
Microglia-Mediated Neurovascular Unit Dysfunction in Alzheimer’s Disease
Abstract
The neurovascular unit (NVU) is involved in the pathological changes in Alzheimer’s disease (AD). The NVU is a structural and functional complex that maintains microenvironmental homeostasis and metabolic balance in the central nervous system. As one of the most important components of the NVU, microglia not only induce blood-brain barrier breakdown by promoting neuroinflammation, the infiltration of peripheral white blood cells and oxidative stress but also mediate neurovascular uncoupling by inducing mitochondrial dysfunction in neurons, abnormal contraction of cerebral vessels, and pericyte loss in AD. In addition, microglia-mediated dysfunction of cellular components in the NVU, such as astrocytes and pericytes, can destroy the integrity of the NVU and lead to NVU impairment. Therefore, we review the mechanisms of microglia-mediated NVU dysfunction in AD. Furthermore, existing therapeutic advancements aimed at restoring the function of microglia and the NVU in AD are discussed. Finally, we predict the role of pericytes in microglia-mediated NVU dysfunction in AD is the hotspot in the future.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease characterized by decreased cognitive function and is the most common type of dementia. The classic pathological changes in AD include amyloid-β peptide (Aβ) accumulation in the brain parenchyma and around brain vessels and neurofibrillary tangles formed by hyperphosphorylated microtubule-related protein tau in neurons [1, 2]. The number of people suffering from AD is more than 50 million globally, which makes AD the fifth leading cause of death in the world [2]. With the trend in global ageing, the prevalence of AD will greatly increase in the coming years, and economic and social burdens will also increase heavily [3]. Therefore, it is urgent to reveal new pathological mechanisms and discover therapeutic targets for AD.
The neurovascular unit (NVU) is a closely connected structural and functional complex composed of microglia, astrocytes, oligodendrocytes, neurons, interneurons, vascular smooth cells (VSMCs), pericytes, brain microvascular endothelial cells (BMECs), basement membrane and extracellular matrix. In fact, the NVU is responsible for regulating microenvironmental homeostasis and metabolic balance [4]. BMECs, VSMCs, pericytes, and perivascular astrocytic end-feet make up the blood-brain barrier (BBB), which selectively controls the exchange of substances between the central nervous system (CNS) and peripheral blood. Moreover, the NVU maintains the balance of metabolism in the CNS by providing sufficient blood fluids and nutrients. The NVU regulates cerebral blood fluid (CBF) according to neuronal activity depending on neurovascular coupling [5].
NVU dysfunction has been found in the early stage of AD [4]. It includes BBB destruction, neurovascular uncoupling, and abnormal communication among NVU cells [5]. In fact, we are mainly focusing on the role of NVU dysfunction in hippocampus in AD. BBB breakdown begins and aggravates in the hippocampus and its CA1 and dentate gyrus regions in normal aging and in mild cognitive impairment (MCI) individuals [6]. Impaired neurovascular coupling has been found in the hippocampus, parahippocampal gyrus, precuneus, and posterior cingulate cortex in the MCI patients and in the hippocampus in AD patients [7]. Moreover, NVU dysfunction significantly accelerating the progression of AD. First, BBB breakdown exacerbates Aβ accumulation [8] and which destroys the microenvironmental homeostasis of the CNS [9]. Second, neurovascular uncoupling, which is characterized by a decreased response of blood vessels to neuronal activity, decreases CBF [5], increases BBB permeability [10], exacerbates Aβ deposition [11] and tau hyperphosphorylation [12], and induces neuroinflammation, oxidative stress [13], and neuronal damage [5].
As one of the most important constituents of the NVU, microglia are the main resident immune cells in the CNS, and excessive microglial activation is one of the most important changes in the early stage of AD [14]. Though early recruitment of microglia may be neuroprotective by Aβ clearance, microglial engulfment, and degradation of Aβ is suppressed and proinflammatory cytokines are increased as disease progresses [15]. In fact, Aβ activated M1 microglia depending on P2X7 receptor (P2X7R) and the poly (ADP-ribose) polymerase-1 (PARP-1) pathways while spleen tyrosine kinase pathway is critical in neuroprotective microglia in AD [16–18]. And Aβ activates NLRP3 inflammasome and induce lysosomal damage in microglia in the process of engulfment [19]. Aβ and tau induce mitochondrial dysfunction in AD brain, while mitophagy impairment in microglia upregulates neuroinflammation and suppresses phagocytosis of Aβ and tau [20, 21]. Moreover, microglia mediate NVU dysfunction in many diseases, including AD. Excessive activation of microglia destroys BBB integrity and the regulation of CBF and contributes to neurodegeneration [16, 22, 23]. However, the exact mechanism is still unclear.
In this review, we examined microglia to clarify the mechanism of NVU dysfunction in AD, including BBB destruction, neurovascular uncoupling, and impairment in constituent cells. Furthermore, we summarized the existing AD therapies targeting microglia and NVU dysfunction to inspire researchers to go in new directions to identify medications for microglia-mediated NVU impairment.
MICROGLIA MEDIATE NVU DYSFUNCTION IN AD
Microglia mediate NVU dysfunction via BBB impairment
The influence of microglia on the BBB is largely dependent on the activation phenotypes of microglia. Aβ-activated M1 microglia have increased secretion of inflammatory factors and chemokines, including tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), interleukin-6 (IL-6), interleukin-12 (IL-12), CCL-2, CXCL-10, reactive oxygen species (ROS), and nitric oxide (NO), which exacerbate damage to the BBB [24–27]. In contrast, M2 microglia are characterized by alternative activation and acquired inactivation and play an important role in the protection and repair of the BBB by suppressing neuroinflammation in the CNS [25].
Microglia affect tight junctions (TJs) and adhesion junctions (AJs) between BMECs to modulate the permeability of the BBB. Under physiological conditions, resting microglia maintain the integrity of the BBB. These cells improve the expression of TJ factors such as zonula occludens 1 (ZO-1) and occludin on BMECs in vitro [26]. Moreover, microglia in a resting state directly provide the essential proteins for TJs, such as claudin-5, to protect the integrity of the BBB [28].
It has reported age-dependent BBB breakdown in hippocampus which is significantly aggravated in MCI patients [6]. M1 microglial activation is also found in the early stage of AD, and evidence indicates it is involved in BBB dysfunction in the early stage of AD [29]. First, proinflammatory factors derived from M1 microglia, including IL-1β, TNF-α, and CCL2, affect BMECs and directly destroy the BBB in AD. Aβ-activated microglia secrete IL-1β by upregulating the expression of P2X7 receptor (P2X7R) [16]. IL-1β increases the expression of IL-6, interleukin-8 (IL-8), and TNF-α and decreases the expression of ZO-1, enhancing BBB paracellular permeability. A study also showed that IL-1β upregulated the expression of intracellular adhesion molecule-1 and vascular cell adhesion molecule-1 to increase the transmigration of peripheral white blood cells (WBCs) into the CNS [30]. Aβ-activated M1 microglia release NO and TNF-α via PARP-1 to downregulate the expression of TJ proteins, including ZO-1 and occludin, increasing BBB paracellular permeability in vitro [26]. CCL2 decreases AJs and increases platelet endothelial cell adhesion molecule-1 (PECAM-1) on BMECs by inducing the phosphorylation and transference of β-Catenin from AJs to PECAM-1, which may contribute to BBB dysfunction and increase WBC recruitment [31]. Aβ-activated M1 microglia also release matrix metalloproteinases-9 (MMP-9) and ROS to damage BMECs and TJs [32]. Second, Aβ-activated microglia activate astrocytes by releasing inflammatory factors, further contributing to the indirect destruction of the BBB [33–35]. Third, Aβ-activated microglia secrete proinflammatory factors to induce pericyte loss [26, 36–38], potentially leading to BBB destruction [39–41]. Overall, M1 microglia can destroy TJs and AJs between BMECs by mediating neuroinflammation, astrocyte dysfunction and pericyte loss, leading to BBB and NVU impairment (Fig. 1).
Fig. 1
Microglia mediate BBB dysfunction. Firstly, microglia induced by Aβ secrete IL-1β, TNF-α, MMP-9, and CCL2 to destroy TJs and AJs between BMECs and increase the expression of leukocyte adhesion molecules (LAMs) on BMECs to attract peripheral WBCs. Secondly, activated microglia secrete C1q, TNF-α and IL-1β to activate astrocytes. The activated astrocytes promote transmigration of peripheral WBCs across BBB by secreting CCL2 and induce BBB breakdown by secreting C3 and vascular endothelial growth factors (VEGF). Thirdly, activated microglia mediate pericyte dysfunction by secreting TNF-α and presenting Aβ to Th1 cells. Then, activated pericytes increase the release of MMP-9 to destroy TJs leading to BBB impairment.
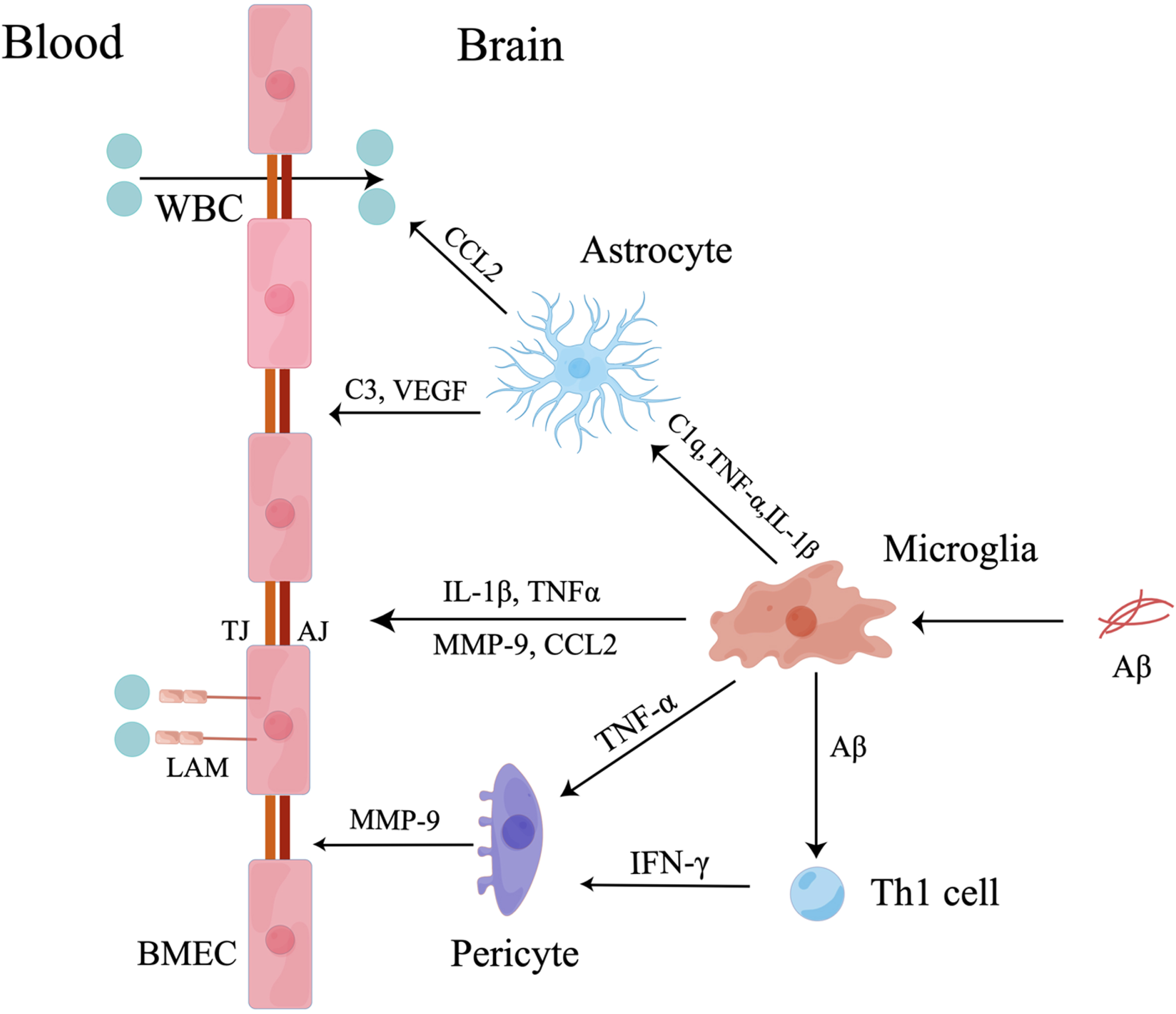
Increased infiltration of peripheral WBCs in the CNS exacerbates neuroinflammation and BBB dysfunction. Aβ-activated microglia and cerebral macrophages secrete chemokines and cytokines to induce peripheral monocytes to accumulate in the CNS and transform into resident macrophages to exacerbate BBB and NVU dysfunction [42]. Microglial inflammatory factors mediate peripheral WBC migration in complex ways. Proinflammatory factors (IL-1β, TNF-α, IL-6, and IL-12) and chemokines (monocyte chemoattractant protein-1, macrophage inflammatory protein-1α, macrophage inflammatory protein-1β, and IL-8) secreted by Aβ-activated M1 microglia increase the permeability of the BBB and the infiltration of monocytes into the CNS in vitro [42]. Specifically, IL-1β induced the migration of peripheral WBCs and exacerbated the destruction of the BBB by increasing the release of proinflammatory factors, including CCL2, CCL20, and CXCL2 [43]. Then, CCL2 was shown to cooperate with monocytes and lymphocytes to reduce the expression of TJs [44] and induce the disruption of AJs, destroying the integrity of the BBB. Increased PECAM-1 was also shown to recruit peripheral WBCs [31]. Finally, microglia-derived TNF-α plays an important role in peripheral WBC migration. Aβ-activated microglia secrete TNF-α, which is essential for CXCR2-dependent transmigration of T cells across the BBB [45]. These cells induced T cells and monocytes to transmigrate into the CNS by upregulating the expression of major histocompatibility complex and vascular cell adhesion molecule-1 on BMECs [46, 47] (Fig. 1).
Microglia mediate NVU dysfunction via neurovascular uncoupling
Neurovascular uncoupling is one of the most important reasons for CBF reductions in AD [48, 49]. Reduced CBF is a sign of the early stage of AD, which leads to neuronal death and neurodegeneration through metabolic dysfunction in neurons and glial cells [48, 49]. Microglia-mediated neuroinflammation has been observed in the early stages of AD [29, 50], and studies have shown a close correlation between microglia and neurovascular uncoupling in AD [51]. Multiple lines of evidence have shown that microglia modulate neurovascular coupling via the P2Y12 receptor (P2Y12R) under physiological conditions. The expression of P2Y12R on microglia is significantly decreased in AD, impairing cellular communication and the response of cerebral vessels to neuronal activity [22, 52].
Damage to neuronal mitochondria has been proven to contribute to metabolic dysfunction in neurons and neurovascular uncoupling [53]. In fact, gp91phox, a catalytic subunit of NADPH oxidase (NOX), is significantly upregulated in microglia and neurons in MCI patients [54]. A study showed that ROS derived from NOX containing gp91phox were essential for Aβ-induced neurovascular uncoupling [55]. Therefore, the interaction between microglia and neurons plays a crucial role in neurovascular uncoupling in AD. It was reported that Aβ-activated microglia promote the release of IL-1β, TNF-α, and NO and the activation of NOX by upregulating P2X7R and PARP-1 pathways in an AD mouse model [16, 17]. Increases in H2O2 and NO and proinflammatory factors led to structural and functional damage to neuronal mitochondria, contributing to increased ROS. TNF-α induced neuronal mitochondrial dysfunction via neuron-derived extracellular vesicles (EVs), leading to increased oxygen consumption and gradual ROS release from neurons [56]. As a result, increased ROS activate NOD-like receptor protein 3 (NLRP3) inflammasomes to exacerbate oxidative stress, neuroinflammation, insulin resistance and metabolic abnormalities [48, 53], leading to neurovascular uncoupling in AD.
Recent studies have shown that the primary mechanism of neurovascular uncoupling in AD is cerebrovascular component dysfunction [57]. First, activated microglia promote VSMC and pericyte dysfunction, leading to neurovascular uncoupling. On the one hand, microglia can directly mediate pericyte degeneration. Activated microglia can secrete proinflammatory factors to induce pericyte loss [26, 36–38]. LPS-activated microglia directly increased the release of ROS from pericytes by secreting TNF-α and IL-1β, leading to death in rat retinal microglia and pericytes [58]. Then, pericyte degeneration reduces the capillary response to neuronal activity, resulting in neurovascular uncoupling [59]. We think a similar mechanism by which activated microglia induce pericyte loss leading to neurovascular uncoupling is present in AD. On the other hand, activated microglia indirectly induce cerebral vessels to contract by exacerbating Aβ accumulation. Aβ-activated microglia upregulate cyclooxygenase 2 to increase the expression of IL-1β. Then, IL-1β upregulates β-site APP-cleaving enzyme (BACE-1) in neuron-like cells (SH-SY5Y cells) to exacerbate Aβ accumulation [60]. Increased Aβ deposition mediates the hypercontractive phenotype of VSMCs in small arteries in the CNS and decreases CBF via high levels of serum response factor (SRF)/myocardin (MYOCD) [61, 62]. In human and mouse models of AD, Aβ also induced pericytes around brain capillaries to contract through NADPH-oxidase-4 (NOX4)-derived ROS [63]. However, some researchers believe that VSMCs can directly contract cerebral vessels, while pericytes cannot. In contrast, pericytes influence CBF only by indirectly regulating cellular interactions [64]. Finally, the end-feet of astrocytes on cerebral vessels are very important for communication between blood vessels and neurons. The expression of aquaporin-4 (AQP-4) in astrocytic end-feet on the vessels is significantly reduced in AD and is accompanied by a prominent increase in microglial activation and proliferation [65]. Excessive levels of activated microglia may be involved in astrocyte-mediated neurovascular uncoupling, but the exact mechanism is still unknown. Overall, microglia can mediate neurovascular uncoupling in AD by downregulating the expression of P2Y12R, increasing ROS and proinflammatory factors to induce neuronal mitochondrial impairment, exacerbating Aβ accumulation to attenuate the response of cerebral vessels to neuronal activity and inducing dysfunction in pericytes and astrocytes (Fig. 2).
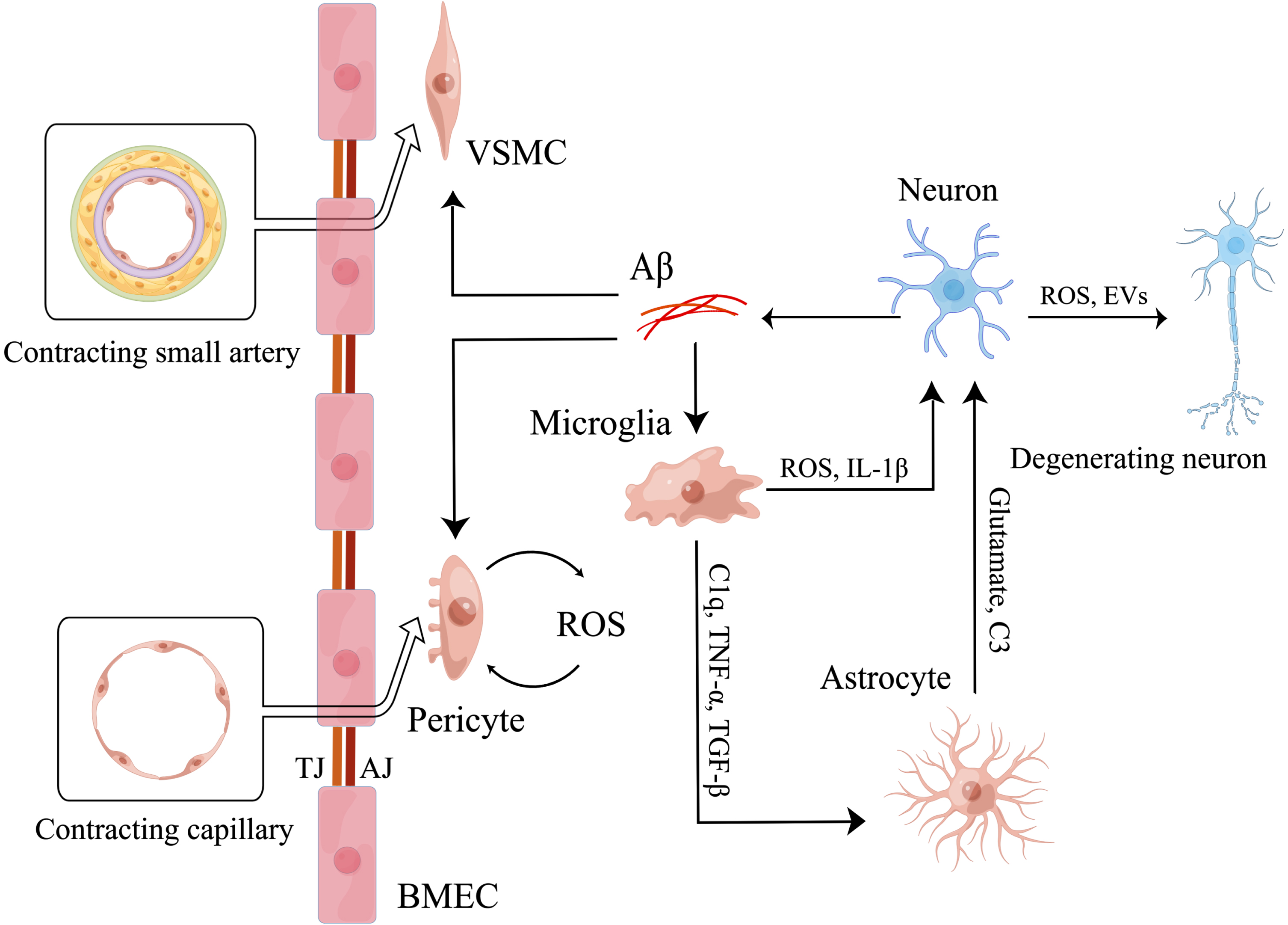
Fig. 2
Microglia mediate neurovascular uncoupling. Firstly, microglia induced by Aβ secrete ROS and IL-1β to induce neuronal mitochondrial dysfunction directly. Then, activated microglia also secrete C1q, TNF-α, TGF-β to activate astrocytes. And activated astrocytes increase the release of glutamate and C3 to induce neuronal mitochondrial dysfunction. As a result, neuronal mitochondrial dysfunction induces degeneration of more surrounding neurons by releasing ROS and EVs, finally leading to neurovascular uncoupling. Meanwhile, microglia are involved in the regulation of neurovascular uncoupling by affecting the function of cerebral vessels. Aβ induces abnormal contraction of small arteries by promoting the conversion of VSMCs to hypercontractive phenotype. And Aβ also induces abnormal contraction of capillaries by increasing the release of ROS of pericytes. Eventually, microglia aggravate the effects of Aβ by secreting IL-1β to increase the release of Aβ of neurons.
Microglia mediate NVU dysfunction through impairment of the cellular components of the NVU
The NVU is a structural and functional complex [4]. Dysfunction of the cellular components of the NVU has been observed in AD, including changes in the distribution of capillaries, pericyte diminishment, astrocyte activation and proliferation, and the relocation of microphages [66]. Evidence support that microglia is involved in this process and potentially lead to NVU dysfunction.
Microglia mediate NVU dysfunction via astrocyte impairment
As an important constituent of the NVU, astrocytes are reactive cells in the CNS that can rapidly and simultaneously exchange information with several synapses. Astrocytes maintain the integrity of the BBB and regulate CBF with BMECs, pericytes and VSMCs [67]. Additionally, reactive astrocytes accelerate AD progression by significantly affecting Aβ and tau pathology via BACE-1 and IL-3 pathways [68, 69].
Microglia induce excitotoxicity in astrocytes and disturb the communication among astrocytes and surrounding cells to mediate astrocyte impairment, ultimately leading to NVU dysfunction in AD [70]. First, microglia play an important role in severe neuronal excitotoxicity induced by the release of calcium ion-dependent gliotransmitters such as glutamate from astrocytes [71]. TNF-α, which is mainly secreted by microglia, was shown to be essential for calcium-dependent release of glutamate by astrocytes [72, 73]. TNF-α mediates calcium ion-dependent glutamate release by activating prostaglandin E2 in astrocytes, which leads to neuronal apoptosis in an AD mouse model [74]. The expression of intermediate conductance calcium-activated potassium channels (KCa3.1 channels) on reactive astrocytes and activated microglia was enhanced in AD patients and SAMP8 mice [75, 76]. Some researchers found that microglia-derived TGF-β upregulated astrocyte KCa3.1 channels to induce astrogliosis and increased the level of calcium ions to mediate the release of glutamate during AD [71, 75, 77]. Second, a high number of gap junctions is important for intercellular communication among astrocytes. In particular, Aβ-activated microglia suppress the expression of astrocyte gap junctions by secreting TNF-α and IL-1β, contributing to astrogliosis [78]. Third, activated microglia are involved in the regulation of astrocyte complement-dependent intercellular communication leading to NVU dysfunction. Specifically, activated microglia release C1q to activate the nuclear factor-kappa B (NF-κB) pathway in astrocytes, which release C3 [33, 79]. C3 has been reported to compromise neuronal synapses and cognitive function by acting on neuronal C3R and destroying the BBB by acting on BMECs [35, 80]. Finally, C3 attenuated microglial phagocytosis of Aβ [81].
Aβ-activated microglia induce NVU dysfunction by mediating astrocyte activation and neuroinflammation. Microglia are more sensitive to pathological changes than astrocytes [25], and the activation of astrocytes is highly dependent on microglia [82]. This finding indicates the initial role of microglia and the amplified role of astrocytes in neuroinflammation [83, 84]. In addition, microglia can mediate the phenotypic conversion of astrocytes. Similar to microglia, there are A1 and A2 astrocytes in AD. Aβ-activated M1 microglia secrete IL-1β, TNF-α, C1q, and NO to induce A1 astrocytes [27, 33]. A1 astrocytes have increased release of proinflammatory factors, chemokines and complement to amplify neuroinflammation, leading to BBB destruction and neuronal injury [33–35, 85]. Moreover, IL-1β indirectly enhances the permeability of the BBB in vitro by suppressing the expression of sonic hedgehog in astrocytes, which was neuroprotective [43]. IL-1β also mediates the destruction of the BBB by increasing the expression of proinflammatory chemokines and vascular endothelial growth factors in astrocytes [43, 85]. Moreover, it was reported that chronic neuroinflammation induced degeneration and death in astrocytes [86]. However, as M1 microglia are converted to M2 microglia, they secrete IL-10 to induce the activation of A2 astrocytes, which increases the expression of anti-inflammatory factors [25, 87] (Fig. 3).
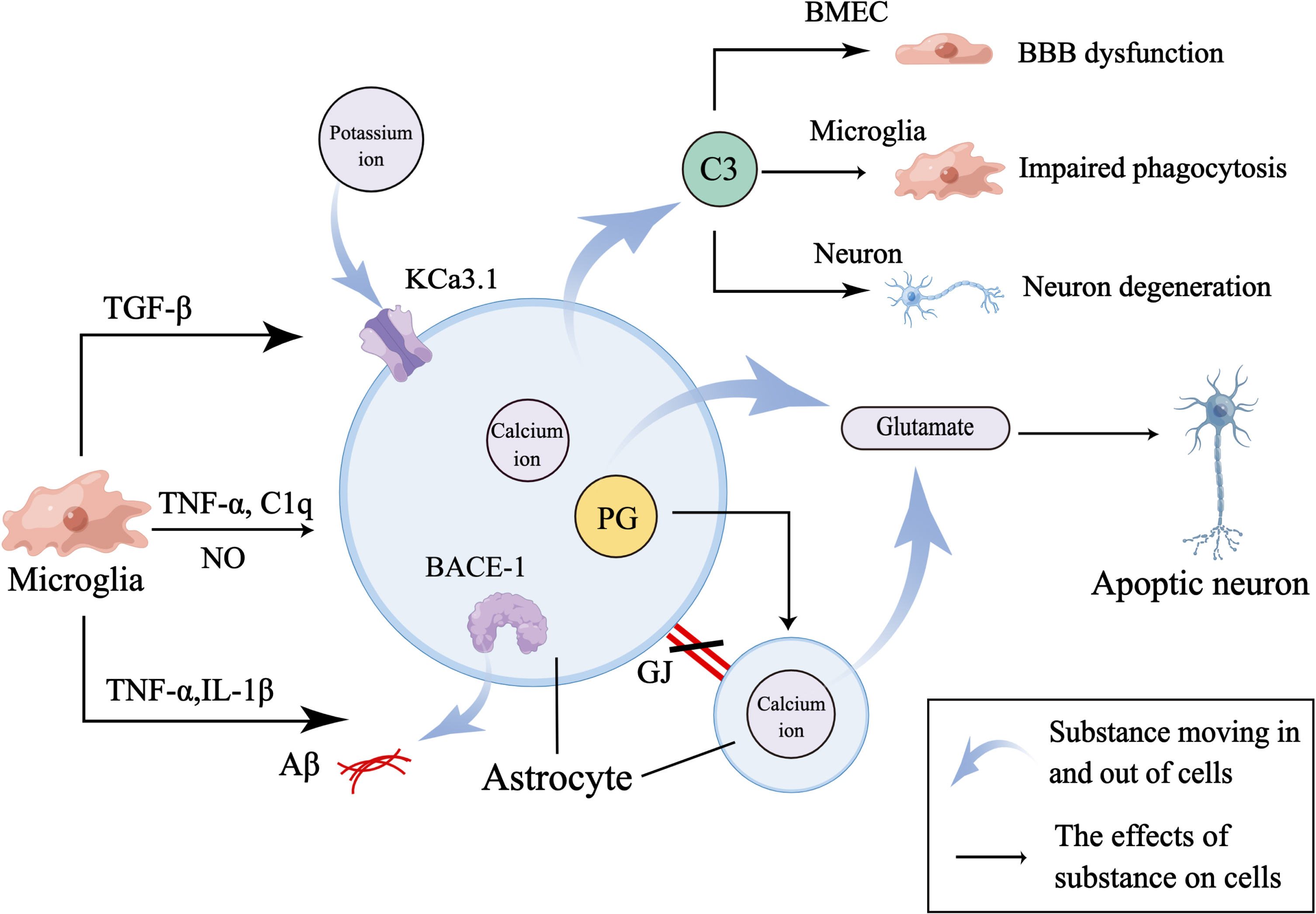
Fig. 3
Microglia mediate astrocyte dysfunction. Firstly, activated microglia secrete TNF-α, C1q, and NO to activate astrocytes. And activated astrocytes increase the release of C3 to mediate NVU dysfunction by regulating the function of BMEC, microglia, and neuron. Secondly, microglia secrete TNF-α and IL-1β to increase the expression of BACE-1 in astrocyte to aggravate Aβ accumulation and destroy GJs between astrocytes to affect normal cellular communication between astrocytes. And TNF-α also increases the expression of PG in astrocyte to evoke calcium ion-mediated release of glutamate which induces the apoptosis of neuron. Thirdly, microglia-derived TGF-β increases the expression of KCa3.1 channel which works together with elevated calcium ion in astrocytes to promote release of glutamate.
Microglia mediate NVU dysfunction via pericyte impairment
Brain pericytes, which are important cellular components of cerebral capillaries, have features similar to those of VSMCs and form the BBB with BMECs, astrocytic end-feet and the basement membrane [88]. The coverage ratio of pericytes was proven to be associated with the permeability of the BBB [39]. Evidence has demonstrated that microglia and neuroinflammation can mediate pericyte impairment and loss in various ways, leading to NVU dysfunction in AD. First, Aβ-activated microglia induce BBB destruction via pericyte-derived MMP-9. Aβ-activated microglia secrete TNF-α in AD [26], and TNF-α promotes pericytes to release MMP-9, which leads to BBB breakdown by destroying TJs and extracellular matrix between BMECs and promoting the migration of pericytes surrounding vessels to other regions in the brain, leading to pericyte loss in vitro [36]. Second, microglial cytokines participate in pathways that lead to pericyte loss from cerebral vessels in AD. Pericyte loss contributes to neurovascular uncoupling and BBB dysfunction. It was reported that microglia presented Aβ to Th1 cells and promoted Th1 cells to secrete IFN-γ in vitro [37]. Then, IFN-γ promoted the phosphorylation, internalization, and degradation of platelet-derived growth factor receptor-β (PDGFR-β) in pericytes to prevent PDGF-BB-associated proliferation and migration [38], leading to the loss of pericytes. Moreover, TGF-β, which is mainly derived from microglia, induced pericyte loss and NVU dysfunction [73, 89]. TGF-β attenuated the phagocytic capacity of pericytes and increased the expression of classic proinflammatory factors, leading to pericyte loss [90]. The loss of pericytes reduced the cerebral capillary response to neuronal activity and increased basal blood flow rates in cerebral capillaries, providing less nutrients and oxygen for neurons [59, 91] and destroying the metabolic balance and neurovascular coupling in AD. Moreover, pericyte loss enhanced the permeability of the BBB in several ways. In PDGFR-β –/–mice, the loss of pericytes increased paracellular permeability and the migration of peripheral WBCs by downregulating TJs and upregulating leukocyte adhesion molecules [39]. Then, in PDGFR-β variant mice, the loss of pericyte increased BBB permeability by enhancing the phagocytosis of BMECs and dysfunction in astrocytic end-feet [40]. Moreover, in PDGFR-β ret/ret mice, pericyte loss mediated endothelial arteriovenous zonation, angiogenic quiescence and impaired functions of the BBB [92]. Third, it was reported that activated microglia exacerbated Aβ-induced abnormal pericyte contraction and neurovascular uncoupling in AD, which has been elucidated [60, 63, 93].
Overall, it has been proven that Aβ-activated microglia induce pericyte dysfunction and loss by releasing TNF-α, IFN-γ, and TGF-β. Pericyte dysfunction and loss lead to BBB breakdown and neurovascular uncoupling in several ways. However, evidence is still needed to determine whether microglia directly mediate NVU dysfunction by regulating pericyte function in AD.
Microglia mediate NVU dysfunction via injury to neurons and interneurons
A decrease in neurons is one of the important factors in the progressively declining cognitive and memory function of AD patients, which is mediated by microglia through phagocytosis and the secretion of inflammatory factors, EVs and ROS. First, abnormal microglial phagocytosis induces neuronal damage in AD. It was reported that enhanced microglial phagocytosis damaged neuronal synapses in the early stage of AD in TREM2-variant mice, but phagocytosis was neuroprotective in the late stage of AD by engulfing Aβ [94]. Aβ-induced microglial phagocytosis of synapses was proven to be dependent on the complement pathway [79, 95]. M1 microglia secrete inflammatory factors and C1q to increase the release of C3 by A1 astrocytes, leading to the rapid death of neurons [33, 79]. Increased C3 enhances microglial phagocytosis of synapses by binding to C3R on microglia [95]. Second, microglia-mediated neuroinflammation and oxidative stress regulate neuronal function. Aβ-activated microglia have increased release of TNF-α, glutamate, and ROS, leading to excessive neuronal activation and death [16, 96–98]. In addition, Aβ-activated microglia damage neurons via Toll-like receptor-2/3 (TLR-2/3) by releasing IL-6, TNF-α, and ROS [99, 100]. Third, ATP-activated microglia produce EVs that convert aggregated Aβ to soluble Aβ, which is more neurotoxic [101]. Finally, activated microglia affect the function of interneurons and neural networks. In 5XFAD and 3x-TgAD mice, activated microglia were closely associated with damage to interneurons in AD [102]. In addition, damage to interneurons leads to dysfunction of neural networks and cognition by inducing imbalance between suppression and excitation in the CNS [103].
Microglia mediate NVU dysfunction via impairment of other cellular components in the NVU
First, microglia mediate dysfunction in BMECs and the BBB in AD by destroying TJs and AJs and increasing the entry of peripheral WBCs into the CNS. Second, microglia exacerbate Aβ accumulation [93] to affect the communication between BMECs and astrocytic end-feet [41], which contributes to neurovascular uncoupling in AD and alters calcium signaling in BMECs [104]. Third, microglia mediate the hypercontractive phenotype of VSMCs by exacerbating Aβ accumulation, leading to neurovascular uncoupling in AD. Finally, oligodendrocytes, which are the myelin-forming cells in the CNS, play a significant role in the conduction of action potentials and neuronal protection [105]. In 5XFAD mice, TREM2-variant microglia were associated with the phagocytosis of myelin and lipid metabolism in the CNS, and reactive oligodendrocytes were always accompanied by damage to axonal myelin and metabolic imbalance [106–108]. This finding indicates the potential correlation between microglia and oligodendrocytes in AD, although there is no direct evidence to support it.
POTENTIAL THERAPIES FOR RESTORING THE FUNCTION OF MICROGLIA AND THE NVU IN AD
Both AD pathologies and NVU dysfunction are present much earlier than clinical manifestations therefore the principle of medication treatment timing is as early as possible. After innumerable failure of studies of anti-Aβ, aducanumab was approved as the first disease-modifying drug of AD by FAD in 2021. However, there is still much controversy concerning the clinical benefits of aducanumab in AD [122, 123]. Because of various risk factors and complex mechanisms of AD, we believe different therapeutic approaches would be helpful to AD with different causes [124, 125]. The important roles of microglia and NVU in AD, and quickly advanced technologies make them valuable choices [4, 126, 127]. The potential therapeutics aiming to alleviate AD progression and improve cognitive function by restoring microglial and NVU function have been described in Table 1.
Table 1
Potential therapies for regulating microglia and NVU function
| Outcomes | Subject and group division | Reference | |
| Regulating microglia function | |||
| TREM2McAb | Improve cognitive function | 5xFAD mice | [109] |
| •Promote acute activation of protective microglia. | •hTREM2-Ab group (n = 9):1 mg/kg hTREM2-Ab once 2 weeks for 3 months. | ||
| •Increase phagocytic Aβ and apoptotic neurons. | •hTREM2-Ab group (n = 10):10 mg/kg hTREM2-Ab once 2 weeks for 3 months. | ||
| •Inhibit chronic neuroinflammation. | •hIgG group (n = 9):10 mg/kg hIgG once 2 weeks for 3 months. | ||
| •PBS sham control group (n = 9): PBS once 2 weeks for 3 months. | |||
| Pantethine | Improve the results of behavior test | 1.5-month-old Tg (5XFAD) and age-matched WT male mice | [110] |
| •Suppress activation of microglia and astrocyte. | •Tg + pantethine group (n = 10): saline + 15 mg pantethine i.p. 3 times a week for 5.5 months. | ||
| •Change the expression of genes including inflammation, complement and phagocytosis. | •Tg + vehicle group (n = 10): saline i.p. 3 times a week for 5.5 months. | ||
| •WT + vehicle group (n = 10): saline i.p. 3 times a week for 5.5 months. | |||
| •Alleviate Aβ pathology. | •WT + pantethine group (n = 10): saline + 15 mg pantethine i.p. 3 times a week for 5.5 months. | ||
| AdipoRon | Improve spatial memory function | 5.5-month-old (±2 weeks) 5xFAD mice and age-matched WT C57BL/6N mice, age-matched APN-deficient 5xFAD (5xFAD; APN– /–) | [111] |
| •Lowered plaque and Aβ levels. | •WT group:vehicle daily by oral gavage for 3 months. | ||
| •Restore microglial phagocytic capacity. | •5xFAD + AdipoRon group: 50 mg/kg adipoRon daily by oral gavage for 3 months. | ||
| •Suppress activation of microglia and astrocyte and release of proinflammatory factors. | •5xFAD + Vehicle group: vehicle daily by oral gavage for 3 months. | ||
| •(5xFAD; APN– /–) + AdipoRon group: 50 mg/kg adipoRon daily by oral gavage for 3 months. | |||
| •Rescued neuronal and synaptic loss. | •(5xFAD; APN– /–) + Vehicle group: vehicle daily by oral gavage for 3 months. | ||
| Methystictin | Attenuate the long-term memory decline | 25-week-old APP/PS1 mice and age-matched WT mice | [112] |
| •Suppress activation of microglia and astrocyte via Nrf2 pathway. | •APP/PS1mice + Methysticin group (n = 6): 6 mg/kg methysticin by oral gavage for 27 weeks. | ||
| •Reduce microgliosis, astrogliosis and secretion of the proinflammatory cytokines. | •APP/PS1 mice + Placebo group (n = 6): placebo by oral gavage for 27 weeks. | ||
| •WT mice + Placebo group (n = 6): placebo by oral gavage for 27 weeks. | |||
| •Reduce oxidative damage. | |||
| Regulating NVU function | |||
| Anserine | Improve cognitive function of AD mice | Over 18-months old APPswe/PSEN1dE9 mice and age-matched WT mice | [113] |
| •Suppress neuroinflammation. | •WT group (n = 10): regular autoclaved drinking water for 8 weeks. | ||
| •Increase the vascular coverage ratio of pericytes. | •WT + Anserine group (n = 11): autoclaved drinking water + 2.0 g/L anserine for 8 weeks. | ||
| •AD group (n = 10): regular autoclaved drinking water for 8 weeks. | |||
| •AD + Anserine group (n = 10): autoclaved drinking water + 2.0 g/L anserine for 8 weeks. | |||
| Sildenafil | Improve CBF, CMRO2 and declines CVR | 62– 87-years old AD patients of the early stage | [114] |
| •Increase CBF. | •Self before and after the control. | ||
| •Restore NVU function. | •A single oral administration of 50 mg. | ||
| Dendrimer-tesaglitazar | •Promote microglia M1 phenotype conversion to M2. | BV2 murine microglial cell line | [115] |
| •Reduce ROS production. | |||
| •Increase Aβ phagocytosis and degradation. | |||
| Dabigatran | Improve cognition, brain perfusion and BBB function | 2-month-old female TgCRNB8 mice and age-matched WT mice | [116] |
| •Reduce phagocytic microglia and infiltrating T cell. | •TgCRNB8 etexilate group (n = 13– 18): 5 mg/g etexilate (prodrug of dabigatran) for 22/52 weeks. | ||
| •Suppress astrogliosis and neuroinflammation. | •WT etexilate group (n = 13– 18): 5 mg/g etexilate (prodrug of dabigatran) for 22/52 weeks. | ||
| •Maintain pericyte function. | •TgCRNB8 placebo group (n = 13– 18): placebo for 22/52 weeks. | ||
| •Restore AQP-4 in astrocyte end feet to protect BBB. | •WT placebo group (n = 13– 18): placebo for 22/52 weeks. | ||
| •Alleviate Aβ pathology. | |||
| L-Norvaline | Decrease BBB permeability | 4-month-old male 3×Tg-AD mice and age-matched male C57Bl/6 mice | [117] |
| •Upregulate iNOS. | •WT + control group (n = 3): animals’ drinking water for 10 weeks. | ||
| •Suppress microglial activation and astrocyte degeneration. | •WT + L-Norvaline group (n = 3): animals’ drinking water + 250 mg/L L-Norvaline for 10 weeks. | ||
| •Decrease permeability of BBB. | •3xTg AD + control group (n = 6): animals’ drinking water for 10 weeks. | ||
| •3xTg AD + L-Norvaline group (n = 6): animals’ drinking water + 250 mg/L L-Norvaline for 10 weeks. | |||
| Rapamycin | Rescue vascular, metabolic and learning deficits | 1-month-old female apoE4 transgenic and age-matched WT mice | [118] |
| •Restores CBF. | •WT control group (n = 10): control diet for 6 months. | ||
| •Restores the integrity of BBB. | •Tg control group (n = 10): control diet + microencapsulating materials for 6 months. | ||
| •Restores glucose metabolism. | •Tg rapamycin group (n = 10): control diet + 2.24 mg/kg/d microencapsulated rapamycin for 6 months. | ||
| Statins | Improve memory function and lessen SPs | 12-month-old APP transgenic mice and age-matched WT mice | [119] |
| •Decline activity of MMP-9. | •WT mice group (n = 20): 0.5% methylcellulose (MC). | ||
| •Alleviate destruction of basement membrane. | •APP vehicle group (n = 17): 0.5% MC. | ||
| •Suppress astrogliosis and detachment of astrocyte end feet and BMECs. | •APP atorvastatin group (n = 19): 0.5% MC + 30 mg/kg/day atorvastatin. | ||
| •APP pitavastatin group (n = 18): 0.5% MC + 3 mg/kg/day pitavastatin | |||
| Pinocembrin | Improve cognitive function | 4-month-old APP/PS1 mice and age-matched WT mice | [120] |
| •Suppress glial neuroinflammation. | •WT controls group (n = 9): 20% hydroxypropyl-b-cyclodextrin by oral gavage 5 days per week for 12 weeks. | ||
| •Protect cholinergic system to maintain the function of NVU. | •WT pinocembrin group (n = 9): 40 mg/kg pinocembrin in 20% hydroxypropyl-b-cyclodextrin by oral gavage 5 days per week for 12 weeks. | ||
| •APP/PS1 controls group (n = 9):20% hydroxypropyl-b-cyclodextrin by oral gavage 5 days per week for 12 weeks. | |||
| •APP/PS1 pinocembrin group (n = 9): 40 mg/kg pinocembrin in 20% hydroxypropyl-b-cyclodextrin by oral gavage 5 days per week for 12 weeks. | |||
| Quercetin | Improve study and memory ability and protect NVU integrity. | Aβ (25– 35)-induced amnestic mice | [121] |
| •Suppress the activation of RAGE pathway to protect BBB. | •The sham group:distilled water containing 20% hydroxypropyl-beta-cyclodextrin without quercetin once per day for 8 days by oral gavage. | ||
| •Maintain cholinergic neuronal regulation of CBF. | •Aβ (25– 35)-treated group:distilled water containing 20% hydroxypropyl-beta-cyclodextrin without quercetin once per day for 8 days by oral gavage | ||
| •Reduce the expression of ERK1/2 and nuclear p65 to protect BBB. | •Quercetin group (n = 19): 5 mg/kg quercetin + distilled water containing 20% hydroxypropyl-beta-cyclodextrin once per day for 8 days by oral gavage. | ||
| •Inhibit RAGE pathway to restore neurovascular coupling. | •Quercetin group (n = 19): 10 mg/kg quercetin in distilled water containing 20% hydroxypropyl-beta-cyclodextrin once per day for 8 days by oral gavage | ||
| •Quercetin group (n = 19): 20 mg/kg quercetin + distilled water containing 20% hydroxypropyl-beta-cyclodextrin once per day for 8 days by oral gavage. | |||
| •Quercetin group (n = 19): 40 mg/kg quercetin + distilled water containing 20% hydroxypropyl-beta-cyclodextrin once per day for 8 days by oral gavage. | |||
TREM2McAb, triggering receptor expressed in myeloid cells monoclonal antibody; Aβ, amyloid-β; PBS, phosphate-buffered saline; Tg mice, transgenic mice; WT mice, wildtype mice; AdipoRon, adiponectin receptor agonist; Nrf2,nuclear factor erythroid 2-related factor 2; i.p, intraperitoneal injection; AD, alzheimer’s disease; CBF, cerebral blood fluid; CMRO2, cerebral metabolic rate of oxygen consumption; CVR, cerebrovascular resistance; NVU, neurovascular unit dysfunction; ROS, reactive oxygen species; BBB, blood-brain barrier; AQP-4, aquaporins 4; iNOS, inducible nitric oxide synthase; apoE4, apolipoprotein E4; SP, senile plaque; MMP-9, matrix metalloproteinase-9; BMEC, brain microvascular endothelial cell; APP, amyloid precursor protein; APP/PS1 mice, amyloid precursor protein/presenilin 1 mice; MC, methylcellulose; RAGE, the receptor for advanced glycation end products; ERK1/2, extracellular signal regulated kinase1/2.
First, inhibiting neuroinflammation is most likely helpful in improving BBB function to restore NVU function in AD. Annexin A1 (ANXA1) not only inhibits neuroinflammation by increasing IL-10 and decreasing TNF-α [128] but also alleviates Aβ-induced BBB breakdown by suppressing the RhoA-Rock pathway [129]. Second, multiple-target drugs, including dabigatran, L-norvaline and quercetin, regulate the function of several types of cells in the NVU to reduce BBB permeability in AD mouse models [116, 117, 121]. Third, a clinical trial demonstrated that resveratrol reduced the concentration of MMP-9 in CSF to preserve BBB integrity and improved the cognitive function of AD patients [130]. Finally, rapamycin (mTOR inhibitor) was proven to upregulate TJs and inhibit the activity of MMP-9 to maintain BBB integrity in an AD mouse model [131].
Medications have also been designed to restore neurovascular coupling and increase CBF in AD. First, NAD+ intermediates have been shown to restore NAD+ concentrations to alleviate ageing-associated neurovascular uncoupling. Nicotinamide mononucleotide (NMN) rescued cerebral vessel responses to neuronal activity and cognitive function in a mouse model of aging by increasing endothelial NO-derived relaxation in cerebral microvessels and alleviating mitochondrial dysfunction [132]. NMN has also been reported to be beneficial for the cognitive function in AD mice by attenuating Aβ accumulation, synaptic loss, and neuroinflammation [133]. Second, the PARP-1 inhibitor PJ-34 was shown to improve neurovascular coupling and cognitive function by regulating endothelial-mediated vasodilation in an aging mouse model [134]. Third, twendee X has been proven to alleviate tau and α-synuclein pathology and neurovascular uncoupling in APP23+ CCH mice [135]. Furthermore, sildenafil increased CBF and improved NVU function in AD patients in clinical trials [114, 136].
Instead of restoring BBB function and neurovascular coupling alone, medications have also been designed to simultaneously regulate the function of multiple cells in the NVU to improve NVU function in AD. First, anserine can improve NVU function in AD. It was reported that anserine and carnosine improved brain perfusion and verbal episodic memory in elderly people [137, 138], although the effect of anserine on AD patients is still unclear. A recent study demonstrated the role of anserine in maintaining NVU integrity by suppressing astrocyte-mediated neuroinflammation and improving the coverage ratio of pericytes in a mouse model of AD [113]. Second, statins, including atorvastatin and pitavastatin, restored NVU function by inhibiting the activity of MMP-9 in neurons and attenuating astrogliosis, leading to improved behavioral memory and a reduction in senile plaques in an AD mouse model [119]. Third, pinocembrin was reported to suppress neuroinflammation and protect the cholinergic system to restore the function of the NVU [120]. Fourth, adipoRon, an adiponectin receptor agonist, enhanced the effects of adiponectin, whose deficiency was proven to increase Aβ-induced microglial activation and neuroinflammation. AdipoRon treatment suppressed neuroinflammation, restored microglial phagocytosis of Aβ, rescued neuronal and synaptic function, and improved spatial memory function in AD mice [111, 139]. Finally, rapamycin plays a crucial role in NVU protection. Rapamycin increases CBF, decreases BBB permeability and maintains neuronal function in vivo. Moreover, rapamycin alleviates Aβ pathology and normalizes glucose metabolism. Most importantly, rapamycin was demonstrated to improve the cognitive function of AD mice [118, 131, 140].
Due to the close correlation between microglia and astrocytes, many medications have been designed to regulate the function of microglia and astrocytes to improve NVU function and delay AD progression. First, suppressing the activation of microglia and astrocytes and neuroinflammation may be helpful in delaying AD progression. For example, methystictin activates the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway; Nrf2 is an anti-inflammatory transcription factor that significantly reduces microgliosis, astrogliosis, proinflammatory cytokines, and oxidative stress in APP/PS1 mice [112]. Low molecular weight glucocorticoid-induced leucine zipper (GILZ) analogues, which are the peptide analogues of the p65 binding domain of GILZ, suppress the NF-κB pathway. GILZ analogues decrease Aβ accumulation, reduces activated microglia and astrocytes, and inhibits inflammatory cytokines and TLR expression in 5xFAD mice [141]. TPPU, a specific small-molecule soluble epoxide hydrolase inhibitor, improves the concentration and anti-inflammatory activity of epoxy fatty acids. TPPU significantly reversed microglia and astrocyte activation and suppressed neuroinflammation in 5xFAD mice [142]. Second, pantethine not only suppressed glial neuroinflammation in mice but also suppressed the expression of complement genes and other inflammatory genes in astrocytes [110]. Third, boldine suppresses the activity of hemichannels between microglia and astrocytes without influencing the function of gap junctions to reduce calcium ion and ATP-induced glutamate release [143]. Recently, NLY01, a GLP-1R agonist, is reported specifically to inhibit Aβ-induced microglial activation and the conversion of reactive astrocytes as well as protecting neurons in AD mice, leading to the improvement of cognitive function [144].
Medications regulate microglial function to treat AD in various ways. First, medications suppress microglia-associated inflammatory pathways and cytokines to prevent the progression of AD. For example, dapansutrile and edaravone suppress microglia-mediated neuroinflammation by inhibiting the NLRP3 inflammasome in AD [145, 146]. Artemether and tiliroside suppress microglia-mediated neuroinflammation in AD by activating the anti-inflammatory Nrf2 pathway [147, 148]. Second, some medications alleviated the accumulation of Aβ and apoptotic cells to maintain NVU homeostasis by enhancing microglial phagocytic capacity. For instance, K161 improves microglial phagocytosis of Aβ and apoptotic neurons by suppressing the Pan-SHIP1/2 pathway [149]. Magnolol improves microglial phagocytosis and the degradation of Aβ and suppresses microglia-mediated neuroinflammation [150]. TREM2 monoclonal antibodies improve cognitive function by promoting acute microglial activation and microglial phagocytosis of Aβ and suppressing chronic microglia-mediated inflammation [109]. Third, microglial phenotypic transformation from M1 to M2 is also important in AD. For example, DHCR24, TPP-MoS2QDs, naringenin, and Dendrimer-tesaglitazar were reported to induce M1 microglia to convert to M2 microglia to suppress neuroinflammation and improve microglial phagocytosis [115, 151–153]. Eventually, there are many mutant genes in microglia in AD, and some have been proven to be associated with AD, including TREM2, CD33, PILRA, and CR [154]. Interestingly, the TREM2-activated antibody AL002c has been proven to be efficient and safe for delaying the AD in mouse models and phase 1 clinical trials [155]. The TREM2 monoclonal antibody promoted the acute activation of protective microglia and suppressed microglia-associated chronic neuroinflammation to attenuate AD pathology in vivo and in vitro. CLEC7A antibody alleviated microglial excessive activation and enhance microglial engulfment of Aβ by activating Syk which delays the disease process and improves cognitive function in AD mice [156].
CONCLUSIONS: CHALLENGES AND PERSPECTIVES
In this review, we have discussed the existing mechanism of microglia-mediated NVU dysfunction and the advancements in therapeutics for restoring the function of microglia and the NVU in AD. Microglia, which are critical in neuroinflammation in AD, mediate NVU dysfunction via various pathways. Specifically, Aβ-activated microglia not only induce BBB breakdown by mediating neuroinflammation, the infiltration of peripheral WBCs and oxidative stress but also impair neurovascular coupling by mediating direct damage to neuronal mitochondria, inducing abnormal contraction of cerebral vessels and pericyte loss in AD [16, 60, 93]. Moreover, microglia-mediated impairment of cellular components of the NVU destroys the integrity of the NVU and leads to NVU dysfunction [4]. Specifically, microglia-mediated activation and dysfunction of astrocytes regulates neuroinflammation and the destruction of the NVU. Pericyte dysfunction and loss is mediated by microglia and affect the function of the NVU by destroying the BBB and neurovascular coupling. Furthermore, microglia induce neuronal impairment and loss via abnormal phagocytosis, neuroinflammation, oxidative stress, the complement pathway and EVs, subsequently leading to NVU dysfunction. As a result, microglia mediate NVU dysfunction by destroying homeostasis in the CNS and inducing an imbalance in metabolism and neurodegeneration in the early stage of AD. Additionally, evidence support the role of microglia-mediated NVU dysfunction in AD. For example, microglia modulate neurovascular coupling via P2Y12 pathway under physiological condition while this process may be disrupted as significantly decreased expression of P2Y12 in activated microglia has been found in brain of AD patients using multispectral immunofluorescence [22, 52]. Recently, NLY01, a GLP-1R agonist, is reported specifically to inhibit Aβ-induced microglial activation and the conversion of reactive astrocytes as well as protecting neurons in AD mice, leading to the improvement of cognitive function [144].
Pericytes play an indispensable role in BBB impairment and neurovascular uncoupling in AD, which are important characteristics of NVU dysfunction [59, 157]. It was proven that microglia can increase the permeability of the BBB by inducing pericyte dysfunction and loss. Furthermore, microglia are involved in neurovascular coupling and metabolic balance by inducing pericyte impairment and loss and exacerbating Aβ accumulation. However, we know little about the mechanism of this process, and it is still unclear whether pericytes can contract cerebral capillaries and impair neurovascular coupling in AD [64]. In addition, we find that some experiments on the crucial steps in the mechanism were not performed under AD conditions. For example, a study used mouse retinal-derived microglia and pericytes to show that microglia secreted proinflammatory factors to induce pericytes to release ROS and abnormally contract capillaries, leading to decreased CBF and neurovascular uncoupling [58]. However, we do not know if this process would be the same in AD. Therefore, in vivo studies in the state of AD are urgently needed to determine the mechanisms. As pericytes are involved in AD-associated BBB destruction and neurovascular uncoupling and the mechanism is still unclear, we predict that a future research hotspot of AD is likely the mechanism of microglia mediate NVU dysfunction by regulating pericyte function.
ACKNOWLEDGMENTS
We would like to thank our supporters: National Natural Science Foundation of China (81701078), Natural Science Foundation of Heilongjiang Province of China (Outstanding Youth Foundation, YQ2022H003), China postdoctoral science foundation (2016M600261, 2018T110317), Heilongjiang Postdoctoral Financial Assistance (LBH-Z15163), Innovation and Entrepreneurship Training Program for College Students in Harbin Medical University (201910226322), Postgraduate Scientific Research and Practice Innovation Project of Harbin Medical University (YJSCX2020-03HYD).
Figures were created by Figdraw (www.figdraw.com).
FUNDING
This work was supported in part by grants from National Natural Science Foundation of China (81701078), Natural Science Foundation of Heilongjiang Province of China (Outstanding Youth Foundation, YQ2022H003), China postdoctoral science foundation (2016M600261, 2018T110317), Heilongjiang Postdoctoral Financial Assistance (LBH-Z15163), Innovation and Entrepreneurship Training Program for College Students in Harbin Medical University (201910226322), Postgraduate Scientific Research and Practice Innovation Project of Harbin Medical University (YJSCX2020-03HYD).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Alzheimer A ((1907) ) Uber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Z Psychiatr 64: , 146–148. |
[2] | Hodson R ((2018) ) Alzheimer’s disease. Nature 559: , S1. |
[3] | ((2020) ) 2020 Alzheimer’s disease facts and figures. Alzheimers Dement 16: , 391–460. |
[4] | Soto-Rojas LO , Pacheco-Herrero M , Martínez-Gómez PA , Campa-Córdoba BB , Apátiga-Pérez R , Villegas-Rojas MM , Harrington CR , de la Cruz F , Garcés-Ramírez L , Luna-Muñoz J ((2021) ) The neurovascular unit dysfunction in Alzheimer’s disease. Int J Mol Sci 22: , 2022. |
[5] | Nelson AR , Sweeney MD , Sagare AP , Zlokovic BV ((2016) ) Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta 1862: , 887–900. |
[6] | Montagne A , Barnes SR , Sweeney MD , Halliday MR , Sagare AP , Zhao Z , Toga AW , Jacobs RE , Liu CY , Amezcua L , Harrington MG , Chui HC , Law M , Zlokovic BV ((2015) ) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85: , 296–302. |
[7] | Sweeney MD , Kisler K , Montagne A , Toga AW , Zlokovic BV ((2018) ) The role of brain vasculature in neurodegenerative disorders. Nat Neurosci 21: , 1318–1331. |
[8] | Storck SE , Meister S , Nahrath J , Meißner JN , Schubert N , Di Spiezio A , Baches S , Vandenbroucke RE , Bouter Y , Prikulis I , Korth C , Weggen S , Heimann A , Schwaninger M , Bayer TA , Pietrzik CU ((2016) ) Endothelial LRP1 transports amyloid-β(1-42) across the blood-brain barrier. J Clin Invest 126: , 123–136. |
[9] | Noe CR , Noe-Letschnig M , Handschuh P , Noe CA , Lanzenberger R ((2020) ) Dysfunction of the blood-brain barrier-a key step in neurodegeneration and dementia. Front Aging Neurosci 12: , 185. |
[10] | van de Haar HJ , Jansen JFA , van Osch MJP , van Buchem MA , Muller M , Wong SM , Hofman PAM , Burgmans S , Verhey FRJ , Backes WH ((2016) ) Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging 45: , 190–196. |
[11] | Salvadores N , Searcy JL , Holland PR , Horsburgh K ((2017) ) Chronic cerebral hypoperfusion alters amyloid-β peptide pools leading to cerebral amyloid angiopathy, microinfarcts and haemorrhages in Tg-SwDI mice. Clin Sci (Lond) 131: , 2109–2123. |
[12] | Korte N , Nortley R , Attwell D ((2020) ) Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol 140: , 793–810. |
[13] | Daulatzai MA ((2017) ) Cerebral hypoperfusion and glucose hypometabolism: Key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer’s disease. J Neurosci Res 95: , 943–972. |
[14] | Subhramanyam CS , Wang C , Hu Q , Dheen ST ((2019) ) Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin Cell Dev Biol 94: , 112–120. |
[15] | Hickman SE , Allison EK , El Khoury J ((2008) ) Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci 28: , 8354–8360. |
[16] | Lee HG , Won SM , Gwag BJ , Lee YB ((2011) ) Microglial P2X7 receptor expression is accompanied by neuronal damage in the cerebral cortex of the APPswe/PS1dE9 mouse model of Alzheimer’s disease. Exp Mol Med 43: , 7–14. |
[17] | Kauppinen TM , Suh SW , Higashi Y , Berman AE , Escartin C , Won SJ , Wang C , Cho SH , Gan L , Swanson RA ((2011) ) Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid β. J Neuroinflammation 8: , 152. |
[18] | Ennerfelt H , Frost EL , Shapiro DA , Holliday C , Zengeler KE , Voithofer G , Bolte AC , Lammert CR , Kulas JA , Ulland TK , Lukens JR ((2022) ) SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell 185: , 4135–4152.e4122. |
[19] | Halle A , Hornung V , Petzold GC , Stewart CR , Monks BG , Reinheckel T , Fitzgerald KA , Latz E , Moore KJ , Golenbock DT ((2008) ) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9: , 857–865. |
[20] | Eshraghi M , Adlimoghaddam A , Mahmoodzadeh A , Sharifzad F , Yasavoli-Sharahi H , Lorzadeh S , Albensi BC , Ghavami S ((2021) ) Alzheimer’s disease pathogenesis: Role of autophagy and mitophagy focusing in microglia. Int J Mol Sci 22: , 3330. |
[21] | Lautrup S , Lou G , Aman Y , Nilsen H , Tao J , Fang EF ((2019) ) Microglial mitophagy mitigates neuroinflammation in Alzheimer’s disease. Neurochem Int 129: , 104469. |
[22] | Császár E , Lénárt N , Cserép C , Környei Z , Fekete R , Pósfai B , Balázsfi D , Hangya B , Schwarcz AD , Szabadits E , Szöllosi D , Szigeti K , Máthé D , West BL , Sviatkó K , Brás AR , Mariani JC , Kliewer A , Lenkei Z , Hricisák L , Benyó Z , Baranyi M , Sperlágh B , Menyhárt Á , Farkas E , Dénes Á ((2022) ) Microglia modulate blood flow, neurovascular coupling, and hypoperfusion via purinergic actions. J Exp Med 219: , e20211071. |
[23] | Zhao M , Jiang XF , Zhang HQ , Sun JH , Pei H , Ma LN , Cao Y , Li H ((2021) ) Interactions between glial cells and the blood-brain barrier and their role in Alzheimer’s disease. Ageing Res Rev 72: , 101483. |
[24] | Ronaldson PT , Davis TP ((2020) ) Regulation of blood-brain barrier integrity by microglia in health and disease: A therapeutic opportunity. J Cereb Blood Flow Metab 40: , S6–S24. |
[25] | Liu LR , Liu JC , Bao JS , Bai QQ , Wang GQ ((2020) ) Interaction of microglia and astrocytes in the neurovascular unit. Front Immunol 11: , 1024. |
[26] | Mehrabadi AR , Korolainen MA , Odero G , Miller DW , Kauppinen TM ((2017) ) Poly(ADP-ribose) polymerase-1 regulates microglia mediated decrease of endothelial tight junction integrity. Neurochem Int 108: , 266–271. |
[27] | Luciūnaitė A , McManus RM , Jankunec M , Rácz I , Dansokho C , Dalgėdienė I , Schwartz S , Brosseron F , Heneka MT ((2020) ) Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J Neurochem 155: , 650–661. |
[28] | Haruwaka K , Ikegami A , Tachibana Y , Ohno N , Konishi H , Hashimoto A , Matsumoto M , Kato D , Ono R , Kiyama H , Moorhouse AJ , Nabekura J , Wake H ((2019) ) Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat Commun 10: , 5816. |
[29] | Nordengen K , Kirsebom BE , Henjum K , Selnes P , Gísladóttir B , Wettergreen M , Torsetnes SB , Grøntvedt GR , Waterloo KK , Aarsland D , Nilsson LNG , Fladby T ((2019) ) Glial activation and inflammation along the Alzheimer’s disease continuum. J Neuroinflammation 16: , 46. |
[30] | Labus J , Häckel S , Lucka L , Danker K ((2014) ) Interleukin-1β induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J Neurosci Methods 228: , 35–45. |
[31] | Roberts TK , Eugenin EA , Lopez L , Romero IA , Weksler BB , Couraud PO , Berman JW ((2012) ) CCL2 disrupts the adherens junction: Implications for neuroinflammation. Lab Invest 92: , 1213–1233. |
[32] | Dudvarski Stankovic N , Teodorczyk M , Ploen R , Zipp F , Schmidt MHH ((2016) ) Microglia-blood vessel interactions: A double-edged sword in brain pathologies. Acta Neuropathol 131: , 347–363. |
[33] | Liddelow SA , Guttenplan KA , Clarke LE , Bennett FC , Bohlen CJ , Schirmer L , Bennett ML , Münch AE , Chung WS , Peterson TC , Wilton DK , Frouin A , Napier BA , Panicker N , Kumar M , Buckwalter MS , Rowitch DH , Dawson VL , Dawson TM , Stevens B , Barres BA ((2017) ) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: , 481–487. |
[34] | Michinaga S , Koyama Y ((2019) ) Dual roles of astrocyte-derived factors in regulation of blood-brain barrier function after brain damage. Int J Mol Sci 20: , 571. |
[35] | Propson NE , Roy ER , Litvinchuk A , Köhl J , Zheng H ((2021) ) Endothelial C3a receptor mediates vascular inflammation and blood-brain barrier permeability during aging. J Clin Invest 131: , e140966. |
[36] | Takata F , Dohgu S , Matsumoto J , Takahashi H , Machida T , Wakigawa T , Harada E , Miyaji H , Koga M , Nishioku T , Yamauchi A , Kataoka Y ((2011) ) Brain pericytes among cells constituting the blood-brain barrier are highly sensitive to tumor necrosis factor-α, releasing matrix metalloproteinase-9 and migrating in vitro. J Neuroinflammation 8: , 106. |
[37] | Monsonego A , Imitola J , Zota V , Oida T , Weiner HL ((2003) ) Microglia-mediated nitric oxide cytotoxicity of T cells following amyloid beta-peptide presentation to Th1 cells. J Immunol 171: , 2216–2224. |
[38] | Jansson D , Scotter EL , Rustenhoven J , Coppieters N , Smyth LC , Oldfield RL , Bergin PS , Mee EW , Graham ES , Faull RL , Dragunow M ((2016) ) Interferon-gamma blocks signalling through PDGFRbeta in human brain pericytes. J Neuroinflammation 13: , 249. |
[39] | Daneman R , Zhou L , Kebede AA , Barres BA ((2010) ) Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468: , 562–566. |
[40] | Armulik A , Genové G , Mäe M , Nisancioglu MH , Wallgard E , Niaudet C , He L , Norlin J , Lindblom P , Strittmatter K , Johansson BR , Betsholtz C ((2010) ) Pericytes regulate the blood-brain barrier. Nature 468: , 557–561. |
[41] | Kimbrough IF , Robel S , Roberson ED , Sontheimer H ((2015) ) Vascular amyloidosis impairs the gliovascular unit in a mouse model of Alzheimer’s disease. Brain 138: , 3716–3733. |
[42] | Fiala M , Zhang L , Gan X , Sherry B , Taub D , Graves MC , Hama S , Way D , Weinand M , Witte M , Lorton D , Kuo YM , Roher AE ((1998) ) Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med 4: , 480–489. |
[43] | Wang Y , Jin S , Sonobe Y , Cheng Y , Horiuchi H , Parajuli B , Kawanokuchi J , Mizuno T , Takeuchi H , Suzumura A ((2014) ) Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS One 9: , e110024. |
[44] | Stamatovic SM , Shakui P , Keep RF , Moore BB , Kunkel SL , Van Rooijen N , Andjelkovic AV ((2005) ) Monocyte chemoattractant protein-1 regulation of blood-brain barrier permeability. J Cereb Blood Flow Metab 25: , 593–606. |
[45] | Liu YJ , Guo DW , Tian L , Shang DS , Zhao WD , Li B , Fang WG , Zhu L , Chen YH ((2010) ) Peripheral T cells derived from Alzheimer’s disease patients overexpress CXCR2 contributing to its transendothelial migration, which is microglial TNF-alpha-dependent. Neurobiol Aging 31: , 175–188. |
[46] | Yang YM , Shang DS , Zhao WD , Fang WG , Chen YH ((2013) ) Microglial TNF-α-dependent elevation of MHC class I expression on brain endothelium induced by amyloid-beta promotes T cell transendothelial migration. Neurochem Res 38: , 2295–2304. |
[47] | Kallmann BA , Hummel V , Lindenlaub T , Ruprecht K , Toyka KV , Rieckmann P ((2000) ) Cytokine-induced modulation of cellular adhesion to human cerebral endothelial cells is mediated by soluble vascular cell adhesion molecule-1. Brain 123: (Pt 4), 687–697. |
[48] | Yoo SM , Park J , Kim SH , Jung YK ((2020) ) Emerging perspectives on mitochondrial dysfunction and inflammation in Alzheimer’s disease. BMB Rep 53: , 35–46. |
[49] | Kisler K , Nelson AR , Montagne A , Zlokovic BV ((2017) ) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18: , 419–434. |
[50] | Rangaraju S , Dammer EB , Raza SA , Rathakrishnan P , Xiao H , Gao T , Duong DM , Pennington MW , Lah JJ , Seyfried NT , Levey AI ((2018) ) Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol Neurodegener 13: , 24. |
[51] | Johnson ECB , Dammer EB , Duong DM , Ping L , Zhou M , Yin L , Higginbotham LA , Guajardo A , White B , Troncoso JC , Thambisetty M , Montine TJ , Lee EB , Trojanowski JQ , Beach TG , Reiman EM , Haroutunian V , Wang M , Schadt E , Zhang B , Dickson DW , Ertekin-Taner N , Golde TE , Petyuk VA , De Jager PL , Bennett DA , Wingo TS , Rangaraju S , Hajjar I , Shulman JM , Lah JJ , Levey AI , Seyfried NT ((2020) ) Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med 26: , 769–780. |
[52] | Kenkhuis B , Somarakis A , Kleindouwel LRT , van Roon-Mom WMC , Höllt T , van der Weerd L ((2022) ) Co-expression patterns of microglia markers Iba1, TMEM119 and P2RY12 in Alzheimer’s disease. Neurobiol Dis 167: , 105684. |
[53] | Yin F , Sancheti H , Patil I , Cadenas E ((2016) ) Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radic Biol Med 100: , 108–122. |
[54] | Bruce-Keller AJ , Gupta S , Parrino TE , Knight AG , Ebenezer PJ , Weidner AM , LeVine H , 3rd, Keller JN , Markesbery WR ((2010) ) NOX activity is increased in mild cognitive impairment. Antioxid Redox Signal 12: , 1371–1382. |
[55] | Park L , Anrather J , Zhou P , Frys K , Pitstick R , Younkin S , Carlson GA , Iadecola C ((2005) ) NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci 25: , 1769–1777. |
[56] | Russell AE , Jun S , Sarkar S , Geldenhuys WJ , Lewis SE , Rellick SL , Simpkins JW ((2019) ) Extracellular vesicles secreted in response to cytokine exposure increase mitochondrial oxygen consumption in recipient cells. Front Cell Neurosci 13: , 51. |
[57] | Lourenco CF , Ledo A , Barbosa RM , Laranjinha J ((2017) ) Neurovascular uncoupling in the triple transgenic model of Alzheimer’s disease: Impaired cerebral blood flow response to neuronal-derived nitric oxide signaling. Exp Neurol 291: , 36–43. |
[58] | Ding X , Zhang M , Gu R , Xu G , Wu H ((2017) ) Activated microglia induce the production of reactive oxygen species and promote apoptosis of co-cultured retinal microvascular pericytes. Graefes Arch Clin Exp Ophthalmol 255: , 777–788. |
[59] | Kisler K , Nelson AR , Rege SV , Ramanathan A , Wang Y , Ahuja A , Lazic D , Tsai PS , Zhao Z , Zhou Y , Boas DA , Sakadzić S , Zlokovic BV ((2017) ) Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci 20: , 406–416. |
[60] | Wang P , Guan PP , Wang T , Yu X , Guo JJ , Wang ZY ((2014) ) Aggravation of Alzheimer’s disease due to the COX-2-mediated reciprocal regulation of IL-1β and Aβ between glial and neuron cells. Aging Cell 13: , 605–615. |
[61] | Chow N , Bell RD , Deane R , Streb JW , Chen J , Brooks A , Van Nostrand W , Miano JM , Zlokovic BV ((2007) ) Serum response factor and myocardin mediate arterial hypercontractility and cerebral blood flow dysregulation in Alzheimer’s phenotype. Proc Natl Acad Sci U S A 104: , 823–828. |
[62] | Reynolds MR , Singh I , Azad TD , Holmes BB , Verghese PB , Dietrich HH , Diamond M , Bu G , Han BH , Zipfel GJ ((2016) ) Heparan sulfate proteoglycans mediate Aβ-induced oxidative stress and hypercontractility in cultured vascular smooth muscle cells. Mol Neurodegener 11: , 9. |
[63] | Nortley R , Korte N , Izquierdo P , Hirunpattarasilp C , Mishra A , Jaunmuktane Z , Kyrargyri V , Pfeiffer T , Khennouf L , Madry C , Gong H , Richard-Loendt A , Huang W , Saito T , Saido TC , Brandner S , Sethi H , Attwell D ((2019) ) Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science 365: , eaav9518. |
[64] | Hill RA , Tong L , Yuan P , Murikinati S , Gupta S , Grutzendler J ((2015) ) Regional blood flow in the normal and ischemic brain is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 87: , 95–110. |
[65] | Duncombe J , Lennen RJ , Jansen MA , Marshall I , Wardlaw JM , Horsburgh K ((2017) ) Ageing causes prominent neurovascular dysfunction associated with loss of astrocytic contacts and gliosis. Neuropathol Appl Neurobiol 43: , 477–491. |
[66] | Kirabali T , Rust R , Rigotti S , Siccoli A , Nitsch RM , Kulic L ((2020) ) Distinct changes in all major components of the neurovascular unit across different neuropathological stages of Alzheimer’s disease. Brain Pathol 30: , 1056–1070. |
[67] | Carter SF , Herholz K , Rosa-Neto P , Pellerin L , Nordberg A , Zimmer ER ((2019) ) Astrocyte biomarkers in Alzheimer’s disease. Trends Mol Med 25: , 77–95. |
[68] | Yamamoto M , Kiyota T , Horiba M , Buescher JL , Walsh SM , Gendelman HE , Ikezu T ((2007) ) Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol 170: , 680–692. |
[69] | McAlpine CS , Park J , Griciuc A , Kim E , Choi SH , Iwamoto Y , Kiss MG , Christie KA , Vinegoni C , Poller WC , Mindur JE , Chan CT , He S , Janssen H , Wong LP , Downey J , Singh S , Anzai A , Kahles F , Jorfi M , Feruglio PF , Sadreyev RI , Weissleder R , Kleinstiver BP , Nahrendorf M , Tanzi RE , Swirski FK ((2021) ) Astrocytic interleukin-3 programs microglia and limits Alzheimer’s disease. Nature 595: , 701–706. |
[70] | Liu CY , Yang Y , Ju WN , Wang X , Zhang HL ((2018) ) Emerging roles of astrocytes in neuro-vascular unit and the tripartite synapse with emphasis on reactive gliosis in the context of Alzheimer’s disease. Front Cell Neurosci 12: , 193. |
[71] | Agulhon C , Sun MY , Murphy T , Myers T , Lauderdale K , Fiacco TA ((2012) ) Calcium signaling and gliotransmission in normal vs. reactive astrocytes. Front Pharmacol 3: , 139. |
[72] | Domercq M , Brambilla L , Pilati E , Marchaland J , Volterra A , Bezzi P ((2006) ) P2Y1 receptor-evoked glutamate exocytosis from astrocytes: Control by tumor necrosis factor-alpha and prostaglandins. J Biol Chem 281: , 30684–30696. |
[73] | Welser-Alves JV , Milner R ((2013) ) Microglia are the major source of TNF-α and TGF-β1 in postnatal glial cultures; regulation by cytokines, lipopolysaccharide, and vitronectin. Neurochem Int 63: , 47–53. |
[74] | Rossi D , Brambilla L , Valori CF , Crugnola A , Giaccone G , Capobianco R , Mangieri M , Kingston AE , Bloc A , Bezzi P , Volterra A ((2005) ) Defective tumor necrosis factor-alpha-dependent control of astrocyte glutamate release in a transgenic mouse model of Alzheimer disease. J Biol Chem 280: , 42088–42096. |
[75] | Yi M , Yu P , Lu Q , Geller HM , Yu Z , Chen H ((2016) ) KCa3.1 constitutes a pharmacological target for astrogliosis associated with Alzheimer’s disease. Mol Cell Neurosci 76: , 21–32. |
[76] | Maezawa I , Jenkins DP , Jin BE , Wulff H ((2012) ) Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer’s disease. Int J Alzheimers Dis 2012: , 868972. |
[77] | Yu Z , Yu P , Chen H , Geller HM ((2014) ) Targeted inhibition of KCa3.1 attenuates TGF-β-induced reactive astrogliosis through the Smad2/3 signaling pathway. J Neurochem 130: , 41–49. |
[78] | Même W , Calvo CF , Froger N , Ezan P , Amigou E , Koulakoff A , Giaume C ((2006) ) Proinflammatory cytokines released from microglia inhibit gap junctions in astrocytes: Potentiation by beta-amyloid. FASEB J 20: , 494–496. |
[79] | Jiang S , Bhaskar K ((2017) ) Dynamics of the complement, cytokine, and chemokine systems in the regulation of synaptic function and dysfunction relevant to Alzheimer’s disease. J Alzheimers Dis 57: , 1123–1135. |
[80] | Lian H , Yang L , Cole A , Sun L , Chiang AC , Fowler SW , Shim DJ , Rodriguez-Rivera J , Taglialatela G , Jankowsky JL , Lu HC , Zheng H ((2015) ) NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85: , 101–115. |
[81] | Lian H , Litvinchuk A , Chiang AC , Aithmitti N , Jankowsky JL , Zheng H ((2016) ) Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci 36: , 577–589. |
[82] | Holm TH , Draeby D , Owens T ((2012) ) Microglia are required for astroglial Toll-like receptor 4 response and for optimal TLR2 and TLR3 response. Glia 60: , 630–638. |
[83] | Krasnow SM , Knoll JG , Verghese SC , Levasseur PR , Marks DL ((2017) ) Amplification and propagation of interleukin-1β signaling by murine brain endothelial and glial cells. J Neuroinflammation 14: , 133. |
[84] | Kirkley KS , Popichak KA , Afzali MF , Legare ME , Tjalkens RB ((2017) ) Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J Neuroinflammation 14: , 99. |
[85] | Wang QS , Ding HG , Chen SL , Liu XQ , Deng YY , Jiang WQ , Li Y , Huang LQ , Han YL , Wen MY , Wang MQ , Zeng HK ((2020) ) Hypertonic saline mediates the NLRP3/IL-1β signaling axis in microglia to alleviate ischemic blood-brain barrier permeability by downregulating astrocyte-derived VEGF in rats. CNS Neurosci Ther 26: , 1045–1057. |
[86] | van Kralingen C , Kho DT , Costa J , Angel CE , Graham ES ((2013) ) Exposure to inflammatory cytokines IL-1β and TNFα induces compromise and death of astrocytes; implications for chronic neuroinflammation. PLoS One 8: , e84269. |
[87] | Norden DM , Fenn AM , Dugan A , Godbout JP ((2014) ) TGFβ produced by IL-10 redirected astrocytes attenuates microglial activation. Glia 62: , 881–895. |
[88] | Winkler EA , Sagare AP , Zlokovic BV ((2014) ) The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol 24: , 371–386. |
[89] | Morgan TE , Nichols NR , Pasinetti GM , Finch CE ((1993) ) TGF-beta 1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp Neurol 120: , 291–301. |
[90] | Rustenhoven J , Aalderink M , Scotter EL , Oldfield RL , Bergin PS , Mee EW , Graham ES , Faull RL , Curtis MA , Park TI , Dragunow M ((2016) ) TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J Neuroinflammation 13: , 37. |
[91] | Watson AN , Berthiaume AA , Faino AV , McDowell KP , Bhat NR , Hartmann DA , Shih AY ((2020) ) Mild pericyte deficiency is associated with aberrant brain microvascular flow in aged PDGFRβ(+/-) mice. J Cereb Blood Flow Metab 40: , 2387–2400. |
[92] | Mäe MA , He L , Nordling S , Vazquez-Liebanas E , Nahar K , Jung B , Li X , Tan BC , Chin Foo J , Cazenave-Gassiot A , Wenk MR , Zarb Y , Lavina B , Quaggin SE , Jeansson M , Gu C , Silver DL , Vanlandewijck M , Butcher EC , Keller A , Betsholtz C ((2021) ) Single-cell analysis of blood-brain barrier response to pericyte loss. Circ Res 128: , e46–e62. |
[93] | Buxbaum JD , Oishi M , Chen HI , Pinkas-Kramarski R , Jaffe EA , Gandy SE , Greengard P ((1992) ) Cholinergic agonists and interleukin 1 regulate processing and secretion of the Alzheimer beta/A4 amyloid protein precursor. Proc Natl Acad Sci U S A 89: , 10075–10078. |
[94] | Sheng L , Chen M , Cai K , Song Y , Yu D , Zhang H , Xu G ((2019) ) Microglial Trem2 induces synaptic impairment at early stage and prevents amyloidosis at late stage in APP/PS1 mice. FASEB J 33: , 10425–10442. |
[95] | Hong S , Beja-Glasser VF , Nfonoyim BM , Frouin A , Li S , Ramakrishnan S , Merry KM , Shi Q , Rosenthal A , Barres BA , Lemere CA , Selkoe DJ , Stevens B ((2016) ) Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352: , 712–716. |
[96] | Bhaskar K , Maphis N , Xu G , Varvel NH , Kokiko-Cochran ON , Weick JP , Staugaitis SM , Cardona A , Ransohoff RM , Herrup K , Lamb BT ((2014) ) Microglial derived tumor necrosis factor-α drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol Dis 62: , 273–285. |
[97] | Floden AM , Li S , Combs CK ((2005) ) Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci 25: , 2566–2575. |
[98] | Liu N , Zhuang Y , Zhou Z , Zhao J , Chen Q , Zheng J ((2017) ) NF-κB dependent up-regulation of TRPC6 by Aβ in BV-2 microglia cells increases COX-2 expression and contributes to hippocampus neuron damage. Neurosci Lett 651: , 1–8. |
[99] | Schilling S , Chausse B , Dikmen HO , Almouhanna F , Hollnagel JO , Lewen A , Kann O ((2021) ) TLR2- and TLR3-activated microglia induce different levels of neuronal network dysfunction in a context-dependent manner. Brain Behav Immun 96: , 80–91. |
[100] | Kwon HS , Koh SH ((2020) ) Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl Neurodegener 9: , 42. |
[101] | Joshi P , Turola E , Ruiz A , Bergami A , Libera DD , Benussi L , Giussani P , Magnani G , Comi G , Legname G , Ghidoni R , Furlan R , Matteoli M , Verderio C ((2014) ) Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ 21: , 582–593. |
[102] | Crapser JD , Spangenberg EE , Barahona RA , Arreola MA , Hohsfield LA , Green KN ((2020) ) Microglia facilitate loss of perineuronal nets in the Alzheimer’s disease brain. EBioMedicine 58: , 102919. |
[103] | Verret L , Mann EO , Hang GB , Barth AM , Cobos I , Ho K , Devidze N , Masliah E , Kreitzer AC , Mody I , Mucke L , Palop JJ ((2012) ) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149: , 708–721. |
[104] | Guerra G , Lucariello A , Perna A , Botta L , De Luca A , Moccia F ((2018) ) The role of endothelial Ca(2+) signaling in neurovascular coupling: A view from the lumen. Int J Mol Sci 19: , 938. |
[105] | Peferoen L , Kipp M , van der Valk P , van Noort JM , Amor S ((2014) ) Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 141: , 302–313. |
[106] | Nugent AA , Lin K , van Lengerich B , Lianoglou S , Przybyla L , Davis SS , Llapashtica C , Wang J , Kim DJ , Xia D , Lucas A , Baskaran S , Haddick PCG , Lenser M , Earr TK , Shi J , Dugas JC , Andreone BJ , Logan T , Solanoy HO , Chen H , Srivastava A , Poda SB , Sanchez PE , Watts RJ , Sandmann T , Astarita G , Lewcock JW , Monroe KM , Di Paolo G ((2020) ) TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105: , 837–854.e839. |
[107] | Jadhav VS , Lin PBC , Pennington T , Di Prisco GV , Jannu AJ , Xu G , Moutinho M , Zhang J , Atwood BK , Puntambekar SS , Bissel SJ , Oblak AL , Landreth GE , Lamb BT ((2020) ) Trem2 Y38C mutation and loss of Trem2 impairs neuronal synapses in adult mice. Mol Neurodegener 15: , 62. |
[108] | Zhou Y , Song WM , Andhey PS , Swain A , Levy T , Miller KR , Poliani PL , Cominelli M , Grover S , Gilfillan S , Cella M , Ulland TK , Zaitsev K , Miyashita A , Ikeuchi T , Sainouchi M , Kakita A , Bennett DA , Schneider JA , Nichols MR , Beausoleil SA , Ulrich JD , Holtzman DM , Artyomov MN , Colonna M ((2020) ) Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat Med 26: , 131–142. |
[109] | Fassler M , Rappaport MS , Cuño CB , George J ((2021) ) Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in Alzheimer’s disease models. J Neuroinflammation 18: , 19. |
[110] | Baranger K , van Gijsel-Bonnello M , Stephan D , Carpentier W , Rivera S , Khrestchatisky M , Gharib B , De Reggi M , Benech P ((2019) ) Long-term pantethine treatment counteracts pathologic gene dysregulation and decreases Alzheimer’s disease pathogenesis in a transgenic mouse model. Neurotherapeutics 16: , 1237–1254. |
[111] | Ng RC , Jian M , Ma OK , Bunting M , Kwan JS , Zhou GJ , Senthilkumar K , Iyaswamy A , Chan PK , Li M , Leung KM , Kumar Durairajan SS , Lam KS , Chu LW , Festenstein R , Chung SK , Chan KH ((2021) ) Chronic oral administration of adipoRon reverses cognitive impairments and ameliorates neuropathology in an Alzheimer’s disease mouse model. Mol Psychiatry 26: , 5669–5689. |
[112] | Fragoulis A , Siegl S , Fendt M , Jansen S , Soppa U , Brandenburg LO , Pufe T , Weis J , Wruck CJ ((2017) ) Oral administration of methysticin improves cognitive deficits in a mouse model of Alzheimer’s disease. Redox Biol 12: , 843–853. |
[113] | Kaneko J , Enya A , Enomoto K , Ding Q , Hisatsune T ((2017) ) Anserine (beta-alanyl-3-methyl-L-histidine) improves neurovascular-unit dysfunction and spatial memory in aged AβPPswe/PSEN1dE9 Alzheimer’s-model mice. Sci Rep 7: , 12571. |
[114] | Sheng M , Lu H , Liu P , Li Y , Ravi H , Peng SL , Diaz-Arrastia R , Devous MD , Womack KB ((2017) ) Sildenafil improves vascular and metabolic function in patients with Alzheimer’s disease. J Alzheimers Dis 60: , 1351–1364. |
[115] | DeRidder L , Sharma A , Liaw K , Sharma R , John J , Kannan S , Kannan RM ((2021) ) Dendrimer-tesaglitazar conjugate induces a phenotype shift of microglia and enhances β-amyloid phagocytosis. Nanoscale 13: , 939–952. |
[116] | Cortes-Canteli M , Kruyer A , Fernandez-Nueda I , Marcos-Diaz A , Ceron C , Richards AT , Jno-Charles OC , Rodriguez I , Callejas S , Norris EH , Sanchez-Gonzalez J , Ruiz-Cabello J , Ibanez B , Strickland S , Fuster V ((2019) ) Long-term dabigatran treatment delays Alzheimer’s disease pathogenesis in the TgCRND8 mouse model. J Am Coll Cardiol 74: , 1910–1923. |
[117] | Polis B , Gurevich V , Assa M , Samson AO ((2019) ) Norvaline restores the BBB integrity in a mouse model of Alzheimer’s disease. Int J Mol Sci 20: , 4616. |
[118] | Lin AL , Jahrling JB , Zhang W , DeRosa N , Bakshi V , Romero P , Galvan V , Richardson A ((2017) ) Rapamycin rescues vascular, metabolic and learning deficits in apolipoprotein E4 transgenic mice with pre-symptomatic Alzheimer’s disease. J Cereb Blood Flow Metab 37: , 217–226. |
[119] | Kurata T , Kawai H , Miyazaki K , Kozuki M , Morimoto N , Ohta Y , Ikeda Y , Abe K ((2012) ) Statins have therapeutic potential for the treatment of Alzheimer’s disease, likely via protection of the neurovascular unit in the AD brain. J Neurol Sci 322: , 59–63. |
[120] | Liu R , Li JZ , Song JK , Zhou D , Huang C , Bai XY , Xie T , Zhang X , Li YJ , Wu CX , Zhang L , Li L , Zhang TT , Du GH ((2014) ) Pinocembrin improves cognition and protects the neurovascular unit in Alzheimer related deficits. Neurobiol Aging 35: , 1275–1285. |
[121] | Liu R , Zhang TT , Zhou D , Bai XY , Zhou WL , Huang C , Song JK , Meng FR , Wu CX , Li L , Du GH ((2013) ) Quercetin protects against the Aβ(25-35)-induced amnesic injury: Involvement of inactivation of rage-mediated pathway and conservation of the NVU. Neuropharmacology 67: , 419–431. |
[122] | Lalli G , Schott JM , Hardy J , De Strooper B ((2021) ) Aducanumab: A new phase in therapeutic development for Alzheimer’s disease? EMBO Mol Med 13: , e14781. |
[123] | Alexander GC , Emerson S , Kesselheim AS ((2021) ) Evaluation of Aducanumab for Alzheimer disease: Scientific evidence and regulatory review involving efficacy, safety, and futility. JAMA 325: , 1717–1718. |
[124] | Silva MVF , Loures CMG , Alves LCV , de Souza LC , Borges KBG , Carvalho MDG ((2019) ) Alzheimer’s disease: Risk factors and potentially protective measures. J Biomed Sci 26: , 33. |
[125] | van Bokhoven P , de Wilde A , Vermunt L , Leferink PS , Heetveld S , Cummings J , Scheltens P , Vijverberg EGB ((2021) ) The Alzheimer’s disease drug development landscape. Alzheimers Res Ther 13: , 186. |
[126] | Lewcock JW , Schlepckow K , Di Paolo G , Tahirovic S , Monroe KM , Haass C ((2020) ) Emerging microglia biology defines novel therapeutic approaches for Alzheimer’s disease. Neuron 108: , 801–821. |
[127] | Takata K , Ginhoux F , Shimohama S ((2021) ) Roles of microglia in Alzheimer’s disease and impact of new findings on microglial heterogeneity as a target for therapeutic intervention. Biochem Pharmacol 192: , 114754. |
[128] | Ries M , Watts H , Mota BC , Lopez MY , Donat CK , Baxan N , Pickering JA , Chau TW , Semmler A , Gurung B , Aleksynas R , Abelleira-Hervas L , Iqbal SJ , Romero-Molina C , Hernandez-Mir G , d’Amati A , Reutelingsperger C , Goldfinger MH , Gentleman SM , Van Leuven F , Solito E , Sastre M ((2021) ) Annexin A1 restores cerebrovascular integrity concomitant with reduced amyloid-β and tau pathology. Brain 144: , 1526–1541. |
[129] | Park JC , Baik SH , Han SH , Cho HJ , Choi H , Kim HJ , Choi H , Lee W , Kim DK , Mook-Jung I ((2017) ) Annexin A1 restores Aβ(1-42)-induced blood-brain barrier disruption through the inhibition of RhoA-ROCK signaling pathway. Aging Cell 16: , 149–161. |
[130] | Moussa C , Hebron M , Huang X , Ahn J , Rissman RA , Aisen PS , Turner RS ((2017) ) Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflammation 14: , 1. |
[131] | Van Skike CE , Jahrling JB , Olson AB , Sayre NL , Hussong SA , Ungvari Z , Lechleiter JD , Galvan V ((2018) ) Inhibition of mTOR protects the blood-brain barrier in models of Alzheimer’s disease and vascular cognitive impairment. Am J Physiol Heart Circ Physiol 314: , H693–h703. |
[132] | Tarantini S , Valcarcel-Ares MN , Toth P , Yabluchanskiy A , Tucsek Z , Kiss T , Hertelendy P , Kinter M , Ballabh P , Süle Z , Farkas E , Baur JA , Sinclair DA , Csiszar A , Ungvari Z ((2019) ) Nicotinamide mononucleotide (NMN) supplementation rescues cerebromicrovascular endothelial function and neurovascular coupling responses and improves cognitive function in aged mice. Redox Biol 24: , 101192. |
[133] | Yao Z , Yang W , Gao Z , Jia P ((2017) ) Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci Lett 647: , 133–140. |
[134] | Tarantini S , Yabluchanskiy A , Csipo T , Fulop G , Kiss T , Balasubramanian P , DelFavero J , Ahire C , Ungvari A , Nyúl-Tóth Á , Farkas E , Benyo Z , Tóth A , Csiszar A , Ungvari Z ((2019) ) Treatment with the poly(ADP-ribose) polymerase inhibitor PJ-34 improves cerebromicrovascular endothelial function, neurovascular coupling responses and cognitive performance in aged mice, supporting the NAD+depletion hypothesis of neurovascular aging. Geroscience 41: , 533–542. |
[135] | Liu X , Yamashita T , Shang J , Shi X , Morihara R , Huang Y , Sato K , Takemoto M , Hishikawa N , Ohta Y , Abe K ((2019) ) Twendee X ameliorates phosphorylated tau, α-synuclein and neurovascular dysfunction in Alzheimer’s disease transgenic mice with chronic cerebral hypoperfusion. J Stroke Cerebrovasc Dis 28: , 104310. |
[136] | Samudra N , Motes M , Lu H , Sheng M , Diaz-Arrastia R , Devous M , Hart J , Womack KB ((2019) ) A pilot study of changes in medial temporal lobe fractional amplitude of low frequency fluctuations after sildenafil administration in patients with Alzheimer’s disease. J Alzheimers Dis 70: , 163–170. |
[137] | Hisatsune T , Kaneko J , Kurashige H , Cao Y , Satsu H , Totsuka M , Katakura Y , Imabayashi E , Matsuda H ((2016) ) Effect of anserine/carnosine supplementation on verbal episodic memory in elderly people. J Alzheimers Dis 50: , 149–159. |
[138] | Szcześniak D , Budzeń S , Kopeć W , Rymaszewska J ((2014) ) Anserine and carnosine supplementation in the elderly: Effects on cognitive functioning and physical capacity. Arch Gerontol Geriatr 59: , 485–490. |
[139] | Jian M , Kwan JS , Bunting M , Ng RC , Chan KH ((2019) ) Adiponectin suppresses amyloid-β oligomer (AβO)-induced inflammatory response of microglia via AdipoR1-AMPK-NF-κB signaling pathway. J Neuroinflammation 16: , 110. |
[140] | Lin AL , Zheng W , Halloran JJ , Burbank RR , Hussong SA , Hart MJ , Javors M , Shih YY , Muir E , Solano Fonseca R , Strong R , Richardson AG , Lechleiter JD , Fox PT , Galvan V ((2013) ) Chronic rapamycin restores brain vascular integrity and function through NO synthase activation and improves memory in symptomatic mice modeling Alzheimer’s disease. J Cereb Blood Flow Metab 33: , 1412–1421. |
[141] | Lindsay A , Hickman D , Srinivasan M ((2021) ) A nuclear factor-kappa B inhibiting peptide suppresses innate immune receptors and gliosis in a transgenic mouse model of Alzheimer’s disease. Biomed Pharmacother 138: , 111405. |
[142] | Ghosh A , Comerota MM , Wan D , Chen F , Propson NE , Hwang SH , Hammock BD , Zheng H ((2020) ) An epoxide hydrolase inhibitor reduces neuroinflammation in a mouse model of Alzheimer’s disease. Sci Transl Med 12: , eabb1206. |
[143] | Yi C , Ezan P , Fernández P , Schmitt J , Sáez JC , Giaume C , Koulakoff A ((2017) ) Inhibition of glial hemichannels by boldine treatment reduces neuronal suffering in a murine model of Alzheimer’s disease. Glia 65: , 1607–1625. |
[144] | Park JS , Kam TI , Lee S , Park H , Oh Y , Kwon SH , Song JJ , Kim D , Kim H , Jhaldiyal A , Na DH , Lee KC , Park EJ , Pomper MG , Pletnikova O , Troncoso JC , Ko HS , Dawson VL , Dawson TM , Lee S ((2021) ) Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol Commun 9: , 78. |
[145] | Wang HM , Zhang T , Huang JK , Xiang JY , Chen JJ , Fu JL , Zhao YW ((2017) ) Edaravone attenuates the proinflammatory response in amyloid-β-treated microglia by inhibiting NLRP3 inflammasome-mediated il-1β secretion. Cell Physiol Biochem 43: , 1113–1125. |
[146] | Lonnemann N , Hosseini S , Marchetti C , Skouras DB , Stefanoni D , D’Alessandro A , Dinarello CA , Korte M ((2020) ) The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 117: , 32145–32154. |
[147] | Okorji UP , Velagapudi R , El-Bakoush A , Fiebich BL , Olajide OA ((2016) ) Antimalarial drug artemether inhibits neuroinflammation in BV2 microglia through Nrf2-dependent mechanisms. Mol Neurobiol 53: , 6426–6443. |
[148] | Velagapudi R , El-Bakoush A , Olajide OA ((2018) ) Activation of Nrf2 pathway contributes to neuroprotection by the dietary flavonoid tiliroside. Mol Neurobiol 55: , 8103–8123. |
[149] | Pedicone C , Fernandes S , Dungan OM , Dormann SM , Viernes DR , Adhikari AA , Choi LB , De Jong EP , Chisholm JD , Kerr WG ((2020) ) Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J Cell Sci 133: . |
[150] | Xie Z , Zhao J , Wang H , Jiang Y , Yang Q , Fu Y , Zeng H , Hölscher C , Xu J , Zhang Z ((2020) ) Magnolol alleviates Alzheimer’s disease-like pathology in transgenic C. elegans by promoting microglia phagocytosis and the degradation of beta-amyloid through activation of PPAR-γ. Biomed Pharmacother 124: , 109886. |
[151] | Zu HB , Liu XY , Yao K ((2020) ) DHCR24 overexpression modulates microglia polarization and inflammatory response via Akt/GSK3β signaling in Aβ(25)(-)(35) treated BV-2 cells. Life Sci 260: , 118470. |
[152] | Yang Z , Kuboyama T , Tohda C ((2019) ) Naringenin promotes microglial M2 polarization and Aβ degradation enzyme expression. Phytother Res 33: , 1114–1121. |
[153] | Ren C , Li D , Zhou Q , Hu X ((2020) ) Mitochondria-targeted TPP-MoS(2) with dual enzyme activity provides efficient neuroprotection through M1/M2 microglial polarization in an Alzheimer’s disease model. Biomaterials 232: , 119752. |
[154] | Anwar S , Rivest S ((2020) ) Alzheimer’s disease: Microglia targets and their modulation to promote amyloid phagocytosis and mitigate neuroinflammation. Expert Opin Ther Targets 24: , 331–344. |
[155] | Wang S , Mustafa M , Yuede CM , Salazar SV , Kong P , Long H , Ward M , Siddiqui O , Paul R , Gilfillan S , Ibrahim A , Rhinn H , Tassi I , Rosenthal A , Schwabe T , Colonna M ((2020) ) Anti-human TREM2 induces microglia proliferation and reduces pathology in an Alzheimer’s disease model. J Exp Med 217: , e20200785. |
[156] | Wang S , Sudan R , Peng V , Zhou Y , Du S , Yuede CM , Lei T , Hou J , Cai Z , Cella M , Nguyen K , Poliani PL , Beatty WL , Chen Y , Cao S , Lin K , Rodrigues C , Ellebedy AH , Gilfillan S , Brown GD , Holtzman DM , Brioschi S , Colonna M ((2022) ) TREM2 drives microglia response to amyloid-β via SYK-dependent and -independent pathways. Cell 185: , 4153–4169.e4119. |
[157] | Machida T , Takata F , Matsumoto J , Takenoshita H , Kimura I , Yamauchi A , Dohgu S , Kataoka Y ((2015) ) Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9-releasing cell type constituting the blood-brain barrier in vitro. Neurosci Lett 599: , 109–114. |


