
Alpha-, Beta-, and Gamma-Secretase, Amyloid Precursor Protein, and Tau Protein Genes in the Hippocampal CA3 Subfield in an Ischemic Model of Alzheimer’s Disease with Survival up to 2 Years
Abstract
Background:
Understanding the phenomena underlying the non-selective susceptibility to ischemia of pyramidal neurons in the CA3 is important from the point of view of elucidating the mechanisms of memory loss and the development of dementia.
Objective:
The aim of the study was to investigate changes in genes expression of amyloid precursor protein, its cleaving enzymes and tau protein in CA3 post-ischemia with survival of 12–24 months.
Methods:
We used an ischemic model of Alzheimer’s disease to study the above genes using an RT-PCR protocol.
Results:
The expression of the amyloid precursor protein gene was above the control values at all times post-ischemia. The expression of the α-secretase gene also exceeded the control values post-ischemia. The expression of the β-secretase gene increased 12 and 24 months post-ischemia, and 18 months was below control values. Presenilin 1 and 2 genes expression was significantly elevated at all times post-ischemia. Also, tau protein gene expression was significantly elevated throughout the observation period, and peak gene expression was present 12 months post-ischemia.
Conclusions:
The study suggests that the genes studied are involved in the non-amyloidogenic processing of amyloid precursor protein. Additionally data indicate that brain ischemia with long-term survival causes damage and death of pyramidal neurons in the CA3 area of the hippocampus in a modified tau protein-dependent manner. Thus defining a new and important mechanism of pyramidal neuronal death in the CA3 area post-ischemia. In addition expression of tau protein gene modification after brain ischemia is useful in identifying ischemic mechanisms occurring in Alzheimer’s disease.
INTRODUCTION
The hippocampus, a brain structure central to memory encoding, exhibits numerous neurochemical, electrophysiological, neuropathophysiological, neuropathological, structural and cognitive changes in neurodegenerative diseases such as ischemia [1, 2] and Alzheimer’s disease (AD) [3–6]. All these changes underlie the memory loss typical of advanced age, ischemia, and AD [4–6]. The hippocampus is a unidirectional net-work with a trisynaptic pathway that originates in the entorhinal cortex, connecting the dentate gyrus with pyramidal neurons of the CA3 and CA1 areas. Area CA3 receives signals from the dentate gyrus by mossy fibers and connects to the ipsilateral CA1 region through the Schaffer collateral pathway and further connects to the contralateral CA1 area by the commissural association pathway [4]. Additionally, the CA1 area receives signals from the entorhinal cortex by the perforant pathway and connects to the subiculum. Moreover, there is a monosynaptic connection of the prefrontal cortex with the CA3 and CA1 areas of the hippocampus that is able to induce contextual memory retrieval. Areas CA1 and CA3 form a continuum in the hippocampus and have parallel inputs, but these areas have different output pathways and different network architectures [4]. Therefore, these two regions play different roles and process different information [4]. CA1 pyramidal neurons are important for mediating associations with temporal components and have the ability to maintain short-term memory, while CA3 neurons are involved in processes related to the rapid formation of spatial or contextual memory [4, 7]. The hippocampal CA3 and CA1 regions, despite being linked by Schaffer collaterals and forming a continuum, have been shown to respond differently to ischemic episodes [2, 3, 8, 9].
In this article, we focus on changes in the CA3 area as one part of a specific hippocampal pathway. Studies have shown acute and chronic alterations in the CA3 subfield of pyramidal neurons in animals surviving after ischemia for up to 2 years [2, 10], which were identical to changes in AD [4–6, 11]. Elevated extracellular glutamate levels have been demonstrated by microdialysis in this structure immediately after ischemia, indicating a disorder in the neuronal or astrocytic glutamate uptake system [12, 13]. The increase in extracellular glutamate after ischemia led to the activation of glutamate receptors and neurotoxicity with a 74% decrease in basal calcium levels in this space [11]. In addition, persistent dysfunction of the blood-brain barrier caused extravasation of blood elements such as T lymphocytes, platelets, amyloid, tau protein, immunoglobulins, complement, fibrinogen, and pro-inflammatory factors and affected the production of free radicals [4, 14]. Fibrinogen with pro-inflammatory pathway activating properties significantly activated astrocytes and microglia in the CA3 area after ischemia in experimental studies with survival up to 2 years [4, 15]. Experimental studies have shown that neuroinflammatory processes are supported by the activity of astrocytes and microglia even up to 2 years after cerebral ischemia [11, 15]. Damage to the blood-brain barrier results in impaired communication between astrocytes and vessels, which in turn leads to changes in the cytoarchitecture of the blood-brain barrier, changes in regional homeostasis and energy deficiency, which in turn leads to an energy crisis [4, 16, 17]. In case of increased energy demand after ischemia as it is in CA3 area of hippocampus, damage to the blood-brain barrier affects the functionality of energy-intensive neurons [18]. In addition, immunohistochemical studies after experimental cerebral ischemia with survival up to 1 year showed deposits of the C-terminal of the amyloid precursor protein and amyloid-β peptide in the extracellular space around the brain vessels [19].
Recent studies of the CA3 area of the hippocampus showed significant changes in gene expression associated with the death of pyramidal neurons after cerebral ischemia with post-ischemic survival of 2 to 30 days [4, 20, 21]. Studies have shown that on day 2 during recirculation, the expression of the apoptosis gene caspase 3 was under the control limit. But, it was significantly expressed over the next 7–30 days [4, 20, 21]. In the CA3 subfield, autophagy-related genes have also been studied [22], which play an important role in physiological and pathological conditions through increased or decreased activity and can lead to cell death [17, 21, 23–26]. Autophagy is the main lysosomal pathway for the removal of proteins, organelles, and microbes in eukaryotic cells. Autophagy failure is indicated to occur in after ischemia [17, 26–29] and AD [21, 23–25]. The dysfunction manifests itself as a reduced ability of cells such as neurons, astrocytes and microglia to remove abnormal protein aggregates for instance tau protein and amyloid [21, 23–25]. The consequence of this phenomenon is excessive accumulation of modified tau protein and amyloid-β peptide [21, 23–25]. BECN1 gene in the CA3 region remained below the control value for 2–7 days and increased significantly on day 30 after ischemia [20]. In contrast, the BNIP3 gene was reduced throughout the follow-up period after ischemia [20]. In addition, gene expression studies in the CA3 area with survival from 2 to 30 days post-ischemia related to the processing of amyloid precursor protein and tau protein showed significant changes in expression, too [30]. Two and 30 days after ischemia, amyloid precursor protein gene expression was near control values, but 7 days after ischemia, amyloid precursor protein gene expression was maximal. Throughout the observation period, the α-secretase gene in this region was reduced. The β-secretase gene was reduced during 2–7 days following ischemia, and 30 days after ischemia it was above the control values. On the other hand, the expression of the presenilin 1 gene after 2 and 7 days was above the control values and fluctuated around the control values on the 30th day after ischemia. On the 2nd day post-ischemia, the expression of the presenilin 2 gene oscillated around the control values, on the 7th day it fell under the control values, and at the 30th day it significantly exceeded the control values. Expression of tau protein gene was reduced 2 days post-ischemia, and there was a significant maximal increase in its expression after 7–30 days of survival [30].
The data presented showed numerous and abundant amyloid deposits in hippocampal tissue and adjacent to or around blood-brain barrier vessels spreading to adjacent tissue [19]. This indicates disruption of the blood-brain barrier for amyloid after ischemia [4, 19]. There is also evidence that, like amyloid, tau protein crosses the blood-brain barrier from the systemic circulation to the hippocampus after ischemia [31]. Blood-brain barrier permeability occurred 1 year after ischemia and was associated with the accumulation of amyloid, tau protein and progressive death of pyramidal neurons and increased numbers of activated neuroglial cells in hippocampal regions [4, 10, 15, 19]. The development of neuroinflammation was observed in all sectors of the hippocampus, even 2 years after ischemia [15]. Following ischemia, there is progressive and chronic death of neurons in all sectors of the hippocampus [2]. Available data indicate that neurodegenerative processes in the hippocampus are chronic. These processes end in the formation of amyloid plaques and neurofibrillary tangles and ultimately resemble the proteinopathy of AD with full-blown dementia [32–34]. The observed changes after ischemia in hippocampus are remarkably similar to changes in AD hippocampus. Therefore, understanding new neurodegenerative mechanisms causing post-ischemic hippocampal neurodegeneration resembling AD proteinopathy may be a significant step in elucidating the etiology of AD.
Currently, we are observing an increase in interest in research on brain ischemia in the context of the development of a neurodegenerative disease such as AD and the possibilities of its prevention or treatment [2, 25, 35–40]. A large amount of research into the causes, molecular processes and treatments of AD has not yielded any breakthroughs [25]. This is because the previously proposed etiology of the disease based solely on amyloid and tau protein simply did not explain the etiology of the disease [25]. However, a growing body of literature indicates that ischemia may play a key role in driving amyloid and tau protein in the etiology of AD [2, 25, 35–40]. Therefore, the aim of our research is to understand the specific mechanisms driving amyloid and tau protein after ischemia at different times and structures, which may provide interesting clues related to the development of AD dementia.
This work is part of an ongoing series of experimental studies focusing on the quantification of AD-related genes using the RT-PCR protocol, such as, β-secretase, presenilin 1 and 2, α-secretase, amyloid precursor protein, and tau protein in the CA3 area of the hippocampus in rats that survived 12, 18, and 24 months after an ischemic episode.
METHODS AND MATERIALS
Animals
Female Wistar rats (n = 29) 2 months old, body weighing 120–150 g were subjected to 10-minute cerebral ischemia by cardiac arrest with survival after the ischemic episode at 12, 18, and 24 months [41]. The first group of rats tested (n = 10) consisted of rats weighing 150–180 g that survived 12 months after ischemia. The second group of rats tested (n = 9) consisted of rats weighing 180–200 g, which survived 18 months after ischemia. The third group of tested rats (n = 10) consisted of rats weighing more than 200 g that survived 24 months post-ischemia. Female Wistar rats (n = 29), 2 months old, weighing 120–150 g, after sham surgery, without inducing complete brain ischemia, served control groups. The first control group consisted of rats (n = 10) weighing 150–180 g, which survived 12 months after sham-operation. The second control group consisted of rats (n = 9) weighing 180–200 g, which survived 18 months after sham-operation. The third control group consisted of rats (n = 10) weighing over 200 g that survived 24 months after sham-operation.
Experimental procedures
2.0% isoflurane carried via oxygen was used to anesthetize the animals [30]. Anesthesia was discontinued shortly before the initiation of the cardiac arrest procedure. A hook made of an L-shaped steel needle was introduced into the chest through the right parasternal line and the third intercostal space. Next the hook was then gently moved towards the spine until slight resistance was felt. In the next stage, the hook was gently tilted 10–20° towards the tail. This meant that the hook in this position was under the bundle of heart vessels. The hook was then pulled to the sternum, which led to compression and closure of the heart vessel bundle through the sternum. In order to prevent chest movements and ensure complete closure of the vessels, external pressure was applied to the sternum with the index and middle fingers, which resulted in complete hemostasis and subsequent cardiac arrest. After 3.5 min, the hook was removed from the chest and the rats remained in this state for next 6.5 min until resuscitation starts [17]. Resuscitation started of artificial ventilation and external heart massage until cardiac function returned and breathing occurred. During this time, air was administered using a respirator through a polyethylene tube inserted into the trachea. The heart massage frequency range was from 150 to 240/min [41].
Before and after ischemia, rats were housed in the animal house under a 12-h light/dark cycle. All studies were done within the day, and rats were handled in accordance with the NIH recommendations for the Care and Use of experimental animals and the Directive of the Council of the European Community. In addition, the Local Ethics Committee for Animal Experiments (31.12.2017, Nr 339/2017) approved all planned experimental procedures. Every effort has been made to minimize animal suffering and reduce the number of rats used.
Immediately before collecting hippocampal samples from the CA3 area, brains were perfused through the left ventricle with cold 0.9% NaCl to flush blood from blood vessels. The brains were then removed from the skulls and transferred on ice-cooled Petri dishes. Samples with a volume of approximately 1 mm3 have been taken from the CA3 area of the hippocampus after ischemia and from controls with a narrow scalpel from both sides and immediately placed in RNAlater-ICE solution (Life Technologies, USA) [31].
The method described by Chomczyński and Sacchi [42] was used to isolate cellular RNA. Assessment of RNA quantity and quality was performed using a Nano Drop 2000 spectrophotometer (Thermo Scientific, USA) [20, 30]. The isolated RNA was stored in 80% ethanol at −20°C for further analyses [21, 31]. In further studies, 1μg of RNA was reverse transcribed into cDNA. Veriti Dx (Applied Biosystems, USA) was used for cDNA synthesis using the manufacturer’s SDS software [20, 30]. The cDNA obtained was amplified by real-time gene expression analysis (qPCR) using the manufacturer’s SDS software [20, 30]. The tested gene was assessed in relation to the control gene (Rpl13a), and the relative amount (RQ) of the tested gene was presented using ΔCT, and the final value was presented as RQ = 2−rmDeltarmDeltaCT [30, 43]. The final result is presented after logarithmic conversion of the RQ values (LogRQ) [30]. LogRQ = 0 means that the expression of the tested gene after ischemia and in the control did not change. LogRQ < 0 meant decreased gene expression after ischemia, and LogRQ > 0 indicated increased gene expression after ischemia compared to the control.
Statistica v. 12 was used to statistically evaluate the data using the Kruskal-Wallis test with the “z” test. Data in graphs are means±SD. p≤0.05 means significant statistical changes in genes expression.
RESULTS
Expression of the amyloid precursor protein gene post-ischemia
In the studied CA3 area, the expression of the amyloid precursor protein gene (APP) after ischemia with a survival of 12, 18, and 24 months was higher than the control values. On the 12th month post-ischemia, the minimum was 0.688-fold change and maximum 2.354-fold change with a median of 1.607-fold change. On the 1.5 year post-ischemia, the minimum was 0.192-fold change and maximum 2.185-fold change with a median of 1.877-fold change. On the 2 year post-ischemia, the minimum was 0.268-fold change and maximum 0.888-fold change with a median of 0.476-fold change. Changes of the expression of the APP gene illustrates Fig. 1. The reduction in APP expression was statistically significant between 1 and 2 (z = 2.469, p = 0.04) years after ischemia (Fig. 1). However, between 12 and 18 months and 18 and 24 months after ischemia, no statistically significant changes in gene expression after ischemia were observed.
Fig. 1
The mean gene levels of APP gene expression in the CA3 region 12 (n = 10), 18 (n = 9), and 24 (n = 10) months following brain ischemia. Marked SD, standard deviation. Kruskal-Wallis test. *p≤0.05.
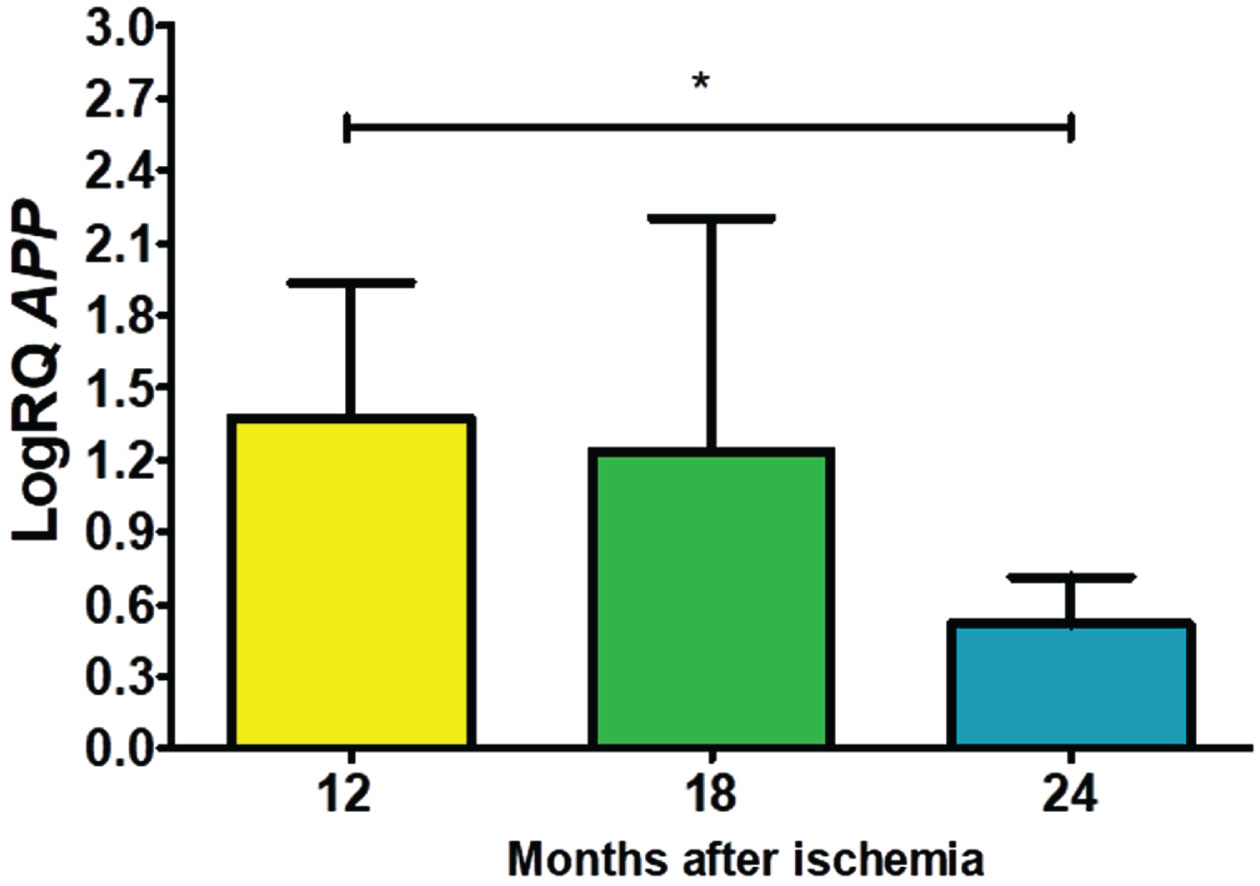
Expression of the α-secretase gene post-ischemia
In the CA3 subfield, the α-secretase gene (ADAM10) expression after ischemia at 12, 18 and 24 months of recirculation remained over control values throughout the entire observation period. On the 12th month after ischemia, a median was 2.317-fold change, the minimum was 0.423-fold change with maximum 2.493-fold change. On the 18th month post-ischemia, the maximum was 0.389-fold change and minimum was 0.089-fold change and with a median of 0.137-fold change. At the 2 years after ischemia, the minimum was 0.112-fold change and maximum 0.577-fold change with a median of 0.175-fold change. Figure 2 demonstrates results of the α-secretase gene expression. There were statistically significant differences in the gene expression reduction between 12 and 18 (z = 4.113, p = 0.0001) and 12 and 24 (z = 3.204, p = 0.01) months post-ischemia (Fig. 2). However, between 18 and 24 months after ischemia, there were no statistically significant changes in gene expression after ischemia.
Fig. 2
The mean gene levels of ADAM10 expression in the hippocampus CA3 region 12 (n = 10), 18 (n = 9), and 24 (n = 10) months after ischemia. Marked SD, standard deviation. Kruskal-Wallis test. **p≤0.01, ***p≤0.0001.
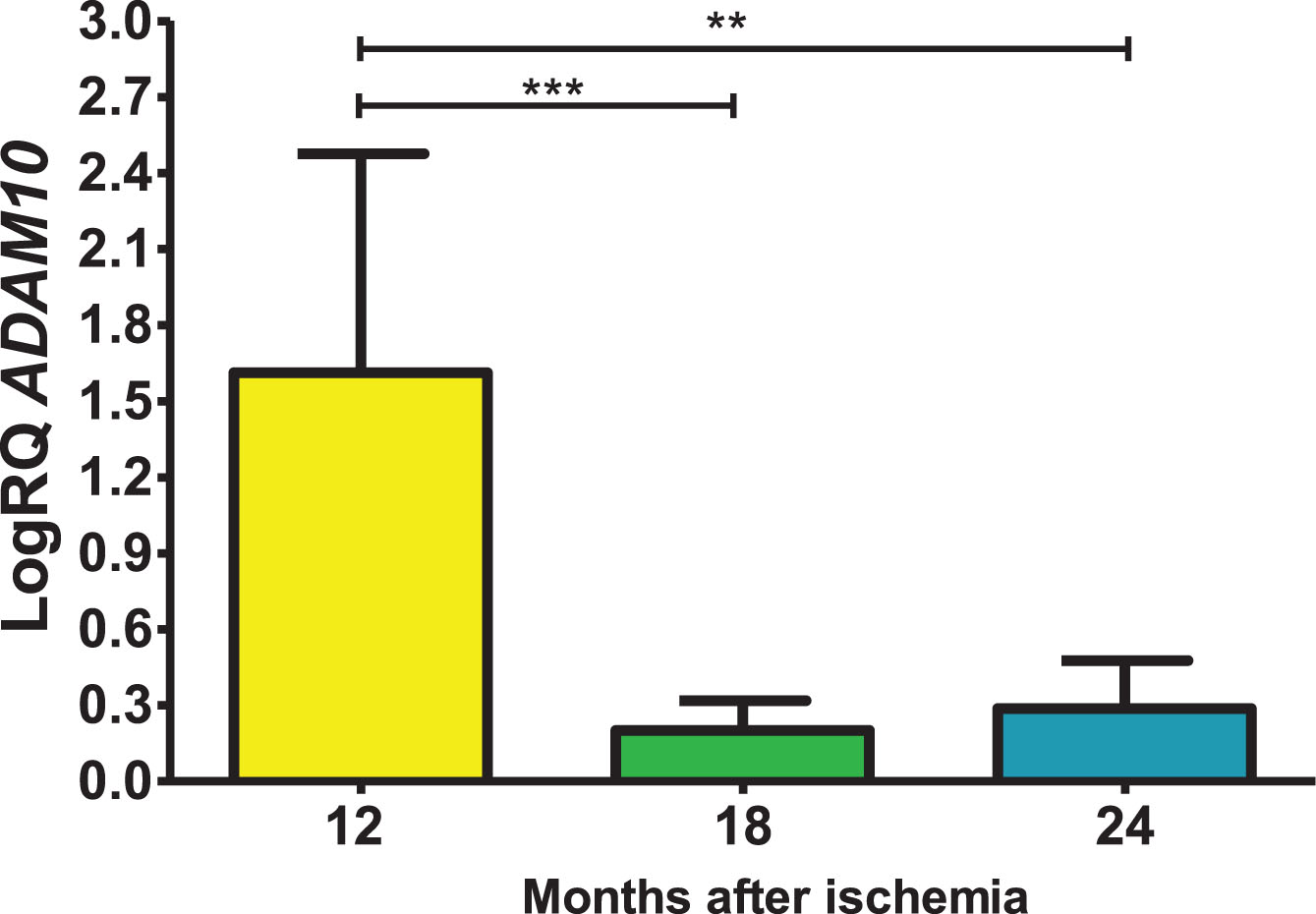
Expression of the β-secretase gene post-ischemia
In the structure studied, the level of the β-secretase (BACE1) gene expression after ischemia was over control values after 12 and 24 months of survival, while after 18 months the values were below control. On the 12th month after ischemia, maximum was 2.120-fold change and the minimum was 0.314-fold change and with a median of 1.432-fold change. On the 1.5 year post-ischemia, the minimum was −0.487-fold change and maximum −0.121-fold change with a median of −0.195-fold change. At the 2 years after ischemia, the minimum was 0.105-fold change, maximum 0.576-fold change and a median of 0.256-fold change. Figure 3 presents changes in the level of the BACE1 gene expression. There were statistically significant differences in the reduction of gene levels between 12 and 18 (z = 4.908, p = 0.00001) and 12 and 24 (z = 2.469, p = 0.04) months post-ischemia. Moreover, statistically significant differences were found in the increase in gene expression levels between 18 and 24 (z = 2.505, p = 0.04) months after ischemia.
Fig. 3
The mean gene levels of BACE1 expression in the hippocampus CA3 region 12 (n = 10), 18 (n = 9), and 24 (n = 10) months following brain ischemia. Marked SD, standard deviation. Kruskal-Wallis test. *p≤0.05, ***p≤0.00001.
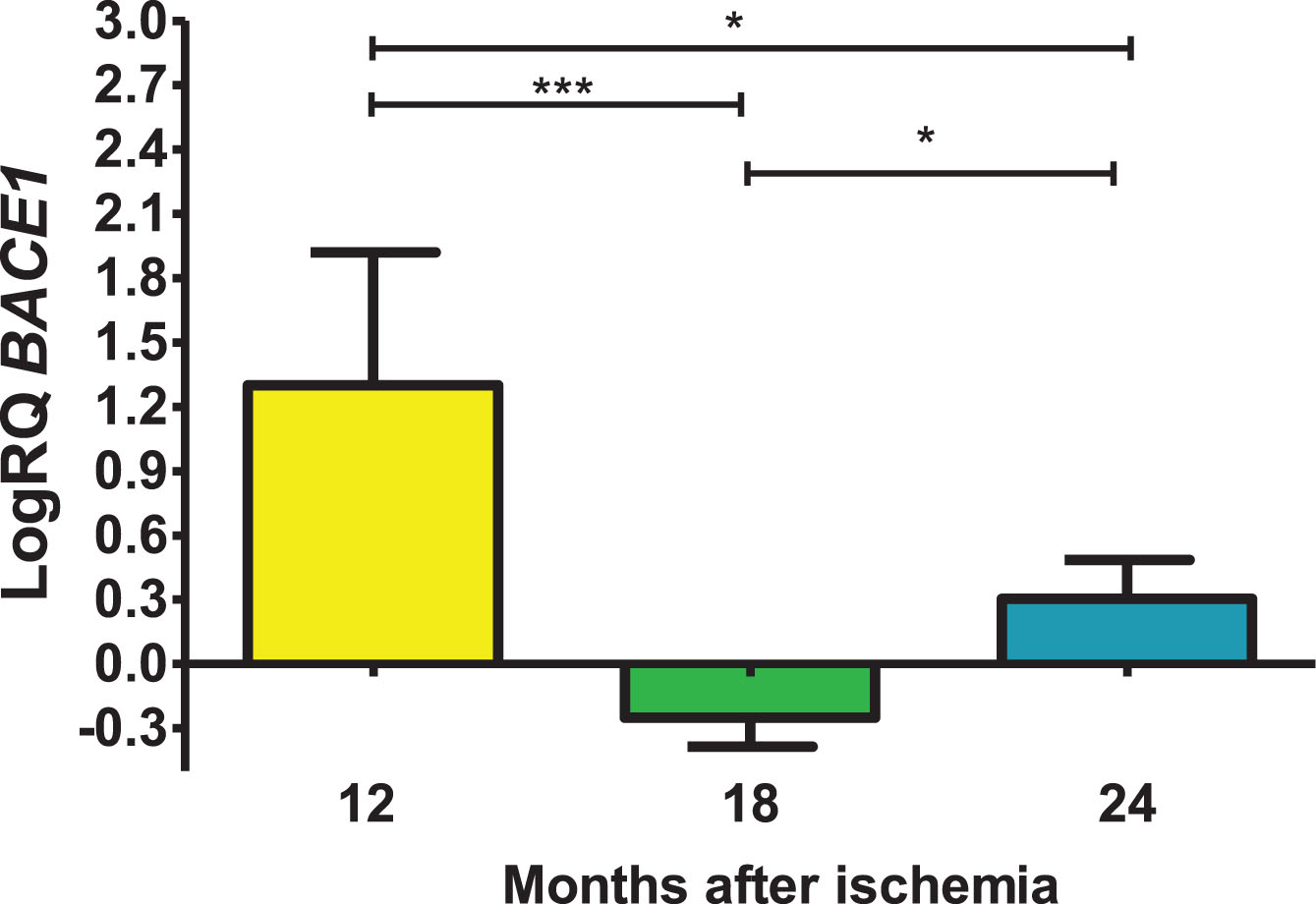
Expression of the presenilin 1 gene post-ischemia
In the presented region, the expression of the presenilin 1 gene (PSEN1) after ischemia with a survival of 1, 1.5, and 2 years was higher compared to the control values. On the 1 year following ischemia, the minimum was 0.249-fold change, maximum 2.264-fold change and a median of 2.038-fold change. On the 1.5 year post-ischemia, the minimum was 0.063-fold change, maximum 0.242-fold change and a median of 0.142-fold change. On the 2 years following ischemia, the minimum was 0.088-fold change, maximum 0.587-fold change and a median of 0.185-fold change. Figure 4 presents data changes in the level of the presenilin 1 gene expression. There were statistically significant differences in the levels of gene expression reduction between 12 and 18 (z = 4.042, p = 0.0001) and between 12 and 24 (z = 2.889, p = 0.01) months after ischemia. However, between 18 and 24 months after ischemia, there were no statistically significant changes in gene expression post-ischemia.
Fig. 4
The mean gene levels of PSEN1 expression in the hippocampus CA3 region 12 (n = 10), 18 (n = 9), and 24 (n = 10) months following brain ischemia. Marked SD, standard deviation. Kruskal-Wallis test. *p≤0.01, ***p≤0.0001.
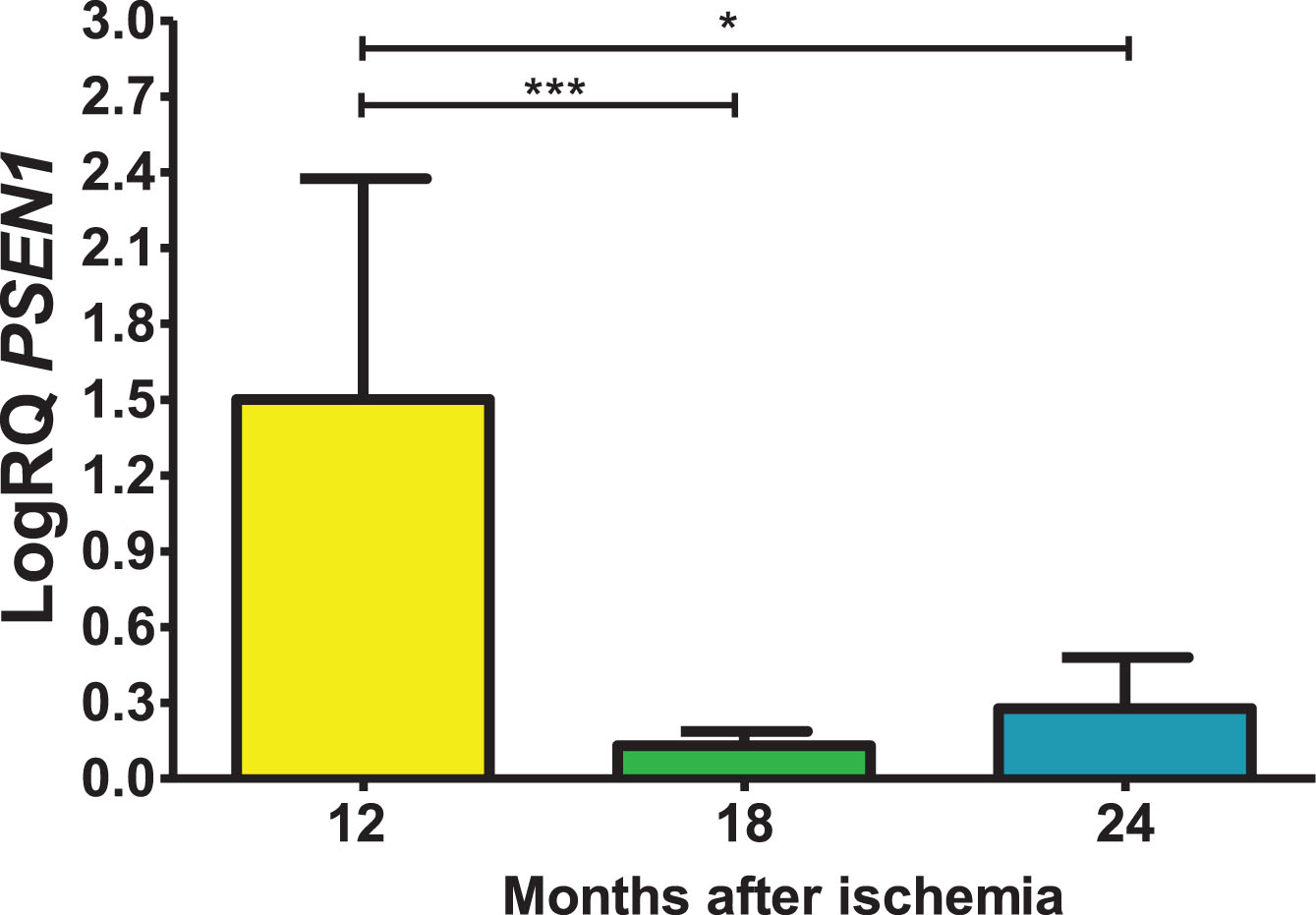
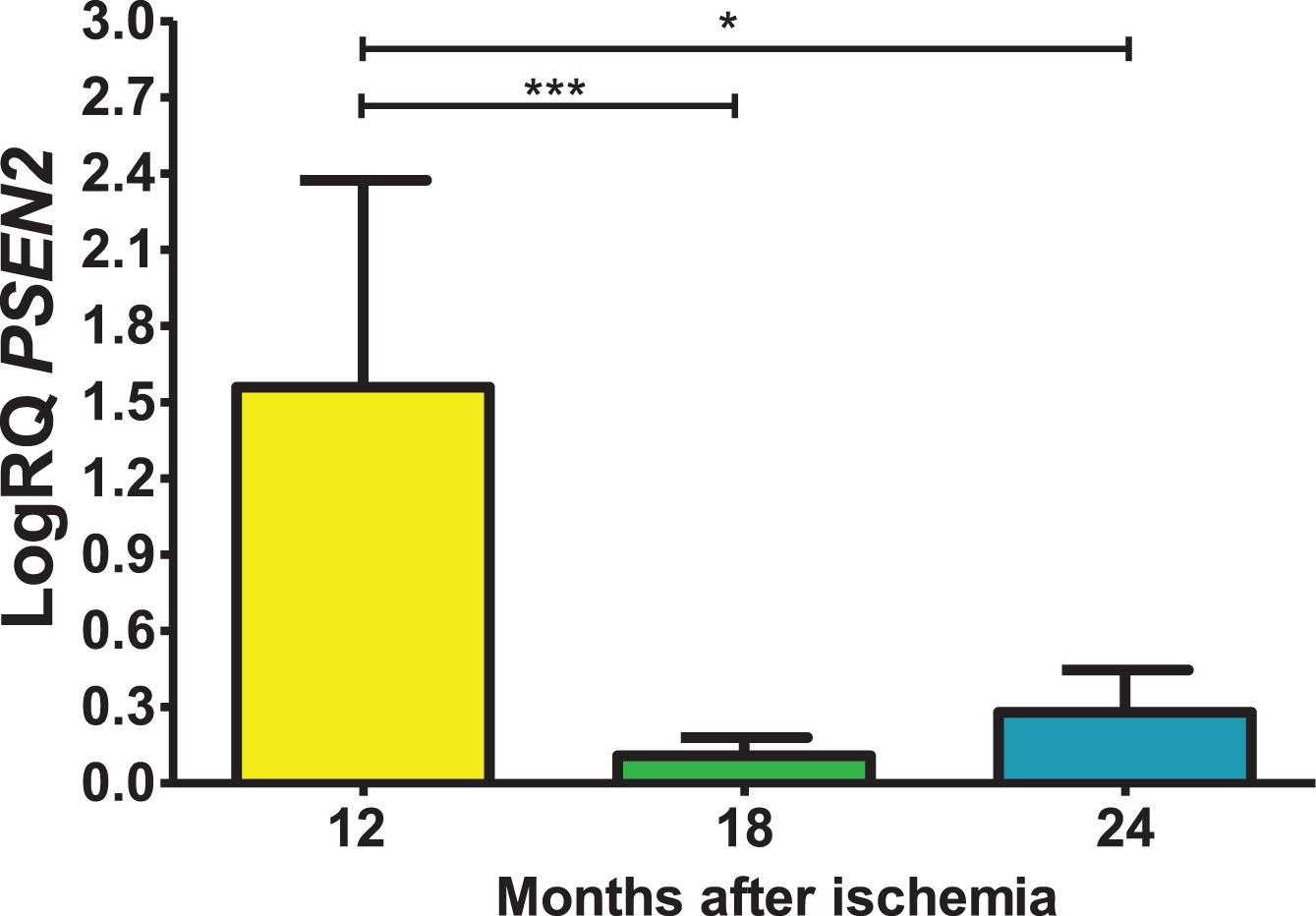
Expression of the presenilin 2 gene post-ischemia
In the CA3 area, presenilin 2 (PSEN2) gene expression post-ischemia with recirculation after 12, 18 and 24 months was above control values. On the 12 months post-ischemia, the minimum was 0.449-fold change, maximum 2.390-fold change and a median of 1.996-fold change. On the 18 month post-ischemia, the minimum was 0.015-fold change, maximum 0.200-fold change and a median of 0.125-fold change. On the 24 months following ischemia, the minimum was 0.044-fold change, maximum 0.619-fold change and a median of 0.273-fold change. Figure 5 presents levels of the presenilin 2 gene expression. There were statistically significant differences in the levels of gene expression reduction between 12 and 18 (z = 4.482, p = 0.0001) and between 12 and 24 (z = 2.863, p = 0.01) months post-ischemia. However, between 18 and 24 months after ischemia, there were no statistically significant changes in gene expression after ischemia.
Fig. 5
The mean gene levels of PSEN2 expression in the hippocampus CA3 subfield 12 (n = 10), 18 (n = 9), and 24 (n = 10) months following brain ischemia. Marked SD, standard deviation. Kruskal-Wallis test. *p≤0.01, ***p≤0.0001.
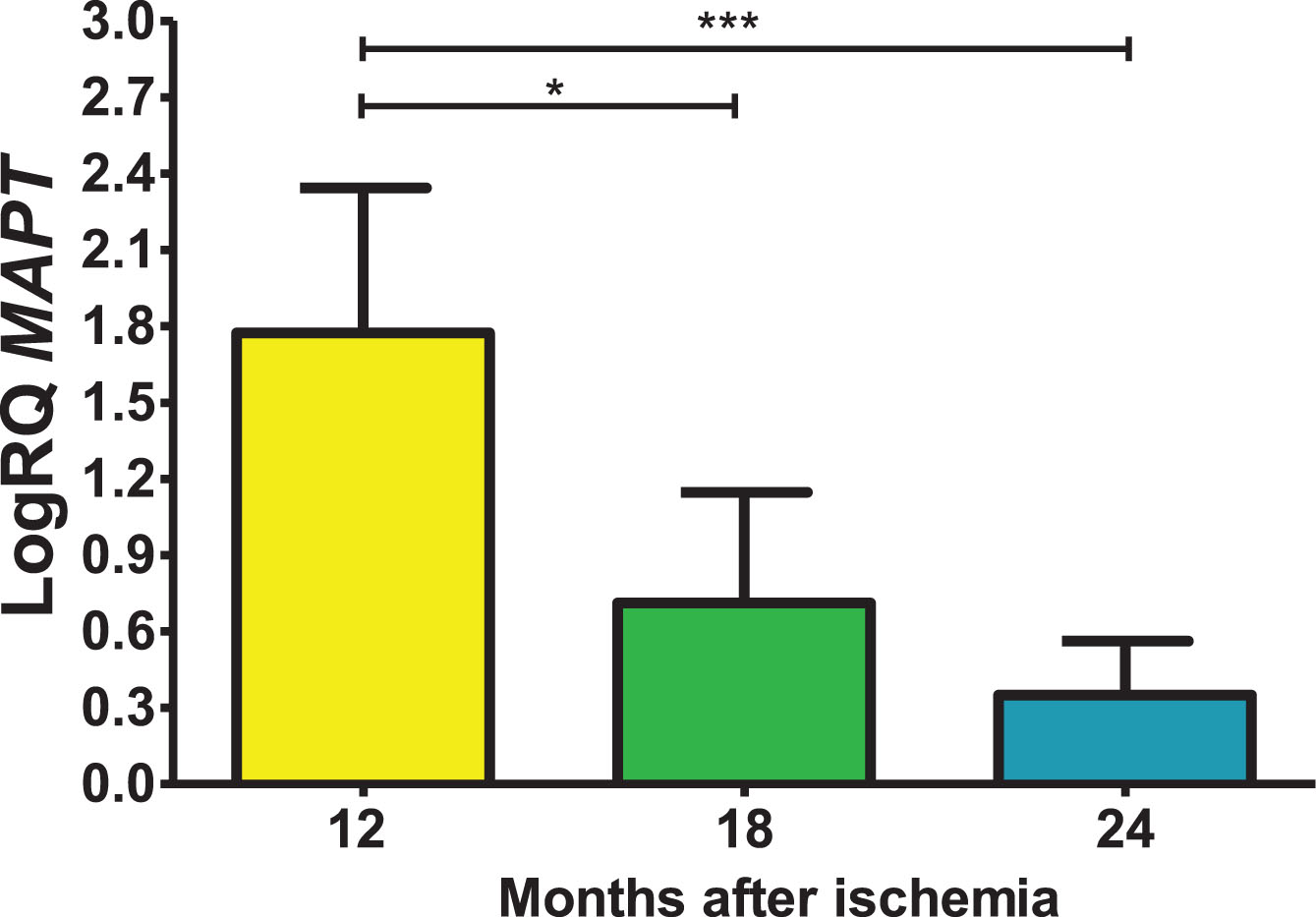
Expression of the tau protein gene post-ischemia
In the studied region, tau protein (MAPT) gene expression post-ischemia with recirculation after 12, 18 and 24 months was increased above control values. On the 1 year post-ischemia, the minimum was 1.100-fold change, maximum 2.338-fold change and a median of 2.017-fold change. On the 1.5 year following ischemic episode, the minimum was 0.143-fold change, maximum 1.270-fold change and a median of 0.934-fold change. On the 2 year following ischemic injury, the minimum was 0.096-fold change, maximum 0.750-fold change and a median of 0.295-fold change. Figure 6 shows changes of the tau protein gene expression. There were statistically significant differences in the levels of gene expression reduction between 12 and 18 months (z = 2.823, p = 0.01) and between 12 and 24 months (z = 4.320, p = 0.0001) post-ischemia. However, there was no statistically significant difference in the level of gene expression reduction between 18 and 24 months after ischemia.
Fig. 6
The mean gene levels of MAPT expression in the hippocampus CA3 region 12 (n = 10), 18 (n = 9), and 24 (n = 10) months following brain ischemia. Marked SD, standard deviation. Kruskal-Wallis test. *p≤0.01, ***p≤0.0001.
DISCUSSION
In this work, we continue studies to investigate temporal changes in the expression of amyloid precursor protein processing and tau protein genes in an ischemic model of AD with survival up to 2 years. Our data provide for the first time, changes of genes expression in: ADAM10 and BACE1, PSEN1 and PSEN2, and APP and MAPT connected with AD in the CA3 subfield following 10-min ischemic brain injury in animals with 1, 1.5, and 2 years of survival. First of all, we observed post-ischemic, non-amyloidogenic processing of amyloid precursor protein in the CA3 area. Furthermore, our data demonstrate that brain ischemia with recirculation triggers neuronal cells alterations and loss in the CA3 area in a tau protein-dependent manner, thus identifying a novel way of regulating the survival or loss of ischemic neurons at very late stages after ischemia.
In the present study, ADAM10 gene expression was over control values throughout the observation period. However, the maximum level of the BACE1 gene expression was found after 12 months, but after 18 months it was below control values, and its another increase was observed 2 years after ischemia. PSEN1 gene expression was significantly increased over control values through 2 years of recirculation. PSEN2 gene expression showed the same trend as PSEN1 gene expression. Expression of the APP gene was consistently over control values following ischemia, with massive, significant overexpression at 12 and 18 months. These data suggest non-amyloidogenic processing of amyloid precursor protein in the CA3 area of the hippocampus after long-term post-ischemia, and this observation contrasts markedly with amyloid production in this region during 30 days after ischemia [30]. Overall, the response of hippocampal CA3 genes expression, such as amyloid precursor protein, α-secretase, β-secretase, and presenilin 1 and 2, found in this study was opposite to the response of these genes expression in animals surviving after ischemia for up to 30 days [30]. The data show that after ischemia in animals with a long-term survival up to two years, at the same time three secretases responsible for the metabolism of amyloid precursor protein, i.e., alpha-, beta-, and gamma-secretase, showed increased expression. These data suggest that after long-term post-ischemic survival there is no production and deposition of amyloid-β peptide in hippocampal tissue; if amyloid does appear, it probably comes from the systemic circulation [14]. As evidenced by a chronically open blood-brain barrier and the presence of amyloid in the serum after cerebral ischemia in humans and animals [19].
Additionally, MAPT gene expression was above control values throughout the post-ischemic follow-up period, with a maximum value after 1 year of recirculation. Another study found an increase in phosphorylated tau protein in the CA3 area of the hippocampus after bilateral common carotid artery ligation [44]. Excessively phosphorylated tau protein has a reduced ability to bind to microtubules, leading to microtubule depolymerization [44]. Previous studies have shown that tau protein and glycogen synthase kinase-3β (GSK-3β) are also promoters of secondary brain damage after ischemia [45, 46] as in the case of AD [47]. Moreover, the increase in GSK-3β level after focal brain ischemia in rats resulted in hyperphosphorylation of tau protein with hippocampal neuronal death, increased blood-brain barrier permeability, brain edema, increased neurological deficits, and increased infarct size [48]. Consistent with the above reports, a significant increase in the level of tau protein phosphorylation (S404) and interaction with GSK-3β was observed after transient local cerebral ischemia [49]. This suggests that tau protein S404 phosphorylation is an indicator of neuronal damage after ischemia [49]. The increase in the level of total tau protein, which was assessed by brain microdialysis after ischemia, has to be also mentioned [50], and this increase correlated with immunostaining [51] and with our study on tau protein gene expression. Numerous studies have shown that after ischemia, tau protein is hyperphosphorylated in neurons and is closely related to the development of their death through the mechanism of apoptosis [52, 53]. Finally, hyperphosphorylation of tau protein causes the formation of paired helical filaments after brain ischemia [54], neurofibrillary tangles-like [55], and neurofibrillary tangles [33, 34] characteristic of AD.
The current data may help, at least in part, to elucidate the molecular mechanism(s) for the slower and later appearance of neuronal cell alterations and loss in the ischemic hippocampal region CA3 than in CA1 [2]. This phenomenon correlated with the appearance of acute and chronic neuronal cell alterations in the CA3 subfield 1–2 years after ischemia [2]. Data indicate that 2 years after ischemia in our ischemic model of AD, neurodegenerative damage is slower and more complicated in CA3 compared with CA1 [2]. Progressive damage to the CA3 region is associated with the development of irreversible memory impairment [20, 21]. Memory deficiency is an early symptom of AD –our data indicate that neuronal death in the early stages in the CA3 region after ischemia contributes to memory impairment in an amyloid- and tau protein-dependent manner [30], and in the late stages post-ischemia only through modification of tau protein [47].
The degree of neuronal loss, especially in the hippocampus, is thought to influence the development and clinical manifestations of AD [56]. In AD, the number of pyramidal neurons is reduced by as much as 45%, which correlates with the density of neurofibrillary tangles and senile amyloid plaques [56]. In recent years, the tau protein and amyloid hypotheses have become the dominant hypotheses explaining the pathogenesis of AD [56]. However, a growing body of literature supports the view that ischemia plays a major role in driving tau protein and amyloid in the neuropathophysiology and neuropathology of AD [35, 38–40, 52].
Thus, our model represents a progressive, time- and area-specific development of neuropathology in CA3 with genes expression of amyloid precursor protein processing and tau protein and dementia symptoms such as those found in patients with AD [2, 15, 19, 30, 32, 57]. In spite of this, additional study is necessary to settle whether the injury and loss of neuronal cells in the CA3 area are causal phenomena or self-regulating consequences post-ischemia happening in parallel and resulting in the development of dementia due to ischemia. Lastly, our model appears to be suitable for determining the role of AD-related genes expression. By extensively examining the common genetic processes involved in these two diseases, these data may help unravel phenomena associated with the development of AD and neurodegeneration after brain ischemia, and may lead future studies on AD or brain ischemia in new directions. Thus, characterization of an ischemic animal model may help us better understand the mechanisms of AD and its neuropathogenesis. The possibility of survival of rats for up to 2 years in the presented model ensures continuous research and long-term observation of processes potentially influencing the development of AD at the genomic, proteomic, neuropathological, structural, functional and behavioral levels.
Our study has some strengths and limitations. First, the strengths of our study include the ability of rats to survive up to two years after an episode of complete cerebral ischemia, which allowed for the first time to assess selected genes expression associated with AD. Second, this analysis allowed us to discover that tau protein modification is important in the development of neuronal death in CA3 after long-term survival. Third, our study used three long observation periods, i.e., 12, 18, and 24 months, which increased the accuracy of assessing changes in the expression of selected genes. Four, for the first time we assessed the association of the studied genes expression with the progression of neurodegenerative changes after ischemia, in association with long survival times. The main limitation of our study, however, was the small size of the experimental groups, which limited the availability of studied material from such a small structure as the CA3 area of the hippocampus. The last weak point was the use of young animals in experiments, when it is known that brain ischemia is rather age-related [58]. On the other hand, a factor limiting the use of old animals was the long survival time after ischemia. Moreover, our results require confirmation in more extensive studies, especially at the protein level, in order to link them to gene expression changes. Western blot or immunohistochemistry would help interpret better the data. This would provide a comprehensive assessment of the dysregulation of genes expression and their proteins associated with AD and their impact in determining the rate of progression from acute to chronic changes after brain ischemia.
AUTHOR CONTRIBUTIONS
Ryszard Pluta (Conceptualization; Project administration; Writing – original draft; Writing – review & editing); Stanisław J Czuczwar (Writing – original draft; Writing – review & editing); Janusz Kocki (Data curation; Investigation; Methodology); Barbara Miziak (Data curation; Formal analysis; Investigation; Visualization); Jacek Bogucki (Software; Visualization); Anna Bogucka-Kocka (Formal analysis; Investigation; Methodology).
ACKNOWLEDGMENTS
The authors acknowledge the support of the Medical University of Lublin, Poland (DS 721/23-SJC). Moreover, the authors would like to thank Mr. Sławomir Januszewski for technical assistance.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data of this study are available on request from the corresponding author.
REFERENCES
[1] | Liu HX , Zhang JJ , Zheng P , Zhang Y ((2005) ) Altered expression of MAP-2, GAP-43, and synaptophysin in the hippocampus of rats with chronic cerebral hypoperfusion correlates with cognitive impairment. Mol Brain Res 139: , 169–177. |
[2] | Pluta R , Ułamek M , Jabłoński M ((2009) ) Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec (Hoboken) 292: , 1863–1881. |
[3] | Bartsch T , Döhring J , Reuter S , Finke C , Rohr A , Brauer H , Deuschl G , Jansen O ((2015) ) Selective neuronal vulnerability of human hippocampal CA1 neurons: Lesion evolution, temporal course, and pattern of hippocampal damage in diffusion weighted MR imaging. J Cereb Blood Flow Metab 35: , 1836–1845. |
[4] | Lana D , Ugolini F , Giovannini MG ((2020) ) An overview on the differential interplay among neurons-astrocytes-microglia in CA1 and CA3 hippocampus in hypoxia/ischemia. Front Cell Neurosci 14: , 585833. |
[5] | Babcock KR , Page JS , Fallon JR , Webb AE ((2021) ) Adult hippocampal neurogenesis in aging and Alzheimer’s disease. Stem Cell Rep 16: , 681–693. |
[6] | Tang Y , Yan Y , Mao J , Ni J , Qing H ((2023) ) The hippocampus associated GABAergic neural network impairment in early-stage of Alzheimer’s disease. Ageing Res Rev 86: , 101865. |
[7] | van Staalduinen EK , Zeineh MM ((2022) ) Medial temporal lobe anatomy. Neuroimaging Clin N Am 32: , 475–489. |
[8] | Kirino T ((2000) ) Delayed neuronal death. Neuropathology 20: , 95–97. |
[9] | Lee TK , Kim DW , Sim H , Lee JC , Kim HI , Shin MC , Cho JH , Park JH , Lee CH , Won MH , Ahn JH ((2022) ) Hyperthermia accelerates neuronal loss differently between the hippocampal CA1 and CA2/3 through different HIF 1α expression after transient ischemia in gerbils. Int J Mol Med 49: , 55. |
[10] | Sekeljic V , Bataveljic D , Stamenkovic S , Ułamek M , Jabłonski M , Radenovic L , Pluta R , Andjus PR ((2012) ) Cellular markers of neuroinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct Funct 217: , 411–420. |
[11] | Padurariu M , Ciobica A , Mavroudis I , Fotiou D , Baloyannis S ((2012) ) Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer’s disease patients. Psychiatr Danub 24: , 152–158. |
[12] | Pluta R , Salínska E , Puka M , Stafiej A , Lazarewicz JW ((1988) ) Early changes in extracellular amino acids andcalcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 16: , 193–210. |
[13] | Rossi DJ , Oshima T , Attwell D ((2000) ) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403: , 316–321. |
[14] | Pluta R , Barcikowska M , Januszewski S , Misicka A , Lipkowski AW ((1996) ) Evidence of blood-brain barrier permeability/leakage for circulating human Alzheimer’s beta-amyloid-(1-42)-peptide. Neuroreport 7: , 1261–1265. |
[15] | Radenovic L , Nenadic M , Ułamek-Kozioł M , Januszewski S , Czuczwar Andjus SJPR , Pluta R ((2020) ) Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging (Albany NY) 12: , 12251–12267. |
[16] | Sweeney MD , Sagare AP , Zlokovic BV ((2018) ) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14: , 133–150. |
[17] | Beccari S , Sierra-Torre V , Valero J , Pereira-Iglesias M , García-Zaballa M , Soria FN , De Las Heras-Garcia L , Carretero-Guillen A , Capetillo-Zarate E , Domercq M , Huguet PR , Ramonet D , Osman A , Han W , Dominguez C , Faust TE , Touzani O , Pampliega O , Boya P , Schafer D , Mariño G , Canet-Soulas E , Blomgren K , Plaza-Zabala A , Sierra A ((2023) ) Microglial phagocytosis dysfunction in stroke is driven by energy depletion and induction of autophagy. Autophagy 19: , 1952–1981. |
[18] | Muddapu VR , Dharshini SAP , Chakravarthy VS , Gromiha MM ((2020) ) Neurodegenerative diseases –Is metabolic deficiency the root cause? Front Neurosci 14: , 213. |
[19] | Pluta R ((2003) ) Blood-brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia-reperfusion brain injury with 1-year survival. Acta Neurochir Suppl 86: , 117–122. |
[20] | Ułamek-Kozioł M , Czuczwar SJ , Kocki J , Januszewski S , Bogucki J , Bogucka-Kocka A , Pluta R ((2019) ) Dysregulation of autophagy, mitophagy, and apoptosis genes in the CA3 region of the hippocampus in the ischemic model of Alzheimer’s disease in the rat. J Alzheimers Dis 72: , 1279–1286. |
[21] | Lee JH , Nixon RA ((2022) ) Autolysosomal acidification failure as a primary driver of Alzheimer disease pathogenesis. Autophagy 18: , 2763–2764. |
[22] | Yamamoto H , Zhang S , Mizushima N ((2023) ) Autophagy genes in biology and disease. Nat Rev Genet 24: , 382–400. |
[23] | Litwiniuk A , Juszczak GR , Stankiewicz AM , Urbańska K ((2023) ) The role of glial autophagy in Alzheimer’s disease. Mol Psychiatry. doi:10.1038/s41380-023-02242-5. |
[24] | Mary A , Eysert F , Checler F , Chami M ((2023) ) Mitophagy in Alzheimer’s disease: Molecular defects and therapeutic approaches. Mol Psychiatry 28: , 202–216. |
[25] | Yamashima T , Seike T , Mochly-Rosen D , Chen CH , Kikuchi M , Mizukoshi E ((2023) ) Implication of the cooking oil-peroxidation product “hydroxynonenal” for Alzheimer’s disease. Front Aging Neurosci 15: , 1211141. |
[26] | Yu X , Luo Y , Yang L , Duan X ((2023) ) Plasma metabonomic study on the effect of Para-hydroxybenzaldehyde intervention in a rat model of transient focal cerebral ischemia. Mol Med Rep 28: , 224. |
[27] | Ułamek-Kozioł M , Kocki J , Bogucka-Kocka A , Petniak A , Gil-Kulik P , Januszewski S , Bogucki J , Jabłoński M , Furmaga-Jabłońska W , Brzozowska J , Czuczwar SJ , Pluta R ((2016) ) Dysregulation of autophagy, mitophagy, and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J Alzheimers Dis 54: , 113–121. |
[28] | Ułamek-Kozioł M , Kocki J , Bogucka-Kocka A , Januszewski S , Bogucki J , Czuczwar SJ , Pluta R ((2017) ) Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol Rep 69: , 1289–1294. |
[29] | Mahemuti Y , Kadeer K , Su R , Abula A , Aili Y , Maimaiti A , Abulaiti S , Maimaitituerxun M , Miao T , Jiang S , Axier A , Aisha M , Wang Y , Cheng X ((2023) ) TSPO exacerbates acute cerebral ischemia/reperfusion injury by inducing autophagy dysfunction. Exp Neurol 369: , 114542. |
[30] | Pluta R , Ułamek-Kozioł M , Kocki J , Bogucki J , Januszewski S , Bogucka-Kocka A , Czuczwar SJ ((2020) ) Expression of the tau protein and amyloid protein precursor processing genes in the CA3 Area of the hippocampus in the ischemic model of Alzheimer’s disease in the Rat. Mol Neurobiol 57: , 1281–1290. |
[31] | Banks WA , Kovac A , Majerova P , Bullock KM , Shi M , Zhang J ((2017) ) Tau proteins cross the blood-brain barrier. J Alzheimers Dis 55: , 411–419. |
[32] | Kiryk A , Pluta R , Figiel I , Mikosz M , Ulamek M , Niewiadomska G , Jablonski M , Kaczmarek L ((2011) ) Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res 219: , 1–7. |
[33] | Kato T , Hirano A , Katagiri T , Sasaki H , Yamada S ((1988) ) Neurofibrillary tangle formation in the nucleus basalis of Meynert ipsilateral to a massive cerebral infarct. Ann Neurol 23: , 620–623. |
[34] | Hatsuta H , Takao M , Nogami A , Uchino A , Sumikura H , Takata T , Morimoto S , Kanemaru K , Adachi T , Arai Hasegawa TM , Murayama S ((2019) ) Tau and TDP-43 accumulation of the basal nucleus of Meynert in individuals with cerebral lobar infarcts or hemorrhage. Acta Neuropathol Commun 7: , 49. |
[35] | Salminen A , Kauppinen A , Kaarniranta K ((2017) ) Hypoxia/ischemia activate processing of amyloid precursor protein: Impact of vascular dysfunction in the pathogenesis of Alzheimer’s disease. J Neurochem 140: , 536–549. |
[36] | Eskandari S , Sajadimajd S , Alaei L , Soheilikhah Z , Derakhshankhah H , Bahrami G ((2021) ) Targeting common signaling pathways for the treatment of stroke and Alzheimer’s: A comprehensive review. Neurotox Res 39: , 1589–1612. |
[37] | Kriska J , Hermanova Z , Knotek T , Tureckova J , Anderova M ((2021) ) On the common journey of neural cells through ischemic brain injury and Alzheimer’s disease. Int J Mol Sci 22: , 9689. |
[38] | Lecordier S , Pons V , Rivest S , ElAli A ((2022) ) Multifocal cerebral microinfarcts modulate early Alzheimer’s disease pathology in a sex-dependent manner. Front Immunol 12: , 813536. |
[39] | Elman-Shina K , Efrati S ((2022) ) Ischemia as a common trigger for Alzheimer’s disease. Front Aging Neurosci 14: , 1012779. |
[40] | Das TK , Ganesh BP , Fatima-Shad K ((2023) ) Common signaling pathways involved in Alzheimer’s disease and stroke: Two faces of the same coin. J Alzheimers Dis Rep 7: , 381–398. |
[41] | Pluta R , Lossinsky AS , Mossakowski MJ , Faso L , Wiśniewski HM ((1991) ) Reassessment of new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol 83: , 1–11. |
[42] | Chomczynski P , Sacchi N ((1987) ) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: , 156–159. |
[43] | Livak KJ , Schmittgen TD ((2001) ) Analysis of relative gene expression data using real time quantitative PCR and the 2-rmDeltarmDeltaCT method. Methods 25: , 402–408. |
[44] | Wang X , Shi YJ , Niu TY , Chen TT , Li HB , Wu SH , Li GL ((2023) ) Neuroprotective effect of 20 (S)-Protopanaxadiol (PPD) attenuates NLRP3 inflammasome-mediated microglial pyroptosis in vascular dementia rats. Neurosci Lett 814: , 137439. |
[45] | Bi M , Gladbach A , van Eersel J , Ittner A , Przybyla M , van Hummel A , Chua SW , van der Hoven J , Lee WS , Müller J , Parmar J , Jonquieres GV , Stefen H , Guccione E , Fath T , Housley GD , Klugmann M , Ke YD , Ittner LM ((2017) ) Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat Commun 8: , 473. |
[46] | Venna VR , Benashski SE , Chauhan A , McCullough LD ((2015) ) Inhibition of glycogen synthase kinase-3β enhances cognitive recovery after stroke: The role of TAK1. Learn Mem 22: , 336–343. |
[47] | Fujii H , Takahashi T , Mukai T , Tanaka S , Hosomi N , Maruyama H , Sakai N , Matsumoto M ((2016) ) Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer’s disease –Hyperphosphorylation and cleavage of 4- and 3-repeat tau. Br J Pharmacol 37: , 2441–2457. |
[48] | Wang W , Li M , Wang Y , Wang Z , Zhang W , Guan F , Chen Q , Wang J ((2017) ) GSK-3β as a target for protection against transient cerebral ischemia. Int J Med Sci 14: , 333–339. |
[49] | Mehta SL , Kim T , Chelluboina B , Vemuganti R ((2023) ) Tau and GSK-3β are critical contributors to α-synuclein-mediated post-stroke brain damage. Neuromolecular Med 25: , 94–101. |
[50] | Schiefecker AJ , Putzer G , Braun P , Martini J , Strapazzon G , Antunes AP , Mulino M , Pinggera D , Glodny B , Brugger H , Paal P , Mair P , Pfausler B , Beer R , Humpel C , Helbok R ((2021) ) Total tau protein as investigated by cerebral microdialysis increases in hypothermic cardiac arrest: A pig study. Ther Hypothermia Temp Manag 11: , 28–34. |
[51] | Geddes JW , Schwab C , Craddock S , Wilson JL , Pettigrew LC ((1994) ) Alterations in tau immunostaining in the rat hippocampus following transient cerebral ischemia. J Cereb Blood Flow Metab 14: , 554–564. |
[52] | Pluta R , Januszewski S , Czuczwar SJ ((2021) ) Brain ischemia as a prelude to Alzheimer’s disease. Front Aging Neurosci 13: , 636653. |
[53] | Pluta R , Januszewski S , Jabłoński M ((2022) ) Acetylated tau protein: A new piece in the puzzle between brain ischemia and Alzheimer’s diseaseInt J Mol Sci 23: , 9174. |
[54] | Khan S , Yuldasheva NY , Batten TFC , Pickles AR , Kellett KAB , Saha S ((2018) ) Tau pathology and neurochemical changes associated with memory dysfunction in an optimized murine model of global cerebral ischaemia –A potential model for vascular dementia? Neurochem Int 118: , 134–144. |
[55] | Wen Y , Yang SH , Liu R , Perez EJ , Brun-Zinkernagel AM , Koulen P , Simpkins JW ((2007) ) Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta 1772: , 473–483. |
[56] | Wu Y , Eisel ULM ((2023) ) Microglia-astrocyte communication in Alzheimer’s disease. J Alzheimers Dis 95: , 785–803. |
[57] | Cohan CH , Neumann JT , Dave KR , Alekseyenko A , Binkert M , Stransky K , Lin HW , Barnes Wright CACB , Perez Pinzon MA ((2015) ) Effect of cardiac arrest on cognitive impairment and hippocampal plasticity in middle-aged rats. PLoS One 10: , e0124918. |
[58] | Hermann DM , Popa-Wagner A , Kleinschnitz C , Doeppner TR ((2019) ) Animal models of ischemic stroke and their impact on drug discovery. Expert Opin Drug Discov 14: , 315–326. |


