
The Role of Interferon-α in Neurodegenerative Diseases: A Systematic Review
Abstract
Background:
Neurodegenerative diseases (NDs) impose significant financial and healthcare burden on populations all over the world. The prevalence and incidence of NDs have been observed to increase dramatically with age. Hence, the number of reported cases is projected to increase in the future, as life spans continues to rise. Despite this, there is limited effective treatment against most NDs. Interferons (IFNs), a family of cytokines, have been suggested as a promising therapeutic target for NDs, particularly IFN-α, which governs various pathological pathways in different NDs.
Objective:
This systematic review aimed to critically appraise the currently available literature on the pathological role of IFN-α in neurodegeneration/NDs.
Methods:
Three databases, Scopus, PubMed, and Ovid Medline, were utilized for the literature search.
Results:
A total of 77 journal articles were selected for critical evaluation, based on the inclusion and exclusion criteria. The studies selected and elucidated in this current systematic review have showed that IFN-α may play a deleterious role in neurodegenerative diseases through its strong association with the inflammatory processes resulting in mainly neurocognitive impairments. IFN-α may be displaying its neurotoxic function via various mechanisms such as abnormal calcium mineralization, activation of STAT1-dependent mechanisms, and increased quinolinic acid production.
Conclusion:
The exact role IFN-α in these neurodegenerative diseases have yet to be determine due to a lack in more recent evidence, thereby creating a variability in the role of IFN-α. Future investigations should thus be conducted, so that the role played by IFN-α in neurodegenerative diseases could be delineated.
INTRODUCTION
Neurodegenerative diseases (NDs) represent a diverse set of neurological disorders characterized by progressive degeneration of the structure and function of the nervous system. NDs may manifest differently (weakness, tremors, neurocognitive impairment, etc.) in people depending on the type and extend of the neuronal degeneration [1, 2]. Examples of NDs include Alzheimer’s disease (AD), multiple sclerosis, Huntington’s disease (HD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). In the United States alone, a 2017 report suggested that 6.08 million people suffered from AD while 1.04 million people suffered from PD in 2017 [3, 4]. A study by Tsolaki et al. found that it took caregivers up to 16 months to seek for medical advice as many of them misinterpreted dementia as a normal process of aging [5], thus suggesting the number of dementia cases may be underreported. As NDs are likely to result in long-term disability, they impose significant medical, financial, and public health burdens not only on patients and their caregivers, but also on a country’s economy [6]. Thus, with the growing number of neurodegenerative cases worldwide, there may be an urgent need for a valuable treatment avenue for neurodegenerative disease, especially since the progression of these neurological diseases may drastically reduce ones’ quality of life as well as their caregivers.
Current treatment strategies against most NDs predominantly lies through the aid of managing or slowing the progression/severity of the neurodegenerative symptoms (e.g., levodopa-carbidopa, MAO-B inhibitors, etc., for PD, and donepezil for AD) [7–9]. The pathological mechanisms behind these diseases have yet to be fully elucidated, but genetic and environmental factors have been explored previously [10, 11]. In addition, neuroinflammation and its mediators have also been suggested as a common pathology among most NDs [12], thus may serve as a promising area for intervention studies against NDs.
Interferons (IFNs), a family of cytokines produced by cells after infected by a virus, have the ability to inhibit viral growth and have shown to exhibit potent anti-viral properties [1, 13]. There are three types of human IFNs, namely interferon- α (IFN-α), interferon-beta (IFN-β), and interferon-gamma (IFN-γ), produced by the white blood cells, fibroblasts, and lymphocytes, respectively. Both IFN-α and IFN-β are type I interferons, while IFN-γ is a type II interferon [2, 3, 14, 15]. Previous studies have demonstrated that IFNs in general may exert its signals by binding to its interferon alpha/beta receptor which activates the Janus family tyrosine kinases (JAK) - signal transducer and activator of transcription (STAT) signaling pathway, leading to the phosphorylation of STAT1 and STAT2. These phosphorylated transcription factors may further move into the nucleus and induce the expression of IFN-stimulated genes, thereby mediating IFN-α (Fig. 1) [16].
Fig. 1
Type I IFNs, such as IFN-α, may bind to cell surface receptor containing two subunits: IFNAR1 and IFNAR2. This complex triggers the phosphorylation of the JAK-STAT proteins. The IFN regulatory factor 9 (IRF9), will form complexes with STAT dimers, and will translocate into the nucleus. Upon binding to interferon-sensitive response element (ISRE), IFN-induced genes would be expressed to induce IFN-mediated biological effects.
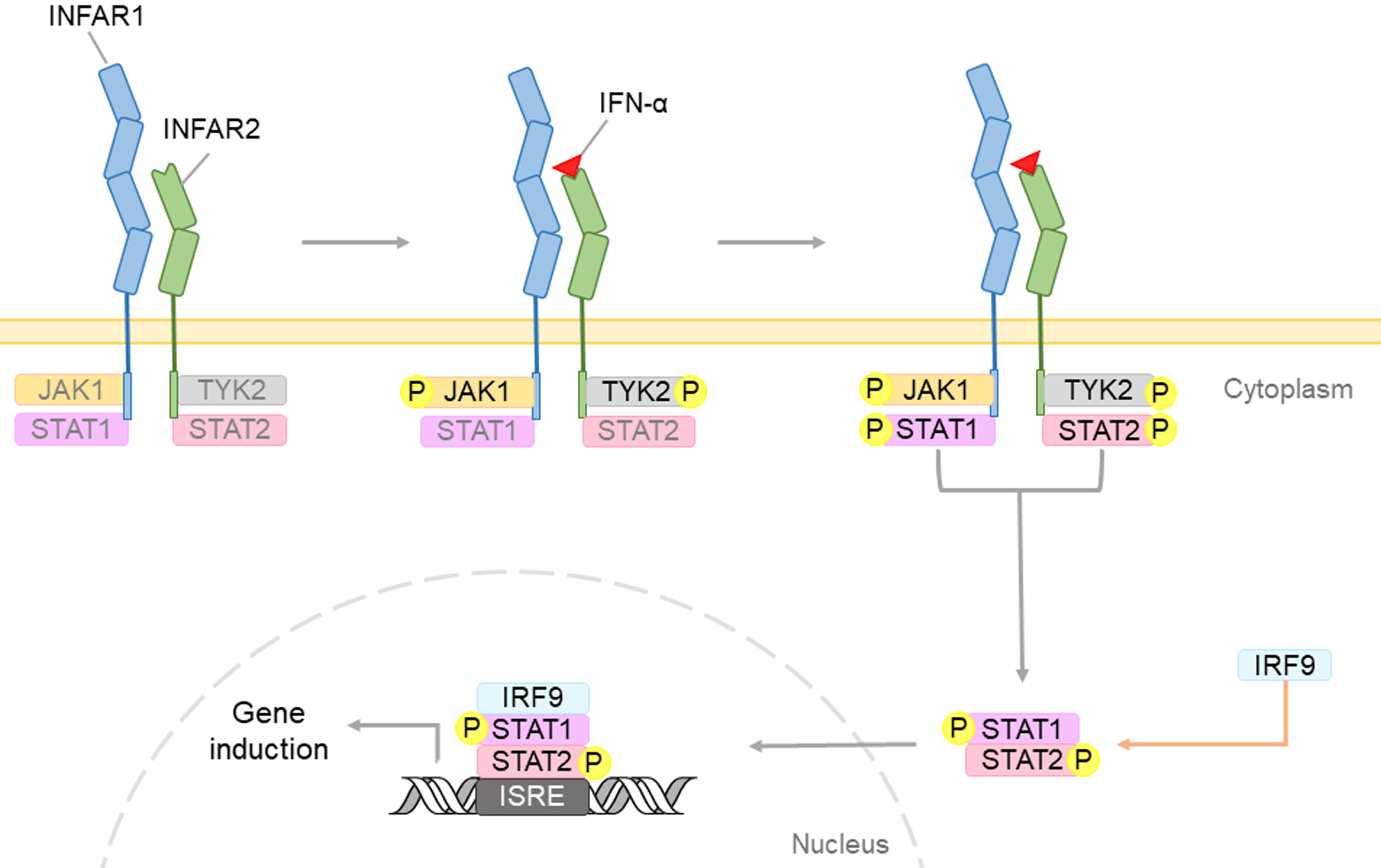
The JAK-STAT signaling pathway is implicated in neuroinflammation and is one of the IFN-regulated pathways which acts as a critical checkpoint in neurodegenerative diseases. Dysregulation of the JAK-STAT pathway has been shown to be implicated in most NDs including AD, by modulating synaptic plasticity of neurons and the overall neuronal survival [17].
Broadly, Type 1 IFN signaling have shown to demonstrate different outcomes, both protective and deleterious roles in different diseases [18], depending on the type of IFN. In the central nervous system (CNS), IFN-β has been used as first line treatment for multiple sclerosis and was found to be effective in decreasing the relapse rate in patients [19, 20]. Meanwhile, effects like depression and neurocognitive impairment have been observed in individuals receiving IFN-α in the context of neurodegenerative diseases such as AD and HIV-associated neurocognitive disorder (HAND) [21–23]. Similarly, deficiencies of interferon α/β receptor (IFNAR) have been reported to cause reduced ALS disease progression in a SOD1(G93A) ALS model and decreased dopaminergic cell death in the MPTP model of PD [24, 25]. Taken together, this suggests that the IFN-1 may serve as a good therapeutic target for various NDs.
IFN-β was approved by FDA on 1993 as a treatment for MS due to its ability to decrease the rate of exacerbation of MS by 35% [26]. Considering that IFN-α and IFN-β are both type I interferons, target the same receptor (IFNAR) and are actively involved in regulation of immune response, it may be inferred that IFN-α may also govern various pathological pathways in various neurodegenerative diseases. Thus, this systematic review aimed to critically review the literature concerning the diverse role of IFN-α in the numerous pathological pathways of these neurodegenerative diseases.
METHODS
Literature search
An extensive literature search was done to identify all currently existing articles related to IFN-α and neurodegenerative diseases using three electronic databases: PubMed, Ovid Medline, and Scopus. The search terms employed in this systematic review are “Interferon-alpha”, “Neurodegenerative diseases”, “Alzheimer’s disease”, “Dementia”, “Parkinson disease”, “Huntington”, “Amyotrophic Lateral Sclerosis”, and “Multiple Sclerosis”. A title, abstract, and keyword search was performed using these search terms. The terms from these databases were linked via the Boolean operator “AND”. Articles were initially vetted through their titles and abstracts before focusing on full-text screening of relevant articles.
Literature selection
The literature selection was carried out independently by two researchers using the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) guidelines [27]. Duplicates were initially removed, and the results were then categorized into original and non-original research papers. The chosen studies were limited to original research papers as they provided better information for evaluation and analysis in this systematic review. The inclusion criteria employed were 1) articles reporting the association between IFN-α and neurodegenerative diseases, 2) peer-reviewed original research articles, and 3) articles available with full text. Exclusion criteria were 1) non-English papers, 2) duplicated articles, and 3) non-original research articles such as letters, conference papers, systematic reviews, meta-analysis, book chapters, symposiums, editorials, case reports. Both the researchers screened through titles and abstracts of the studies individually to identify the relevant studies.
Quality appraisal
Two tools were used to appraise the eminence of the selected relevant articles. The Quality Assessment Tool for Quantitative Studies by the Effective Public Health Practice Project (EPHPP) (Project, 1998) (Supplementary Table 1) was employed for clinical studies while the Systematic Review Centre for Laboratory Animal Experimentation Risk of Bias (SYRCLE RoB tool) (Supplementary Table 2) was used for preclinical animal studies. However, for cell-based studies, the quality analysis was not able to be carried out as there are no appraisal tools available. The whole methodological process was independently performed by two researchers, who reached consensus before the selection of the final number of articles for critical evaluation.
RESULTS
A total of 1,547 articles were retrieved from the initial search where 785 duplicate articles were excluded leaving 762 articles to be screened following the PRISMA guidelines (Fig. 2). Based on the inclusion and exclusion criteria, out of the 762 articles screened, 390 articles were omitted leaving 372 research articles that were vetted for relevancy towards the aim of this systematic review which was to examine the role of IFN-α in neurodegenerative diseases. Out of the 372 articles assessed for relevancy to aim, 295 studies were found irrelevant, leaving 77 research articles which was included in this review and critical appraised (Fig. 2). These studies consisted of 61 clinical studies and 16 preclinical studies, of which 12 articles were of animal studies and the other 4 preclinical studies were of mainly in vitro cells. The overall 77 articles were further segregated into 7 categories; 8 articles were on AD, 3 articles were on PD, 1 article was on HD, 3 articles were on ALS, 49 articles of multiple sclerosis, 11 articles were on HAND, and lastly 2 articles were on neurodegeneration in general. Table 1 summarizes the important findings from these articles.
Fig. 2
PRISMA flowchart showing article selection for systematic review.
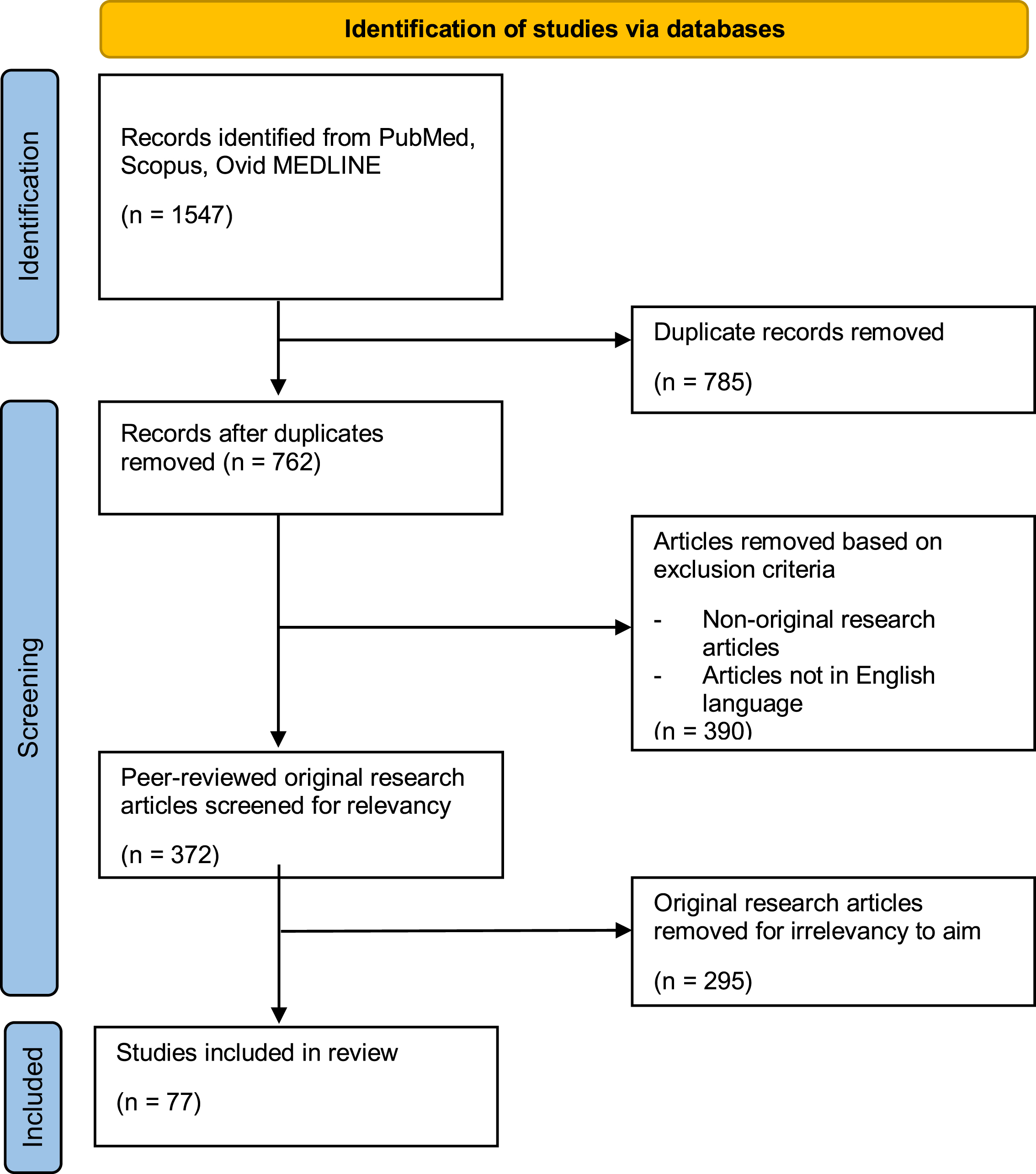
Table 1
Sample characteristics and important findings of selected clinical and preclinical studies.
| Type of study | Sample characteristics | Key findings on the role of IFN-α | References |
| Alzheimer’s Disease (AD) | |||
| Preclinical | Intervention group: MyD88–/– mice on C57Bl/6 background Control group: Wildtype C57Bl/6 mice Gender: Female Age = Unknown | •The IFN-α response to soluble amyloid in AD was mediated via the toll-like receptors. | [28] |
| Intervention group: APP/PS1 mice on a dual C57Bl/6 &C3H/Hej mice Control group: Age-matched littermate wild type control mice Gender: Unknown Age = 9–12 months | •Two-fold increase in IFNα in pre-frontal cortex in APP/PS1 brains compared with control brains •Type-1 interferons has a role in the pro-inflammatory response and neuronal cell death in AD •Blocking the type-1 IFN-α receptor 1 could limit the disease progression | [29] | |
| Clinical | Intervention group: N = 50, M = 7, F = 43 Age = 78.5±6.4 years Control group: N = 37, M = 7, F = 30 Age = 77.0±7.3 years | •There was no significant difference in the levels of serum IFN-α between the AD and control group. | [30] |
| AD group: N = 45 Gender: Unknown Age = 79.7±0.9 years MCI group: N = 34 Gender: Unknown Age = 76.6±1.4 years Control group: N = 28 Gender: Unknown Age = 80.4±2.0 years | •Plasma levels of IFN-α significantly increased in patients with MCI versus control group and patients with AD. | [31] | |
| AD group: N = 22, M = 10, F = 12 Age = 73±10 years Control group: N = 17, M = 5, F = 12 Age = 64±13 years | •The plasma concentration of IFN-α in AD patients were within normal range. | [32] | |
| AD group: N = 12, M = 6, F = 6 Age = 67–88 years Control group: N = 5, M = 4, F = 1 Age = 61–82 years | •A subset of reactive microglia in AD cortex stained positive for IFN-α. •Microglial aggregates in senile plaque were stained intensively. | [33] | |
| AD group: N = 39, M = 12, F = 27 Age = 57–99 years | •Most AD brains showed decreased mRNA levels of IFN-α while a small group of AD patients had increased levels in the hippocampus. | [34] | |
| AD group: N = 8 Gender: Unknown Age = 67–81 years Control group: N = 6 Gender: Unknown Age = 65–82 years | •In AD brains, white matter microglia were intensely labelled for IFN-α. | [35] | |
| Parkinson’s Disease (PD) | |||
| Clinical | PD group: N = 8, M = 2, F = 6 Age = 66.8 years Control group: N = 4, M = 1, F = 3 Age = 70.2 years | •The induction of MxA mRNA by IFN-α in PD patients is higher compared with that of the normal controls. | [36] |
| PD group: N = 20, M = 11, F = 9 Age = 63.3±5.0 years Control group: N = 19, M = 12, F = 8 Age = 61.1±3.2 years | •The production of IFN-α2 is not different from values in the control group. | [37] | |
| Intervention group: N = 23,647, M = 13,074, F = 10,573 Age = 50.8±11.2 years Control group: N = 23,647, M = 13,007, F = 10,640 Age = 50.8±11.3 years | •Treatment with pegylated IFN α-2a or IFN α-2b, with ribavirin (Peg IFN/RBV) results in a significantly lower cumulative incidence of parkinsonism in patients with chronic hepatitis C. •IFN-α treatment (PEG IFN/RBV) reduces PD risk in patients with chronic hepatitis C. | [38] | |
| Huntington’s Disease (HD) | |||
| Pre-clinical | Intervention group: Littermate transgenic HD minipigs Control group: Wild-type minipigs Gender: Unknown Age = 36 months | •Significant decline in CSF IFN-α at early pre-symptomatic time intervals of HD. •IFN-α as a potential biomarker in HD mutation carriers. | [39] |
| Amyotrophic Lateral Sclerosis (ALS) | |||
| Clinical | Intervention group: N = 5 Control group: N = 5 Gender: Unknown Age = Unknown | •Significantly reduced mean serum calcium level, osmolality, and a reduction in mean urinary magnesium excretion in IFN-α group. •IFN-α induces water and electrolyte imbalance in ALS individuals. Dose used: Continuous intravenous infusion of IFN-α, 100 x 106 IU/day during days 1-2 &200 x 106 IU/day during days 3–5. | [40] |
| Intervention group: N = 12 Gender: Unknown Age = 49.4±11.8 years Gender: Unknown Age = 52.3±8.1 years Control group: N = 3 | •Treatment with intravenous IFN-α infusion resulted in a reversible deterioration in logical verbal memory task, digit-span-backwards task, writing time as well as calculation ability. •IFN-α induces a reversible cognitive decline in ALS. Dose used: Continuous intravenous infusion of IFN-α, 100 x 106 IU/day during days 1-2 &200 x 106 IU/day during days 3–5. | [41] | |
| Intervention group: N = 10, M = 5, F = 5 Age = Unknown | •Treatment with intrathecal administration of IFN-α did not affect the rate of ALS deterioration but resulted in reversible EEG slowing and mental status changes. •IFN-α does not confer therapeutic benefits in ALS. Dose used: Intrathecal injection of partial purified native human IFN-α, 1 million units/week for 7–24 weeks. | [42] | |
| Neurodegeneration | |||
| Pre-Clinical | Human primary macrophages derived from CD14+ blood monocytes | •IFN-α suppressed the pro-inflammatory and tissue-destructive activities of IL-1. •IFN-α may play a homeostatic role in neurodegeneration. | [43] |
| Intervention group: GIFN-39 and GIFN-12 transgenic mice Control group: Wild-type littermates Gender: Unknown Age = Unknown | •Transgenic expression of IFN-α in mice CNS resulted in a progressive inflammatory encephalopathy, marked neurodegeneration, calcium mineralization and gliosis. •A number of mice were ataxic, had a terminal decline in functionality and were susceptible to seizures. •CNS IFN-α induces neurodegeneration by abnormal calcium mineralization and morphologic endoplasmic reticulum changes. | [44] | |
| HIV-associated Neurocognitive Disorder | |||
| Pre-clinical | Primary human astrocytes (no. 1800) and human microglia (no. 1900) types | •IFN-α increased microglia SAMHD1 mRNA and protein expression, and reduced miR-181a levels. •Interplay between IFN-α and SAMHD1 contributes to the pathogenesis of HAND. | [45] |
| Primary human monocytes | •IFN-α stimulates an increase in macrophage production of quinolinic acid. Dose used: Stimulation with IFN-α (10 IU/mL, 50 IU/mL) | [46] | |
| Human monocytes and HEK 293T (ATCC, Manassas, VA, USA) cells | •IFN-α treatment increased STAT1 phosphorylation, glutaminase mRNA as well as protein levels. •Stimulation of GLS1 promoter activity by IFN- α correlated to STAT1 phosphorylation. •IFN-α treatment may propagate neuronal excitotoxicity during HIV infection. | [47] | |
| Intervention group: B6.CB17-Prkdc/Szj (SCID) mice Control group: Wildtype B6.CB17-Prkdc/Szj (SCID) mice Gender: Male Age = 5 weeks | •B18R with a common combined antiretroviral therapy (cART) improve histopathological markers better than cART alone. •B18R reverses Objective Recognition Test behavioral abnormalities in HAND mice. •IFN-α-binding protein (B18R) reverses neurotoxicity in HAND mouse model. | [48] | |
| Intervention group: C57BL/6J mice Control group: Wildtype C57BL/6J mice Gender: Male Age = 4 weeks | •Intensity of IFN-α staining was increased in mice with HIV. •IFN-α levels in the brain correlated directly with working memory errors. | [49] | |
| Intervention group: B6.CB17 mice Control group: Wildtype B6.CB17 mice Gender: Male Age = 4 weeks | •HIVE mice with IFN-α neutralizing antibodies resulted in improved behavioral performance, with significant reductions in microgliosis and loss of dendritic arborization. •CNS IFN-α induces neurodegeneration. | [50] | |
| Clinical | Intervention (HIV) group: N = 15, M = 12, F = 3 Age = 49 years Control group: N = 2, M = 0, F = 2 Age = 54.5 years | •CSF IFN-α levels was significantly higher in HIV-infected participants compared to participants without HIV. •Negative correlation between CSF IFN-α with executive function and processing speed. | [51] |
| Intervention (HIV) group: N = 46, M > F Age = 50% <45 years Control group: N = 54 Age = 44% <50 years | •Alterations in CSF metabolites correlated with plasma markers of systemic inflammation like IFN-α. | [52] | |
| Intervention (HIV) group: N = 22, M = 19, F = 3, Age = 39 years (median) | •The level of HIV-1 RNA correlates with the level of IFN-α in CSF. | [53] | |
| Intervention (HIV) group: N = 35, M = 35, F = 0 Control group: N = 8, M = 8, F = 0 Age = 50.2±7.5 years | •Association between high IFN-α-driven peripheral monocyte transcripts and lower levels of N-acetylaspartatein in frontal white matter. | [54] | |
| Intervention (HIV with dementia) group: N = 21 Intervention (HIV without dementia) group: N = 23Control group: N = 48 | •HIV-infected individuals with dementia have significantly higher levels of CSF IFN-α compared to HIV-infected individuals without dementia &HIV-negative controls. | [55] | |
| Multiple Sclerosis (MS) | |||
| Preclinical | Intervention group: C57BL/6 mice Control group: Wildtype C57BL/6 mice Gender: Female Age = 8 weeks N = 16 | •Peg-IFN α-2a attenuates the severity of experimental •autoimmune encephalomyelitis (EAE) and delays its onset. •IFN-α reduces inflammation. | [56] |
| Intervention group: SJL/J mice Control group: Wildtype SJL/J mice Gender: Female Age = 7–10 weeks | •Oral administration of human or murine IFN-α reduces relapses in EAE &reduces adoptive transfer in EAE. | [57] | |
| Intervention group: Marmoset monkeys (C. jacchus) Control group: Wildtype Marmoset monkeys (C. jacchus), M = 3, F = 3 Age = 3–5 | •IFN-α, IFN-γ, TNF-α, IL-1, IL-2, IL-4 and IL-12 were expressed by mononuclear cells in brain infiltrate in EAE monkey. | [58] | |
| Intervention group: SJL/J mice Control group: Wildtype SJL/J mice Gender: Female Age = 5-6 weeks | •IFN-β reduced myelin-specific CD4 T cell responses and demyelinating process but not IFN-α. | [59] | |
| Intervention group: C57BL/6 mice Control group: Wildtype C57BL/6 mice Gender: Female Age = 6–8 weeks | •Low dose IFN-α protected mice from developing EAE. | [60] | |
| Clinical | MS group: N = 41, M = 8, F = 33 NMO group: N = 36, M = 11, F = 25 | •IFN-α was less detectable in serum from MS patients (2/41), than in the NMO group (9/36). | [61] |
| IFN group: N = 60, M = 25, F = 35 Age = 19–50 years TF group: N = 61, M = 25, F = 36 Age = 20–49 years Placebo group: N = 61, M = 20, F = 41 Age = 18–48 years | •IFN-α did not have any significant effects on the rate of progression of MS compared to controls. | [62] | |
| MS group: N = 12, M = 7, F = 5 Control group: N = 9, M = 2, F = 7 Age = Unknown | •After stimulation of TLR-9, IFN-α production by pDCs was significantly lower in MS compared to controls. | [63] | |
| MS group: N = 51, M = 18, F = 33 Age = 28–71 years Unaffected family members: N = 75, M = 41, F = 34 Age = 27–87 years Control group: N = 141, M = 63, F = 78 | •No genetic linkage between multiple sclerosis and the IFN-α & -β locus. | [64] | |
| MS group: N = 20, M = 9, F = 11 Age = Unknown | •Recombinant IFN-α therapy associated with reduction of IFN-γ &TNF-α production by peripheral blood lymphocytes. •IgG level was systemically increased. •Serum β2 microglobulin level increased as well. | [65] | |
| MS group: N = 6 Age = 38–74 years OND patient group: N = 3 Age = 11–74 years Misc patient group: N = 9 Age = 16–87 years | •Expression of IFN-α was detected in most patients with MS. •However, it was also detectable in patients with other illnesses. | [66] | |
| MS group: N = 10 Control group: N = 18 Age = Unknown | •In MS patients who ingested 10000 or 30000 units IFN-α RRMS, there was a significant decrease in Con A-mediated proliferation and sICAM-I, a marker for disease activity in MS. | [67] | |
| RRMS patient group: N = 33, M = 5, F = 28 Mean age = 41.5±8.4 years | •Effect on lesions were not significantly seen. •A possible treatment effect may be suggested in the 10,000 IU group. | [68] | |
| MS Group: N = 24 Age = 18–55 years | •Oral IFN-α represses TNF-α mRNA in MS. | [69] | |
| RRMS patients: N = 41, M = 10, F = 31 Mean age = 43.5 years Control group: N = 37, M = 9, F = 28 Mean age = 42.9 years | •In response to IFN-α stimulation, there was an extensive upregulation in levels of phosphorylated STAT proteins in MS patients. •It was found across all cell subsets but especially within the NK cell population. | [70] | |
| MS group: N = 23, M = 10, F = 13
Age = 21–48 years | •Elevated IFN-α levels were found in the CSF of stable phase MS patients when compared to those in active phase. •MS patients and controls showed no significant serum level differences. •MS patients in remission has a significant correlation in CSF IFN-α levels. | [71] | |
| MS group: N = 10, M = 4, F = 6 Age = 18–42 years Healthy donor group: N = 6, M = 2, F = 4 Age = 18–48 years | •Non-purified auto-IFN-α and auto-IFN-γ did not stimulate NK activity of peripheral blood lymphocytes in healthy donors. | [72] | |
| MS group: N = 15, M = 6, F = 9 Mean age = 42.2 years | •The priming effects of IFN preparations were not related to IFN-α production by peripheral blood lymphocytes. | [73] | |
| IFN-β-untreated MS group: N = 236, M = 89, F = 147 Mean age = 42.3±9.8 years Healthy adult group: N = 25, M = 7, F = 18 Mean age = 39.8 years | •IFN-α/MxA system was activated in MS patients. | [74] | |
| MS group: N = 48, M = 14, F = 34 Age = 18–68 years Control group: N = 28, M = 10, F = 18 Age = 18–68 years | •IFN-α– secreting pDCs were found to be 2-fold lower inpatients with MS than in HSs. •There was a reduction in the production of IFN-α by pDCs in the cells taken from MS patients, regardless of the phase of the disease and of the donor’s gender. | [75] | |
| MS Group: N = 71 Mean age = 34 years Untreated MS group: N = 160 Mean age = 36 years Healthy control group: N = 54 Mean age = 35 years | •IFN-α- and IFN-β-specific response programs vary between MS patients. •No conclusion could be made from this study. | [76] | |
| MS group: N = 16, M = 5, F = 11 Age = 25–60 years Control group: N = 28, M = 9, F = 19 Age = 26–60 years | •There was no significant difference in production of IFN-α after stimulated by TLR ligand. •However, there was a lower steady-state release of IFN-α in MS patients. | [77] | |
| MS group: N = 8, M = 3, F = 5 Age = 26–56 years Control group: N = 8 Age = 24–48 years | •IFN-α levels increased after remission of a relapse. | [78] | |
| MS group: N = 12, M = 5, F = 7 Age = 18–46 years Control group: N = 8, M = 4, F = 4 Age = 18–50 years | •High-dose systemic recombinant IFN-α. •Reduced exacerbation rate and MRI signs of disease activity. •Decreased peripheral blood lymphocyte IFN-γ production. •Increased the percent of CD8+ high CD 1 lb+low lymphocytes (T suppressor-effector subset) in CSF. | [79] | |
| Treatment MS group: N = 12, M = 5, F = 7 Age = 18–46 years Placebo MS group: N = 8, M = 4, F = 4 Age = 29–57 years | •Both therapeutic and adverse effects of recombinant IFN-α were temporary. | [80] | |
| Treatment MS group: N = 12, M = 5, F = 7 Age = 18–46 years Placebo MS group: N = 8, M = 4, F = 4 Age = 29–57 years | •Recombinant IFN-α therapy was associated with fatigue, fever, depression, raised liver enzyme, lymphophenia, and autoantibodies. | [81] | |
| MS group: N = 55, M = 20, F = 35 Mean age = 47 years Control group: N = 150, M = 45, F = 105 Mean age = 41 years | •Serum IFN-α activity was lower in therapy-naïve MS compared to healthy and SLE patients. | [4, 82] | |
| MS group: N = 18, M = 2, F = 16 Control group: N = 10 Age = 21–57 years | •Mitogen stimulated peripheral blood mononuclear cells from MS patients are resistant to inhibitory effect of IFN-α and IL-10. | [83] | |
| MS group: N = 42, M = 13, F = 29 Control group: N = 10 Age = 23–37 years | •Intermittent presence of serum IFN- α/β in MS patients were not associated with disease activity or viral infection. | [84] | |
| MS group: N = 15 | |||
| Control group: N = 15 Mean age = 36.2±10.5 years | •IFN-α production from peripheral blood mononuclear cells was significantly lower from MS patients after induced by CpG DNA. | [85] | |
| MS group: N = 100 Mean age = 33.5 years | •IFN-α increased activity of NK cells against K562 myeloid target cell. •However, there was a decline in NK activity when IFN-α were given for a longer duration. | [86] | |
| Healthy peripheral blood mononuclear cells donors Age = 25–35 years | •The study investigated the potency between IFN-α, IFN-β and IFN-γ. •However, the results vary between participants. | [87] | |
| RRMS patients: N = 12 Mean age = 45 years Control group: N = 18 Mean age = 43 years | •IFN-α or EBV increased expression of HLA-E. | [88] | |
| MS group: N = 24 Age = 25–45 years | •IFN-α therapy caused a reduction in production of IFN. | [89] | |
| MS group: N = 6 Age = 33–51 years | •Disease activity score worsened after IFN-α-2b therapy. | [90] | |
| MS group: N = 303, M = 67, F = 236 Age = 22–81 years | •Participants with MS protective HLA alleles had a higher level of IFNAR2 but with a lower response to IFN. | [91] | |
| MS group: N = 43 OND group: N = 13 | •MS patients had fewer IFN-α producing cells than OND or normal controls. | [92] | |
| RRMS patients: N = 32, M = 12, F = 20 Mean age = 42±9 years Control group: N = 33, M = 14, F = 19 Mean age = 38±12 years | •IFN-α production from pDCs was significantly lower from MS patients after induced by TLR7. | [93] | |
| MS group: N = 97 Age = 21–48 years | •IFN-α injection decreased the number of MRI active lesson without affecting the disease progression and activity clinically. | [94] | |
| MS group: N = 21 Age = Unknown | •The levels of sCD35 decreased while TCC and C3bc increased in MS patients treated with IFN-α2a. | [95] | |
| MS group: N = 16 Mean age = 43±8 years Control group: N = 16 Mean age = 28±8 years | •Recombinant IFN-α increased suppressor function in MS patients. | [96] | |
| MS group: N = 327 Control group: N = 698 Mean age = 50 years | •Truncated interferon- α 17 allele was not associated with development of MS. | [97] | |
| MS group: N = 17 Age = Unknown | •Mechanism of action of IFN-α and IFN-β showed to be similar to each other as there was no significant differences in the modulation of gene. | [98] | |
| MS group: N = 49 Age = 18–70 years | •IFN-α reduced the rate of relapse by 80%. | [99] | |
| MS group: N = 76, M = 23, F = 53 Age = 18–59 years Control group: N = 34, M = 12, F = 22 Age = 17–66 years | •The production of IFN-α is lower in MS patients. •However, production of IFN-α was not found to be correlated with the activity of NK cells. | [100] | |
| MS group: N = 11 Age = 32–73 years OND group: N = 3 Age = 12–74 years Control group: N = 3 Age = 25–75 years | •IFN-α was predominately expressed on Ia-positive macrophages and not on astrocytes at the site of MS lesions. | [101] | |
| MS group: N = 11 Age = 11–73 years | •IFN-α was mainly detected on macrophages in active chronic MS lesions but not in control groups or silent chronic plaques. | [102] | |
| RRMS patient group: N = 97 Age = Unknown | •IFN-α induced a significant decrease in lymphocyte concentration and a significant increase in IgG. •However, there was no significant changes in immune parameters and disease scoring. | [103] | |
| MS group: N = 21, M = 11, F = 10 | •Stable MS patients had a lower production of IFN-α and amount of NK cells compared to control but higher as compared to active MS patients. | [104] | |
| Age = 20–57 years | |||
AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; CNS, central nervous system; CSF, cerebrospinal fluid; EAE, experimental autoimmune encephalitis; EBV, Epstein-Barr virus; F, female; HD, Huntington’s disease; HIVE, HIV encephalitis; Ig, immunoglobulin; IL, interleukin; IFN-α, interferon-alpha; IFN-β, interferon beta; IFN-γ, interferon gamma; IFNAR2, Interferon-alpha/beta receptor; M, male; MS, multiple sclerosis; N, sample size; NMO, neuromyelitis optica; NK cell, natural killer cell; OND, other neurological disease; pDCs, plasmacytoid dendritic cells; PD, Parkinson’s disease; RRMS, relapse remitting multiple sclerosis; SLE, systemic lupus erythematosus; TLR, toll-like receptor; TNF-α, tumor necrosis factor alpha.
Overview of selected study characteristics
For the clinical studies, most patients were recruited within the age group of 30–90 years old and there was no gender biasness observed. However, other demographic factors like ethnicity were not highlighted in these studies. Additionally, most of the clinical studies were of either controlled clinical trial or case control studies, that investigated both the role and levels of IFN-α as well as the therapeutic potential of IFN-α in patients with neurodegenerative diseases. The quality appraisal for the clinical studies suggested that most of the studies were rated as strong in quality where the sample sizes remained within the statistical power and most of the studies had a low patient withdrawal rate (Supplementary Table 1). For the preclinical studies, the rodent model were predominantly used with a slight variation in the age (adult and aged) and strain (C57Bl/6, APP/PS1, C3H/Hej, B6.CB17) employed. Contrastingly to the clinical studies, there seems to be a gender biasness in the preclinical studies as male rodents were largely employed. The preclinical studies also investigated the pathological role of IFN-α and its therapeutic potential in relation to the various neurodegenerative diseases which could not have been investigated in the clinical setting. Based on the study quality assessment, most of the preclinical studies appeared to be unbiased as the sample sizes fall within the statistical power and selective reporting of results was not detected (Supplementary Table 2).
DISCUSSION
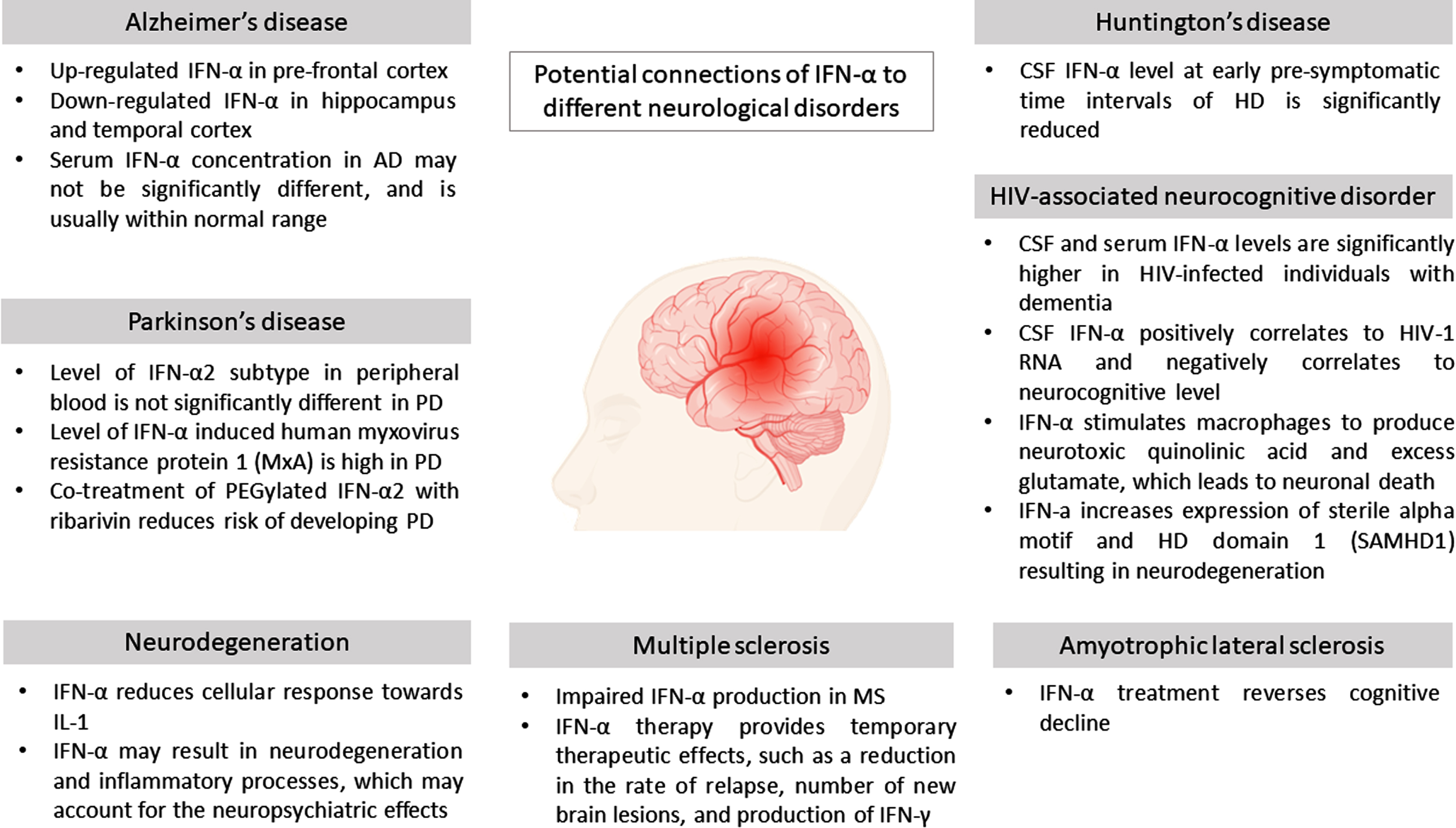
Based on Table 1, it is evident that IFN-α has a potential deleterious role in most NDs which aggravates the inflammatory processes. However, IFN-α, may also pose a protective effect post-treatment in several other NDs, similar to IFN-β which displays this dual opposing effects. The potential affiliations of IFN-α to NDs are summarized in Fig. 3.
Fig. 3
Potential connections of IFN-α to different neurological disorders.
Alzheimer’s disease
AD is a neurodegenerative disease characterized predominantly by gradual cognitive impairments and behavioral changes that may significantly disrupt everyday functioning [2]. Based on Table 1, there were two preclinical and six clinical studies investigating the role of IFN-α in the context of AD.
Several postmortem studies have reported an upregulation of IFN-α in the brain tissues of AD patients [29, 35]. Similarly, Akiyama et al. reported positive staining for IFN-α in the reactive microglia of a subset of AD samples, with greater intensity in staining of microglial aggregates in senile plaques [33]. Considering the neuropsychiatric effects associated with IFN-α, its presence in the AD brain tissue may help explain the cognitive decline commonly observed in individuals with AD. In a separate study, it was found that though there was an increase in the hippocampal mRNA expression of IFN-α in a small group of AD patients, most AD patients exhibited a decrease in IFN-α mRNA in the hippocampus and temporal cortex instead [34]. These differences may represent the different clinical stages of AD in the patient sample of this study as well as when compared with the previous study sample. It may also be possible that different brain regions respond differently throughout the course of AD, as the inflammatory process may be evolving across these regions with time.
Viral infections have also been implicated in the pathogenesis of AD [105, 106] through the impairment of an individual’s innate anti-microbial gene expression profile or genetic make-up. For instance, APOE ɛ4 allele AD carriers and those with rs6598008 polymorphism of IRF7 have been associated with decreased hippocampal IFN-α mRNA expression and an increase risk towards AD [34]. Thus, having an efficient immune system may be pivotal in attenuating AD neurodegeneration, but the role of IFN-α in this relationship requires further investigation. Although there were considerable differences in the level of IFN-α expression in the brain tissue of individuals with AD compared to that observed in the brain tissue of healthy individuals, the serum concentration of IFN-α may not be significantly different between them [30]. In fact, Singh et al. reported that the serum concentration of IFN-α in AD patients were usually within normal range, and thus were not appropriate biomarkers for AD diagnosis [32]. Studies comparing AD with mild cognitive impairment (MCI) showed that the serum concentration of IFN-α was significantly higher in patients with MCI when compared to that observed in patients with AD [31], suggesting that the IFN-α may have greater systemic inflammation association compared to AD. Taken together, these evidences suggested that even though IFN-α may not serve as a good diagnosis biomarker for AD, these findings corroborate that amyloidosis does prompt type-I IFN-mediated neuroinflammation which in turn further coerces neurodegeneration and contributes to AD progression. However, there seems to still be contrasting results also reported in terms of the exact role of IFN-α in relieving neuroinflammation. Thus, it still remains inconclusive to warrant the effects of IFN-α and more research will definitely be crucial to further comprehend the exact role of IFN-α. Comprehensive understanding of the neuroinflammatory as well as the immunoregulatory processes in AD will aid in the development of anti-neuroinflammatory approaches, which may serve as a novel potential therapeutic target for the management/treatment of AD.
Parkinson’s disease
PD is an age-related neurodegenerative condition that affects predominately dopamine-producing (“dopaminergic”) neurons in the substantia nigra [107]. PD may eventually lead to the clinical manifestations of bradykinesia, resting tremor, muscle rigidity, and abnormal posture and pace [108]. Table 1 suggests that there has not been any preclinical studies performed on PD disease in relation to IFN-α.
One study found that the level of IFN-α2 in the peripheral blood of PD patients was comparable to that of the control group [37]. IFN- α2 was classified as one the subtypes of IFN-α [2, 15]. While there was no significant difference in the level of IFN-α2 in peripheral blood of individuals with PD versus that of the healthy controls, the level of other subtypes of IFN-α were not investigated. However, given that the pathology in PD mainly stemmed from CNS (i.e., loss of dopaminergic neurons in the substantia niagra), there possibly may be raised levels of IFN-α in the brain tissue of individuals with PD as noted in other neurodegenerative diseases. However, this was not investigated in the current studies and future studies should be performed to further elucidate the role of CNS IFN-α in PD.
MxA protein, an interferon-induced protein, is able to inhibit influenza A viral infection [109]. Studies have reported expression of IFN-α in some microglial cells in the white matter. Additionally, MxA protein may be associated with the Lewy bodies in neurons of PD patients [35]. Interestingly, Kawaguchi et al. reported that the relative level of MxA mRNA in the peripheral blood mononuclear cells (PBMCs) of patients with PD was lower when compared to that of controls [36]. However, when PBMCs were incubated with IFN-α, the relative level of MxA mRNA in patients with PD increased equally to that of the controls, suggesting a higher induction of MxA mRNA by IFN-α in patients with PD. This in turn indicate that IFN-α increases MxA mRNA among the PD patients.
Therapeutically, there may be a role for IFN-α in the prevention of PD in patients with chronic hepatitis C as well. Treatment of chronic hepatitis C with pegylated IFN α-2a or pegylated IFN α-2b, plus ribavirin (Peg IFN/RBV) was found to reduce the risk of developing PD [38]. Similarly, results from a cohort study of 188,152 patients with hepatitis C infection showed that patients treated with Peg IFN/RBV had lower incidence density and risk of developing PD when compared to the untreated group [110]. However, this therapeutic benefit of IFN-α was only within the context of individuals with chronic hepatitis C. On the contrary, a case report from 2002 had highlighted the development of severe PD in a 79-year-old male receiving IFN-α for chronic myeloid leukemia [111]. This case showed that the parkinsonism resolved after termination of IFN-α treatment, which suggested that IFN-α may contribute to the pathogenesis of neurodegenerative disease, particularly PD. Considering the potential PD-aggravating side effects of extracellular IFN-α administration, further investigations should be performed to assess the safety of IFN-α treatment/intervention in reducing the risk of PD, even in patients with chronic hepatitis C.
Huntington’s disease
HD is a progressive neurodegenerative disorder with genetic origins and can be characterized by the gradual development of involuntary muscle movements. These include uncontrollable, irregular movements that may be rapid and jerky (chorea), as well as the development of athetosis, which is seen as a slow, writhing involuntary movement. HD may be transmitted as an autosomal dominant trait caused by mutations of a gene known as huntingtin (HTT), which was shown to be located on the short arm of chromosome 4 [107]. HD may occur due to the expansion of polyglutamine (polyQ) which were present within the HTT protein that leads to a defective huntingtin protein being produced. This defective protein may further misfold and accrues within neurons forming aggregates, thus ultimately affecting normal cellular function [112]. Based on Table 1, only one preclinical study had investigated the role of IFN-α in HD disease. This article reported that the littermate transgenic HD model displayed a significant decline in CSF IFN-α at early pre-symptomatic time intervals of HD compared to wild-type minipigs [5, 6, 39]. Cellular changes in HD may precede the observable clinical symptoms by decades [113]. Thus, this suggest that there may be a role for CSF IFN-α as a potential early biomarker for HD mutation carriers. However, whether symptomatic HD patients may display an increase in IFN-α in the CSF should also be investigated in future studies, so that IFN-α may serve as a therapeutic target of HD intervention.
Amyotrophic lateral sclerosis
ALS, or Lou Gehrig’s disease, is a progressive degeneration of neurons in the motor pathways from the cortex to the anterior horn of the spinal cord. To date there is no available treatment that is effective for ALS due to a poor understanding of its underlying pathological mechanisms.
Though the exact cause of ALS is unknown, patients with herpes simplex virus infection seem to be at higher risk of developing ALS [114]. As interferons may be released by host cells in response to viral infections, there may be a therapeutic role for IFN-α in ALS. However, in a clinical study involving 10 ALS patients, intrathecal administration of native IFN-α at weekly doses of 1 million units for 7 to 24 weeks did not affect the rate of ALS deterioration [42]. This shows that intrathecal IFN-α at the dosage used was not effective. In a placebo-controlled study involving ALS patients, cognitive alteration was observed after 5 days of high-dose IFN-α administration [41]. The treatment group received 100 million IU/day during the first two days and 200 million IU during the next three days. However, the cognitive decline reversed 3 days after cessation of IFN-α infusion. Mora et al. reported a similar pattern of reversible cognitive decline in ALS patients receiving 1 million U IFN-α for 7 to 24 weeks. Neurocognitive impairment may suggest an underlying neurodegeneration induced by IFN-α infusion. However, CSF somatostatin (marker of brain damage) was not raised [42]. In a separate study, patients receiving IFN-α 100 million IU/day during the first two days and 200 million IU on days 3 to 5 demonstrated a significant decrease in serum calcium, serum osmolality and daily urinary excretion of magnesium when compared to the placebo group [40]. The urinary excretion of potassium and calcium were also decreased; these were however not statistically significant. Though the electrolyte disturbances resolved 2 to 4 days after cessation of IFN-α infusion, these parameters should ideally be monitored when administering high-dose IFN-α to patients with ALS. Nevertheless, given that this evidence of IFN-α in ALS was performed more than three decades ago, recent understanding on the complexity of the inflammatory pathways and newer studies manipulating the dosage, administration route and drug combination may provide better insight into IFN-α therapeutic role in the context of ALS.
Multiple sclerosis
Experimental autoimmune encephalomyelitis (EAE) is a commonly used mouse model for MS [115]. Three out of 4 pre-clinical EAE studies showed that IFN-α was protective against EAE [56, 57, 59, 60]. IFN-α may decrease the rate of relapse, immune cell infiltration, demyelination of the spinal cord, and IFN-γ production in EAE mouse.
In clinical studies, most of the MS studies have shown a therapeutic effect of IFN-α therapy. IFN-α therapy was associated with a reduction in numbers of patients with clinical exacerbation, numbers of new MRI brain lesions and baseline IFN-γ production [79]. Secretions of IL-2 and TGF-B were also lower in MS patients treated with IFN-α [57]. Activity of NK cells increased dramatically after IFN-α therapy [86]. Furthermore, Squillacote et al. showed a dose-response effect in IFN-α therapy. IFN-α was shown to be effective in decreasing the rate of relapse and stabilizing patients with MS. Those who were treated with a longer duration and a higher dosage also had a greater reduction in Extended Disability Status Scale (EDSS). In addition, Guillen et al. found that IFN-α was able to enhance the effect of cyclosporin in treating MS [83]. Hence, IFN-α was shown to be beneficial in MS.
All together there have been three types of administration methods that have been used in clinical studies which were oral, subcutaneous injection, and intramuscular injection. More studies with subcutaneous administrated IFN-α failed to show a beneficial effect in MS patients compared to the other two administration methods [62, 90, 94]. However, there were some potential factors that might lead to negative results in these studies such as small sample size and detection methodology biases. Thus, it may not be possible to conclude the therapeutic effect of subcutaneous IFN-α at this point.
Multiple adverse effects were detected from most of the clinical studies. IFN-α therapy was found to be associated with fatigue, fever, depression, raised liver enzyme, lymphopenia, and autoantibodies formation [81]. There was a significant number of participants who withdrew from the studies due to the intolerable adverse effects of IFN-α therapy. Brod et al. showed that oral IFN-α was associated with lesser adverse effects and had similar therapeutic effects to parenteral IFN-α. However, there was a decrease in the number of enhancements in MS patients treated with 10000 IU IFN-α, but not with 30000 IU IFN-α. Brod et al. suspected that pharmacologic tolerance may have been induced in patients treated with higher dose of IFN-α. They also suggested that the mechanism of action of ingested IFN-α might differ from parenteral IFN-α. The therapeutic effects of ingested IFN-α may be associated with lymphoid tissues from Peyer’s patches. However, more studies may be needed in order to prove oral IFN-α to be beneficial for MS patients [69].
All studies have shown that the therapeutic effect of IFN-α was temporary. Improvement in EDSS score and immunological values reverted 6 months after the cessation of the therapy [65, 80, 81]. Moreover, MS patients were found to be more resistant towards IFN-α after treatment. The responsiveness of NK cells towards recombinant IFN-α2 decreased significantly overtime [86]. Kamin-Lewis et al. showed a decrease in IFN-α production in MS patients after treated with IFN therapy. Some studies suggested that patients should be on chronic high dose IFN-α therapy to have a longer-lasting effect, but more studies were needed to investigate the long-term efficacy of IFN-α therapy. In conclusion, multiple evidence has supported the presence of impaired IFN-α production in MS. There was also an increase in response of immune cells observed when stimulated by IFN-α. Studies have also found an association between levels of IFN-α and disease activity of MS. However, more studies may be needed to further discover the underlying mechanism of this association.
HIV-associated neurocognitive disorder
HAND is defined as a range of neurocognitive dysfunction associated with HIV infections. HAND has continued to be a major health concern amongst HIV-infected individuals even with the availability of combination anti-retroviral therapy (ART) [116]. Rho et al. reported that compared to HIV-infected individuals without dementia and HIV-negative controls, HIV-infected individuals with dementia had a significantly higher level of CSF IFN-α [55]. This was in line with later findings suggesting a correlation between HIV-1 RNA and the level of CSF IFN-α [53]. Preclinical studies on C57BL/6J SCID mice injected with HIV-infected macrophages resulted in working memory errors, which was correlated directly to the increased level of CSF IFN-α [49]. Similarly, a study examining the CSF IFN-α in ambulatory HIV-infected individuals found that the level of CSF IFN-α negatively correlated with measures of neurocognitive function [51]. One potential mechanism that have been suggested was that elevated IFN-α levels in the CSF of HIV-infected individuals may stimulate an increase in quinolinic acid production by activated macrophages [46]. Quinolinic acid have shown to induce neuronal death in human brain culture [117]. In support, studies involving animal models and cell culture systems have also observed that quinolinic acid was indeed a neurotoxin[118–120].
In more recent studies, IFN-α has been linked to an increase in SAMHD1 mRNA and protein expression, and to a reduction in miR-181a levels, especially in microglia [6, 7, 45]. SAMHD1 is a host restriction factor for HIV-1. Mutations in SAMHD1 have been known to result in Aicardi–Goutières syndrome (AGS), a rare neurodegenerative disease also characterized by elevated CSF IFN-α [121]. Given the similarities between AGS and HAND, the interplay between CSF IFN-α and SAMHD1 may contribute to the neurodegeneration seen in HAND. Interestingly, IFN-α was not only found to be elevated in the CSF but have also found to be increased in the serum of HIV-infected individuals [52, 122], suggesting IFN-α as an easily accessible biomarker for HAND in HIV patients.
IFN-α has also been shown to activate the binding of STAT1 to the glutaminase 1 promoter in HIV-1-infected macrophages, which resulted in the upregulation of glutaminase and a subsequent rise in glutamate production [47]. Glutamate may play a pivotal role in everyday physiology but when in excess, extracellular glutamate has been implicated in neuronal death [123]. Other possible mechanisms as to how IFN-α may contribute to the pathogenesis of HAND included altered brain waste disposal and chronic peripheral immune activation [52, 54]. Interestingly, a preclinical study on HIV encephalitis mice showed that treatment with IFN-α neutralizing antibodies resulted in an improved cognitive performance, with improved pathological markers [50]. Quite similarly, Koneru et al. demonstrated that IFN-α-induced neurotoxicity observed in HAND mouse model was reversible with B18R, an IFN-α-binding protein [48]. Moreover, this study showed that treatment with combined ART plus B18R improved histopathological markers better than treatment with combined ART alone. These results suggest a potential role for IFN-α-binding/inhibiting therapeutics in the treatment and management of HAND.
Neurodegeneration
Neurodegeneration in general (non-disease specific) have also been associated with IFN-α. Though IFN-α in the CNS may confer protection against neutropenia viral infections, its production may result in neurodegeneration and inflammatory processes [44]. This may account for the neuropsychiatric effects associated with IFN-α; the most prominent of which are depression and neurocognitive impairment [21–23]. On the contrary, IFN-α has been shown to reduce the cellular response towards interleukin-1 [43], a potent inflammatory cytokine that has been implicated in the pathogenesis of various diseases [124]. Thus, there may still be some contradictions to the role of IFN-α in neurodegeneration, which may require more understanding prior to the investigation of its therapeutic potential for neurodegenerative diseases.
Conclusion
The studies selected and elucidated in this current systematic review have showed that IFN-α may play a deleterious role in neurodegenerative diseases through its potential association with the inflammatory processes resulting in mainly neurocognitive impairments. IFN-α may display its neurotoxic function via various mechanisms such as abnormal calcium mineralization, activation of STAT1-dependent mechanisms, and increased quinolinic acid production. However, the exact role IFN-α in neurodegenerative diseases remains controversial due to the contrasting findings across different studies. Moreover, its role in diseases like PD and ALS have yet to be determine due to a lack of more recent evidence, thereby contributing to the variability in the role of IFN-α in these diseases. Future investigations should be conducted, with greater emphasis on 1) determining the levels of IFN-α in serum, CSF, and brain tissue across all neurodegenerative diseases, and 2) understanding the role of IFN-α inhibitors as potential therapeutic options. With this, the role played by IFN-α in neurodegenerative diseases could be delineated. This review suggested that more insightful and updated evidence of IFN-α may provide a new therapeutic avenue for neurodegenerative diseases.
ACKNOWLEDGMENTS
The authors express gratitude to the School of Science and Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia for supporting this research.
FUNDING
The project is funded by the Malaysia Ministry of Higher Education, Fundamental Research Grant Scheme (FRGS/1/2020/SKK0/MUSM/02/6) and Early Career Research (ECR) Grant (Grant number ECR-000023), Jeffrey Cheah School of Medicine and Health Sciences, Monash University Malaysia.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-221081.
REFERENCES
[1] | Kouli A , Torsney KM , Kuan WL ((2018) ) Parkinson’s disease: Etiology, neuropathology, and pathogenesis. s Disease: Pathogenesis and Clinical Aspects, Stoker TB, Greenland JC, eds. Codon Publications, Brisbane. |
[2] | Kumar A , Sidhu J , Goyal A , Tsao JW ((2021) ) Alzheimer disease. In StatPearls Publishing, Treasure Island. StatPearls. |
[3] | Brookmeyer R , Abdalla N , Kawas CH , Corrada MM ((2018) ) Forecasting the prevalence of preclinical and clinical Alzheimer’s disease in the United States. Alzheimers Dement 14: , 121–129. |
[4] | Kowal SL , Dall TM , Chakrabarti R , Storm MV , Jain A ((2013) ) The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord 28: , 311–318. |
[5] | Tsolaki M , Paraskevi S , Degleris N , Karamavrou S ((2009) ) Attitudes and perceptions regarding Alzheimer’s diseasein Greece. Am J Alzheimers Dis Other Demen 24: , 21–26. |
[6] | Fereshtehnejad SM , Vosoughi K , Heydarpour P , Sepanlou SG , Farzadfar F , Tehrani-Banihashemi A , Malekzadeh R , Sahraian MA , Vollset SE , Naghavi M , Vos T , Feigin V , Murray C , Mokdad AH , Moradi-Lakeh M Global Burden of Disease Study 2016 Eastern Mediterranean Region Collaborators - Neurological Diseases Section ((2019) ) Burden of neurodegenerative diseases in the Eastern Mediterranean Region, 1990–2016: Findings from the Global Burden of Disease Study 2016. Eur J Neurol 26: , 1252–1265. |
[7] | Dooley M , Lamb HM ((2000) ) Donepezil: A review of its use in Alzheimer’s disease. Drugs Aging 16: , 199–226. |
[8] | Fernandez HH , Chen JJ ((2007) ) Monoamine oxidase-B inhibition in the treatment of Parkinson’s disease.174S-185S. Pharmacotherapy 27: . |
[9] | Salat D , Tolosa E ((2013) ) Levodopa in the treatment of Parkinson’s disease: Current status and new developments. J Parkinsons Dis 3: , 255–269. |
[10] | Jellinger KA ((2010) ) mechanisms of neurodegeneration: A critical update. J Cell Mol Med 14: , 457–487 Basic. |
[11] | Gan L , Cookson MR , Petrucelli L , La Spada AR ((2018) ) Converging pathways in neurodegeneration, from genetics to mechanisms. Nat Neurosci 21: , 1300–1309. |
[12] | Guzman-Martinez L , Maccioni RB , Andrade V , Navarrete LP , Pastor MG , Ramos-Escobar N ((2019) ) Neuroinflammation as a common feature of neurodegenerative disorders. Front Pharmacol 10: , 1008. |
[13] | Lin FC , Young HA ((2014) ) Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev 25: , 369–376. |
[14] | Samuel CE ((2001) ) Antiviral actions of interferons. Clin Microbiol Rev 14: , 778–809. |
[15] | de Andrea M , Ravera R , Gioia D , Gariglio M , Landolfo S ((2002) ) The interferon system: An overview. Eur JPaediatr Neurol 6: (Suppl A), A41–A46; discussion A55-48. |
[16] | Schreiber G , Piehler J ((2015) ) The molecular basis for functional plasticity in type I interferon signaling. Trends Immunol 36: , 139–149. |
[17] | Nicolas CS , Amici M , Bortolotto ZA , Doherty A , Csaba Z , Fafouri A , Dournaud P , Gressens P , Collingridge GL , Peineau S ((2013) ) The role of JAK-STAT signaling within the CNS. JAKSTAT 2: , e22925. |
[18] | Owens T , Khorooshi R , Wlodarczyk A , Asgari N ((2014) ) Interferons in the central nervous system: A few instruments play many tunes. Glia 62: , 339–355. |
[19] | Dubois BD , Keenan E , Porter BE , Kapoor R , Rudge P , Thompson AJ , Miller DH , Giovannoni G ((2003) ) Interferon beta in multiple sclerosis: Experience in a British specialist multiple sclerosis centre. J Neurol Neurosurg Psychiatry 74: , 946–949. |
[20] | Filipi M , Jack S ((2020) ) Interferons in the treatment of multiple sclerosis: A clinical efficacy, safety, and tolerability update. Int J MS Care 22: , 165–172. |
[21] | Valentine AD , Meyers CA , Kling MA , Richelson E , Hauser P ((1998) ) Mood and cognitive side effects of interferon-alpha therapy. Semin Oncol 25: , 39–47. |
[22] | Raison CL , Demetrashvili M , Capuron L , Miller AH ((2005) ) Neuropsychiatric adverse effects of interferon-alpha: Recognition and management. CNS Drugs 19: , 105–123. |
[23] | Borsini A , Cattaneo A , Malpighi C , Thuret S , Harrison NA , Consortium MI , Zunszain PA , Pariante CM ((2017) ) Interferon-alpha reduces human hippocampal neurogenesis and increases apoptosis via activation of distinct STAT1-dependent mechanisms. Int J Neuropsychopharmacol 21: , 187–200. |
[24] | Wang R , Yang B , Zhang D ((2011) ) Activation of interferon signaling pathways in spinal cord astrocytes from an ALS mouse model. Glia 59: , 946–958. |
[25] | Main BS , Zhang M , Brody KM , Ayton S , Frugier T , Steer D , Finkelstein D , Crack PJ , Taylor JM ((2016) ) Type-1 interferons contribute to the neuroinflammatory response and disease progression of the MPTP mouse model of Parkinson’s disease. Glia 64: , 1590–1604. |
[26] | Cohan SL , Hendin BA , Reder AT , Smoot K , Avila R , Mendoza JP , Weinstock-Guttman B ((2021) ) Interferons and multiple sclerosis: Lessons from 25 years of clinical and real-world experience with intramuscular interferon beta-1a (Avonex). CNS Drugs 35: , 743–767. |
[27] | Moher D , Shamseer L , Clarke M , Ghersi D , Liberati A , Petticrew M , Shekelle P , Stewart LA PRISMA-P Group ((2015) ) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev 4: , 1. |
[28] | Minter MR , Main BS , Brody KM , Zhang M , Taylor JM , Crack PJ ((2015) ) Soluble amyloid triggers a myeloid differentiation factor 88 and interferon regulatory factor 7 dependent neuronal type-1 interferon response. J Neuroinflammation 12: , 71. |
[29] | Taylor JM , Minter MR , Newman AG , Zhang M , Adlard PA , Crack PJ ((2014) ) Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer’s disease. Neurobiol Aging 35: , 1012–1023. |
[30] | Araga S , Kagimoto H , Funamoto K , Takahashi K ((1991) ) Reduced natural killer cell activity in patients with dementia of the Alzheimer type. Acta Neurol Scand 84: , 259–263. |
[31] | Bermejo P , Martín-Aragón S , Benedí J , Susín C , Felici E , Gil P , Ribera JM , Villar AM ((2008) ) Differences of peripheral inflammatory markers between mild cognitive impairment and Alzheimer’s disease. Immunol Lett 117: , 198–202. |
[32] | Singh VK , Guthikonda P ((1997) ) Circulating cytokines in Alzheimer’s disease. J Psychiatr Res 31: , 657–660. |
[33] | Akiyama H , Ikeda K , Katoh M , McGeer EG , McGeer PL ((1994) ) Expression of MRP14, 27E10, interferon-alpha and leukocyte common antigen by reactive microglia in postmortem human brain tissue. J Neuroimmunol 50: , 195–201. |
[34] | Romagnoli M , Porcellini E , Carbone I , Veerhuis R , Licastro F ((2020) ) Impaired innate immunity mechanisms in the brain of Alzheimer’s disease. Int J Mol Sci 21: , 1126. |
[35] | Yamada T , Horisberger MA , Kawaguchi N , Moroo I , Toyoda T ((1994) ) Immunohistochemistry using antibodies to α-interferon and its induced protein, MxA, in Alzheimer’s and Parkinson’s disease brain tissues. Neurosci Lett 181: , 61–64. |
[36] | Kawaguchi N , Yamada T , Takahashi M , Hattori T ((1999) ) Expression of MxA mRNA in peripheral blood mononuclear cells in Parkinson’s disease. Parkinsonism Relat Disord 5: , 43–47. |
[37] | Klüter H , Vieregge P , Stolze H , Kirchner H ((1995) ) Defective production of interleukin-2 in patients with idiopathic Parkinson’s disease. J Neurol Sci 133: , 134–139. |
[38] | Su TH , Yang HC , Tseng TC , Chou SW , Lin CH , Liu CH , Liu CJ , Chen CL , Kao JH ((2019) ) Antiviral therapy in patients with chronic hepatitis C is associated with a reduced risk of parkinsonism. Mov Disord 34: , 1882–1890. |
[39] | Valekova I , Jarkovska K , Kotrcova E , Bucci J , Ellederova Z , Juhas S , Motlik J , Gadher SJ , Kovarova H ((2016) ) Revelation of the IFNα, IL-10, IL-8 and IL-1β as promising biomarkers reflecting immuno-pathological mechanisms in porcine Huntington’s disease model. J Neuroimmunol 293: , 71–81. |
[40] | Färkkilä AM , Iivanainen MV , Färkkilä MA ((1990) ) Disturbance of the water and electrolyte balance during high-dose interferon treatment. J Interferon Res 10: , 221–227. |
[41] | Poutiainen E , Hokkanen L , Niemi ML , Färkkilä M ((1994) ) Reversible cognitive decline during high-dose α-interferon treatment. Pharmacol Biochem Behav 47: , 901–905. |
[42] | Mora JS , Munsat TL , Kao KP , Finison LJ , Hedlund W , Bradley GA , Scheife R , Georgiades JA ((1986) ) Intrathecal administration of natural human interferon alpha in amyotrophic lateral sclerosis. Neurology 36: , 1137–1140. |
[43] | Hu X , Ho HH , Lou O , Hidaka C , Ivashkiv LB ((2005) ) Homeostatic role of interferons conferred by inhibition of IL-1-mediated inflammation and tissue destruction. J Immunol 175: , 131–138. |
[44] | Akwa Y , Hassett DE , Eloranta ML , Sandberg K , Masliah E , Powell H , Whitton JL , Bloom FE , Campbell IL ((1998) ) Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 161: , 5016–5026. |
[45] | Jin C , Peng X , Liu F , Cheng L , Xie T , Lu X , Wu H , Wu N ((2016) ) Interferon-induced sterile alpha motif and histidine/aspartic acid domain-containing protein 1 expression in astrocytes and microglia is mediated by microRNA-181a. AIDS 30: , 2053–2064. |
[46] | Pemberton LA , Kerr SJ , Smythe G , Brew BJ ((1997) ) Quinolinic acid production by macrophages stimulated with IFN-γ, TNF- α, and IFN-α . J Interferon Cytokine Res 17: , 589–595. |
[47] | Zhao L , Huang Y , Tian C , Taylor L , Curthoys N , Wang Y , Vernon H , Zheng J ((2012) ) Interferon-α regulates glutaminase 1 promoter through STAT1 phosphorylation: Relevance to HIV-1 associated neurocognitive disorders. PLoS One 7: , e32995. |
[48] | Koneru R , Bimonte-Nelson H , Ciavatta V , Haile W , Elmore K , Ward J , Maroun L , Tyor WR ((2018) ) Reversing interferon-alpha neurotoxicity in a HIV-associated neurocognitive disorder mouse model. AIDS 32: , 1403–1411. |
[49] | Sas AR , Bimonte-Nelson HA , Tyor WR ((2007) ) Cognitive dysfunction in HIV encephalitic SCID mice correlates with levels of Interferon-α in the brain. AIDS 21: , 2151–2159. |
[50] | Sas AR , Bimonte-Nelson H , Smothers CT , Woodward J , Tyor WR ((2009) ) Interferon-α causes neuronal dysfunction in encephalitis. J Neurosci 29: , 3948–3955. |
[51] | Anderson AM , Lennox JL , Mulligan MM , Loring DW , Zetterberg H , Blennow K , Kessing C , Koneru R , Easley K , Tyor WR ((2017) ) Cerebrospinal fluid interferon alpha levels correlate with neurocognitive impairment in ambulatory HIV-Infected individuals. J Neurovirol 23: , 106–112. |
[52] | Cassol E , Misra V , Dutta A , Morgello S , Gabuzda D ((2014) ) Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. AIDS 28: , 1579–1591. |
[53] | Krivine A , Force G , Servan J , Cabée A , Rozenberg F , Dighiero L , Marguet F , Lebon P ((1999) ) Measuring HIV-1 RNAand interferon-alpha in the cerebrospinal fluid of AIDS patients: Insights into the pathogenesis of AIDS DementiaComplex. J Neurovirol 5: , 500–506. |
[54] | Pulliam L , Rempel H , Sun B , Abadjian L , Calosing C , Meyerhoff DJ ((2011) ) A peripheral monocyte interferon phenotype in HIV infection correlates with a decrease in magnetic resonance spectroscopy metabolite concentrations. AIDS 25: , 1721–1726. |
[55] | Rho MB , Wesselingh S , Glass JD , McArthur JC , Choi S , Griffin J , Tyor WR ((1995) ) A potential role for interferon-α in the pathogenesis of hiv-associated dementia. Brain Behav Immun 9: , 366–377. |
[56] | Afraei S , Sedaghat R , Zavareh FT , Aghazadeh Z , Ekhtiari P , Azizi G , Mirshafiey A ((2018) ) Therapeutic effects of pegylated-interferon-α2a in a mouse model of multiple sclerosis. Cent Eur J Immunol 43: , 9–17. |
[57] | Brod SA , Khan M , Kerman RH , Pappolla M ((1995) ) Oral administration of human or murine interferon alpha suppresses relapses and modifies adoptive transfer in experimental autoimmune encephalomyelitis. J Neuroimmunol 58: , 61–69. |
[58] | Laman JD , Van Meurs M , Schellekens MM , de Boer M , Melchers B , Massacesi L , Lassmann H , Claassen E , T Hart BA ((1998) ) Expression of accessory molecules and cytokines in acute EAE in marmoset monkeys (Callithrix jacchus). J Neuroimmunol 86: , 30–45. |
[59] | Olson JK , Miller SD ((2009) ) The innate immune response affects the development of the autoimmune response in Theiler’s virus-induced demyelinating disease. J Immunol 182: , 5712–5722. |
[60] | Vasquez M , Consuegra-Fernandez M , Aranda F , Jimenez A , Tenesaca S , Fernandez-Sendin M , Gomar C , Ardaiz N , Di Trani CA , Casares N , Lasarte JJ , Lozano F , Berraondo P ((2019) ) Treatment of experimental autoimmune encephalomyelitis by sustained delivery of low-dose IFN-alpha. J Immunol 203: , 696–704. |
[61] | Asgari N , Voss A , Steenstrup T , Kyvik KO , Stenager E , Thue Lillevang S ((2013) ) Interferon alpha association with neuromyelitis optica. Clin Dev Immunol 2013: , 713519. |
[62] | ((1989) ) Interferon-alpha and transfer factor in the treatment of multiple sclerosis: A double-blind, placebo-controlled trial. AUSTIMS Research Group. J Neurol Neurosurg Psychiatry 52: , 566–574. |
[63] | Bayas A , Stasiolek M , Kruse N , Toyka KV , Selmaj K , Gold R ((2009) ) Altered innate immune response of plasmacytoid dendritic cells in multiple sclerosis. Clin Exp Immunol 157: , 332–342. |
[64] | Bergkvist M , Martinsson T , Åman P , Sandberg-Wollheim M ((1996) ) No genetic linkage between multiplesclerosis and the interferon α/β locus. J Neuroimmunol 65: , 163–165. |
[65] | Bongioanni MR , Durelli L , Ferrero B , Imperiale D , Oggero A , Verdun E , Aimo G , Pagni R , Geuna M , Bergamasco B ((1996) ) Systemic high-dose recombinant-alpha-2a-interferon therapy modulates lymphokine production in multiple sclerosis. J Neurol Sci 143: , 91–99. |
[66] | Brandt ER , Mackay IR , Hertzog PJ , Cheetham BF , Sherritt M , Bernard CC ((1993) ) Molecular detection of interferon-α expression in multiple sclerosis brain. J Neuroimmunol 44: , 1–5. |
[67] | Brod SA , Kerman RH , Nelson LD , Marshall Jr GD , Henninger EM , Khan M , Jin R , Wolinsky JS ((1997) ) Ingested IFN-alpha has biological effects in humans with relapsing-remitting multiple sclerosis. Mult Scler 3: , 1–7. |
[68] | Brod SA , Lindsey JW , Vriesendorp FS , Ahn C , Henninger E , Narayana PA , Wolinsky JS ((2001) ) Ingested IFN-alpha: Results of a pilot study in relapsing-remitting MS. Neurology 57: , 845–852. |
[69] | Brod SA , Nguyen M , Hood Z , Shipley GL ((2006) ) Ingested (oral) IFN-α represses TNF-α mRNA in relapsing-remitting multiple sclerosis. J Interferon Cytokine Res 26: , 150–155. |
[70] | Canto E , Isobe N , Didonna A , MS-EPIC Study Group; Hauser SL , Oksenberg JR ((2018) ) Aberrant STAT phosphorylation signaling in peripheral blood mononuclear cells from multiple sclerosis patients. J Neuroinflammation 15: , 72. |
[71] | Carrieri PB , Maiorino A , Provitera V , Perrella A , Perrella O ((1996) ) Evaluation of interleukin 10 and interferon-α in the cerebrospinal fluid and serum of patients with multiple sclerosis. Eur J Neurol 3: , 544–547. |
[72] | Cheknev SB , Ashmanova YG , Pritsker AD , Latysheva OL , Yershov FI , Kulberg AJ ((1994) ) Autologous enhancement by interferon of natural killer activity of human peripheral blood lymphocytes. Mediators Inflamm 3: , 341–346. |
[73] | Cheknev SB , Mezentseva MV , Narovlyansky AN , Kobyakina NA , Ershov FI ((2001) ) Correlations betweeneffects of preparations of interferon and its inducers on blood cells in patients with multiple sclerosis. Bull Exp Biol Med 131: , 382–384. |
[74] | Chieux V , Chehadeh W , Hautecoeur P , Harvey J , Wattr P , Hober D ((2001) ) Increased levels of antiviral MxA protein in peripheral blood of patients with a chronic disease of unknown etiology. J Med Virol 65: , 301–308. |
[75] | Chiurchiù V , Cencioni MT , Bisicchia E , de Bardi M , Gasperini C , Borsellino G , Centonze D , Battistini L , Maccarrone M ((2013) ) Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide inmultiple sclerosis. Ann Neurol 73: , 626–636. |
[76] | de Jong TD , Vosslamber S , Mantel E , de Ridder S , Wesseling JG , van der Pouw Kraan TC , Leurs C , Hegen H , Deisenhammer F , Killestein J , Lundberg IE , Vencovsky J , Nurmohamed MT , van Schaardenburg D , Bultink IE , Voskuyl AE , Pegtel DM , van der Laken CJ , Bijlsma JW , Verweij CL ((2016) ) Physiological evidence for diversification ofIFNα- and IFNβ-mediated response programs in different autoimmune diseases. Arthritis ResTher 18: , 49. |
[77] | Deckx N , Willekens B , Wens I , Eijnde BO , Goossens H , Van Damme P , Berneman ZN , Cools N ((2016) ) Altered molecular expression of TLR-signaling pathways affects the steady-state release of IL-12p70 and IFN-alpha in patients with relapsing-remitting multiple sclerosis. Innate Immun 22: , 266–273. |
[78] | Dettke M , Scheidt P , Prange H , Kirchner H ((1997) ) Correlation between interferon production and clinical disease activity in patients with multiple sclerosis. J Clin Immunol 17: , 293–300. |
[79] | Durelli L , Bongioanni MR , Cavallo R , Ferrero B , Ferri R , Ferrio MF , Bradac GB , Riva A , Vai S , Geuna M , et al. ((1994) ) Chronic systemic high-dose recombinant interferon alfa-2a reduces exacerbation rate, MRI signs of disease activity, and lymphocyte interferon gamma production in relapsing-remitting multiple sclerosis. Neurology 44: , 406–413. |
[80] | Durelli L, Bongioanni MR, Ferrero B, Ferri R, Imperiale D, Bradac GB, Bergui M, Geuna M, Bergamini L, Bergamasco B (1996a) Interferon alpha-2a treatment of relapsing-remitting multiple sclerosis: Disease activity resumes after stopping treatment. Neurology 47: , 123–129. |
[81] | Durelli L , Bongioanni MR , Ferrero B , Imperiale D , Verdun E , Oggero A , Gentile E , Bradac GB , Bergui M , Bergamini L , Bergamasco B ((1996) ) Long term recombinant interferon alpha treatment in MS with special emphasis to side effects. Mult Scler 1: , 366–371. |
[82] | Feng X , Reder NP , Yanamandala M , Hill A , Franek BS , Niewold TB , Reder AT , Javed A ((2012) ) Type I interferon signature is high in lupus and neuromyelitis optica but low in multiple sclerosis. J Neurol Sci 313: , 48–53. |
[83] | Guillen C , Prieto A , Alvarez-Cermeño JC , de la Piedra M , Gimeno A , Alvarez-Mon M ((1999) ) Regulatory effects of cytokines and cyclosporine A on peripheral blood mononuclear cells from stable multiple sclerosis patients. Immunopharmacol Immunotoxicol 21: , 527–549. |
[84] | Hertzog PJ , Wright A , Harris G , Linnane AW , Mackay IR ((1991) ) Intermittent interferonemia and interferon responses in multiple sclerosis. Clin Immunol Immunopathol 58: , 18–32. |
[85] | Hirotani M , Niino M , Fukazawa T , Yaguchi H , Nakamura M , Kikuchi S , Sasaki H ((2012) ) Decreased interferon-alpha production in response to CpG DNA dysregulates cytokine responses in patients with multiple sclerosis. Clin Immunol 143: , 145–151. |
[86] | Hirsch RL , Johnson KP ((1985) ) Natural killer cell activity in multiple sclerosis patients treated with recombinantinterferon-α2. Clin Immunol Immunopathol 37: , 236–244. |
[87] | Jansen M , Reinhard Jr JF ((1999) ) Interferon response heterogeneity: Activation of a pro-inflammatory response by interferon α and β. A possible basis for diverse responses to interferon β in MS. J Leukoc Biol 65: , 439–443. |
[88] | Jørgensen PB , Livbjerg AH , Hansen HJ , Petersen T , Höllsberg P ((2012) ) Epstein-Barr virus peptide presented by HLA-E is predominantly recognized by CD8 (bright) cells in multiple sclerosis patients. PLoS One 7: , e46120. |
[89] | Kamin Lewis RM , Panitch HS , Merigan TC , Johnson KP ((1984) ) Decreased interferon synthesis and responsiveness to interferon by leukocytes from multiple sclerosis patients given natural alpha interferon. J Interferon Res 4: , 423–432. |
[90] | Kinnunen E , Timonen T , Pirttilä T , Kalliomäki P , Ketonen L , Matikainen E , Sepponen R , Juntunen J ((1993) ) Effects of recombinant α-2b-interferon therapy in patients with progressive MS. Acta Neurol Scand 87: , 457–460. |
[91] | Lundtoft C , Pucholt P , Imgenberg-Kreuz J , Carlsson-Almlöf J , Eloranta ML , Syvänen AC , Nordmark G , Sandling JK , Kockum I , Olsson T , Rönnblom L , Hagberg N ((2020) ) Function of multiple sclerosis-protective HLA class I alleles revealed by genome-wide protein-quantitative trait loci mapping of interferon signalling. PLoS Genet 16: , e1009199. |
[92] | Merrill JE , Ellison GW , Myers LW ((1984) ) Cytotoxic activity of peripheral blood and cerebrospinal fluid lymphocytes from patients with multiple sclerosis and other neurological diseases: Analysis at the single cell level of the relationship of cytotoxic effectors and interferon-producing cells. Clin Immunol Immunopathol 31: , 390–402. |
[93] | Mycko MP , Cwiklinska H , Cichalewska M , Matysiak M , Lewkowicz P , Sliwinska B , Selmaj I , Selmaj KW ((2014) ) Plasmocytoid dendritic cell deficit of early response to toll-like receptor 7 agonist stimulation in multiple sclerosis patients pDC deficit of early response to toll-like receptor 7 agonist stimulation. Clin Immunol 153: , 211–219. |
[94] | Myhr KM , Riise T , Lilleås FEG , Beiske TG , Celius EG , Edland A , Jensen D , Larsen JP , Nilsen R , Nortvedt MW , Smievoll AI , Vedeler C , Nyland HI ((1999) ) Interferon-α2a reduces MRI disease activity in relapsing-remitting multiple sclerosis. Neurology 52: , 1049–1056. |
[95] | Myhr KM , Sadallah S , Mollnes TE , Meri S , Nyland HI , Schifferli J , Vedeler CA ((2000) ) Interferon-α2a effects on complement activation and regulation in MS patients. Acta Neurol Scand 101: , 30–35. |
[96] | Noronha A , Toscas A , Jensen MA ((1992) ) Contrasting effects of alpha, beta, and gamma interferons on nonspecific suppressor function in multiple sclerosis. Ann Neurol 31: , 103–106. |
[97] | Nyström M , Ruuth K , Lundgren E , Sundström P ((2007) ) No support for a truncated interferon-α17 allele as risk factor for MS. Eur J Neurol 14: , 1302–1304. |
[98] | Pentón-Rol G , Cervantes-Llanos M , Cabrera-Gómez JA , Alonso-Ramírez R , Valenzuela-Silva C , Rodríguez-Lara R , Montero-Casimiro E , Bello-Rivero I , López-Saura P ((2008) ) Treatment with type I interferons induces a regulatory T cell subset in peripheral blood mononuclear cells from multiple sclerosis patients. Int Immunopharmacol 8: , 881–886. |
[99] | Squillacote D , Martinez M , Sheremata W ((1996) ) Natural alpha interferon in multiple sclerosis: Results of three preliminary series. J Int Med Res 24: , 246–257. |
[100] | Stöger I , Tálas M , Benczúr M , Gyódi E , Petrányi GG , Pálffy G , Kotsy B ((1982) ) Lack ofcorrelation between impaired interferon production and natural killer activity of lymphocytes in multiplesclerosis. Arch Virol 71: , 259–265. |
[101] | Traugott U , Lebon P ((1988) ) Multiple sclerosis: Involvement of interferons in lesion pathogenesis. Ann Neurol 24: , 243–251. |
[102] | Traugott U , Lebon P ((1988) ) Interferon-γ and Ia antigen are present on astrocytes in active chronic multiple sclerosis lesions. J Neurol Sci 84: , 257–264. |
[103] | Ulvestad E , Aarseth JH , Vedeler C , Nyland H , Myhr KM ((2004) ) The effects of interferon-alpha2a on concentrations of immunoglobulins, complement and lymphocytes in patients with multiple sclerosis. Scand J Immunol 59: , 103–108. |
[104] | Wandinger KP , Wessel K , Neustock P , Siekhaus A , Kirchner H ((1997) ) Diminished production of type-I interferons and interleukin-2 in patients with multiple sclerosis. J Neurol Sci 149: , 87–93. |
[105] | Carbone I , Lazzarotto T , Ianni M , Porcellini E , Forti P , Masliah E , Gabrielli L , Licastro F ((2014) ) Herpes virus in Alzheimer’s disease: Relation to progression of the disease. Neurobiol Aging 35: , 122–129. |
[106] | Readhead B , Haure-Mirande JV , Funk CC , Richards MA , Shannon P , Haroutunian V , Sano M , Liang WS , Beckmann ND , Price ND , Reiman EM , Schadt EE , Ehrlich ME , Gandy S , Dudley JT ((2018) ) Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99: , 64–82. |
[107] | Dauer W , Przedborski S ((2003) ) Parkinson’s disease: Mechanisms and models. Neuron 39: , 889–909. |
[108] | Herrero MT , Estrada C , Maatouk L , Vyas S ((2015) ) Inflammation in Parkinson’s disease: Role of glucocorticoids. Front Neuroanat 9: , 32. |
[109] | Xiao H , Killip MJ , Staeheli P , Randall RE , Jackson D ((2013) ) The human interferon-induced MxA protein inhibits early stages of influenza A virus infection by retaining the incoming viral genome in the cytoplasm. J Virol 87: , 13053–13058. |
[110] | Lin WY , Lin MS , Weng YH , Yeh TH , Lin YS , Fong PY , Wu YR , Lu CS , Chen RS , Huang YZ ((2019) ) Association of antiviral therapy with risk of Parkinson disease in patients with chronic hepatitis C virus infection. JAMA Neurol 76: , 1019–1027. |
[111] | Sarasombath P , Sumida K , Kaku DA ((2002) ) Parkinsonism associated with interferon alpha therapy for chronic myelogenous leukemia.48. Hawaii Med J 61: , 57. |
[112] | Williams AJ , Paulson HL ((2008) ) Polyglutamine neurodegeneration: Protein misfolding revisited. Trends Neurosci 31: , 521–528. |
[113] | Aylward EH , Sparks BF , Field KM , Yallapragada V , Shpritz BD , Rosenblatt A , Brandt J , Gourley LM , Liang K , Zhou H , Margolis RL , Ross CA ((2004) ) Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology 63: , 66–72. |
[114] | Cermelli C , Vinceti M , Beretti F , Pietrini V , Nacci G , Pietrosemoli P , Bartoletti A , Guidetti D , Sola P , Bergomi M , Vivoli G , Portolani M ((2003) ) Risk of sporadic amyotrophic lateral sclerosis associated with seropositivity for herpesviruses and echovirus-7. Eur J Epidemiol 18: , 123–127. |
[115] | Constantinescu CS , Farooqi N , O’Brien K , Gran B ((2011) ) Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 164: , 1079–1106. |
[116] | Clifford DB , Ances BM ((2013) ) HIV-associated neurocognitive disorder. Lancet Infect Dis 13: , 976–986. |
[117] | Kerr SJ , Armati PJ , Brew BJ ((1995) ) Neurocytotoxity of quinolinic acid in human brain cultures. J Neurovirol 1: , 375–380. |
[118] | Schwarcz R , Whetsell WO Jr , Mangano RM ((1983) ) Quinolinic acid: An endogenous metabolite that produces -sparing lesions in rat brain. Science 219: , 316–318 axon. |
[119] | Whetsell Jr WO , Schwarcz R ((1989) ) Prolonged exposure to submicromolar concentrations of quinolinic acid causes excitotoxic damage in organotypic cultures of rat corticostriatal system. Neurosci Lett 97: , 271–275. |
[120] | Reinhard Jr JF , Erickson JB , Flanagan EM ((1994) ) Quinolinic acid in neurological disease: Opportunities for novel drug discovery. Adv Pharmacol 30: , 85–127. |
[121] | Lebon P , Badoual J , Ponsot G , Goutières F , Hémeury-Cukier F , Aicardi J ((1988) ) Intrathecal synthesis ofinterferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci 84: , 201–208. |
[122] | Von Sydow M , Sönnerborg A , Gaines H , Strannegård O ((1991) ) Interferon-alpha and tumor necrosis factor-alpha in serum of patients in various stages of HIV-1 infection. AIDS Res Hum Retroviruses 7: , 375–380. |
[123] | Newcomb R , Sun X , Taylor L , Curthoys N , Giffard RG ((1997) ) Increased production of extracellular glutamate by the mitochondrial glutaminase following neuronal death. J Biol Chem 272: , 11276–11282. |
[124] | Dinarello CA ((1996) ) Biologic basis for interleukin-1 in disease. Blood 87: , 2095–2147. |


