
Pathological and Therapeutic Advances in Parkinson’s Disease: Mitochondria in the Interplay
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative illness majorly affecting the population between the ages of 55 to 65 years. Progressive dopaminergic neuronal loss and the collective assemblage of misfolded alpha-synuclein in the substantia nigra, remain notable neuro-pathological hallmarks of the disease. Multitudes of mechanistic pathways have been proposed in attempts to unravel the pathogenesis of PD but still, it remains elusive. The convergence of PD pathology is found in organelle dysfunction where mitochondria remain a major contributor. Mitochondrial processes like bioenergetics, mitochondrial dynamics, and mitophagy are under strict regulation by the mitochondrial genome and nuclear genome. These processes aggravate neurodegenerative activities upon alteration through neuroinflammation, oxidative damage, apoptosis, and proteostatic stress. Therefore, the mitochondria have grabbed a central position in the patho-mechanistic exploration of neurodegenerative diseases like PD. The management of PD remains a challenge to physicians to date, due to the variable therapeutic response of patients and the limitation of conventional chemical agents which only offer symptomatic relief with minimal to no disease-modifying effect. This review describes the patho-mechanistic pathways involved in PD not only limited to protein dyshomeostasis and oxidative stress, but explicit attention has been drawn to exploring mechanisms like organelle dysfunction, primarily mitochondria and mitochondrial genome influence, while delineating the newer exploratory targets such as GBA1, GLP, LRRK2, and miRNAs and therapeutic agents targeting them.
INTRODUCTION
Parkinson’s disease (PD) is the second most common form of neurodegenerative disease, following Alzheimer’s disease (AD) affecting an estimated population of 10 million worldwide [1], and about 0.58 million people in India alone [2]. This extrapyramidal syndrome has shown debilitating effects on the quality of life of patients due to the associated synucleinopathy, affecting their motor activity along with cognitive functions in patients. PD motor presentations include tremor, dystonia, rigidity, and bradykinesia while the non-motor symptoms include bowel disturbances, fatigue, anxiety, cognitive decline, and apathy. Sporadic PD etiology is attributed to predisposing factors like age, gender, social, and lifestyle factors. Population with age >60 years are at higher risk of developing parkinsonism where again females remain slightly protected than the counter-balanced males due to estrogen’s neuroprotective effect on the dopaminergic (DA) system. Furthermore, smoking, sedentary lifestyle, and pesticide exposure also cause aggravated disease pathology. Co-morbid conditions like diabetes, hypertension, chronic pulmonary disease, cerebrovascular disease, and paralysis also contribute to PD pathology [3]. Although a plethora of proposed hypothesis and theories exist for PD pathogenesis and manifestation, aging and organelle (mitochondria) dysfunction remain the foremost notable contributors. Mitochondria is an important organelle of the cell regulating the energy homeostasis, principally and is prone to oxidative damage leading to its dysregulated function. Mitochondria perform a wide array of activities beyond simple ATP synthesis including fatty acid breakdown, amino acid and nucleotide metabolism, lipid, quinone and steroid biosynthesis, iron-sulfur cluster biogenesis, calcium and iron homeostasis, and also apoptosis [4]. Its supposed interaction with various other organelles such as the endoplasmic reticulum, lysosome, cytoskeleton, peroxisomes, and the nucleus helps in the maintenance of cellular homeostasis. The dysfunction of mitochondrial dynamics such as biogenesis, fission, fusion, mitophagy, and its motility play a vital role in the oxidative damage mediated pathogenic pathway activation [5]. Furthermore, the alterations in the mitochondrial DNA (mtDNA) at the genetic and epigenetic level have been linked with the manifestation of several neurodegenerative diseases including AD and PD [6]. Therefore, its improper functioning has become a principal attraction for pathological research related to diseases ranging from neurodegeneration to metabolic disorders and cardiovascular dysfunction [7]. Several small molecule modulators have been recognized for the regulation of various disturbed pathways in PD, including GBA1 agonists, PPAR agonists, GLP1 agonists, LRRK2 inhibitors, ion channel modulators of glutamate and calcium, as well as antioxidant and anti-inflammasome agents. This review includes an overview of the pathological perturbations in PD with an emphasis on its relationship with altered mitochondrial functioning, and further describes agents undergoing preclinical and clinical screening for their efficacy in PD treatment and management.
PATHOLOGICAL PERTUBATIONS IN PD
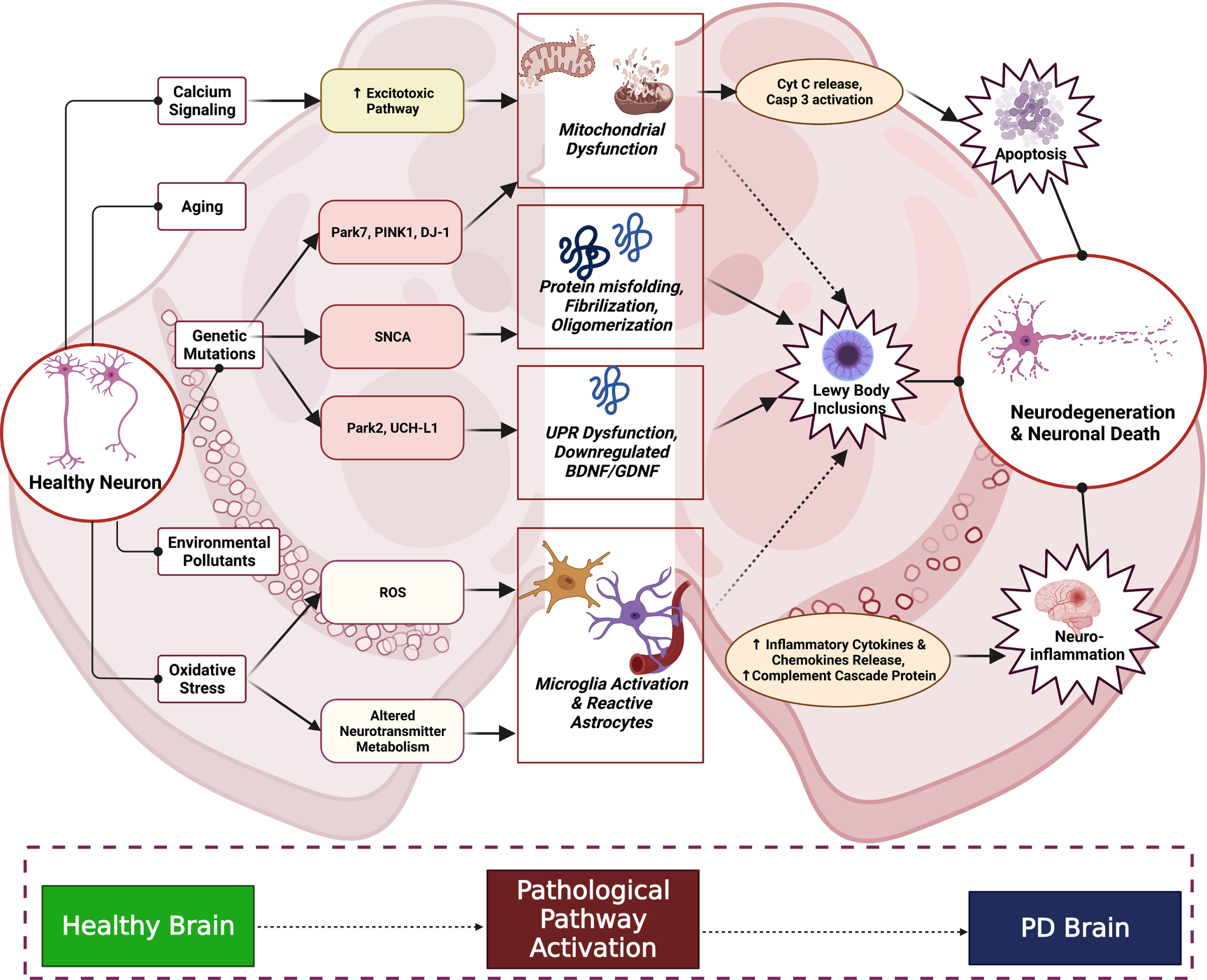
As we look back to the studies exploring PD pathogenesis in past few decades, we can say that PD is a disorder beyond just proteinopathies. The complex pathway and the factors associated with the clinical phenotypes of PD are explained in Fig. 1. Several mechanisms involved in the pathogenesis of PD leads to neuronal death. Key players such as mitochondrial dysfunction, glial cells induction, aging, environmental pollutants (MPTP-N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, Rotenone, paraquat (PQ), and pesticides), elevated cytosolic calcium levels leading to neuronal excitotoxicity, oxidative stress, and the like, contribute to the loss of DA neurons. Apart from these, genetic mutations of SNCA, PARK7, DJ1 (Parkin 7), PINK1 (PTEN Induced Kinase 1), LRRK2 (leucine-rich repeat kinase 2), and HTRA2 (high temperature requirement A2) impairs mitochondrial function and thereby activates the caspase pathway and induces apoptosis. Mutations in the α-Synuclein gene (SNCA) cause protein misfoldings, protofibril formation, and oligomerization. Mutations of PARK2 and UCHL1 (ubiquitin carboxy-terminal hydrolase L1) are connected with the ubiquitin-proteasome system pathway and proteotoxic stress. PARK2 mutations significantly alter the mitochondrial activity, potentially impairing proteasomal function and produces downregulation of brain-derived neurotrophic factor and glial cell-derived neurotropic factor. Increase in the reactive oxygen species (ROS) because of mitochondrial dysfunction and altered neurotransmitter metabolism leads to activation of reactive astrocyte and microglial cells, ultimately increasing the release of pro-inflammatory cytokines such as tumor necrosis factor (TNF-α), interleukin-1β, interleukin-6 (IL-6), and Interferon-γ, which potentiate the chemokine release and trigger the activation of the complement cascade. All these cellular mechanisms are the pathological hallmarks of PD which are individually explained in sections ahead [8].
Fig. 1
PD Pathological Cascade in Substantia Nigra. The above complex cascade shows the interplay of factors like aging, oxidative stress, environmental factors, and altered physiological signaling between cells. Neuroinflammatory process begins majorly due to cellular oxidative damage leading to microglial activation and cytokine infiltration and further inflammatory damage. Proteostasis is altered owing to genetic mutations (SNCA, PINK1, etc.), environmental stressors and oxidative damage leading to Lewy body inclusions. The apoptotic signaling is activated by excitotoxic damage to the neurons and mitochondrial dysfunction leading to caspase activation occurs. These processes cumulatively result in the manifestation of neurodegeneration and hence neuronal death, aggravating the PD condition.
Proteinopathy in PD
Proteinopathy in PD is extensively linked to α-synuclein (α-Syn) misfolding, a 140 amino acid long protein. α-Syn normally exists in an unfolded state and regulates the normal neuronal physiology like synaptic vesicle trafficking and subsequent neurotransmitter release. It also aids in the neuronal interactions of the mitochondria and the endoplasmic reticulum (ER). But certain environmental triggers causes its conformational change from a monomer into oligomers, leading to its accumulation in the neurons. These misfolding lead to β-sheet-rich Lewy body formation which later deposits in various cytoplasmic brain regions imposing major detrimental effects on the basal ganglia. Dynamic balance of this protein is maintained by phosphorylation (involved in activation and aggregation, majorly) and dephosphorylation of its various subunits. The phosphorylation of α-Syn at serine 129 is one of the majorly detected features in the PD pathology which leads to neuronal Lewy body inclusion [9]. α-Syn accumulation in the mitochondria contributes to the disruption of the neuronal function in the Substantia Nigra Pars Compacta (SNpc) [10]. Certain forms of α-Syn accumulate in the non-DA areas of the CNS causing the non-motor symptoms and Lewy bodies in the brain regions, including the olfactory bulb, the dorsal motor nucleus of the vagal nerve, locus coeruleus, raphe nucleus, basal nucleus of Meynert, and pedunculopontine nucleus [11]. Multitude of autopsy studies, of PD patients, showed the deposition of Lewy neurites and Lewy body inclusion in the DA neurons situated in the SNpc region of the midbrain as well as in the striatum and amygdala regions [12, 13]. α-Syn aggregates not only localize in cytoplasmic and synaptic fluids but its localization in mitochondria has also been strongly linked factor for PD pathogenesis. It aggregates in the mitochondria associated membranes (MAM) principally, affecting the mitochondrial bioenergetics (through interaction with Complex I and Complex IV), mitochondrial dynamics and trafficking (via interaction with Miro and Milton proteins), and also mitophagy [14]. There are studies reporting overexpression of α-Syn in cells cause translocation of the aggregates to mitochondria which ultimately disrupts cellular function [15]. Mitochondrial membrane lipid, cardiolipin, is said to interact with the α-Syn protein leading to its refolding into α-helical form by pulling out monomers from fibrils. It has also been reported to activate LC3 response which further regulates autophagy [16]. The toxic cellular effects of α-Syn aggregation in mitochondria were reversed by caspase-inhibition in-vitro [17]. The exact mechanism regulating α-Syn mediated mitochondrial damage is yet to be explored, but still AMPK, SIRT3, LC3, VDAC (Voltage-Dependent Anion Channel) and cardiolipin have been explored as potential mediators [18]. Also, synaptic pile-up of toxic α-Syn impairs the synaptic molecular trafficking by altering the morphology of synaptic vesicles, dysregulates presynaptic SNARE complex leading to variable priming and fusion of synaptic vesicles which further causes impaired synaptic signaling and neuro-communication. Toxic α-Syn induced synaptopathy leads to aberrant network activity and dying-back neurodegeneration leading to prodormal symptomology. Although α-Syn is ubiquitously present in the brain and is accumulated in presynaptic terminals universally, only the mid-brain DA neurons remain highly susceptible to it since it inhibits the enzymes responsible for DA synthesis. Additionally, it also interacts with DAT and VMAT2, and alters α-Syn levels. In a nutshell, deposition of α-Syn triggers DA dysregulation which has the potential to cause synaptic malfunctioning later leading to impaired neuronal networking and thereby, progressive neurodegeneration like seen in case of PD [19].
Gut microbial link
Not just centrally aggregated peptide but even the peripheral clusters of misfolded α-Syn in the enteric nervous system (ENS) are said to contribute to the motor symptoms observed in the disease. The gut microbial link was well explored by Braak et al. where the olfactory (peripheral nervous system) and vagal nerve (enteric nervous system) were reported to be involved in disease initiation in case of sporadic PD. PD patients experience non-motor symptoms due to gastrointestinal (GI) dysmotility (constipation), increased intestinal permeability and movement of bacteria and their products, all resulting in oxidative stress observed in human studies. The metabolites produced by gut microbiota including short chain fatty acids (SCFAs), niacinamide (NAM), neurotransmitters and bile salts interferes with the pathological pathways involved in PD. These metabolites travel from intestinal mucosa through the intestinal nerve plexus to the vagal nerve and finally to CNS. The other possible pathway of the interconnection is through blood circulation [20]. Braak’s hypothesis states that the spread of misfolded α-Syn occurs from ENS to CNS in a reverse pattern via the vagus nerve. The symptomatology-based staging of disease progression as per Braak’s observation states that at early stage (stages 1 and 2- involving lewy neurite formation in lower brain stem and medulla oblangata) the non-motor symptoms are noted in clinical cases while the motor symptoms are visible at intermediate stage (stage 3 and 4- lewy body lesions observed in SNpc, basal forebrain, amygdala, thalamus with progression into the cortex) of disease and the later stage (stage 5 and 6-DA cell death in cortex as well as other involved regions) marks the cognitive decline [11, 21]. PD patients showed decreased contents of the bacterial strains like Dorea, Bacteroides, Prevotella, Faecalibacterium, Bacteroides massiliensis, Stoquefichusmassiliensis, Bacteroides coprocola, Blautiaglucerasea, Dorealongicatena, Bacteroides dorei, Bacteroides plebeius, Prevotellacopri, Coprococcuseutactus, and Ruminococcuscallidus, and increased contents of Christensenella, Catabacter, Lactobacillus, Oscillospira, Bifidobacterium, Christensenellaminuta, Catabacterhongkongensis, Lactobacillusmucosae, Ruminococcusbromii, and Papillibactercinnamivorans [22]. Formation of a type of amyloid fiber known as curli by the gut microbes affects the α-Syn aggregation process and contributes to PD pathology. Intraperitoneal administration of lipopolysaccharide causes hyper-permeability of large intestine and abnormal clump of phosphorylated α-Syn. H. Pylori infection may lead to neurodegeneration and cause PD as levodopa absorption increases after elimination of H. Pylori infection. The metabolites formed by gut microbes can be directly linked to mitochondrial dysfunction in PD, as metabolites like NAM act as a precursor for nicotinamide adenine dinucleotide (NAD) formation which is an important component of mitochondrial respiration. Similarly, bile salts like tauroursodesoxycholic acid and ursodesoxycholic acid participate in mitophagy regulation. SCFA’s are important component of the membrane lipids and aid in mitochondrial dynamics. Targeting such microbial dyshomeostasis may aid in countering PD etiological contributors including mitochondrial dysmetabolism [20]. Neuroprotective effect by fecal microbiota transplantation on MPTP-induced PD mice subdue the (TLR 4)/ TNF-α signaling pathway via microbiota-gut brain axis [23].
Protein clearance failure
Inefficient clearance of protein aggregates are also major contributors of PD pathology besides excessive peptide secretion and protein misfolding, where an inability to clear the aggregated proteins become toxic to the cell. Toxic aggregates of abnormally folded proteins are cleared either through autophagy- lysosome pathway or the ubiquitin-proteasome system (UPS). The autophagy-lysosomal pathway involves three main processes, namely macrophagy, microphagy and chaperon-mediated autophagy (CMA) [24]. In case of PD, autophagic failure contributes to the continuous accumulation of the misfolded α-Syn protein in neuronal cells. These misfolded proteins later induce the apoptosis of DA neurons by causing mitochondrial dysfunction. This is achieved by, passing into the mitochondria and attaching to the inner membrane and thereby injuring the electron transport chain (ETC) complexes and also reduces the voltage-dependent anion channel 1 (VDAC1) expression. A study hypothesized that the α-Syn damages the DA neuronal mitochondria by upregulating the expression of MT-CO2 gene. MT-CO2 is rumored to regulate the release of cytochrome c, BCL2 family proteins, and Smac/DIABLO which act as an apoptogenic factors leading to ETC dysfunction and cell death. α-Syn induced mitochondrial dysfunction via cytochrome c oxidase subunit 2 in SH-SY5Y cells [25]. The aggregates of α-Syn decrease lysosomal degradation activity by degrading hydrolases trafficking, such as of glucocerebrosidase 1 (GCase1), from the endoplasmic reticulum to the lysosome. Mutations in GBA1 gene causes dysfunction of lysosome autophagy system, which is involved in the genetic risk of PD. Mutations of GCase1, a lysosomal hydrolase enzyme responsible for disrupting the lipid glucosylceramide into ceramide and glucose, leads to an autosomal-recessive or autosomal-dominant atypical Parkinsonian syndrome [26, 27]. Recent studies have established a bidirectional link between PD and lysosomal storage diseases (LSD). The presence of α-Syn inclusions in the brain regions of patients with LSD and the further identification of several lysosomal genes involved in LSD as a genetic risk-factors to develop PD support the established link between the two diseases [28].
UPS follows the principle of ubiquitination and further proteasomal degradation of the troublesome misfolded protein, the proteolytic activities of 26/20S proteasomes are weakened in the SNpc. 20S proteasome α-subunit and other molecules involved in the normal function of the UPS, like PA700 and PA28 (proteasome activators), are decreased in sporadic PD. Mutations in the PARK2 gene that encodes the E3-ubiquitin ligase parkin, is observed in early onset PD (EOPD) since it is actually involved in the clearance of abnormally misfolded proteins via ubiquitin-proteasome system. As PARK gene is a neuropathological hallmark not involved in Lewy body formation, the role of Parkin as an E3-ubiquitin ligase with extensive neuroprotective functions including the maintenance of mitochondrial metabolism and the ubiquitin-proteasome system (where parkin plays a crucial role in the ubiquitin-mediated degradation of misfolded or damaged proteins and also in deletion of dysfunctional mitochondria via mitophagy) has been widely explored [29].
CMA-related proteins such as Hsc70 and LAMP2A found in the post-mortem SNpc and amygdala of PD patient’s brain, further support this theory [24]. Bioactive constituents such as Dihydromyricetin (DHA) and Salvianolic acid B (SAL B) were found to induce the CMA pathway and reduce the microglia and astroglia-directed neuroinflammation in a PD mouse model expressing human wildtype α-Syn fused with the green fluorescent protein (GFP) (BAC-α-syn-GFP transgenic mice) [30]. The selective deletion of autophagy-related gene 7 (Atg7) resulted in α-Syn accumulation and further DA neurodegeneration in SNpc, pointing out the role of dysregulated autophagy as a potential cause of neurodegeneration [31]. Excessive expression of α-Syn was found to inhibit autophagy regulators such as Rab1a, Lmx1b, transcription factor EB (TFEB) contributing to autophagy/lysosomal pathway disruption [32]. In a recent study, autophagy was observed to mediate the clearance of α-Syn and TPPP/p25A in certain atrophy models. Tubulin polymerization-promoting protein, via exophagy, promotes the secretion of α-Syn which is said to meddle with the autophagosome-lysosome fusion process [33].
ER stress and calcium dyshomeostasis
ER stress and autophagy are highly intertwined components protecting cells from stress by inducing death of damaged ones. Inositol-requiring protein-1 (IRE1), protein kinase RNA like ER kinase (PERK) and the activating transcription factor-6 (ATF6) are the three main stress sensors that recognize misfolded, faulty proteins [34]. In a study it was reported that, the neuroprotective action of ATF6 was compromised by the over-expression of α-Syn [35]. Impaired ATF6 signaling along with dysregulated ER associated protein degradation (ERAD) pathway leads to apoptosis, thus, disrupting the UPR pathway. In α-Syn loaded nigral DA neurons, the PERK–eIF2α pathway was also found to be over-activated [36]. In a very recent study involving MPTP-lesioned PD model, Ghrelin’s (hunger hormone) neuroprotective activity by halting the MPP+-induced oxidative stress was explored [37]. Dawson et al. [36] have also linked the mutations of Parkin gene with the dysregulations of the ERAD pathways of misfolded ER proteins [38]. Accumulation of faulty proteins in the ER lumen also triggers the activation of ER chaperones such as Bip and PDIp. DA degeneration induced by α-Syn accumulation in the nematode C. elegans was found to decrease due to the over-expression of LRRK2 by indirect upregulation of the ER chaperone Bip. Lack of LRRK2 made the neurons susceptible to ER-stress induced neurodegeneration [39].
ER stress also affects calcium homeostasis in cell. Neuronal membranes are loaded with voltage-gated calcium (CaV) channels; CaV1 (L-type), CaV2 (P-, N- and R-type) and CaV3 (T-Type). The CaV1 and CaV2 channel isoforms activate at a high depolarization voltage and produce long-lasting calcium currents, while CaV3 channels open at a lower depolarization voltage and produce brief intermittent rise in Ca2 + currents [40]. Such aberrant Ca2 + currents over a period of time triggers mitochondrial oxidative stress. The transient rise of DA neuronal calcium level triggers oxidative stress of the mitochondria, which results in compromised cellular bioenergetics and ultimately pushes the cell to its demise [41]. This irregular calcium current induced oxidative stress gained strong support upon finding out that, population on L type Ca2 + channel blockers were at a low risk for developing PD. Isradipine, a CaV1.3 channel antagonist, showed promising neuroprotective effects in 6-OHDA induced PD mouse model but when administered to a large clinical early-stage PD population, it failed to slow down the disease progression [42, 43]. In another study, it was demonstrated that the α-Syn aggregates bind to the ER calcium pump SERCA and activates it. This results in the efflux of calcium from the cytosol into the ER [44]. Mutations of the kinase protein LRRK2 imbalances the calcium levels, its buffering capacity and also induces mitochondrial degradation and neurite aggregation, which could further potentially precipitate neurodegeneration [45]. It was observed by Nath et al. [46, 47] that, human cell lines expressing α-Syn gene over-expressed the corresponding protein when intracellular calcium levels increased, though the exact mechanism behind this sudden over-expression could not be precisely elucidated.
ER and mitochondria are both dynamic organelles that regulate cellular apoptosis. Both these organelles physically come together to form structures referred as mitochondria-associated ER membranes (MAMs) through which vital cellular processes such as lipid and calcium (Ca2 +) homeostasis, mitochondrial dynamics, autophagy, inflammation, apoptosis, and the like transpire. ER is prone to stress and such stressful conditions promote the excessive release of calcium into the cytosol which then triggers the production of mitochondrial ROS (mtROS) due to the depolarization of the inner mitochondrial membrane. This very mtROS aids in the accumulation of misfolded proteins within ER which further leads to ER stress and organelle dilation. The transient rise of DA neuronal calcium level triggers oxidative stress of the mitochondria, which results in compromised cellular bioenergetics and ultimately pushes the cell to its demise by releasing the cytochrome c apoptotic protease activating factor-1 (Apaf-1), for the formation of apoptosome and thereby activates caspase 9, which further activates caspase 3, which induces cellular death cues. This type of mitochondria-derived cellular death induction via cytochrome c release can also be triggered when there is an accumulation of aberrant proteins in the ER lumen as seen in the case of several neurodegenerative diseases [48].
2.2Neuroinflammation
Chronic neuroinflammation is one of the hallmarks of PD wherein innate immunity of the nervous system is overactivated. This subsequently leads to the release of inflammatory mediators such as NO, inducible nitric oxide synthase (iNOS), and COX-2 due to aging, pollution, oxidative stress, and infection. Inflammatory mediators further cause activation of NF-κB, MAPK (mitogen-activated protein kinase), and PI3K/AKT pathways which trigger neuroinflammation to promote neuronal damage and cell death [49].
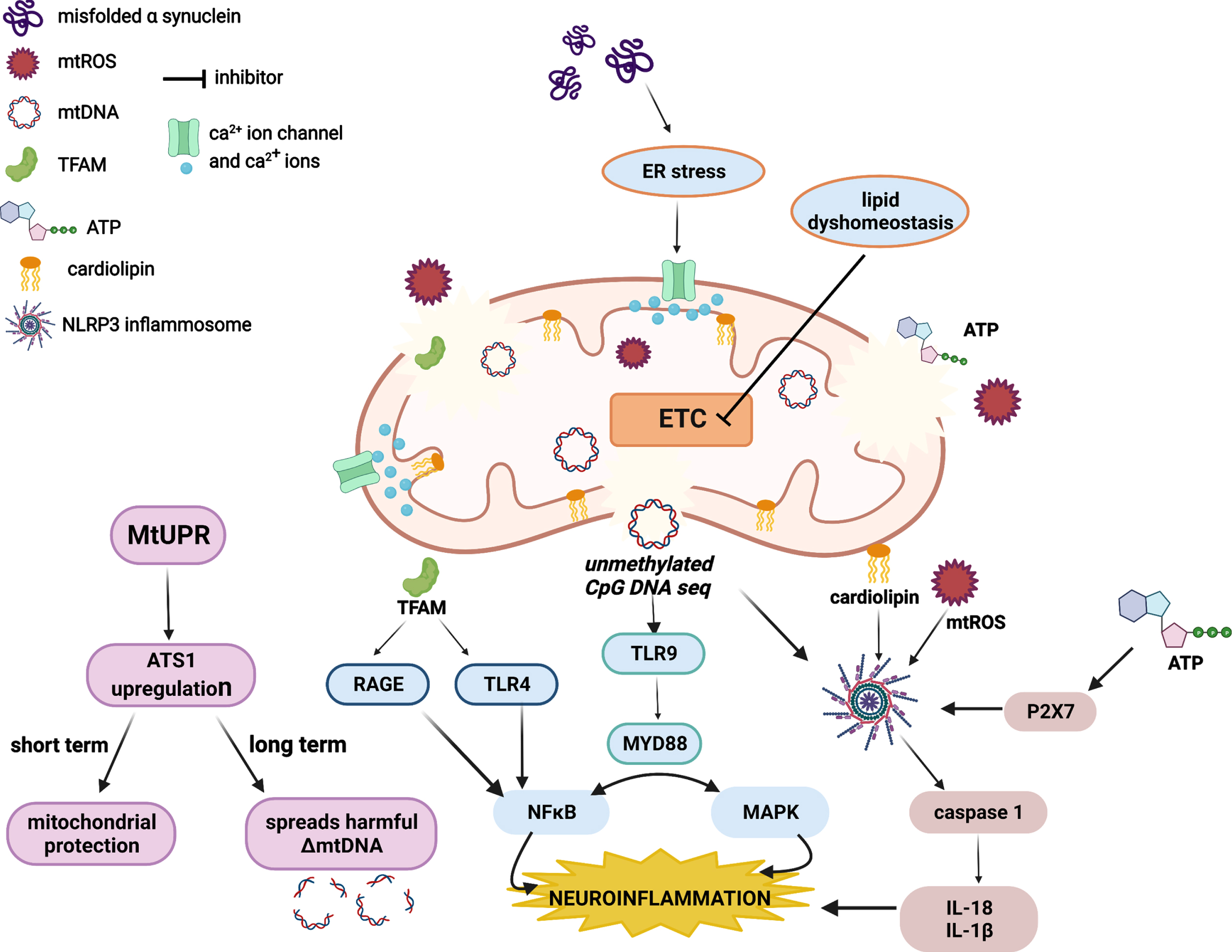
This state of neuroinflammation can be augmented by mitochondrial-derived damage-associated molecular patterns (mtDAMPs) such as mtDNA, mtROS, mitochondrial unfolded protein response (mtUPR), transcription factor A mitochondria (TFAM), adenosine triphosphate (ATP), cardiolipin, iron, cytochrome c, and mitochondrial Ca2 +. The linked pathways have been described in Fig. 2 linking neuroinflammation to mitochondrial dysregulation. The unmethylated CpG-DNA sequence of mtDNA serves as a ligand for TLR9, which upon activation, induces the NF-κB and MAPK pathway leading to neuroinflammation. mtDNA also has the potential to trigger NLRP3 inflammasome which mediates inflammation by the release of IL-18 and IL-1β. mtUPR upregulates the expression of ATS1, a transcription factor that governs the mitochondrial-nucleus crosstalk during UPR. Initial ATS1 accumulation is found beneficial but has long term detrimental effects by enhancing the percentage of harmful heterogenous mtDNA in the neurons. mtROS (produced majorly by mitochondrial ETC and NADPH oxidase, NOX) and cardiolipin serve as NLRP3 inflammasome activators and drive the progression of neuronal inflammation. ATP leaking out from damaged neurons or glial cells also triggers the NLRP3 inflammasome via P2X7 activation. TFAM has been found to interact with TLR4, RAGE, and Mac-1 to activate the proinflammatory microglia [50, 51].
Fig. 2
Neuroinflammation associated to mitochondrial-DAMPs.
Lipid dyshomeostasis
Linolenic acid (LA) is well-known for its antioxidant properties and its dampened concentration may contribute to an excess of oxidative stress, especially in PD patients [52]. The fatty acids like α-linolenic acid (ALA), arachidonic acid (AA), and docosahexaenoic acid (DHA) were found altered in PD patients, where lower levels of LA and ALA were associated to motor symptoms while non-motor symptoms were linked to higher plasma levels of DHA and AA [52]. This phenomenon together with the decrease of antioxidant vitamins, namely B3 and B5, could contribute to the proinflammatory and prooxidant environment that likely advance some of the mechanisms underlying neurodegeneration. Excess monomers of α-Syn interact with oleic acid and leads to the formation of monounsaturated fatty acids which makes α-Syn more neurotoxic [53]. The mitochondria serve as an important site for intracellular lipid metabolism since this is the organelle where beta oxidation of fats occurs. Accumulation of lipids inside the mitochondria leads to its decreased biogenesis and also hampers the ETC leading to excessive ROS accumulation. The very core of Lewy bodies comprises of numerous lipids surrounded by dystrophic mitochondria linking lipid abnormality and mitochondrial malfunctioning [54].
Gene mutations
The α-Syn protein is encoded by NG_011851 (SNCA) gene. Genetic mutation in the N-terminal of this gene (A53E/T, A30P, E46K H50Q, and G51D) increases the production of ROS, mitochondrial respiration decreases and changes the mitochondrial membrane permeability [55]. The first pA53 mutation was identified in the phenotype of PD in 20 to 85 years and the p.A30 gene variation in 54 to 76 years [56]. The mutant form of SNCA gene encodes for abnormal α-Syn which causes apoptosis via triggering caspase pathway and calcium signaling [10]. In recent studies of human postmortem PD tissue and experimental models, the interaction of the α-Syn and translocase of the outer mitochondrial membrane (TOM20) are being widely explored. It is suggested that certain forms of α-Syn bind to TOM20 thereby inhibiting the binding interaction between the TOM20 and TOM22, which ultimately affects the mitochondrial functioning and the mitochondrial protein import mechanism. In PD patients, TOM40 another component of mitochondrial import machinery, has been found to be reduced [57].
Mutations in the LRRK2 gene (PARK8) are very strongly linked with late-onset autosomal dominant type of PD. LRRK2 with the mutation Gly2019Ser, are detected in patients with autosomal dominant PD. In preclinical animal models of PD, the mutation in this gene includes an increase in the LRRK2 kinase activity. Arg1441Gly mutation is also commonly observed in PD. It may also increase expression of ATP13A2, a gene related to autosomal recessive PD (ARPD) which encodes the lysosomal P-type transport ATPase ATP13A2, providing further suggestion that LRRK2 mutations can cause PD phenotype by disturbing neuronal lysosomal function [58, 59]. LRRK2 G2019S mutation lead to significant changes in DAT and VMAT2 levels in a recent study involving mice models of PD [60]. A decade’s worth of study indicates the underlying role of LRRK2 mutants in causing neurodegeneration via mitochondrial dysfunction. Though the specific mechanisms are unknown and yet to be unfurled, it can be said with confidence that LRKK2 mutations play an intricate role in affecting diverse functions of the mitochondria by increasing oxidative stress, depolarizing the mitochondrial membrane, impairing ETC and mitophagy, morphologically changing the organelle etc.
The DJ-1 is a causative gene for recessive early-onset familial PD. DJ-1 linked PD (L172Q) that has been detected so far, shows Lewy body pathology, suggesting the existence of a connection between the DJ1 and synucleiopathy. This protein is highly expressed in the astrocytes of the frontal cortex and SNpc nigra in idiopathic PD cases. The protective effect against the oxidative stress is provided by DJ1 protein. It binds directly to the F1FO ATP synthase β subunit and declines the mitochondrial uncoupling and increases the ATP production while mutations of DJ-1 contrastingly increase mitochondrial uncoupling and depolarizes neuronal mitochondria. DJ-1 governing ATP synthase regulation potentially enhances DA neuron metabolism [61]. The mutations in DJ1 gene (M26I, E64D, A104T, D149A, and L166P) have been identified in the PD [62, 63]. PARK7 functions as reactive oxygen chaperone [64]. PRKN, PINK1, and DJ-1, involved in mitochondria and mitophagy related functions, are usually responsible for PD forms, slow evolution, and upright retort to DA therapy [65].
Apart from these nuclear gene mutations, the variations in the genetics of mitochondrial complex 1 has also been explored in the pathogenesis of various neurodegenerative disorders, as it is the very first step of the mammalian ETC and any malfunctioning in the activity of this complex is at the center of pathological ROS release which are detrimental for ideal cellular functioning. Since complex 1 is encoded by both nuclear DNA (nDNA) and mtDNA, there is reason to believe that mutations in either of these genes manifest as severely devastating neuromuscular disorders due to the compromise in ATP production. 11778G>A mutation within the gene for the ND4 subunit is commonly found in LHON cases and an additional 3866T>C mutation within the ND1 gene of complex 1 was seen to worsen the progression of LHON [66]. A recent cybrid cell studies have also established a relation between MT-ND1m.3460G>A mutation and varying degrees of LHON due to impaired oxidative phosphorylation (OXPHOS) [67]. In a present-day study conducted in a Tamil Nadu population, missense mutations of ND1 and ND5 were linked with autism spectrum disorder, showing that complex 1 genes may play a vital role in the onset of autism spectrum disorder [68]. The ND6-P25L mtDNA mutation, that leads to the substitution of leucine instead of proline at position 25, was found to offer protection against cardiac IR injury by blocking the reverse electron transfer (RET) phenomenon [69]. The 10237T>C in MT-ND3 gene heteroplasmic mutation, and the homoplasmic mutations such as 14484T>C in MT-ND6 gene and 5437C>T in MT-ND2 gene have been rumored to impact the structural integrity and functioning of the complex 1 enzyme and thereby play a role in the pathogenesis of multiple sclerosis [70]. Excessive mutations of m.14597A>G in MT-ND6 has been reported to cause leigh syndrome [71]. Certain mutations were found to be beneficial, for example, the MT-ND2m.5178C>A gene mutation has proven to be protective in case of various diseases, but the underlying mechanism through which this protection is offered is yet to be known. A lymphocyte cell line carrying this mutation was seen to produce more ATP and less ROS. The transcription level of Bcl-2 gene was also high and so was the protein translation rate. It also showed decreased activity of caspase 3/7 and decreased early and late apoptosis. The overall mitochondrial complex 1 activity of the mutant group was found to be much higher in comparison to the control [72]. A whole-genome sequencing of skeletal muscle DNA revealed a unique previously unreported mutation- m.10372A>G in MT-ND3 causing adult-onset sensorimotor axonal polyneuropathy due to decreased ATP production, which results from hindered activity of complex 1 [73]. Another point mutation in ND3, i.e., m.10191T>C observed in patients with leigh syndrome, was found to be strongly correlated with epilepsy [74]. An extremely high-resolution technique-Mitochondrial DNA Structural Variation Sequencing (MitoSV-seq) was developed by Jaberi et al., for screening the structural variations and single-nucleotide variations in individual neurons. Certain common SVs (structural variations) discovered in MT-ND5 sequence were common between hematological cancers and Ifnar1–/– DA neurons (murine PD model), hinting that its mutations might have high clinical value as respective disease biomarkers [75].
The brain cells are majorly composed of neurons, glia, and astrocytes which help in homeostatic functioning of the brain. Healthy functioning of vital organs like brain and liver requires high energy supply along with increased oxygen. As more energy is required for their optimum performance, mitochondria the “powerhouse of the cell” becomes a vital organelle [76]. The mitochondria play an important role in energy production through the operation of ETC. Aging causes improper functioning of mitochondria leading to its dysfunction and other related amalgamations with the pathogenesis of PD, these include impairment of mitochondrial biogenesis, increased ROS production, defective mitophagy, ETC dysfunction, changes in mitochondrial dynamics, calcium homeostasis as well as oxytosis/ferroptosis inhibition which are strongly linked to neurodegeneration. Hence, mitochondrial link to PD pathogenesis needs to be explored in order to understand PD pathological pathway.
MITOCHONDRIA: THE CENTERFOLD OF THE CELL
Approximately 1.45 billion years ago, the mitochondria (close descendants of the purple nonsulfur bacteria) and the animal cell entered into a fateful endosymbiosis and this hypothesis was put forth by Dr. Lynn Margulis, in the 1970 s [77]. Despite the presence of overwhelming circumstantial evidence supporting this proposal, the origin and antiquity of the mitochondria has been a subject of much debate. While the animal cell offered protection, the mitochondria favored it with cellular respiration [78, 79]. What started as a simple interdependent relation quickly escalated and presently, the mitochondria are at the center of various complex cellular signaling pathways, making it one of the most crucial components of cell survival. Being the most dynamic, double-membrane bound organelle which is most commonly referred to as the ‘powerhouse’ or ‘engine’ of the cell, the mitochondria, serves as a ground for complex biochemical reactions including the invaluable OXPHOS reactions. The production of ATP, which is the quintessential cellular fuel, is one of the most important outcomes of these reactions [80]. More recently the role of mitochondria in the immune system has been explored endlessly. It is believed to have an explicit role in the growth and function of immune cells, also their activation, proliferation, and in phagocytosis. As a result of ROS production, these immune cells are capable of performing complicated processes such as apoptosis, pyroptosis, and NETosis. ROS also aids in the activation of adaptive immunity, B cell antibody synthesis, and T cell stimulation. Apart from this, Yu-Jih Su and his team [81] have very recently reviewed the interesting involvement of the mitochondria in the immune cell mediated tissue regeneration and aging. Mitochondrial genome has henceforth come to light for its involvement in various forms of diseases and disorders.
Housing its own maternally inherited genome, the mitochondria is further involved in a plethora of other functions including fatty acid breakdown, amino acid and nucleotide metabolism, lipid, quinone, and steroid biosynthesis, iron-sulphur cluster biogenesis, apoptosis, calcium and iron homeostasis; and also makes functionally relevant contacts with other organelles [82]. The mammalian mtDNA was sequenced for the very first time in 1981 [83]. Unlike the nDNA, mtDNA is histone free and also lacks introns in humans [83]. It is packed with 37 genes in a sequence of only 16,549 bps and has a double-stranded structure containing a light strand (L) and a heavy strand (H). The strands are differentiated based on their buoyant density differences owing to their varying G+T nitrogenous base composition [84]. The heavy strand being information rich codes for 2 ribosomal RNAs (rRNAs) (12 s and 16 s), 14 transfer RNAs (tRNAs), and 12 polypeptides while the light strand codes for eight tRNAs and one polypeptide. This constitutes a total of 11 messenger RNAs (mRNAs) (translated into 13 proteins), 2 rRNAs (12S and 16S rRNA), and 22 tRNAs. The OXPHOS reactions are accomplished with the involvement of about 90 proteins of which only 13 are encoded by the mtDNA and the rest by the nDNA. Nevertheless, these 13 proteins form the heart of the system performing the OXPHOS reactions and hence any compromise in them would ultimately affect the cellular aerobic respiration and in turn the health of the cell [83]. The presence of a triple stranded non-coding region (NCR) called the displacement loop or D-loop has also been observed. Comprising of 520 to 700 base pairs, it is complimentary to the L strand and also contains the origin of replication of the H strand which happens to be the leading strand. Further, it houses two committed promoter sequences, light-strand promoter (LSP) and heavy-strand promoter (HSP) along with other regulatory domains that govern the process of replication and transcription of the mitogenome. Each mitochondrion contains 2 to 10 copies of the mitogenome, and these numbers may even go up to 1000 in some cases. Organelles with identical copies of mtDNA display homoplasmy while the ones with mutated copies display heteroplasmy [85]. Generally, these mutations are recessive and not harmful. They become a threat to the respiratory processes when they overpass a certain threshold. The root of these mutations are found be due to the lack of histone proteins [80], low fidelity of the replication enzymes such as DNA polymerase gamma [85], and oxidative stress as a result of ROS generation [85, 86]. The free radical tenet is undoubtedly the most debated among the others. While some groups state that a higher mitochondrial potential is the source of H2O2 generation, other groups counter that low membrane potential triggers H2O2 release [87].
mtDNA, totally comprises of 37 genes, housing 28 in the H strand and the remaining 9 on the L strand. Out of the 28 genes residing on the H strand, 6 code for the subunits of the complex 1 (NADH: ubiquinone oxidoreductase) enzyme of the respiratory chain, namely, ND1, ND2, ND3, ND4L, ND4, and ND5; 3 genes encode the subunits of cytochrome oxidase c enzyme including COX1, COX2, and COX3; 1 gene encodes for cytochrome b subunit and; 2 genes code for ATPase subunits: ATP6 and ATP8. These two genes notably have overlapping open reading frames on the genome. Apart from this, the H strand also has 2 genes that encode for the rRNAs namely, 16S rRNA and 12S rRNA and 14 genes for the 14 different tRNAs of the amino acids, i.e., F, V, L, I, M, W, D, K, G, R, H, S, L, and T. The L strand is not information dense unlike the H strand and thus has an ample amount of NCR. One of its gene, ND6 codes for a specific subunit of the complex 1 enzyme and the remaining 8 genes code for tRNAs of the following amino acids Q, A, N, C, Y, S, E, and P [63, 64, 68]. The genes regulating mitochondrial ETC has been enlisted in Table 1. Though this organelle is pretty much autonomous and self-regulating, it still needs guidance from the nucleus for its chronic well-being [91].
Table 1
Genes regulating mitochondrial ETC
| Genes | Protein encoded | Complex | Location |
| ND1 | NADH: ubiquinoneoxidoreductase | I | H |
| ND2 | NADH: ubiquinone oxidoreductase | I | H |
| ND3 | NADH: ubiquinone oxidoreductase | I | H |
| ND4L | NADH: ubiquinone oxidoreductase | I | H |
| ND4 | NADH: ubiquinone oxidoreductase | I | H |
| ND5 | NADH: ubiquinone oxidoreductase | I | H |
| COX1 | cytochrome oxidase c | IV | H |
| COX2 | cytochrome oxidase c | IV | H |
| COX3 | cytochrome oxidase c | IV | H |
| CYB | cytochrome b | III | H |
| ATP6 | ATP synthase 6 | V | H |
| ATP8 | ATP synthase 8 | V | H |
| MT-RNR1 | 12S rRNA | H | |
| MT-RNR2 | 16S rRNA | H | |
| tRNAF, tRNAV, tRNAL, tRNAI, tRNAM, tRNAW, tRNAD, tRNAK, tRNAG, tRNAR, tRNAH, tRNAS, tRNAL and tRNAT | F, V, L, I, M, W, D, K, G, R, H, S, L and T | H | |
| ND6 | NADH: ubiquinoneoxidoreductase | I | L |
| tRNAQ, tRNAA, tRNAN, tRNAC, tRNAY, tRNAS, tRNAE and tRNAP | Q, A, N, C, Y, S, E and P | L |
THE NUCLEO-MITO CROSSTALK
Time and time again, it has been established through innumerable research that there exists, a constant crosstalk between the nucleus and the mitochondria. In the nucleus, epigenetic changes occur in the form of chemical alterations of the histone proteins and these changes have a prominent hold on gene expression. The frequently occurring modifications include histone acetylation, deacetylation, lysine methylation, phosphorylation, ubiquitylation, sumoylation, and PARylation [91, 92]. Unlike the nucleus, the mitochondria do not have histone proteins wrapped around the mtDNA. That being said, the epigenetic changes of the nucleoid proteins does, however, have a role in regulation of gene expression in the organelle [91].
DNA methylation is considered as a cardinal epigenetic modification that regulates gene expression and preserves genetic stability. As the name suggests, methylation refers to the addition of a methyl group onto the 5th position of the cytosine ring (C to 5mC) and is catalyzed by a group of enzymes known as DNA methyltransferases (DNMT), which pick up the methyl group from S-adenosylmethionine (SAM). Methylation of cytosine is most common in eukaryotes whereas adenine methylation is common in prokaryotes [93]. There are studies that propose that altered DNA methylation levels could be responsible for the pathology of various diseases ranging from cancer, obesity, diabetes to neurodegeneration (PD and AD) due to irreversible alteration in the DNA sequences [94]. Another family of enzymes involved in epigenomic changes includes the TET, i.e., ten-eleven translocation methyl cytosine dioxygenases that convert 5mC to 5hmC (5-hydroxymethylcytosine) [92]. The activities of both DNMT and TET is significantly swayed by environmental cues such as tobacco smoke, toxic pollutants, etc. [95].
Mitochondrial processes including bioenergetics, dynamics, biogenesis, and mitochondrial autophagy are under strict regulatory control for maintaining a homeostatic balance in cellular processes. Mitochondrial biogenesis, autophagy, fission and fusion as well as mtDNA replication are dependent on the integrated interaction between the organelle and the nucleus. It has been found that variations in the mtDNA could potentially lead to differential expression of nuclear transcription factors that are essential for the mtDNA replication. The mito-nuclear crosstalk directs the retrograde response that is usually triggered when the membrane potential of the mitochondria drops below an unhealthy threshold, seen typically in case of mitochondrial dysfunction due to aging [96]. NAD+/NADH mediated signaling, energy homeostasis mediated by AMP-activated protein kinase signaling, redox signaling, and calcium signaling are examples of some distinct pathways that decide the very fate of the cell [97]. Though there is overwhelming data to show that nuclear genes influence the mitogenome, the explicit science behind how it is achieved is still in the infancy and yet to be elucidated precisely.
MITOCHONDRIAL PROCESSES IN THE CELL AND THEIR REGULATION
Mitochondrial bioenergetics
Bioenergetic process in mitochondria comprises of the ETC. The ETC encompasses complex I–V and two free electron carrier CoQ (ubiquinone) and cytochrome c involved in generation of ATP in the inner mitochondrial membrane through OXPHOS [98]. Respiratory chain complex proteins are encoded by both nDNA and mtDNA. Affliction of the ETC leads to insufficient formation of ATP, ergo mitochondrial dysfunction, excessive ROS production, reactive nitrogen species from nitric oxide synthase, which are all concerned with neurodegenerative disorders [99]. MPTP (1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine), PQ, and rotenone are neurotoxins that inhibit the complex 1 of ETC upon exposure and facilitate its collapse. These pesticides promote the deposition of α-Syn clusters and Lewy body inclusions in brain, owing to the pathogenesis of PD. Though there exists a close inter-relation between ETC complex break down and oxidative stress overload, the temporal aspect of this relation has not yet been precisely ventured. Whether ETC impairment causes ROS production or ROS production leads to ETC malfunctioning is still a debatable topic. Better understanding of such temporal aspects could potentially lead to the treatment of at-risk PD patients at the prodormal stage itself, thereby halting or decelerating the disease progression. Coenzyme Q10 (CoQ10) and creatine have been widely studied for their role in bioenergetics and have also proceeded to clinical trials in de novo PD patients holding a promise for the future [100]. Hence recovering the physiological ATP production via modulating the mitochondrial bioenergetics could potentially work as a good disease modifying approach.
Mitochondrial biogenesis
The biogenesis of mitochondria is a critically regulated process wherein the peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α) is stimulated by the AMPK (2-Adenosine monophosphate activated protein kinase) signaling. TFAM in the nucleus relocates to the mtDNA site and stimulates mitochondrial biogenesis by mtDNA transcription and translation. Damaged mitochondria cause activation of this pathway helping in production of mtDNA and protein synthesis. PGC-1α pathway dysregulation leads to impaired mitochondrial biogenesis causing neuronal loss in PD through parkin interacting substrate (PARIS). Decline in the mRNA levels of nuclear-encoded RC genes that are reactive to PGC-1α has been acknowledged in individual DA neurons of PD patients, and in neurons in regions with subclinical PD-related Lewy body neuropathology. In addition to mitochondrial biogenesis, PGC-1α is also capable of triggering the appearance of antioxidant enzymes in retort to oxidative stress and defending DA neurons against neuroinflammation. In PC-12 cells, efforts to study the effect of PINK1 on the mitochondrial biogenesis were made. To evaluate the consequence of the PINK1 gene on mitochondrial biogenesis, the genes directly involved in mitochondrial biogenesis (PGC-1α and mtTFA/TFAM) were restrained in answer to downregulation or upregulation of PINK1 expression. Downregulation of PINK1 led to a reduction of Parkin phosphorylated form (p-Parkin), ensuring an increase of PGC-1α and TFAM. In count, mtDNA copy number was significantly increased when PINK1 was downregulated, and mtDNA copy number was suddenly decreased when PINK1 was upregulated, describing an inverse relation between PINK1 and mitochondrial biogenesis process [101]. While biogenesis is essential for the formation of new mitochondria, the timely clearance of malfunctioning mitochondria from the cell is also equally important. Mitophagy is a specific autophagic process that gets rid of damaged mitochondria. In order to achieve mitochondrial homeostasis, it is necessary for the process of biogenesis and mitophagy to have a balanced coordination. These two critical processes are co-regulated by SKN-1. Prolonged pile up of malfunctioning mitochondria leads to the induction of a retrograde response that is mediated by SKN-1 which further results in the activation of mitochondrial biogenesis and mitophagy genes. Additional critical assessment of these molecular mechanisms would pave the way for future therapeutics that would regulate mitochondrial homeostasis [102].
The making of mtDNA: Replication
The DNA of the mitochondria does not exist as a single naked entity, rather they form a complex with multiple proteins (that regulate the genome’s maintenance and transcription) to form a structure known as nucleoid. Any flaw in the replication enzymes (DNA polymerase gamma, TWINKLE helicase, primase polymerase, and RNA polymerase) or dNTP supply or any malfunctioning of the mitochondrial dynamics would lead to a disrupt DNA replication process, birthing a group of multisystem heterogenous disorders due to massive energy failure. Such disorders are known as mtDNA maintenance defects. Few diseases that supposedly occur due to POLG deletion or depletion include Childhood myocerebrohepatopathy spectrum, Alpers–Huttenlocher syndrome, myoclonic epilepsy-myopathy-sensory ataxia, ataxia neuropathy spectrum, progressive external ophthalmoplegia, and mitochondrial neuro-gastrointestinal encephalopathy [103–107]. The dysregulation of mtDNA homeostasis is a major contributor to the neuronal loss in PD. This makes the DA neurons of the SNpc collectively more prone to age-dependent mutagenesis of the mitochondria. DA neurons of PD individuals showed increased levels of somatic mtDNA deletions in comparison to the healthy population. mtDNA copy number increases in healthy individuals, maintaining the pool of wild-type DNA while in PD patients the copy number does not increase leading to the diminution in the wild-type number [108].
Transcription of the mitogenome
One of the earliest in vivo studies of the mtDNA transcription was observed in HeLa cells by Aloni et al. in 1971. It was shown that the human mitogenome was transcribed in a polycistronic manner symmetrical to the bacterial transcription. The entire complex process of transcription can be narrowed and broken down under a few simple steps such as promoter recognition, initiation, elongation and termination. In order to initiate transcription TFAM binds to the region upstream of the HSP or LSP resulting in conformational changes which allows the effective binding of the RNA polymerase enzyme also known by the gene that encodes it, POLRMT [84]. MTERF1 binds with certain selective tRNALeu gene sequences, as a result of which the DNA strands unwind, and the nucleotide base capsize or flip the opposite way leading to the termination of transcription. In the literature, this is classically referred to as the DNA base flipping [89, 109].
The mRNA undergoes polyadenylation by mitochondrial poly (A) polymerase to render the finally processed mRNA. Any changes in the length of the poly (A) end leads to the impairment of post transcriptional mitochondrial gene expression and dysregulation of the ETC. The mitochondrial RNA polymerase that plays a pivotal role in the initiation of transcription is directly regulated by the interactions it has with the nucleoid proteins and these nucleoid proteins are encoded by the nuclear genes and not the mitochondrial genes. Hence it is appropriate to conclude that the nucleus of the cell indirectly regulates the process of transcription in the mitochondria. Leucine rich pentatricopeptide repeat containing protein (LRPPRC), mitochondrial ribosomal protein L12 (MRPL12), TEFM, and MTERF1 are some of the nucleoid proteins that are involved in homeostatic regulation of mitochondrial transcription. LRPPRC forms a complex with SLIRP which later acts as an RNA chaperone, binds to and stabilizes mitochondrial mRNAs, and exposes the appropriate sites for polyadenylation. Apart from regulating mitochondrial poly (A) polymerase, it also plays a role in regulating complex IV. Knock-down of LRPPRC in mammalian cells was found to cause an imbalance in the complex IV subunits encoded by the mitochondrial and nuclear genome. This imbalance later triggers the mitochondrial unfolded protein response in order to maintain the mitochondrial proteostasis. Dedicated minigene assays for PD-linked genetic variants show that LRPPRC intronic variants can affect the normal splicing of pre mRNA and this may influence disease risk in PD and related disorders [110]. For example, in cell lines and mice deficient of LRPPRC, mitochondrial transcription was seen to be decreased and mitochondrial dysfunction was also observed [111]. LRPPRC also has a substantial amount of contribution in the pathogenesis of PD, neurofibromatosis 1, viral infections, and venous thromboembolism [110]. Increased levels of LRPPRC have shown to maintain Bcl-2 levels, inhibit mitophagy, and prevent autophagy mediated mitochondrial degradation via interacting with Parkin and stabilizing it. Unmasking the exact interacting domains of LRPPRC and Parkin may lead to the characterization and development of novel molecules that can precisely regulate such neuroprotective interactions and would be beneficial in neurological disorders like PD [112]. MRPL12 is the first mitochondrial ribosomal protein to be characterized in mammals and its knockout models revealed decreased levels of mitochondrial transcripts due to the instability of mitochondrial RNA polymerase enzyme [113]. Mutated MRPL12 protein was found to play a potential role in retardation, neurological deterioration and mitochondrial translation deficiency [114]. MRPL12 also positively regulates the mitochondrial OXPHOS and mtDNA copy number indirectly through SQSTM1/p62 (autophagy adaptor protein) regulation [115].
Mitochondrial RNA modifications
A plethora of RNA modifications have been reported in all the three domains of cellular life which includes the Bacteria, Archaea, and Eukarya, and more than 150 such modifications have been detected using MODOMICS [116]. The different chemical modifications arising in the coding as well as non-coding RNAs at a covalent level, due to the contribution of external or internal environmental factors have been studied overtime and are united under a broader term called ‘epitranscriptome’ [117]. Such epitranscriptomic changes are associated with neurological disorders, cancer, and metabolic disorders as well [117, 118]. For example, G ms2i6A (2-methylthio-N6-isopentenyl adenosine) and A I (inosine) modifications were associated with faulty CDK5RAP1 and ADAR2 enzymes respectively, resulting in AD [119]. The RNA metabolism and modifications associated with the pathogenesis of motor neuron diseases were reviewed in depth by Lemmens et al. [120].
An important type of RNA is the microRNA (miRNA). miRNAs are non-coding 22 nucleotide sequence long, single-stranded structures that are involved in post transcriptional modification of gene expression via gene silencing that targets the RNA interference silencing complex (RISC). miRNA formed right after transcription is called the primary miRNA and undergoes cleavage by both DROSHA nuclease in the nucleus and later DICER nuclease in the cytoplasm to form pre-miRNA and mature miRNA respectively. miRNAs are important bioregulators of the mitochondria [121]. Different types of miRNAs govern unique functions associated with regular activity of the organelle. Few of the known quintessential epitranscriptomic modifications include the m6A methylation of the pri-miRNAs, m7G methylation of G-quadruplex structures in pri-miRNA and the methylation of the 5’ monophosphate of the pre-miRNAs [121–124]. Additional light on specific epitranscriptomic modifications is yet to be shed.
The mito-ribosome is made up of two subunits, a larger 39S subunit and a smaller 28S subunit. These subunits encode the 12 s and 16 s rRNAs, respectively. All the modifications are catalyzed by nuclear encoded enzymes just as in the case of tRNAs. Two highly conserved modifications include the adenosine dimethylation in m62A of A936 and A937 [117]. In vivo analysis depicted these modifications in 12 s rRNA are catalyzed by TFB1M which is determined as mammalian SAM-dependent methyltransferase. Genetic analyses has unveiled that loss of TFB1M could potentially cause mitochondrial dysfunction induced type-2 diabetes mellitus and mitochondria-associated deafness [92, 125]. Another significant modification that is responsible for a harmonized mitochondrial translation is the methylation of m1A in A947 of 16 s rRNA by tRNA methyltransferase 61B (TRMT61B) [126]. The therapeutic applicability of RNAs is described in later parts of this review.
Mitochondrial dynamics
Various metabolic and environmental cues regulate the timely fusion, fragmentation, and destruction of the mitochondria. These processes are more collectively referred to as the mitochondrial dynamics. It serves as a quality control system in maintaining the mitochondrial homeostasis, thereby, monitoring the cell survival [127]. Since the mitochondria are double-membrane structures, the fusion process will typically comprise of two main steps: fusion of the outer mitochondrial membrane (OMM) followed by the fusion of inner mitochondrial membrane. The mitochondrial fusion is accomplished with the help of three enzymes possessing GTPase activity (belonging to the dynamin superfamily of proteins) [128, 129]. The ones responsible for OMM fusion are mitofusins: Mfn1 and Mfn2 which dwell on the OMM itself. Trans interactions occurs between mitofusins of the two fusing mitochondria to achieve tethering. Optic Atrophy 1 (OPA1) protein governs the inner membrane fusion [129]. Apart from this, OPA1 also governs the activity of OXPHOS complexes and maintains the structural integrity of the mitochondrial cristae [130]. The terminal outcome of the fusion process is the interflow of matrix elements, after two individual mitochondria come together and merge into a larger one. In certain special cases of fusion, for example, kiss-and-run fusion, only the matrix information is exchanged and there is no permanent morphological switch [127]. Fusion also ensures the equitable inheritance of healthy mtDNA by compromising the damaged DNA with wild type ones, keeping the heteroplasmy at bay [130].
In humans, mutations in the Mfn2 protein are associated with Charcot–Marie–Tooth disease type 2A (CMT2A), which is a sensory cum motor neuropathic disorder. About 100 different mutations in Mfn2 have been reported in the cases CMT2A [131]. Although not super specific, mutations of Mfn2 have recently been linked with several other neurodegenerative conditions like AD, PD, and Huntington’s chorea as well [132]. The expression levels of Mfn1, Mfn2, OPA1, and Dynamin-related protein 1 (Drp1) was reported to be less in cases of AD [133]. The reason behind decreased Mfn2 levels was correlated with the upregulation of miR195 by Zhang et al. in 2016 [134]. In the very same study, it was also observed that, the use of miR195 inhibitor or antagomir lead to increased levels of Mfn2, making these molecules a potential treatment drug for AD in the long run. The protein Parkin is responsible for the ubiquitination of Mfn2, and it is a well-known fact that heterozygous Parkin mutations are closely linked with sporadic and familial forms of PD. Parkin also conciliates the expression of a transcriptional coactivator: PGC-1α, which is the supreme governor of mitochondrial biogenesis. Under the conditions of stress, PGC-1α controls the expression levels of Mfn2 [135]. Affixment of the Parkin-ubiquitination machinery to the mitochondrial membrane by Mfn2 deflects the mitochondrial fusion to mitophagy [136]. All of the above data bear witness to the idea that Mfn2 regulation is somehow knit with PD. Overexpression of the HTT mutant with unstable CAG repeats on exon 1 of the HTT gene (chromosome 4p16.3) inflates the mitochondrial fission process, but the upregulation of the Mfn2 protein subdued such undesired phenotypes [137]. Under rare circumstances, it has been reported that homozygous mutations in Mfn2 causes multiple symmetric lipomatosis associated with neuropathy [138]. PQ induced mitochondrial fragmentation of human DA neuroblastoma SH-SY5Y cells was reversed by the overexpression of Mfn2. Oxidative damage and stress signaling induced by PQ as well as selective degeneration of DA neurons in the SNpc and striatal axonal terminals was also inhibited in transgenic mice overexpressing hMfn2 (human mitofusin2) proving mitofusin 2 as a rich therapeutic target in PD and other related disorders. Probucol treatment of cybrid cells having mitochondria of AD/mild cognitive impairment patients significantly increased Mfn2 and decreased Drp1 expression [139]. In another PD study, it was seen that probucol offered protection against 6-OHDA-induced hyperlocomotion and striatal lipid peroxidation, catalase upregulation, and decrease of TH levels [140]. Icariin, isolated from the Chinese herb epimendium reduced Drp1 and increased Mfn2 levels in AD neurons by modulation of mitochondrial dynamics. Besides this, icariin was also found to offer neuroprotection for DA neurons by inhibiting microglia-mediated neuroinflammation through the suppression of nuclear factor-κB (NF-κB) pathway activation in an in vitro study [141].
Heterozygous mutations of OPA1 are considered as the foundation for the genesis of dominant optic atrophy, which is the most frequently inherited optic neuropathy. Other manifestations of OPA1 mutations include hearing loss, myopathy, and peripheral neuropathy. Stress-induced OPA1 activation causes mitochondrial fragmentation, resulting in mitophagy, through the upregulation of OMA1 activity. Korwitz et al. observed that the loss of OMA1 activity detains the progression of neurodegeneration by downregulating this stress-induced OPA1 signaling [142]. A very recent yeast-based screening of pre-approved FDA drugs was performed and later validated using mouse embryonic fibroblasts and patient’s fibroblasts, carrying the OPA1 mutations. These agents were found to ameliorate mitochondrial morphology and the ATP content and hence improved the cell viability. Salicylanilide, one of the compounds used in the study delivered optimistic results in neurodegenerative disorders like AD [143].
Mitochondrial fission is the process by which a single mitochondrion subsequently divides into two new daughter mitochondria. First observed in Caenorhabditis elegans, Drp1 is the key executioner of this event and it happens to be evolutionary conserved. Dynamin2 (Dnm2) along with Drp1 performs the severing and constriction of the organelle respectively [144]. Drp1 is a cytosolic protein, and it is recruited to the mitochondria during the event of fission. It spirals the organelle accurately along the constriction sites and promotes the hydrolysis of GTP to effectuate mitochondrial scission [144, 145]. Impaired mitochondrial fission due to the loss of Drp1 causes neurodegeneration in in vivo model of drosophila with hereditary spastic paraplegia [146]. In a very recent study conducted by Portz et al. [147], it was observed that transgenic mouse model with familial PD mutant A53T α-Syn had decreased Drp1 activity in the mitochondria, hinting the involvement of Drp1 dysregulation in the PD pathogenesis. Pathological increase of Drp1 was observed in case of MPP+/MPTP-induced PD models and Carboxyl terminus of Hsp70-interacting protein (CHIP) was found to inhibit this disastrous increase. CHIP is a co-chaperone that regulates chaperones such as HSP70, HSP90, and other heat shock proteins during protein folding to render a functionally and conformationally active protein. It binds to HSP70 and HSP90 through its TPR (Tetratrico Peptide Repeat) domain and functions as an E3 ubiquitin ligase and degrades aberrant proteins in the body, for instance, misfolded α-Syn found in case of PD. Besides being an important co-chaperone it is also involved in the maintenance of mitochondrial integrity [148].
Fate of a malfunctioning mitochondrion: Mitophagy
Contrary to biogenesis, which involves the birth of a new mitochondrion, mitophagy is the process of destruction of the existing damaged mitochondria. It can either be driven by PINK/PARKIN dependent pathway or via mitochondrial mitophagy receptors (independent pathways) such as NIX, BNIP3, FUNDC1, Bcl2L13, FKBP8, NLRX1, and the like. The latter pathways have not been elucidated properly while the former one is the best characterized. Therefore, recent studies revolve around molecules that target the PINK/PARKIN dependent mitophagy [149].
When the ratio of pathogenic mtDNA to wild type mtDNA becomes unruly, disruptions in the mitochondrial dynamics occur. These malfunctioning mitochondria are subjected to a destructive process called mitophagy. It essentially refers to the autophagosomal clearance of the damaged mitochondria. An increase in the rate of mitophagy due to faulty mitodynamics will compromise the bioenergetics of the cell, leading to the manifestation of mitochondrial diseases, including PD. The defective bioenergetics of the organelle leads to the depolarization of the mitochondrial membrane, triggering the recruitment of PINK1. This protein starts to accumulate and stabilize on the OMM. It phosphorylates the ser 65 residues of the already existing ubiquitin molecules. These changing chemical dynamics, eventually, attract a cytosolic auto-inhibited protein called parkin (an E3 ubiquitin ligase). RING1 domain of parkin almost immediately binds to the phosphorylated ubiquitin ser65 residue (pSer65Ub). The entire complex gets activated now, and parkin starts to ubiquitinate the proteins expressed on the OMM. This triggers their phosphorylation at ser 65 residues by PINK1 and this in turn results in further recruitment of parkin. The cycle continues until a maximally active ubiquitin chain is achieved. Then the tagged proteins head for proteosomal destruction and clearance. The PINK1/parkin dependent mitophagy can be triggered by a combination of oligomycin (ATP synthase inhibitor) and antimycin A (complex 3 inhibitor) or by cyanide m-chlorophenyl hydrazine. An increase in the intracellular ROS levels also promotes this process [5].
Mutations in the PINK1 and parkin genes have been endlessly reported in monogenic forms of PD, indicating the malfunctioning of mitophagy in its pathogenesis [150]. Apart from this, Parkin or Pink1 deficient mice were found to develop inflammation upon subjection to vigorous exercise due to the activation of stimulator of interferon genes (STING) pathway, that upregulates cytokine release in response to intracellular DNA [151]. Many long-associated PD-risk genes such as SNCA, LRRK2, CHCHD2 (PARK22), GCH1, and VPS35 (PARK17) display dysregulated PINK1/parkin mitophagy in in vitro and in vivo PD studies as well as in PD postmortem patients [152]. All these studies indicate that there exists a strong relation between impaired mitophagy and PD. Several mitophagy-inducing novel compounds have been explored for enhancing the mitochondrial clearance in PD. Few of the notable examples include nilotinib, FT385, p62-mediated mitophagy inducer, sulforaphane, deferiprone, urothilin A, GYY4137, gemcitabine, kinetin, USP30i, and T271. The compounds listed are positive regulators of mitophagy and act by modulating various upstream proteins that regulate PINK1 or Parkin [153].
NOVEL STRATEGIES FOR PD MANAGEMENT
Mitochondrial mediators
Mitochondria targeted drugs are designed in order to regulate the imbalanced mitochondrial homeostasis where ETC targeted compounds still remain a major class. The toxic metabolite, 1-methyl-4-phenylpyridinium (MPP+), is formed by monoamine oxidase (type B)-catalyzed oxidation of MPTP. The MPP+ transported into the brain by the DA transporter accumulates in the mitochondria, resulting in adenosine triphosphate (ATP) depletion through inhibition of complex I activity, altered mitochondrial membrane potential, increased ROS, and apoptotic cell death. Recent research studies explored several potential antioxidant molecules like Mito-Q, Mito-honokiol, Mito-metformin (Mito-Met), and Mito-apocynin (Mito-Apo) for their potential application in PD management. Mitochondrial-targeting was induced by adding triphenylphosphonium (TPP+) group attached via varying alkyl chain lengths coupled to a naturally occurring antioxidant molecule or an FDA-approved drug. Further studies also developed drugs targeting mitophagy, as PINK1/Parkin mutations are majorly linked to PD. PGC-1α is also a potential target for the PD management [154].
Mito Q is produced by modification of coenzyme Q10, a factor in respiratory chain. In neurodegenerative disorders, there is a decrease in enzyme COQ10 in the DA neuron. Neuroprotective action on the DA neuron by Mito Q10 supplementation was observed in MPTP induced PD model. Mito Q helps in reduction of tyrosine hydroxylase produced by the neurotoxin, DA depletion and offers protection against the locomotor dysfunction. Mito Q activates AMPK signaling via depolarization of mitochondria and initiation of autophagy, thereby promoting cell survival. The effect of Mito Q on disease progression was investigated in a large phase III randomized, placebo-controlled, double blind clinical trial [155]. Similarly, Mito-Apo had neuroprotective action upon oral administration, in a genetically engineered preclinical PD model Mitopark expressing the phenotypic features (mitochondrial dysfunction, microglial activation, DA degeneration and depletion of DA, progressive neuronal deficits, and protein occlusion) of human PD. Knocking out TFAM in DA neurons of animal model created disability of mitochondrial respiratory chain. Mito-Apo helped in restoring the motor deficit, ameliorated the nigrostriatal tyrosine hydroxylase loss, reinstated DA levels in the striatum and also restored the non-motor functions including the loss of smell. The neuroprotective mechanism of Mito-Apo was extrapolated to blockage of the NOX (NADPH oxidase) enzyme and iNOS. The mutation in LRRK2 genes in mouse model significantly prolonged treatment with Mito-Apo used to stave off the early non-motor symptoms of PD [156].
Ubiquinol, acting promptly up on the lipid peroxidation induced oxidative stress, entered phase III trial and was safe and tolerable in PD patients but it failed to show any significant clinical sign improvement in patients [157]. Ursonorm/ursodeoxycholic acid is a bile salt approved for hepatic cirrhosis treatment but when studied for its restorative role on mitochondrial function in AD and PD fibroblasts, it showed a positive effect and also reduced the Drp1 levels [158]. It is hence under phase II clinical screening (NCT03840005). Recent studies show that metformin helps undo TRAPS (TNF receptor-associated periodic syndrome)-mutation-associated change in the mitochondrial function in PD mice models, partially rebuild the DA level, ameliorate the neurochemical loss and behavioral alteration in vivo.
Mito-met GPI 1485 is a neurotrophic immunophilin-ligand that has shown the property to repair neuronal degeneration and avert cell death in animal models. In a 6-month phase II clinical trial of GPI 1485, there was a raze towards favorable effect on imaging but not clinical measures in patients with PD [160]. The National Institute of Neurological Disorders and Stroke Neuroprotection Exploratory Trials (NINDS NET) for PD, which enrolled and randomly assigned 213 participants, 71 to the CoQ10, 71 to the GPI 1485, and 71 to the placebo arm, was not emphatic regarding the disease-modifying effects of the tested compounds and further studies were required to confirm efficacy of the compound [161]. Decreased glutathione reduced form (GSH) was found in early progression stage of PD, and glutathione expansion was proposed as a therapeutic strategy in PD. In a phase IIb study, intranasal glutathione was not primary to placebo. Glutathione/N-Acetylcysteine (NAC) or EPI-589 used as safety and biomarker study in PD (NCT02462603). Inosine increases levels of urate, a dormant antioxidant, and was proved safe and tolerable in patients with early PD (SURE-PD trial). SURE-PD III is in progress and plans to accept 270 patients with early untreated PD. NAC has role to decrease the oxidative stress and regain the level of glutathione. Mitochondrial Rho GTPase has also been studied for mitochondrial structural integrity and mitophagy homeostasis restoration. Creatinine, a natural antioxidant, had neuroprotective effect against MPTP-induced DA depletion in mice model [162].
Calcium modulators
As discussed previously, Calcium (Ca2 +) plays a key role in the normal functioning of neurons and is also involved in many cellular processes, for example, oxidative stress, mitochondrial impairment, proteasomal dysfunction, excitotoxicity, neuroinflammation, and apoptosis that can eventually lead to cell death in PD. Neurodegenerative conditions like PD generally affect mitochondria and the bioenergetic status of the cell, where mitochondrial Ca2 +dysregulation plays a key role in pathogenesis. However, the underlying molecular mechanism(s) are still under investigation due to which Ca2 + homeostasis and mitochondrial bioenergetics have received more attention in recent years. Several studies have reported calcium dysregulation as the underlying mechanism for the elevated cytosolic calcium level. For example, under normal physiological conditions, voltage-dependent Ca2 + channels (L-type) are opened by neuronal plasma membrane depolarization during an action potential and closed during repolarization. In midbrain neurons, stimulation of the cells with DA lead to activation of these channels. In PD, L-type Ca2 + channels are autonomously active leading to an increased cytosolic Ca2 + influx. Furthermore, α-Syn itself has also been reported to increase cytosolic Ca2 + levels by forming plasma membrane pores allowing extracellular Ca2 + to pass. The elevated intracellular Ca2 + levels require higher ATPase activity to transfer Ca2 + across the plasma membrane, resulting in an extra burden to the already bioenergetically impaired neurons in PD. Increased intracellular Ca2 + levels also affect Ca2 + handling in intracellular organelles such as the endoplasmic reticulum and mitochondria which might potentiate pathological effects. Zonisamide, anticonvulsant agent used for epilepsy, is a T-type calcium channel blocker, blocks recurring firing by voltage gated sodium channel blockage or by allosterically binding to GABA receptor. It aids in increasing striatal DA levels in animal model of PD and hence, entered clinical trial as an adjunctive therapy for advanced stage PD management (NCT04182399). Suvecaltamide/ CX-8998, T-type Ca2 + channel blocker clinical trial phase II has been withdrawn which aimed at assessing its effect on tremor associated with PD (NCT03436953). Isradipine, L-type Ca2 + channel blocker, is currently under phase III screening for PD management (NCT02168842) but it failed to slow the PD progression clinically [42].
PPAR agonist
PPAR is a kind of ligand-activated transcription factor belonging to the class of nuclear receptor super family and are actively engaged in the regulation of mitochondrial functioning, inflammatory processes, redox balance, wound healing, and metabolism of blood sugar and lipids. The three significant isoforms were identified and cloned so far includes, PPAR-α, PPAR-β/δ, and PPAR-γ, and are widely distributed in various body regions [163]. Activation of the PPARs takes place with the help of lipophilic molecules, which further form heterodimers with retinoid X receptors in order to carry out cytoplasmic stimulation. This heterodimer interacts with DNA sequence elements known as peroxisome proliferator response elements to modulate the transcription of genes that are actively involved in several biological processes, such as, insulin sensitization, inflammatory processes, and neuronal protection. PPAR agonists protects neuron via suppression of autophagy-lysosome system dysfunction, UPS dysfunction, mitochondrial dysfunction, oxidative stress, neuroinflammation, and programmed cell death. PPAR agonists, suppress oxidative stress by elevating the levels of SOD and CAT. Additionally, these agonists inhibit the activation of microglia, triggered by toxin exposure. Moreover, PPAR agonists, non-steroidal anti-inflammatory drugs, and montelukast declines the expression of iNOS, NF-κB inflammatory mediators, ROS, microglia activation, and neuroinflammation. Besides, a PPAR-γ-coactivator, namely PGC-1α decreases oxidative stress by elevating the levels of GSH, SOD, and CAT, and provokes the NRF1 and NRF2 expression associated with mitochondrial biogenesis, thereby furnish neuronal protection. Apart from this, resveratrol, an antioxidant also exhibits neuroprotective action by activating PGC-1α [164]. In a recent study, a novel PPAR α/γ dual agonist MHY908, has shown to suppress inflammatory response and diminish insulin resistance in aged rats and db/db mice. MHY908 attenuates MPTP-induced glial activations in the nigrostriatal pathway, and MHY908 effectively blocked MPP+ induced cell death and production of ROS in SH-SY5Y neuroblastoma cells. The Study also unraveled inhibition of MPP+-induced Astro-glial activation by suppressing NF-κB signaling in primary astrocytes [163].
Glucagon-like peptide 1 receptor agonist
Glucagon-like peptide-1 (GLP-1) and glucose dependent insulinotropic peptide are synthesized and released from gut enteroendocrine cells within small intestine that controls meal-related glycemic excursions by promoting release of insulin and inhibition of glucagon secretion [165]. These two hormones have been reported to be responsible for the majority of insulin production, with GLP-1 showing more intense effects than GIP in type 2 diabetes mellitus. According to epidemiological studies, type 2 diabetes mellitus and related insulin resistance conditions are considered as risk factors for PD and other age-related neurological disorders, suggesting the existence of a connection between dysregulated communication of the gut-brain axis and progression of degeneration of neurons. Furthermore, metabolic and neurodegenerative disorders share common pathophysiological attributes that includes inflammation, oxidative stress, abnormal protein aggregation and disrupted cognition apart from insulin resistance.
Activation of GLP-1 receptor (GLP-1R) on neuron and glial cells, leads to initiation of downstream pathways that alleviate the effects of PD pathology, the major upstream secondary messenger is cyclic adenosine monophosphate (cAMP). Upregulation of cAMP triggers downstream effector proteins that ameliorate inflammation, oxidative stress, and apoptosis, which provides neuroprotection and proliferative capabilities for neurite outgrowth. Additionally, restoration of insulin signaling through the upregulation of active insulin receptor substrate-1 and downstream proteins also provide neuroprotection. Increased production of brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor, contributes to combat PD [166]. According to studies, GLP1 and exenatide has been shown to possess neuroprotective property in SH-SY5Y cell against 6-OHDA induced cell death. 6-OHDA is a neurotoxin that kills DA neurons. In ventral mesencephalic neuronal cultures, treatment with exenatide did not only show to protect DA neurons but also induced 60% elevation in the level of TH positive cells over control values. It also lowers proapoptotic proteins [167]. Semaglutide has shown neuroprotective activity by suppressing the α-Syn aggregates in mouse PD model [168] and further advanced to phase II clinical trial (NCT03659682). Liraglutide improved non-motor symptoms in PD patients in phase II clinical trial (NCT02953665).
GBA1
GBA encodes glucocerebrosidase, a membrane associated protein comprises of 497 amino acids with 39 amino acid leader sequence and 5 glycosylation sites. It is synthesized in endoplasmic reticulum and glycosylated but the activation takes place in acidic environment of lysosome upon binding with co-factor saposin C [169]. It performs the hydrolysis of the lipid glucosylceramide into glucose and ceramide. Reports suggest the existence of an inverse relationship between glucocerebrosidase and α-Syn set by lysosome, which is responsible for degradation of proteins, lipids and organelles. Lysosomal degradation of α-Syn is carried out by both chaperone mediated autophagy and macroautophagy.
Studies have shown that an elevation in α-Syn leads to inhibition of translocation of glucocerebrosidase from ER to lysosome. Reduction in glucocerebrosidase results in gradual increase of glucocerebroside inside the lysosome. Accumulated glucocerebroside can trigger oligomerization and aggregation of α-Syn in lysosome making it dysfunctional and impairing autophagy mediated α-Syn degradation resulting in aggregation of α-Syn in cytoplasm [170]. One more mechanism by which GBA mutations lead to PD is the impairment of ERAD and cellular demise. The aggregation of α-Syn will induce ER tension, prevent the ER-related substrate degradation, and also obstruct the ER-to-Golgi traffic. GBA mutations can cause alteration in structure of the GCase protein, leading to the loss or reduced enzymatic activity. It has been postulated that, these effects may emerge through any of the following pathways: inability of GCase to escape the ER, failure of GCase to bind to its trafficking transporter LIMP2, failure of GCase to leave the Golgi, breakdown of unstable and misfolded GCase by the proteasome system, inactivation of GCase due to the mutations at the active site, and degradation of GCase by the proteasome system [171].
Activation of wild-type GCase indicates an emerging approach for the PD treatment. A novel small-molecule GCase modulator called S-181, increased wild type GCase activity in iPSC-derived DA neurons from sporadic PD patients as well as patients carrying 84GG mutation in GBA1, or other mutations which results in reduction of GCase activity. Upon treatment with S-181, lysosomal function got partially restored and a decreased accumulation of oxidized DA, glucosylceramide, and α-Syn was also observed [172]. According to data obtained from in vivo and in vitro studies performed, it has been reported that ambroxol increases GCase enzymatic activity and declines α-Syn levels. This indicates a vital role of ambroxol in modifying a relevant pathogenetic pathway in PD [173]. LY3884961 is being clinically screened for its safety and efficacy in patients with at least 1 GBA1 gene mutation (NCT04127578).
Glutamate receptors
Glutamatergic neurotransmission accounts several sensory, motor, autonomic, and cognitive activities. So, to ensure proper neuronal transmission it is essential to maintain extracellular glutamate levels in physiological range [174]. Glutamate receptors are primarily located on pre- and post-synaptic neuron and distributed in all areas of the central nervous system. Based on their pharmacological activity they have been categorized into two classes: ionotropic receptors (iGluRs) and metabotropic receptors (mGluRs). Ionotropic glutamate receptors carry out fast excitatory transmission in the mammalian CNS and are further classified into N-methyl-D- aspartate (NMDA) receptors, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, and kainate receptors. While metabotropic glutamate receptor mediates slow glutamate responses and are further classified into 3 groups consisting of mGluR1-8. Stimulation of NMDA receptors by agonists, such as glutamate and glycine, could induce the depolarization of neurons and allow the influx of Ca2 +, by opening the ion channel pore. Excessive accumulation of Ca2 + in cytoplasmic matrix triggers a series of Ca2 +-dependent enzymes and cause pathophysiological changes in intracellular biochemistry, which eventually leads to neuronal death. Therefore, NMDA receptor has been considered to potential target for parkinsonism [175]. Memantine, a partial antagonist of NMDA receptor, approved for treatment of moderate to severe AD has been reported to possess neuroprotective activity. It acts by blocking the NMDA glutamate receptor, normalizing glutamatergic system and improve cognitive activity [176].
AMPA receptors are weakly permeable to Ca2 + ions as compared to NMDA receptor, and mediate influx of Na+ in neuron when stimulated by glutamate. Activation of AMPA receptors leads to increased influx of extracellular Zn2 + in DA neuron causing motor dysfunction. An elevation in the expressions of AMPA receptors has been observed in Parkinsonian monkeys (induced using MPTP) and Dopa-mimetic treatment in subregions of striatum. Thus, AMPA receptor antagonists are likely to show antiparkinsonian activity [175]. In monoamine depleted rats, 2,3- dihydroxy-6-nitro-sulfamoyl-benzo(f)quinoxaline, a selective AMPA receptor antagonist has shown to suppress muscular rigidity but had no effect on akinesia while there was a clinically apparent improvement in akinesia, tremor, posture, and other motor activity in MPTP induced parkinsonism in aged rhesus monkeys [177].
16 preclinical studies of mGluR5 antagonists have been reported in animal models of L-Dopa induced dyskinesias and also 7 clinical studies in patients with PD and L-Dopa induced dyskinesia were included. Anti-dyskinetic activity by mGluR5 blockade (MPEP, MTEP, fenobam, or MRZ-8676) was observed in dyskinetic 6-hydroxydopamine-lesioned rats. Studies also reported anti-dyskinetic effects of MPEP, MTEP, mavoglurant (AFQ056), and fenobam in MPTP-lesioned non-human primates. In three randomized double blind clinical trials, anti-dyskinetic effects of mavoglurant were observed with no effects on antiparkinsonian therapy [178]. In a recent study, it has been shown that mGluR5 plays a critical role in counteracting α-Syn induced neuroinflammation. The study has provided a novel mechanism which involves dissociation of mGluR5-α-Syn complex in microglia by enhanced mGluR5 expression, and it may be a potential strategy for neuroprotection [179]. Studies also reported role of mGluR4 in motor dysfunction and L-DOPA induced dyskinesia in primate model of PD. LSP1-2111 an mGluR4 agonist has been shown to produce an anti-parkinsonian effect without provoking dyskinesia in L-DOPA primed MPTP-treated marmosets, however, it was unable to reduce the severity of established LID [180].
LRRK2
LRRK2 is a large protein composed of 2527- amino acids with multiple functional domains: (1) N- to –C terminus armadillo ankyrin; (2) a Ras-of-complex (ROC) GTPase; (3) a COR dimerization domain; (4) Leucin-rich repeats; (5) a serine/threonine kinase domain [181]. LRRK2 plays numerous role which includes cytoskeletal remodeling to protein expression, synaptic transmission, and membrane trafficking, via phosphorylation of diverse substrates such as β-tubulin, tau, microtubule affinity-regulating kinase 1 (MARK1), etc. [182]. More than 30 mutations of LRRK2 have been described, most common ones belong to ROC-COR GTPase (R1441G/CH, Y1669C) and kinase domains (G2019S, l2020T), representing the crucial role of both enzymatic domains in pathogenicity of PD driven by LRRK2 [181].
The G2019S mutation has been indicated as the prominent genetic determinant of PD. And it is believed that G2019S mutation is responsible for the upregulation of LRRK2 kinase activity, which eventually leads to neuronal loss [183]. Rab GTPase family consists of more than 60 members in human genome, and carryout regulation of intracellular trafficking. It has been reported that LRRK2 can phosphorylate 14 Rab GTPases (Rab3a/b/c/d, Rab5a/b/c, Ra/b, Rab10, Rab12, Rab29, Rab35, and Rab43) [184]. LRRK2 regulate the endo-lysosomal pathway via interacting physically or functionally at several steps, and that manipulation in LRRK expression levels or kinase activity resulting into pathological dysfunction in vesicle trafficking and protein degradation [185]. Reports suggests that binding and hydrolysis of GTP affects LRRK2 kinase activity, and that G2019S mutation-driven neurodegeneration relies on both GTPase and kinase activity. Preclinical studies performed in transgenic rodent models of PD have shown that mice overexpressing kinase activating LRRK2 G2019S showed progressive degeneration of DA neurons, and it can be reduced by LRRK2 inhibitors, they also reduced trans-axonal α-Syn aggregates and α-Syn propagation, reverse neurite shortening in primary neuronal culture from transgenic mice over expressing LRRK2 G2019S or R1441G, inhibition of LRRK2 kinase reduced proinflammatory microglial signaling in rats [186].
Curcumin is a potent antioxidant, in one study curcumin pre-treatment has been shown to improve mitochondrial dysfunction triggered by paraquat in fibroblasts from LRRK2-mutation positive PD individuals [187]. BIIB122/ DNL151, a potent LRRK2 inhibitor is under phase IIb clinical screening for its efficacy to delay the worsening of pathology for early-stage PD patients (NCT05348785). DNL 201, LRRK2 protein inhibitor, is under phase I clinical trial for safety and efficacy evaluation in PD (NCT03710707).
microRNAs
miRNAs reported to regulate the α-Syn gene expression, i.e., SNCA gene include miR-7, miR-153, and miR-203a-3p. Overexpression of miR-7 generally decreases the protein expression levels of the SNCA gene. In PD patients, there occurs an overexpression of the α-Syn protein owing to the low expression levels of miR-7 [188]. miR-153 degrades the mRNA responsible for the translation of α-Syn protein, keeping them healthy. miR-203a-3p levels were downregulated in human DA neuroblastoma cells treated with MMP+. Upregulation of miR-203a-3p inhibited the SNCA expression and also, was found to cease the cleavage of caspase-3, thus, preventing neuronal cell death [189].
Parkin (E3 Ubiquitin Ligase) is a protein that plays an explicit role in mitophagy. It was observed that the Parkin levels were downregulated with the overexpression of miR-103a-3p and miR-181a. The miR-103a-3p knockout lead to an increased parkin expression proving that this miRNA regulates the expression of parkin mRNA by binding to the 3’UTR region [189, 190]. In rotenone-treated SH-SY5Y cells, the transcription of miR-146a increases due to the induction of NF-κB, as a result of which, the parkin protein levels drop low. Apart from regulating parkin levels, in SH-SY5Y cells, miR-181a also controlled the expression of neurite growth genes such as SMAD1 and SMAD5 transcription factors. The downregulation of this miRNA increased the SMAD1 and 5 levels causing an increase in neurite length and neurite branching [191]. From a study conducted by Guo and Hua in 2020, it was inferred that the downregulation of miR-181 offered neuroprotection to MPP+ treated cells by increasing viability and inhibiting apoptosis, thereby, making it a promising therapeutic agent for posterity [189, 191]. Additionally, the miR-218 was found to negatively regulate pink1/parkin induced mitophagy, meaning, its decreased expression would trigger the mitophagy pathway intended for destroying malfunctioning mitochondria [192]. PINK1 gene encodes for an enzyme situated on the mitochondria known as PTEN-induced serine threonine kinase 1. Defective mutations of this gene would trigger the collapse of the ETC and affect the bioenergetics of the cell. PINK1 mutations are associated with early onset of PD [150]. Both miR-27a and miR-27b downregulate the expression of PINK1 by binding onto 3’ UTR region. Lack of PINK1 signifies a lack of parkin recruitment onto the OMM and hence PINK1/parkin induced mitophagy is ceased [193].
The LRRK2 gene codes for the kinase protein dardarin and its mutations have led to the manifestation of sporadic and familial forms of PD. The expression of LRRK2 gene and miR-205 has a converse correlation with one another. Upregulation of one would downregulate the other. On a similar note, miR-599 expression is extremely downregulated in various PD models, augmenting the levels of LRRK2 protein and vice versa [194].
Additionally, a few miRNAs have been reported to show a potential protective action for DA neuron survival. miR34-b and miR34-c were both found to be expressed at low levels in different brain regions of PD patients. Their downregulation led to increased ROS production, decreased cell viability, and induced mitochondrial dysfunction. A decrease in levels of protective proteins such as Parkin and DJ1 was also observed in tandem [195]. On the contrary, miR-126 was found to be upregulated in DA neurons, making them liable to 6-OHDA and staurosporine (non-selective protein kinase inhibitor) [195]. DA neuronal cells over expressed with miR-126 were susceptible to damage induced by amyloid-β protein associated with AD [196]. An increase in miR-126 expression is most likely to downregulate insulin/IGF-1/phosphatidylinositol-3-kinase (PI3K)/AKT and ERK signaling pathways, thereby, promoting the death of DA neurons [197]. The expression levels of miR-326 and miR-128 was found to be very low in MPTP-induced PD mice model and on the contrary, their overexpression was found to improve motor dysfunction and the overall health of SNpc and increase the expression of excitatory amino acid transporter 4, preventing apoptosis of DA neurons respectively. miR-326 played a role in the induction of autophagy in an in vivo mouse model of PD by triggering the JNK pathway by binding to the X-box binding protein 1 [198]. In addition to this, miR-326 upregulation was found to inhibit immunomodulatory factors such as IL-1, IL-6, TNF-α, and INF-γ [199–201]. Collectively, the levels of miR-16, miR-326, and miR-128 were found to be considerably low in multiple sclerosis patients receiving a formulation of nano-curcumin. Nano-curcumin was found to decrease the levels of Th-17 in these patients. Emerging evidences suggest that peripheral T lymphocytes, especially, Th-17 plays a key role in PD inflammation and hence targeting it via miRNA alteration could potentially satisfy the unmet needs of PD patients [202].
NLRP3 inflammasome
NLRP3 is a complex inflammasome sensor protein that has an abundant role in inflammatory diseases and is currently the talk of the scientific community. The very basic understanding of its working is as follows: activation of this inflammasome by DAMPs or PAMPs causes the recruitment of ASC speck (adaptor protein) and pro-caspase 1 (zymogen). This triggers autoproteolysis of pro-caspase 1 into active caspase 1 (effector), which in turn cleaves pro-IL-1β into IL-1β, pro-IL-18 into IL-18, and gasdermin-D into N-terminal gasdermin-D. N-terminal gasdermin-D forms pores in the membrane which further leads to pyroptosis and secretion of the pro-inflammatory cytokines extracellularly. The evidence calling out on NLRP3 inflammasome driven inflammaging is overwhelming. It sits at the very roots of pathogenic mechanisms involved in the inflammation-driven diseases such as arthritis, inflammatory bowel disease, cryopyrin-associated periodic syndromes, and neurodegenerative disorders including AD and PD [203].
Recently in 2019, Ising et al. [204] established an immense interconnection that the tau protein pathology observed most commonly in AD as well as in some cases of PD, is driven by the activation of NLRP3 inflammasome. A research conducted by Richard Gordon and his team proved the inhibition of this inflammasome beneficial in preventing α-Syn pathology and DA neurodegeneration in mice [205]. Much after the ground-breaking discovery of MCC950 (a small molecule direct inhibitor of NLRP3 inflammasome) by Cooper and O’Neil in 2015, numerous clinical R and D set-ups were established by major pharma giants like Pfizer and Novartis to develop inhibitors of NLRP3 inflammasome. Various manufacturers of these selective drugs include IFM Tre (currently sold to Novartis), Inflazome (recently acquired by Roche), Nodthera, Olatec, Quench bio, ZyVersa, and the like. Dapansutrile from olatec and Somalix and inzomelid from Inflazome are currently undergoing phase II clinical trials for various inflammatory diseases including PD and AD. Canakinumab, the IL-b1 inhibitor from Novartis, is also in phase II trials for mild cognitive impairment and AD. A novel drug, IC-100, developed by ZyVersa therapeutics is a monoclonal antibody aimed against ASC protein of the inflammasome complex. NLRP3 inhibitors of Nodthera and IFM Tre are also in clinical trials for inflammatory diseases such as inflammatory bowel disease, Crohn’s disease, fibrosis, arthritis, and the like [203]. Erinacine A, extracted from Hericiumerinaceus mycelium, have shown potential activity to attenuate the DA neuronal loss, motor function impairment, and apoptosis in neurons in PD rat model. Hence, this agent was further carried to clinical trial to evaluate the effectiveness of the compound on non-motor symptoms in early-stage PD patients (NCT04428983). Rotigotine/LY03003, a DA D2 receptor agonist formulated as an extended-release microsphere, is under phase III clinical screening for symptomatic management of early-stage PD patients (NCT04455555). Another DA receptor modulator, CJH1/CLR4001, is undergoing phase II clinical screening (NCT01684475). Tavapadon/PF-06649751, a D1/D5 receptor agonist, is under phase II trial for PD management (NCT02847650). Pramipexole/P2B001, D2 receptor agonist, is undergoing phase II/III trial for symptomatic management of PD (NCT01968460). Ropinirole, a D2 receptor agonist, was undergoing phase III clinical trial but was terminated (NCT01929317). ABBV-0805, a α-Syn aggregate solubilizing antibody, has shown its efficacy to reduce the α-Syn load in mouse model of PD and hence advanced to clinical trial phase I, but due to strategic reconsideration, the trial was halted [206]. SAGE-217/Zuranolone, a positive allosteric modulator of GABA-A receptor, is currently in phase II clinical trials for PD management (NCT03000569). Cannabinoid receptor CB1/2 agonist, nabilone, has undergone phase III trial (NCT03773796). Trehalose, an autophagy stimulant which also act to inhibit protein aggregation, is under phase IV clinical screening for its efficacy in PD (NCT05355064). Table 2 lists the agents under clinical screening.
Table 2
Agents under clinical screening for PD
| S. N. | Name | Mode of Action | Clinical Trial Phase |
| Mitochondrial Agents | |||
| 1. | Ubiquinol | Lipid Peroxidation inhibition | III |
| 2. | Ursonorm/ Ursodeoxycholic acid (NCT03840005) | Restorative role on mitochondrial function in AD and PD fibroblasts also reduced Drp-1 level | II |
| 3. | EPI-589 (NCT02462603) | Safety and biomarker study | II |
| Calcium Acting | |||
| 1. | Zonisamide (NCT04182399) | T-type calcium channel blocker | |
| 2. | Suvecaltamide/ CX-8998 (NCT03436953) | T-type Ca2 + channel blocker | II (withdrawn) |
| 3. | Isradipine (NCT02168842) | L-type calcium channel blocker | III |
| LRRK2 | |||
| 1. | BIIB122/ DNL151(NCT05348785) | LRRK2 inhibitor | IIb |
| 2. | DNL 201 (NCT03710707) | LRRK2 inhibitor | I |
| 3. | BIIB094/ Ion-849 (NCT03976349) | LRRK2 inhibitor | I |
| GBA1 | |||
| 1. | LY3884961 (NCT04127578) | Screened for safety and efficacy | I/II |
| 2. | Ambroxol HCl | Disease modifying | II |
| GLP1 | |||
| 1. | Semaglutide (NCT03659682) | Neuroprotective action, suppress α-syn aggregates | II |
| 2. | Liraglutide (NCT02953665) | Improve non-motor symptoms | II |
| NLRP3 Inflammasome | |||
| 1. | Canakinumab | IL-b inhibitor | II |
| 2. | IC-100 | Act against ASC protein of inflammasome complex | |
| Dopamine Receptor Modulators | |||
| 1. | Erinacrine A (NCT04428983) | Attenuate DA neuronal loss, motor function impairment and Apoptosis | |
| 2. | Rotigotine/LY03003 (NCT04455555) | DA D2 receptor agonist | III |
| 3. | CJH1/ CLR4001 (NCT01684475) | DA receptor modulator | II |
| 4. | Tavapadon/ PF-06649751 (NCT02847650) | D1/D5 receptor agonist | II |
| 5. | Pramipexole/P2B001 (NCT02847650) | D2 receptor agonist | II/III |
| 6. | Ropinirole | D2 receptor agonist | III (terminated) |
| Others | |||
| 1. | ABBV-0805 (NCT04127695) | Syn aggregates solubilizing antibody, reduce α-Syn load | I |
| 2. | SAGE-217/ Zuranolone (NCT03000569) | Positive allosteric modulator of GABA-A receptor | II |
| 3. | Nabilone (NCT03773796) | Cannabinoid receptor CB1/2 agonist | III |
| 4. | Trehalose (NCT05355064) | Autophagy stimulant, also inhibit protein aggregation | IV |
CONCLUSION
PD is a complex progressive neurodegenerative disorder involving several pathological changes including inflammation, oxidative damage, excitotoxicity, and epigenetic modulation leading to transcriptional modulation of gene expression and dysregulated proteostasis. Decades of research in PD has led to the unraveling of numerous pathological proteins and pathways where recently, mitochondrial dysregulation has grabbed the spotlight due to its influential role in homeostatic cellular processes. The small molecule inhibitors discovered for such dysregulated proteins include agents in advanced phases (phase III/IV trials) like isradipine, ubiquinol, and trehalose while some agents just entered clinical screening like DNL201, ABBV-0805, and erinacrine. In addition to these small molecule inhibitors, antibodies have also caught attention as disease-modifying agents for neurodegenerative disease, like the recently approved aducanumab that has an ability to inhibit amyloid peptide aggregation. Similarly, a few peptides like 4554 W [207], BIIB054 [208], and scFv [209] are studied for their efficacy to inhibit synuclein aggregation, which remains the pathological hallmark for PD. This signifies the need for a more strategical discovery that can be implemented for designing optimized pathological modulators. Elusiveness of PD pathology raises an urgent need to find out the affected protein and ergo, the disease pathway. Our work provides a brief description of the recently discovered target proteins and the agents studied against them, but a lot more must be consciously understood and unraveled in order to come up with ideal and exquisite alternatives for the PD remedy.
ACKNOWLEDGMENTS
The authors would like to thank the facility and resources made available by National Institute of Pharmaceutical Education and Research (NIPER)-Hyderabad. The figures in this manuscript were created at Biorender.com.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/22-0682r1).
REFERENCES
[1] | Surguchov A ((2021) ) Invertebrate models untangle the mechanism of neurodegeneration in parkinson’s disease. Cells 10: , 407. |
[2] | Rajan R , Divya KP , Kandadai RM , Yadav R , Satagopam VP , Madhusoodanan UK , Agarwal P , Kumar N , Ferreira T , Kumar H , Prasad AVS , Shetty K , Mehta S , Desai S , Kumar S , Prashanth LK , Bhatt M , Wadia P , Ramalingam S , Wali GM , Pandey S , Bartusch F , Hannussek M , Krüger J , Kumar-Sreelatha A , Grover S , Lichtner P , Sturm M , Roeper J , Busskamp V , Chandak GR , Schwamborn J , Seth P , Gasser T , Riess O , Goyal V , Pal PK , Borgohain R , Krüger R , Kishore A , Sharma M ((2020) ) Genetic architecture of parkinson’s disease in the indian population: Harnessing geneticdiversity to address critical gaps in parkinson’s disease research. Front Neurol 11: , 524. |
[3] | Horta SM , Kasem HB , Barba AH , Sedano BP , García DS , Fonticoba TDD , Jesús S , Aguilar M , Planellas L ((2021) ) Identifyingcomorbidities and lifestyle factors contributing to the cognitiveprofile of early Parkinson’s disease. BMC Neurol 21: , 477. |
[4] | Brand MD , Orr AL , Perevoshchikova I V , Quinlan CL , Brand MD ((2013) ) The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br J Dermatol 169: (Suppl 2), 1–8. |
[5] | Malpartida AB , Williamson M , Narendra DP , Wade-Martins R , Ryan BJ ((2021) ) Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem Sci 46: , 329–343. |
[6] | Smidt MP ((2019) ) Entanglement of genetics and epigenetics in Parkinson’s. Front Neurosci 13: , 277. |
[7] | Prasuhn J , Davis RL , Kumar KR ((2021) ) Targeting mitochondrial impairment in Parkinson’s disease: Challenges and opportunities. Front Cell Dev Biol 8: , 615461. |
[8] | Singh S , Sen Rai SN , Birla H , Zahra W , Rathore AS , Singh SP ((2020) ) NF-κB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox Res 37: , 491–507. |
[9] | Bendor JT , Logan TP , Edwards RH ((2013) ) The function of α-synuclein. Neuron 79: , 1044–1066. |
[10] | Meade RM , Fairlie DP , Mason JM ((2019) ) Alpha-synuclein structure and Parkinson’s disease. Mol Neurodegener 14: , 29. |
[11] | Gómez-Benito M , Granado N , García-Sanz P , Michel A , Dumoulin M , Moratalla R ((2020) ) Modeling Parkinson’s disease with thealpha-synuclein protein. Front Pharmacol 11: , 356. |
[12] | Braak H , Del K , Rüb U , De Vos RAI , Jansen ENH , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[13] | Stefanis L ((2012) ) α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2: , a009399. |
[14] | Guardia-Laguarta C , Area-Gomez E , Schon EA , Przedborski S ((2015) ) Novel subcellular localization for α-synuclein, ossible functional consequencesFront Neuroanat 9: , 17. |
[15] | Shavali S , Brown-borg HM , Ebadi M , Porter J ((2008) ) Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett 439: , 125–128. |
[16] | Ryan T , Bamm V V , Stykel MG , Coackley CL , Humphries KM , Jamieson-williams R , Ambasudhan R , Mosser DD , Lipton SA , Harauz G , Ryan SD ((2018) ) Cardiolipin exposure on the outer mitochondrial membrane modulates α-synuclein. Nat Commun 9: , 817. |
[17] | Ganjam GK , Bolte K , Matschke LA , Neitemeier S , Dolga AM , Höllerhage M , Höglinger GU , Adamczyk A , Decher N , Oertel WH ((2019) ) Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis 10: , 865. |
[18] | Risiglione P , Zinghirino F , Carmela M , Rosa D , Magr A , Messina A ((2021) ) Alpha-synuclein and mitochondrial dysfunction in Parkinson’s disease: The emerging role of VDAC. Biomolecules 11: , 718. |
[19] | Bridi JC , Hirth F ((2018) ) Mechanisms of α-synuclein induced synaptopathy in Parkinson’s disease. Front Neurosci 12: , 80. |
[20] | Liang Y , Cui L , Gao J , Zhu M , Zhang Y , Zhang H ((2021) ) Gut microbial metabolites in Parkinson’s disease: Implications of mitochondrial dysfunction in the pathogenesis and treatment. Mol Neurobiol 58: , 3745–3758. |
[21] | Rietdijk CD , Perez-Pardo P , Garssen J , Van Wezel RJA ((2017) ) Exploring Braak’s hypothesis of Parkinson’s disease. Front Neurol 8: , 37. |
[22] | UYAR GÖ ((2019) ) A nutritional approach to microbiota in Parkinson’s disease. Biosci Microbiota Food Health 38: , 115–127. |
[23] | Munoz-Pinto MF , Empadinhas N , Cardoso SM ((2021) ) The neuromicrobiology of Parkinson’s disease: A unifying theory. Ageing Res Rev 70: , 101396. |
[24] | Alvarez-Erviti L , Rodriguez-Oroz MC , Cooper JM , Caballero C , Ferrer I , Obeso JA , Schapira AH ((2010) ) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67: , 1464–1472. |
[25] | Danyu L , Yanran L , Xiuna J , Ying C , Sudan P , Tianen Z , Zhifen Z , Dezhi Z , Kaixun H , Yingyu X , Enxiang T ((2019) ) α-Synuclein induced mitochondrial dysfunction via cytochrome c oxidase subunit 2 in SH-SY5Y cells. Exp Cell Res 378: , 57–65. |
[26] | Kouli A , Torsney KM , Kuan W-L (2018) Parkinson’s disease: Etiology, neuropathology, and pathogenesis Parkinson’s disease: Etiology, neuropathology, and pathogenesis. In Pathogenesis and Clinical Aspects, Stoker TB, reenland JC, eds. Codon Publications, Brisbane, pp. 3-26. |
[27] | Pang SYY , Lo RCN , Ho PWL , Liu HF , Chang EES , Leung CT , Malki Y , Choi ZYK , Wong WY , Kung MHW , Ramsden DB , Ho SL ((2022) ) LRRK2, GBA and their interaction in the regulation of autophagy: Implications on therapeutics inParkinson’s disease. Transl Neurodegener 11: , 5. |
[28] | Navarro-Romero A , Montpeyó M , Martinez-Vicente M ((2020) ) The emerging role of the lysosome in Parkinson’s disease. Cells 9: , 2399. |
[29] | Madsen DA , Schmidt SI , Blaabjerg M , Meyer M ((2021) ) Interaction between parkin and α-synuclein in park2-mediated Parkinson’s disease. Cells 10: , 283. |
[30] | Wu J , Ardah M , Haikal C , Svanbergsson A , Diepenbroek M , Vaikath NN , Li W , Wang Z , Outeiro TF , El-agnaf OM , Li J ((2019) ) Dihydromyricetin and Salvianolic acid B inhibit alpha-synuclein aggregation and enhance chaperone-mediated autophagy. Transl Neurodegener 8: , 18. |
[31] | Ahmed I , Liang Y , Schools S , Dawson VL , Dawson TM , Savitt JM ((2012) ) Development and characterization of a new Parkinson’s disease model resulting from impaired autophagy. J Neurosci 32: , 16503–16509. |
[32] | Decressac M , Mattsson B , Weikop P , Lundblad M , Jakobsson J , Björklund A ((2013) ) TFEB-mediated autophagy rescues midbraindopamine neurons from α-synuclein toxicity. Proc NatlAcad Sci U S A 110: , E1817–E1826. |
[33] | Ejlerskov P , Rasmussen I , Nielsen TT , Bergström A , Tohyama Y , Jensen PH , Vilhardt F ((2013) ) Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem 288: , 17313–17335. |
[34] | Smith M , Wilkinson S ((2017) ) ER homeostasis and autophagy. Essays Biochem 61: , 625–635. |
[35] | Credle JJ , Forcelli PA , Delannoy M , Oaks AW , Permaul E , Berry DL , Duka V , Wills J , Sidhu A ((2015) ) α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol Dis 76: , 112–125. |
[36] | Cherubini M , Wade-martins R ((2018) ) Convergent pathways in Parkinson’s disease. Cell Tissue Res 373,: , 79–90. |
[37] | Wang H , Dou S , Zhu J , Shao Z , Wang C , Cheng B ((2020) ) Peptides Ghrelin mitigates MPP+-induced cytotoxicity: Involvement of ERK1/2- mediated Nrf2 / HO-1 and endoplasmic reticulum stress PERK signaling pathway. Peptides 133: , 170374. |
[38] | Dawson TM , Dawson VL ((2010) ) The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord 25: (Suppl 1), S32–S3939. |
[39] | Yuan Y , Cao P , Smith MA , Kramp K , Huang Y , Hisamoto N , Matsumoto K , Hatzoglou M , Jin H , Feng Z ((2011) ) Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS One 6: , e22354. |
[40] | Nanou E , Catterall WA ((2018) ) calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 98: , 466–481. |
[41] | Guzman JN , Sanchez-padilla J , Wokosin D , Kondapalli J , Schumacker PT , Surmeier DJ ((2015) ) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468: , 696–700. |
[42] | ((2020) ) Isradipine versus placebo in early Parkinson disease. Ann Intern Med 172: . doi: 10.7326/P20-0004. |
[43] | Ilijic E , Guzman JN , Surmeier DJ ((2011) ) The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol Dis 43: , 364–371. |
[44] | Betzer C , Lassen LB , Olsen A , Kofoed RH , Reimer L , Gregersen E , Zheng J , Calì T , Gai W , Chen T , Moeller A , Brini M , Fu Y , Halliday G , Brudek T , Aznar S , Pakkenberg B , Andersen JP , Jensen PH ((2018) ) Alpha-synucleinaggregates activate calcium pump SERCA leading to calcium dysregulation. Cell Mol Life Sci 79: , 1–21. |
[45] | Schwab AJ , Ebert AD ((2015) ) Neurite aggregation and calcium dysfunction in iPSC-derived sensory neurons with Parkinson’s disease-related LRRK2 G2019S mutation. Stem Cell Rep 5: , 1039–1052. |
[46] | Follett J , Darlow B , Wong MB , Goodwin J , Pountney DL ((2012) ) Potassium depolarization and raised calcium induces α-synuclein aggregates. Neurotox Res 23: , 378–392. |
[47] | Nath S , Goodwin J , Engelborghs Y , Pountney DL ((2011) ) Raised calcium promotes α-synuclein aggregate formation. Mol Cell Neurosci 46: , 516–526. |
[48] | Martucciello S , Masullo M , Cerulli A , Piacente S ((2020) ) Natural products targeting ER stress, and the functional link to mitochondria. Int J Mol Sci 21: , 1905. |
[49] | Wang Q , Liu Y , Zhou J ((2015) ) Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener 4: , 19. |
[50] | Lin MM , Liu N , Qin ZH , Wang Y (2022) Mitochondrialderived damage-associated molecular patterns amplify neuroinflammation in neurodegenerative diseases. Acta Pharmacol Sin. doi: 10.1038/s41401-022-00879-6. |
[51] | Dai XG , Li Q , Li T , Huang WB , Zeng ZH , Yang Y , Duan ZP , Wang YJ , Ai YH ((2020) ) The interaction between C/EBPβ and TFAM promotes acute kidney injury via regulating NLRP3 inflammasome-mediated pyroptosis. Mol Immunol 127: , 136–145. |
[52] | Yoo D , Lim Y , Ahn TB ((2021) ) Dietary intake and plasma levels of polyunsaturated fatty acids in early-stage Parkinson’s disease. Sci Rep 11: , 12489. |
[53] | Fanning S , Selkoe D , Dettmer U ((2020) ) Parkinson’s disease: Proteinopathy or lipidopathy? . NPJ Parkinsons Dis 6: , 3. |
[54] | Erskine D , Koss D , Korolchuk VI , Outeiro TF , Attems J , McKeith I ((2021) ) Lipids, lysosomes and mitochondria: Insights into Lewy body formation from rare monogenic disorders. Acta Neuropathol 141: , 511–526. |
[55] | Mcglinchey RP , Ni X , Shadish JA , Jiang J , Lee JC ((2021) ) The N terminus of α-synuclein dictates fibril formation. Proc Natl Acd Sci U S A 118: , e2023487118. |
[56] | Rouaud T , Corbillé AG , Leclair-Visonneau L , de Guilhem de Lataillade A , Lionnet A , Preterre C , Damier P , Derkinderen P ((2021) ) Pathophysiology of Parkinson’s disease: Mitochondria, alpha-synuclein and much more. . .. Rev Neurol (Paris) 177: , 260–271. |
[57] | Monzio Compagnoni G , Di Fonzo A , Corti S , Comi GP , Bresolin N , Masliah E ((2020) ) The role of mitochondria in neurodegenerative diseases: The lesson from Alzheimer’s disease and Parkinson’s disease. Mol Neurobiol 57: , 2959–2980. |
[58] | Deng H , Wang P , Jankovic J ((2017) ) The genetics of Parkinson disease. Ageing Res Rev 42: , 72–85. |
[59] | Tolosa E , Vila M , Klein C , Rascol O ((2020) ) LRRK2 in Parkinson disease: Challenges of clinical trials. Nat Rev Neurol 16: , 97–107. |
[60] | Domenicale C , Mercatelli D , Albanese F , Novello S , Vincenzi F , Varani K , Morari M ((2022) ) Dopamine transporter, phosphoserine129 α-synuclein and α-synuclein levels in aged LRRK2 G2019S knock-in and knock-out mice. Biomedicines 10: , 881. |
[61] | Chen R , Park H , Mnatsakanyan N , Niu Y , Licznerski P , Wu J , Miranda P , Graham M , Tang J , Boon AJW , Cossu G , Mandemakers W , Bonifati V , Smith PJS , Alavian KN , Jonas EA ((2019) ) Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis 10: , 469. |
[62] | Mukherjee D , Satpati P ((2019) ) Mutations in Parkinson’s disease associated protein DJ-1 alter the energetics of DJ-1 dimerization. J Chem Inf Model 59: , 1497–1507. |
[63] | Repici M , Giorgini F ((2019) ) DJ-1 in Parkinson’s disease: Clinical insights and therapeutic perspectives. J Clin Med 8: , 1377. |
[64] | Stephenson S , Djaldetti R , Rafehi H , Wilson GR , Bahlo M , Lockhart P ((2019) ) Familial early onset Parkinson’s disease caused by a homozygous frameshift variant in PARK7: Clinical features and literature update. Parkinsonsim Relat Disord 64: , 308–311. |
[65] | Lunati A , Lesage S , Brice A ((2018) ) The genetic landscape of Parkinson’s disease. Rev Neurol (Paris) 174: , 628–643. |
[66] | Ji Y , Zhang J , Lu Y , Yi Q , Chen M , Xie S , Mao X , Xiao Y , Meng F , Zhang M , Yang R , Guan MX ((2020) ) Complex I mutations synergize to worsen the phenotypic expression of Leber’s hereditary optic neuropathy. J Biol Chem 295: , 13224–13238. |
[67] | Zhang J , Ji Y , Chen J , Xu M , Wang G , Ci X , Lin B , Mo JQ , Zhou X , Guan M ((2021) ) Assocation between Leber’s hereditary optic neuropathy and MT-ND1 3460G>A mutation-induced alterations in mitochondrial function, apoptosis, and mitophagy. Invest Ophthalmol Vis Sci 62: , 38. |
[68] | Mahalaxmi I , Subramaniam MD , Gopalakrishnan AV , Vellingiri B ((2021) ) Dysfunction in mitochondrial electron transport chain complex i, pyruvate dehydrogenase activity, and mutations in ND1 and ND4 gene in autism spectrum disorder subjects from Tamil Nadu population, India. Mol Neurobiol 58: , 5303–5311. |
[69] | Yin Z , Burger N , Kula-alwar D , Aksentijevi D , Bridges HR , Prag HA , Grba DN , Viscomi C , James AM , Mottahedin A , Krieg T , Murphy MP , Hirst J ((2021) ) Structural basis for a complex I mutation that blocks pathological ROS production. Nat Commun 12: , 707. |
[70] | Id GA , Bakheit HF , Alali F , Fattah M , Alhajeri S , Alharbi A , Daif A , Mahmood M , Id A , Alwehaidah S , Bakhiet M ((2022) ) Next-generation sequencing of the whole mitochondrial genome identifies functionally deleterious mutations in patients with multiple sclerosis. . PLoS One 17: , e0263606. |
[71] | Kishita Y , Ishikawa K , Nakada K , Hayashi JI , Fushimi T , Shimura M , Kohda M , Ohtake A , Murayama K , Okazaki Y ((2021) ) A high mutation load of m.14597A>G in MT-ND6 causes Leigh syndrome. Sci Rep 11: , 11123. |
[72] | Tian L , Zhu C , Yang H , Li Y , Liu Y ((2021) ) Protective effect of mitochondrial ND2 C5178A gene mutation on cell and mitochondrial functions. Oxid Med Cell Longev 2021: , 4728714. |
[73] | Bruhn H , Samuelsson K , Schober FA , Engvall M , Lesko N , Wibom R , Nennesmo I , Calvo-Garrido J , Press R , Stranneheim H , Freyer C , Wedell A , Wredenberg A ((2021) ) Novel mutation m.10372A>G in MT-ND3 causing sensorimotor axonal polyneuropathy. Neurol Genet 7: , e566. |
[74] | Na JH , Lee MJ , Lee CH , Lee YM ((2021) ) Association between epilepsy and leigh syndrome with MT-ND3 mutation, particularly the m.10191T>C point mutation. Front Neurol 12: , 752467. |
[75] | Jaberi E , Tresse E , Grønbæk K , Weischenfeldt J , Issazadeh-navikas S ((2020) ) Identification of unique andshared mitochondrial DNA mutations in neurodegeneration and cancer by single-cell mitochondrial DNA structuralvariation sequencing (MitoSV-seq). EBioMedicine 57: , 102868. |
[76] | Golpich M , Amini E , Mohamed Z , Ali RA , Ibrahim NM ((2017) ) Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogenesis and treatment. CNS Neurosci Ther 23: , 5–22. |
[77] | Cox CJ , Foster PG , Hirt RP , Harris SR , Embley TM ((2008) ) The archaebacterial origin of eukaryotes. Proc Natl Acad Sci U S A 105: , 20356–20361. |
[78] | Ballard JWO , Whitlock MC ((2004) ) The incomplete natural history of mitochondria. Mol Ecol 13: , 729–744. |
[79] | Martin WF , Garg S , Zimorski V ((2015) ) Endosymbiotic theories for eukaryote origin. Philos Trans R Soc B Biol Sci 370: , 20140330. |
[80] | Nunnari J , Suomalainen A ((2012) ) Mitochondria: In sickness and in health. Cell 148: , 1145–1159. |
[81] | Su YJ , Wang PW , Weng SW ((2021) ) The role of mitochondria in immune-cell-mediated tissue regeneration and ageing. Int J Mol Sci 22: , 2668. |
[82] | Lackner LL ((2019) ) The expanding and unexpected functions of mitochondria contact sites. Trends Cell Biol 29: , 580–590. |
[83] | Anderson S , Bankier AT , Barrell BG , De Bruijn MHL , Coulson AR , Drouin J , Eperon IC , Nierlich DP , Roe BA , Sanger F , Schreier PH , Smith AJH , Staden R , Young IG ((1981) ) Sequence and organization of the human mitochondrial genome. Nature 290: , 457–465. |
[84] | Taanman JW ((1999) ) The mitochondrial genome: Structure, transcription, translation and replication. Biochim Biophys Acta 1410: , 103–123. |
[85] | Filograna R , Mennuni M , Alsina D , Larsson NG ((2021) ) Mitochondrial DNA copy number in human disease: The more the better? . FEBS Lett 595: , 976–1002. |
[86] | Muller FL , Liu Y , Remmen HV ((2004) ) Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem 279: , 49064–49073. |
[87] | Nohl H , Gille L , Staniek K ((2005) ) Intracellular generation of reactive oxygen species by mitochondria. Biochem Pharmacol 69: , 719–723. |
[88] | Gustafsson CM , Falkenberg M ((2016) ) Maintenance and expression of mammalian mitochondrial DNA. Annu Rev Biochem 85: , 133–160. |
[89] | Kotrys A V , Szczesny RJ ((2020) ) Mitochondrial gene expression and beyond — novel aspects of cellular physiology. Cells 9: , 17. |
[90] | Chinnery PF , Lightowlers RN ((1999) ) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23: , 3965. |
[91] | Nidhi S , Monica SP , AP ((2019) ) Mitochondrial DNA: Epigenetics and environment. Environ Mol Mutagen 60: , 668–682. |
[92] | Wiese M , Bannister AJ ((2020) ) Two genomes, one cell: Mitochondrial-nuclear coordination via epigenetic pathways. Mol Metab 38: , 100942. |
[93] | Lopes AFC ((2020) ) Mitochondrial metabolism and DNA methylation: A review of the interaction between two genomes. Clin Epigenetics 12: , 182. |
[94] | Borchiellini M , Ummarino S , Di Ruscio A ((2019) ) The bright and dark side of DNA methylation: A matter of balance. Cells 8: , 1243. |
[95] | Martin EM , Fry RC ((2018) ) Environmental influences on the epigenome: Exposure- associated DNA methylation in human populations. Annu Rev Public Health 39: , 309–333. |
[96] | Lechuga-Vieco AV , Justo-Méndez R , Enríquez JA ((2021) ) Notall mitochondrial DNAs are made equal and the nucleus knows it. IUBMB Life 73: , 511–529. |
[97] | Mohammad Saki AP ((2017) ) DNA damage related crosstalk between the nucleus and mitochondria. Physiol Behav 176: , 139–148. |
[98] | Li J , Lin T , Chen P , Guo T , Huang S , Chen C , Lin C , Chan C ((2021) ) Mitochondrial function and Parkinson’s disease: From the perspective of the electron transport chain. Front Mol Neurosci 14: , 797833. |
[99] | Rango M , Bresolin N ((2018) ) Brain mitochondria, aging, and Parkinson’s disease. Genes (Basel) 9: , 250. |
[100] | Thomas B , Beal MF ((2010) ) Mitochondrial therapies for Parkinson’s disease. , S155-S. Mov Disord 25: , 160. |
[101] | Peng K , Xiao J , Yang L , Ye F , Cao J , Sai Y ((2019) ) Mutual antagonism of PINK1/Parkin and PGC-1α contributes to maintenance of mitochondrial homeostasis in rotenone-induced neurotoxicity. Neurotox Res 35: , 331–343. |
[102] | Palikaras K , Lionaki E , Tavernarakis N ((2015) ) Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ 22: , 1399–1401. |
[103] | Varma H , Faust PL , Iglesias AD , Lagana SM , Wou K , Hirano M , DiMauro S , Mansukani MM , Hoff KE , Nagy PL , Copeland WC , Naini AB ((2016) ) Whole exome sequencing identifies a homozygous POLG2 missense variant in an infant with fulminant hepatic failure and mitochondrial DNA depletion. Eur J Med Genet 59: , 540–545. |
[104] | Saneto RP , Cohen BH , Copeland WC , Naviaux RK ((2013) ) Alpers-huttenlocher syndrome. Pediatr Neurol 48: , 167–178. |
[105] | Lin Y , Du J , Wang W , Ren H , Zhao D , Liu F , Lin P , Ji K , Zhao Y , Yan C ((2021) ) Novel biallelic mutations in POLG gene: Large deletion and missense variant associated with PEO. Neurol Sci 42: , 4271–4280. |
[106] | Hikmat O , Tzoulis C , Chong WK , Chentouf L , Klingenberg C , Fratter C , Carr LJ , Prabhakar P , Kumaraguru N , Gissen P , Cross JH , Jacques TS , Taanman JW , Bindoff LA , Rahman S ((2017) ) The clinical spectrum and natural history of early-onset diseases due to DNA polymerase gamma mutations. Genet Med 19: , 1217–1225. |
[107] | Almannai M , El-Hattab AW , Scaglia F ((2018) ) Mitochondrial DNA replication: Clinical syndromes. Essays Biochem 62: , 297–308. |
[108] | Dölle C , Flønes I , Nido GS , Miletic H , Osuagwu N , Kristoffersen S , Lilleng PK , Larsen JP , Tysnes OB , Haugarvoll K , Bindoff LA , Tzoulis C ((2016) ) Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 7: , 13548. |
[109] | Yakubovskaya E , Mejia E , Byrnes J , Hambardjieva E , Garcia-Diaz M ((2011) ) Helix unwinding and base flipping enable human MTERF1 to terminate mitochondrial transcription. Cell 141: , 982–993. |
[110] | Cui J , Wang L , Ren X , Zhang Y , Zhang H ((2019) ) LRPPRC: A multifunctional protein involved in energy metabolism and human disease. Front Physiol 10: , 595. |
[111] | Harmel J , Ruzzenente B , Terzioglu M , Spåhr H , Falkenberg M ((2013) ) The leucine-rich pentatricopeptide repeat-containing protein(LRPPRC) does not activate transcription in mammalian mitochondria. J Biol Chem 288: , 15510–15519. |
[112] | Zou J , Yue F , Li W , Song K , Jiang X , Yi J , Liu L ((2014) ) Autophagy inhibitor LRPPRC suppresses mitophagy through interaction with mitophagy initiator Parkin. PLoS One 9: , e94903. |
[113] | Wang Z , Cotney J , Shadel GS ((2007) ) Human mitochondrial ribosomal protein MRPL12 interacts directly with mitochondrial RNA polymerase to modulate mitochondrial gene expression. J Biol Chem 282: , 12610–12618. |
[114] | Serre V , Rozanska A , Beinat M , Chretien D , Boddaert N , Munnich A , Rötig A , Chrzanowska-lightowlers M ((2013) ) Mutations in mitochondrial ribosomal protein MRPL12 leads to growth retardation, neurological deterioration and mitochondrial translation deficiency. Biochim Biophys Acta 1832: , 1304–1312. |
[115] | Ma Y , Zhu S , Lv T , Gu X , Feng H , Zhen J , Xin W , Wan Q ((2020) ) SQSTM1/p62 controls mtDNA expression and participates in mitochondrial energetic adaption via MRPL12. iScience 23: , 101428. |
[116] | Boccaletto P , Machnicka MA , Purta E , Wirecki TK , Ross R , Limbach A , Kotter A , Helm M , Bujnicki JM ((2018) ) MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res 46: , 303–307. |
[117] | Bohnsack MT , Sloan KE ((2018) ) The mitochondrial epitranscriptome: The roles of RNA modifications in mitochondrial translation and human disease. Cell Mol Life Sci 75: , 241–260. |
[118] | Sarkar A , Gasperi W , Begley U , Nevins S , Huber SM , Dedon PC , Begley TJ ((2021) ) Detecting the epitranscriptome. , e. Wiley Interdiscip Rev RNA 12: , 1663. |
[119] | Jonkhout N , Tran J , Smith MA , Schonrock N , Mattick JS , Novoa EVAM ((2017) ) The RNA modification landscape in human disease. RNA 23: , 1754–1769. |
[120] | Lemmens R , Moore MJ , Al-chalabi A Jr , RHB Robberecht W ((2010) ) RNA metabolism and the pathogenesis of motor neuron diseases. Trends Neurosci 33: , 249–258. |
[121] | Bartel DP ((2018) ) Metazoan microRNAs. Cell 173: , 20–51. |
[122] | Lee H , Goodarzi H , Halberg N , Tavazoie SF ((2015) ) N6-methyl-adenosine (m6A) marks primary microRNAs for processing. Nature 519: , 482–485. |
[123] | Xhemalce B , Robson SC , Kouzarides T ((2012) ) Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell 151: , 278–288. |
[124] | Pandolfini L , Barbieri I , Bannister AJ , Onofrio M , Balasubramanian S , Kouzarides T ((2019) ) METTL1 promotes let-7 microRNA processing via article METTL1 promotes let-7 microRNA processing via m7G methylation.. Mol Cell 74: , 1278–1290.e9. |
[125] | Sharoyko V V , Abels M , Sun J , Nicholas LM , Mollet IG , Malmgren S , Storm P , Spe P , Stamenkovic A , Go I , Eliasson L , Wierup N , Mulder H , Metodiev MD ((2014) ) Loss of TFB1M results in mitochondrial dysfunction that leads to impaired insulin secretion and diabetes. Hum Mol Genet 23: , 5733–5749. |
[126] | Bar-yaacov D , Frumkin I , Yashiro Y , Chujo T , Ishigami Y ((2016) ) Mitochondrial 16S rRNA is methylated by tRNA methyltransferase TRMT61B in all vertebrates.. PLoS Biol 14: , e1002557. |
[127] | Wang S , Xiao W , Shan S , Jiang C , Chen M , Zhang Y , Chen J , Zhang C , Chen Q , Long M , Lu S ((2012) ) Multi-patterned dynamics of mitochondrial fission and fusion in a living cell. PLoS One 7: , e19879. |
[128] | Chappie JS , Acharya S , Leonard M , Schmid SL , Dyda F , Jolla L ((2010) ) G domain dimerization controls dynamin’s assembly-stimulated GTPase activity. Nature 465: , 435–440. |
[129] | Gao S , Hu J ((2021) ) Mitochondrial fusion: The machineries in and out. Trends Cell Biol 31: , 62–74. |
[130] | Chan DC ((2020) ) Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol 15: , 235–259. |
[131] | Stuppia G , Rizzo F , Riboldi G , Del Bo R , Nizzardo M , Simone C , Comi GP , Bresolin N , Corti S ((2015) ) MFN2-related neuropathies: Clinical features, molecular pathogenesis and therapeutic perspectives. J Neurol Sci 356: , 7–18. |
[132] | Bereiter-Hahn J , Jendrach M ((2010) ) Mitochondrial dynamics. Int Rev Cell Mol Biol 284: , 1–65. |
[133] | Wang X , Su B , Zheng L , Perry G , Smith MA , Zhu X ((2009) ) Mitochondrial dynamics: A delicate balance of fusion and fission Abnormal mitochondrial dynamics in AD. J Neurochem 109: , 153–159. |
[134] | Zhang R , Zhou H , Jiang L , Mao Y ((2016) ) MiR-195 dependent roles of mitofusin2 in the mitochondrial dysfunction of hippocampal neurons in SAMP8 mice. Brain Res 1652: , 135–143. |
[135] | Soriano FX , Liesa M , Bach D , Chan DC , Palacı M , Zorzano A ((2006) ) Evidence for a mitochondrial regulatory pathway defined by peroxisome proliferator–activated. Diabetes 55: , 1783–1791. |
[136] | Dorn GW ((2021) ) Mitofusins as mitochondrial anchors and tethers. J Mol Cell Cardiol 142: , 146–153. |
[137] | Wang H , Lim PJ , Karbowski M , Monteiro MJ ((2009) ) Effects of overexpression of Huntingtin proteins on mitochondrial integrity. Hum Mol Genet 18: , 737–752. |
[138] | Dyment DA , Tetreault M , Consortium CC ((2015) ) Homozygous mutations in MFN2 cause multiple symmetric lipomatosis associated with neuropathy. Hum Mol Genet 24: , 5109–5114. |
[139] | Sita G , Hrelia P , Graziosi A , Morroni F ((2020) ) Back to the fusion: Mitofusin-2 in Alzheimer’s disease. J Clin Med 9: , 126. |
[140] | Ribeiro RP , Moreira ELG , Santos DB , Colle D , Dos Santos AA , Peres KC , Figueiredo CP , Farina M ((2013) ) Probucolaffords neuroprotection in a 6-OHDA mouse model of Parkinson’s disease. Neurochem Res 38: , 660–668. |
[141] | Wang G , Li D , Huang C , Lu D , Zhang C , Zhou S ((2018) ) Icariin reducesdopaminergic neuronal loss and microglia-mediated inflammation in vivo and in vitro. Front Mol Neurosci 10: , 441. |
[142] | Korwitz A , Merkwirth C , Dennerlein RR , Tröder SE , Sprenger HG , Quirós PM , Otín CL , Rugarli EI , Langer T ((2015) ) Loss ofOMA1 delays neurodegeneration by preventing stress-induced OPA1processing in mitochondria. J Cell Biol 212: , 157–166. |
[143] | Aleo SJ , Dotto V Del , Fogazza M , Maresca A , Lodi T , Goffrini P , Ghelli A , Rugolo M , Carelli V , Baruffini E , Zanna C ((2020) ) Drug repositioning as a therapeutic strategy for neurodegenerations associated with OPA1 mutations. Hum Mol Genet 29: , 3631–3645. |
[144] | Kraus F , Ryan MT ((2017) ) The constriction and scission machineries involved in mitochondrial fission. J Cell Sci 130: , 2953–2960. |
[145] | Ingerman E , Perkins EM , Marino M , Mears JA , Mccaffery JM , Hinshaw JE , Nunnari J ((2005) ) Dnm1 forms spirals that are structurally tailored to fit mitochondria. J Cell Biol 170: , 1021–1027. |
[146] | Fowler PC , Byrne DJ , Blackstone C ((2020) ) Loss of the mitochondrial fission GTPase Drp1 contributes to neurodegeneration in a Drosophila model of hereditary spastic paraplegia. Brain Sci 10: , 646. |
[147] | Portz P , Lee MK ((2021) ) Changes in Drp1 function and mitochondrial morphology are associated with the α-synuclein pathology in a transgenic mouse model of Parkinson’s disease. Cells 13: , 885. |
[148] | Hu Z , Mao C , Wang H , Zhang Z , Zhang S , Tang M , Yang J , Yuan Y , Wang Y , Liu Y , Fan L , Yao D , Liu F , Schisler JC , Shi C , Xu Y ((2021) ) CHIP protects against MPP+/MPTP-induced damage by regulating Drp1 in two models of Parkinson’s disease. Aging (Albany NY) 13: , 1458–1472. |
[149] | Killackey SA , Philpott DJ , Girardin SE ((2020) ) Mitophagy pathways in health and disease. J Cell Biol 219: , e202004029. |
[150] | Hernandez DG , Reed X , Singleton AB ((2016) ) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139: , 59–74. |
[151] | Sliter DA , Martinez J , Hao L , Chen X , Sun N , Fischer TD , Burman JL , Li Y , Zhang Z , Narendra DP , Cai H , Klein C , Youle RJ , Branch SN , Institutes N , Heart N ((2020) ) Parkin and PINK1 mitigate STING-induced inflammation. Nature 561: , 258–262. |
[152] | Ma KY , Fokkens MR , Reggiori F , Mari M , Verbeek DS ((2021) ) Parkinson’s disease–associated VPS35 mutant reduces mitochondrial membrane potential and impairs PINK1/Parkin-mediated mitophagy. Transl Neurodegener 10: , 19. |
[153] | Clark EH , de la Torre AV , Hoshikawa T , Briston T ((2021) ) Targeting mitophagy in Parkinson’s disease. J Biol Chem 296: , 100209. |
[154] | Fernández-Moriano C , González-Burgos E , Gómez-serranillos MP ((2015) ) Mitochondria-targeted protectivecompounds in Parkinson’s and Alzheimer’s diseases. Oxid Med Cell Longev 2015: , 408927. |
[155] | Snow BJ , Rolfe FL , Lockhart MM , Frampton CM , Sullivan JDO , Fung V , Smith RAJ , Murphy MP , Taylor KM , Study P ((2010) ) A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord 25: , 1670–1674. |
[156] | Ghosh A , Langley MR , Harischandra D , Neal ML , Anantharam V , Joseph J , Brenza T , Narasimhan B , Kalyanaraman B , Kanthasamy AG ((2016) ) Mitoapocynin treatment protects against neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of PD. J Neuroimmune Pharmacol 11: , 259–278. |
[157] | Parkinson Study Group QE3 Investigators, Beal MF , Oakes D , Shoulson I , Henchcliffe C , Galpern WR , Haas R , Juncos JL , Nutt JG , Voss TS , et al. ((2015) ) A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol 71: , 543–552. |
[158] | Bell SM , Barnes K , Clemmens H , Al-Rafiah AR , Al-Ofi EA , Leech V , Bandmann O , Shaw PJ , Blackburn DJ , Ferraiuolo L , Mortiboys H ((2018) ) Ursodeoxycholic acid improves mitochondrial function and redistributes Drp1 in fibroblasts from patients with either sporadic or familial Alzheimer’s disease. J Mol Biol 430: , 3942–3953. |
[159] | Fitzgerald JC , Zimprich A , Carvajal Berrio DA , Schindler KM , Maurer B , Schulte C , Bus C , Hauser AK , Kübler M , Lewin R , Bobbili DR , Schwarz LM , Vartholomaiou E , Brockmann K , Wüst R , Madlung J , Nordheim A , Riess O , Martins LM , Glaab E , May P , Schenke-Layland K , Picard D , Sharma M , Gasser T , Krüger R ((2017) ) Metformin reverses TRAP1 mutation-associated alterations in mitochondrial function in Parkinson’s disease. Brain 140: , 2444–2459. |
[160] | Poulter MO , Payne KB , Steiner JP ((2004) ) Neuroimmunophilins: A novel drug therapy for the reversal ofneurodegenerative disease? . Neuroscience 128: , 1–6. |
[161] | Elkouzi A , Vedam-Mai V , Eisinger RS , Okun MS ((2019) ) Emerging therapies in Parkinson disease - repurposed drugs and new approaches. Nat Rev Neurol 15: , 204–223. |
[162] | Kalyanaraman B ((2020) ) Teaching the basics of repurposing mitochondria-targeted drugs: From Parkinson’s disease to cancer and back to Parkinson’s disease. Redox Biol 36: , 101665. |
[163] | Lee Y , Cho J , Lee S , Lee W , Chang S ((2019) ) Neuroprotective effects of MHY908, a PPAR α/γ dual agonist, in a MPTP-induced Parkinson’s disease model. Brain Res 1704: , 47–58. |
[164] | Inchingolo AD , Malcangi G , Inchingolo AM , Piras F , Settanni V , Garofoli G , Palmieri G , Ceci S , Patano A , Leonardis De N , Pede Di C , Montenegro V , Azzollini D , Garibaldi MG , Kruti Z , Tarullo A , Coloccia G , Mancini A , Rapone B , Semjonova A , Hazballa D , Oria MTD , Jones M , Macchia L , Bordea IR , Scarano A , Lorusso F , Tartaglia GM , Maspero C , Fabbro Del M , Nucci L , Ferati K , Ferati AB ((2022) ) Benefits and implications of resveratrol supplementation on microbiota modulations: A systematic review of the literature. Int J Mol Sci 23: , 4027. |
[165] | Müller TD , Finan B , Bloom SR , Alessio DD , Drucker DJ , Flatt PR , Fritsche A , Gribble F , Grill HJ , Habener JF , Holst JJ , Langhans W , Meier JJ , Nauck MA , Pocai A , Reimann F , Sandoval DA , Schwartz TW , Seeley RJ , Woods SC , Dimarchi RD , Tschöp MH , Gribble F , Grill H , Jf H , Holst J , Langhans W , Meier J , Nauck M , Pocai A , Reimann F , Sandoval D , Schwartz T , Seeley R , Stemmer K , Woods S , Tschöp M ((2019) ) Glucagon-like peptide 1 (GLP-1). Mol Metab 30: , 72–130. |
[166] | Glotfelty EJ , Olson L , Karlsson TE , Li Y , Greig NH ((2020) ) Glucagon-like peptide-1 (GLP-1)-based receptor agonists as a treatment for Parkinson’s disease. Expert Opin Investig Drugs 29: , 595–602. |
[167] | Kim DS , Choi H , Wang Y , Luo Y , Hoffer BJ , Greig NH ((2017) ) A new treatment strategy for Parkinson’s disease through the gut–brain axis: The glucagon-like peptide-1 receptor pathway. Cell Transplant 26: , 1560–1571. |
[168] | Zhang L , Zhang L , Li L , Christian H ((2019) ) Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of Parkinson’s disease. J Parkinsons Dis 9: , 157–171. |
[169] | Do J , Mckinney C , Sharma P , Sidransky E ((2019) ) Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegen 14: , 36. |
[170] | Bae E , Yang NY , Lee C , Lee H , Kim S , Sardi SP , Lee S ((2015) ) Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α -synuclein aggregation. , e. Exp Mol Med 47: , 153–8. |
[171] | Behl T , Kaur G , Fratila O , Buhas C , Judea-pusta CT , Negrut N , Bustea C , Bungau S ((2021) ) Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl Neurodegener 10: , 4. |
[172] | Burbulla LF , Jeon S , Zheng J , Song P ((2019) ) A modulator of wild-type glucocerebrosidase improves pathogenic phenotypes in dopaminergic neuronal models of Parkinson’s disease. , eaau. Sci Transl Med 11: , 6870. |
[173] | Mullin S , Smith L , Lee K , D’Souza G , Woodgate P , Elflein J , Hällqvist J , Toffoli M , Streeter A , Hosking J , Heywood WE , Khengar R , Campbell P , Hehir J , Cable S , Mills K , Zetterberg H , Limousin P , Libri V , Foltynie T , Schapira AHV ((2020) ) Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neurol 77: , 427–434. |
[174] | Zhou Y , Danbolt NC ((2014) ) Glutamate as a neurotransmitter in the healthy brain. J Neural Transm (Vienna) 121: , 799–817. |
[175] | Verma M , Lizama BN , Chu CT ((2022) ) Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl Neurodegener 7: , 3. |
[176] | Olivares D , Deshpande VK , Shi Y , Lahiri DK , Greig NH , Rogers JT , Huang X ((2012) ) N-Methyl D-Aspartate (NMDA) receptor antagonists and memantine treatment for Alzheimer’s disease, vascular dementia and Parkinson’s disease. Curr Alzheimer Res 9: , 746–758. |
[177] | Klockgether T , Turski L , Honoré T , Zhang ZM , Gash DM , Kurlan R , Greenamyre JT ((1991) ) The AMPA receptorantagonist NBQX has antiparkinsonian effects in monoamine-depleted rats and MPTP-treated monkeys. AnnNeurol 30: , 717–723. |
[178] | Rascol O , Fox S , Gasparini F , Kenney C , Di T , Gomez-Mancilla B ((2014) ) Use of metabotropic glutamate 5-receptorantagonists for treatment of levodopa-induced dyskinesias. Parkinsonism Relat Disord 20: , 947–956. |
[179] | Zhang Y , Fan J , Gu L , Yang H , Zhan S , Zhang H ((2021) ) Metabotropic glutamate receptor 5 inhibits α -synuclein-induced microglia inflammation to protect from neurotoxicity in Parkinson’s disease. J Neuroinflammation 18: , 23. |
[180] | Iderberg H , Maslava N , Thompson AD , Bubser M , Niswender CM , Hopkins CR , Lindsley CW , Conn PJ , Jones CK , Cenci MA ((2016) ) Pharmacological stimulation of metabotropic glutamate receptor type 4 in a rat model of Parkinson’s disease and L-DOPA-induced dyskinesia: Comparison between a positive allosteric modulator and an orthosteric agonist. Neuropharmacology 95: , 121–129. |
[181] | Tsika E , Moore DJ ((2013) ) Contribution of GTPase activity to LRRK2-associated Parkinson disease. Small GTPases 4: , 37–41. |
[182] | Jeong GR , Lee BD ((2020) ) Pathological functions of LRRK2 in Parkinson’s disease. Cells 9: , 2565. |
[183] | Thaler A , Ash E , Gan-Or Z , Orr-Urtreger A , Giladi N ((2009) ) The LRRK2 G2019S mutation as the cause of Parkinson’s disease in Ashkenazi Jews. J Neural Trans (Vienna) , 116: , 1473–1482. |
[184] | Kiral FR , Kohrs FE , Jin EJ , Hiesinger PR ((2018) ) Rab GTPases and membrane trafficking in neurodegeneration. Curr Biol 28: , 471–486. |
[185] | Erb ML , Moore DJ ((2020) ) LRRK2 and the endolysosomal system in Parkinson’s disease. J Parkinsons Dis 10: , 1271–1291. |
[186] | Bieri G , Brahic M , Bousset L , Couthouis J , Kramer NJ , Ma R ((2019) ) LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol 137: , 961–980. |
[187] | Abrahams S , Miller HC , Lombard C , Van Der Westhuizen FH , Bardien S ((2021) ) Curcumin pre-treatment may protect against mitochondrial damage in LRRK2-mutant Parkinson’s disease and healthy control fibroblasts. Biochem Biophys Rep 27: , 101035. |
[188] | Mcmillan KJ , Murray TK , Bengoa-vergniory N , Cordero-llana O , Cooper J , Buckley A , Wade-martins R , Uney JB , Neill MJO , Wong LF , Caldwell MA ((2017) ) Loss of microRNA-7 regulation leads toα-synuclein accumulation and dopaminergic neuronal lossin vivo. Mol Ther 25: , 2404–2414. |
[189] | Nies YH , Haliza N , Najib M , Lim WL ((2021) ) microRNA dysregulation in Parkinson’s disease: A narrative review. Front Neurosci 15: , 660379. |
[190] | Zhou J , Zhao Y , Li Z , Zhu M , Wang Z , Li Y , Xu T , Feng D , Zhang S , Tang F , Yao J ((2020) ) miR-103a-3p regulates mitophagy in Parkinson’s disease through Parkin/Ambra1 signaling. Pharmacol Res 160: , 105197. |
[191] | Hegarty S V , Sullivan AM , Keeffe GWO ((2018) ) Inhibition of miR-181a promotes midbrain neuronal growth through a Smad1/5-dependent mechanism: Implications for Parkinson’s disease. , NS. Neuronal Signal 2: , 20170181. |
[192] | Di Rita A , Maiorino T , Bruqi K , Volpicelli F , Bellenchi GC , Strappazzon F ((2020) ) miR-218 inhibits mitochondrial clearance by targeting PRKN E3 ubiquitin ligase. Int J Mol Sci 21: , 355. |
[193] | Nelson PT , Springer W , Kim J ((2016) ) clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1) miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol Neurodegener 11: , 55. |
[194] | Chen Q , Huang X , Li R ((2018) ) lncRNA MALAT1/miR-205-5p axis regulates MPP+-induced cell apoptosis in MN9D cells by directly targeting LRRK2. Am J Transl Res 10: , 563–572. |
[195] | Miñones-Moyano E , Porta S , Escaramís G , Rabionet R , Iraola S , Kagerbauer B , Espinosa-Parrilla Y , Ferrer I , Estivill X , Martí E ((2011) ) MicroRNA profiling of Parkinson’s disease brainsidentifies early downregulation of miR-34b/c which modulatemitochondrial function. Hum Mol Genet 20: , 3067–3078. |
[196] | Kim W , Noh H , Lee Y , Jeon J , Shanmugavadivu A , Mcphie DL , Kim K , Cohen BM , Seo H , Sonntag KC ((2014) ) miR-126 regulates growth factor activities and vulnerability to toxic insult in neurons. Mol Neurobiol 53: , 95–108. |
[197] | Kim W , Lee Y , Mckenna ND , Yi M , Simunovic F , Wang Y , Kong B , Rooney RJ , Seo H , Stephens RM , Sonntag KC ((2014) ) miR-126 contributes to Parkinson’s disease by dysregulating the insulin-like growth factor/phosphoinositide 3-kinase signaling. Neurobiol Aging 35: , 1712–1721. |
[198] | Zhao XH , Wang YB , Yang J , Liu HQ , Wang LL ((2019) ) microRNA-326 suppresses iNOS expression and promotes autophagy of dopaminergic neurons through the JNK signaling by targeting XBP1 in a mouse model of Parkinson’s disease. J Cell Biochem 120: , 14995–15006. |
[199] | Bai X , Tang Y , Yu M , Wu L , Liu F , Ni J , Wang Z ((2017) ) Downregulation of blood serum microRNA 29 family in patients with Parkinson’s disease. Sci Rep 7: , 5411. |
[200] | Kanagaraj N , Beiping H , Dheen ST , Tay SSW ((2014) ) Downregulation of miR-124 in MPTP-treated mouse model of Parkinson’s disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience 272: , 167–179. |
[201] | Zhang J , Liu W , Wang Y , Zhao S , Chang N ((2017) ) miR-135b plays a neuroprotective role by targeting GSK3 β in MPP+-intoxicated SH-SY5Y cells. Dis Markers 2017: , 5806146. |
[202] | Yavarpour-Bali H , Pirzadeh M , Ghasemi-Kasman M ((2019) ) Curcumin-loaded nanoparticles: A novel therapeutic strategy in treatment of central nervous system disorders. Int J Nanomedicine 14: , 4449–4460. |
[203] | Schwaid AG , Spencer KB ((2020) ) Strategies for targeting the NLRP3 inflammasome in the clinical and preclinical space. J Med Chem 64: , 101–122. |
[204] | Ising C , Venegas C , Zhang S , Scheiblich H , Schmidt S V , Vieira-saecker A , Schwartz S , Albasset S , Mcmanus RM , Tejera D , Griep A , Santarelli F , Brosseron F , Opitz S , Stunden J , Merten M , Kayed R , Golenbock DT , Blum S , Latz E , Buée L , Heneka MT ((2019) ) NLRP3 inflammasome activation drives tau pathology. Nature 575: , 669–673. |
[205] | Gordon R , Albornoz EA , Christie DC , Langley MR , Kumar V , Mantovani S , Robertson AAB , Butler MS , Rowe DB , Neill LAO , Kanthasamy AG , Schroder K , Cooper MA , Woodruff TM ((2018) ) Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. , eaah. Sci Transl Med 10: , 4066. |
[206] | Eriksson F , Sigvardson J , Johannesson M , Nordstr E , Kasrayan A , Jones-kostalla M , Appelkvist P , Linda S , Nygren P , Blom M , Rachalski A , Nordenankar K , Zachrisson O , Amandius E , Osswald G , Moge M , Ingelsson M , Bergstr J , Giorgetti M , Johanna F , Lannfelt L , Christer M ((2021) ) ABBV-0805, a novel antibody selective for soluble aggregated α -synuclein, prolongs lifespan and prevents buildup of α -synuclein pathology in mouse models of Parkinson’s disease. Neurobiol Dis 161: , 105543. |
[207] | Torpey JH , Meade RM , Mistry R , Mason JM , Madine J ((2020) ) Insights into peptide inhibition of alpha-synuclein aggregation. Front Neurosci 14: , 561462. |
[208] | Weihofen A , Liu YT , Arndt JW , Huy C , Quan C , Smith BA , Baeriswyl JL , Cavegn N , Senn L , Su L , Marsh G , Auluck PK , Montrasio F , Nitsch RM , Hirst WD , Cedarbaum JM , Pepinsky RB , Grimm J , Weinreb PH ((2019) ) Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol Dis 124: , 276–288. |
[209] | Lynch SM , Zhou C , Messer A ((2008) ) An scFv intrabody against the nonamyloid component of α-synuclein reduces intracellular aggregation and toxicity. J Mol Biol 377: , 136–147. |


