
Progranulin Gene Mutations in Chinese Patients with Frontotemporal Dementia: A Case Report and Literature Review
Abstract
Background:
Progranulin (GRN) mutations in frontotemporal dementia (FTD) have been less frequently reported in China than in Western countries.
Objective:
This study reports a novel GRN mutation and summarizes the genetic and clinical features of patients with GRN mutations in China.
Methods:
Comprehensive clinical, genetic, and neuroimaging examinations were conducted on a 58-year-old female patient diagnosed with semantic variant primary progressive aphasia. A literature review was also conducted and clinical and genetic features of patients with GRN mutations in China were summarized.
Results:
Neuroimaging revealed marked lateral atrophy and hypometabolism in the left frontal, temporal, and parietal lobes. The patient was negative for pathologic amyloid and tau deposition by positron emission tomography. A novel heterozygous 45-bp deletion (c.1414-14_1444delCCCTTCCCCGCCAGGCTGTGTGCTGCGAGGATCGCCAGCACTGCT) was detected by whole-exome sequencing of the patient’s genomic DNA. Nonsense-mediated mRNA decay was presumed to be involved in the degradation of the mutant gene transcript. The mutation was deemed pathogenic according to American College of Medical Genetics and Genomics criteria. The patient had a reduced plasma GRN level. In the literature, there were reports of 13 Chinese patients – mostly female – with GRN mutations; the prevalence was 1.2% –2.6% and patients mostly had early disease onset.
Conclusion:
Our findings expand the mutation profile of GRN in China, which can aid the diagnosis and treatment of FTD.
INTRODUCTION
Frontotemporal dementia (FTD) is a spectrum of neurodegenerative disorders that includes behavioral variant FTD (bvFTD), progressive non-fluent aphasia, and semantic dementia [1]. The clinical and genetic features of FTD can overlap with those of neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD), cortical basal syndrome (CBS), and progressive supranuclear palsy (PSP) [2–4]. Approximately one-third of FTD cases harbor heritable pathogenic genes such as chromosome 9 open reading frame 72 (C9orf72), granulin (GRN), and microtubule-associated protein tau (MAPT) [5]. FTD caused by GRN mutations is rare in China compared with Western countries [5–8].
The GRN gene on chromosome 17q21 encodes progranulin (PGRN), a multifunctional 593-amino acid protein that promotes the development and survival of neurons and microglia [9]. Loss of PGRN function is a monogenic cause of FTD [10]. Approximately 114 pathogenic mutations in the GRN gene have been identified, including some that cause haploinsufficiency [8, 11]. GRN mutations that reduce the level of PGRN increase the risk of dementia, whereas increasing and/or restoring PGRN level is an effective therapeutic strategy [11]. Therefore, from the epidemiologic, clinical, and healthcare resource management standpoints, it is important to document the full spectrum of GRN mutations in China and their associated clinical manifestations.
In this study, we report a novel GRN mutation in a Chinese patient with FTD. We also summarize the genetic and clinical features of all Chinese patients with GRN mutations reported in the literature.
MATERIALS AND METHODS
Clinical and neuropsychological workup
The patient was evaluated at Xuanwu Hospital in Beijing, China. We collected a range of clinical data relating to initial symptoms, disease progression, and family history. A professional neurologist conducted neurologic testing. Cognitive function was assessed with the Mini-Mental State Examination (MMSE), Montreal Cognitive Assessment (MoCA). Disease severity was tested by Frontotemporal Lobar Degeneration-Clinical Dementia Rating scale (FTLD-CDR). Language function was evaluated with the Boston Naming Test (BNT). Behavioral abnormalities were assessed with the Frontal Behavior Inventory (FBI), and the ability to perform daily activities was evaluated with the Activities of Daily Living (ADL) scale.
The study was approved by the Ethics Committees of the Xuanwu Hospital of Capital Medical University, China, and was conducted according to the principles outlined in the Declaration of Helsinki. Written informed consent was obtained from the patient.
Neuroimaging
Positron emission tomography (PET) and magnetic resonance imaging (MRI) were performed at Xuanwu Hospital using a 3.0 T time-of-flight hybrid PET/MRI scanner (Signa PET/MR, GE Healthcare, Madison, WI, USA) with a 19-channel head and neck union coil. We also performed amyloid PET (AV45), tau PET (PI2620), and 18F fluorodeoxyglucose (18F-FDG)-PET. PET data were reconstructed using an ordered subset expectation maximization algorithm with weighted attenuation.
Whole-exome sequencing (WES)
The patient’s genomic DNA (gDNA) was isolated from peripheral blood leukocytes (QIAamp DNA Blood Kits; Qiagen, Valencia, CA, USA). Her families refused genetic testing. We performed exome capture with a SureSelect Human All Exon V6+UTR (89 Mb) Kit (Agilent Technologies, Santa Clara, CA, USA). Paired-end sequencing was performed on a HiSeq2500 system (Illumina, San Diego, CA, USA) using HiSeq SBS Kit V4 (Illumina), which generated 100-bp reads. The average and minimum sequencing depths were 125× and 20×, respectively. The following reference databases were used: hg38 (GRCh38) (https://genome.ucsc.edu), Human Gene Mutation Database (https://www.hgmd.cf.ac.uk), Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and Single Nucleotide Polymorphism Database (https://www.ncbi.nlm.nih.gov/SNP). WES data were analyzed for single-nucleotide variants and insertion/deletions in dementia-related genes (Supplementary Table 1). Minor allele frequency, conservation, predicted pathogenicity, and disease association were analyzed, and the results were confirmed by Sanger sequencing.
GRN gene sequencing
The gDNA sample of the patient was used for standard polymerase chain reaction (PCR) amplification of the deleted region flanking the intron 10/exon 11 boundary. All primers used in this study are listed in Supplementary Table 2. Amplification products were purified and sequenced in both directions. Sanger sequencing was performed by Eurofins Genomics (Louisville, KY, USA), and trace files were analyzed with Seq Scanner 2 software (Thermo Fisher Scientific, Waltham, MA, USA).
RNA isolation, reverse transcription, and quantitative real-time PCR (qRT-PCR)
Reverse transcription, Sanger sequencing, and qRT-PCR were performed using total RNA from blood samples of the proband and 3 wild-type control subjects. Total RNA was extracted from leukocytes using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. Reverse transcription was performed using the iScript gDNA Clear cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA) and cDNA was amplified by long-range PCR using a primer set designed to amplify from the untranslated region (UTR) of exon 0 to exon 12 of GRN. qRT-PCR was performed with SoAdvanced Universal SYBR Green Supermix (Bio-Rad). Relative quantification of GRN gene expression levels was performed using the ΔCT method, with the level of the ribosomal protein S17 (RPS17) gene in leukocytes used for normalization.
Biomarker study and enzyme-linked immunosorbent assay (ELISA)
We analyzed PGRN expression levels in the patient carrying the Val473fs mutation, 5 FTD patients without GRN mutations, and 5 healthy control subjects. Plasma was obtained within 90 days from peripheral blood samples and was centrifuged, aliquoted, and stored within 2 h at –80°C until analysis. PGRN expression level in the samples was measured with the human Progranulin ELISA kit (Adipogen, Incheon, Seoul, Korea) according to the manufacturer’s instructions using a 1:100 dilution of sample in 1× diluent. Samples were added to the wells of a 96-well plate and incubated overnight at 4°C; recombinant human PGRN provided with the ELISA kit was used as the standard. The wash solution was aspirated after each third wash to ensure that all residual wash solution was removed. This was followed by incubation with secondary antibody for 2 h at room temperature. The reaction substrate was then added, followed by a termination solution. Color intensity was measured at 450 nm within 10 min.
PGRN expression levels in FTD patients without GRN mutations and healthy controls subjects were compared with the Mann–Whitney U test. Results were considered statistically significant at p≤0.05.
Literature review
Two researchers (MC and HTN) performed a literature search in PubMed and EMBASE databases from inception to November 2022 for studies describing the clinical features and genotypes of patients with FTD in China. We included all patients who had been diagnosed with FTD and harbored GRN mutations. From each publication, we extracted the first author’s name and year of publication as well as the patients’ age of FTD onset, sex, family history, genotype, and phenotype.
RESULTS
Clinical and neuroimaging data
The patient was a 58-year-old woman with 14 years of education who used to work as a tour guide. At the age of 57 years, she underwent a change in personality, which manifested as a loss of interest in daily activities and a depressive state. One month later, she became apathetic and developed difficulty in finding words and in single-word comprehension; her speech output decreased, and she refused to communicate with her families or friends. She denied any history of drug use. The patient’s pedigree is shown in Fig. 2A. Her mother had developed dementia at the age of 65 years and died at 70 years, but no other family member had a history of dementia, motor dysfunction, or neurodegenerative diseases. The patient’s test scores were as follows: MMSE, 11; MoCA, 6; FTLD-CDR sum of the box, 6; BNT, 10; FBI, 19; and ADL, 25. We observed marked lateral atrophy (Fig. 1A) and hypometabolism (Fig. 1B) in the left cortex in the frontal, temporal, and parietal lobes by MRI and 18F-FDG-PET.
Fig. 1
Neuroimaging of the patient with GRN mutation. A) MRI showing left-side atrophy in the frontal, temporal, and parietal lobes. B) FDG-PET showing left-side hypometabolism in the frontal, temporal, and parietal lobes. C) AV45-PET showing absence of Aβ deposition. D) APN1607 PET showing absence of tau deposition.
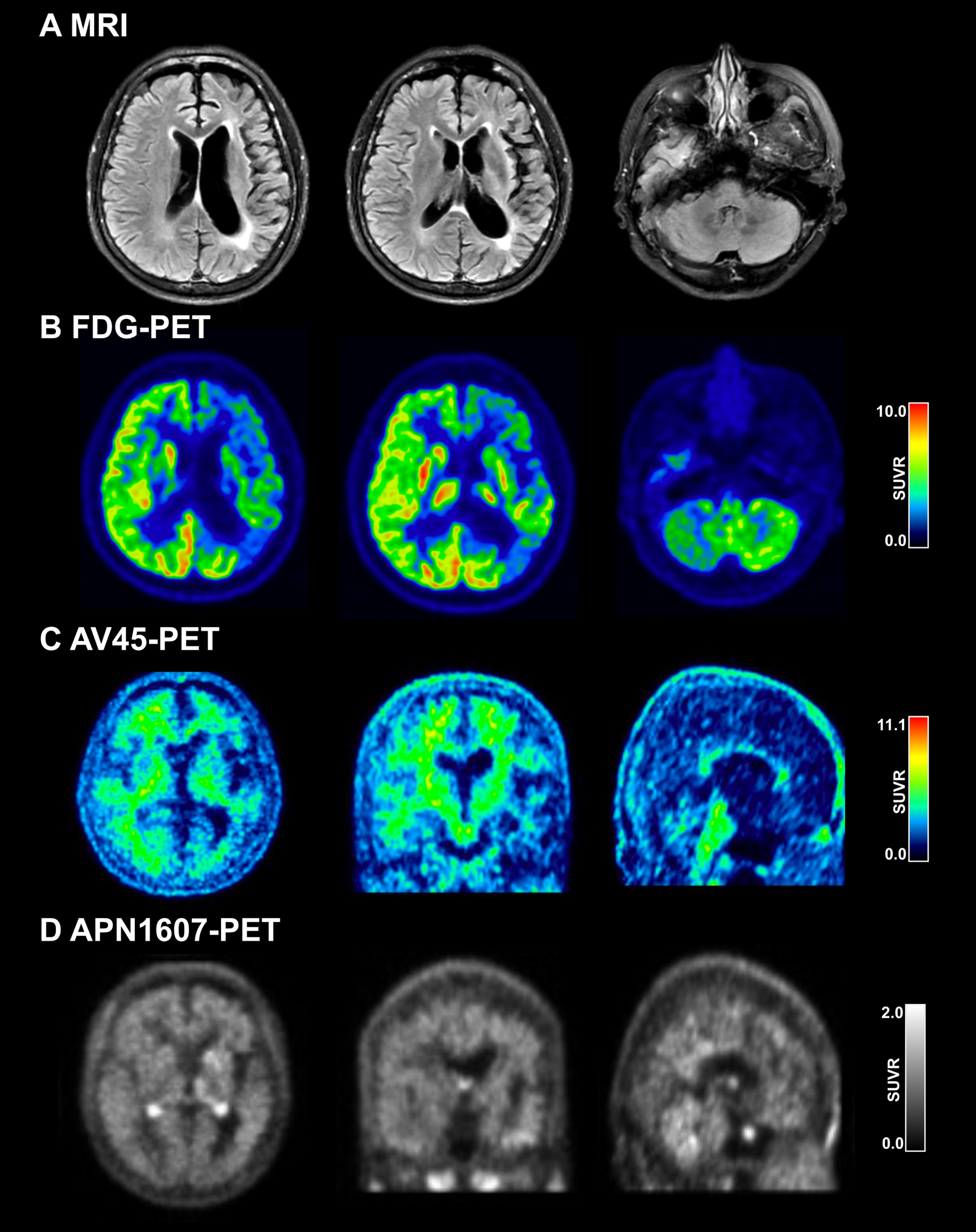
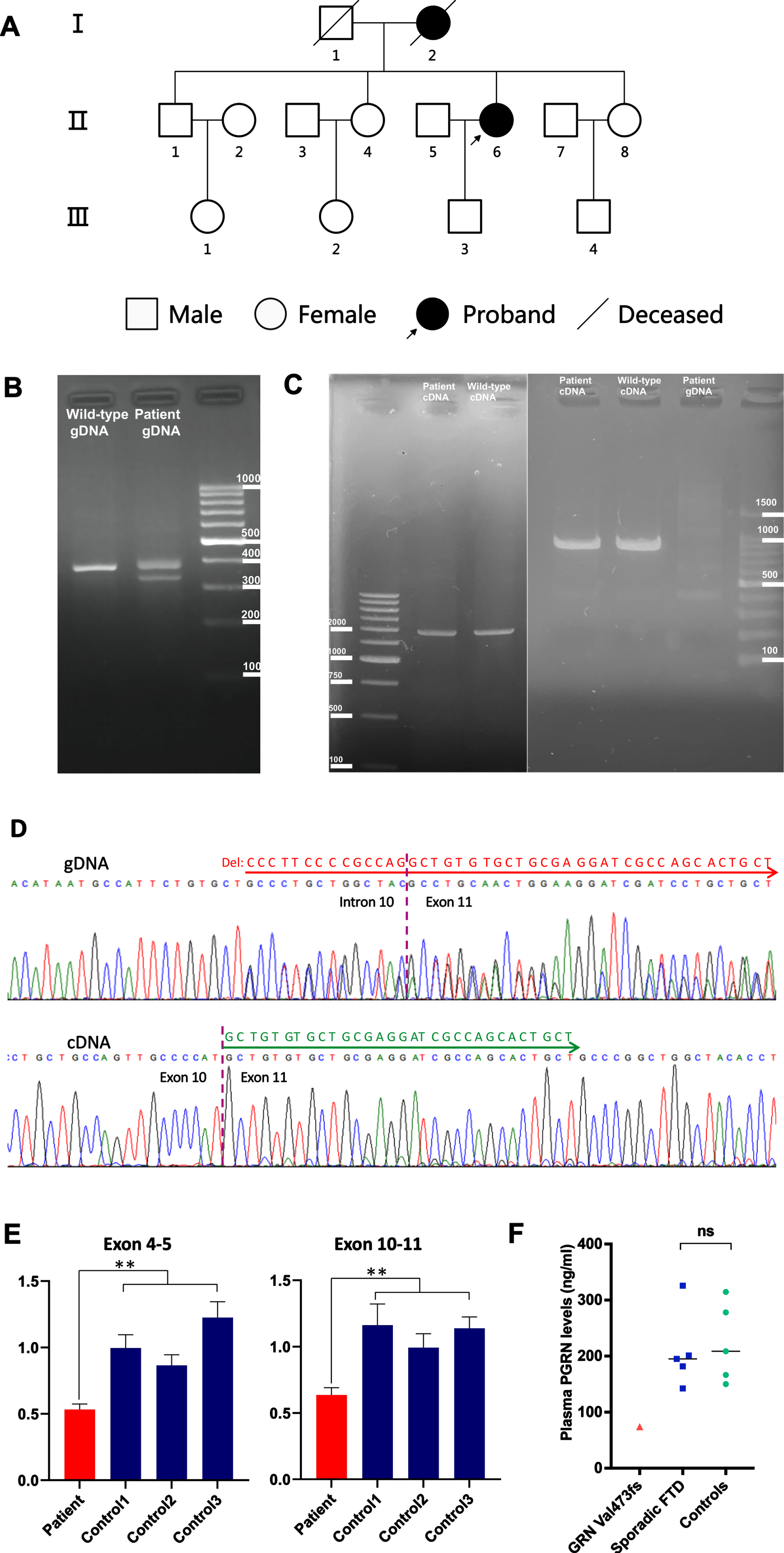
Fig. 2
Genetic analysis of the GRN gene. A) Pedigree of the patient with GRN mutation. B) PCR amplification products across the deletion at the intron 10/exon 11 boundary of GRN from the patient and a wild-type control were examined by agarose gel electrophoresis. The 367-bp band corresponds to the wild-type allele. A 322-bp fragment corresponding to the heterozygous mutated allele with the 45-bp deletion was amplified from the gDNA of the patient. C) Left panel: Agarose gel electrophoresis of the long-range RT-PCR products revealing only the 2081-bp cDNA fragment corresponding to the predicted canonical transcripts in both the patient and control subject. Right panel: Agarose gel electrophoresis of shorter RT-PCR products showing only the 1071-bp band expected in both the patient and control. cDNA, complementary DNA; gDNA, genomic DNA. D) Upper lane: Sequence analysis of the gDNA PCR product of the patient revealed a c.1414-14_1444delCCCTTCCCCGCCAGGCTGTGTGCTGCGAGGATCGCCAGCACTGCT mutation in heterozygous state. Lower lane: Sanger sequencing of the RT-PCR products of the patient revealed the canonical transcripts. We found a substantial reduction in the amount of mutant allele which is virtually absent. E) GRN gene expression was analyzed by qRT-PCR using total RNA extracted from peripheral leukocytes of the patient with primer sets corresponding to sequences within exons 4–5 (left panel) and exons 10–11 (right panel). A –∼50% decrease in the GRN product (normalized to RPS17 level) was observed in the patient compared with the same product in the 3 wild-type control subjects. Data are expressed as Mean±SD of 3 independent experiments. **p < 0.01 (Mann–Whitney U test). F) Plasma PGRN levels (ng/ml) in the patient carrying the GRN Val473fs mutation (red triangle), 5 FTD patients without GRN mutations (blue squares), and 5 healthy control subjects (green circles). Each data point represents an individual. Black horizontal lines indicate the median plasma PGRN levels of FTD patients without GRN mutations and healthy controls.
Based on the patient’s clinical and neuroimaging data, we considered a diagnosis of semantic variant primary progressive aphasia (svPPA) [12], which includes impaired naming, single-word comprehension, and object recognition for low-frequency items while repetition, grammar, and motor functions are spared. To exclude early-onset Alzheimer’s disease (AD), we conducted amyloid (AV45; Fig. 1C) and tau (APN1607; Fig. 1D) PET scanning but found no evidence of pathologic protein deposition.
WES, GRN gene sequencing, and genetic analysis
We first carried out WES of gDNA from the patient. We examined gene mutations known to be involved in AD, FTD, or other dementia-related neurodegenerative diseases. We identified a heterozygous 45-bp deletion (c.1414-14_1444delCCCTTCCCCGCCAGGCTGTGTGCTGCGAGGATCGCCAGCACTGCT [GenBank accession no. NM_002087.3]) at the boundary between intron 10 and exon 11 of the GRN gene. There were no additional disease-causing mutations in any other genes.
We next examined the 45-bp deletion in the GRN gene in the patient; a fragment encompassing the deletion at the intron 10/exon 11 boundary of the GRN gene was amplified by PCR. We examined PCR products from the patient and a wild-type control by agarose gel electrophoresis and Sanger sequencing. The results confirmed a heterozygous 45-bp deletion in the GRN gene in the proband (Fig. 2B, D).
The GRN deletion was absent from the HGMD, ClinVar, gnomAD, and 1000 Genomes databases (last access date of all databases: November 22, 2022). We, therefore, concluded that it was a novel causative mutation of FTD. Bioinformatic analyses using Mutation Taster (https://www.mutationtaster.org) predicted that this is a disease-causing mutation, and the mutation is pathogenic according to American College of Medical Genetics and Genomics (ACMG) criteria (PVS1+PS3+PM2+PP3+PP4) [13].
Transcription analysis
The translation of gene transcripts can be disrupted by mutations in splicing elements or splicing signal sequences or by the introduction of premature termination codons (Supplementary Figure 1A) [14]. The 45-bp deletion at the intron 10/exon 11 boundary of the GRN gene was expected to affect the 5′ splice site of exon 11 and result in exon skipping (Supplementary Figure 1B). The results of the bioinformatic analysis showed that the deletion may also lead to a frameshift and introduction of a premature stop codon (p.Val473Argfs*8) (SupplementaryFigure 1C). In both ways, the degradation of the mutated mRNA by nonsense-mediated mRNA decay (NMD) is implicated.
To confirm that the GRN mRNA expression level was affected by the deletion, we performed RT-PCR using total RNA from the peripheral leukocytes of the patient. Sanger sequencing of the RT-PCR products from both directions showed only canonical transcripts (Fig. 2D lower lane). Agarose gel electrophoresis of long-range RT-PCR products (exon 0 UTR–exon 12 UTR) revealed only the expected 2081-bp band in both the patient and control, with no abnormal bands (Fig. 2C). The same results were obtained using a different set of primers targeting a shorter fragment (exon 4-exon 11). Agarose gel electrophoresis of these RT-PCR products revealed only the expected 1071-bp band in both the patient and control and Sanger sequencing in both directions confirmed only canonical transcripts in the patient (Fig. 2C). Thus, we found no additional GRN isoforms, and the truncated transcript which is virtually absent was reduced substantially. To corroborate these results, we quantified mRNA levels in the patient’s leukocytes by qRT-PCR; the results showed that the GRN transcript level was approximately 0.5-fold lower in the patient compared with the 3 wild-type controls (Fig. 2E).
Plasma levels of PGRN
Plasma PGRN levels in the patient carrying the Val473fs mutation, 5 FTD patients without GRN mutations, and 5 healthy control subjects were measured by ELISA (Fig. 2F). PGRN level in the patient with the Val473fs mutation (74.15 ng/ml) was one-third of the level in the 10 non-mutation carriers (Mean±SD, 216.40±21.01 ng/ml, and range, 142.47–325.80 ng/ml). There was no significant difference in median plasma PGRN levels of the 5 FTD patients without GRN mutations (201.24 ng/ml; range, 142.47–325.80 ng/ml) and 5 control subjects (208.61 ng/ml; range, 150.10–314.63 ng/ml) (p = 0.76).
Literature review
Nine publications that reported GRN mutations in a total of 13 Chinese patients with 12 different mutations were reviewed, including 6 missense, 4 frameshifts, 1 nonsense, and 1 splice site mutation. The GRN mutations were in an intron (IVS7+1G>A), exon 2 (P50fs), exon 4 (415T>C), exon 5 (560delT), exon 6 (659C>T), exon 7 (750C>A), exon 8 (898C>G), exon 10 (1352C>T and P439fs), and exon 11 (1460C>T and 1498G>A). The phenotype of GRN mutation carriers in China varied and included 4 bvFTD, 2 cortical basal syndromes (CBS), 1 PPA, 1 atypical PD, and 1 ALS case. The frequency of GRN mutation in an FTD cohort at a single center ranged from 1.2% (1/82) to 2.6% (1/38). The age of onset was <65 years and among patients whose sex was reported, there was a predominance of women (6/9, 75%). Figure 3 illustrates the GRN mutations identified in China, and related clinical and genetic information is shown in Table 1. Other GRN frameshift mutations that have been reported outside of China are listed in Supplementary.
Fig. 3
Schematic representation of GRN mutations in China. The schematic representation of the 12 coding exons (green boxes) and UTR of GRN shows all pathogenic mutations identified in Chinese patients reported to date. Most of the mutations cause premature termination of the coding sequence, leading to the degradation of the mutant GRN mRNA by nonsense-mediated decay. The c.1414-14_1444del mutation identified in this study is indicated in red.
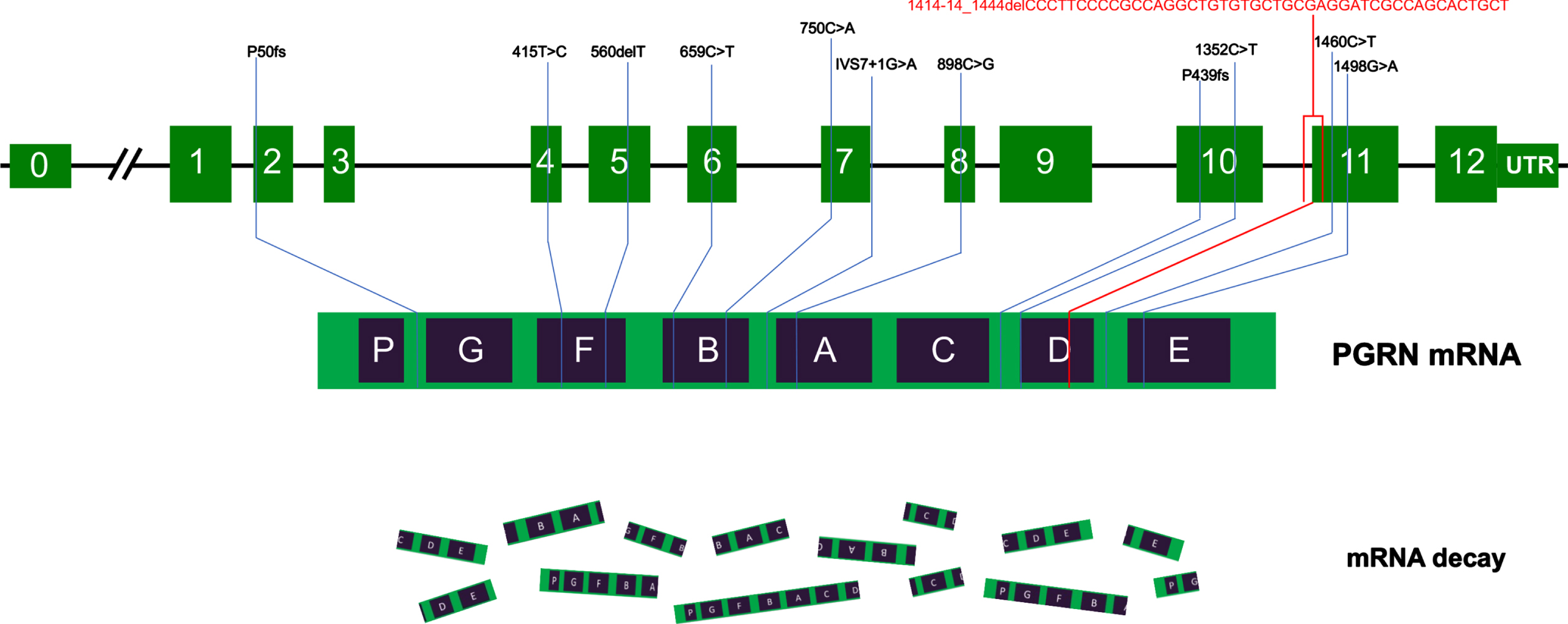
Table 1
Summary of the GRN mutations reported in Chinese patients with FTD and related disease
| Author year [ref] | Exon | Amino acid change | Protein change | Phenotype | Frequency | Onset age | Sex | Family history |
| Masellis 2006 [30] | Splicing | IVS7+1G>A | Truncationa | CBS | Case | 62 | F | Yes |
| IVS7+1G>A | Truncationa | CBS | Case | 57 | F | Yes | ||
| Shi 2016 [15] | 5 | 560delT | L187fsa | PPA | 1/38 | 64 | F | No |
| Tang 2016 [17] | 8 | 898C>G | Q300Xa | bvFTD | 1/52 | 63 | M | No |
| Che 2017 [18] | 7 | 750C>A | D250Ea | FTD | 1/82 | 62 | F | NA |
| Chang 2018 [31] | 11 | 1460C>T | T487Ia | Atypical PDc | 1/98 | 45 | F | No |
| Zhang 2020 [32] | 4 | 415T>C | C139Rb,d | ALS | Case | 43 | F | No |
| Deng 2021 [33] | 11 | 1498G>A | V500Ia | bvFTD | Case | 55 | M | No |
| Liu 2021 [19] | 10 | c.1352C>T | P451La | bvFTD | 1/49 | 52 | M | No |
| Gao 2022 [16] | 10 | NA | P439fsa | FTD | 3/204 | NA | NA | NA |
| 2 | NA | P50fsa | FTD | / | NA | NA | NA | |
| 6 | 659C>T | T220Ia | FTD | / | NA | NA | NA | |
| This study | 11 | c.1414-14_1444delCCCT TCCCCGCCAGGCT GTGTGCTGCGAGG ATCGCCAGCACT GCT | V473fsa | bvFTD | Case | 57 | F | Yes |
ALS, amyotrophic lateral sclerosis; CBS, cortical-basal syndrome; bvFTD, behavioral variant frontotemporal dementia; F, female; FTD, frontotemporal dementia; M, male; NA, not available; PD, Parkinson’s disease; PPA, primary progressive aphasia. aNot reported in Western countries. bAlso reported in Western countries. GRN p.C139R was reported in Italy [36] and Spain [37, 38]. cThis patient presented with psychotic behavior at the age of 45 years. dThis patient carried 2 pathogenic mutations including the NEK1 nonsense mutation p.K1210* and GRN mutation p.C139R.
DISCUSSION
In this study, we identified a novel pathogenic V473fs mutation of the GRN gene in a Chinese woman with FTD. We also summarized all patients with GRN mutations reported in China and found a prevalence of 1.2% –2.6%, early disease onset, and female predominance in this population. The results of this study expand the GRN mutation spectrum in China. As increasing PGRN levels has been shown to be an effective treatment for FTD [11], screening for GRN mutations can potentially guide treatment strategies for FTD.
We determined that our patient had haploinsufficiency due to a GRN mutation that caused reduced GRN mRNA expression and plasma PGRN level. The novel GRN mutation was deemed pathogenic according to the ACMG and Association for Molecular Pathology standards and guidelines [13] based on the following criteria. 1) Loss of function is a known mechanism of the disease (PVS1). 2) GRN mRNA level was reduced in the patient compared with healthy control subjects (PS3). 3) The mutation was located in a functional domain (PM1). 4) The mutation was absent in the healthy population (PM2). 5) The gene is highly conserved across species and computational predictions support a deleterious effect (PP3). 6) The patient’s phenotype indicated a disease with single-gene etiology (PP4).
GRN mutations are rare in China [7], with a prevalence of just 1.2% –2.6% [15–19], and have been infrequently reported in other Asian countries including Japan, Philippines, India, and Korea (where they have never been observed) [20–24]. In contrast, GRN mutations are common in Western countries, accounting for 34.6% of mutations in patients with FTD [5]. Other gene mutations differing in prevalence in Western countries and China are C9, which is more common in the former, and MAPT, is more common in the latter [5, 7]. Such differences are mainly due to ethnicity and environmental factors. The most common GRN mutations in Western countries are Thr272fs, Arg493X, IVS7–1G⟶A, Cys31fs, Gly35fs, and Ala9Asp [5], but these are not present in the Chinese population. In our review, we identified mutations in exons 2, 5, 6, 7, and 8, 2 in exon 10, and 3 in exon 11; there were no specific clustering tendencies compared with mutations reported in Western countries [25].
We observed atrophy and hypometabolism in the left frontal, temporal, and parietal lobes in our patient, which is consistent with the asymmetric atrophy reported in GRN mutation carriers [25–27]. The white matter hyperintense signal was found in our patient, which has also been previously reported in other patients with GRN mutations [20, 28]. Amyloid and tau PET were negative, although one case of FTD with GRN mutation in the literature had TAR DNA-binding protein 43 (TDP-43) protein inclusions [29].
The clinical phenotypes for GRN mutations vary; most patients in our review presented with bvFTD and others with CBS, PSP, atypical PD, or ALS [30–33]. Left-side temporal lobe atrophy may contribute to language deficits whereas right-side frontal lobe atrophy has been linked to psychoses and behavioral deficits in previous cases [25, 34]. Our patient mainly presented with semantic variant PPA, which was associated with functional involvement of left-side brain regions as determined by 18F-FDG-PET/MRI. Additionally, we observed female predominance among Chinese patients with GRN mutations, consistent with the findings of a meta-analysis showing a higher prevalence of GRN-related FTD among women [35].
Our study had some limitations. First, the sample size of the review was small because GRN-related FTD is rare in China. Second, there may have been publication bias in the review because genetic screening is not common in some rural areas of China. Larger, multicenter genetic studies are needed to confirm the frequency of GRN mutations in China.
Conclusion
In conclusion, we identified a novel pathogenic GRN gene mutation (V473fs) in a patient with FTD. The prevalence of the mutation was 1.2% –2.6% among Chinese patients with FTD, who were mostly female and had an early disease onset. Our findings expand the mutation spectrum of GRN in FTD and provide a basis for the development of targeted therapies.
ACKNOWLEDGMENTS
The authors are grateful to the patient for participating in the study.
FUNDING
This work was supported by grants from the National Natural Science Foundation of China (nos. 82271464 and 81971011).
CONFLICT OF INTEREST
Liyong Wu is an Editorial Board Member of this journal but was not involved in the peer review process, nor had access to any information regarding its peer review.
DATA AVAILABILITY
The datasets used and analyzed in the current study are available from the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-230052
REFERENCES
[1] | Seelaar H , Rohrer JD , Pijnenburg YA , Fox NC , van Swieten JC ((2011) ) Clinical, genetic and pathological heterogeneity of frontotemporal dementia: A review. J Neurol Neurosurg Psychiatry 82: , 476–486. |
[2] | Snowden JS (2022) Changing perspectives on fron totemporal dementia: A review. J Neuropsychol. doi: 10.1111/jnp.12297. |
[3] | Ferrari R , Kapogiannis D , Huey ED , Momeni P ((2011) ) FTD and ALS: A tale of two diseases. Curr Alzheimer Res 8: , 273–294. |
[4] | Liu L , Liu S , Chu M , Wang J , Xie K , Cui Y , Ma J , Nan H , Cui C , Qiao H , Rosa-Neto P , Chan P , Wu L ((2022) ) Involvement of striatal motoric subregions in familial frontotemporal dementia with parkinsonism harboring the C9orf72 repeat expansions. NPJ Parkinsons Dis 8: , 128. |
[5] | Moore KM , Nicholas J , Grossman M , McMillan CT , Irwin DJ , Massimo L , Van Deerlin VM , Warren JD , Fox NC , Rossor MN , Mead S , Bocchetta M , Boeve BF , Knopman DS , Graff-Radford NR , Forsberg LK , Rademakers R , Wszolek ZK , van Swieten JC , Jiskoot LC , Meeter LH , Dopper EG , Papma JM , Snowden JS , Saxon J , Jones M , Pickering-Brown S , Le Ber I , Camuzat A , Brice A , Caroppo P , Ghidoni R , Pievani M , Benussi L , Binetti G , Dickerson BC , Lucente D , Krivensky S , Graff C , Öijerstedt L , Fallström M , Thonberg H , Ghoshal N , Morris JC , Borroni B , Benussi A , Padovani A , Galimberti D , Scarpini E , Fumagalli GG , Mackenzie IR , Hsiung GR , Sengdy P , Boxer AL , Rosen H , Taylor JB , Synofzik M , Wilke C , Sulzer P , Hodges JR , Halliday G , Kwok J , Sanchez-Valle R , Lladó A , Borrego-Ecija S , Santana I , Almeida MR , Tábuas-Pereira M , Moreno F , Barandiaran M , Indakoetxea B , Levin J , Danek A , Rowe JB , Cope TE , Otto M , Anderl-Straub S , de Mendonça A , Maruta C , Masellis M , Black SE , Couratier P , Lautrette G , Huey ED , Sorbi S , Nacmias B , Laforce R Jr. , Tremblay ML , Vandenberghe R , Damme PV , Rogalski EJ , Weintraub S , Gerhard A , Onyike CU , Ducharme S , Papageorgiou SG , Ng ASL , Brodtmann A , Finger E , Guerreiro R , Bras J , Rohrer JD ((2020) ) Age atsymptom onset and death and disease duration in geneticfrontotemporal dementia: An international retrospective cohortstudy. Lancet Neurol 19: , 145–156. |
[6] | Ramos EM , Dokuru DR , Van Berlo V , Wojta K , Wang Q , Huang AY , Deverasetty S , Qin Y , van Blitterswijk M , Jackson J , Appleby B , Bordelon Y , Brannelly P , Brushaber DE , Dickerson B , Dickinson S , Domoto-Reilly K , Faber K , Fields J , Fong J , Foroud T , Forsberg LK , Gavrilova R , Ghoshal N , Goldman J , Graff-Radford J , Graff-Radford N , Grant I , Grossman M , Heuer HW , Hsiung GR , Huey E , Irwin D , Kantarci K , Karydas A , Kaufer D , Kerwin D , Knopman D , Kornak J , Kramer JH , Kremers W , Kukull W , Litvan I , Ljubenkov P , Lungu C , Mackenzie I , Mendez MF , Miller BL , Onyike C , Pantelyat A , Pearlman R , Petrucelli L , Potter M , Rankin KP , Rascovsky K , Roberson ED , Rogalski E , Shaw L , Syrjanen J , Tartaglia MC , Tatton N , Taylor J , Toga A , Trojanowski JQ , Weintraub S , Wong B , Wszolek Z , Rademakers R , Boeve BF , Rosen HJ , Boxer AL , Coppola G ((2020) ) Genetic screening of a large series of North American sporadic and familial frontotemporal dementia cases. Alzheimers Dement 16: , 118–130. |
[7] | Jiang Y , Jiao B , Xiao X , Shen L ((2021) ) Genetics of frontotemporal dementia in China. Amyotroph Lateral Scler Frontotemporal Degener 22: , 321–335. |
[8] | Greaves CV , Rohrer JD ((2019) ) An update on genetic frontotemporal dementia. J Neurol 266: , 2075–2086. |
[9] | Baker M , Mackenzie IR , Pickering-Brown SM , Gass J , Rademakers R , Lindholm C , Snowden J , Adamson J , Sadovnick AD , Rollinson S , Cannon A , Dwosh E , Neary D , Melquist S , Richardson A , Dickson D , Berger Z , Eriksen J , Robinson T , Zehr C , Dickey CA , Crook R , McGowan E , Mann D , Boeve B , Feldman H , Hutton M ((2006) ) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442: , 916–919. |
[10] | Mukherjee O , Wang J , Gitcho M , Chakraverty S , Taylor-Reinwald L , Shears S , Kauwe J , Norton J , Levitch D , Bigio E , Hatanpaa K , White C , Morris J , Cairns N , Goate A ((2008) ) Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat 29: , 512–521. |
[11] | Rhinn H , Tatton N , McCaughey S , Kurnellas M , Rosenthal A ((2022) ) Progranulin as a therapeutic target in neurodegenerative diseases. Trends Pharmacol Sci 43: , 641–652. |
[12] | Gorno-Tempini ML , Hillis AE , Weintraub S , Kertesz A , Mendez M , Cappa SF , Ogar JM , Rohrer JD , Black S , Boeve BF , Manes F , Dronkers NF , Vandenberghe R , Rascovsky K , Patterson K , Miller BL , Knopman DS , Hodges JR , Mesulam MM , Grossman M ((2011) ) Classification of primary progressive aphasia and its variants. Neurology 76: , 1006–1014. |
[13] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[14] | Wang GS , Cooper TA ((2007) ) Splicing in disease: Disruption of the splicing code and the decoding machinery. Nat Rev Genet 8: , 749–761. |
[15] | Shi Z , Liu S , Xiang L , Wang Y , Liu M , Liu S , Han T , Zhou Y , Wang J , Cai L , Gao S , Ji Y ((2016) ) Frontotemporal dementia-related gene mutations in clinical dementia patients from a Chinese population. J Hum Genet 61: , 1003–1008. |
[16] | Dong L , Wang J , Liu C , Li J , Mao C , Huang X , Chu S , Peng B , Cui L , Gao J ((2022) ) Genetic spectrum and clinical heterogeneity of chinese frontotemporal dementia patients: Data from PUMCH Dementia Cohort. J Alzheimers Dis 89: , 893–901. |
[17] | Tang M , Gu X , Wei J , Jiao B , Zhou L , Zhou Y , Weng L , Yan X , Tang B , Xu J , Shen L ((2016) ) Analyses MAPT, GRN, and C9orf72 mutations in Chinese patients with frontotemporal dementia, Neurobiol Aging 46: , 235.e211–235. |
[18] | Che XQ , Zhao QH , Huang Y , Li X , Ren RJ , Chen SD , Wang G , Guo QH ((2017) ) Genetic features of MAPT, GRN, C9orf72 and CHCHD10 gene mutations in Chinese patients with frontotemporal dementia. Curr Alzheimer Res 14: , 1102–1108. |
[19] | Liu L , Cui B , Chu M , Cui Y , Jing D , Li D , Xie K , Kong Y , Xia T , Wang C , Wu L ((2021) ) The frequency of genetic mutations associated with behavioral variant frontotemporal dementia in Chinese Han patients. Front Aging Neurosci 13: , 699836. |
[20] | Dominguez J , Ng A , Yu J , Guevarra AC , Daroy ML , Alfon A , Catindig JA , Dizon M , Santiago J , Del Moral MC , Yu J , Jamerlan A , Ligsay A , Bagyinszky E , An SS , Kim S ((2020) ) Autosomal dominant frontotemporal lobar degeneration in a Filipino family with progranulin mutation. Dement Geriatr Cogn Disord 49: , 557–564. |
[21] | Kim EJ , Kwon JC , Park KH , Park KW , Lee JH , Choi SH , Jeong JH , Kim BC , Yoon SJ , Yoon YC , Kim S , Park KC , Choi BO , Na DL , Ki CS , Kim SH ((2014) ) Clinical and genetic analysis of MAPT, GRN, and C9orf72 genes in Korean patients with frontotemporal dementia. Neurobiol Aging 35: , 1213.e1213–1217. |
[22] | Kim HJ , Jeon BS , Yun JY , Seong MW , Park SS , Lee JY ((2010) ) Screening for MAPT and PGRN mutations in Korean patients with PSP/CBS/FTD. Parkinsonism Relat Disord 16: , 305–306. |
[23] | Ogaki K , Li Y , Takanashi M , Ishikawa K , Kobayashi T , Nonaka T , Hasegawa M , Kishi M , Yoshino H , Funayama M , Tsukamoto T , Shioya K , Yokochi M , Imai H , Sasaki R , Kokubo Y , Kuzuhara S , Motoi Y , Tomiyama H , Hattori N ((2013) ) Analyses of the MAPT, PGRN, and C9orf72 mutations in Japanese patients with FTLD, PSP, and CBS. Parkinsonism Relat Disord 19: , 15–20. |
[24] | Aswathy PM , Jairani PS , Raghavan SK , Verghese J , Gopala S , Srinivas P , Mathuranath PS ((2016) ) Progranulin mutation analysis: Identification of one novel mutation in exon 12 associated with frontotemporal dementia. Neurobiol Aging 39: , 218.e211–213. |
[25] | Beck J , Rohrer JD , Campbell T , Isaacs A , Morrison KE , Goodall EF , Warrington EK , Stevens J , Revesz T , Holton J , Al-Sarraj S , King A , Scahill R , Warren JD , Fox NC , Rossor MN , Collinge J , Mead S ((2008) ) A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 131: , 706–720. |
[26] | Whitwell JL , Weigand SD , Boeve BF , Senjem ML , Gunter JL , DeJesus-Hernandez M , Rutherford NJ , Baker M , Knopman DS , Wszolek ZK , Parisi JE , Dickson DW , Petersen RC , Rademakers R , Jack CR Jr. , Josephs KA ((2012) ) Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 135: , 794–806. |
[27] | Rohrer JD , Ridgway GR , Modat M , Ourselin S , Mead S , Fox NC , Rossor MN , Warren JD ((2010) ) Distinct profiles of brain atrophy in frontotemporal lobar degeneration caused by progranulin and tau mutations. Neuroimage 53: , 1070–1076. |
[28] | Caroppo P , Le Ber I , Camuzat A , Clot F , Naccache L , Lamari F , De Septenville A , Bertrand A , Belliard S , Hannequin D , Colliot O , Brice A ((2014) ) Extensive white matter involvement in patients with frontotemporal lobar degeneration: Think progranulin. JAMA Neurol 71: , 1562–1566. |
[29] | Gliebus G , Bigio EH , Gasho K , Mishra M , Caplan D , Mesulam MM , Geula C ((2010) ) Asymmetric TDP-43 distribution in primary progressive aphasia with progranulin mutation. Neurology 74: , 1607–1610. |
[30] | Masellis M , Momeni P , Meschino W , Heffner R Jr. , Elder J , Sato C , Liang Y , StGeorge-Hyslop P , Hardy J , Bilbao J , Black S , Rogaeva E ((2006) ) Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain 129: , 3115–3123. |
[31] | Chang KH , Lee GC , Huang CC , Kuo HC , Chen CM , Hsiao YC , Hsu HC , Hsu KJ , Lin CH , Chang CW , Lee-Chen GJ , Wu YR ((2018) ) Genetic and functional characters of GRN p.T487I mutation in Taiwanese patients with atypical parkinsonian disorders. Parkinsonism Relat Disord 51: , 61–66. |
[32] | Zhang K , Lu Y , Chen J , Li J , Yadav KK , Yin J , Yang X ((2020) ) NEK1 and GRN mutations coexist in a sporadic Chinese Hui descent ALS patient. Amyotroph Lateral Scler Frontotemporal Degener 21: , 624–626. |
[33] | Deng B , Zheng Z , Zheng J , Yang W , Huang Y , Luo Y , Jin D , Shen L , Jin K , Wang Q ((2021) ) FTD-PSP is an unusual clinical phenotype in a frontotemporal dementia patient with a novel progranulin mutation. Aging Dis 12: , 1741–1752. |
[34] | Mendez MF ((2018) ) Manic behavior and asymmetric right frontotemporal dementia from a novel progranulin mutation. Neuropsychiatr Dis Treat 14: , 657–662. |
[35] | Curtis AF , Masellis M , Hsiung GR , Moineddin R , Zhang K , Au B , Millett G , Mackenzie I , Rogaeva E , Tierney MC ((2017) ) Sex differences in the prevalence of genetic mutations in FTD and ALS: A meta-analysis. Neurology 89: , 1633–1642. |
[36] | Sassi C , Capozzo R , Hammer M , Zecca C , Federoff M , Blauwendraat C , Bernstein N , Ding J , Gibbs JR , Price T , Singleton A , Logroscino G ((2021) ) Exploring dementia and neuronal ceroid lipofuscinosis genes in 100 FTD-like patients from 6 towns and rural villages on the Adriatic Sea cost of Apulia. Sci Rep 11: , 6353. |
[37] | Gómez-Tortosa E , Guerrero-López R , Gil-Neciga E , Franco E , del Ser T , Jiménez Escrig A , Pérez-Pérez J , Sainz MJ ((2013) ) Plasma progranulin levels in cortical dementia phenotypeswith asymmetric perisylvian atrophy. Eur J Neurol 20: , 1319–1324. |
[38] | Antonell A , Gil S , Sánchez-Valle R , Balasa M , Bosch B , Prat MC , Chiollaz AC , Fernández M , Yagüe J , Molinuevo JL , Lladó A ((2012) ) Serum progranulin levels in patients with frontotemporallobar degeneration and Alzheimer’s disease: Detection of GRNmutations in a Spanish cohort. J Alzheimers Dis 31: , 581–591. |


