
Glycation in Huntington’s Disease: A Possible Modifier and Target for Intervention
Abstract
Glycation is the non-enzymatic reaction between reactive dicarbonyls and amino groups, and gives rise to a variety of different reaction products known as advanced glycation end products (AGEs). Accumulation of AGEs on proteins is inevitable, and is associated with the aging process. Importantly, glycation is highly relevant in diabetic patients that experience periods of hyperglycemia. AGEs also play an important role in neurodegenerative diseases including Alzheimer’s (AD) and Parkinson’s disease (PD). Huntington’s disease (HD) is a hereditary neurodegenerative disease caused by an expansion of a CAG repeat in the huntingtin gene. The resulting expanded polyglutamine stretch in the huntingtin (HTT) protein induces its misfolding and aggregation, leading to neuronal dysfunction and death. HD patients exhibit chorea and psychiatric disturbances, along with abnormalities in glucose and energy homeostasis. Interestingly, an increased prevalence of diabetes mellitus has been reported in HD and in other CAG triplet repeat disorders. However, the mechanisms underlying the connection between glycation and HD progression remain unclear. In this review, we explore the possible connection between glycation and proteostasis imbalances in HD, and posit that it may contribute to disease progression, possibly by accelerating protein aggregation and deposition. Finally, we review therapeutic interventions that might be able to alleviate the negative impact of glycation in HD.
INTRODUCTION
The progressive age-associated accumulation of advanced glycation end products (AGEs) results in structural and functional alterations in proteins, possibly increasing the risk of impairments in proteostasis and, thereby, increased risk for the development of age-associated disorders [1]. However, whether AGEs are a cause or consequence of aging and age-related diseases is still a matter of debate. The controversy is strengthened by the growing number of known AGE targets and their gradual accumulation during life. The study of glycation has been impaired due to limitations in models and in tools and sensitive techniques to quantify and to detect AGEs in biological samples.
The formation of AGEs is elevated in individuals with altered carbohydrate metabolism [2]. Sporadic or hereditary neurodegenerative diseases, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), or Friedreich’s ataxia (FRDA), are often associated with impairments in carbohydrate metabolism, such as those caused by diabetes mellitus [3]. In particular, increased prevalence of diabetes mellitus has been reported in Huntington’s disease (HD) patients and in patients affected by other CAG-triplet repeat disorders [4–6]. The patients often display increased carbohydrate intake [7], hyperinsulinemia and insulin resistance [8, 9].However, the impact of AGE formation in disease progression remains largely unknown.
In this review, we explore the putative mechanistic links between AGE formation and progression of HD. Furthermore, we discuss several drugs known for targeting different aspects of the glycation process, and posit they may prove useful for mitigating detrimental effects of AGEs in HD.
HUNTINGTON’S DISEASE: GENETICS AND PATHOBIOLOGY
HD is a progressive neurodegenerative disorder normally manifesting during adulthood [10]. Psychiatric manifestations, such as personality and behavioural changes, often precede the onset of motor dysfunction by several years [11–13]. The motor disturbances include chorea and dystonia, and are followed by cognitive decline. Furthermore, degeneration of the striatum (caudate nucleus and putamen), and a general shrinkage of the brain are observed in post mortem studies [14]. Loss of cortical mass is another early event in the pathology progression [15]. Patients recurrently show progressive weight loss and muscle deterioration [16–18], features that are also characteristic in several transgenic mouse models of the disease [19–22].
The disease is caused by an abnormal expansion of a CAG repeat sequence in exon 1 of the huntingtin gene (HTT) [23–25]. This is translated into an elongated polyglutamine (polyQ) tract in the N-terminal region of the huntingtin (HTT) protein [24, 26]. The CAG repeat length correlates with both age of onset and severity of the disease. In non-affected individuals, the CAG repeat length is between 9–35. More than 35 repeats causes disease, although incomplete penetrance has been reported for CAG repeats between 36–39. Tracts above 60 result in juvenile onset HD [24]. Importantly, and in contrast to the vast majority of AD and PD cases, genetic testing enables the identification of individuals that will develop HD decades prior to the onset of motor symptoms.
Long polyglutamine tracts lead to the accumulation of intranuclear and cytoplasmic mutant HTT aggregates [27–29], sequestration of glutamine-rich proteins [28, 30], and cell damage in the striatum and cerebral cortex [31–33]. However, hypothalamic atrophy and cell death can also occur [34–37]. Since the discovery of the HTT gene, strong efforts have been undertaken to decipher the function of wild-type HTT. The protein expressed in most tissues, but its physiological function is still unclear. Intracellularly, HTT is associated with various organelles as Golgi complex, endoplasmic reticulum (ER) and nucleus [38–41]. Previous studies suggest that HTT interacts with clathrin-coated vesicles, endosomal compartments and microtubules in the neurites and at synapses [42]. Furthermore, wild-type HTT interacts with several partners [24, 43] and it may be involved in transcriptional regulation, and in mitochondrial function [24, 25, 44, 45].
The molecular underpinnings of disease are also still unclear, but it is likely that both a loss of protein function and a toxic gain of function are involved [25, 46, 47]. The age of HD onset correlates inversely with the CAG repeats length. However, repeat length only accounts for approximately 50% of variation in age of onset [48]. Both genetic modifiers and life-style play a role in the observed variability of the initial clinical symptoms, strengthening the need for identifying additional modifiers of pathology [49–51].
Several studies showed a high prevalence of glucose intolerance and diabetes mellitus in patients with neurodegenerative disorders, such as AD, PD, FRDA, and also in HD [3, 6, 52]. Peripheral abnormalities in glucose metabolism might considerably affect the quality of life of HD patients and the neurodegenerative process [53, 54], possibly due to hypothalamic dysfunction and peripheral defects in glucose and fat metabolism [16, 52, 54]. In addition, an altered glucose metabolism might accelerate glycation reactions, a relevant non-enzymatic process that interferes with protein folding and proteostasis (Fig. 1).
DIABETES MELLITUS: A CULPRIT IN NEURODEGENERATION
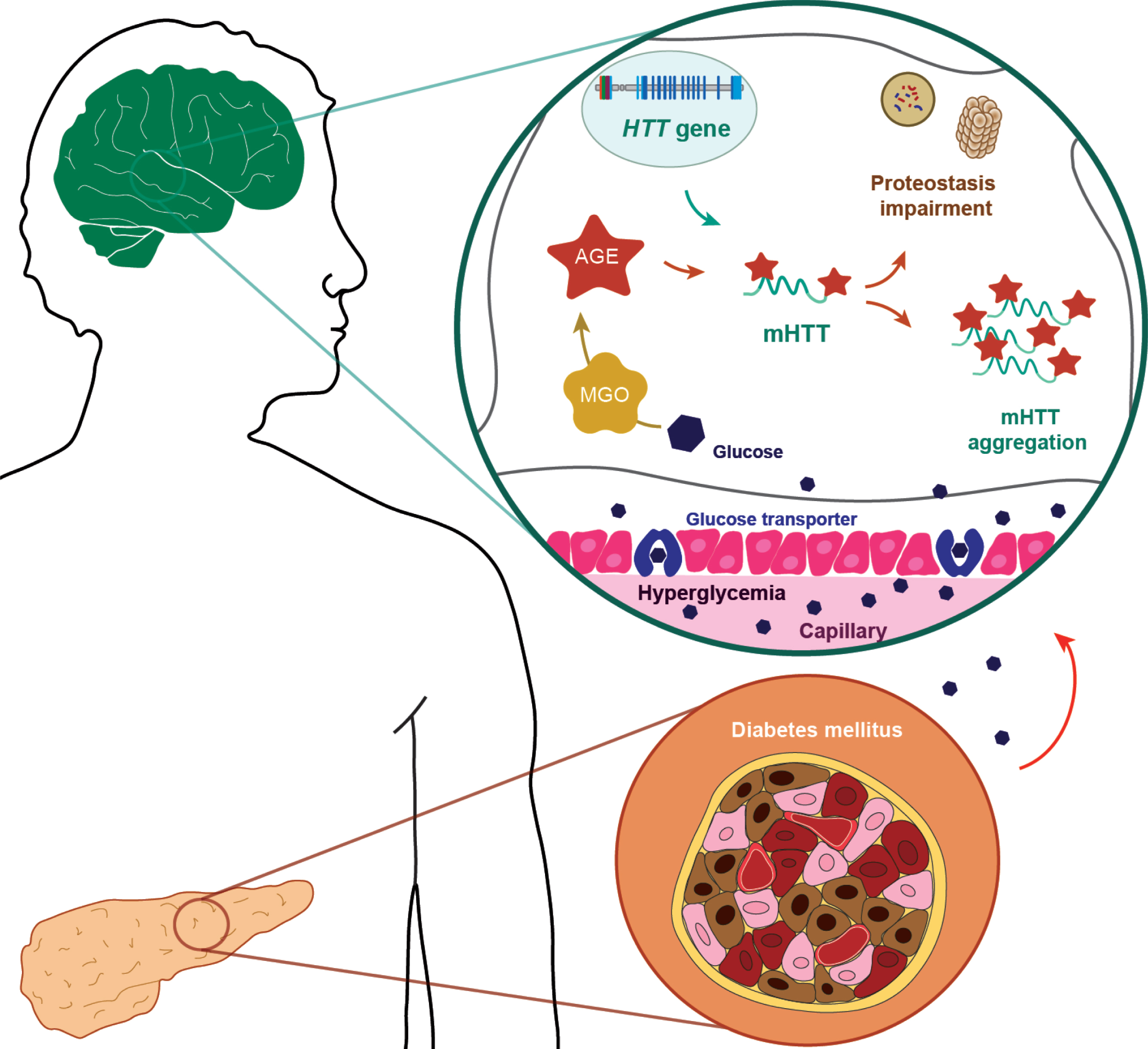
Fig.1
Glycation in Huntington’s disease. The metabolic disease diabetes mellitus results from either the inability of the pancreas to produce sufficient insulin or from cell failure to respond to insulin. Abnormalities in glucose homeostasis and higher prevalence of diabetes mellitus, have been reported in HD patients. During hyperglycemic conditions, glucose transporters increase intracerebral glucose levels, leading increased glycation. AGEs (advanced glycation end products) can be produced intracellularly by multiple pathways: Methylglyoxal (MGO)-generated AGEs are particularly relevant in neuronal cells. In HD, an abnormal elongation of CAG repeats in the huntingtin gene (HTT) results in the production of mutant huntingtin protein with an extended polyglutamine tract (mHTT), causing its aggregation. The cytoplasmic mHTT aggregates impair autophagic and proteasomal pathways. Additionally, glycation can further contribute to the aggregation of mHTT, potentiating deficits in proteostasis pathways and, ultimately, leading to cell death.
Increased levels of AGEs and reactive dicarbonyls are a hallmark of diabetic patients. Due to the association between HD and diabetes, it is important to understand the relevance of AGEs in the context of HD [2, 55]. Diabetes mellitus is a metabolic disorder characterized by elevated glucose levels in the blood (hyperglycemia). It is caused either by the inability of the pancreas to produce enough insulin (type I) or due to the body cells being insensitive to circulating insulin (type II). Diabetes mellitus also causes several complications in the central nervous system (CNS): both hyperglycemia and insulin-deficiency contribute to neuronal dysfunction. Increased sugar levels lead to a number of metabolic changes including oxidative stress, antioxidant depletion, neuro-inflammation, electrophysiological deficits and hormonal responses [56]. Importantly, the levels of methylglyoxal (MGO), a potent glycating agent, are elevated in diabetic patients, most likely due to hyperglycemia [57]. Consistently, higher AGE levels were detected in diabetic patients and in animal models of diabetes [58, 59].
Glucose is the preferred energy source of the brain and, since neurons can barely store glucose intracellularly, a continuous glucose supply from the blood is critical for normal brain function [60]. The glucose transporters GLUT1 and GLUT3 that are mainly expressed in blood-brain barrier and neurons, respectively, are insulin-independent. Thus, although the insulin dependent glucose transporter GLUT4 is expressed in some brain areas, it is thought that most glucose uptake in the brain is insulin-independent [60]. In line with this, several studies suggest that the intracerebral glucose solely depends on plasma glucose levels in murine models [61–63]. Diabetic animal models have brain glucose levels that are approximately 4-fold higher than those in plasma. Even those diabetes patients that are stabilised on proper medication frequently experience hypo- and hyperglycemic episodes [64]. Magnetic resonance spectroscopy in diabetic patients revealed that the glucose levels in the brain’s interstitial fluid follow those in the blood plasma, albeit in a dampened and delayed manner [65]. Furthermore, diabetic and non-diseased individuals display altered brain glucose responses to changes in the blood plasma glucose [65]. Whether the transport capacities of the blood-brain barrier adapt to acute or chronic hyperglycemia to prevent high glucose levels in the brain is still debated [66]. In summary, chronic or temporary hyper- and hypoglycemia in the central nervous system are important feature in diabetes, and their impact should be carefully analysed in the context of neurodegenerative disorders.
THE MAILLARD REACTION: INITIATORS, MECHANISM AND PROPAGATORS
Glycation, also referred to as non-enzymatic glycosylation, is the non-enzymatic reaction of sugars or other reducing carbohydrates with amino acids or nucleotides. In contrast, enzymatic glycosylation is a post translational modification (PTM) that involves the active attachment of sugar molecules at defined sites and, usually, takes place in the ER and Golgi. Glycation reactions were first described in 1912 in the context of food processing by Louis Camille Maillard [67]. The reaction starts with the condensation of a carbonyl group with the amino or thiol group of amino acids, nucleic acids or amino lipids, leading to the formation of early glycation products including Schiff Base and Amadori products. Initiators of the reactions include glucose, fructose and other sugars, as well as reactive dicarbonyls – such as glyoxal and MGO [68]. MGO is a byproduct of glycolysis and, due to its strong glycation ability, is often used to model glycation reactions [69]. In the second step, intermediate compounds are rearranged and break in one of various chemical pathways to form AGEs [68, 70]. It is important to emphasize that glycation is not a single reaction but, instead, a complex network of related reactions that may begin with different initiators and can result in diverse products [71]. Abundant AGEs include the MGO-derived carboxyethyl lysine (CEL), MGO hydroimidazolone, and glyoxal-derived carboxymethyl lysine (CML). Altogether, AGEs are a heterogenous group of compounds: they can occur bound to proteins or exist in a free state. Many AGEs are intrinsically fluorescent and cause cross links between proteins [70, 72, 73]. Furthermore, AGEs are stable compounds that accumulate during aging [1]. Thus, it is not surprising that cells developed mechanisms to detoxify reactive carbonyls. Glyoxalases are evolutionary conserved enzymes that catabolize dicarbonyls to non-toxic metabolites in glutathione-dependent reactions [74]. Single nucleotide polymorphisms (SNPs) in glyoxalases increase the incidence for the diabetic complications nephropathy and retinopathy [75]. Furthermore, dicarbonyls can be catabolized via nicotinamide adenine dinucleotide phosphate (NAPDH)-dependent aldo-keto reductases [76].
A number of studies reported age-associated accumulation of AGEs on crystallins, collagens and other long-lived proteins [1, 2, 77–79]. Whether increased AGE levels are a cause or a consequence the aging process remains a matter of debate [80]. Interestingly, higher levels of CML, a highly abundant AGE, in adults older than 65, were found to be associated with a higher risk of all-cause or cardiovascular disease mortality [81]. In addition, studies in Drosophilla melanogaster and Caenorhabditis elegans showed that overexpression of glyoxalases increase lifespan, suggesting that glycation reactions contribute to the aging process [82–84].
The receptor for advanced glycation end products (RAGE), is a major pro-inflammatory AGEs receptor and is involved in several neurodegenerative diseases [85, 86]. RAGE ligands in the CNS include CML, beta-amyloid (Aβ), S100 calcium-binding protein B (S100B) and High Mobility Group Box 1 [87–90]. Upon ligand binding, RAGE signals via the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), phosphatidylinositol-3 kinase (PI3K) and mitogen-activated protein kinases (MAPKs) [91, 92].
In addition, AGEs are known to play a role in the context of several neurodegenerative diseases including AD, PD and diabetic complications [2, 93–95]. The mechanisms connecting AGE formation and these disorders include inflammation, dopamine glycation, and decreased degradation of the aggregated and cross-linked proteins.
The loss of dopaminergic cells in the substantia nigra of PD patients is a hallmark of the disease. Interestingly, the catecholamine dopamine itself can undergo several chemical transformations that produce toxic molecules. MGO-mediated modification of dopamine leads to the formation of 1-acetyl-6, 7- dihydroxyl-1, 2, 3, 4-tetrahydroisoquinoline (ADTIQ), a toxin that is also present in the brains of PD patients [96]. Enzymatic oxidation of dopamine by monoamine oxidase produces 3,4-dihydrox-yphenylacetaldehyde (DOPAL), a highly reactive molecule that leads to the oligomerization of alpha-synuclein (aSyn), a central player in the pathology of PD and other neurodegenerative disorders known as synucleinopathies [97]. Furthermore, glycation increases aSyn oligomerization in vitro and glycated aSyn is present in postmortem tissue from individuals with PD and Lewy body disease [84, 98, 99].
Interestingly, several studies suggested that deregulation of glucose metabolism, as in diabetes mellitus, is associated with an increased risk for PD, and with more severe PD features [93]. In AD, AGEs promote aggregation and cross-linking of tau and Aβ. These patients also show higher levels of AGEs in amyloid plaques [100–103]. In addition, levels of amyloid precursor protein (APP) are upregulated through glycation, which ultimately also increases Aβ levels [104]. In summary, intensified research on glycation reaction is highlighting its importance in the pathology of several neurodegenerative diseases.
GLYCATION AS A POTENTIAL PROMOTER OF HD PATHOLOGY
In contrast to AD or PD, HD is dominantly inherited. Thus, the question is not whether altered carbohydrate metabolism and increased AGE formation increase the risk to develop HD. Instead, one may hypothesize that factors that potentiate glycation, such as alterations in carbohydrate metabolism and increased AGE formation, might modify the age of onset and progression of HD (Fig. 1). Despite many advances on the investigation of the roles of AGEs in neurodegenerative diseases, the contribution of AGE formation to HD pathogenesis remain elusive. However, although several studies suggest that altered glucose metabolism may be an important feature of HD, others found no correlation, and this controversy dates back to the 1970 s.
Interestingly, epidemiologic studies reported an increased incidence of diabetes in HD patients [5, 105]. A study on a Chinese family over five generations reported a drastically increased incidence of diabetes mellitus among family members affected by HD [106]. In addition, decreased insulin sensitivity and increased insulin levels were found in non-diabetic HD patients [107].
In a proteomic analysis of HD versus control brains, three proteins involved in glycolysis were found to be differentially expressed [108]. However, other studies found no altered carbohydrate metabolism or increased diabetes mellitus incidence among HD patients [109–115]. Therefore, additional epidemiologic studies, preferably longitudinal, on well-characterized cohorts are needed in order to further establish a connection between HD and altered glucose metabolism or diabetes mellitus.
Studies in animal models suggest that HD affects the pancreatic function. The R6/2 mouse model of HD, which expresses exon 1 of the HTT gene containing 150 CAG repeats, develops insulin-responsive diabetes [116]. In addition, increased glucose plasma levels have also been observed in different models expressing HTT with shorter polyQ repeats (82Q and 89Q) [117, 118]. HTT inclusions were found in the Langerhans islets in R6/2 mice [19, 119]. The pathological mechanisms causing diabetes in these mice include decreased beta-cell replication and insulin secretion, and reduced insulin messenger RNA (mRNA) and protein levels [7, 19]. In contrast, pancreatic tissue from HD patients revealed that insulin mRNA and protein distribution and beta-cell area are identical in control and diseased brains [120].
As mentioned above, RAGE is involved in a variety of cellular processes including homeostasis, inflammation, neurodegeneration, development and neurite outgrowth. Interestingly, RAGE levels are elevated both in brains of HD patients and in mouse models [121, 122]. RAGE is expressed in medium spiny neurons and astrocytes in the caudate nucleus and subependymal layer of HD brains [122]. RAGE co-localizes with CML and S100B, mainly in astrocytes and, interestingly, its levels increase with disease severity [123].
DJ-1, the product of the Parkinson-associated PARK7 gene, was shown to act as a deglycase on early glycation products of cysteines, lysines and arginines [124, 125]. Interestingly, DJ-1 levels are elevated in the frontal cortex of HD brain tissue, R6/2 mice, and in cell models, and overexpression of DJ-1 protects yeast and fly HD models against HTT-induced pathology. In contrast, in other cell models, DJ-1 overexpression leads to increased HTT aggregation and toxicity [126].
Atrophy of skeletal muscle is observed in HD patients and in the R6/2 mouse model [119, 127]. The mechanisms underlying muscle wasting in HD are unknown but may be the consequence of inclusion body formation in muscle cells of HD patients and mouse models [119, 128, 129]. Furthermore, skeletal muscle gene expression changes were observed both in mouse models and human HD [130, 131]. Interestingly, the muscle expression in HD shares some features with those induced by diabetes or fasting [130].
In a recent study from our group, we found a correlation between glycation and pathogenesis in models of HD. Using yeast and mammalian cell models, we found that increasing glycation increases HTT aggregation and toxicity, and impairs HTT clearance. Furthermore, increasing glycation causes neurotoxicity and impairs lifespan and development in a fly model of HD, confirming in vivo effects [132]. In summary, even though additional research is needed to ultimately demonstrate the contribution of AGE formation towards HD age of onset and progression, several lines of evidence point to a critical role of glycation.
TARGETING GLYCATION AS A THERAPEUTIC APPROACH IN HD
Although no disease-modifying therapies are available for HD [25, 133], patients often receive standard drug treatments to alleviate some of the symptoms of the disease [134, 135].
The association between HD and glucose metabolism alterations has been explored as a possible target for strategies using different hypoglycemic agents [112, 136, 137]. Compounds such as exendin-4, resveratrol, glibenclamide, rosiglitazone, insulin and the fusion of the glucagon-like peptide 1 with a non-glycosylated form of human transferrin (GLP- 1Tf) were previously tested in patients and animal models [136, 138–141]. The administration of glibenclamide, exendin-4, GLP-1Tf and resveratrol in animal models resulted in a decrease of the glucose levels in the blood. Interestingly, mice respond to glibenclamide (which induces insulin exocytosis) but not to rosiglitazone (which induces sensitization to insulin) [142]. This supports the hypothesis that diabetes mellitus in the HD mouse model may be caused by an impairment in insulin release rather than by insulin insensitivity. Exendin-4 was the only treatment able to increase the insulin sensitivity [20, 143, 144]. Furthermore, insulin and GLP-1Tf increase plasma insulin levels, in contrast to exendin-4 [145]. Interestingly, both exendin-4 and GLP-1Tf improve motor coordination and life span in HD animal models. Despite the improvement in diabetes mellitus symptoms, chronic treatment with these hypoglycaemic agents has no effect on either the course of diabetes or the progression of HD in mice. More recently, administration of metformin resulted in reduced translation of mutant HTT protein and, therefore, decreased the protein load in vitro and in animal models [146]. This drug is regularly used in patients with diabetes mellitus, and was previously tested in another study where it increased the lifespan of mice [147]. However, the effects on glycation pathways or AGEs were not analysed in this study, and remain to be investigated.
Given the evidence implicating AGEs in diabetes, drugs capable of detoxifying the reactive compounds might constitute an important approach in the treatment of diabetes and also of age-associated neurodegenerative disorders [148–151]. Aminoguanidine, also known as Pimagedine, inhibits AGE formation in animal models [152–154], but was discontinued from the clinical trial in humans due to side effects [155]. The rationale behind this clinical trial is a proof of concept that inhibiting AGE formation may be important for attenuating the serious complications of diabetes mellitus [156]. In this context, strategies aimed at lowering MGO levels are an additional possibility [157–159]. D-penicillamine, aminoguanidine and metformin trap dicarbonyl compounds (e.g. glyoxal and MGO) to form substituted triazines [160]. Another strategy to lower MGO levels is to stimulate the anaerobic pentose phosphate pathway of glycolysis [158, 161]. Tenilsetam, another dicarbonyl compound, inhibits protein cross-linking and cell death [100, 162, 163].
Oxidative stress and neuroinflammation are additional factors known to play an important role in the progression of the neurodegenerative process. Interestingly, AGE production is significantly augmented under oxidative stress [148, 164, 165]. Therefore, therapies combining the use of antioxidants and protein glycation inhibitors may be a more effective approach in neurodegenerative diseases [166–168].
Several synthetic compounds inhibit AGE formation [169]. However, these compounds were withdrawn from the clinical trials because of their low efficacy, unsatisfactory safety, and poor pharmacokinetics [170, 171]. Alternatively, natural products have been proven to be safe for human consumption and many plant extracts have been tested for their anti-glycation activity [172]. Furthermore, previous studies hypothesise that enrichment of diet in natural anti-glycating agents, as polyphenols and other natural antioxidants, may halt the aging process and neurological problems [173].
Finally, as described above, RAGE is as an important subject of research [174], and in vitro and in vivo studies have demonstrated the potential of RAGE as a therapeutic target in neurodegeneration [150, 151, 175–177]. Additional studies will be important to confirm this.
In summary, future clinical trials aimed at lowering AGE formation and the downstream effect of such species will unveil whether the promising results in animal models translate into clinical application.
CONCLUSIONS
Currently, there is a limited understanding of the causal effects of both diet and AGEs on aging and age-related diseases. More importantly, aspects of AGE formation, accumulation and detoxification in several neurodegenerative diseases remain poorly understood. A relative low number of studies explored the connection between diabetes mellitus and HD, suggesting this is a field that deserves additional studies. We have recently shown that glycation potentiates mutant HTT aggregation and toxicity in cell and animal models, highlighting the importance of this modification in HD pathology. Investigating common molecular mechanisms underlying these pathways might reveal novel targets for the development of disease-modifying therapies. Interestingly, metformin, a drug commonly used for type II diabetes, recently showed positive effects in HD models. The studies showed a reduction in mutant HTT levels and a reversion of other pathological features characteristic of HD. In conclusion, we hypothesize that the development of therapeutic strategies targeting glycation may serve as an orthogonal approach to treat both diabetic complications as well as neurodegenerative diseases, such as HD.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
TFO is supported by an EU Joint Programme – Neurodegenerative Disease Research (JPND) project (aSynProtec) and by the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 721802.
REFERENCES
[1] | Chaudhuri J , Bains Y , Guha S , Kahn A , Hall D , Bose N , et al. The role of advanced glycation end products in aging and metabolic diseases: Bridging association and causality. Cell Metab. (2018) ;28: :337–52. |
[2] | Singh VP , Bali A , Singh N , Jaggi AS . Advanced glycation end products and diabetic complications. Korean J Physiol Pharmacol. (2014) ;18: :1–14. |
[3] | Ristow M . Neurodegenerative disorders associated with diabetes mellitus. J Mol Med. (2004) ;82: :510–29. |
[4] | Farrer LA . Diabetes mellitus in Huntington disease. Clin Genet. (1985) ;27: :62–7. |
[5] | Podolsky S , Leopold NA , Sax DS . Increased frequency of diabetes mellitus in patients with Huntington’s chorea. Lancet. (1972) ;1: :1356–8. |
[6] | Montojo MT , Aganzo M , González N . Huntington’s disease and diabetes: Chronological sequence of its association. J Huntingtons Dis. (2017) ;6: :179–88. |
[7] | Andreassen OA , Dedeoglu A , Stanojevic V , Hughes DB , Browne SE , Leech CA , et al. Huntington’s disease of the endocrine pancreas: Insulin deficiency and diabetes mellitus due to impaired insulin gene expression. Neurobiol Dis. (2002) ;11: :410–24. |
[8] | Finocchiaro G , Baio G , Micossi P , Pozza G , di Donato S . Glucose metabolism alterations in Friedreich’s ataxia. Neurology. (1988) ;38: :1292–6. |
[9] | Krentz AJ , Clark PM , Cox L , Williams AC , Nattrass M . Hyperproinsulinaemia in patients with myotonic dystrophy. Diabetologia. (1992) ;35: :1170–2. |
[10] | McColgan P , Tabrizi SJ . Huntington’s disease: A clinical review. Eur J Neurol. (2018) ;25: :24–34. |
[11] | Ambrose CM , Duyao MP , Barnes G , Bates GP , Lin CS , Srinidhi J , et al. Structure and expression of the Huntington’s disease gene: Evidence against simple inactivation due to an expanded CAG repeat. Somat Cell Mol Genet. (1994) ;20: :27–38. |
[12] | Morales LM , Estevez J , Suarez H , Villalobos R , Chacin de Bonilla L , Bonilla E . Nutritional evaluation of Huntington disease patients. Am J Clin Nutr. (1989) ;50: :145–50. |
[13] | Trejo A , Boll MC , Alonso ME , Ochoa A , Velasquez L . Use of oral nutritional supplements in patients with Huntington’s disease. Nutrition. (2005) ;21: :889–94. |
[14] | Reiner A , Albin RL , Anderson KD , D’Amato CJ , Penney JB , Young AB . Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. (1988) ;85: :5733–37. |
[15] | Rosas HD , Liu AK , Hersch S , Glessner M , Ferrante RJ , Salat DH , et al. Regional and progressive thinning of the cortical ribbon in Huntington’s disease. Neurology. (2002) ;58: :695–701. |
[16] | Aziz NA , Swaab DF , Pijl H , Roos RA . Hypothalamic dysfunction and neuroendocrine and metabolic alterations in Huntington’s disease: Clinical consequences and therapeutic implications. Rev Neurosci. (2007) ;18: :223–51. |
[17] | Aziz NA , van der Marck MA , Pijl H , Olde Rikkert MG , Bloem BR , Roos RA . Weight loss in neurodegenerative disorders. J Neurol. (2008) ;255: :1872–80. |
[18] | Trejo A , Tarrats RM , Alonso ME , Boll MC , Ochoa A , Velasquez L . Assessment of the nutrition status of patients with Huntington’s disease. Nutrition. (2004) ;20: :192–6. |
[19] | Björkqvist M , Fex M , Renström E , Wierup N , Petersén Å , Gil J , et al. The R6/2 transgenic mouse model of Huntington’s disease develops diabetes due to deficient β-cell mass and exocytosis. Hum Mol Genet. (2005) ;14: :565–74. |
[20] | Martin B , Golden E , Carlson OD , Pistell P , Zhou J , Kim W , et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes. (2009) ;58: :318–28. |
[21] | Smith R , Bacos K , Fedele V , Soulet D , Walz HA , Obermüller S , et al. Mutant huntingtin interacts with beta-tubulin and disrupts vesicular transport and insulin secretion. Hum Mol Genet. (2009) ;18: :3942–54. |
[22] | van der Burg JM , Bacos K , Wood NI , Lindqvist A , Wierup N , Woodman B , et al. Increased metabolism in the R6/2 mouse model of Huntington’s disease. Neurobiol Dis. (2008) ;29: :41–51. |
[23] | A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. (1993) ;72: :971–83. |
[24] | Li SH , Li XJ . Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. (2004) ;20: :146–54. |
[25] | Jimenez-Sanchez M , Licitra F , Underwood BR , Rubinsztein DC . Huntington’s disease: Mechanisms of pathogenesis and therapeutic strategies. Cold Spring Harb Perspect Med. (2017) ;7: :a024240. |
[26] | Sharp AH , Loev SJ , Schilling G , Li SH , Li XJ , Bao J , et al. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. (1995) ;14: :1065–74. |
[27] | Onodera O , Burke JR , Miller SE , Hester S , Tsuji S , Roses AD , et al. Oligomerization of expanded-polyglutamine domain fluorescent fusion proteins in cultured mammalian cells. Biochem Biophys Res Commun. (1997) ;238: :599–605. |
[28] | Preisinger E , Jordan BM , Kazantsev A , Housman D . Evidence for a recruitment and sequestration mechanism in Huntington’s disease. Philos Trans R Soc L B Biol Sci. (1999) ;354: :1029–34. |
[29] | DiFiglia M , Sapp E , Chase KO , Davies SW , Bates GP , Vonsattel JP , et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. (1997) ;277: :1990–3. |
[30] | Kazantsev A , Preisinger E , Dranovsky A , Goldgaber D , Housman D . Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci U S A. (1999) ;96: :11404–9. |
[31] | Nagai Y , Onodera O , Strittmatter WJ , Burke JR , Polyglutamine domain proteins with expanded repeats bind neurofilament, altering the neurofilament network. Ann N Y Acad Sci. (1999) ;893: :192–202. |
[32] | Senut MC , Suhr ST , Kaspar B , Gage FH . Intraneuronal aggregate formation and cell death after viral expression of expanded polyglutamine tracts in the adult rat brain. J Neurosci. (2000) ;20: :219–29. |
[33] | Vonsattel JP , Myers RH , Stevens TJ , Ferrante RJ , Bird ED , Richardson EP Jr . Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. (1985) ;44: :559–77. |
[34] | Kremer HP , Roos RA , Dingjan G , Marani E , Bots GT . Atrophy of the hypothalamic lateral tuberal nucleus in Huntington’s disease. J Neuropathol Exp Neurol. (1990) ;49: :371–82. |
[35] | Kremer HP , Roos RA , Dingjan GM , Bots GT , Bruyn GW , Hofman MA . The hypothalamic lateral tuberal nucleus and the characteristics of neuronal loss in Huntington’s disease. Neurosci Lett. (1991) ;132: :101–4. |
[36] | Kassubek J , Gaus W , Landwehrmeyer GB . Evidence for more widespread cerebral pathology in early HD: An MRI-based morphometric analysis. Neurology. (2004) ;62: :523–4. |
[37] | Petersen A , Gil J , Maat-Schieman ML , Bjorkqvist M , Tanila H , Araujo IM , et al. Orexin loss in Huntington’s disease. Hum Mol Genet. (2005) ;14: :39–47. |
[38] | DiFiglia M , Sapp E , Chase K , Schwarz C , Meloni A , Young C , et al. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. (1995) ;14: :1075–81. |
[39] | Velier J , Kim M , Schwarz C , Kim TW , Sapp E , Chase K , et al. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp Neurol. (1998) ;152: :34–40. |
[40] | Hilditch-Maguire P , Trettel F , Passani LA , Auerbach A , Persichetti F , MacDonald ME . Huntingtin: An iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet. (2000) ;9: :2789–97. |
[41] | Kegel KB , Meloni AR , Yi Y , Kim YJ , Doyle E , Cuiffo BG , et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. (2002) ;277: :7466–76. |
[42] | Li J-Y , Plomann M , Brundin P . Huntington’s disease: A synaptopathy? Trends Mol Med. (2003) ;9: :414–20. |
[43] | Harjes P , Wanker EE . The hunt for huntingtin function: Interaction partners tell many different stories. Trends Biochem Sci. (2003) ;28: :425–33. |
[44] | Cattaneo E , Rigamonti D , Goffredo D , Zuccato C , Squitieri F , Sipione S . Loss of normal huntingtin function: New developments in Huntington’s disease research. Trends Neurosci. (2001) ;24: :182–8. |
[45] | Cattaneo E , Zuccato C , Tartari M . Normal huntingtin function: An alternative approach to Huntington’s disease. Nat Rev Neurosci. (2005) ;6: :919–30. |
[46] | Greenamyre JT . Huntington’s disease: Getting closer. Am J Psychiatry. (2007) ;164: :1318. |
[47] | Benn CL , Sun T , Sadri-Vakili G , McFarland KN , DiRocco DP , Yohrling GJ , et al. Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J Neurosci. (2008) ;28: :10720–33. |
[48] | Gusella JF , MacDonald ME , Lee J-M . Genetic modifiers of Huntington’s disease. Mov Disord. (2014) ;29: :1359–65. |
[49] | Garcia-Gorro C , Garau-Rolandi M , Escrichs A , Rodriguez-Dechicha N , Vaquer I , Subira S , et al. An active cognitive lifestyle as a potential neuroprotective factor in Huntington’s disease. Neuropsychologia. (2019) ;122: :116–24. |
[50] | de Diego-Balaguer R , Schramm C , Rebeix I , Dupoux E , Durr A , Brice A , et al. COMT Val158Met polymorphism modulates Huntington’s disease progression. PLoS One. (2016) ;11: :e0161106. |
[51] | Nithianantharajah J , Barkus C , Vijiaratnam N , Clement O , Hannan AJ . Modeling brain reserve: Experience-dependent neuronal plasticity in healthy and Huntington’s disease transgenic mice. Am J Geriatr Psychiatry. (2009) ;17: :196–209. |
[52] | Petersen A , Bjorkqvist M . Hypothalamic-endocrine aspects in Huntington’s disease. Eur J Neurosci. (2006) ;24: :961–7. |
[53] | Craft S , Watson GS . Insulin and neurodegenerative disease: Shared and specific mechanisms. Lancet Neurol. (2004) ;3: :169–78. |
[54] | van der Burg JM , Bjorkqvist M , Brundin P . Beyond the brain: Widespread pathology in Huntington’s disease. Lancet Neurol. (2009) ;8: :765–74. |
[55] | Henning C , Liehr K , Girndt M , Ulrich C , Glomb MA . Extending the spectrum of α-dicarbonyl compounds in vivo . J Biol Chem. (2014) ;289: :28676–88. |
[56] | Tomlinson DR , Gardiner NJ . Glucose neurotoxicity. Nat Rev Neurosci. (2008) ;9: :36–45. |
[57] | Shamsaldeen YA , Mackenzie LS , Lione LA , Benham CD . Methylglyoxal, A metabolite increased in diabetes is associated with insulin resistance, vascular dysfunction and neuropathies. Curr Drug Metab. (2016) ;17: :359–67. |
[58] | Dalfó E , Portero-Otín M , Ayala V , Martínez A , Pamplona R , Ferrer I . Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J Neuropathol Exp Neurol. (2005) ;64: :816–30. |
[59] | Ashraf JM , Ahmad S , Choi I , Ahmad N , Farhan M , Tatyana G , et al. Recent advances in detection of AGEs: Immunochemical, bioanalytical and biochemical approaches. IUBMB Life. (2015) ;67: :897–913. |
[60] | Camandola S , Mattson MP . Brain metabolism in health, aging, and neurodegeneration. EMBO J. (2017) ;36: :1474–92. |
[61] | de Vries MG , Arseneau LM , Lawson ME , Beverly JL . Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes. (2003) ;52: :2767–73. |
[62] | Silver IA , Erecińska M . Extracellular glucose concentration in mammalian brain: Continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci. (1994) ;14: :5068–76. |
[63] | Abi-Saab WM , Maggs DG , Jones T , Jacob R , Srihari V , Thompson J , et al. Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: Effects of hyperglycemia and hypoglycemia. J Cereb Blood Flow Metab. (2002) ;22: :271–9. |
[64] | Tomlinson DR , Gardiner NJ . Glucose neurotoxicity. Nat Rev Neurosci. (2008) ;9: :36–45. |
[65] | Hwang JJ , Jiang L , Hamza M , Sanchez Rangel E , Dai F , Belfort-DeAguiar R , et al. Blunted rise in brain glucose levels during hyperglycemia in adults with obesity and T2DM. JCI insight. (2017) ;2: :95913. |
[66] | Hasselbalch SG , Knudsen GM , Capaldo B , Postiglione A , Paulson OB . Blood-brain barrier transport and brain metabolism of glucose during acute hyperglycemia in humans. J Clin Endocrinol Metab. (2001) ;86: :1986–90. |
[67] | Maillard LC . Action des acides amines sur les sucre: Formation des melanoidines par voie methodique. C R Hebd Seances Acad Sci. (1912) ;66–8. |
[68] | Henning C , Glomb MA . Pathways of the Maillard reaction under physiological conditions. Glycoconj J. (2016) ;33: :499–512. |
[69] | Thornalley PJ , Langborg A , Minhas HS . Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J. (1999) ;344 Pt 1: :109–16. |
[70] | Singh R , Barden A , Mori T , Beilin L . Advanced glycation end-products: A review. Diabetologia. (2001) ;44: :129–46. |
[71] | Hemmler D , Roullier-Gall C , Marshall JW , Rychlik M , Taylor AJ , Schmitt-Kopplin P . Evolution of complex Maillard chemical reactions, resolved in time. Sci Rep. (2017) ;7: :3227. |
[72] | Brings S , Fleming T , Freichel M , Muckenthaler M , Herzig S , Nawroth P . Dicarbonyls and advanced glycation end-products in the development of diabetic complications and targets for intervention. Int J Mol Sci. (2017) ;18: :984. |
[73] | Vistoli G , De Maddis D , Cipak A , Zarkovic N , Carini M , Aldini G . Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic Res. (2013) ;47: (Suppl 1):3–27. |
[74] | Thornalley PJ . The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem J. (1990) ;269: :1–11. |
[75] | Wu JC , Li XH , Peng YD , Wang JB , Tang JF , Wang YF . Association of two glyoxalase I gene polymorphisms with nephropathy and retinopathy in Type 2 diabetes. J Endocrinol Invest. (2011) ;34: :e343–8. |
[76] | Vander Jagt DL1 , Robinson B , Taylor KK , Hunsaker LA . Reduction of trioses by NADPH-dependent aldo-keto reductases. Aldose reductase, methylglyoxal, and diabetic complications. J Biol Chem. (1992) ;267: :4364–9. |
[77] | Sell DR , Lapolla A , Odetti P , Fogarty J , Monnier VM . Pentosidine formation in skin correlates with severity of complications in individuals with long-standing IDDM. Diabetes. (1992) ;41: :1286–92. |
[78] | Nagaraj RH , Linetsky M , Stitt AW . The pathogenic role of Maillard reaction in the aging eye. Amino Acids. (2012) ;42: :1205–20. |
[79] | Monnier VM , Kohn RR , Cerami A . Accelerated age-related browning of human collagen in diabetes mellitus. Proc Natl Acad Sci U S A. (1984) ;81: :583–7. |
[80] | Monnier VM . Toward a Maillard reaction theory of aging. Prog Clin Biol Res. (1989) ;304: :1–22. |
[81] | Semba RD , Bandinelli S , Sun K , Guralnik JM , Ferrucci L . Plasma carboxymethyl-lysine, an advanced glycation end product, and all-cause and cardiovascular disease mortality in older community-dwelling adults. J Am Geriatr Soc. (2009) ;57: :1874–80. |
[82] | Morcos M , Du X , Pfisterer F , Hutter H , Sayed AAR , Thornalley P , et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. (2008) ;7: :260–9. |
[83] | Chaudhuri J , Bose N , Gong J , Hall D , Rifkind A , Bhaumik D , et al. A Caenorhabditis elegans model elucidates a conserved role for TRPA1-Nrf signaling in reactive α-dicarbonyl detoxification. Curr Biol. (2016) ;26: :3014–25. |
[84] | VicenteMiranda H , Szegő ÉM , Oliveira LMA , Breda C , Darendelioglu E , de Oliveira RM , et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain. (2017) ;140: :1399–419. |
[85] | Schmidt AM , Vianna M , Gerlach M , Brett J , Ryan J , Kao J , et al. Isolation and characterization of two binding proteins for advanced glycosylation end products from bovine lung which are present on the endothelial cell surface. J Biol Chem. (1992) ;267: :14987–97. |
[86] | Daffu G , del Pozo C , O’Shea K , Ananthakrishnan R , Ramasamy R , Schmidt A , et al. Radical roles for RAGE in the pathogenesis of oxidative stress in cardiovascular diseases and beyond. Int J Mol Sci. (2013) ;14: :19891–910. |
[87] | Kislinger T , Fu C , Huber B , Qu W , Taguchi A , Du Yan S , et al. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for receptor for advanced glycation end products that activate cell signaling pathways and modulate gene expression. J Biol Chem. (1999) ;274: :31740–9. |
[88] | Taguchi A , Blood DC , del Toro G , Canet A , Lee DC , Qu W , et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. (2000) ;405: :354–60. |
[89] | Hofmann MA , Drury S , Fu C , Qu W , Taguchi A , Lu Y , et al. RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell. (1999) ;97: :889–901. |
[90] | Yan SD , Zhu H , Zhu A , Golabek A , Du H , Roher A , et al. Receptor-dependent cell stress and amyloid accumulation in systemic amyloidosis. Nat Med. (2000) ;6: :643–51. |
[91] | Yan SF , Ramasamy R , Schmidt AM . The RAGE axis. Circ Res. (2010) ;106: :842–53. |
[92] | Ramasamy R , Yan SF , Schmidt AM . Receptor for AGE (RAGE): Signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci. (2011) ;1243: :88–102. |
[93] | König A , Vicente Miranda H , Outeiro TF . Alpha-synuclein glycation and the action of anti-diabetic agents in Parkinson’s disease. J Parkinsons Dis. (2018) ;8: :33–43. |
[94] | Vicente Miranda H , El-Agnaf OMA , Outeiro TF . Glycation in Parkinson’s disease and Alzheimer’s disease. Mov Disord. (2016) ;31: :782–90. |
[95] | Castellani R , Smith MA , Richey PL , Perry G . Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. (1996) ;737: :195–200. |
[96] | Xie B , Lin F , Ullah K , Peng L , Ding W , Dai R , et al. A newly discovered neurotoxin ADTIQ associated with hyperglycemia and Parkinson’s disease. Biochem Biophys Res Commun. (2015) ;459: :361–6. |
[97] | Plotegher N , Berti G , Ferrari E , Tessari I , Zanetti M , Lunelli L , et al. DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci Rep. (2017) ;7: :40699. |
[98] | Lee D , Park CW , Paik SR , Choi KY . The modification of α-synuclein by dicarbonyl compounds inhibits its fibril-forming process. Biochim Biophys Acta. (2009) ;1794: :421–30. |
[99] | Padmaraju V , Bhaskar JJ , Prasada Rao UJS , Salimath PV , Rao KS . Role of advanced glycation on aggregation and DNA binding properties of α-synuclein. J Alzheimers Dis. (2011) ;24: (Suppl 2):211–21. |
[100] | Munch G , Mayer S , Michaelis J , Hipkiss AR , Riederer P , Muller R , et al. Influence of advanced glycation end-products and AGE-inhibitors on nucleation-dependent polymerization of beta-amyloid peptide. Biochim Biophys Acta. (1997) ;1360: :17–29. |
[101] | Vitek MP , Bhattacharya K , Glendening JM , Stopa E , Vlassara H , Bucala R , et al. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci U S A. (1994) ;91: :4766–70. |
[102] | Ledesma MD , Bonay P , Colaço C , Avila J . Analysis of microtubule-associated protein tau glycation in paired helical filaments. J Biol Chem. (1994) ;269: :21614–9. |
[103] | Woltjer RL , Maezawa I , Ou JJ , Montine KS , Montine TJ . Advanced glycation endproduct precursor alters intracellular amyloid-beta/A beta PP carboxy-terminal fragment aggregation and cytotoxicity. J Alzheimers Dis. (2003) ;5: :467–76. |
[104] | Ko S-Y , Lin Y-P , Lin Y-S , Chang S-S . Advanced glycation end products enhance amyloid precursor protein expression by inducing reactive oxygen species. Free Radic Biol Med. (2010) ;49: :474–80. |
[105] | Farrer LA . Diabetes mellitus in Huntington disease. Clin Genet. (1985) ;27: :62–7. |
[106] | Hu Y , Liang J , Yu S . High prevalence of diabetes mellitus in a five-generation Chinese family with Huntington’s disease. J Alzheimer’s Dis. (2014) ;40: :863–8. |
[107] | Lalić NM , Marić J , Svetel M , Jotić A , Stefanova E , Lalić K , et al. Glucose homeostasis in Huntington disease. Arch Neurol. (2008) ;65: :476. |
[108] | Schönberger SJ , Jezdic D , Faull RLM , Cooper GJS . Proteomic analysis of the human brain in Huntington’s Disease indicates pathogenesis by molecular processes linked to other neurodegenerative diseases and to type-2 diabetes. J Huntingtons Dis. (2013) ;2: :89–99. |
[109] | Kremer HP , Roos RA , Frölich M , Radder JK , Nieuwenhuijzen Kruseman AC , Van der Velde A , et al. Endocrine functions in Huntington’s disease. A two-and-a-half years follow-up study. J Neurol Sci. (1989) ;90: :335–44. |
[110] | Lavin PJ , Bone I , Sheridan P . Studies of hypothalamic function in Huntington’s chorea. J Neurol Neurosurg Psychiatry. (1981) ;44: :414–8. |
[111] | Keogh HJ , Johnson RH , Nanda RN , Sulaiman WR . Altered growth hormone release in Huntington’s chorea. J Neurol Neurosurg Psychiatry. (1976) ;39: :244–8. |
[112] | Nambron R , Silajdžić E , Kalliolia E , Ottolenghi C , Hindmarsh P , Hill NR , et al. A metabolic study of Huntington’s disease. PLoS One. (2016) ;11: :e0146480. |
[113] | Russo C V , Salvatore E , Saccá F , Tucci T , Rinaldi C , Sorrentino P , et al. Insulin sensitivity and early-phase insulin secretion in normoglycemic Huntington’s disease patients. J Huntingtons Dis. (2013) ;2: :501–7. |
[114] | Zarowitz BJ , O’Shea T , Nance M . Clinical, demographic, and pharmacologic features of nursing home residents with Huntington’s disease. J Am Med Dir Assoc. (2014) ;15: :423–8. |
[115] | Boesgaard TW , Nielsen TT , Josefsen K , Hansen T , Jørgensen T , Pedersen O , et al. Huntington’s disease does not appear to increase the risk of diabetes mellitus. J Neuroendocrinol. (2009) ;21: :770–6. |
[116] | Hurlbert MS , Zhou W , Wasmeier C , Kaddis FG , Hutton JC , Freed CR . Mice transgenic for an expanded CAG repeat in the Huntington’s disease gene develop diabetes. Diabetes. (1999) ;48: :649–51. |
[117] | Josefsen K , Nielsen MD , Jørgensen KH , Bock T , Nørremølle A , Sørensen SA , et al. Impaired glucose tolerance in the R6/1 transgenic mouse model of Huntington’s disease. J Neuroendocrinol. (2008) ;20: :165–72. |
[118] | Duan W , Guo Z , Jiang H , Ware M , Li X-J , Mattson MP . Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A. (2003) ;100: :2911–6. |
[119] | Sathasivam K , Hobbs C , Turmaine M , Mangiarini L , Mahal A , Bertaux F , et al. Formation of polyglutamine inclusions in non-CNS tissue. Hum Mol Genet. (1999) ;8: :813–22. |
[120] | Bacos K , Björkqvist M , Petersén A , Luts L , Maat-Schieman MLC , Roos RAC , et al. Islet beta-cell area and hormone expression are unaltered in Huntington’s disease. Histochem Cell Biol. (2008) ;129: :623–9. |
[121] | Anzilotti S , Giampá C , Laurenti D , Perrone L , Bernardi G , Melone MAB , et al. Immunohistochemical localization of receptor for advanced glycation end (RAGE) products in the R6/2 mouse model of Huntington’s disease. Brain Res Bull. (2012) ;87: :350–8. |
[122] | Ma L , Nicholson LF . Expression of the receptor for advanced glycation end products in Huntington’s disease caudate nucleus. Brain Res. (2004) ;1018: :10–7. |
[123] | Kim J , Waldvogel HJ , Faull RLM , Curtis MA , Nicholson LFB . The RAGE receptor and its ligands are highly expressed in astrocytes in a grade-dependant manner in the striatum and subependymal layer in Huntington’s disease. J Neurochem. (2015) ;134: :927–42. |
[124] | Richarme G , Mihoub M , Dairou J , Bui LC , Leger T , Lamouri A . Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J Biol Chem. (2015) ;290: :1885–97. |
[125] | Lee J , Song J , Kwon K , Jang S , Kim C , Baek K , et al. Human DJ-1 and its homologs are novel glyoxalases. Hum Mol Genet. (2012) ;21: :3215–25. |
[126] | Sajjad MU , Green EW , Miller-Fleming L , Hands S , Herrera F , Campesan S , et al. DJ-1 modulates aggregation and pathogenesis in models of Huntington’s disease. Hum Mol Genet. (2014) ;23: :755–66. |
[127] | Ribchester RR , Thomson D , Wood NI , Hinks T , Gillingwater TH , Wishart TM , et al. Progressive abnormalities in skeletal muscle and neuromuscular junctions of transgenic mice expressing the Huntington’s disease mutation. Eur J Neurosci. (2004) ;20: :3092–114. |
[128] | Ciammola A , Sassone J , Alberti L , Meola G , Mancinelli E , Russo MA , et al. Increased apoptosis, huntingtin inclusions and altered differentiation in muscle cell cultures from Huntington’s disease subjects. Cell Death Differ. (2006) ;13: :2068–78. |
[129] | Moffitt H , McPhail GD , Woodman B , Hobbs C , Bates GP . Formation of polyglutamine inclusions in a wide range of non-CNS tissues in the HdhQ150 knock-in mouse model of Huntington’s disease. PLoS One. (2009) ;4: :e8025. |
[130] | Strand AD , Aragaki AK , Shaw D , Bird T , Holton J , Turner C , et al. Gene expression in Huntington’s disease skeletal muscle: A potential biomarker. Hum Mol Genet. (2005) ;14: :1863–76. |
[131] | Luthi-Carter R , Hanson SA , Strand AD , Bergstrom DA , Chun W , Peters NL , et al. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: Parallel changes in muscle and brain. Hum Mol Genet. (2002) ;11: :1911–26. |
[132] | Vicente Miranda H , Gomes MA , Branco-Santos J , Breda C , Lázaro DF , Lopes LV , et al. Glycation potentiates neurodegeneration in models of Huntington’s disease. Sci Rep. (2016) ;6: :36798. |
[133] | Frank S . Treatment of Huntington’s disease. Neurotherapeutics. (2014) ;11: :153–60. |
[134] | Potkin KT , Potkin SG . New directions in therapeutics for Huntington disease. Futur Neurol. (2018) ;13: :101–21. |
[135] | Caron NS , Dorsey ER , Hayden MR . Therapeutic approaches to Huntington disease: From the bench to the clinic. Nat Rev Drug Discov. (2018) ;17: :729–50. |
[136] | Lalic NM , Maric J , Svetel M , Jotic A , Stefanova E , Lalic K , et al. Glucose homeostasis in Huntington disease: Abnormalities in insulin sensitivity and early-phase insulin secretion. Arch Neurol. (2008) ;65: :476–80. |
[137] | Wang R , Ross CA , Cai H , Cong W-N , Daimon CM , Carlson OD , et al. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front Physiol. (2014) ;5: :231. |
[138] | Montojo MT , Aganzo M , Gonzalez N . Huntington’s disease and diabetes: Chronological sequence of its association. J Huntingtons Dis. (2017) ;6: :179–88. |
[139] | Kim BJ , Zhou J , Martin B , Carlson OD , Maudsley S , Greig NH , et al. Transferrin fusion technology: A novel approach to prolonging biological half-life of insulinotropic peptides. J Pharmacol Exp Ther. (2010) ;334: :682–92. |
[140] | Ho DJ , Calingasan NY , Wille E , Dumont M , Beal MF . Resveratrol protects against peripheral deficits in a mouse model of Huntington’s disease. Exp Neurol. (2010) ;225: :74–84. |
[141] | Chaudhury A , Duvoor C , Reddy Dendi VS , Kraleti S , Chada A , Ravilla R , et al. Clinical review of antidiabetic drugs: Implications for type 2 diabetes mellitus management. Front Endocrinol. (2017) ;8: :6. |
[142] | Hunt MJ , Morton AJ . Atypical diabetes associated with inclusion formation in the R6/2 mouse model of Huntington’s disease is not improved by treatment with hypoglycaemic agents. Exp Brain Res. (2005) ;166: :220–9. |
[143] | Egan JM , Clocquet AR , Elahi D . The insulinotropic effect of acute exendin-4 administered to humans: Comparison of nondiabetic state to type 2 diabetes. J Clin Endocrinol Metab. (2002) ;87: :1282–90. |
[144] | Elfers CT , Simmons JH , Roth CL . Glucagon-like peptide-1 agonist exendin-4 leads to reduction of weight and caloric intake in a rat model of hypothalamic obesity. Horm Res Paediatr. (2012) ;78: :47–53. |
[145] | Martin B , Chadwick W , Cong WN , Pantaleo N , Daimon CM , Golden EJ , et al. Euglycemic agent-mediated hypothalamic transcriptomic manipulation in the N171-82Q model of Huntington disease is related to their physiological efficacy. J Biol Chem. (2012) ;287: :31766–82. |
[146] | Arnoux I , Willam M , Griesche N , Krummeich J , Watari H , Offermann N , et al. Metformin reverses early cortical network dysfunction and behavior changes in Huntington’s disease. Elife. e. (2018) ;7: :38744. |
[147] | Ma TC , Buescher JL , Oatis B , Funk JA , Nash AJ , Carrier RL , et al. Metformin therapy in a transgenic mouse model of Huntington’s disease. Neurosci Lett. (2007) ;411: :98–103. |
[148] | Li J , Liu D , Sun L , Lu Y , Zhang Z . Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J Neurol Sci. (2012) ;317: :1–5. |
[149] | Rocken C , Kientsch-Engel R , Mansfeld S , Stix B , Stubenrauch K , Weigle B , et al. Advanced glycation end products and receptor for advanced glycation end products in AA amyloidosis. Am J Pathol. (2003) ;162: :1213–20. |
[150] | Ramasamy R , Vannucci SJ , Yan SS , Herold K , Yan SF , Schmidt AM . Advanced glycation end products and RAGE: A common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology. (2005) ;15: :16R–28R-. |
[151] | Ray R , Juranek JK , Rai V . RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci Biobehav Rev. (2016) ;62: :48–55. |
[152] | Li YM , Steffes M , Donnelly T , Liu C , Fuh H , Basgen J , et al. Prevention of cardiovascular and renal pathology of aging by the advanced glycation inhibitor aminoguanidine. Proc Natl Acad Sci U S A. (1996) ;93: :3902–7. |
[153] | Bucala R , Vlassara H . Advanced glycosylation end products in diabetic renal and vascular disease. Am J Kidney Dis. (1995) ;26: :875–88. |
[154] | Freedman BI , Wuerth JP , Cartwright K , Bain RP , Dippe S , Hershon K , et al. Design and baseline characteristics for the aminoguanidine clinical trial in overt type 2 diabetic nephropathy (ACTION II). Control Clin Trials. (1999) ;20: :493–510. |
[155] | Thornalley PJ . Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys. (2003) ;419: :31–40. |
[156] | Bolton WK , Cattran DC , Williams ME , Adler SG , Appel GB , Cartwright K , et al. Randomized trial of an inhibitor of formation of advanced glycation end products in diabetic nephropathy. Am J Nephrol. (2004) ;24: :32–40. |
[157] | Wu S , Ren J . Benfotiamine alleviates diabetes-induced cerebral oxidative damage independent of advanced glycation end-product, tissue factor and TNF-alpha. Neurosci Lett. (2006) ;394: :158–62. |
[158] | Thornalley PJ . The potential role of thiamine (vitamin B1) in diabetic complications. Curr Diabetes Rev. (2005) ;1: :287–98. |
[159] | Dukic-Stefanovic S , Schinzel R , Riederer P , Munch G . AGES in brain ageing: AGE-inhibitors as neuroprotective and anti-dementia drugs? Biogerontology. (2001) ;2: :19–34. |
[160] | Krautwald M , Munch G . Advanced glycation end products as biomarkers and gerontotoxins - A basis to explore methylglyoxal-lowering agents for Alzheimer’s disease? Exp Gerontol. (2010) ;45: :744–51. |
[161] | Rabbani N , Alam SS , Riaz S , Larkin JR , Akhtar MW , Shafi T , et al. High-dose thiamine therapy for patients with type 2 diabetes and microalbuminuria: A randomised, double-blind placebo-controlled pilot study. Diabetologia. (2009) ;52: :208–12. |
[162] | Munch G , Taneli Y , Schraven E , Schindler U , Schinzel R , Palm D , et al. The cognition-enhancing drug tenilsetam is an inhibitor of protein crosslinking by advanced glycosylation. J Neural Transm Park Dis Dement Sect. (1994) ;8: :193–208. |
[163] | Webster J , Urban C , Berbaum K , Loske C , Alpar A , Gärtner U , et al. The carbonyl scavengers aminoguanidine and tenilsetam protect against the neurotoxic effects of methylglyoxal. Neurotox Res. (2005) ;7: :95–101. |
[164] | Videira PAQ , Castro-Caldas M . Linking glycation and glycosylation with inflammation and mitochondrial dysfunction in Parkinson’s disease. Front Neurosci. (2018) ;12: :381. |
[165] | Verdile G , Keane KN , Cruzat VF , Medic S , Sabale M , Rowles J , et al. Inflammation and oxidative stress: The molecular connectivity between insulin resistance, obesity, and Alzheimer’s disease. Mediators Inflamm. (2015) ;2015: :105828. |
[166] | Bavkar LN , Patil RS , Rooge SB , Nalawade ML , Arvindekar AU . Acceleration of protein glycation by oxidative stress and comparative role of antioxidant and protein glycation inhibitor. Mol Cell Biochem. (2019) . doi: 10.1007/s11010-019-03550-7. |
[167] | Borriello M , Iannuzzi C , Sirangelo I . Pinocembrin protects from AGE-induced cytotoxicity and inhibits non-enzymatic glycation in human insulin. Cells. (2019) ;8: :385. |
[168] | Forni C , Facchiano F , Bartoli M , Pieretti S , Facchiano A , D’Arcangelo D , et al. Beneficial role of phytochemicals on oxidative stress and age-related diseases. Biomed Res Int. (2019) ;2019: :1–16. |
[169] | Reddy VP , Beyaz A . Inhibitors of the Maillard reaction and AGE breakers as therapeutics for multiple diseases. Drug Discov Today. (2006) ;11: :646–54. |
[170] | Ueda H , Kuroiwa E , Tachibana Y , Kawanishi K , Ayala F , Moriyasu M . Aldose reductase inhibitors from the leaves of Myrciaria dubia (H.B. & K.) McVaugh. Phytomedicine. (2004) ;11: :652–6. |
[171] | Manzanaro S , Salva J , de la Fuente JA . Phenolic marine natural products as aldose reductase inhibitors. J Nat Prod. (2006) ;69: :1485–7. |
[172] | Lee GY , Jang DS , Lee YM , Kim JM , Kim JS . Naphthopyrone glucosides from the seeds of Cassia tora with inhibitory activity on advanced glycation end products (AGEs) formation. Arch Pharm Res. (2006) ;29: :587–90. |
[173] | Ahmad S , Farhan M . Impact of non-enzymatic glycation in neurodegenerative diseases: Role of natural products in prevention. Adv Neurobiol. (2016) ;12: :125–51. |
[174] | Bongarzone S , Savickas V , Luzi F , Gee AD . Targeting the receptor for advanced glycation endproducts (RAGE): A medicinal chemistry perspective. J Med Chem. (2017) ;60: :7213–32. |
[175] | Deane R , Singh I , Sagare AP , Bell RD , Ross NT , LaRue B , et al. A multimodal RAGE-specific inhibitor reduces amyloid beta-mediated brain disorder in a mouse model of Alzheimer disease. J Clin Invest. (2012) ;122: :1377–92. |
[176] | Cary BP , Brooks AF , Fawaz MV , Drake LR , Desmond TJ , Sherman P , et al. Synthesis and evaluation of [(18)F]RAGER: A first generation small-molecule PET radioligand targeting the receptor for advanced glycation endproducts. ACS Chem Neurosci. (2016) ;7: :391–8. |
[177] | Zhu CW , Sano M , Ferris SH , Whitehouse PJ , Patterson MB , Galasko D , et al. Alzheimer’s Disease Cooperative Study Prevention Instrument Project assessing resource use and volunteer and paid work in healthy elders: A longitudinal study. J Am Geriatr Soc. (2014) ;62: :985–8. |


