
Untangling the Role of TREM2 in Conjugation with Microglia in Neuronal Dysfunction: A Hypothesis on a Novel Pathway in the Pathophysiology of Alzheimer’s Disease
Abstract
Alzheimer’s disease (AD) is a complex neurodegenerative disorder involving heterogenous pathophysiological characteristics, which has become a challenge to therapeutics. The major pathophysiology of AD comprises amyloid-β (Aβ), tau, oxidative stress, and apoptosis. Recent studies indicate the significance of Triggering receptor expressed on myeloid cells 2 (TREM2) and its mutant variants in AD. TREM2 are the transmembrane receptors of microglial cells that performs a broad range of physiological cell processes. Phagocytosis of Aβ is one of the physiological roles of TREM2, which plays a pivotal role in AD progression. R47H, a mutant variant of TREM2, increases the risk of AD by impairing TREM2–Aβ binding. Inconclusive evidence regarding the TREM2 signaling cascade mechanism of Aβ phagocytosis motivates the current review to propose a new hypothesis. The review systematically assesses the cross talk between TREM2 and other AD pathological domains and the influence of TREM2 on amyloid and tau seeding. Disease associated microglia (DAM), a novel state of microglia with unique transcriptional and functional signatures reported in neurodegenerative conditions, also depend on the TREM2 pathway for its differentiation. DAM is suggested to have a neuroprotective role. We hypothesize that TREM2, along with its signaling adaptors and endogenous proteins, play a key role in ameliorating Aβ clearance. We indicate that TREM2 has the potential to ameliorate the Aβ burden, though with differential clearance ability and may act as a potential therapeutic target.
INTRODUCTION
Alzheimer’s disease (AD), an insidious neurodegenerative disease, severely impairs the cognitive abilities of the affected individual [1]. The complex pathophysiology of AD includes amyloid-β (Aβ), tau, oxidative stress, and apoptosis, which pose a challenge to therapeutics [2]. Currently, 50 million people across the globe are affected with dementia, and by 2050, the number may accelerate 3 times to 152 million with one new dementia case every 3 seconds [3]. The majority of AD cases are sporadic, and the critical interaction among its heterogenous pathological domains is still obscure and its mechanisms are not known [4]. Recent studies indicate the significance of triggering receptor expressed on myeloid cells 2 (TREM2) and its mutant variants in AD and is emerging as the novel dimension with potential therapeutic scope [5, 6]. TREM2 is a trans-membrane receptor belonging to the super family of immunoglobulin and is exclusively expressed by microglial cells in the central nervous system (CNS) [7]. Studies have identified that, for Aβ, TREM2 is considered as a receptor, and a mutant variant of TREM2, R47H, impairs the binding of Aβ [8]. Microglia deficient in TREM2 exhibited anomalies in glycolysis, anabolic metabolism, levels of ATP, and activation of mammalian target of rapamycin (mTOR) pathway, and the role of TREM2–DNAX activation protein 12 (DAP12) interaction in microglial function is still not clear [9–12]. The above studies reported conflicting results with respect to the role of TREM2 in microglial activation. Additionally, there is an increase in Aβ plaques in the 5XFAD model, with deficiency of TREM2 [13], and the contradictory results are reported in different studies [14, 15]. All these studies are inconclusive and contradictory, raising the need to focus on and conduct TREM2 and microglial-based research. Despite certain contradictory findings, so far, all the tested Aβ models of AD suggest that TREM2 is imperative for activation and clustering of disease associated microglia (DAM) and also further enhancing signaling of the mTOR pathway [16]. Our review is focused on the structure of TREM2 to understand its ligand binding domains and various mutations of the TREM2 structural domains leading to deleterious pathological features. The mechanism of TREM2 downstream signaling and phagocytosis of Aβ has been emphasized in the review. Further, the understanding of the complex pathological interactions among TREM2 and other AD pathological domains has been extended on the cross talk of TREM2 with Aβ, tau, Apolipoprotein E (APOE), and phospholipase Cγ2 (PLCγ2). The role of TREM2 in transforming homeostatic microglia to a DAM state also has been discussed in the review. In the backdrop of inconclusive evidence regarding the TREM2 signaling cascade, a new hypothesis has been proposed by us to understand the novel complex pathophysiology of AD.
STRUCTURE OF TREM2
The solution NMR structure of the TREM2 wild type was recently elucidated by Steiner [17]. 13Cα and 13Cβ chemical shifts were employed for measurement of the secondary structure of U2–H, 13C, 15N-labelled TREM2–TM WT (amino acids 161–206). The α-helix was observed in amino acids ranging from S174 to W198. The study also assessed the magnitude of chemical shifts; 13Cα and 13Cβ and demonstrated that the C-terminal portion of TREM2–TM exhibited reduced α-helical propensity. Further, nuclear Overhauser effect (NOE) studies of NMR suggested that there is no secondary structure between the amino acids A189 and L193. Therefore, this unstructured kink region results in a differential orientation of the short stretch of the C-terminal helix with regard to the N-terminal α-helix. In order to eliminate structural effects caused by tiny spherical detergent micelle, researchers performed insertion of TREM2 into lipid bilayer nano discs. The results suggest a similar secondary α-helical content devoid of NOE contact between the amino acid ranges A189-L193. Due to the unfavorable location of lysine—186 in hydrophobic lipid environment, it assumes an important role in determination of TREM2–TM WT structural dynamics. Accordingly, NMR studies of the TREM2–TM in complex with DAP12 structure indicates that the α-helical structure exhibited continuously from S174–W198 without the amorphic kink region.
Native TREM2 embodies three domains: 1) Extracellular domain comprising of 1–172 amino acids, 2) transmembrane domain comprising of 173–195 amino acids, 3) intracellular domain comprising of 196–230 amino acids [14] (Fig. 1). Mature TREM2 protein comprising of 212 amino acids (aa 19–230) after trimming aa 1–18, which serve as a membrane targeting peptide, where in, extracellular component of TREM2 assumes V-like immunoglobulin fold and a transmembrane domain adopts helical configuration [18]. Two disulfide bonds, C36 ↔ C110 and C51 ↔ C60, were located in the extracellular domain, and asparagine at amino acid positions 20 and 79 were subjected to N-glycosylation post [19].
Fig. 1
Structure of TREM2–DAP adaptor complex: The figure explains the composition of TREM2 in Plasma membrane and its complex formation with DAP 10 and 12 complex. TREM2, Triggering receptor expressed on myeloid cells 2; DAP 12 and 10, DNAX activation proteins 12 and 10; ITAM, Immunoreceptor tyrosine based activation motif; YINM, tyrosine-isoleucine-asparagine-methionine; SYK, Spleen tyrosine kinase; PI3K, Phosphatidyl inositol 3-kinase; sTREM2, Soluble TREM2; ADAM 10 and 17, α-secretase-disintegrin and metalloproteinase domain containing proteins 10 and 17.
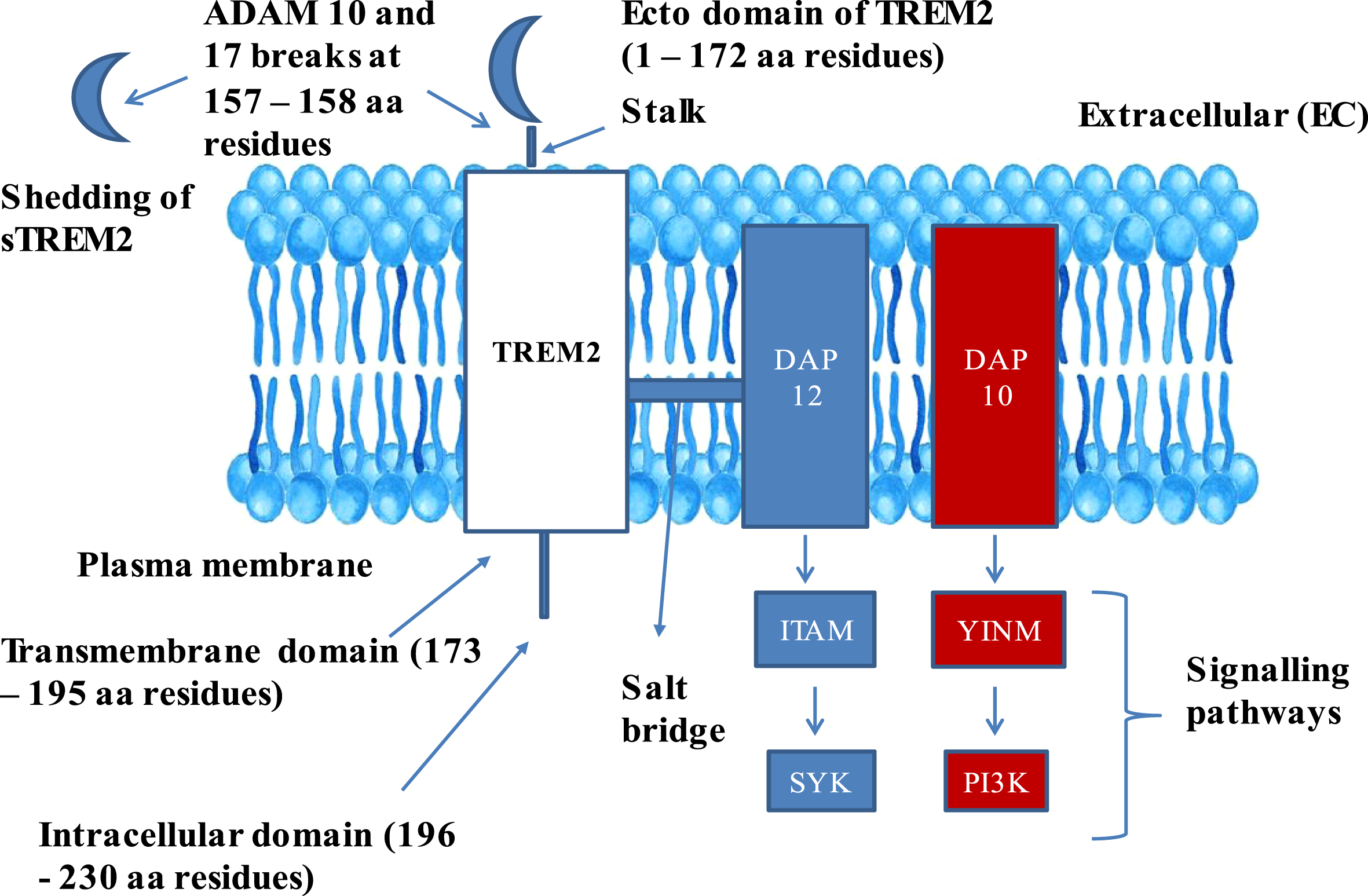
The extracellular domain of TREM2 also comprises a short stalk region, whereas the intracellular domain is devoid of any signaling motifs. The transmembrane domain of TREM2 is associated with two adaptor proteins: DAP12, also called TYRO protein tyrosine kinase-binding protein, and DNAX activation protein 10 (DAP10), also called hematopoietic cell signal transducer [16]. DAP12 is functionally required for TREM2 cell surface expression, and for intracellular transmission signals via spleen tyrosine kinase (SYK) [20]. DAP10 facilitates the phosphatidyl inositol 3 kinase (PI3K) recruitment [21]. Triggering receptor expressed on myeloid cells 1 (TREM1) also belongs to the super family of immune globulin and stimulates neutrophils and monocytes by DAP12 signaling. Additionally, TREM1 facilitates the secretion of pro-inflammatory cytokines and chemokines in response to fungal and bacterial invasions [22].
TREM2 FACILITATES Aβ PLAQUE PHAGOCYTOSIS
TREM2 facilitates phagocytosis of Aβ through its signaling cascade. Initially, Aβ fibrils associate with the ecto domain of TREM2, which contains V-type Ig like region with which different ligands interact [23]. The study reported that amino acid residues 31–91 located in the ecto domain of TREM2 interacts with Aβ, and the group has identified the ligand binding sequence by constructing a set of shortened C-terminal recombinant TREM2 proteins and by conducting Aβ1 - 42 binding assays [24]. In vitro studies suggests that different Aβ filaments exhibited varied affinities with ecto domain of TREM2, where in, Aβ oligomers exhibited highest affinity for TREM2 ectodomain than the Aβ40 monomers, Aβ42 monomers, and Aβ42 fibrils [25]. Further, it results in the formation of a TREM2-Aβ complex, and activation of microglia takes place to facilitate programmed phagocytosis of cell, so as to clear the protein aggregates [15]. Simultaneously, both TREM2 and sTREM2 accumulate around Aβ plaques there by prohibiting the dispersal of plaques towards the proximal healthy tissues [8].
TREM2/DAP12 facilitates microglial survival, proliferation, phagocytosis, and motility through signaling cascade molecules. Initially, TREM2/DAP12 triggers a cascade of events, in the way of activation of various downstream molecules which comprises PI3K, extracellular signal regulated protein kinase (ERK), PLCγ2, and VAV, which in turn activates Akt (Serine/threonine kinase Akt (protein kinase B)), diacylglycerol (DAG), and inositol 1,4,5-trisphosphate (IP3) resulting to mTOR pathway activation. Similarly, TREM2 facilitates microglial survival and inflammation through activation of downstream signaling molecules, PI3K, NFκB, and ERK, which activate the Akt pathway [26]. With regard to neuronal inflammation, the majority of the experimental results confirmed the anti-inflammatory role of TREM2 by suppressing Toll-like receptor (TLR)-induced release of pro inflammatory cytokines: Interleukin (IL)-6 and Tumor necrosis factor (TNF)-α [27, 28]. Based on the current studies, it can be concluded that TREM2 has an imperative role in Aβ phagocytosis and has a potential role in modifying the progression of AD.
DOWNSTREAM SIGNALING MECHANISMS OF TREM2
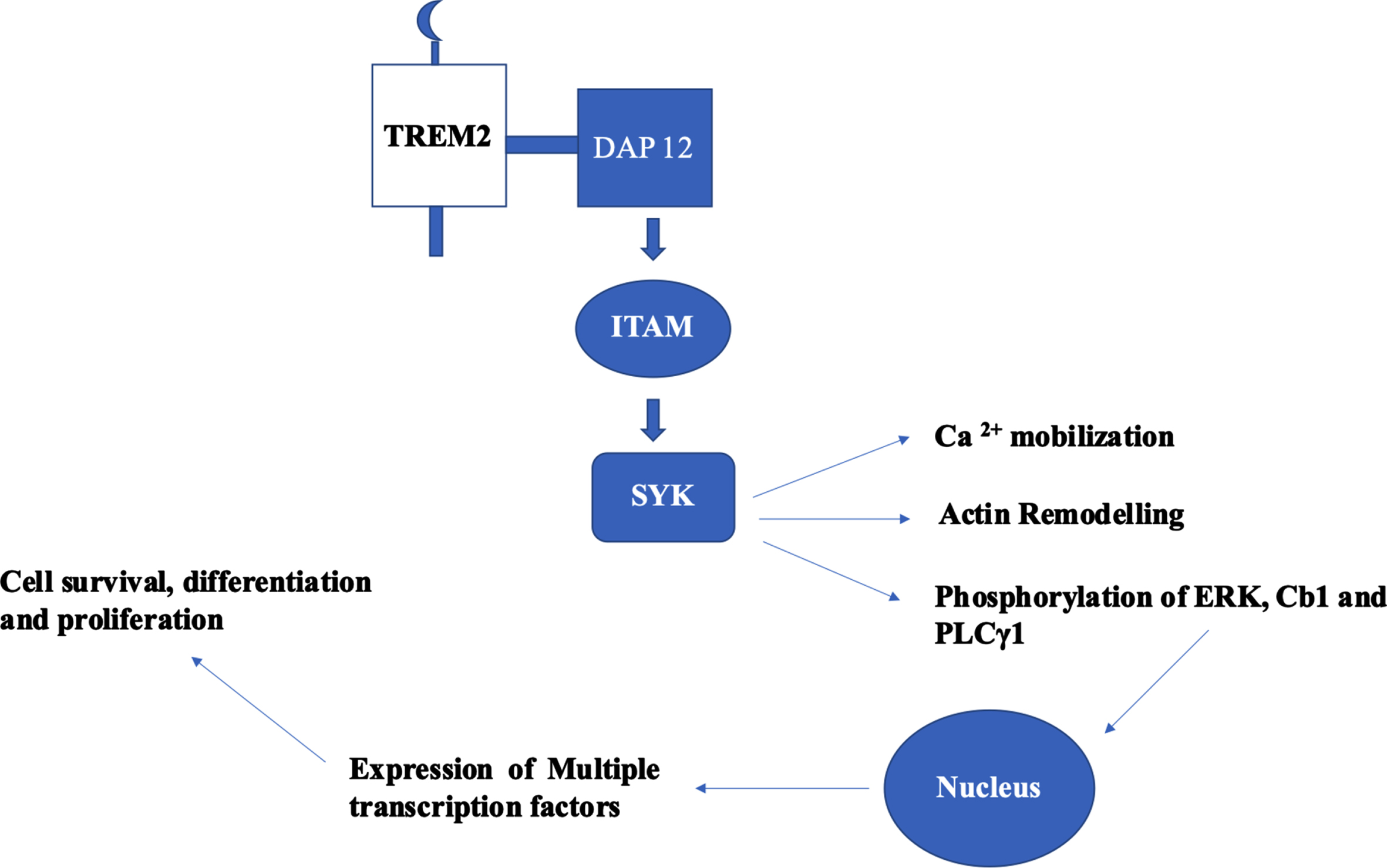
TREM2-based signal transduction based on the adaptor proteins DAP12 and DAP10 are a) TREM2-DAP12-ITAM-SYK and b) TREM2-DAP10-YINM-PI3K. In TREM2-DAP12 signaling cascade, DAP12 is the adaptor for TREM2 and several other single transmembrane receptors. The consensus sequence of immunoreceptor tyrosine-based activation motif (ITAM) of DAP12 is YxxI/Lx (6–12) YxxI/L. Upon activation of TREM2 by ligands, DAP12 mediates TREM2 signaling which leads to phosphorylation of ITAM which culminates to Ca2 + mobilization, phosphorylation of ERK, and remodeling of actin [29]. The Ly-49 receptors expressed on natural killer cells and some subsets of T cells. These receptors are homo-dimers and are lectin like type II transmembrane receptors [30]. LY49D model based in natural killer cells explain the canonical signaling pathway of TREM2-DAP12 [31]. Initially, binding of LY49D leads to the phosphorylation of tyrosine residues in ITAM of DAP12 by SRC kinases, which act as binding sites for SH2 domain of SYK. SYK activation results in the phosphorylation of endogenous signaling proteins—ERK 1/2, Cb1, and PLCγ1. This signaling cascade initiates the hallmark TREM2 signaling through ERK, activating RSK. Both translocate to the nucleus and activates multiple transcription factors to synthesize effector proteins (Fig. 2). The signaling cascade mechanisms are implicated in cell survival, differentiation, and proliferation [29]. In haemopoietic cells, ITAM phosphorylation explores scaffold proteins such as Linkers for activation of T and B cells (LAT and LAB) to recruit Grb2, cb1, vav, and PI3K [32]. In osteoclasts and bone marrow derived macrophages, TREM2-DAP12 use LAB to stimulate calcium mobilization [33]. The signaling cascade mechanism of TREM2 serves as a core platform in clearing the Aβ burden. Keeping in view of the obscure information on the kind and number of ligands in the TREM2 cascade, further research has to be conducted to reveal and to understanding the specific ligands involved which will serve as potential therapeutic targets.
Fig. 2
Canonical signaling pathway of TREM2–DAP12 cascade and its functional role. Phosphorylation of tyrosine residues of ITAM in DAP 12 leads to the activation of SYK which results to phosphorylation of endogenous signaling proteins: ERK 1/2, Cb1, and PLCγ1. These proteins translocate to nucleus and initiates the expression of multiple transcription factors which had implications in cell survival, differentiation, and proliferation.
TREM2 VARIANTS AND ITS PATHOLOGICAL ROLE IN AD
Different TREM2 variants lead to deleterious pathological consequences. For instance, impairment of TREM2 and Aβ binding and disruption of signaling mechanism. Loss of TREM2 functional activity attributes to biallelic mutations which is rare and causing Nasu-Hakola disease (NHD) [34] and in certain cases causes frontotemporal dementia (FTD) [35]. The mutations in TREM2 that result in NHD and FTD originate from the ecto domain (Y38C, T66M) [36] and transmembrane domain (D134G, K186N). Even mutations in early stop codons [37] or splice sites [38] may result in NHD and FTD. With regard to AD, R47H, a mutant variant of TREM2, increases the risk for AD as suggested by two independent groups analyzing European and North American cohorts [5] and Icelandic cohorts [6]. Further, researchers suggested a considerable association of R47H and R62H TREM2 variants with late onset AD (LOAD) in cohorts of African Americans, but these mutations contributing to AD risk were extremely rare in Caucasian cohorts.
Kober and Brett [29], during mapping of TREM2 electrostatic surface, recognized a large basic patch which was found to have a unique functional role in TREM2. Interestingly, this unique domain was not located in other family members of TREM. Further, TREM2 mutants R47H and R62H reduce the basic patch size, which lowers the binding ability of TREM2, thereby impairing its function. Another variant T96K increases the basic patch size resulting to functional gain in TREM2.
R47H, the crux TREM2 variant
R47H TREM2 variant is the most commonly responsible for LOAD [39]. The genome wide association studies (GWAS) indicated that the R47H mutational variant triples the AD risk with 23 % faster dementia rate compared with non-variant carriers [40]. With regard to the functional disruption role of the R47H TREM2 variant, it disrupts the TREM2 ligand binding domain, thereby severely impairing ligand-receptor interactions [41]. Comparative studies on the structures of R47H and wild type TREM2 based on high resolution unearths that Arg47 was a key amino acid residue in 2 loops of complementarity determining region which determines the functional specificity in binding to ligands like lipids and others [42]. Impaired TREM2 and Aβ fibrils binding leads to the accumulation of Aβ plaques, besides weakening of immunity. In addition, immunofluorescence studies in the brain of R47H carriers suggest that fewer microglia have been activated, which results to the distortion of barrier function of microglia [43]. Considering all these findings, it can be inferred that R47H increases the risk of AD. Different variants of TREM2 and its effects on TREM2 ligand binding affinity has been tabulated in Table 1. Results suggested that TREM2 variants, R47H, R62H, G145T, T66M, Y38C, and Q33X, lower the TREM2-ligand binding affinity. But T96K increases the ligand binding affinity, whereas W191X, E151K, and L211P did not exhibit any noticeable effect [23].
Table 1
TREM2 Variants. Different TREM2 variants and its effects on the TREM2-Ligand affinity
| S. No | TREM2 Variants | Effect on TREM2 –Ligand affinity |
| 1 | R47H | Lowers the binding affinity |
| 2 | R62H | Lowers the binding affinity |
| 3 | W191X | NA |
| 4 | E151K | NA |
| 5 | L211P | NA |
| 6 | G145T | Lowers the binding affinity |
| 7 | T96K | Increase the binding affinity |
| 8 | T66M | Lowers the binding affinity |
| 9 | Y38C | Lowers the binding affinity |
| 10 | Q33X | Lowers the binding affinity |
GENESIS AND FUNCTIONAL ROLE OF SOLUBLE TREM2
In vitro and in vivo studies suggest that through the catabolic activities of the sheddases, the extracellular domain of TREM2 can be broken down to generate an independent soluble TREM2 (sTREM2) which regulates the interaction between surrounding microenvironment and neurons [44]. The α-secretase-disintegrin and metalloproteinase domain containing proteins 10 and 17 (ADAM 10 and ADAM 17) [16] are the kind of sheddases which belonged to the metalloproteinase family, which have been exhibited to cleave stalk region of TREM2 resulting to the liberation of immunoglobulin (Ig) domain thereby generating sTREM2 [45]. Mass spectrometry studies of human cultured macrophages suggests that His 157–Ser 158 are the cleavage portions of TREM which are catabolized by ADAM 10 and ADAM 17 respectively [46]. Significantly, a TREM2 gene mutation, H157Y, favors the shedding of TREM2 to generate sTREM2 by ADAM enzymes and can be correlated with an increase in AD risk [47]. After the ADAMs enzymatic activities, the remaining transmembrane domain of TREM2 is further subjected to γ-secretase proteolytic activity [44]. In addition to ADAM-based sTREM2 generation, sTREM2 also generated from alternate splicing of TREM2 transcript, which produces two isoforms, which have been identified in CNS of humans [48].
Generally, after sTREM2 is generated, it is secreted into extracellular/luminal space. Further, it can also settle in varied extracellular spaces, for instance, sTREM2 is generated from microglial TREM2 released into brain parenchyma [49], whereas sTREM2 of monocytes released into the blood.
Pathological role of sTREM2 in AD
The shedding of the ectodomain portion of TREM2 is promoted by various ligands [29]. As indicated earlier, ADAMs-based enzymatic cleavage releases sTREM2, whereas the remnant transmembrane peptide subjected to γ-secretase cleavage which triggers intracellular signal transduction wherein both these processes facilitate microglial activation in the CNS [50]. Upon activation, microglial cells secrets number of cytokines which are both pro- and anti-inflammatory and in turn impact the innate immune system [51]. They exhibited a correlation between sTREM2 and CSF tau/p-tau levels [52, 53]. Intriguingly, these studies suggest a strong correlation between sTREM2 and CSF tau, but not Aβ, therefore, it can be inferred that TREM2 correlates with tauopathy in AD progression. Nevertheless, a research study suggests that levels of CSF sTREM2 correlate with both tau and Aβ, reaching peak levels during the initial stages of clinical impairment. Based on these prevailing studies, it can be suggested that accumulation of sTREM2 during activation of microglia as a response to the pathological events, but not a cause of these events. The current studies significantly indicate that sTREM2 is a potential biomarker with strong therapeutic potential for future exploration.
CROSS TALK OF TREM2 WITH AD PATHOLOGICAL DOMAINS
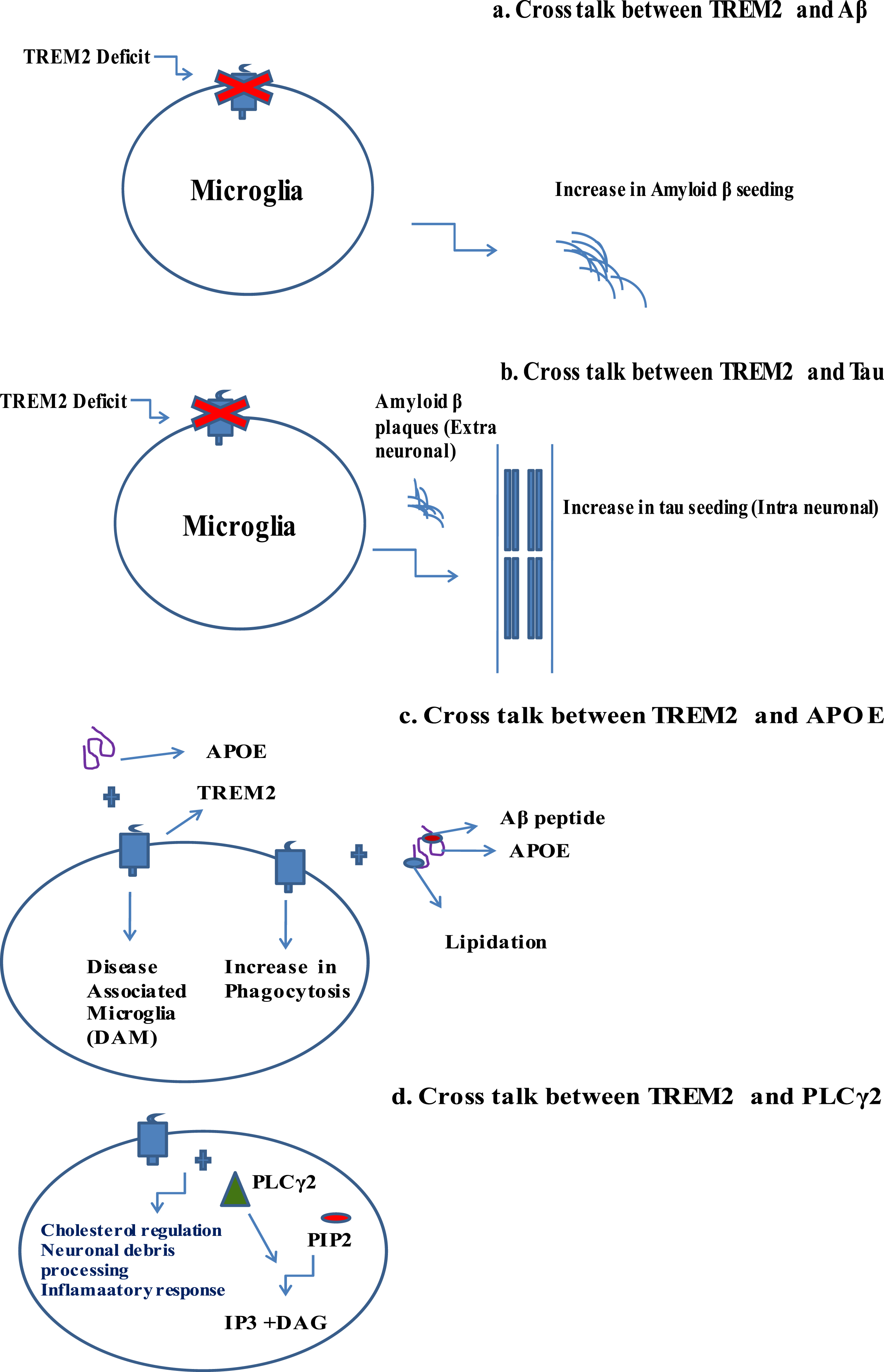
TREM2 interacts with other pathological domains of AD: Aβ, tau, APOE, and PLCγ2. This section of the paper intrinsically assesses the interplay of TREM2 with other domains. The study suggests that the absence of functional TREM2 raises the amyloid plaques in experimental mouse models, where TREM2 deficiency leads to tau seeding and spreading. Both TREM2 and APOE are needed for the differentiation of classical homogenate microglial state to DAM state. Pro522Arg, a rare variant of PLCγ2, lowers the AD risk [54–57]. Pathological implications in cross talk between TREM2 and other AD domains have been analyzed in detail (Fig. 3).
Does TREM2 influence amyloid seeding?
Fig. 3
Pathological implications in cross talk between TREM2 and other AD domains. a) Cross talk between TREM2 and Aβ. b) Cross talk between TREM2 and tau. c) Cross talk between TREM2 and APOE. d) Cross talk between TREM2 and PLCγ2. All these cross talk features play a crucial role in novel pathophysiology leading to neuronal cell dysfunction.
Proliferation and dispersion of misfolded amyloidogenic proteins through varied regions of the brain is the chief feature of many neurodegenerative disorders, and a redolent of prion-like disorders [58]. Under experimental conditions, amyloid seeding and spreading can be triggered by intracerebral injection of homogenates originating from mouse models for varied neurodegenerative disorders or human brain with respective disorders [59]. The functional role of TREM2 is diverse ranging from chemotaxis, dead cells engulfment [60], phagocytosis of Aβ fibrils, and energy metabolism maintenance [61]. The studies based on different mouse models suggests that by suppressing homeostatic mRNA, homeostatic microglial cells transform into DAM by enhancing its transcriptional profile [56]. To ascertain the functional and pathological association between TREM2 and amyloid seeding, recently Parhizkar et al. [54] conducted research studies in different themes. To exemplify the TREM2 role during the early genesis of amyloid cascade, the researchers have used APP/PS1 transgenic mice and conducted the intrahippocampal injection of brain extracts comprising of Aβ. The studies have generated important findings and progressive outcomes. It was reported that lack of functional TREM2 raises the amyloid plaques in experimental mouse models. APP mice, which were non seeded and uninjected, do not exhibited the development of amyloid plaque seeding, but in young age, plaque formation is quite unpredictable. Intense seeding of amyloid plaques was exhibited in the hippocampi of APP/PS1/Trem2+/+ mice, which has been injected with Aβ brain homogenate. Similarly, lack of TREM2 functioning not only led to increase of amyloid plaque seeding but also exhibited more diffuse type Aβ fibrils. TREM2 function also influences the microglial clustering which enriches the amyloid plaque seeding. Contradicting the same, in the case of APP/PS1/Trem2–/– or APP/PS1/Trem2pT66M, clustering of microglia was severely reduced. These findings clearly indicate that microglial clustering around amyloid plaque seeding was inevitably dependent on TREM2 functioning; otherwise, it may lead to deleterious pathophysiological conditions like poor immune defense against the amyloid plaques as the disease associated microglia cannot be transformed in the absence of TREM2 functioning. Parhizkar and colleagues [54] also reported the functional connection between TREM2 and amyloid plaque associated ApoE. Co-deposition of ApoE in amyloid plaques comes under the influence of microglial cells in the way that after the homeostatic microglial conversion to DAM, the same will begin the microglial ApoE expression wherein TREM2 functioning is essential for DAM progression [56]. Corroborating the same, devoid of TREM2, despite the existence of high amyloid plaque load in hippocampal plaques exhibited considerable reduction of amyloid plaque associated ApoE. Based on these findings, it can be analyzed that in mice with loss of TREM2 function, leads to the reduction of microglial cluster around the amyloid seeding, as the former influences the latter’s DAM state, which is the pathophysiological defense in varied neurodegenerative disorders, therefore, reducing the clearing of amyloid plaques by phagocytosis. It also lowers the levels of Aβ associated ApoE. Finally, the significant outcome of this research is loss of TREM2 function promotes amyloid seeding at the early phase of the disease and amyloid plaque fibrillation is highly influenced by the activation of microglial cells, i.e., DAM stage.
Does TREM2 influence tau seeding?
The characteristic pathophysiology of AD is formation of neuritic plaques (NPs), its chief features encompass swollen dystrophic neuronal processes consisting of phosphorylated-tau (p-tau) aggregation, which encircles Aβ deposits [55]. Neurofibrillary tangles (NFTs), which are also one of the pathological features of AD, also encompasses p-tau and are mostly formed in the cerebrum’s medial temporal lobe in persons with healthy cognitive levels. He et al. [62] exhibited that Aβ plaques facilitated seedings of local tau in dystrophic neurites, which in turn favors the genesis of p-tau in NFTs and NPs. This consolidated the idea that Aβ plaques favor the formation of p-tau in NFTs and NPs, and there by progressing of AD. At this juncture, earlier research work concretizes the idea that microgliosis impedes the peri-plaque neuritic dystrophy [63]. Leyns et al. [55] tested that TREM2 loss or reduced microgliosis would accentuate NP tau formation, furthering its aggregation and spreading. They also examined the effects of deficiency of TREM2 and its AD variant TREM2R47H on neuritic plaque tau pathology. The researchers have used a novel mouse model (APP/PS1–21) in different modes with endogenous tau-inducing AD-like features. The research has generated valuable outcomes and insights. The work reported widespread seeding of NP tau aggregates in both ipsilateral and contralateral cortex of APP/PS1–21 mice wherein, the levels of NP tau were higher in T2KO and T2R47H mice comparing to the T2CV and T2WT mice. Significantly, aggregation of neuritic plaques were not reported in the absence of Aβ plaques. The point of inference is Aβ plaques creates conditions favoring neuritic plaques. The studies also examined connection between TREM2 function and NP tau seeding, based on the investigations, they have suggested that Aβ plaques triggers neuronal toxicity there by facilitating the manifestation of a favorable environment which prompts the formation of pathological tau seeding. Researchers also examined the possibility of human AD cases in carriers of AD associated variants of TREM2. In prefrontal cortex tissue, variants of TREM2, R47H and R62H, are risky carriers of LOAD. These findings reported considerably higher p-tau levels which encircles the amyloid plaques in AD risk TREM2 variants. In this work, a novel mouse model has been developed to investigate the pathological effects of TREM2 on both the Aβ and tau aggregation, which was a significant development. The key findings of the work suggests that higher vulnerability to tau seeding and spreading in dystrophic neurons encircling Aβ plaques in cases of TREM2 deficiency and AD risk TREM2 variant, R47H. Finally, the significant outcome of this research suggests that TREM2 facilitates microgliosis around Aβ plaques, thereby limiting the damage inflicting on adjoining neuronal processes, most probably by restricting toxic Aβ42 levels.
TREM2 and APOE
As per the GWAS and whole exome sequencing, over 30 genetic loci were identified as risk factors for AD, wherein APOE and TREM2 were considered as the important genetic attributers of AD [64]. Expressing in glial cells, TREM2 and APOE constituted as part of the major chunk of genes which are associated with LOAD [65]. The functional connection between TREM2 and APOE is reported by Atagi and colleagues [66]. The research group demonstrated that APOE enhances the phagocytosis of apoptotic neurons by dint of TREM2 pathway, whereas the TREM2 R47H variant, which is an AD risk variant was exhibited to lower the affinity of TREM2 binding to APOE. Significantly, lipidation of APOE increases its binding ability to TREM2 and formation of complexes between Aβ and APOE, low density lipoprotein (LDL) or clusterin which enhances the efficiency of microglia in Aβ uptake. Corroboratingly, the same study reported that microglia deficient in TREM2 have a low ability to uptake of Aβ-APOE or Aβ-LDL complexes [67]. Jendresen and colleagues [68] exhibited that amino acid residues between 130–149 of human APOE constituted as the TREM2 binding site and also suggested that specificity in APOE isoforms determines binding with TREM2. TREM2 deficient microglial cells in the way of haplo deficient, gene knockout or TREM2 R47H reported dose dependent declining of microglial activation encircling the Aβ plaques leading to the formation of highly diffuse and less compact Aβ plaques [8].
Consistent to these findings, TREM2 overexpression leads to considerable reduction in Aβ plaques and amelioration of cognitive impairment in 5xFAD mice [69]. Transformation from homeostatic microglia to DAM requires unique transcriptional signatures, wherein APOE and TREM2 were part of this domain. This finding is supported by APP mice deficient in APOE/TREM2 failing to transform into DAM [56]. Therefore, failure in converting to DAM results in impairment of essential immune defenses such as phagocytosis, proliferation, survival, and chemotaxis.
TREM2 and PLCγ2
PLCγ2 is a crucial signaling molecule for different cell surface receptors comprising of immunoreceptor tyrosine-based activation motif (ITAM) as an intracellular domain [70]. GWAS has revealed that rare variant of PLCγ2 gene–Pro522Arg (P522R) found to be associated with lowering AD risk [57]. P522R not only exhibited protective action against AD but also other neurological disorders like Lewy body dementia and FTD. It is very much interesting that people carrying the P522R variant may have a longer longevity at least 90 years of age and with healthy cognitive levels. Surprisingly, the variant of PCLγ2 had no significant neuroprotective effect in certain neurological disorders, for instance, amyotrophic lateral sclerosis, progressive supranuclear palsy, Parkinson’s disease, or multiple sclerosis [71].
PLCγ2 catabolizes the membrane phospholipid phosphatidylinositol 4,5-bisphosphate releasing IP3 and DAG [72]. Andreone and colleagues reported that PLCγ2 acts as a significant node for functioning of TREM2 and inflammatory response in human microglial cells. Signaling of PLCγ2 downstream of TREM2 facilitates induced pluripotent stem cell-derived microglial like (iMG) cells. The study demonstrated that based on the quantification of phosphorylated SYK levels at amino acid residues Y525 and Y526 in cells of WT, TREM2, and PLCγ2 KO iMG. The researchers also suggested that PLCγ2 is needed for TREM2 dependent metabolism of cholesterol regulation. Other important findings included that TREM2 and PLCγ2 were needed for neuronal debris processing and, most importantly, that PLCγ2 facilitates inflammatory response by dint of TREM2 independent signaling. These research findings open new horizons in AD pathophysiology through TREM2 and PLCγ2. Catabolic activity of PLCγ2 depends on its phosphorylation; ITAM performs this task by recruiting kinases such as Bruton’s tyrosine kinase, SYK, and B cell linker. The resultant DAG and IP3 mediate secondary signaling process for both membrane bound immunological receptor, and growth factor receptor [69] also embodies PKC activation and release of intracellular calcium [73]. The secondary messages of PLCγ2 are needed for varied membrane functions, for instance, endocytosis, cellular proliferation, and calcium flux [70].
Increased expression of PLCγ2 was reported based on comparative gene expression profiling with regard to established genetic risks in AD [74]. Similarly, sequencing of single microglia pertaining to two different AD disease models of mice exhibited either Aβ or tau pathology also suggested the AD risk gene expression which also comprises of PLCγ2 gene [75]. Of late, recombinant human induced pluripotent stem cell-derived microglial like cells exhibited that activity of PLCγ2 in microglial cells regulates a wide array of processes downstream of TREM2 comprising of myelin phagocytosis, survival, lipid metabolism, and processing of inflammation downstream of TLRs [76]. Summation of all these findings suggests that PLCγ2 may play a significant role in pathophysiology of AD.
BIOLOGY OF MICROGLIA
Microglial cells are the resident phagocytic cells of the CNS exhibit neuronal inflammation [77]. Microglial cells attain two morphological forms: inactive form, which is meant for surveillance; and active form, which in turn, exists in two states, M1 and M2 [78]. Pro-inflammatory cytokines, for instance, Interleukin-1β (IL-1β), Interleukin-1 (IL-1), and TNF are secreted by M1 form, whereas M2 active stage can functions as anti-inflammatory component, repairing of tissues and ingestion of cellular debris [79]. Disproportionation between pro- and anti-inflammatory states of microglial cells results to chronic inflammation, which is a severe pathological state and different from that of acute inflammation, which is normal defense response of the brain against pathological abnormalities and injuries [80]. Adding to the AD pathophysiological heterogeneity, microglia and TREM2 receptor emerged as novel pathway in AD pathogenesis.
The percentage of microglia is less in total population of glial cells within the brain and in healthy CNS microglial cells prevails in resting state [81]. The activated microglial cells during pathological conditions undergo morphological alterations and secrets chemokines and cytokines, there by affecting adjoining cells. The microglial activities in AD comprises clearing of Aβ deposits, stimulating phagocytosis and secretion of cytotoxic mediators. Activation of microglia by Aβ under in vitro conditions triggers the expression of pro-inflammatory cytokines comprising of interleukins: IL-6, IL-8, IL-1β, TNF-α, reactive nitrogen and oxygen species, and chemokines, all contributes to neuronal damage [82].
Microglia expresses different kinds of receptors such as receptor for advanced glycosylation end products, CD36, TREM2, TLRs, Scavenger receptors (SR - A I/II), Fc receptors and complement receptors, all these receptors function in a cooperative manner and are involved in the recognition process, internalization and clearance of Aβ, and activation of cells [83].
DISEASE ASSOCIATED MICROGLIA AND TREM2
A novel and unique subset of microglial cells of CNS was reported by Keren-Shaul et al. [56] as DAM, and their findings were based on comprehensive single cell RNA analysis of immune cells in CNS in neurodegenerative pathological conditions. DAM exhibited distinct transcriptional and functional signatures. Under neurodegenerative conditions, microglial cells transform into DAM [43] and may play a neuroprotective role [56]. Many of the genes which were expressing in the DAM were identified in human GWAS to be linked to AD and other kinds of neurodegenerative conditions, which also comprise TREM2 membrane receptor, needed for activation of DAM [84]. Neurodegenerative studies conducted in mouse models deficient in TREM2 receptor exhibited that TREM2 signaling is imperative for microglial cells to recognize and respond to neurodegenerative conditioning. Based on the microglial analysis from TREM2 deficient mouse model (5xFAD and APP/PS1), differentiation of DAM is a dual sequential process. In stage 1, which is a TREM2 independent process, transition of homeostatic microglia into stage 1 DAM is facilitated by unknow signals, whereas stage 1 DAM differentiates into stage 2 DAM through TREM2 signal [85]. TREM2 dependent signaling step embodies upregulation of phagocytic, lysosomal, and lipid metabolic pathways, for instance, Lpl, Itgax, Axl, and Cst7. TREM2 signaling pathway which facilitates the differentiation from stage 1 DAM to stage 2 DAM is still obscure. Different hypotheses were proposed to describe how signaling process of TREM2 transform stage 1 DAM to stage 2 DAM. Research studies which exhibited that TREM2 tend to be pro survival and pro proliferative through the secretion of β-catenin, PI3K, and mTOR pathways [9, 12, 21]. The finding supported the hypothesis that TREM2 may sustain microglial activation triggered by other receptors in stage 1 DAM. Molecular characteristics of DAM includes expression of Iba1, Hexb, and Cst3, which are typical microglial markers [22].
OUR HYPOTHESIS
Microglial TREM2 receptors and its associated DAP adaptors driven signaling pathways ameliorates amyloid-β aggregates in Alzheimer’s disease (Fig. 4)
Fig. 4
Hypothetical mechanism of amelioratory role of TREM2 microglial receptors in Amyloid β degradation in Alzheimer’s disease. TREM2, Triggering receptor expressed on myeloid cells 2; DAP 12 and 10, DNAX activation proteins 12 and 10; ITAM, Immunoreceptor tyrosine based activation motif; YINM, tyrosine-isoleucine-asparagine-methionine; SYK, Spleen tyrosine kinase; PI3K, Phosphatidyl inositol 3-kinase; PLCγ2, Phosphatidyl inositol specific phospholipase Cγ2
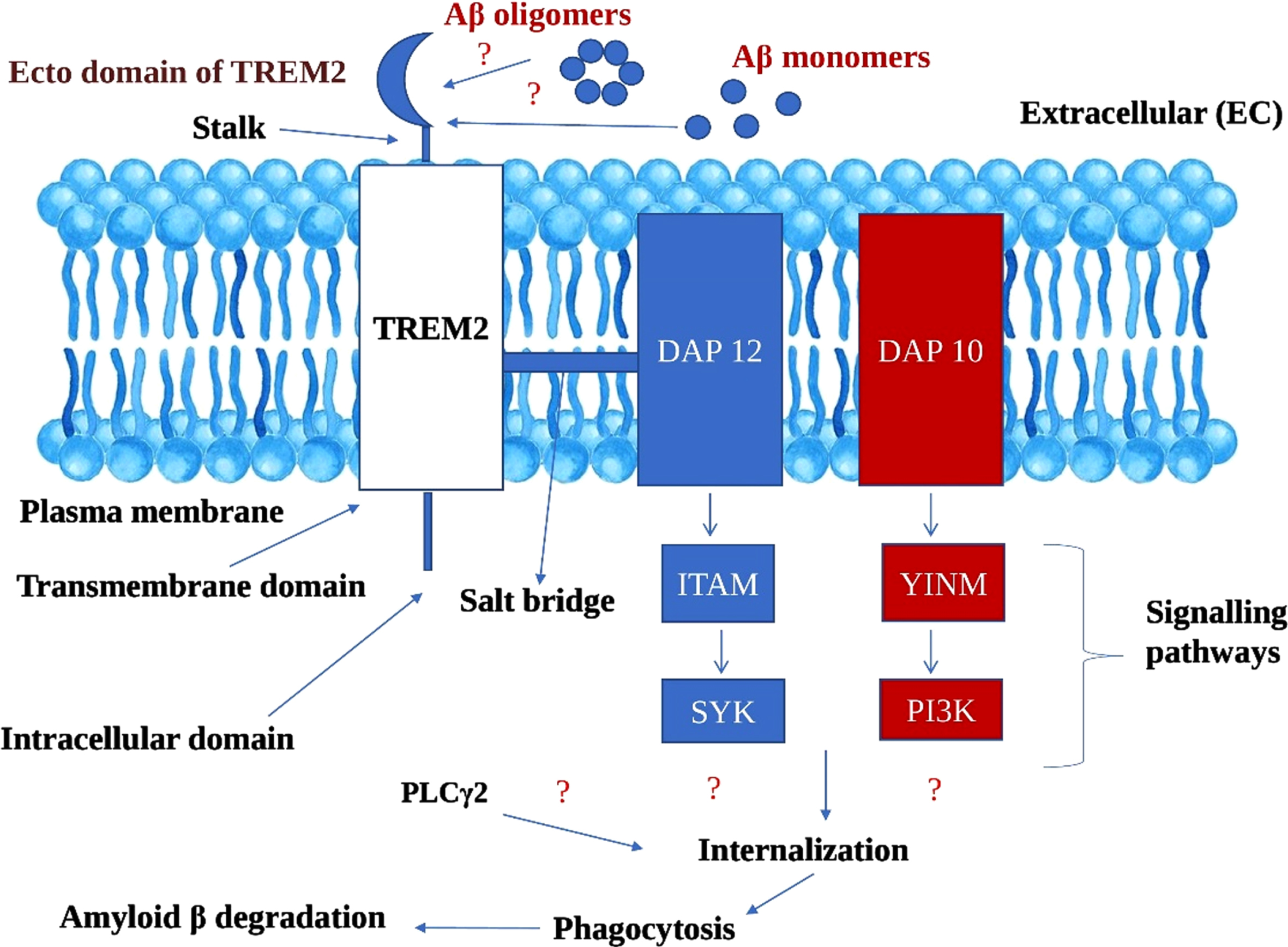
We propose a hypothesis that ecto domain of TREM2 binds to both Aβ oligomers and monomers, though it may be with differential efficacy. We assume that the binding ability of Aβ oligomers with TREM2 ecto domain is high owing to its soluble nature, and therefore imperative in obstructing the fibrillation Aβ which is highly neurotoxic and furthering the Aβ plaque formation. Dual signaling cascading mechanisms derived from DAP adaptor proteins (DAP12 and DAP10) which are linked to TREM2. The signaling pathways derived from DAP12 and DAP10 encompasses: a) DAP12 ⟶ ITAM ⟶ SYK; b) DAP10 ⟶ Tyrosine-Isoleucine-Asparagine-Methionine (YINM) ⟶ PI3K [16].
We explain that signaling pathways of two adaptor proteins, i.e., DAP12 and DAP10, either collaboratively or independently recruit other endogenous compounds like PLCγ2, leading to the chain of events. During internalization, through endocytosis process, Aβ were drawn inside the cell followed by lysosomal phagocytosis. The catabolic activities of lysosomal phagocytosis culminates in Aβ degradation. Though TREM2 differentially binds to different forms of Aβ, we argue that the TREM2-based signaling cascade has the potentiality to ameliorate the Aβ burden. As of now, the exact mechanism of Aβ phagocytosis by microglial cells is still obscure and complex, but our proposed hypothesis will give a new direction in understanding the novel pathophysiological features of AD.
CONCLUSION
The review systematically assesses the functional and pathological role of TREM2 and microglial cells in AD. This articulates that Aβ binding of TREM2 receptor activates the microglial cells, thereby phagocytizing the Aβ fibrils, which may otherwise lead to the accumulation of Aβ plaques, in turn culminating in AD. Most importantly, TREM2 is indispensable for microglial cells to acquire DAM state, which exhibits neuroprotective role in neurological disorders like AD. R47H, a deleterious mutant variant of TREM2, severely impairs the TREM2 ligand binding domain (ecto domain) resulting in accumulation of Aβ plaques. Chronic inflammation is also a matter of serious pathological concern in AD, which is the resultant of disproportionation between pro- and anti-inflammatory molecules. Considering the imperative functional and pathological role of TREM2 and microglial cells in AD, the review suggests the TREM2 and microglial cells centric therapeutic measures for future direction to face AD.
FUTURE THERAPEUTIC DIRECTIONS
1. Overexpression of TREM2 in microglial cells enhances Aβ phagocytosis. The crux point for this therapeutic measure is overexpression of TREM2 gene resulting in the generation of a greater number of TREM2 receptors which facilitates the phagocytosis of accumulating loads of Aβ plaques.
2. Administration of nanoparticles can act as intermediaries in phagocytotic signaling pathway, thereby enhancing the Aβ phagocytosis by microglial cells. In addition, administration of engineered cytokines induces the clump formation of microglial cells around Aβ plaques, which secretes anti-plaque signaling, therefore prohibiting proliferation of Aβ plaques complexes.
3. Administration of anti-inflammatory cytokines in AD patients may also serve as a prudent therapeutic strategy that prohibits the chronic neuronal inflammation, which have deleterious pathological consequences in AD.
Finally, the review suggests that owing to the complex and heterogenous pathophysiology of AD, holistic and combinational therapeutic measures coupled with early diagnosis has to be framed and worked out against AD.
ACKNOWLEDGMENTS
SK Chand Basha is thankful and grateful to KLEF [KLU] Post-doctoral fellowship, to my mother, though she left me, but her blessings are always with me and also grateful to Almighty for guiding and showing the right path. M. Janaki Ramaiah is thankful to KLEF management for constant encouragement and Rao KS is thankful to SNI –SENACYT, Panama and KLEF-India for the support.
FUNDING
The current work was supported by KLEF [KLU] Post-doctoral fellowship awarded to SK Chand Basha.
CONFLICT OF INTEREST
Rao KS is an Editorial board member of this journal but was not involved in the peer review process nor had access to any information regarding its peer-review.
REFERENCES
[1] | Sirkis DW , Bonham LW , Johnson TP , La Joie R , Yokoyama JS ((2022) ) Dissecting the clinical heterogeneity of early-onset Alzheimer’s disease. Mol Psychiatry 27: , 2674–2688. |
[2] | Tiwari S , Atluri V , Kaushik A , Yndart A , Nair M ((2019) ) Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int J Nanomed 19: , 5541–5554. |
[3] | Patterson C (2018)World Alzheimer Report 2018. The State of the Art of Dementia Research: New Frontiers. An Analysis of Prevalence, Incidence, Cost and Trends. Alzheimer’s Disease International, London. |
[4] | Lane CA , Hardy J , Schott JM ((2018) ) Alzheimer’s disease. Eur J Neurol 25: , 59–70. |
[5] | Guerreiro R , Wojtas A , Bras J , Carrasquillo M , Rogaeva E , Majounie E , Cruchaga C , Sassi C , Kauwe JS , Younkin S , Hazrati L , Collinge J , Pocock J , Lashley T , Williams J , Lambert JC , Amouyel P , Goate A , Rademakers R , Morgan K , Powell J , StGeorge-Hyslop P , Singleton A , Hardy J ; Alzheimer Genetic Analysis Group ((2013) ) TREM2 variants in Alzheimer’s disease. N Engl JMed 368: , 117–127. |
[6] | Jonsson T , Stefansson H , Steinberg S , Jonsdottir I , Jonsson PV , Snaedal J , Bjornsson S , Huttenlocher J , Levey AI , Lah JJ , Rujescu D , Hampel H , Giegling I , Andreassen OA , Engedal K , Ulstein I , Djurovic S , Ibrahim-Verbaas C , Hofman A , Ikram MA , van Duijn CM , Thorsteinsdottir U , Kong A , Stefansson K ((2013) ) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368: , 107–116. |
[7] | Schmid CD ((2002) ) Heterogeneous expression of the triggering receptor expressed on myeloid cells-2 on adult murine microglia. J Neurochem 83: , 1309–1320. |
[8] | Zhao Y , Wu X , Li X , Jiang LL , Gui X , Liu Y , Sun Y , Zhu B , Piña-Crespo JC , Zhang M , Zhang N , Chen X , Bu G , An Z , Huang TY , Xu H ((2018) ) TREM2 is a receptor for β-amyloid that mediatesmicroglial function. Neuron 97: , 1023–1031. |
[9] | Ulland TK , Song WM , Huang SC , Ulrich JD , Sergushichev A , Beatty WL , Loboda AA , Zhou Y , Cairns NJ , Kambal A , Loginicheva E , Gilfillan S , Cella M , Virgin HW , Unanue ER , Wang Y , Artyomov MN , Holtzman DM , Colonna M ((2017) ) TREM2 maintains microglial metabolic fitness in Alzheimer’s disease. Cell 170: , 649–663. |
[10] | Klünemann HH , Ridha BH , Magy L , Wherrett JR , Hemelsoet DM , Keen RW , De Bleecker JL , Rossor MN , Marienhagen J , Klein HE , Peltonen L , Paloneva J ((2005) ) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2 Neurology 64: , 1502–1507. |
[11] | Hakola HP ((1972) ) Neuropsychiatric and genetic aspects of a new hereditary disease characterized by progressive dementia and lipomembranous polycystic osteodysplasia. Acta Psychiatr Scand 232: , 1–173. |
[12] | Otero K , Turnbull IR , Poliani PL , Vermi W , Cerutti E , Aoshi T , Tassi I , Takai T , Stanley SL , Miller M , Shaw AS , Colonna M ((2009) ) Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol 10: , 734–743. |
[13] | Wang Y , Cella M , Mallinson K , Ulrich JD , Young KL , Robinette ML , Gilfillan S , Krishnan GM , Sudhakar S , Zinselmeyer BH , Holtzman DM , Cirrito JR , Colonna M ((2015) ) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160: , 1061–1071. |
[14] | Jay TR , Hirsch AM , Broihier ML , Miller CM , Neilson LE , Ransohoff RM , Lamb BT , Landreth GE ((2017) ) Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J Neurosci 37: , 637–647. |
[15] | Leyns CEG , Ulrich JD , Finn MB , Stewart FR , Koscal LJ , Remolina Serrano J , Robinson GO , Anderson E , Colonna M , Holtzman DM ((2017) ) TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc Natl Acad Sci U S A 114: , 11524–11529. |
[16] | Ulland TK , Colonna M ((2018) ) TREM2 - a key player in microglial biology and Alzheimer disease. Nat Rev Neurol 14: , 667–675. |
[17] | Andrea Steiner (2020) NMR studies on the disease-linked single-pass membrane proteins TREM2 and TNFa. Technische Universität München, Munich, Germany (Ph.D. Thesis). |
[18] | Colonna M ((2003) ) TREMs in the immune system and beyond. Nat Rev Immunol 3: , 445–453. |
[19] | Kober DL , Alexander-Brett JM , Karch CM , Cruchaga C , Colonna M , Holtzman MJ , Brett TJ ((2016) ) Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. eLife 5: , e20391. |
[20] | Xing J , Titus A , Humphrey MB ((2015) ) The TREM2-DAP12 signaling pathway in Nasu–Hakola disease: A molecular genetics perspective. Res Rep Biochem 5: , 89–100. |
[21] | Peng Q , Malhotra S , Torchia JA , Kerr WG , Coggeshall KM , Humphrey MB ((2010) ) TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal 3: , ra38. |
[22] | Butovsky O , Jedrychowski MP , Moore CS , Cialic R , Lanser AJ , Gabriely G , Koeglsperger T , Dake B , Wu PM , Doykan CE , Fanek Z , Liu L , Chen Z , Rothstein JD , Ransohoff RM , Gygi SP , Antel JP , Weiner HL ((2014) ) Identification of a unique TGF-b-dependent molecular and functional signature in microglia. Nat Neurosci 17: , 131–143. |
[23] | Yang J , Fu Z , Zhang X , Xiong M , Meng L , Zhang Z ((2020) ) TREM2 ectodomain and its soluble form in Alzheimer’s disease. J Neuroinflammation 17: , 204. |
[24] | Zhong L , Wang Z , Wang D , Wang Z , Martens YA , Wu L , Xu Y , Wang K , Li J , Huang R , Can D , Xu H , Bu G , Chen XF ((2018) ) Amyloid-beta modulates microglial responses by binding to the triggering receptor expressed on myeloid cells 2 (TREM2). Mol Neurodegener 13: , 15. |
[25] | Lessard CB , Malnik SL , Zhou Y , Ladd TB , Cruz PE , Ran Y , Mahan TE , Chakrabaty P , Holtzman DM , Ulrich JD , Colonna M , Golde TE ((2018) ) High-affinity interactions and signal transduction between Aβ oligomers and TREM2. EMBO Mol Med 10: , e9027. |
[26] | Konishi H , Kiyama H ((2018) ) Microglial TREM2/DAP12 signaling: A double-edged sword in neural diseases. Front Cell Neurosci 12: , 206. |
[27] | Turnbull IR , Gilfillan S , Cella M , Aoshi T , Miller M , Piccio L , Hernandez M , Colonna M ((2006) ) Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 177: , 3520–3524. |
[28] | Hamerman JA , Tchao NK , Lowell CA , Lanier LL ((2005) ) Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol 6: , 579–586. |
[29] | Kober DL , Brett TJ ((2017) ) TREM2-Ligand Interactions in Health and Disease. J Mol Biol 429: , 1607–1629. |
[30] | Kane KP , Lavender KJ , Ma BJ ((2004) ) Ly-49 receptors and their functions. Crit Rev Immunol 24: , 321–348. |
[31] | McVicar DW , Taylor LS , Gosselin P , Willette-Brown J , Mikhael AI , Geahlen RL , Nakamura MC , Linnemeyer P , Seaman WE , Anderson SK , Ortaldo JR , Mason LH ((1998) ) DAP12-mediated signal transduction in natural killer cells. A dominant role for the Syk protein-tyrosine kinase. J Biol Chem 273: , 32934–32942. |
[32] | Orr SJ , McVicar DW ((2011) ) LAB/NTAL/Lat2: A force to be reckoned with in all leukocytes?J Leukoc Biol 89: , 11–19. |
[33] | Whittaker GC , Orr SJ , Quigley L , Hughes L , Francischetti IM , Zhang W , McVicar DW ((2010) ) The linker for activation of B cells (LAB)/non-T cell activation linker (NTAL) regulates triggering receptor expressed on myeloid cells (TREM)-2 signaling and macrophage inflammatory responses independently of the linker for activation of T cells. J Biol Chem 285: , 2976–2985. |
[34] | Paloneva J , Manninen T , Christman G , Hovanes K , Mandelin J , Adolfsson R , Bianchin M , Bird T , Miranda R , Salmaggi A , Tranebjaerg L , Konttinen Y , Peltonen L ((2002) ) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 71: , 656–662. |
[35] | Cuyvers E , Bettens K , Philtjens S , Van Langenhove T , Gijselinck I , van der Zee J , Engelborghs S , Vandenbulcke M , Van Dongen J , Geerts N , Maes G , Mattheijssens M , Peeters K , Cras P , Vandenberghe R , De Deyn PP , Van Broeckhoven C , Cruts M , Sleegers K ; BELNEU consortium ((2014) ) Investigating the role of rare heterozygous TREM2 variants in Alzheimer’s disease and frontotemporal dementia. Neurobiol Aging 35: , 726.e11–726.e19. |
[36] | Le Ber I , De Septenville A , Guerreiro R , Bras J , Camuzat A , Caroppo P , Lattante S , Couarch P , Kabashi E , Bouya-Ahmed K , Dubois B , Brice A ((2014) ) Homozygous TREM2 mutation in a family with atypical frontotemporal dementia. Neurobiol Aging 35: , 2419.e2423–2419.e2425. |
[37] | Paloneva J , Mandelin J , Kiialainen A , Bohling T , Prudlo J , Hakola P , Haltia M , Konttinen YT , Peltonen L ((2003) ) DAP12/TREM2 deficiency results in impaired osteoclast differentiation and osteoporotic features. J Exp Med 198: , 669–675. |
[38] | Numasawa Y , Yamaura C , Ishihara S , Shintani S , Yamazaki M , Tabunoki H , Satoh JI ((2011) ) Nasu-Hakola disease with a splicing mutation of TREM2 in a Japanese family. Eur J Neurol 18: , 1179–1183. |
[39] | Luis EO , Ortega-Cubero S , Lamet I , Razquin C , Cruchaga C , Benitez BA , Lorenzo E , Irigoyen J ; Alzheimer’s Disease Neuroimaging Initiative (ADNI), Pastor MA , Pastor P ((2014) ) Frontobasal gray matter loss is associated with the TREM2 p.R47H variant. Neurobiol Aging 35: , 2681–2690. |
[40] | Del-Aguila JL , Fernández MV , Schindler S , Ibanez L , Deming Y , Ma S , Saef B , Black K , Budde J , Norton J , Chasse R ; Alzheimer’s DiseaseNeuroimaging Initiative (ADNI), Harari O , Goate A , Xiong C , Morris JC , Cruchaga C ((2018) ) Assessment of the genetic architecture ofAlzheimer’s disease risk in rate of memory decline. JAlzheimers Dis 62: , 745–756. |
[41] | Sudom A , Talreja S , Danao J , Bragg E , Kegel R , Min X , Richardson J , Zhang Z , Sharkov N , Marcora E , Thibault S , Bradley J , Wood S , Lim AC , Chen H , Wang S , Foltz IN , Sambashivan S , Wang Z ((2018) ) Molecular basis for the loss-of-function effects of the Alzheimer’s disease-associated R47H variant of the immune receptor TREM2. J Biol Chem 293: , 12634–12646. |
[42] | Park JS , Ji IJ , Kim DH , An HJ , Yoon SY ((2016) ) The Alzheimer’s disease-associated R47H variant of TREM2 has an altered glycosylation pattern and protein stability. Front Neurosci 10: , 618. |
[43] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[44] | Wunderlich P , Glebov K , Kemmerling N , Tien NT , Neumann H , Walter J ((2013) ) Sequential proteolytic processing of the triggering receptor expressed on myeloid cells-2 (TREM2) protein by ectodomain shedding and gamma secretase-dependent intramembranous cleavage. J Biol Chem 288: , 33027–33036. |
[45] | Lichtenthaler SF , Lemberg MK , Fluhrer R ((2018) ) Proteolytic ectodomain shedding of membrane proteins in mammals-hardware, concepts, and recent developments. EMBO J 37: , e99456. |
[46] | Schlepckow K , Kleinberger G , Fukumori A , Feederle R , Lichtenthaler SF , Steiner H , Haass C ((2017) ) An Alzheimer-associated TREM2 variant occurs at the ADAM cleavage site and affects shedding and phagocytic function. EMBO Mol Med 9: , 1356–1365. |
[47] | Thornton P , Sevalle J , Deery MJ , Fraser G , Zhou Y , Ståhl S , Franssen EH , Dodd RB , Qamar S , Gomez Perez-Nievas B , Nicol LS , Eketjäll S , Revell J , Jones C , Billinton A , St George-Hyslop PH , Chessell I , Crowther DC ((2017) ) TREM2 shedding by cleavage at the H157-S158 bond is accelerated for the Alzheimer’s disease-associated H157Y variant. EMBO Mol Med 9: , 1366–1378. |
[48] | Del-Aguila JL , Benitez BA , Li Z , Dube U , Mihindukulasuriya KA , Budde JP , Farias FHG , Fernández MV , Ibanez L , Jiang S , Perrin RJ , Cairns NJ , Morris JC , Harari O , Cruchaga C ((2019) ) TREM2 braintranscript-specific studies in AD and TREM2 mutation carriers. Mol Neurodegener 14: , 18. |
[49] | Tan YJ , Ng ASL , Vipin A , Lim JKW , Chander RJ , Ji F , Qiu Y , Ting SKS , Hameed S , Lee TS , Zeng L , Kandiah N , Zhou J ((2017) ) Higher peripheral TREM2 mRNA levels relate to cognitive deficits and hippocampal atrophy in Alzheimer’s disease and amnestic mild cognitive impairment. J Alzheimers Dis 58: , 413–423. |
[50] | Finelli D , Rollinson S , Harris J , Jones M , Richardson A , Gerhard A , Snowden J , Mann D , Pickering-Brown S ((2015) ) TREM2 analysis and increased risk of Alzheimer’s disease. Neurobiol Aging 36: , 546.e9–13. |
[51] | Wes PD , Sayed FA , Bard F , Gan L ((2016) ) Targeting microglia for the treatment of Alzheimer’s disease. Glia 64: , 1710–1732. |
[52] | Piccio L , Deming Y , Del-Águila JL , Ghezzi L , Holtzman DM , Fagan AM , Fenoglio C , Galimberti D , Borroni B , Cruchaga C ((2016) ) Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease andassociated with mutation status. Acta Neuropathol 131: , 925–933. |
[53] | Suárez-Calvet M , Kleinberger G , Araque Caballero MÁ , Brendel M , Rominger A , Alcolea D , Fortea J , Lleó A , Blesa R , Gispert JD , Sánchez-Valle R , Antonell A , Rami L , Molinuevo JL , Brosseron F , Traschütz A , Heneka MT , Struyfs H , Engelborghs S , Sleegers K , Van Broeckhoven C , Zetterberg H , Nellgård B , Blennow K , Crispin A , Ewers M , Haass C ((2016) ) sTREM2 cerebrospinal fluid levels are apotential biomarker for microglia activity in early-stageAlzheimer’s disease and associate with neuronal injury markers. EMBO Mol Med 8: , 466–476. |
[54] | Parhizkar S , Arzberger T , Brendel M , Kleinberger G , Deussing M , Focke C , Nuscher B , Xiong M , Ghasemigharagoz A , Katzmarski N , Krasemann S , Lichtenthaler SF , Müller SA , Colombo A , Monasor LS , Tahirovic S , Herms J , Willem M , Pettkus N , Butovsky O , Bartenstein P , Edbauer D , Rominger A , Ertürk A , Grathwohl SA , Neher JJ , Holtzman DM , Meyer-Luehmann M , Haass C ((2019) ) Loss of TREM2 functionincreases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci 22: , 191–204. |
[55] | Leyns CEG , Gratuze M , Narasimhan S , Jain N , Koscal LJ , Jiang H , Manis M , Colonna M , Lee VMY , Ulrich JD , Holtzman DM ((2019) ) TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci 22: , 1217–1222. |
[56] | Keren-Shaul H , Spinrad A , Weiner A , Matcovitch-Natan O , Dvir-Szternfeld R , Ulland TK , David E , Baruch K , Lara-Astaiso D , Toth B , Itzkovitz S , Colonna M , Schwartz M , Amit IA ((2017) ) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169: , 1276–1290. |
[57] | Sims R , van der Lee SJ , Naj AC , Bellenguez C , Badarinarayan N , Jakobsdottir J , Kunkle BW , Boland A , Raybould R , Bis JC , et al. ((2017) ) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49: , 1373–1384. |
[58] | Jucker M , Walker LC ((2013) ) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: , 45–51. |
[59] | Meyer-Luehmann M , Coomaraswamy J , Bolmont T , Kaeser S , Schaefer C , Kilger E , Neuenschwander A , Abramowski D , Frey P , Jaton AL , Vigouret JM , Paganetti P , Walsh DM , Mathews PM , Ghiso J , Staufenbiel M , Walker LC , Jucker M ((2006) ) Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 22: , 1781–1784. |
[60] | Mazaheri F , Snaidero N , Kleinberger G , Madore C , Daria A , Werner G , Krasemann S , Capell A , Trümbach D , Wurst W , Brunner B , Bultmann S , Tahirovic S , Kerschensteiner M , Misgeld T , Butovsky O , Haass C ((2017) ) TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep 18: , 1186–1198. |
[61] | Kleinberger G , Brendel M , Mracsko E , Wefers B , Groeneweg L , Xiang X , Focke C , Deußing M , Suárez-Calvet M , Mazaheri F , Parhizkar S , Pettkus N , Wurst W , Feederle R , Bartenstein P , Mueggler T , Arzberger T , Knuesel I , Rominger A , Haass C ((2017) ) The FTD-likesyndrome causing TREM2 T66M mutation impairs microglia function,brain perfusion, and glucose metabolism. EMBO J 36: , 1837–1853. |
[62] | He Z , Guo JL , McBride JD , Narasimhan S , Kim H , Changolkar L , Zhang B , Gathagan RJ , Yue C , Dengler C , Stieber A , Nitla M , Coulter DA , Abel T , Brunden KR , Trojanowski JQ , Lee VM-Y ((2018) ) Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 24: , 29–38. |
[63] | Yuan P , Condello C , Keene CD , Wang Y , Bird TD , Paul SM , Luo W , Colonna M , Baddeley D , Grutzendler J ((2016) ) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 92: , 252–264. |
[64] | Shi Y , Holtzman DM ((2018) ) Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol 18: , 759–772. |
[65] | Villegas-Llerena C , Phillips A , Garcia-Reitboeck P , Hardy J , Pocock JM ((2016) ) Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol 36: , 74–81. |
[66] | Atagi Y , Liu CC , Painter MM , Chen XF , Verbeeck C , Zheng H , Li X , Rademakers R , Kang SS , Xu H , Younkin S , Das P , Fryer JD , Bu G ((2015) ) Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem 290: , 26043–26050. |
[67] | Yeh FL , Wang Y , Tom I , Gonzalez LC , Sheng M ((2016) ) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron 91: , 328–340. |
[68] | Jendresen C , Årskog V , Daws MR , Nilsson LNG ((2017) ) The Alzheimer’s disease risk factors apolipoprotein E and TREM2 are linked in a receptor signaling pathway. J Neuroinflammation 14: , 59. |
[69] | Nishizuka Y ((1995) ) Protein kinase C and lipid signaling for sustained cellular responses. FASEB J 9: , 484–496. |
[70] | Jackson JT , Mulazzani E , Nutt SL , Masters SL ((2021) ) The role of PLCγ2 in immunological disorders, cancer, and neurodegeneration. J Biol Chem 297: , 100905. |
[71] | Van der Lee SJ , Conway OJ , Jansen I , Carrasquillo MM , Kleineidam L , van den Akker E , Hernández I , van Eijk KR , Stringa N , Chen JA , et al. ((2019) ) A nonsynonymous mutation in PLCG2 reduces the risk of Alzheimer’s disease, dementia with Lewy bodies and frontotemporal dementia, and increases the likelihood of longevity. Acta Neuropathol 138: , 237–250. |
[72] | Berridge MJ ((1993) ) Inositol trisphosphate and calcium signalling. Nature 361: , 315–325. |
[73] | Hernandez D , Egan SE , Yulug IG , Fisher EM ((1994) ) Mapping the gene that encodes phosphatidylinositol-specific phospholipase C-gamma 2 in the human and the mouse. Genomics 23: , 504–507. |
[74] | Castillo E , Leon J , Mazzei G , Abolhassani N , Haruyama N , Saito T , Saido T , Hokama M , Iwaki T , Ohara T , Ninomiya T , Kiyohara Y , Sakumi K , LaFerla FM , Nakabeppu Y ((2017) ) Comparative profiling of cortical gene expression in Alzheimer’s disease patients and mouse models demonstrates a link between amyloidosis and neuroinflammation. Sci Rep 7: , 17762. |
[75] | Sierksma A , Lu A , Mancuso R , Fattorelli N , Thrupp N , Salta E , Zoco J , Blum D , Buée L , De Strooper B , Fiers M ((2020) ) Novel Alzheimerrisk genes determine the microglia response to amyloid-β butnot to TAU pathology. EMBO Mol Med 12: , e10606. |
[76] | Andreone BJ , Przybyla L , Llapashtica C , Rana A , Davis SS , van Lengerich B , Lin K , Shi J , Mei Y , Astarita G , Di Paolo G , Sandmann T , Monroe KM , Lewcock JW ((2020) ) Alzheimer’s associated PLCγ2 is a signaling node required for both TREM2 function and the inflammatory response in human microglia. Nat Neurosci 23: , 927–938. |
[77] | Sarma JD ((2014) ) Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J Neurovirology 20: , 122–136. |
[78] | Davalos D , Grutzendler J , Yang G , Kim JV , Zuo Y , Jung S , Littman DR , Dustin ML , Gan W-B ((2005) ) ATP mediates rapid microglial response tolocal brain injury in vivo. Nat Neurosci 8: , 752–758. |
[79] | Stansley B , Post J , Hensley K ((2012) ) A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J Neuroinflammation 9: , 115. |
[80] | Ferreira ST , Clarke JR , Bomfim TR , De Felice FG ((2014) ) Inflammation, defective insulin signaling, and neuronal dysfunction in Alzheimer’s disease. Alzheimers Dement 10: , S76–S83. |
[81] | Lawson LJ , Perry VH , Gordon S ((1992) ) Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48: , 405–415. |
[82] | Fernandez PL , Britton GB , Rao KS ((2013) ) Potential immunotargets for Alzheimer’s disease treatment strategies. J Alzheimers Dis 33: , 297–312. |
[83] | Doens D , Fernández PL ((2014) ) Microglia receptors and theirimplications in the response to amyloid β for Alzheimer’sdisease pathogenesis. J Neuroinflammation 11: , 48. |
[84] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , et al. P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452–1458. |
[85] | Deczkowska A , Keren-Shaul H , Weiner A , Colonna M , Schwartz M , Amit I ((2018) ) Disease-associated microglia: A universal immune sensor of neurodegeneration. Cell 173: , 1073–1081. |


