baldo B , Weiss A , Parker CN , Bibel M , Paganetti P , Kaupmann K , A screen for enhancers of clearance identifies huntingtin as a heat shock protein 90 (Hsp90) client protein, J Biol Chem (2012) ;287: (2):1406–14. doi: 10.1074/jbc.M111.294801. |
[26] | Bao J , Sharp AH , Wagster MV , Becher M , Schilling G , Ross CA , et al., Expansion of polyglutamine repeat in huntingtin leads to abnormal protein interactions involving calmodulin, Proc Natl Acad Sci U S A (1996) ;93: (10):5037–42. doi: 10.1073/pnas.93.10.5037. |
[27] | Barbaro BA , Lukacsovich T , Agrawal N , Burke J , Bornemann DJ , Purcell JM , et al., Comparative study of naturally occurring huntingtin fragments in Drosophila points to exon 1 as the most pathogenic species in Huntington’s disease, Hum Mol Genet (2015) ;24: (4):913–25. doi: 10.1093/hmg/ddu504. |
[28] | Bardai FH , Verma P , Smith C , Rawat V , Wang L , D’Mello SR , Disassociation of histone deacetylase-3 from normal huntingtin underlies mutant huntingtin neurotoxicity, J Neurosci, (2013) ;33: (29):11833–8. doi: 10.1523/JNEUROSCI.5831-12.2013. |
[29] | Bauer PO , Hudec R , Goswami A , Kurosawa M , Matsumoto G , Mikoshiba K , et al., ROCK-phosphorylated vimentin modifies mutant huntingtin aggregation via sequestration of IRBIT, Mol Neurodegener (2012) ;7: :43. doi: 10.1186/1750-1326-7-43. |
[30] | Bauer PO , Wong HK , Oyama F , Goswami A , Okuno M , Kino Y , et al., Inhibition of Rho kinases enhances the degradation of mutant huntingtin, J Biol Chem (2009) ;284: (19):13153–64. doi: 10.1074/jbc.M809229200. |
[31] | Bertaux F , Sharp AH , Ross CA , Lehrach H , Bates GP , Wanker E , HAP1-huntingtin interactions do not contribute to the molecular pathology in Huntington’s disease transgenic mice, FEBS Lett (1998) ;426: (2):229–32. doi: 10.1016/s0014-5793(98)00352-4. |
[32] | Borgonovo JE , Troncoso M , Lucas JJ , Sosa MA , Mutant huntingtin affects endocytosis in striatal cells by altering the binding of AP-2 to membranes, Exp Neurol (2013) ;241: :75–83. doi: 10.1016/j.expneurol.2012.11.025. |
[33] | Boutell JM , Thomas P , Neal JW , Weston VJ , Duce J , Harper PS , et al., Aberrant interactions of transcriptional repressor proteins with the Huntington’s disease gene product, huntingtin, Hum Mol Genet (1999) ;8: (9):1647–55. doi: 10.1093/hmg/8.9.1647. |
[34] | Boutell JM , Wood JD , Harper PS , Jones AL , Huntingtin interacts with cystathionine beta-synthase, Hum Mol Genet (1998) ;7: (3):371–8. doi: 10.1093/hmg/7.3.371. |
[35] | Bradley SV , Holland EC , Liu GY , Thomas D , Hyun TS , Ross TS , Huntingtin interacting protein 1 is a novel brain tumor marker that associates with epidermal growth factor receptor, Cancer Res (2007) ;67: (8):3609–15. doi: 10.1158/0008-5472.CAN-06-4803. |
[36] | Broncel M , Serwa RA , Bunney TD , Katan M , Tate EW , Global Profiling of Huntingtin-associated protein E (HYPE)-mediated AMPylation through a chemical proteomic approach, Mol Cell Proteomics (2016) ;15: (2):715–25. doi: 10.1074/mcO115.054429. |
[37] | Burke JR , Enghild JJ , Martin ME , Jou YS , Myers RM , Roses AD , et al., Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH, Nat Med (1996) ;2: (3):347–50. doi: 10.1038/nm0396-347. |
[38] | Butland SL , Sanders SS , Schmidt ME , Riechers SP , Lin DT , Martin DD , et al., The palmitoyl acyltransferase HIP14 shares a high proportion of interactors with huntingtin Implications for a role in the pathogenesis of Huntington’s disease.. Hum Mol Genet (2014) ;23: (15):4142–60. doi: 10.1093/hmg/ddu137 |
[39] | Caviston JP , Ross JL , Antony SM , Tokito M , Holzbaur EL , Huntingtin facilitates dynein/dynactin-mediated vesicle transport, Proc Natl Acad Sci U S A (2007) ;104: (24):10045–50. doi: 10.1073/pnas.0610628104. |
[40] | Chakraborty J , Pandey M , Navneet AK , Appukuttan TA , Varghese M , Sreetama SC , et al., Profilin-2 increased expression and its altered interaction with β-actin in the striatum of 3-nitropropionic acid-induced Huntington’s disease in rats, Neuroscience (2014) ;281: :216–28. doi: 10.1016/j.neuroscience.2014.09.035. |
[41] | Charles V , Mezey E , Reddy PH , Dehejia A , Young TA , Polymeropoulos MH , et al., Alpha-synuclein immunoreactivity of huntingtin polyglutamine aggregates in striatum and cortex of Huntington’s disease patients and transgenic mouse models, Neurosci Lett (2000) ;289: (1):29–32. doi: 10.1016/s0304-3940(00)01247-7. |
[42] | Chaudhary RK , Patel KA , Patel MK , Joshi RH , Roy I , Inhibition ofaggregation of mutant Huntingtin by nucleic acid aptamers invitro and in a yeast model of Huntington’s disease, Mol Ther. (2015) ;23: (12):1912–26. doi: 10.1038/mt.2015.157. |
[43] | Chen CY , Brodsky FM , Huntingtin-interacting protein 1 (Hip1) andHip1-related protein (Hip1R) bind the conserved sequence of clathrinlight chains and thereby influence clathrin assembly in vitroand actin distribution in vivo, J Biol Chem (2005) ;280: (7):6109–17. doi: 10.1074/jbc.M408454200. |
[44] | Cho KJ , Lee BI , Cheon SY , Kim HW , Kim HJ , Kim GW , Inhibition of apoptosis signal-regulating kinase 1 reduces endoplasmic reticulum stress and nuclear huntingtin fragments in a mouse model of Huntington disease, Neuroscience (2009) ;163: (4):1128–34. doi: 10.1016/j.neuroscience.2009.07.048. |
[45] | Chopra VS , Metzler M , Rasper DM , Engqvist-Goldstein AE , Singaraja R , Gan L , et al., HIP12 is a non-proapoptotic member of a gene family including HIP1, an interacting protein with huntingtin, Mamm Genome (2000) ;11: (11):1006–15. doi: 10.1007/s003350010195. |
[46] | Choudhury KR , Bhattacharyya NP , Chaperone protein HYPK interacts with the first 17 amino acid region of Huntingtin and modulates mutant HTT-mediated aggregation and cytotoxicity, Biochem Biophys Res Commun (2015) ;456: (1):66–73. doi: 10.1016/j.bbrc.2014.11.035. |
[47] | Chow WN , Luk HW , Chan HY , Lau KF . Degradation of mutant huntingtin via the ubiquitin/proteasome system is modulated by FE65, Biochem J (2012) ;443: (3):681–9. doi: 10.1042/BJ20112175. |
[48] | Chun W , Lesort M , Tucholski J , Faber PW , MacDonald ME , Ross CA , et al., Tissue transglutaminase selectively modifies proteins associated with truncated mutant huntingtin in intact cells, Neurobiol Dis (2001) ;8: (3):391–404. doi: 10.1006/nbdi.2001.0390. |
[49] | Colomer V , Engelender S , Sharp AH , Duan K , Cooper JK , Lanahan A , et al., Huntingtin-associated protein 1 (HAP1) binds to a Trio-like polypeptide, with a rac1 guanine nucleotide exchange factor domain, Hum Mol Genet (1997) ;6: (9):1519–25. |
[50] | Cornett J , Cao F , Wang CE , Ross CA , Bates GP , Li SH , et al., Polyglutamine expansion of huntingtin impairs its nuclear export, Nat Genet (2005) ;37: (2):198–204. doi: 10.1038/ng1503. |
[51] | Corradini E , Burgers PP , Plank M , Heck AJ , Scholten A , Huntingtin-associated protein 1 (HAP1) is a cGMP-dependent kinase anchoring protein (GKAP) specific for the cGMP-dependent protein kinase Iβ isoform, J Biol Chem (2015) ;290: (12):7887–96. doi: 10.1074/jbc.M114.622613. |
[52] | Culver BP , Savas JN , Park SK , Choi JH , Zheng S , Zeitlin SO , et al., Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis, J Biol Chem (2159) ;287: (26):9–614. doi: 10.1074/jbc.M112.359307. |
[53] | Dai Y , Dudek NL , Li Q , Fowler SC , Muma NA , Striatal expression of a calmodulin fragment improved motor function, weight loss, and neuropathology in the R6/2 mouse model of Huntington’s disease, J Neurosci (2009) ;29: (37):11550–9. doi: 10.1523/JNEUROSCI.3307-09.2009. |
[54] | Danson CM , Pearson N , Heesom KJ , Cullen PJ , Sorting nexin-21 is a scaffold for the endosomal recruitment of huntingtin, J Cell Sci (2018) ;131: (17):jcs211672. doi: 10.1242/jcs.21167. |
[55] | Darrow MC , Sergeeva OA , Isas JM , Galaz-Montoya JG , King JA , Langen R , et al., Structural mechanisms of mutant Huntingtin aggregation suppression by the synthetic chaperonin-like CCT5 complex explained by cryoelectron tomography, J Biol Chem (2015) ;290: (28):17451–61. doi: 10.1074/jbc.M115.655373. |
[56] | Datta M , Bhattacharyya NP , Regulation of RE1 protein silencing transcription factor (REST) expression by HIP1 protein interactor (HIPPI), J Biol Chem (2011) ;286: (39):33759–69. doi: 10.1074/jbc.M111.265173. |
[57] | Davranche A , Aviolat H , Zeder-Lutz G , Busso D , Altschuh D , Trottier Y , et al., Huntingtin affinity for partners is not changed by polyglutamine length: Aggregation itself triggers aberrant interactions, Hum Mol Genet (2011) ;20: (14):2795–806. doi: 10.1093/hmg/ddr178. |
[58] | Desplats PA , Lambert JR , Thomas EA , Functional roles for the striatal-enriched transcription factor, Bcl11b, in the control of striatal gene expression and transcriptional dysregulation in Huntington’s disease, Neurobiol Dis (2008) ;31: (3):298–308. doi: 10.1016/j.nbd.2008.05.005. |
[59] | Díaz-Hernández M , Valera AG , Morán MA , Gómez-RamosP , Alvarez-Castelao B , Castaño JG , et al., Inhibition of 26Sproteasome activity by huntingtin filaments but not inclusion bodiesisolated from mouse and human brain, J Neurochem (2006) ;98: (5):1585–96. doi: 10.1111/j.1471-4159.2006.03968.x. |
[60] | Dong G , Callegari EA , Gloeckner CJ , Ueffing M , Wang H , Prothymosin-α interacts with mutant huntingtin and suppresses its cytotoxicity in cell culture, J Biol Chem (2012) ;287: (2):1279–89. doi: 10.1074/jbc.M111.294280. |
[61] | Dudek NL , Dai Y , Muma NA , Neuroprotective effects of calmodulin peptide 76-121aa: Disruption of calmodulin binding to mutant huntingtin, Brain Pathol (2010) ;20: (1):176–89. doi: 10.1111/j.1750-3639.2008.00258.x. |
[62] | Dudek NL , Dai Y , Muma NA . Protective effects of interrupting the binding of calmodulin to mutant huntingtin, J Neuropathol Exp Neurol (2008) ;67: (4):355–65. doi: 10.1097/NEN.0b013e31816a9e60. |
[63] | Dunah AW , Jeong H , Griffin A , Kim YM , Standaert DG , Hersch SM , et al., Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease, Science. (2002) ;296: (5576):2238–43. doi: 10.1126/science.1072613. |
[64] | Eriguchi M , Mizuta H , Luo S , Kuroda Y , Hara H , Rubinsztein DC , alpha Pix enhances mutant huntingtin aggregation, J Neurol Sci (2010) ;290: (1-2):80–5. doi: 10.1016/j.jns.2009.11.003. |
[65] | Faber PW , Barnes GT , Srinidhi J , Chen J , Gusella JF , MacDonald ME , Huntingtin interacts with a family of WW domain proteins, Hum Mol Genet (1998) ;7: (9):1463–74. doi: 10.1093/hmg/7.9.1463. |
[66] | Fan J , Cowan CM , Zhang LY , Hayden MR , Raymond LA , Interaction of postsynaptic density protein-95 with NMDA receptors influences excitotoxicity in the yeast artificial chromosome mouse model of Huntington’s disease, J Neurosci (2009) ;29: (35):10928–38. doi: 10.1523/JNEUROSCI.2491-09.2009. |
[67] | Federspiel JD , Greco TM , Lum KK , Cristea IM , Hdac4 interactions in Huntington’s disease viewed through the prism of multiomics, Mol Cell Proteomics (2019) ;18: (8 Suppl 1):S92–S113. doi: 10.1074/mcRA118.001253. |
[68] | Fuchs M , Poirier DJ , Seguin SJ , Lambert H , Carra S , Charette SJ , et al., Identification of the key structural motifs involved in HspB8/HspB6-Bag3 interaction, Biochem J (2009) ;425: (1):245–55. doi: 10.1042/BJ20090907. |
[69] | Fujita K , Nakamura Y , Oka T , Ito H , Tamura T , Tagawa K , et al., A functional deficiency of TERA/VCP/p97 contributes to impaired DNA repair in multiple polyglutamine diseases, Nat Commun (2013) ;4: :1816. doi: 10.1038/ncomms2828. |
[70] | Futter M , Diekmann H , Schoenmakers E , Sadiq O , Chatterjee K , Rubinsztein DC , Wild-type but not mutant huntingtin modulates the transcriptional activity of liver X receptors, J Med Genet (2009) ;46: (7):438–46. doi: 10.1136/jmg.2009.06639. |
[71] | Gao YG , Yang H , Zhao J , Jiang YJ , Hu HY , Autoinhibitory structure of the WW domain of HYPB/SETD2 regulates its interaction with the proline-rich region of huntingtin, Structure (2014) ;22: (3):378–86. doi: 10.1016/j.str.2013.12.005. |
[72] | Garcia M , Charvin D , Caboche J , Expanded huntingtin activates the c-Jun terminal kinase/c-Jun pathway prior to aggregate formation in striatal neurons in culture, Neuroscience (2004) ;127: (4):859–70. doi: 10.1016/j.neuroscience.2004.05.054. |
[73] | Ghosh DK , Roy A , Ranjan A . The ATPase VCP/p97 functions as a disaggregase against toxic Huntingtin-exon1 aggregates, FEBS Lett (2018) ;592: (16):2680–92. doi: 10.1002/1873-3468.13213. |
[74] | Goehler H , Lalowski M , Stelzl U , Waelter S , Stroedicke M , Worm U , et al., A protein interaction network links GIT1, an enhancer of huntingtin aggregation, to Huntington’s disease, Mol Cell (2004) ;15: (6):853–65. doi: 10.1016/j.molcel.2004.09.016. |
[75] | Gopalakrishnan C , Jethi S , Kalsi N , Purohit R , Biophysical aspect of Huntingtin protein during polyQ: An in silico insight, Cell Biochem Biophys (2016) ;74: (2):129–39. doi: 10.1007/s12013-016-0728-7. |
[76] | Grison A , Mantovani F , Comel A , Agostoni E , Gustincich S , Persichetti F , et al., Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin, Proc Natl Acad Sci U S A (2011) ;108: (44):17979–84. doi: 10.1073/pnas.1106198108. |
[77] | Guo X , Sun X , Hu D , Wang YJ , Fujioka H , Vyas R , et al., VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease, Nat Commun (2016) ;7: :12646. doi: 10.1038/ncomms12646. |
[78] | Guzhova IV , Lazarev VF , Kaznacheeva AV , Ippolitova MV , Muronetz VI , Kinev AV , et al., Novel mechanism of Hsp70 chaperone-mediated prevention of polyglutamine aggregates in a cellular model of huntington disease, Hum Mol Genet (2011) ;20: (20):3953–63. doi: 10.1093/hmg/ddr314. |
[79] | Hackam AS , Yassa AS , Singaraja R , Metzler M , Gutekunst CA , Gan L , et al., Huntingtin interacting protein 1 induces apoptosis via a novel caspase-dependent death effector domain, J Biol Chem (2000) ;275: (52):41299–308. doi: 10.1074/jbc.M008408200. |
[80] | Hattula K , Peränen J , FIP-2, a coiled-coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis, Curr Biol (2000) ;10: (24):1603–6. doi: 10.1016/s0960-9822(00)00864-2. |
[81] | Havel LS , Wang CE , Wade B , Huang B , Li S , Li XJ , Preferential accumulation of N-terminal mutant huntingtin in the nuclei of striatal neurons is regulated by phosphorylation, Hum Mol Genet (2011) ;20: (7):1424–37. doi: 10.1093/hmg/ddr023. |
[82] | Hoffner G , Kahlem P , Djian P . Perinuclear localization of huntingtin as a consequence of its binding to microtubules through an interaction with beta-tubulin: Relevance to Huntington’s disease. J Cell Sci (2002) ;115: (Pt 5):941–8. doi: 10.1242/jcs.115.5.941 |
[83] | Holbert S , Dedeoglu A , Humbert S , Saudou F , Ferrante RJ , Néri C , Cdc42-interacting protein 4 binds to huntingtin: Neuropathologic and biological evidence for a role in Huntington’s disease Proc Natl Acad Sci U S A (2003) ;100: (5):2712–7. doi: 10.1073/pnas.0437967100 |
[84] | Holbert S , Denghien I , Kiechle T , Rosenblatt A , Wellington C , Hayden MR , et al., The Gln-Ala repeat transcriptional activator CA150 interacts with huntingtin: Neuropathologic and genetic evidence for a role in Huntington’s disease pathogenesis Proc Natl Acad Sci U S A (2001) ;98: (4):1811–6. doi: 10.1073/pnas.041566798. |
[85] | Holmberg CI , Staniszewski KE , Mensah KN , Matouschek A , Morimoto RI , Inefficient degradation of truncated polyglutamine proteins by the proteasome, EMBO J. (2004) ;23: (21):4307–18. doi: 10.1038/sj.emboj.7600426. |
[86] | Horn SC , Lalowski M , Goehler H , Dröge A , Wanker EE , Stelzl U , Huntingtin interacts with the receptor sorting family protein GASP2, J Neural Transm (Vienna) (2006) ;113: (8):1081–90. doi: 10.1007/s00702-006-0514-6. |
[87] | Hosp F , Vossfeldt H , Heinig M , Vasiljevic D , Arumughan A , Wyler E , et al., Quantitative interaction proteomics of neurodegenerative disease proteins, Cell Rep (2015) ;11: (7):1134–46. doi: 10.1016/j.celre2015.04.030. |
[88] | Huang CC , Faber PW , Persichetti F , Mittal V , Vonsattel JP , MacDonald ME , et al., Amyloid formation by mutant huntingtin: Threshold, progressivity and recruitment of normal polyglutamine proteins. Somat Cell Mol Genet (1998) ;24: (4):217–33. doi: 10.1023/b:scam.0000007124.19463.e5 |
[89] | Huang K , Sanders SS , Kang R , Carroll JB , Sutton L , Wan J , et al., Wild-type HTT modulates the enzymatic activity of the neuronal palmitoyl transferase HIP14, Hum Mol Genet (2011) ;20: (17):3356–65. doi: 10.1093/hmg/ddr242. |
[90] | Humbert S , Bryson EA , Cordelières FP , Connors NC , Datta SR , Finkbeiner S , et al., The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt, Dev Cell (2002) ;2: (6):831–7. doi: 10.1016/s1534-5807(02)00188-0. |
[91] | Irmak D , Fatima A , Gutiérrez-Garcia R , Rinschen MM , Wagle P , Altmüller J , et al., Mechanism suppressing H3K9 trimethylation in pluripotent stem cells and its demise by polyQ-expanded huntingtin mutations, Hum Mol Genet (2018) ;27: (23):4117–34. doi: 10.1093/hmg/ddy304. |
[92] | Jana NR , Nukina N , BAG-1 associates with the polyglutamine-expanded huntingtin aggregates, Neurosci Lett (2005) ;378: (3):171–5. doi: 10.1016/j.neulet.2004.12.031. |
[93] | Jana NR , Tanaka M , Wang Gh , Nukina N , Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: Their role in suppression of aggregation and cellular toxicity, Hum Mol Genet (2000) ;9: (13):2009–18. doi: 10.1093/hmg/9.13.2009. |
[94] | Jiang YJ , Che MX , Yuan JQ , Xie YY , Yan XZ , Hu HY , Interaction with polyglutamine-expanded huntingtin alters cellular distribution and RNA processing of huntingtin yeast two-hybrid protein A (HYPA), J Biol Chem (2011) ;286: (28):25236–45. doi: 10.1074/jbc.M110.216333. |
[95] | Kalchman MA , Graham RK , Xia G , Koide HB , Hodgson JG , Graham KC , et al., Huntingtin is ubiquitinated and interacts with a specific ubiquitin-conjugating enzyme, J Biol Chem (1938) ;271: (32):5–94. doi: 10.1074/jbc.271.32.19385. |
[96] | Kalchman MA , Koide HB , McCutcheon K , Graham RK , Nichol K , Nishiyama K , et al., HIP1, a human homologue of S, cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet (1997) ;16: (1):44–53. doi: 10.1038/ng0597-44. |
[97] | Kaltenbach LS , Romero E , Becklin RR , Chettier R , Bell R , Phansalkar A , et al., Huntingtin interacting proteins are genetic modifiers of neurodegeneration, PLoS Genet (2007) ;3: (5):e82. doi: 10.1371/journal.pgen.0030082. |
[98] | Kaminosono S , Saito T , Oyama F , Ohshima T , Asada A , Nagai Y , et al., Suppression of mutant Huntingtin aggregate formation by Cdk5/p35 through the effect on microtubule stability, J Neurosci (2008) ;28: (35):8747–55. doi: 10.1523/JNEUROSCI.0973-08.2008. |
[99] | Kang JE , Choi SA , Park JB , Chung KC , Regulation of the proapoptotic activity of huntingtin interacting protein 1 by Dyrk1 and caspase-3 in hippocampal neuroprogenitor cells, J Neurosci Res (2005) ;81: (1):62–72. doi: 10.1002/jnr.20534. |
[100] | Kegel KB , Meloni AR , Yi Y , Kim YJ , Doyle E , Cuiffo BG , et al., Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription, J Biol Chem (2002) ;277: (9):7466–76. doi: 10.1074/jbc.M103946200. |
[101] | Khoshnan A , Ko J , Watkin EE , Paige LA , Reinhart PH , Patterson PH , Activation of the IkappaB kinase complex and nuclear factor-kappaB contributes to mutant huntingtin neurotoxicity, J Neurosci.- (2004) ;24: (37):7999–8008. doi: 10.1523/JNEUROSCI.2675-04.2004. |
[102] | Kim MO , Chawla P , Overland RP , Xia E , Sadri-Vakili G , Cha JH , Altered histone monoubiquitylation mediated by mutant huntingtin induces transcriptional dysregulation, J Neurosci (2008) ;28: (15):3947–57. doi: 10.1523/JNEUROSCI.5667-07.2008. |
[103] | Kittler JT , Thomas P , Tretter V , Bogdanov YD , Haucke V , Smart TG , et al., Huntingtin-associated protein 1 regulates inhibitory synaptic transmission by modulating gamma-aminobutyric acid type A receptor membrane trafficking, Proc Natl Acad Sci U S A (2004) ;101: (34):12736–41. doi: 10.1073/pnas.0401860101. |
[104] | Labbadia J , Novoselov SS , Bett JS , Weiss A , Paganetti P , Bates GP , et al., Suppression of protein aggregation by chaperone modification of high molecular weight complexes, Brain (2012) ;135: (Pt 4):1180–96. doi: 10.1093/brain/aws022. |
[105] | Lee H , Ahn HH , Lee W , Oh Y , Choi H , Shim SM , et al., ENC1 modulates the aggregation and neurotoxicity of mutant huntingtin through p62 under ER stress, Mol Neurobiol (2016) ;53: (10):6620–34. doi: 10.1007/s12035-015-9557-8. |
[106] | Lee JA , Yerbury JJ , Farrawell N , Shearer RF , Constantinescu P , Hatters DM , et al., SerpinB2 (PAI-2) Modulates proteostasis via binding misfolded proteins and promotion of cytoprotective inclusion formation, PLoS One. (2015) ;10: (6):e0130136. doi: 10.1371/journal.pone.0130136. |
[107] | Legendre-Guillemin V , Metzler M , Charbonneau M , Gan L , Chopra V , Philie J , et al., HIP1 and HIP12 display differential binding to F-actin, AP2, and clathrin, Identification of a novel interaction with clathrin light chain. J Biol Chem (1989) ;277: (22):7–904. doi: 10.1074/jbc.M112310200. |
[108] | Legendre-Guillemin V , Metzler M , Lemaire JF , Philie J , Gan L , Hayden MR , et al., Huntingtin interacting protein 1 (HIP1) regulates clathrin assembly through direct binding to the regulatory region of the clathrin light chain, J Biol Chem. (2005) ;280: (7):6101–8. doi: 10.1074/jbc.M408430200. |
[109] | Li SH , Gutekunst CA , Hersch SM , Li XJ , Interaction of huntingtin-associated protein with dynactin P150Glued, J Neurosci (1998) ;18: (4):1261–9. doi: 10.1523/JNEUROSCI.18-04-01261.1998. |
[110] | Li XJ , Li SH , Sharp AH , Nucifora FC Jr , Schilling G , Lanahan A , et al., A huntingtin-associated protein enriched in brain with implications for pathology, Nature (1995) ;378: (6555):398–402. doi: 10.1038/378398a0. |
[111] | Li Y , Chin LS , Levey AI , Li L , Huntingtin-associated protein 1 interacts with hepatocyte growth factor-regulated tyrosine kinase substrate and functions in endosomal trafficking, J Biol Chem. (2002) ;277: (31):28212–21. doi: 10.1074/jbc.M111612200. |
[112] | Liu T , Im W , Lee ST , Ban JJ , Chai YJ , Lee M , et al., Modulation of mitochondrial function by stem cell-derived cellular components, Biochem Biophys Res Commun (2014) ;448: (4):403–8. doi: 10.1016/j.bbrc.2014.04.129. |
[113] | Liu YF , Expression of polyglutamine-expanded Huntingtin activates the SEK1-JNK pathway and induces apoptosis in a hippocampal neuronal cell line, J Biol Chem (1998) ;273: (44):28873–7. doi: 10.1074/jbc.273.44.28873. |
[114] | Liu YF , Deth RC , Devys D , SH3 domain-dependent association of huntingtin with epidermal growth factor receptor signaling complexes, J Biol Chem (1997) ;272: (13):8121–4. doi: 10.1074/jbc.272.13.8121. |
[115] | Liu YF , Dorow D , Marshall J , Activation of MLK2-mediated signaling cascades by polyglutamine-expanded huntingtin, J Biol Chem (1903) ;275: (25):5–40. doi: 10.1074/jbc.C000180200. |
[116] | Luo H , Cao L , Liang X , Du A , Peng T , Li H . Herp promotes degradation of mutant Huntingtin: Involvement of the proteasome and molecular chaperones, Mol Neurobiol (2018) ;55: (10):7652–68. doi: 10.1007/s12035-018-0900-8 |
[117] | Luo S , Mizuta H , Rubinsztein DC , p21-activated kinase 1 promotes soluble mutant huntingtin self-interaction and enhances toxicity, Hum Mol Genet (2008) ;17: (6):895–905. doi: 10.1093/hmg/ddm362. |
[118] | Luo S , Vacher C , Davies JE , Rubinsztein DC , Cdk5 phosphorylation of huntingtin reduces its cleavage by caspases: Implications for mutant huntingtin toxicity, J Cell Biol (2005) ;169: (4):647–56. doi: 10.1083/jcb.200412071. |
[119] | Ma B , Liu B , Cao W , Gao C , Qi Z , Ning Y , et al., The Wnt signalingantagonist Dapper1 accelerates Dishevelled2 degradation viapromoting its ubiquitination and aggregate-induced autophagy, J BiolChem (2015) ;290: (19):12346–54. doi: 10.1074/jbc.M115.654590. |
[120] | Mackenzie KD , Lim Y , Duffield MD , Chataway T , Zhou XF , Keating DJ , Huntingtin-associated protein-1 (HAP1) regulates endocytosis and interacts with multiple trafficking-related proteins, Cell Signal (2017) ;35: :176–87. doi: 10.1016/j.cellsig.2017.02.023. |
[121] | Mackenzie KD , Lumsden AL , Guo F , Duffield MD , Chataway T , Lim Y , et al., Huntingtin-associated protein-1 is a synapsin I-binding protein regulating synaptic vesicle exocytosis and synapsin I trafficking, J Neurochem (2016) ;138: (5):710–21. doi: 10.1111/jnc.13703. |
[122] | Maiuri T , Woloshansky T , Xia J , Truant R , The huntingtin N17 domain is a multifunctional CRM1 and Ran-dependent nuclear and cilial export signal, Hum Mol Genet (2013) ;22: (7):1383–94. doi: 10.1093/hmg/dds554. |
[123] | Mano T , Suzuki T , Tsuji S , Iwata A , Differential effect of HDAC3 on cytoplasmic and nuclear huntingtin aggregates, PLoS One. (2014) ;9: (11):e111277. doi: 10.1371/journal.pone.0111277. |
[124] | Månsson C , Kakkar V , Monsellier E , Sourigues Y , Härmark J , Kampinga HH , et al., DNAJB6 is a peptide-bindingchaperone which can suppress amyloid fibrillation of polyglutaminepeptides at substoichiometric molar ratios, Cell Stress Chaperones (2014) ;19: (2):227–39. doi: 10.1007/s12192-013-0448-5. |
[125] | Marcora E , Gowan K , Lee JE , Stimulation of NeuroD activity by huntingtin and huntingtin-associated proteins HAP1 and MLK2, Proc Natl Acad Sci U S A. (2003) ;100: (16):9578–83. doi: 10.1073/pnas.1133382100. |
[126] | McFarland KN , Huizenga MN , Darnell SB , Sangrey GR , Berezovska O , Cha JH , et al.,. MeCP A novel Huntingtin interactor, Hum Mol Genet (2014) ;23: (4):1036–44. doi: 10.1093/hmg/ddt499. |
[127] | McGuire JR , Rong J , Li SH , Li XJ , Interaction of Huntingtin-associated protein-1 with kinesin light chain: Implications in intracellular trafficking in neurons. J Biol Chem (2006) ;281: (6):3552–9. doi: 10.1074/jbc.M509806200 |
[128] | Mealer RG , Murray AJ , Shahani N , Subramaniam S , Snyder SH , Rhes, a striatal-selective protein implicated in Huntington disease, binds beclin-1 and activates autophagy, J Biol Chem (2014) ;289: (6):3547–54. doi: 10.1074/jbc.M113.536912. |
[129] | Mele M , Aspromonte MC , Duarte CB , Downregulation of GABA receptor recycling mediated by HAP1 contributes to neuronal death in in vitro brain ischemia, Mol Neurobiol (2017) ;54: (1):45–57. doi: 10.1007/s12035-015-9661-9. |
[130] | Metzler M , Legendre-Guillemin V , Gan L , Chopra V , Kwok A , McPherson PS , et al., HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2, J Biol Chem (2001) ;276: (42):39271–6. doi: 10.1074/jbc.C100401200. |
[131] | Miller JP , Yates BE , Al-Ramahi I , Berman AE , Sanhueza M , Kim E ,et al., A genome-scale RNA-interference screen identifies RRAS signaling as a pathologic feature of Huntington’s disease, PLoS Genet. (2012) ;8: (11):e1003042. doi: 10.1371/journal.pgen.1003042. |
[132] | Miller SLH , Scappini EL , O’Bryan J , Ubiquitin-interacting motifs inhibit aggregation of polyQ-expanded huntingtin, J Biol Chem (2007) ;282: (13):10096–103. doi: 10.1074/jbc.M611151200. |
[133] | Mishra SK , Agostinelli NR , Brett TJ , Mizukami I , Ross TS , Traub LM , Clathrin- and AP-2-binding sites in HIP1 uncover a general assembly role for endocytic accessory proteins, J Biol Chem. (2001) ;276: (49):46230–6. doi: 10.1074/jbc.M108177200. |
[134] | Mitsui K , Nakayama H , Akagi T , Nekooki M , Ohtawa K , Takio K , et al., Purification of polyglutamine aggregates and identification of elongation factor-1alpha and heat shock protein 84 as aggregate-interacting proteins, J Neurosci (2002) ;22: (21):9267–77. doi: 10.1523/JNEUROSCI.22-21-09267.2002. |
[135] | Modregger J , DiProspero NA , Charles V , Tagle DA , Plomann M , PACSIN 1 interacts with huntingtin and is absent from synaptic varicosities in presymptomatic Huntington’s disease brains, Hum Mol Genet (2002) ;11: (21):2547–58. doi: 10.1093/hmg/11.21.2547. |
[136] | Moharir SC , Bansal M , Ramachandran G , Ramaswamy R , Rawat S , Raychaudhuri S , et al., Identification of a splice variant of optineurin which is defective in autophagy and phosphorylation, Biochim Biophys Acta Mol Cell Res (2018) ;1865: (11 Pt A):1526–38. doi: 10.1016/j.bbamcr.2018.08.009. |
[137] | Monsellier E , Redeker V , Ruiz-Arlandis G , Bousset L , Melki R . Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation, J Biol Chem (2015) ;290: (5):2560–76. doi: 10.1074/jbc.M114.603332. |
[138] | Mueller KA , Glajch KE , Huizenga MN , Wilson RA , Granucci EJ , Dios AM , et al., Hippo signaling pathway dysregulation in human Huntington’s disease brain and neuronal stem cells, Sci Rep (2018) ;8: (1):11355. doi: 10.1038/s41598-018-29319-4. |
[139] | Naphade S , Embusch A , Madushani KL , Ring KL , Ellerby LM , Altered expression of matrix metalloproteinases and their endogenous inhibitors in a human isogenic stem cell model of Huntington’s disease, Front Neurosci (2018) ;11: :736. doi: 10.3389/fnins.2017.00736. |
[140] | O’Brien R , DeGiacomo F , Holcomb J , Bonner A , Ring KL , Zhang N , Integration-independent transgenic Huntington disease fragment mouse models reveal distinct phenotypes and life span in vivo , J Biol Chem (2015) ;290: (31):19287–306. doi: 10.1074/jbc.M114.623561. |
[141] | Ochaba J , Lukacsovich T , Csikos G , Zheng S , Margulis J , Salazar L , et al., Potential function for the Huntingtin protein as a scaffold for selective autophagy, Proc Natl Acad Sci U S A (2014) ;111: (47):16889–94. doi: 10.1073/pnas.1420103111. |
[142] | Okano A , Usuda N , Furihata K , Nakayama K , Bao Tian Q , et al., Huntingtin-interacting protein-1-related protein of rat (rHIP1R) is localized in the postsynaptic regions, Brain Res (2003) ;967: (1-2):210–25. doi: 10.1016/s0006-8993(03)02236-4. |
[143] | Palidwor GA , Shcherbinin S , Huska MR , Rasko T , Stelzl U , Arumughan A , et al., Detection of alpha-rod protein repeats using a neural network and application to huntingtin, PLoS Comput Biol (2009) ;5: (3):e1000304. doi: 10.1371/journal.pcbi.1000304. |
[144] | Pardo R , Molina-Calavita M , Poizat G , Keryer G , Humbert S , Saudou F , pARIS-htt: An optimised expression platform to study huntingtin reveals functional domains required for vesicular trafficking, Mol Brain (2010) ;3: :17. doi: 10.1186/1756-6606-3-17. |
[145] | Parker JA , Metzler M , Georgiou J , Mage M , Roder JC , Rose AM , et al., Huntingtin-interacting protein 1 influences worm and mouse presynaptic function and protects Caenorhabditis elegans neurons against mutant polyglutamine toxicity, J Neurosci (2007) ;27: (41):11056–64. doi: 10.1523/JNEUROSCI.1941-07.2007. |
[146] | Passani LA , Bedford MT , Faber PW , McGinnis KM , Sharp AH , Gusella JF , et al., Huntingtin’s WW domain partners in Huntington’s disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington’s disease pathogenesis, Hum Mol Genet (2000) ;9: (14):2175–82. doi: 10.1093/hmg/9.14.2175. |
[147] | Peters MF , Ross CA . Isolation of a 40-kDa Huntingtin-associated protein, J Biol Chem (2001) ;276: (5):3188–94. doi: 10.1074/jbc.M008099200. |
[148] | Power D , Srinivasan S , Gunawardena S , In-vivo evidence for the disruption of Rab11 vesicle transport by loss of huntingtin, Neuroreport (2012) ;23: (16):970–7. doi: 10.1097/WNR.0b013e328359d990. |
[149] | Pryor WM , Biagioli M , Shahani N , Swarnkar S , Huang WC , Page DT , et al., Huntingtin promotes mTORC1 signaling in the pathogenesis of Huntington’s disease, Sci Signal (2014) ;7: (349):ra103. doi: 10.1126/scisignal.2005633. |
[150] | Qin ZH , Wang Y , Sapp E , Cuiffo B , Wanker E , Hayden MR , et al., Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction, J Neurosci (2004) ;24: (1):269–81. doi: 10.1523/JNEUROSCI.1409-03.2004. |
[151] | Ratovitski T , Arbez N , Stewart JC , Chighladze E , Ross CA , PRMT5- mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington’s disease (HD), Cell Cycle (2015) ;14: (11):1716–29. doi: 10.1080/15384101.2015.103359. |
[152] | Ratovitski T , Chighladze E , Arbez N , Boronina T , Herbrich S , Cole RN , et al., Huntingtin protein interactions altered by polyglutamine expansion as determined by quantitative proteomic analysis, Cell Cycle. (2012) ;11: (10):2006–21. doi: 10.4161/cc.20423. |
[153] | Rawat V , Goux W , Piechaczyk M , D Mello SR , c-Fos protects neurons through a noncanonical mechanism involving HDAC3 interaction: Identification of a 21-amino acid fragment with neuroprotective activity, Mol Neurobiol (2014) ;53: (2):1165–80. doi: 10.1007/s12035-014-9058-1. |
[154] | Rega S , Stiewe T , Chang DI , Pollmeier B , Esche H , Bardenheuer W , et al., Identification of the full-length huntingtin- interacting protein p231HBP/HYPB as a DNA-binding factor, Mol Cell Neurosci (2001) ;18: (1):68–79. doi: 10.1006/mcne.2001.1004. |
[155] | Riechers SP , Butland S , Deng Y , Skotte N , Ehrnhoefer DE , Russ J , et al., Interactome network analysis identifies multiple caspase-6 interactors involved in the pathogenesis of HD, Hum Mol Genet (2016) ;25: (8):1600–18. doi: 10.1093/hmg/ddw036. |
[156] | Ripaud L , Chumakova V , Antonin M , Hastie AR , Pinkert S , Körner R , et al., Overexpression of Q-rich prion-like proteins suppresses polyQ cytotoxicity and alters the polyQ interactome, Proc Natl Acad Sci U S A. (2014) ;111: (51):18219–24. doi: 10.1073/pnas.1421313111. |
[157] | Saleh AA , Gune US , Chaudhary RK , Turakhiya AP , Roy I , Roles of Hsp104 and trehalose in solubilisation of mutant huntingtin in heat shocked Saccharomyces cerevisiae cells, Biochim Biophys Acta (2014) ;1843: (4):746–57. doi: 10.1016/j.bbamcr.2014.01.004. |
[158] | Sancho M , Herrera AE , Orzáez M , Pérez-Payá E , Inactivation of Apaf1 reduces the formation of mutant huntingtin-dependent aggregates and cell death, Neuroscience (2014) ;262: :83–91. doi: 10.1016/j.neuroscience.2013.12.062. |
[159] | Sanders SS , Mui KK , Sutton LM , Hayden MR , Identification of binding sites in Huntingtin for the Huntingtin interacting proteins HIP14 and HIP14L, PLoS One (2014) ;9: (2):e90669. doi: 10.1371/journal.pone.0090669. |
[160] | Sayer JA , Manczak M , Akileswaran L , Reddy PH , Coghlan VM , Interaction of the nuclear matrix protein NAKAP with HypA and huntingtin: Implications for nuclear toxicity in Huntington’s disease pathogenesis Neuromolecular Med (2005) ;7: (4):297–310. doi: 10.1385/NMM:7:4:297. |
[161] | Schaffar G , Breuer P , Boteva R , Behrends C , Tzvetkov N , Strippel N , et al., Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation, Mol Cell (2004) ;15: (1):95–105. doi: 10.1016/j.molcel.2004.06.029. |
[162] | Schwab C , Yu S , McGeer EG , McGeer PL , Optineurin in Huntington’s disease intranuclear inclusions, Neurosci Lett (2012) ;506: (1):149–54. doi: 10.1016/j.neulet.2011.10.070. |
[163] | Sethi R , Patel V , Saleh AA , Roy I , Cellular toxicity of yeast prion protein Rnq1 can be modulated by N-terminal wild type huntingtin, Arch Biochem Biophys. (2016) ;590: :82–89. doi: 10.1016/j.abb.2015.11.036. |
[164] | Shao J , Welch WJ , Diprospero NA , Diamond MI , Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation, Mol Cell Biol (2008) ;28: (17):5196–208. doi: 10.1128/MCB.00079-08. |
[165] | Shen WC , Li HY , Chen GC , Chern Y , Tu PH , Mutations in the ubiquitin-binding domain of OPTN/optineurin interfere with autophagy-mediated degradation of misfolded proteins by a dominant-negative mechanism, Autophagy (2015) ;11: (4):685–700. doi: 10.4161/auto.36098. |
[166] | Shimojo M , Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150 Glued, J Biol Chem (2008) ;283: (50):34880–6. doi: 10.1074/jbc.M804183200. |
[167] | Shirasaki DI , Greiner ER , Al-Ramahi I , Gray M , Boontheung P , Geschwind DH , et al., Network organization of the huntingtin proteomic interactome in mammalian brain, Neuron (2012) ;75: (1):41–57. doi: 10.1016/j.neuron.2012.05.024. |
[168] | Shirendeb UP , Calkins MJ , Manczak M , Anekonda V , Dufour B , McBride JL , et al., Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease, Hum Mol Genet (2012) ;21: (2):406–20. doi: 10.1093/hmg/ddr475. |
[169] | Silva FR , Miranda AS , Santos RPM , Olmo IG , Zamponi GW , Dobransky T , et al., N-type Ca2 + channels are affected by full-length mutant huntingtin expression in a mouse model of Huntington’s disease, Neurobiol Aging (2017) ;55: :1–10. doi: 10.1016/j.neurobiolaging.2017.03.015. |
[170] | Singaraja RR , Hadano S , Metzler M , Givan S , Wellington CL , Warby S , et al., HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis, Hum Mol Genet (2002) ;11: (23):2815–28. doi: 10.1093/hmg/11.23.2815. |
[171] | Sittler A , Wälter S , Wedemeyer N , Hasenbank R , Scherzinger E , Eickhoff H , et al., SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates, Mol Cell (1998) ;2: (4):427–36. doi: 10.1016/s1097-2765(00)80142-2. |
[172] | Smith C , D’Mello SR . Cell and context-dependent effects of the heat shock protein dnajb6 on neuronal survival , Mol Neurobiol (2016) ;53: (8):5628–39. doi: 10.1007/s12035-015-9452-3. |
[173] | Smith R , Bacos K , Fedele V , Soulet D , Walz HA , Obermüller S , et al., Mutant huntingtin interacts with beta-tubulin and disrupts vesicular transport and insulin secretion, Hum Mol Genet (2006) ;18: (20):3942–54. doi: 10.1093/hmg/ddp336. |
[174] | Solans A , Zambrano A , Rodríguez M , Barrientos A , Cytotoxicityof a mutant huntingtin fragment in yeast involves early alterationsin mitochondrial OXPHOS complexes II and III, Hum Mol Genet (2006) ;15: (20):3063–81. doi: 10.1093/hmg/ddl248. |
[175] | Son S , Bowie LE , Maiuri T , Hung CLK , Desmond CR , Xia J , et al., High-mobility group box 1 links sensing of reactive oxygen species by huntingtin to its nuclear entry, J Biol Chem. (2019) ;294: (6):1915–23. doi: 10.1074/jbc.RA117.001440. |
[176] | Song J , Yang Q , Yang J , Larsson L , Hao X , Zhu X , et al., Essential genetic interactors of SIR2 required for spatial sequestration and asymmetrical inheritance of protein aggregates, PLoS Genet (2014) ;10: (7):e1004539. doi: 10.1371/journal.pgen.1004539. |
[177] | Song W , Zsindely N , Faragó A , Marsh JL , Bodai L , Systematic genetic interaction studies identify histone demethylase Utx as potential target for ameliorating Huntington’s disease, Hum Mol Genet (2018) ;27: (4):649–66. doi: 10.1093/hmg/ddx432. |
[178] | Steffan JS , Kazantsev A , Spasic-Boskovic O , Greenwald M , Zhu YZ , Gohler H , et al., The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription, Proc Natl Acad Sci U S A (2000) ;97: (12):6763–8. doi: 10.1073/pnas.100110097. |
[179] | Stroedicke M , Bounab Y , Strempel N , Klockmeier K , Yigit S , Friedrich RP , et al., Systematic interaction network filtering identifies CRMP1 as a novel suppressor of huntingtin misfolding and neurotoxicity, Genome Res (2015) ;25: (5):701–13. doi: 10.1101/gr.182444.114. |
[180] | Sun Y , Savanenin A , Reddy PH , Liu YF , Polyglutamine-expanded huntingtin promotes sensitization of N-methyl-D-aspartate receptors via post-synaptic density 95, J Biol Chem. (2001) ;276: (27):24713–8. doi: 10.1074/jbc.M103501200. |
[181] | Sutton LM , Sanders SS , Butland SL , Singaraja RR , Franciosi S , Southwell AL , et al., Hip14l-deficient mice develop neuropathological and behavioural features of Huntington disease, Hum Mol Genet (2013) ;22: (3):452–65. doi: 10.1093/hmg/dds441. |
[182] | Swayne LA , Chen L , Hameed S , Barr W , Charlesworth E , Colicos MA , et al., Crosstalk between huntingtin and syntaxin 1A regulates N-type calcium channels, Mol Cell Neurosci (2005) ;30: (3):339–51. doi: 10.1016/j.mcn.2005.07.016. |
[183] | Tam S , Geller R , Spiess C , Frydman J . The chaperonin TRiC controls polyglutamine aggregation and toxicity through subunit-specific interactions, Nat Cell Biol (2006) ;8: (10):1155–62. doi: 10.1038/ncb1477. |
[184] | Tang TS , Tu H , Chan EY , Maximov A , Wang Z , Wellington CL , et al., Huntingtin and huntingtin-associated protein 1 influence neuronal calcium signaling mediated by inositol-(1,4,5) triphosphate receptor type 1, Neuron (2003) ;39: (2):227–39. doi: 10.1016/s0896-6273(03)00366-0. |
[185] | Tourette C , Li B , Bell R , O’Hare S , Kaltenbach LS , Mooney SD , et al., A large scale Huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease, J Biol Chem (2014) ;289: (10):6709–26. doi: 10.1074/jbc.M113.523696. |
[186] | Tousley A , Iuliano M , Weisman E , Sapp E , Zhang N , Vodicka P , et al., Rac1 activity is modulated by Huntingtin and dysregulated in models of Huntington’s disease, J Huntingtons Dis (2019) ;8: (1):53–69. doi: 10.3233/JHD-180311. |
[187] | Tung YT , Hsu WM , Lee H , Huang WP , Liao YF , The evolutionarily conserved interaction between LC3 and p62 selectively mediates autophagy-dependent degradation of mutant huntingtin, Cell Mol Neurobiol (2010) ;30: (5):795–806. doi: 10.1007/s10571-010-9507-y. |
[188] | Velier J , Kim M , Schwarz C , Kim TW , Sapp E , Chase K , et al., Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways, Exp Neurol (1998) ;152: (1):34–40. doi: 10.1006/exnr.1998.6832. |
[189] | Verardi R , Kim JS , Ghirlando R , Banerjee A , Structural basis for substrate recognition by the ankyrin repeat domain of human DHHC17 palmitoyltransferase, Structure (2017) ;25: (9):1337–47.e6. doi: 10.1016/j.str.2017.06.018. |
[190] | Waelter S , Scherzinger E , Hasenbank R , Nordhoff E , Lurz R , Goehler H , et al., The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis, Hum Mol Genet (2001) ;10: (17):1807–17. doi: 10.1093/hmg/10.17.1807. |
[191] | Wang Y , Meriin AB , Zaarur N , Romanova NV , Chernoff YO , Costello CE , et al., Abnormal proteins can form aggresome in yeast: Aggresome-targeting signals and components of the machinery, FASEB J (2009) ;23: (2):451–63. doi: 10.1096/fj.08-117614. |
[192] | Wang Z , Peng T , Wu H , He J , Li H , HAP1 helps to regulate actin-based transport of insulin-containing granules in pancreatic β cells, Histochem Cell Biol (2015) ;144: (1):39–48. doi: 10.1007/s00418-015-1311-9. |
[193] | Wanker EE , Rovira C , Scherzinger E , Hasenbank R , Wälter S , Tait D , et al., HIP-I: A huntingtin interacting protein isolated by the yeast two-hybrid system, Hum Mol Genet (1997) ;6: (3):487–95. doi: 10.1093/hmg/6.3.487. |
[194] | Wheeler VC , White JK , Gutekunst CA , Vrbanac V , Weaver M , Li XJ , et al., Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice, Hum Mol Genet (2000) ;9: (4):503–13. doi: 10.1093/hmg/9.4.503. |
[195] | Wilbur JD , Chen CY , Manalo V , Hwang PK , Fletterick RJ , Brodsky FM , Actin binding by Hip1 (huntingtin-interacting protein 1) and Hip1R (Hip1-related protein) is regulated by clathrin light chain, J Biol Chem (2008) ;283: (47):32870–9. doi: 10.1074/jbc.M802863200. |
[196] | Williamson TE , Vitalis A , Crick SL , Pappu RV , Modulation of polyglutamine conformations and dimer formation by the N-terminus of huntingtin, J Mol Biol (2010) ;396: (5):1295–309. doi: 10.1016/j.jmb.2009.12.017. |
[197] | Woerner AC , Frottin F , Hornburg D , Feng LR , Meissner F , Patra M , et al., Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA, Science (2016) ;351: (6269):173–6. doi: 10.1126/science.aad2033. |
[198] | Wolfe KJ , Ren HY , Trepte P , Cyr DM , Polyglutamine-rich suppressors of huntingtin toxicity act upstream of Hsp70 and Sti1 in spatial quality control of amyloid-like proteins, PLoS One (2014) ;9: (5):e95914. doi: 10.1371/journal.pone.0095914. |
[199] | Wu LL , Fan Y , Li S , Li XJ , Zhou XF , Huntingtin-associated protein-1 interacts with pro-brain-derived neurotrophic factor and mediates its transport and release, J Biol Chem (2010) ;285: (8):5614–23. doi: 10.1074/jbc.M109.073197. |
[200] | Yanai A , Huang K , Kang R , Singaraja RR , Arstikaitis P , Gan L , et al., Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function, Nat Neurosci (2006) ;9: (6):824–31. doi: 10.1038/nn1702. |
[201] | Yang H , Liu C , Zhong Y , Luo S , Monteiro MJ , Fang S , Huntingtin interacts with the cue domain of gp78 and inhibits gp78 binding to ubiquitin and p97/VCP, PLoS One (2010) ;5: (1):e8905. doi: 10.1371/journal.pone.0008905. |
[202] | Yang H , Yue HW , He WT , Hong JY , Jiang LL , Hu HY , PolyQ-expanded huntingtin and ataxin-3 sequester ubiquitin adaptors hHR23B and UBQLN2 into aggregates via conjugated ubiquitin, FASEB J. (2018) ;32: (6):2923–33. doi: 10.1096/fj.201700801RR. |
[203] | Yang H , Zhong X , Ballar P , Luo S , Shen Y , Rubinsztein DC , et al., Ubiquitin ligase Hrd1 enhances the degradation and suppresses the toxicity of polyglutamine-expanded huntingtin, Exp Cell Res (2007) ;313: (3):538–50. doi: 10.1016/j.yexcr.2006.10.031. |
[204] | Yano H , Baranov SV , Baranova OV , Kim J , Pan Y , Yablonska S , et al., Inhibition of mitochondrial protein import by mutant huntingtin, Nat Neurosci (2014) ;17: (6):822–31. doi: 10.1038/nn.3721. |
[205] | Yohrling GJ , Farrell LA , Hollenberg AN , Cha JH , Mutant huntingtin increases nuclear corepressor function and enhances ligand-dependent nuclear hormone receptor activation, Mol Cell Neurosci. (2003) ;23: (1):28–38. doi: 10.1016/s1044-7431(03)00032-0. |
[206] | Yue F , Li W , Zou J , Chen Q , Xu G , Huang H , et al., Blocking the association of HDAC4 with MAP1S accelerates autophagy clearance of mutant Huntingtin, Aging (Albany NY) (2015) ;7: (10):839–53. doi: 10.18632/aging.100818. |
[207] | Zainelli GM , Ross CA , Troncoso JC , Fitzgerald JK , Muma NA , Calmodulin regulates transglutaminase 2 cross-linking of huntingtin, J Neurosci. (2004) ;24: (8):1954–61. doi: 10.1523/JNEUROSCI.4424-03.2004. |
[208] | Zainelli GM , Ross CA , Troncoso JC , Muma NA , Transglutaminase cross-links in intranuclear inclusions in Huntington disease, J Neuropathol Exp Neurol (2003) ;62: (1):14–24. doi: 10.1093/jnen/62.1.14. |
[209] | Zala D , Colin E , Rangone H , Liot G , Humbert S , Saudou F , Phosphorylation of mutant huntingtin at S421 restores anterograde and retrograde transport in neurons, Hum Mol Genet (2008) ;17: (24):3837–46. doi: 10.1093/hmg/ddn281. |
[210] | Zheng S , Ghitani N , Blackburn JS , Liu JP , Zeitlin SO , A series of N-terminal epitope tagged Hdh knock-in alleles expressing normal and mutant huntingtin: Their application to understanding the effect of increasing the length of normal Huntingtin’s polyglutamine stretch on CAG140 mouse model pathogenesis. Mol Brain (2012) ;5: :28. doi: 10.1186/1756-6606-5-28. |
[211] | Zuccato C , Tartari M , Crotti A , Goffredo D , Valenza M , Conti L , et al., Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes, Nat Genet. (2003) ;35: (1):76–83. doi: 10.1038/ng1219. |
[212] | Zucchelli S , Marcuzzi F , Codrich M , Agostoni E , Vilotti S , Biagioli M , et al., Tumor necrosis factor receptor-associated factor 6 (TRAF6) associates with huntingtin protein and promotes its atypical ubiquitination to enhance aggregate formation, J Biol Chem. (2011) ;286: (28):25108–17. doi: 10.1074/jbc.M110.187591. |
[213] | Safran M , Rosen N , Twik M , BarShir R , Iny Stein T , Dahary D , et al., The GeneCards Suite. In: Abugessaisa I, Kasukawa T, editors. Practical Guide to Life Science Databases. Singapore: Springer; (2022) . pp. 27–56. https://doi.org/10.1007/978-981-16-5812-92. |
[214] | UniProt Consortium T. UniProt: The universal protein knowledgebase, Nucleic Acids Res (2018) ;46: (5)::2699. doi: 10.1093/nar/gky092.. |
[215] | Szklarczyk D , Gable AL , Nastou KC , Lyon D , Kirsch R , Pyysalo S , et al., The STRING database in Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets, Nucleic Acids Res (2021) ;49: (D1):D605–D612. doi: 10.1093/nar/gkaa1074. |
[216] | Blondel VD , Guillaume JL , Lambiotte D , Lefebvre E , Fast unfolding of communities in large networks. J Stat Mech. 2008;P10008. |
[217] | Ashburner M , Ball CA , Blake JA , Botstein D , Butler H , Cherry JM , et al., Gene ontology: Tool for the unification of biology, The Gene Ontology Consortium. Nat Genet (2000) ;25: (1):25–9. doi: 10.1038/75556. |
[218] | Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine, Nucleic Acids Res (2021) ;49: (D1):D325–34. doi: 10.1093/nar/gkaa1113. |
[219] | Kanehisa M Post-genome informatics. Oxford: Oxford University Press; (2000) . |
[220] | Chen J , Bardes EE , Aronow BJ , Jegga AG , ToppGene Suite for gene list enrichment analysis and candidate gene prioritization, Nucleic Acids Res (2009) ;37: (Web Server issue):W305–11. doi: 10.1093/nar/gkp427. |
[221] | Schilling G , Becher MW , Sharp AH , Jinnah HA , Duan K , Kotzuk JA , et al., Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin, Hum Mol Genet (1999) ;8: (3):397–407. doi: 10.1093/hmg/8.3.397. |
[222] | Eshraghi M , Karunadharma PP , Blin J , Shahani N , Ricci EP , Michel A , et al., Mutant Huntingtin stalls ribosomes and represses protein synthesis in a cellular model of Huntington disease, Nat Commun (2021) ;12: (1):1461. |
[223] | Kegel-Gleason KB , Huntingtin interactions with membrane phospholipids: Strategic targets for therapeutic intervention?, J Huntingtons Dis (2013) ;2: (3):239–50. doi: 10.3233/JHD-130068. |
[224] | King AC , Wood TE , Rodriguez E , Parpura V , Gray M , Differential effects of SNARE-dependent gliotransmission on behavioral phenotypes in a mouse model of Huntington’s disease, Exp Neurol (2020) ;330: :113358. doi: 10.1016/j.expneurol.2020.113358. |
[225] | Schilling J , Broemer M , Atanassov I , Duernberger Y , Vorberg I , Dieterich C , et al., Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington’s disease, J Mol Biol (2018) ;431: (9):1869–77. doi: 10.1016/j.jmb.2019.01.034. |
[226] | Neueder A , Dumas AA , Benjamin AC , Bates GP . Regulatory mechanisms of incomplete huntingtin mRNA splicing, Nat Commun (2018) ;9: (1):3955. doi: 10.1038/s41467-018-06281-3. |
[227] | Ortega Z , Lucas JJ , Ubiquitin-proteasome system involvement in Huntington’s disease, Front Mol Neurosci (2014) ;7: :77. doi: 10.3389/fnmol.2014.00077. |
[228] | Fiorillo A , Morea V , Colotti G , Ilari A , Huntingtin ubiquitination mechanisms and novel possible therapies to decrease the toxic effects of mutated huntingtin, J Pers Med (2021) ;11: (12):1309. doi: 10.3390/jpm11121309. |
[229] | DiFiglia M , Huntingtin fragments that aggregate go their separate ways, Mol Cell (2002) ;10: (2):224–5. doi: 10.1016/s1097-2765(02)00609-3. |
[230] | Chen MZ , Mok SA , Ormsby AR , Muchowski PJ , Hatters DM , N-Terminal fragments of Huntingtin longer than residue 170 form visible aggregates independently to polyglutamine expansion, J Huntingtons Dis (2017) ;6: (1):79–91. doi: 10.3233/JHD-160207. |
[231] | Gutekunst CA , Levey AI , Heilman CJ , Whaley WL , Yi H , Nash NR , et al., Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies, Proc Natl Acad Sci U S A (1995) ;92: (19):8710–4. doi: 10.1073/pnas.92.19.8710. |
[232] | Gutekunst CA , Li SH , Yi H , Mulroy JS , Kuemmerle S , Jones R , et al., Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology, J Neurosci (1999) ;19: (7):2522–34. doi: 10.1523/JNEUROSCI.19-07-02522.1999. |
[233] | Sapp E , Schwarz C , Chase K , Bhide PG , Young AB , Penney J , et al., Huntingtin localization in brains of normal and Huntington’s disease patients, Ann Neurol (1997) ;42: (4):604–12. doi: 10.1002/ana.410420411. |
[234] | Aaronson J , Beaumont V , Blevins RA , Andreeva V , Murasheva I , Shneyderman A , et al., HDinHD: A rich data portal for Huntington’s disease research, J Huntingtons Dis (2021) ;10: (3):405–12. |
[235] | Haenig C , Atias N , Taylor AK , Mazza A , Schaefer MH , Russ J , et al., Interactome mapping provides a network of neurodegenerative disease proteins and uncovers widespread protein aggregation in affected brains, Cell Rep (2020) ;32: (7):108050. doi: 10.1016/j.celre2020.108050. |
[236] | Sap KA , Guler AT , Bury A , Dekkers D , Demmers JAA , Reits EA , Identification of full-length wild-type and mutant Huntingtin Interacting proteins by crosslinking immunoprecipitation in mice brain cortex, J Huntingtons Dis (2021) ;10: (3):335–47. doi: 10.3233/JHD-210476. |
[237] | Vodicka P , Mo S , Tousley A , Green KM , Sapp E , Iuliano M , et al., Mass spectrometry analysis of wild-type and knock-in Q140/Q140 Huntington’s disease mouse brains reveals changes in glycerophospholipids including alterations in phosphatidic acid and lyso-phosphatidic acid, J Huntingtons Dis (2015) ;4: (2):187–201. doi: 10.3233/JHD-150149. |
[238] | Lee CY , Cantle JP , Yang XW , Genetic manipulations of mutant huntingtin in mice: New insights into Huntington’s disease pathogenesis, FEBS J (2013) ;280: (18):4382–94. doi: 10.1111/febs.12418. |
[239] | Farshim PP , Bates GP . Mouse models of Huntington’s disease, Methods Mol Biol (2018) ;1780: :97–120. doi: 10.1007/978-1-4939-7825-0_6. |


