
Biological Aging and the Cellular Pathogenesis of Huntington’s Disease
Abstract
Huntington’s disease (HD) is a fatal, inherited neurodegenerative disorder caused by a mutation in the huntingtin gene (HTT). While mutant HTT is present ubiquitously throughout life, HD onset typically occurs in mid-life, suggesting that aging may play an active role in pathogenesis. Cellular aging is defined as the slow decline in stress resistance and accumulation of damage over time. While different cells and tissues can age at different rates, 9 hallmarks of aging have emerged to better define the cellular aging process. Strikingly, many of the hallmarks of aging are also hallmarks of HD pathology. Models of HD and HD patients possess markers of accelerated aging, and processes that decline during aging also decline at a more rapid rate in HD, further implicating the role of aging in HD pathogenesis. Furthermore, accelerating aging in HD mouse and patient-derived neurons unmasks HD-specific phenotypes, suggesting an active role for the aging process in the onset and progression of HD. Here, we review the overlap between the hallmarks of aging and HD and discuss how aging may contribute to pathogenesis in HD.
INTRODUCTION
Huntington’s disease (HD) is an autosomal dominant, progressive and eventually fatal neurodegenerative disease caused by an expanded polyglutamine-encoding CAG tract in the Huntingtin (HTT) gene, which gets translated into a mutant HTT (mtHTT) protein [1]. mtHTT disrupts several processes in the cell, eventually leading to dysfunction and death of the caudate and putamen, and the cerebral cortex. HD is characterized by psychiatric, cognitive, and progressive movement abnormalities [2]. Age-of-onset of disease is negatively correlated with the number of CAG repeats [3–6], with motor symptoms beginning between ages 35–45 and death occurring 10–20 years after motor-onset [2]. However, the number of repeats only partially explains age-of-onset; individuals with identical tract lengths can have disease onset many decades apart [7]. This suggests that both genetic and environmental factors outside the CAG repeat can influence disease pathogenesis.
Although there is evidence which suggests that neurodevelopmental abnormalities can occur in adult-onset HD [8, 9], those with the mutation that causes adult-onset HD can live several decades without overt symptoms. This suggests that aging, particularly in adult-onset HD, may play a role in pathogenesis. However, how aging may contribute to HD pathogenesis is controversial. Two primary hypotheses have arisen: (1) Aging passively contributes to HD by the simple passage of time, where mtHTT-induced microdamage accumulates over many decades before reaching a threshold of disease onset, or (2) events that occur during the aging process sensitize neurons/the brain to the mtHTT insult and actively contribute to disease onset and/or progression.
Aging is a risk factor for many diseases, including neurodegenerative disease [10]. While chronological aging is defined as the number of years an organism is alive, biological aging describes the overall health of the organism [11, 12]. Biological aging is defined by 9 hallmarks: telomere attrition, epigenetic alterations, altered intercellular communication, dysregulated nutrient sensing, loss of proteostasis, mitochondrial dysfunction, genomic instability, stem cell exhaustion, and cellular senescence [13]. While both chronological and biological age are risk factors for disease, biological aging seems to play a larger role in disease burden of many age-related diseases [14–16]. This suggests that processes that decline during aging play an active role in disease. In HD, many of these aging hallmarks are also hallmarks of disease pathogenesis. Here, we discuss the evidence for accelerated aging in HD, the overlap between the cellular hallmarks of aging and the cellular pathogenesis of HD, and how aging may exacerbate toxicity in HD.
EVIDENCE FOR ACCELERATED AGING IN HUNTINGTON’S DISEASE
Telomere attrition
Telomeres are repetitive TTAGGG nucleotide sequences that act as a cap at the end of chromosomes that insulate them from attrition during replication [17]. Telomeres are maintained by telomerase, an enzyme that is relatively silent throughout the body shortly after birth. Thus, telomeres undergo successive shortening with age, and biological aging has been linked to shortened telomeres [18]. In addition to biological age, shortened telomeres are associated with several diseases, including mood disorders, cancer, type 2 diabetes, and chronic obstructive pulmonary disease [19–22]. While telomere attrition may not occur is post-mitotic neurons [23], several neurodegenerative diseases have shortened telomeres in leukocytes compared to age and sex-matched control individuals [24, 25]. Interestingly, of all neurodegenerative diseases, HD leukocytes have the shortest telomeres [24]. This was also shown in a follow-up study [26], and could potentially be used as a peripheral blood biomarker for HD progression [27, 28]. In addition, there is an association between age-of-onset of HD and SNPs that influence telomere length [29], further demonstrating an association between telomere shortening, a marker of advanced biological aging, and HD.
Epigenetic alterations
The epigenetic landscape of DNA and histones changes with age. Widespread DNA hypomethylation and loss of heterochromatin have been described [30]. In addition, in the aging brain, distinct patterns of epigenetic and, consequently, transcriptional changes occur. In this way, the epigenetic landscape of DNA and chromatin has come to be a predictor of biological age [31–33]. In the brains of HD patients, accelerated epigenetic aging has been shown in the striatum, the most affected part of the brain in HD [34]. Taken together with data from telomeres in HD, this demonstrates that HD brains also have accelerated biological age compared to age-matched controls. This is in concordance with the findings of others that show accelerated brain epigenetic aging in other neurological disorders, including Alzheimer’s disease, Down’s syndrome, and HIV-associated neurocognitive disorders [35–37].
OTHER HALLMARKS OF AGING INVOLVED IN HUNTINGTON’S DISEASE PATHOGENESIS
Altered inter- and intra-cellular communication and stress signaling
Intercellular communication refers to the non-autonomous signaling between cells and tissues, whereby one tissue can have an effect on the other. One of the most common examples of intercellular communication is inflammation. Immune cells such as neutrophils, B cells, T cells, macrophages, astrocytes and microglia release inflammatory signals to surrounding cells as a defense system against cellular harm [38]. Additionally, non-immune cells under stress can release cytokines and chemokines to surrounding cells to boost defenses against foreign pathogens [39]. Together, this response is meant to be acute and resolved by subsequent release of anti-inflammatory factors and tissue remodeling [40]. Throughout aging, however, pro-inflammatory signaling is elevated, and anti-inflammatory factors become diminished, leading to chronic inflammation, deemed “inflamm-aging.” For a detailed review of inflamm-aging pathways, we refer the reader to [41].
Reactive microglia have been found in HD brains that correlate with HD pathology [42, 43]. Macrophages, monocytes, and microglia isolated from YAC128 and R6/2 mice as well as from peripheral blood of HD patients are hyperactive upon stimulation [44, 45], similar to the stimulus-induced hyperactivity observed in microglia from aged mice [46]. Astrocytes can also be dysfunctional in HD brains, with a decreased ability to take up glutamate and increased glutamate release [47, 48]. Interestingly, glial cells from the white matter of adult brains possess shorter telomeres than grey matter glial cells [23]. This could contribute to the observed white matter loss in HD patients prior to grey matter loss, and may suggest that “older” glial cells contribute to HD pathology at earlier stages. Pro-inflammatory markers such as IL-6 and matrix metalloproteinase 3 (MMP3) are also elevated in the plasma and CSF of HD patients, respectively [44, 45], suggesting increased systemic inflammation in HD. Additionally, in mice with mtHTT expressed only in microglia, there is an increase in neuronal cell death [49]. Moreover, reducing mtHTT in astrocytes slows progression of phenotypes in BACHD mice [50], further providing evidence for the role of non-autonomous contributions from glia to HD pathology.
In addition to glial and peripheral immune cell contributions to inflammation, several intracellular pathways can also lead to increased inflammation and cellular dysregulation in both aging and HD. Elevated levels of reactive oxygen species (ROS), which we discuss in greater detail below, can chronically activate nuclear factor kappa B (NF-κB), a transcription factor that induces expression of pro-inflammatory genes [51]. RNA sequencing of HD patient brain found upregulation of several NF-κB target genes [52], and inhibition of NF-κB signaling in astrocytes is sufficient to delay phenotypes in R6/2 mice [53]. Mammalian sterile 20-like kinase1 (MST1) was also found to be activated in post-mortem HD cortical tissue and CAG knock-in Q111 mice [54]. MST1 promotes the innate immune response, increasing inflammation [55]. Furthermore, wildtype HTT is a stress response protein, serving as a scaffold protein for the recruitment of DNA damage response proteins and early endosomes [56–59]. mtHTT does not recover from these responses as quickly, resulting in aberrant signaling and associated stress responses, which can indirectly activate immune response pathways. Together, these aberrant stress signaling pathways coordinate not only to increase damage in HD, but can also cause damaging chronic inflammation.
Dysregulated nutrient sensing
In cells, the master regulator of nutrient sensing is insulin-like growth factor (IGF-1). IGF-1 is activated by glucose, in a similar manner to insulin, and this pathway, which is the most-conserved pathway in organisms, is referred to as the insulin and IGF-1 signaling (IIS) pathway [60]. In aging, IGF-1 declines and contributes to aging [61], although paradoxically, reducing activity of downstream effectors of IGF-1 such as mTOR, FOXO, or Akt promote longevity [62, 63]. This is hypothesized to be because of the slowed metabolism and growth of organisms with decreased IIS activity [64].
Dysregulated nutrient sensing also occurs in HD patients. However, contrary to the hypometabolism often seen in the elderly, patients with HD most often exhibit hypermetabolism, with high rates of weight loss despite an increase in caloric intake [65]. FOXO, mTOR, and Akt, all downstream effectors of the IIS pathway are dysregulated in HD patient brains and models of HD. Activation of the IIS pathway via modulation of FOXO, mTOR, or Akt is protective in HD models by ameliorating mitochondrial function and metabolic rates [66–71]. Somewhat paradoxically, although IIS activation is protective in HD models, intermittent fasting, which reduces IGF-1 levels, promotes clearance of mtHTT in YAC128 mice [72]. To support this, compounds such as rapamycin, which blocks mTOR activity, and metformin, which inhibits IIS, promote autophagic clearance of mtHTT and improve phenotypes in HD mice [73–75]. Furthermore, plasma IGF-1 levels were found to be positive predictor of cognitive decline in HD patients [76]; thus, how exactly the IIS pathway can be protective in HD is still debated.
Loss of proteostasis
Proteostasis, or ‘protein homeostasis’, refers to the process of quality control regulating protein abundance and conformation. The network of proteins involved in maintaining proteostasis include proteins involved in translation, molecular chaperones, which aid in re-folding misfolded proteins, as well as the ubiquitin-proteasome system (UPS) and lysosome/autophagy machinery, which degrade excess and damaged proteins [77, 78]. Canonical protein degradation involves tagging proteins with mono or poly-ubiquitin chains to be recognized by the proteasome or autophagosomes [79–81]. During the process of aging, the proteostasis network in the brain declines in function, evidenced by the appearance of protein aggregates even in aging brains without apparent neurodegenerative disease [82–84]. This is thought to be due, at least in part, to an increase in errors in translation and protein misfolding as a result of accumulated damage and subsequent oxidative and carbonylation of proteins [84].
Autophagosomes and lysosomes decrease in number and activity with aging in several tissues [85, 86]. Furthermore, inhibition of autophagy leads to premature aging phenotypes [87], and stimulating autophagy by caloric restriction and pharmacologic stimulation of upstream autophagy pathways promotes longevity in nearly all model organisms from C. elegans to rhesus monkeys [88]. Additionally, the UPS and lysosome/autophagy pathway become dysregulated with age, with shifts in the ubiquitylation of the proteome and a decrease in the activity of the proteasome and autophagic machinery, further contributing to the decline in protein integrity observed in the aging brain [77].
A decline in overall proteostasis has been linked to disease progression in models of HD [89, 90]. Aggregates of mutant HTT have been shown to sequester ubiquitin as well as components of the proteasome [91], which could result in decreased proteasome function, although this hasn’t directly been linked to cellular decline. It is possible that proteasome sequestration is dynamic, and may not occur at a high level all the time but may be enough to slow down the proteostasis network as a whole [92]. Interestingly, while primary cortical neurons from HD mice do not exhibit aggregation of mtHTT, we recently found that inducing aging in these neurons caused oligomers of mtHTT to form [93]. Additionally, the E3 ubiquitin ligase WWP1 was found to aberrantly ubiquitinate mutant HTT and impair degradation in an age-dependent manner [94], further suggesting a relationship between age and mtHTT toxicity.
Contrary to what is typically observed during aging, autopsied brains of HD patients have increased size and number of autophagic vesicles [95]. Additionally, staining of LC3II, a marker of autophagic vesicles, is increased in HD striatal neurons [96], and UBR5, a ubiquitin ligase responsible for proteosomal degradation is increased in HD iPSCs [97]. Despite this increase, overexpression of UBR5 is protective in HD iPSCs, suggesting either dysfunction in this pathway or an overwhelming of the proteosomal system in HD cells [97]. Indeed, despite this increase in vesicle size and number, actual flux of some proteins and organelles is decreased, possibly due to defects in cargo recognition [98]. wtHTT can function as a scaffold for autophagy [99], and partial loss of this function by mtHTT may contribute to autophagy defects. While the mechanism of autophagy impairment in aging and HD is slightly different, the result is similar: an accumulation of damaged proteins and organelles. One such organelle is the mitochondria, which are turned over by the autophagy/lysosomal pathway. Damaged mitochondria accrue in aged tissues as well as HD neurons, creating a feed-forward loop for both ROS damage and lysosomal impairment, which are particularly susceptible to ROS damage [100].
Mitochondrial dysfunction
There is extensive evidence for mitochondrial dysfunction in HD patients and in many models of HD. This was originally discovered by in vivo positron emission tomography (PET) imaging of brains of HD patients, where cerebral metabolism defects and atrophy were found [101]. These metabolic defects occurred early and severity was associated with functional decline in HD patients [102–105]. Follow-up studies in animal models confirmed altered metabolic rates and found distinct changes in mitochondrial complexes [106–111], although this is not always the case [112, 113]. Decreased cerebral glucose metabolism is also a characteristic of the aging brain [114–116].
Nicotinamide adenine dinucleotide (NAD+) is a metabolite essential for cellular function that has been shown to decline with age [117–119]. NAD+ can be synthesized de novo from its precursor tryptophan, through the Preiss-Handler pathway, or through the recycling of components from enzymes that consume NAD+ to carry out metabolic functions [120]. This process is dependent on nicotinamide phosphoribosyltransferase (NAMPT), an enzyme that converts salvaged nicotinamide to NAD+ [121]. The main causes of NAD+ decline in aging are a decrease in Nampt expression [122] and an increase in NAD+-dependent activity by the poly-(ADP-ribose) polymerases (PARPs), Sirtuins (SIRTs), and CD38 [123]. In line with this, exogenous nicatinamide upregulates BDNF, and PGC-1α gene expression and improves motor phenotypes in R6/1 HD mice [124]. PARPs are enzymes that create ADP polymer chains using NAD+, which occurs during DNA repair [125]. PARP activity, particularly PARP1 activity, is increased in aging tissue, possibly due to the accumulation of DNA damage [126]. This increased utilization of NAD+ by PARP1 is hypothesized to be the cause of the eventual reduction in SIRT activity [127]. PARP1 hyper-activity has also been observed in post-mortem HD brains [128]. In addition, inhibition of PARP1 activity is neuroprotective in R6/2 HD mice [129]. Similar to findings in aged brains, SIRT1 and SIRT3 expression are decreased in cultured HD neurons and HD brains [130–132], and SIRT1 and SIRT3 activation is beneficial HD neurons as well as R6/1 and YAC128 HD mice [130, 132, 133]. While accelerated aging has not been directly linked to the observed changes in enzymatic activity of NAD+-consuming enzymes, the fact that levels change in a similar manner in both aging and HD is suggestive of an accelerated aging component to HD pathogenesis.
In addition to cellular metabolic defects, many models of HD as well as post-mortem brains from HD patients have shown defects in mitochondrial structure and function in similar ways to mitochondrion from the aging brain. Mitochondria are dynamic, networked organelles, undergoing fission or fusion with the network in response to changing cellular environments [134]. In the aging brain, there is decreased abundance in mitochondria and a change in shape toward smaller, rounded, and less-networked mitochondria in many cell types, including neurons [135–138]. In addition to changing structure, mitochondria from aged tissues have decreased oxidative phosphorylation and ATP production [139–141]. In HD, similar observations have been made in neuronal mitochondria. Abnormal mitochondrial dynamics and increased activity of the GTPase DRP1, responsible for mitochondrial fission, have been reported in HD models and post-mortem brains of HD patients [142–145].
Decreased calcium handling is characteristic of mitochondria from aging neurons, and is also observed in HD brains. Mitochondria take up calcium through the calcium uniporter, which helps to buffer calcium input in neurons [146]. Mitochondria from aging neurons do not do this effectively [147]. Calcium handling defects are also observed in transgenic HD mice and rats as well as lymphoblasts from HD patients [148–150], although this is not seen in some HD mouse neurons until they are challenged with NMDA [151, 152].
ROS are byproducts of cellular metabolism that are typically cleared by endogenous antioxidants in the cell [153, 154]. While ROS and antioxidants are typically in homeostatic balance in the cell, ROS can overwhelm antioxidant systems, causing damage to DNA, RNA, proteins, and organelles [155].Throughout the process of aging, this homeostatic imbalance can become pronounced. Antioxidant protein levels and activity decline [156, 157], and age-related disruptions in mitochondrial activity cause more ROS generation, and subsequent damage to DNA and biomolecules [158]. In a similar manner, HD neurons have decreased antioxidant activity, and increased ROS and ROS-induced damage [159–161]. However, recently, we found that in primary cortical neurons from humanized HD mice, the ROS-induced damage and hypersensitivity to oxidative stress observed in HD neurons is dependent upon biological age [93], suggesting that aging uncovers stress-induced phenotypes in HD. Moreover, through aging ROS can promote cellular senescence [162], and recently, RNA sequencing of brains from HD knock-in mice revealed that striata from symptomatic HD mice possess gene signatures associate with cellular senescence [163].
Cellular senescence
Cellular senescence refers to the cell cycle arrest of replicative cells due to aging-related factors [164]. Senescent cells exist in a pro-inflammatory state and can release cytokines, proteases, and growth factors collectively known as a senescence-associated secretory phenotype (SASP) [165]. While senescence in this sense doesn’t apply to terminally differentiated cells such as neurons, aged neurons have increased expression of pro-inflammatory markers, markers of senescence such as p53 and p16INK4A, tumor suppressors that can also trigger senescence-related transcriptional changes in aging, and an increase in senescence-associated β-galactosidase activity [166]. In HD, mtHTT can bind to p53 and increase protein activity [167]. p16 and p53 gene expression are also increased in the striata of HD knock-in mice [163]. Furthermore, both astrocytes and microglia from aged brains acquire SASPs [168, 169]. Furthermore, there is an increase in pro-inflammatory cytokines and matrix metalloproteinases such as MMP-9 in HD mice as well as post-mortem brains of patients with HD [170].
DNA damage and genomic instability
Genomic instability refers to the frequency of mutations within the genome of cells. Chromosomal changes and DNA mutation rate increase with age [171–174]. Mutations in aging are thought to be caused, at least in part, by failure to repair or improper repair of DNA damage. DNA damage continuously occurs in cells due to free radical damage and environmental agents, such as UV rays. DNA damage comes in several forms, including insertions/deletions/substitutions, bulky adducts, single-strand breakage and double-strand breakage [175–177]. Damage accrued independently of replication can be repaired by one of four known pathways, each with multiple scaffolds and polymerases [178]. While each DNA damage response (DDR) pathway has a canonical lesion to repair, there is significant crosstalk between pathways. For a comprehensive review on DDR pathways, see [178].
Throughout the course of aging, several forms of DNA damage can occur, with the oxidative lesion, 8-oxodeoxyguanine (8-oxo-dG), being one of the most prevalent [179–182]. 8-oxo-dG is repaired primarily through the base-excision repair (BER) pathway [182], although both nuclear excision repair (NER) and mismatch repair (MMR) can also repair oxidative lesions [183]. Repair of oxidative lesions is initiated by 8-oxoguanine glycosylase (OGG1), which removes the lesion [184]. The resulting gap in DNA triggers PARP1, which uses NAD+ to add poly(ADP-ribose) chains around the gap, recruiting polymerases to fill the gap [185]. This is important because, throughout the course of biological aging, there is evidence that OGG1-dependent activity can trigger the aging process [183]. Furthermore, aging-related increases in DNA damage and/or aberrant repair is hypothesized to be a cause of age-related increase in cancer risk. However, if too much damage occurs, cellular senescence or apoptosis can be triggered [186].
While there is currently no evidence for a change in overall genomic instability in HD [187, 188], nuclear DNA modifications and increased double-stranded breaks have been observed [189, 190], and there is somatic instability of the CAG repeat itself. Additionally, there is evidence for disrupted nuclear pore complexes in HD [191], which also occurs in aging [192]. Breakdown of the nuclear pore complex can cause excessive DNA damage and aberrant intracellular communication, another hallmark of aging [193, 194]. These events combined may contribute to or cause instability in HD neurons.
There is an inverse relationship between somatic instability of the CAG repeat and age-of-onset of disease [195–197]. Instability may even be necessary in the brain for pathogenesis [197–199]. This instability seems to be dependent upon DNA repair pathways; crossing mouse models of HD with mice deficient in MMR (Msh2, Msh3, Mlh1, or Mlh3 –/–mice) or BER (Ogg1 or Neil1 –/–mice) attenuates instability of the CAG tract and delays HD-relevant phenotypes [198, 200, 201]. Furthermore, a GWAS performed on tissue from HD patients found that several SNPs which influence age-of-onset of HD were found at or near genes involved in the DDR [202]. In a follow-up GWAS investigating genetic modifiers of HD progression, there was a significant signal in MSH3, an MMR protein [203]. This provides further evidence for the role of DNA damage-associated pathways in HD. Recent work from our group demonstrated that accelerating biological age of HD mouse neurons or brains or neurons derived from HD patient iPSCs results in a dramatic and selective increase in DNA damage that can be exacerbated by cellular stress [93], although it is still currently unclear whether age-associated DNA damage can influence instability. Some DDR pathway proteins show a decline in efficiency with age [204], leaving the possibility that aging contributes to somatic expansion by creating heightened, but inefficient DDR pathways. Alternatively, somatic expansion could be triggered as a direct result of increased DNA damage and associated increases in repair, before age-associated declines in DDR efficiency. Both increased DNA damage and inefficient DDR pathways, specifically in the MMR pathway, contribute to microsatellite instability in aging [205]. However, how these pathways change in the brain with aging is still unknown; therefore, how exactly this hallmark may contribute to somatic instability in HD is still unknown and of great importance to HD research.
CONCLUSION AND FUTURE PERSPECTIVES
Many of the hallmarks of aging are also mechanisms of pathogenesis in HD. While all these processes decline with age, they seem to decline more rapidly in patients with HD, suggesting a synergistic interplay. In many adult-onset neurodegenerative diseases, the role of biological aging in onset and progression of disease is being actively investigated. In Parkinson’s and Alzheimer’s diseases, for example, several groups have found links between aging and neurodegeneration [206–208]. In adult-onset HD, the pathogenesis is similar; although HD patients have the gene mutation from birth, it takes several decades for the mutation to manifest; begging the question of what exactly triggers disease-onset. While it could simply be the passage of time, there is overwhelming overlap between the hallmarks of aging and cellular alteration in HD, creating the hypothesis that delaying biological aging could delay onset or progression of HD symptoms as well. In this hypothesis, the aging process plays an active role in HD pathogenesis; exacerbating the toxicity of mutant HTT (Fig. 1).
Fig.1
Proposed pathway for how aging may contribute to HD pathogenesis.
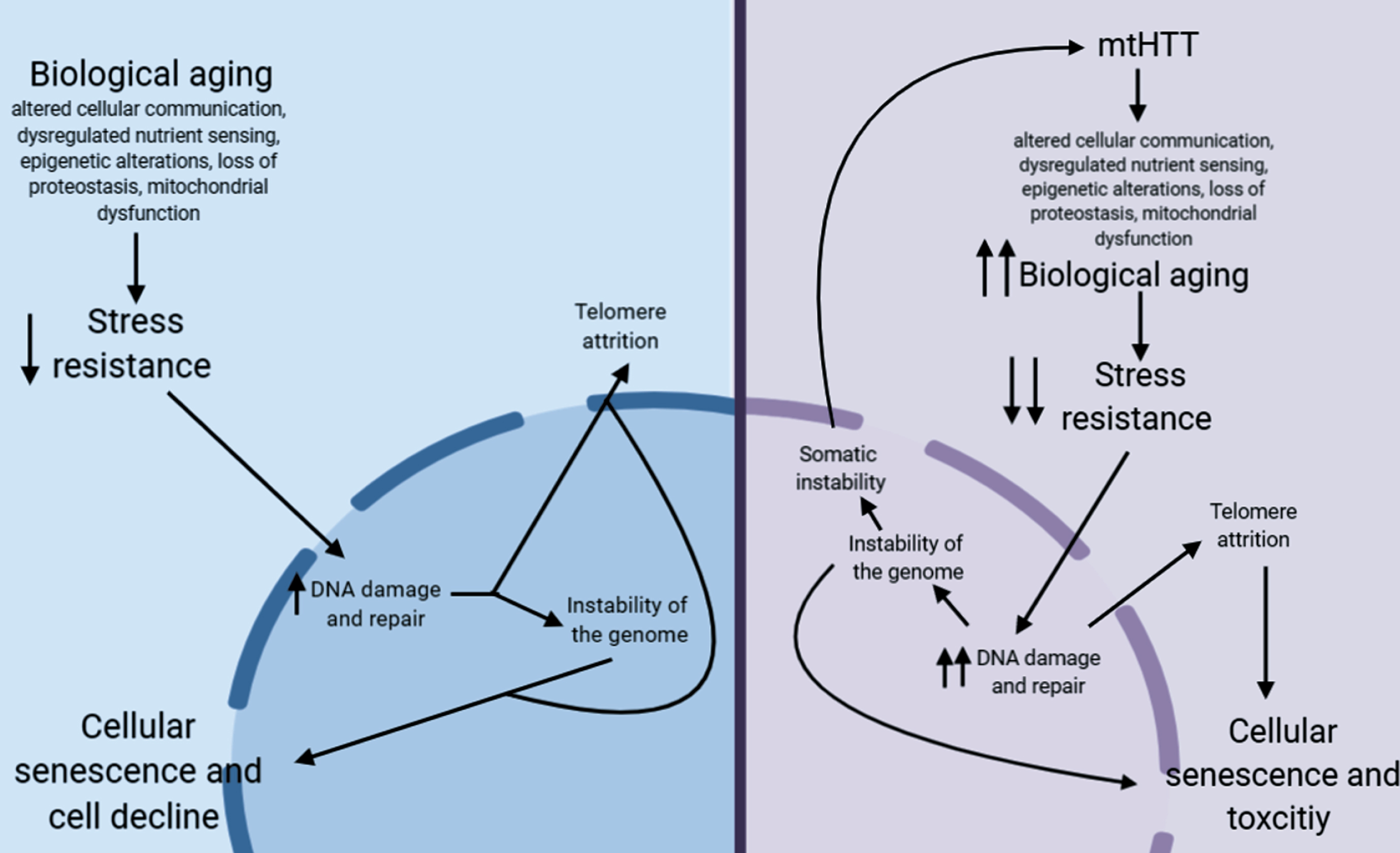
In non-HD neurons, biological aging occurs over time, which eventually decreases stress resistance in cells [209, 210]. This decrease in stress resistance leads to an increase in unrepaired DNA damage, which is hypothesized to cause damage to telomeres leading to telomere attrition [211, 212], and can lead to instability of the genome, particularly at microsatellite repeats [213]. Together, this leads to cellular senescence, another aging hallmark, and eventual cellular decline. In HD neurons, mtHTT accelerates aging by exacerbating several hallmarks of aging. This may lead to the hyper-susceptibility of HD neurons to stress, a further increase in DNA damage, and, as a result, accelerated telomere attrition. This increase in DNA damage could also lead to an increase in somatic instability, further increasing toxicity of mtHTT.
Another point of investigation in the role of aging in HD is how inducing aging in model systems may uncover HD-relevant phenotypes in adult-onset CAG tract length models. Currently, most models of adult-onset HD at the most common pathological repeat lengths display little to no phenotypes. Inducing aging in these models may not only uncover phenotypes, but may provide models systems that are more relevant to the pathology of adult-onset HD.
The overlap between the hallmarks of aging and cellular pathogenesis of HD as well as the accelerated aging found in HD models and patient brain provides rationale for further investigation into the role aging in HD. While we acknowledge the toxicity of mtHTT on its own, in both aging and HD, many of these ‘hallmarks’ of cellular pathogenesis converge on common pathways and can synergistically cause cellular toxicity. Thus, anti-aging therapies could be beneficial for multiple components of HD. Future work should include how modulating aging might affect pathogenesis, including somatic instability.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | MacDonald M , Ambrose C , Duyao M , Datson N . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. (1993) ;72: (6):971–83. |
[2] | Bates GP , Dorsey R , Gusella JF , Hayden MR , Kay C , Leavitt BR , et al. Huntington disease. Nat Rev Dis Primers. (2015) ;1: :15005. |
[3] | Wright GEB , Collins JA , Kay C , McDonald C , Dolzhenko E , Xia Q , et al. Length of uninterrupted CAG, independent of polyglutamine size, results in increased somatic instability, hastening onset of Huntington disease. Am J Hum Genet. (2019) ;104: (6):1116–26. |
[4] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Electronic address: ; Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. CAG repeat not polyglutamine length determines timing of Huntington’s disease onset. Cell. (2019) ;178: (4):887–900 e14. |
[5] | Snell RG , Macmillan JC , Cheadle JP , Fenton I , Lazarou LP , Davies P , et al. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat Genet. (1993) ;4: (4):393–7. |
[6] | Andrew SE , Goldberg YP , Kremer B , Telenius H , Theilmann J , Adam S , et al. The relationship between trinucleotide (Cag) repeat length and clinical-features of Huntington disease. Am J Hum Genet. (1993) ;53: (3):1118. |
[7] | Wexler NS , Lorimer J , Porter J , Gomez F , Moskowitz C , Shackell E , et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc Natl Acad Sci U S A. (2004) ;101: (10):3498–503. |
[8] | Wiatr K , Szlachcic WJ , Trzeciak M , Figlerowicz M , Figiel M . Huntington disease as a neurodevelopmental disorder and early signs of the disease in stem cells. Mol Neurobiol. (2018) ;55: (4):3351–71. |
[9] | Mathkar PP , Suresh D , Dunn J , Tom CM , Mattis VB . Characterization of neurodevelopmental abnormalities in iPSC-derived striatal cultures from patients with Huntington’s disease. J Huntingtons Dis. (2019) ;8: (3):257–69. |
[10] | Hou Y , Dan X , Babbar M , Wei Y , Hasselbalch SG , Croteau DL , et al. Ageing as a risk factor for neurodegenerative disease. Nat Rev Neurol. (2019) ;15: (10):565–81. |
[11] | Jylhava J , Pedersen NL , Hagg S . Biological age predictors. EBioMedicine. (2017) ;21: :29–36. |
[12] | Jazwinski SM , Kim S . Examination of the dimensions of biological age. Front Genet. (2019) ;10: :263. |
[13] | Lopez-Otin C , Blasco MA , Partridge L , Serrano M , Kroemer G . The hallmarks of aging. Cell. (2013) ;153: (6):1194–217. |
[14] | Hayflick L . The not-so-close relationship between biological aging and age-associated pathologies in humans. J Gerontol A Biol Sci Med Sci. (2004) ;59: (6):B547–50; discussion 51-3. |
[15] | Codd V , Nelson CP , Albrecht E , Mangino M , Deelen J , Buxton JL , et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat Genet. (2013) ;45: (4):422–7, 7e1-2. |
[16] | Bulterijs S , Hull RS , Bjork VC , Roy AG . It is time to classify biological aging as a disease. Front Genet. (2015) ;6: :205. |
[17] | Moyzis RK , Buckingham JM , Cram LS , Dani M , Deaven LL , Jones MD , et al. A highly conserved repetitive DNA sequence, (TTAGGG)n, present at the telomeres of human chromosomes. Proc Natl Acad Sci U S A. (1988) ;85: (18):6622–6. |
[18] | Zvereva MI , Shcherbakova DM , Dontsova OA . Telomerase: Structure, functions, and activity regulation. Biochemistry (Mosc). (2010) ;75: (13):1563–83. |
[19] | Simon NM , Smoller JW , McNamara KL , Maser RS , Zalta AK , Pollack MH , et al. Telomere shortening and mood disorders: Preliminary support for a chronic stress model of accelerated aging. Biol Psychiatry. (2006) ;60: (5):432–5. |
[20] | Monickaraj F , Aravind S , Gokulakrishnan K , Sathishkumar C , Prabu P , Prabu D , et al. Accelerated aging as evidenced by increased telomere shortening and mitochondrial DNA depletion in patients with type 2 diabetes. Mol Cell Biochem. (2012) ;365: (1-2):343–50. |
[21] | Savale L , Chaouat A , Bastuji-Garin S , Marcos E , Boyer L , Maitre B , et al. Shortened telomeres in circulating leukocytes of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. (2009) ;179: (7):566–71. |
[22] | Blasco MA . Telomeres and human disease: Ageing, cancer and beyond. Nat Rev Genet. (2005) ;6: (8):611–22. |
[23] | Tomita KI , Aida J , Izumiyama-Shimomura N , Nakamura KI , Ishikawa N , Matsuda Y , et al. Changes in telomere length with aging in human neurons and glial cells revealed by quantitative fluorescence in situ hybridization analysis. Geriatr Gerontol Int. (2018) ;18: (10):1507–12. |
[24] | Kota LN , Bharath S , Purushottam M , Moily NS , Sivakumar PT , Varghese M , et al. Reduced telomere length in neurodegenerative disorders may suggest shared biology. J Neuropsychiatry Clin Neurosci. (2015) ;27: (2):e92–6. |
[25] | Forero DA , Gonzalez-Giraldo Y , Lopez-Quintero C , Castro-Vega LJ , Barreto GE , Perry G . Meta-analysis of telomere length in Alzheimer’s disease. J Gerontol A Biol Sci Med Sci. (2016) ;71: (8):1069–73. |
[26] | Scarabino D , Veneziano L , Peconi M , Frontali M , Mantuano E , Corbo RM . Leukocyte telomere shortening in Huntington’s disease. J Neurol Sci. (2019) ;396: :25–9. |
[27] | Castaldo I , De Rosa M , Romano A , Zuchegna C , Squitieri F , Mechelli R , et al. DNA damage signatures in peripheral blood cells as biomarkers in prodromal huntington disease. Ann Neurol. (2019) ;85: (2):296–301. |
[28] | PerezGrovas-Saltijeral A , Ochoa-Morales A , Miranda-Duarte A , Martinez-Ruano L , Jara-Prado A , Camacho-Molina A , et al. Telomere length analysis on leukocytes derived from patients with Huntington disease. Mech Ageing Dev. (2020) ;185: :111189. |
[29] | Aziz NA , Weydt P . Telomere length as a modifier of age-at-onset in Huntington disease: A two-sample Mendelian randomization study. J Neurol. (2018) ;265: (9):2149–51. |
[30] | Pal S , Tyler JK . Epigenetics and aging. Sci Adv. (2016) ;2: (7):e1600584. |
[31] | Koch CM , Wagner W . Epigenetic-aging-signature to determine age in different tissues. Aging (Albany NY). (2011) ;3: (10):1018–27. |
[32] | Hannum G , Guinney J , Zhao L , Zhang L , Hughes G , Sadda S , et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. (2013) ;49: (2):359–67. |
[33] | Horvath S . DNA methylation age of human tissues and cell types. Genome Biol. (2013) ;14: (10):R115. |
[34] | Horvath S , Langfelder P , Kwak S , Aaronson J , Rosinski J , Vogt TF , et al. Huntington’s disease accelerates epigenetic aging of human brain and disrupts DNA methylation levels. Aging (Albany NY). (2016) ;8: (7):1485–512. |
[35] | Levine AJ , Quach A , Moore DJ , Achim CL , Soontornniyomkij V , Masliah E , et al. Accelerated epigenetic aging in brain is associated with pre-mortem HIV-associated neurocognitive disorders. J Neurovirol. (2016) ;22: (3):366–75. |
[36] | Horvath S , Garagnani P , Bacalini MG , Pirazzini C , Salvioli S , Gentilini D , et al. Accelerated epigenetic aging in Down syndrome. Aging Cell. (2015) ;14: (3):491–5. |
[37] | Levine ME , Lu AT , Bennett DA , Horvath S . Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY). (2015) ;7: (12):1198–211. |
[38] | Fearon DT , Locksley RM . The instructive role of innate immunity in the acquired immune response. Science. (1996) ;272: (5258):50–3. |
[39] | Sadik CD , Luster AD . Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J Leukoc Biol. (2012) ;91: (2):207–15. |
[40] | Serhan CN . Resolution phase of inflammation: Novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu Rev Immunol. (2007) ;25: :101–37. |
[41] | Fulop T , Witkowski JM , Le Page A , Fortin C , Pawelec G , Larbi A . Intracellular signalling pathways: Targets to reverse immunosenescence. Clin Exp Immunol. (2017) ;187: (1):35–43. |
[42] | Rosas HD , Lee SY , Bender AC , Zaleta AK , Vangel M , Yu P , et al. Altered white matter microstructure in the corpus callosum in Huntington’s disease: Implications for cortical “disconnection”. Neuroimage. (2010) ;49: (4):2995–3004. |
[43] | Tai YF , Pavese N , Gerhard A , Tabrizi SJ , Barker RA , Brooks DJ , et al. Microglial activation in presymptomatic Huntington’s disease gene carriers. Brain. (2007) ;130: (Pt 7):1759–66. |
[44] | Connolly C , Magnusson-Lind A , Lu G , Wagner PK , Southwell AL , Hayden MR , et al. Enhanced immune response to MMP3 stimulation in microglia expressing mutant huntingtin. Neuroscience. (2016) ;325: :74–88. |
[45] | Bjorkqvist M , Wild EJ , Thiele J , Silvestroni A , Andre R , Lahiri N , et al. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington’s disease. J Exp Med. (2008) ;205: (8):1869–77. |
[46] | Henry CJ , Huang Y , Wynne AM , Godbout JP . Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1beta and anti-inflammatory IL-10 cytokines. Brain Behav Immun. (2009) ;23: (3):309–17. |
[47] | Estrada-Sanchez AM , Rebec GV . Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington’s disease: Interactions between neurons and astrocytes. Basal Ganglia. (2012) ;2: (2):57–66. |
[48] | Lee W , Reyes RC , Gottipati MK , Lewis K , Lesort M , Parpura V , et al. Enhanced Ca(2+)-dependent glutamate release from astrocytes of the BACHD Huntington’s disease mouse model. Neurobiol Dis. (2013) ;58: :192–9. |
[49] | Crotti A , Benner C , Kerman BE , Gosselin D , Lagier-Tourenne C , Zuccato C , et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. (2014) ;17: (4):513–21. |
[50] | Wood TE , Barry J , Yang Z , Cepeda C , Levine MS , Gray M . Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum Mol Genet. (2019) ;28: (3):487–500. |
[51] | Liu T , Zhang L , Joo D , Sun SC . NF-kappaB signaling in inflammation. Signal Transduct Target Ther. (2017) ;2. |
[52] | Labadorf A , Hoss AG , Lagomarsino V , Latourelle JC , Hadzi TC , Bregu J , et al. RNA Sequence analysis of human Huntington disease brain reveals an extensive increase in inflammatory and developmental gene expression. Plos One. (2015) ;10: (12):e0143563. |
[53] | Hsiao HY , Chen YC , Chen HM , Tu PH , Chern Y . A critical role of astrocyte-mediated nuclear factor-kappaB-dependent inflammation in Huntington’s disease. Hum Mol Genet. (2013) ;22: (9):1826–42. |
[54] | Mueller KA , Glajch KE , Huizenga MN , Wilson RA , Granucci EJ , Dios AM , et al. Hippo signaling pathway dysregulation in human Huntington’s disease brain and neuronal stem cells. Sci Rep. (2018) ;8: (1):11355. |
[55] | Zhou X , Li WY , Wang HY . The roles and mechanisms of MST1/2 in the innate immune response. Yi Chuan. (2017) ;39: (7):642–9. |
[56] | Nath S , Munsie LN , Truant R . A huntingtin-mediated fast stress response halting endosomal trafficking is defective in Huntington’s disease. Hum Mol Genet. (2015) ;24: (2):450–62. |
[57] | Maiuri T , Mocle AJ , Hung CL , Xia JR , van Roon-Mom WMC , Truant R . Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. (2017) ;26: (2):395–406. |
[58] | Munsie L , Caron N , Atwal RS , Marsden I , Wild EJ , Bamburg JR , et al. Mutant huntingtin causes defective actin remodeling during stress: Defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet. (2011) ;20: (10):1937–51. |
[59] | Atwal RS , Truant R . A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy. (2008) ;4: (1):91–3. |
[60] | Laron Z . Insulin-like growth factor 1 (IGF-1): A growth hormone. Mol Pathol. (2001) ;54: (5):311–6. |
[61] | Junnila RK , List EO , Berryman DE , Murrey JW , Kopchick JJ . The GH/IGF-1 axis in ageing and longevity. Nat Rev Endocrinol. (2013) ;9: (6):366–76. |
[62] | Barzilai N , Huffman DM , Muzumdar RH , Bartke A . The critical role of metabolic pathways in aging. Diabetes. (2012) ;61: (6):1315–22. |
[63] | Kenyon CJ . The genetics of ageing. Nature. (2010) ;464: (7288):504–12. |
[64] | Garinis GA , van der Horst GT , Vijg J , Hoeijmakers JH . DNA damage and ageing: New-age ideas for an age-old problem. Nat Cell Biol. (2008) ;10: (11):1241–7. |
[65] | Sanberg PR , Fibiger HC , Mark RF . Body weight and dietary factors in Huntington’s disease patients compared with matched controls. Med J Aust. (1981) ;1: (8):407–9. |
[66] | Vidal RL , Figueroa A , Court FA , Thielen P , Molina C , Wirth C , et al. Targeting the UPR transcription factor XBP1 protects against Huntington’s disease through the regulation of FoxO1 and autophagy. Hum Mol Genet. (2012) ;21: (10):2245–62. |
[67] | Farina F , Lambert E , Commeau L , Lejeune FX , Roudier N , Fonte C , et al. The stress response factor daf-16/FOXO is required for multiple compound families to prolong the function of neurons with Huntington’s disease. Sci Rep. (2017) ;7: (1):4014. |
[68] | Humbert S , Bryson EA , Cordelieres FP , Connors NC , Datta SR , Finkbeiner S , et al. The IGF-1/Akt pathway is neuroprotective in Huntington’s disease and involves Huntingtin phosphorylation by Akt. Dev Cell. (2002) ;2: (6):831–7. |
[69] | Lee JH , Tecedor L , Chen YH , Monteys AM , Sowada MJ , Thompson LM , et al. Reinstating aberrant mTORC1 activity in Huntington’s disease mice improves disease phenotypes. Neuron. (2015) ;85: (2):303–15. |
[70] | Lopes C , Ribeiro M , Duarte AI , Humbert S , Saudou F , Pereira de Almeida L , et al. IGF-1 intranasal administration rescues Huntington’s disease phenotypes in YAC128 mice. Mol Neurobiol. (2014) ;49: (3):1126–42. |
[71] | Abd-Elrahman KS , Ferguson SSG . Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington’s pathology in zQ175 mice. Mol Brain. (2019) ;12: (1):35. |
[72] | Ehrnhoefer DE , Martin DDO , Schmidt ME , Qiu X , Ladha S , Caron NS , et al. Preventing mutant huntingtin proteolysis and intermittent fasting promote autophagy in models of Huntington disease. Acta Neuropathol Commun. (2018) ;6: (1):16. |
[73] | Sanchis A , Garcia-Gimeno MA , Canada-Martinez AJ , Sequedo MD , Millan JM , Sanz P , et al. Metformin treatment reduces motor and neuropsychiatric phenotypes in the zQ175 mouse model of Huntington disease. Exp Mol Med. (2019) ;51: (6):1–16. |
[74] | Arnoux I , Willam M , Griesche N , Krummeich J , Watari H , Offermann N , et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife. (2018) ;7. |
[75] | Ravikumar B , Vacher C , Berger Z , Davies JE , Luo S , Oroz LG , et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. (2004) ;36: (6):585–95. |
[76] | Salem L , Saleh N , Desamericq G , Youssov K , Dolbeau G , Cleret L , et al. Insulin-like growth factor-1 but not insulin predicts cognitive decline in Huntington’s disease. PloS One. (2016) ;11: (9):e0162890. |
[77] | Hipp MS , Kasturi P , Hartl FU . The proteostasis network and its decline in ageing. Nat Rev Mol Cell Biol. (2019) ;20: (7):421–35. |
[78] | Sala AJ , Bott LC , Morimoto RI . Shaping proteostasis at the cellular, tissue, and organismal level. J Cell Biol. (2017) ;216: (5):1231–41. |
[79] | Beal R , Deveraux Q , Xia G , Rechsteiner M , Pickart C . Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc Natl Acad Sci U S A. (1996) ;93: (2):861–6. |
[80] | Grumati P , Dikic I . Ubiquitin signaling and autophagy. J Biol Chem. (2018) ;293: (15):5404–13. |
[81] | Pickart CM . Targeting of substrates to the 26S proteasome. FASEB J. (1997) ;11: (13):1055–66. |
[82] | David DC . Aging and the aggregating proteome. Front Genet. (2012) ;3: :247. |
[83] | Ottis P , Topic B , Loos M , Li KW , de Souza A , Schulz D , et al. Aging-induced proteostatic changes in the rat hippocampus identify ARP3, NEB2 and BRAG2 as a molecular circuitry for cognitive impairment. PloS One. (2013) ;8: (9):e75112. |
[84] | Tanase M , Urbanska AM , Zolla V , Clement CC , Huang L , Morozova K , et al. Role of carbonyl modifications on aging-associated protein aggregation. Sci Rep. (2016) ;6: :19311. |
[85] | Bergamini E , Cavallini G , Donati A , Gori Z . The role of macroautophagy in the ageing process, anti-ageing intervention and age-associated diseases. Int J Biochem Cell Biol. (2004) ;36: (12):2392–404. |
[86] | Cuervo AM , Dice JF . When lysosomes get old. Exp Gerontol. (2000) ;35: (2):119–31. |
[87] | Levine B , Kroemer G . Autophagy in the pathogenesis of disease. Cell. (2008) ;132: (1):27–42. |
[88] | Rubinsztein DC , Marino G , Kroemer G . Autophagy and aging. Cell. (2011) ;146: (5):682–95. |
[89] | Wang J , Wang CE , Orr A , Tydlacka S , Li SH , Li XJ . Impaired ubiquitin-proteasome system activity in the synapses of Huntington’s disease mice. J Cell Biol. (2008) ;180: (6):1177–89. |
[90] | Li XJ , Li S . Proteasomal dysfunction in aging and Huntington disease. Neurobiol Dis. (2011) ;43: (1):4–8. |
[91] | Waelter S , Boeddrich A , Lurz R , Scherzinger E , Lueder G , Lehrach H , et al. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. (2001) ;12: (5):1393–407. |
[92] | Harding RJ , Tong YF . Proteostasis in Huntington’s disease: Disease mechanisms and therapeutic opportunities. Acta Pharmacol Sin. (2018) ;39: (5):754–69. |
[93] | Machiela E , Jeloka R , Caron N , Mehta SR , Schmidt ME , Tom C , et al. The interaction of aging and oxidative stress contributes to pathogenesis in mouse and human Huntington disease neurons. BioRxiv. (2019) ; doi: 10.1101/800268. |
[94] | Lin L , Jin Z , Tan H , Xu Q , Peng T , Li H . Atypical ubiquitination by E3 ligase WWP1 inhibits the proteasome-mediated degradation of mutant huntingtin. Brain Res. (2016) ;1643: :103–12. |
[95] | Martinez-Vicente M , Talloczy Z , Wong E , Tang GM , Koga H , Kaushik S , et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci. (2010) ;13: (5):567-U74. |
[96] | Lee H , Noh JY , Oh Y , Kim Y , Chang JW , Chung CW , et al. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum Mol Genet. (2012) ;21: (1):101–14. |
[97] | Koyuncu S , Saez I , Lee HJ , Gutierrez-Garcia R , Pokrzywa W , Fatima A , et al. The ubiquitin ligase UBR5 suppresses proteostasis collapse in pluripotent stem cells from Huntington’s disease patients. Nat Commun. (2018) ;9: (1):2886. |
[98] | Cortes CJ , La Spada AR . The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov Today. (2014) ;19: (7):963–71. |
[99] | Rui YN , Xu Z , Patel B , Chen Z , Chen D , Tito A , et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. (2015) ;17: (3):262–75. |
[100] | Demers-Lamarche J , Guillebaud G , Tlili M , Todkar K , Belanger N , Grondin M , et al. Loss of mitochondrial function impairs lysosomes. J Biol Chem. (2016) ;291: (19):10263–76. |
[101] | Kuhl DE , Phelps ME , Markham CH , Metter EJ , Riege WH , Winter J . Cerebral metabolism and atrophy in Huntington’s disease determined by 18FDG and computed tomographic scan. Ann Neurol. (1982) ;12: (5):425–34. |
[102] | Hayden MR , Martin WRW , Stoessl AJ , Clark C , Hollenberg S , Adam MJ , et al. Positron emission tomography in the early diagnosis of Huntington’s disease. Neurology. (1986) ;36: (7):888–94. |
[103] | Young AB , Penney JB , Starosta-Rubinstein S , Markel DS , Berent S , Giordani B , et al. PET scan investigations of Huntington’s disease: Cerebral metabolic correlates of neurological features and functional decline. Ann Neurol. (1986) ;20: (3):296–303. |
[104] | Kuwert T , Lange HW , Langen KJ , Herzog H , Aulich A , Feinendegen LE . Cortical and subcortical glucose consumption measured by PET in patients with Huntington’s disease. Brain. (1990) ;113: (Pt 5):1405–23. |
[105] | Powers WJ , Videen TO , Markham J , McGee-Minnich L , Antenor-Dorsey JV , Hershey T , et al. Selective defect of in vivo glycolysis in early Huntington’s disease striatum. Proc Natl Acad Sci U S A. (2007) ;104: (8):2945–9. |
[106] | Seong IS , Ivanova E , Lee JM , Choo YS , Fossale E , Anderson M , et al. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet. (2005) ;14: (19):2871–80. |
[107] | Lim NK , Hung LW , Pang TY , McLean CA , Liddell JR , Hilton JB , et al. Localized changes to glycogen synthase kinase-3 and collapsin response mediator protein-2 in the Huntington’s disease affected brain. Hum Mol Genet. (2014) ;23: (15):4051–63. |
[108] | Tabrizi SJ , Workman J , Hart PE , Mangiarini L , Mahal A , Bates G , et al. Mitochondrial dysfunction and free radical damage in the Huntington R6/2 transgenic mouse. Ann Neurol. (2000) ;47: (1):80–6. |
[109] | Damiano M , Diguet E , Malgorn C , D’Aurelio M , Galvan L , Petit F , et al. A role of mitochondrial complex II defects in genetic models of Huntington’s disease expressing N-terminal fragments of mutant huntingtin. Hum Mol Genet. (2013) ;22: (19):3869–82. |
[110] | Boussicault L , Herard AS , Calingasan N , Petit F , Malgorn C , Merienne N , et al. Impaired brain energy metabolism in the BACHD mouse model of Huntington’s disease: Critical role of astrocyte-neuron interactions. J Cereb Blood Flow Metab. (2014) ;34: (9):1500–10. |
[111] | Olah J , Klivenyi P , Gardian G , Vecsei L , Orosz F , Kovacs GG , et al. Increased glucose metabolism and ATP level in brain tissue of Huntington’s disease transgenic mice. FEBS J. (2008) ;275: (19):4740–55. |
[112] | Tkac I , Henry PG , Zacharoff L , Wedel M , Gong W , Deelchand DK , et al. Homeostatic adaptations in brain energy metabolism in mouse models of Huntington disease. J Cereb Blood Flow Metab. (2012) ;32: (11):1977–88. |
[113] | Guidetti P , Charles V , Chen EY , Reddy PH , Kordower JH , Whetsell WO Jr , et al. Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol. (2001) ;169: (2):340–50. |
[114] | Reiman EM , Caselli RJ , Chen K , Alexander GE , Bandy D , Frost J . Declining brain activity in cognitively normal apolipoprotein E epsilon 4 heterozygotes: A foundation for using positron emission tomography to efficiently test treatments to prevent Alzheimer’s disease. Proc Natl Acad Sci U S A. (2001) ;98: (6):3334–9. |
[115] | Bookheimer SY , Strojwas MH , Cohen MS , Saunders AM , Pericak-Vance MA , Mazziotta JC , et al. Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med. (2000) ;343: (7):450–6. |
[116] | Sokoloff L . Measurement of local cerebral glucose utilization and its relation to local functional activity in the brain. Adv Exp Med Biol. (1991) ;291: :21–42. |
[117] | Johnson S , Imai SI . NAD (+) biosynthesis, aging, and disease. F1000Res. (2018) ;7: :132. |
[118] | Imai S , Guarente L . NAD+ and sirtuins in aging and disease. Trends Cell Biol. (2014) ;24: (8):464–71. |
[119] | Haigis MC , Sinclair DA . Mammalian sirtuins: Biological insights and disease relevance. Annu Rev Pathol. (2010) ;5: :253–95. |
[120] | de Figueiredo LF , Gossmann TI , Ziegler M , Schuster S . Pathway analysis of NAD+ metabolism. Biochem J. (2011) ;439: (2):341–8. |
[121] | Dolle C , Skoge RH , Vanlinden MR , Ziegler M . NAD biosynthesis in humans–enzymes, metabolites and therapeutic aspects. Curr Top Med Chem. (2013) ;13: (23):2907–17. |
[122] | Stein LR , Imai S . Specific ablation of Nampt in adult neural stem cells recapitulates their functional defects during aging. EMBO J. (2014) ;33: (12):1321–40. |
[123] | Bai P , Canto C , Oudart H , Brunyanszki A , Cen Y , Thomas C , et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. (2011) ;13: (4):461–8. |
[124] | Hathorn T , Snyder-Keller A , Messer A . Nicotinamide improves motor deficits and upregulates PGC-1alpha and BDNF gene expression in a mouse model of Huntington’s disease. Neurobiol Dis. (2011) ;41: (1):43–50. |
[125] | Ame JC , Spenlehauer C , de Murcia G . The PARP superfamily. Bioessays. (2004) ;26: (8):882–93. |
[126] | Mendelsohn AR , Larrick JW . The NAD+/PARP1/SIRT1 axis in aging. Rejuvenation Res. (2017) ;20: (3):244–7. |
[127] | Luna A , Aladjem MI , Kohn KW . SIRT1/PARP1 crosstalk: Connecting DNA damage and metabolism. Genome Integr. (2013) ;4: (1):6. |
[128] | Vis JC , Schipper E , de Boer-van Huizen RT , Verbeek MM , de Waal RM , Wesseling P , et al. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropathol. (2005) ;109: (3):321–8. |
[129] | Cardinale A , Paldino E , Giampa C , Bernardi G , Fusco FR . PARP-1 inhibition is neuroprotective in the R6/2 mouse model of Huntington’s disease. Plos One. (2015) ;10: (8):e0134482. |
[130] | Pallas M , Pizarro JG , Gutierrez-Cuesta J , Crespo-Biel N , Alvira D , Tajes M , et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience. (2008) ;154: (4):1388–97. |
[131] | Tulino R , Benjamin AC , Jolinon N , Smith DL , Chini EN , Carnemolla A , et al. SIRT1 activity is linked to its brain region-specific phosphorylation and is impaired in Huntington’s disease mice. PloS One. (2016) ;11: (1):e0145425. |
[132] | Fu J , Jin J , Cichewicz RH , Hageman SA , Ellis TK , Xiang L , et al. trans-(-)-epsilon-Viniferin increases mitochondrial sirtuin 3 (SIRT3), activates AMP-activated protein kinase (AMPK), and protects cells in models of Huntington disease. J Biol Chem. (2012) ;287: (29):24460–72. |
[133] | Naia L , Rosenstock TR , Oliveira AM , Oliveira-Sousa SI , Caldeira GL , Carmo C , et al. Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in Huntington’s disease models. Mol Neurobiol. (2017) ;54: (7):5385–99. |
[134] | Youle RJ , van der Bliek AM . Mitochondrial fission, fusion, and stress. Science. (2012) ;337: (6098):1062–5. |
[135] | Palomera-Avalos V , Grinan-Ferre C , Puigoriol-Ilamola D , Camins A , Sanfeliu C , Canudas AM , et al. Resveratrol protects SAMP8 brain under metabolic stress: Focus on mitochondrial function and Wnt pathway. Mol Neurobiol. (2017) ;54: (3):1661–76. |
[136] | Crane JD , Devries MC , Safdar A , Hamadeh MJ , Tarnopolsky MA . The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. J Gerontol A Biol Sci Med Sci. (2010) ;65: (2):119–28. |
[137] | Shigenaga MK , Hagen TM , Ames BN . Oxidative damage and mitochondrial decay in aging. Proc Natl Acad Sci U S A. (1994) ;91: (23):10771–8. |
[138] | Terman A , Kurz T , Navratil M , Arriaga EA , Brunk UT . Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal. (2010) ;12: (4):503–35. |
[139] | Ames BN , Shigenaga MK , Hagen TM . Mitochondrial decay in aging. Biochim Biophys Acta. (1995) ;1271: (1):165–70. |
[140] | Nicholls DG . Mitochondrial function and dysfunction in the cell: Its relevance to aging and aging-related disease. Int J Biochem Cell Biol. (2002) ;34: (11):1372–81. |
[141] | Melov S . Modeling mitochondrial function in aging neurons. Trends Neurosci. (2004) ;27: (10):601–6. |
[142] | Shirendeb U , Reddy AP , Manczak M , Calkins MJ , Mao P , Tagle DA , et al. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum Mol Genet. (2011) ;20: (7):1438–55. |
[143] | Kim J , Moody JP , Edgerly CK , Bordiuk OL , Cormier K , Smith K , et al. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum Mol Genet. (2010) ;19: (20):3919–35. |
[144] | Shirendeb UP , Calkins MJ , Manczak M , Anekonda V , Dufour B , McBride JL , et al. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet. (2012) ;21: (2):406–20. |
[145] | Song W , Chen J , Petrilli A , Liot G , Klinglmayr E , Zhou Y , et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat Med. (2011) ;17: (3):377–82. |
[146] | Paupe V , Prudent J . New insights into the role of mitochondrial calcium homeostasis in cell migration. Biochem Biophys Res Commun. (2018) ;500: (1):75–86. |
[147] | Toescu EC , Vreugdenhil M . Calcium and normal brain ageing. Cell Calcium. (2010) ;47: (2):158–64. |
[148] | Panov AV , Gutekunst CA , Leavitt BR , Hayden MR , Burke JR , Strittmatter WJ , et al. Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines. Nat Neurosci. (2002) ;5: (8):731–6. |
[149] | Gizatullina ZZ , Lindenberg KS , Harjes P , Chen Y , Kosinski CM , Landwehrmeyer BG , et al. Low stability of Huntington muscle mitochondria against Ca2+ in R6/2 mice. Ann Neurol. (2006) ;59: (2):407–11. |
[150] | Gellerich FN , Gizatullina Z , Nguyen HP , Trumbeckaite S , Vielhaber S , Seppet E , et al. Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats. J Biol Chem. (2008) ;283: (45):30715–24. |
[151] | Brustovetsky N , LaFrance R , Purl KJ , Brustovetsky T , Keene CD , Low WC , et al. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington’s disease. J Neurochem. (2005) ;93: (6):1361–70. |
[152] | Oliveira JM , Jekabsons MB , Chen S , Lin A , Rego AC , Goncalves J , et al. Mitochondrial dysfunction in Huntington’s disease: The bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. (2007) ;101: (1):241–9. |
[153] | Wang Y , Branicky R , Noe A , Hekimi S . Superoxide dismutases: Dual roles in controlling ROS damage and regulating ROS signaling. J Cell Biol. (2018) ;217: (6):1915–28. |
[154] | Beckhauser TF , Francis-Oliveira J , De Pasquale R . Reactive oxygen species: Physiological and physiopathological effects on synaptic plasticity. J Exp Neurosci. (2016) ;10: (Suppl 1):23–48. |
[155] | Di Meo S , Reed TT , Venditti P , Victor VM . Role of ROS and RNS sources in physiological and pathological conditions. Oxid Med Cell Longev. (2016) ;2016: :1245049. |
[156] | Tatone C , Carbone MC , Falone S , Aimola P , Giardinelli A , Caserta D , et al. Age-dependent changes in the expression of superoxide dismutases and catalase are associated with ultrastructural modifications in human granulosa cells. Mol Hum Reprod. (2006) ;12: (11):655–60. |
[157] | Nandi A , Yan LJ , Jana CK , Das N . Role of catalase in oxidative stress- and age-associated degenerative diseases. Oxid Med Cell Longev. (2019) ;2019: :9613090. |
[158] | Guachalla LM , Rudolph KL . ROS induced DNA damage and checkpoint responses: Influences on aging? Cell Cycle. (2010) ;9: (20):4058–60. |
[159] | Browne SE , Ferrante RJ , Beal MF . Oxidative stress in Huntington’s disease. Brain Pathol. (1999) ;9: (1):147–63. |
[160] | Polidori MC , Mecocci P , Browne SE , Senin U , Beal MF . Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci Lett. (1999) ;272: (1):53–6. |
[161] | Sorolla MA , Reverter-Branchat G , Tamarit J , Ferrer I , Ros J , Cabiscol E . Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic Biol Med. (2008) ;45: (5):667–78. |
[162] | Colavitti R , Finkel T . Reactive oxygen species as mediators of cellular senescence. IUBMB Life. (2005) ;57: (4-5):277–81. |
[163] | Bigan E , Sasidharan Nair S , Lejeune FX , Fragnaud H , Parmentier F , Megret L , et al. Genetic cooperativity in multi-layer networks implicates cell survival and senescence in the striatum of Huntington’s disease mice synchronous to symptoms. Bioinformatics. (2020) ;36: (1):186–96. |
[164] | Childs BG , Durik M , Baker DJ , van Deursen JM . Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat Med. (2015) ;21: (12):1424–35. |
[165] | Coppe JP , Desprez PY , Krtolica A , Campisi J . The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol. (2010) ;5: :99–118. |
[166] | Baker DJ , Petersen RC . Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J Clin Invest. (2018) ;128: (4):1208–16. |
[167] | Bae BI , Xu H , Igarashi S , Fujimuro M , Agrawal N , Taya Y , et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. (2005) ;47: (1):29–41. |
[168] | Salminen A , Ojala J , Kaarniranta K , Haapasalo A , Hiltunen M , Soininen H . Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci. (2011) ;34: (1):3–11. |
[169] | Angelova DM , Brown DR . Microglia and the aging brain: Are senescent microglia the key to neurodegeneration? J Neurochem. (2019) ;151: (6):676–88. |
[170] | Chang KH , Wu YR , Chen YC , Chen CM . Plasma inflammatory biomarkers for Huntington’s disease patients and mouse model. Brain Behav Immun. (2015) ;44: :121–7. |
[171] | Moskalev AA , Shaposhnikov MV , Plyusnina EN , Zhavoronkov A , Budovsky A , Yanai H , et al. The role of DNA damage and repair in aging through the prism of Koch-like criteria. Ageing Res Rev. (2013) ;12: (2):661–84. |
[172] | Faggioli F , Wang T , Vijg J , Montagna C . Chromosome-specific accumulation of aneuploidy in the aging mouse brain. Hum Mol Genet. (2012) ;21: (24):5246–53. |
[173] | Forsberg LA , Rasi C , Razzaghian HR , Pakalapati G , Waite L , Thilbeault KS , et al. Age-related somatic structural changes in the nuclear genome of human blood cells. Am J Hum Genet. (2012) ;90: (2):217–28. |
[174] | Brazhnik K , Sun S , Alani O , Kinkhabwala M , Wolkoff AW , Maslov AY , et al. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci Adv. (2020) ;6: (5):eaax2659. |
[175] | Friedberg EC . DNA damage and repair. Nature. (2003) ;421: (6921):436–40. |
[176] | Lans H , Hoeijmakers JHJ , Vermeulen W , Marteijn JA . The DNA damage response to transcription stress. Nat Rev Mol Cell Biol. (2019) ;20: (12):766–84. |
[177] | Petersen LN , Orren DK , Bohr VA . Gene-specific and strand-specific DNA repair in the G1 and G2 phases of the cell cycle. Mol Cell Biol. (1995) ;15: (7):3731–7. |
[178] | Jackson SP , Bartek J . The DNA-damage response in human biology and disease. Nature. (2009) ;461: (7267):1071–8. |
[179] | Nie B , Gan W , Shi F , Hu GX , Chen LG , Hayakawa H , et al. Age-dependent accumulation of 8-oxoguanine in the DNA and RNA in various rat tissues. Oxid Med Cell Longev. (2013) ;2013: :303181. |
[180] | Olinski R , Siomek A , Rozalski R , Gackowski D , Foksinski M , Guz J , et al. Oxidative damage to DNA and antioxidant status in aging and age-related diseases. Acta Biochim Pol. (2007) ;54: (1):11–26. |
[181] | Gan W , Liu XL , Yu T , Zou YG , Li TT , Wang S , et al. Urinary 8-oxo-7,8-dihydroguanosine as a Potential Biomarker of Aging. Front Aging Neurosci. (2018) ;10: :34. |
[182] | Radak Z , Boldogh I . 8-Oxo-7,8-dihydroguanine: Links to gene expression, aging, and defense against oxidative stress. Free Radic Biol Med. (2010) ;49: (4):587–96. |
[183] | Van Houten B , Santa-Gonzalez GA , Camargo M . DNA repair after oxidative stress: Current challenges. Curr Opin Toxicol. (2018) ;7: :9–16. |
[184] | Klungland A , Bjelland S . Oxidative damage to purines in DNA: Role of mammalian Ogg1. DNA Repair (Amst). (2007) ;6: (4):481–8. |
[185] | Noren Hooten N , Kompaniez K , Barnes J , Lohani A , Evans MK . Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J Biol Chem. (2011) ;286: (52):44679–90. |
[186] | Hoeijmakers JH . DNA damage, aging, and cancer. N Engl J Med. (2009) ;361: (15):1475–85. |
[187] | Goellner GM , Tester D , Thibodeau S , Almqvist E , Goldberg YP , Hayden MR , et al. Different mechanisms underlie DNA instability in Huntington disease and colorectal cancer. Am J Hum Genet. (1997) ;60: (4):879–90. |
[188] | Slean MM , Panigrahi GB , Ranum LP , Pearson CE . Mutagenic roles of DNA “repair” proteins in antibody diversity and disease-associated trinucleotide repeat instability. DNA Repair (Amst). (2008) ;7: (7):1135–54. |
[189] | Askeland G , Dosoudilova Z , Rodinova M , Klempir J , Liskova I , Kusnierczyk A , et al. Increased nuclear DNA damage precedes mitochondrial dysfunction in peripheral blood mononuclear cells from Huntington’s disease patients. Sci Rep. (2018) ;8: (1):9817. |
[190] | Gasset-Rosa F , Chillon-Marinas C , Goginashvili A , Atwal RS , Artates JW , Tabet R , et al. Polyglutamine-expanded huntingtin exacerbates age-related disruption of nuclear integrity and nucleocytoplasmic transport. Neuron. (2017) ;94: (1):48–57 e4. |
[191] | Grima JC , Daigle JG , Arbez N , Cunningham KC , Zhang K , Ochaba J , et al. Mutant huntingtin disrupts the nuclear pore complex. Neuron. (2017) ;94: (1):93–107 e6. |
[192] | Lord CL , Timney BL , Rout MP , Wente SR . Altering nuclear pore complex function impacts longevity and mitochondrial function in S.cerevisiae. J Cell Biol. (2015) ;208: (6):729–44. |
[193] | Matsuoka S , Ballif BA , Smogorzewska A , McDonald ER 3rd , Hurov KE , Luo J , et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. (2007) ;316: (5828):1160–6. |
[194] | Lemaitre C , Fischer B , Kalousi A , Hoffbeck AS , Guirouilh-Barbat J , Shahar OD , et al. The nucleoporin 153, a novel factor in double-strand break repair and DNA damage response. Oncogene. (2012) ;31: (45):4803–9. |
[195] | Kennedy L , Evans E , Chen CM , Craven L , Detloff PJ , Ennis M , et al. Dramatic tissue-specific mutation length increases are an early molecular event in Huntington disease pathogenesis. Hum Mol Genet. (2003) ;12: (24):3359–67. |
[196] | Shelbourne PF , Keller-McGandy C , Bi WL , Yoon SR , Dubeau L , Veitch NJ , et al. Triplet repeat mutation length gains correlate with cell-type specific vulnerability in Huntington disease brain. Hum Mol Genet. (2007) ;16: (10):1133–42. |
[197] | Swami M , Hendricks AE , Gillis T , Massood T , Mysore J , Myers RH , et al. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum Mol Genet. (2009) ;18: (16):3039–47. |
[198] | Budworth H , Harris FR , Williams P , Lee DY , Holt A , Pahnke J , et al. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. (2015) ;11: (8):e1005267. |
[199] | Flower M , Lomeikaite V , Ciosi M , Cumming S , Morales F , Lo K , et al. MSH3 modifies somatic instability and disease severity in Huntington’s and myotonic dystrophy type 1. Brain. (2019) . |
[200] | Manley K , Shirley TL , Flaherty L , Messer A . Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat Genet. (1999) ;23: (4):471–3. |
[201] | Pinto RM , Dragileva E , Kirby A , Lloret A , Lopez E , St Claire J , et al. Mismatch repair genes Mlh1 and Mlh3 modify CAG instability in Huntington’s disease mice: Genome-wide and candidate approaches. PLoS Genet. (2013) ;9: (10):e1003930. |
[202] | Genetic Modifiers of Huntington’s Disease (GeM-HD) Consortium. Identification of genetic factors that modify clinical onset of Huntington’s disease. Cell. (2015) ;162: (3):516–26. |
[203] | Moss DJH , Pardinas AF , Langbehn D , Lo K , Leavitt BR , Roos R , et al. Identification of genetic variants associated with Huntington’s disease progression: A genome-wide association study. Lancet Neurol. (2017) ;16: (9):701–11. |
[204] | Gorbunova V , Seluanov A , Mao Z , Hine C . Changes in DNA repair during aging. Nucleic Acids Res. (2007) ;35: (22):7466–74. |
[205] | Ben Yehuda A , Globerson A , Krichevsky S , Bar On H , Kidron M , Friedlander Y , et al. Ageing and the mismatch repair system. Mech Ageing Dev. (2000) ;121: (1-3):173–9. |
[206] | Xia X , Jiang Q , McDermott J , Han JJ . Aging and Alzheimer’s disease: Comparison and associations from molecular to system level. Aging Cell. (2018) ;17: (5):e12802. |
[207] | Cooper JF , Dues DJ , Spielbauer KK , Machiela E , Senchuk MM , Van Raamsdonk JM . Delaying aging is neuroprotective in Parkinson’s disease: A genetic analysis in C. elegans models. NPJ Parkinsons Dis. (2015) ;1: :15022. |
[208] | Collier TJ , Kanaan NM , Kordower JH . Aging and Parkinson’s disease: Different sides of the same coin? Mov Disord. (2017) ;32: (7):983–90. |
[209] | Dues DJ , Andrews EK , Schaar CE , Bergsma AL , Senchuk MM , Van Raamsdonk JM . Aging causes decreased resistance to multiple stresses and a failure to activate specific stress response pathways. Aging (Albany NY). (2016) ;8: (4):777–95. |
[210] | Semenchenko GV , Khazaeli AA , Curtsinger JW , Yashin AI . Stress resistance declines with age: Analysis of data from a survival experiment with Drosophila melanogaster. Biogerontology. (2004) ;5: (1):17–30. |
[211] | Oikawa S , Kawanishi S . Site-specific DNA damage at GGG sequence by oxidative stress may accelerate telomere shortening. FEBS Lett. (1999) ;453: (3):365–8. |
[212] | Jiang H , Schiffer E , Song Z , Wang J , Zurbig P , Thedieck K , et al. Proteins induced by telomere dysfunction and DNA damage represent biomarkers of human aging and disease. Proc Natl Acad Sci U S A. (2008) ;105: (32):11299–304. |
[213] | Jackson AL , Chen R , Loeb LA . Induction of microsatellite instability by oxidative DNA damage. Proc Natl Acad Sci U S A. (1998) ;95: (21):12468–73. |


