
A Novel Heterozygous Missense Variant in the CIAO1 Gene in a Family with Alzheimer’s Disease: The Val67Ile Variant Promotes the Interaction of CIAO1 and Amyloid-β Protein Precursor
Abstract
Familial dementia is a rare inherited disease involving progressive impairment of memory, thinking, and behavior. We report a novel heterozygous pathogenic variant (c.199G > A, p.Val67Ile) in the CIAO1 gene that appears to be co-segregated with Alzheimer’s disease in a Japanese family. Biochemical analysis of CIAO1 protein revealed that the variant increases the interaction of CIAO1 with immature amyloid-β protein precursor (AβPP), but not mature or soluble AβPP, indicating plausible CIAO1 involvement in AβPP processing. Our study indicates that a heterozygous variant in the CIAO1 gene may be closely related to autosomal dominant familial dementia.
INTRODUCTION
Alzheimer’s disease is one of the major causative factors for progressive dementia and is clinically defined by a slowly progressive loss of cognitive function and primarily memory impairment [1]. Early-onset familial Alzheimer’s disease, which is defined as occurring before age 65, is particularly thought to be highly heritable and to run in families [2]. Menelian forms of Alzheimer’s disease tend to present with a similar clinical picture to the other forms, for which three major genetic loci have been identified, i.e., APP (amyloid precursor protein), PSEN1 (presenilin 1), and PSEN2 (presenilin 2) [2, 3].
Meanwhile, CIAO1, which is encoded by the CIAO1 gene, is a subunit of a protein complex involved in the biosynthesis of Fe-S proteins [4]. To date, CIAO1 variants have never been reported to be associated with Alzheimer’s disease physiopathology. Herein, we report a Japanese family with early-onset Alzheimer’s disease with a heterozygous c.199G > A variant in CIAO1.
MATERIALS AND METHODS
See Supplementary Material for details. The study was approved by the Institutional Review Board of the University of Yamanashi. Written informed consent was obtained from all the healthy direct relatives of the participants.
RESULTS
Clinic study
The proband (Fig. 1A, II-4) was a 72-year-old male. He was self-employed, selling water supply equipment until age 70. He presented with a tendency for forgetfulness since age 60. His forgetfulness gradually progressed. At age 70, he started to present personality changes, with fluctuating episodes of confusion and visual hallucinations. He became vacant on questioning and unable to follow instructions. At age 72, he exhibited psychiatric and behavioral problems including episodes of delirium, aggressiveness, self-inflicted injurious behavior, and suicidal behavior. There was no neurological abnormality on examination at age 71. The patient’s score on the revised Hasegawa dementia scale (HDS-R) was 15/30 and that on a Mini-Mental State Examination (MMSE) was 18/30. Brain magnetic resonance imaging (MRI) showed mild atrophy of the entire cerebrum (Fig. 1B). No evident subcortical lesions or microbleeds were detected. Brain 99mTc-ethylcysteinate dimer-single-photon emission computed tomography (SPECT) showed a perfusion decrease in the bilateral occipital lobes and medial temporal lobes including the amygdala, hippocampus, uncal gyrus, and parahippocampus (Fig. 1C). In contrast to dementia with Lewy bodies or frontotemporal dementia (FTD), he presented pronounced memory deficits at an earlier stage instead of personality and behavior changes, and he presented no spontaneous features of parkinsonism. The medial temporal lobe exhibited mild atrophy on MRI scanning with hypoperfusion on SPECT. Moreover, fluorine 18-labeled flutemetamol amyloid positron emission tomography imaging (18F-Flutemetamol PET) was clearly positive with notable Aβ deposits (Fig. 1D), which could be differentiated from the behavioral variant of FTD. Thus, he was clinically diagnosed with Alzheimer’s disease [5].
Fig. 1
Clinical and genetic studies of the present family. A) Pedigree of the present family. The proband is indicated (arrow). Squares indicate males; circles, females; slashes, deceased individuals; shaded (black) symbols, individuals with symptoms of cognitive decline; unshaded symbols, individuals without symptoms of cognitive decline; the proband’s oldest son presented with depression and violence is indicated in half black; individuals that underwent quad WES analysis are denoted by asterisks; and individuals assessed clinically or genetically are denoted by dots. B) Brain MRI of the proband revealed mild atrophy of the entire cerebrum. C) Three-dimensional stereotactic surface projection (3D-SSP) analysis was used to evaluate cerebral perfusion using single-photon emission CT (SPECT). The pixel values of the patient image set are normalized as to the mean global cerebral blood flow (CBF) before the analysis. To quantify perfusion deficits, pixel-by-pixel z scores are used. A positive z score represents reduced CBF in the patient relative to in normal subjects. The proband showed decreased perfusion in the bilateral occipital lobes, temporopolar cortex, and medial temporal lobes including the hippocampus, amygdala, parahippocampal gyrus, and limbic cortex. D) 18F-Flutemetamol PET of the proband showed elevated Aβ deposition in the cerebral cortex, most pronouncedly in the frontal, lateral temporal, parietal regions, as well as the posterior cingulate cortex and the precuneus. E) Brain MRI of the proband’s younger sister revealed mild atrophy of the entire cerebrum, which was most prominent in the bilateral temporal lobes. F) Sanger sequencing revealed the c.199G > A variant in CIAO1 in a heterozygous state in the proband, his older sister, his younger sister, and the second daughter of his older sister. G) The c.199G > A variant in CIAO1 was not detected in the proband’s wife, his brother, his daughter, his younger sister’s daughter, his younger sister’s husband, or the first daughter of his older sister without symptoms. The green arrow indicates the c.199 nucleotide.
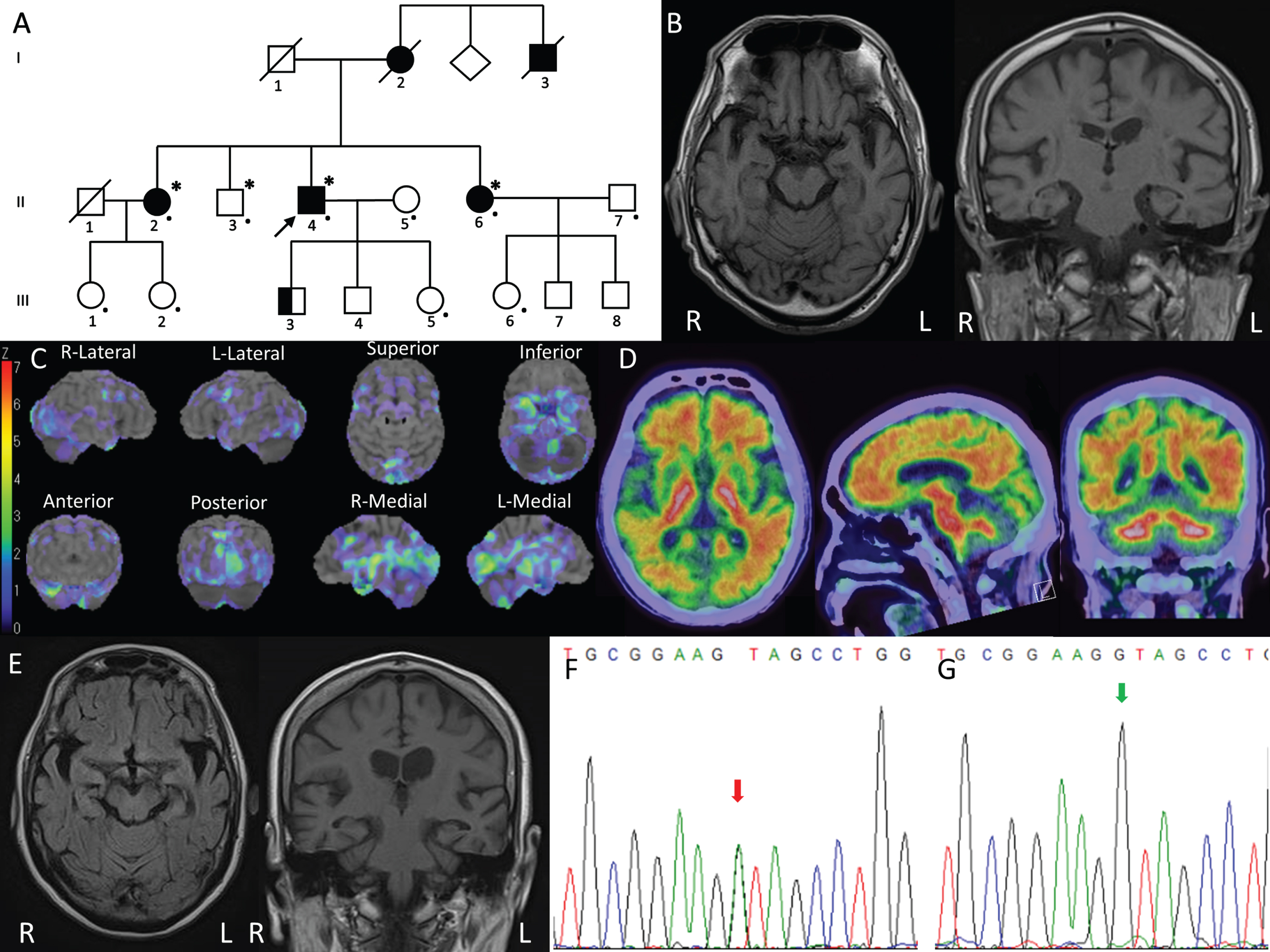
The proband’s younger sister (Fig. 1A, II-6) had been employed as an office assistant in a gas company until age 61 when she was laid off due to frequent mistakes. Her cognitive condition gradually deteriorated, and at age 66 she became disoriented and was unable to find her way home. She also exhibited psychiatric symptoms of mania and aggressiveness. Neurologic examination revealed nothing significant. Brain MRI revealed atrophy of the entire cerebrum, which was most prominent in the bilateral temporal lobes including the hippocampus (Fig. 1E). Subcortical infarcts or microbleeds were absent. Her HDS-R score was 3/30. She was diagnosed with probable Alzheimer’s disease [5], and treated with donepezil and memantine. She is now 69 years old and needs assistance with daily life in the form of in-home care.
The proband’s older sister (Fig. 1A, II-2) was also clinically diagnosed with probable Alzheimer’s disease at age 58 [5]. She is now 76 years old and in the severe late stage of Alzheimer’s disease. The proband’s deceased mother (Fig. 1A, I-2) and uncle (Fig. 1A, I-3) developed “dementia” at around age 60, and they died in their early 80 s. The proband’s oldest son (Fig. 1A, III-3) aged 41 presented with depression and violence from age 20. He received long-term care service at home and could not be assessed clinically or genetically. Other than him, no third-generation family member aged from 36 to 50 has developed cognitive impairment or psychiatric symptoms yet. The two daughters of the proband’s older sister (Fig. 1A, III-1 and III-2) and the daughter of the proband’s younger sister (Fig. 1A, III-6) were clinically evaluated, while the proband’s second son (Fig. 1A, III-4) and the two sons of the proband’s younger sister could not be assessed.
Genetic study
On whole-exome sequencing (WES) for the proband, we could not find any mutations in the causative genes associated with dementia or parkinsonism (Supplementary Material). Moreover, no copy number variations of APP, SNCA, or any other genes mentioned above were detected in the proband on high-density aCGH analysis. To identify the disease-causing gene in this family, WES was then performed using the quadruple diagnostic approach (Fig. 1A, II-2, II-3, II-4, and II-6). The disease-causing variant frequency was set at less than 0.01%, a dominant inheritance mode with full penetrance in both males and females being assumed. Through the analysis, we identified three variants that were in heterozygous states in the proband, his older sister, and his younger sister with cognitive decline, but negative in his brother without symptoms. By analyzing the three variants one by one, we identified a novel missense variant (c.199G > A, p.Val67Ile) in exon 2 of CIAO1 (NCBI NM_004804). The other two variants were excluded by bioinformatic analyses. On Sanger sequencing, we reconfirmed the c.199G > A variant of CIAO1, which was in a heterozygous state in the proband, his older sister, his younger sister, and the second daughter of the proband’s older sister (Fig. 1A, III-2; Fig. 1F). This variant was absent in the proband’s wife (Fig. 1A, II-5), his brother (Fig. 1A, II-3), his daughter (Fig. 1A, III-5), his younger sister’s daughter (Fig. 1A, III-6), his younger sister’s husband, and the first daughter of his older sister (Fig. 1A, III-1) without symptoms (Fig. 1G). Valine was replaced by isoleucine in an area evolutionarily conserved among the human, chimpanzee, rhesus monkey, cat, mouse, pufferfish, dog, elephant, chicken, X. tropicalis, and zebrafish. Bioinformatic analyses using Mutation Taster (http://www.mutationtaster.org), Polyphen2 (http://genetics.bwh.harvard.edu/pph2), M-CAP (http://bejerano.stanford.edu/mcap/), and fathmm-MKL (http://fathmm.biocompute.org.uk/fathmmMKL.htm) predicted that this variant was disease-causing, probably damaging, possibly pathogenic, and deleterious, respectively. This variant has a Combined Annotation Dependent Depletion (CADD v1.6; https://cadd.gs.washington.edu/score) score of 24.62 and is not present in dbSNP, HGMD, GnomAD, or jMorp (a whole-genome variation panel of 8,380 Japanese individuals; https://jmorp.megabank.tohoku.ac.jp/202102/). According to the American College of Medical Genetics and Genomics (ACMG) recommendations [6], this variant is assigned as being likely pathogenic (PS3 + PM2 + PP3 + PP4).
We examined the WES data of 361 patients recruited from the Japanese Alzheimer’s Disease Neuroimaging Initiative (J-ADNI) cohort (140 Alzheimer’s disease cases and 221 mild cognitive impairment (MCI) cases) [7], and the entire genome sequencing (WGS) data of 565 patients with genetically undiagnosed dementia (329 cases) or early-onset Alzheimer’s disease (236 cases) from the Brain Research Institute of Niigata University. No apparently deleterious (frameshift, nonsense, or splice region) variant was identified. However, six missense variants were found (Table 1). We evaluated each missense variant based on the combination of the CADD prediction score and the minor allele frequency (MAF) in the public databases. Two missense variants (c.58T > C and c.623G > A) were identified in two genetically undiagnosed dementia patients with CADD > 25 and MAF < 0.1%. Unfortunately, further information on the two patients with these variants is unavailable but they are unlikely to be familial cases. Finally, to determine whether or not they may be enriched in our dementia cohorts, CIAO1 variants were reviewed in a reference database of genetic variations in the Japanese population (HGVD) as normal controls [8]. However, we did not find any enrichment of CIAO1 missense variants in dementia patients compared with in the normal group (Table 1).
Table 1
CIAO1 variants screened from 926 Japanese patients with idiopathic Alzheimer’s Disease, early-onset dementia, or MCI
| dbSNP ID | Codon change | Amino acid change | J-ADNI/AD | J-ADNI/MCI | CL-NEG | EOAD | CADD | Num_het | Num_hom | allele frequency | MAF | RR |
| rs199793823 | c.56G > C | p.Cys19Ser | 1 | 0 | 1 | 1 | 32.0 | 3 | 0 | 0.162% | 0.289% | 0.56 |
| rs1459911963 | c.58T > C | p.Trp20Arg | 0 | 0 | 1 | 0 | 33.0 | 1 | 0 | 0.054% | 0.032% | 1.69 |
| rs761581891 | c.623G > A | p.Arg208His | 0 | 0 | 1 | 0 | 29.8 | 1 | 0 | 0.054% | 0.041% | 1.31 |
| rs142522111 | c.647G > A | p.Arg216His | 2 | 5 | 1 | 4 | 25.1 | 12 | 0 | 0.648% | 0.661% | 0.98 |
| rs776406200 | c.706G > A | p.Gly236Ser | 1 | 0 | 0 | 0 | 19.2 | 1 | 0 | 0.054% | 0.018% | 2.97 |
| rs191488251 | c.920A > G | p.Asn307Ser | 0 | 0 | 1 | 0 | 26.5 | 1 | 0 | 0.054% | 0.105% | 0.52 |
| Comparison of CIAO1 variants in dementia cohorts with normal population | Dementia | Control | OR | p | ||||||||
| Total Number | 926 | 1210 | ||||||||||
| Number of missense variants (allele frequency) | 19 (0.0103) | 28 (0.0116) | 0.8855 | 0.43 | ||||||||
| Number of missense variants with MAF < 0.001 (allele frequency) | 3 (0.0016) | 9 (0.0037) | 0.4346 | 0.43 | ||||||||
J-ADNI/AD, 140 clinically diagnosed Alzheimer’s disease cases from J-ADNI; J-ADNI/MCI, 221 clinically diagnosed MCI cases from J-ADNI; CL-NEG, 329 clinically diagnosed dementia cases with APP, PSEN1, PSEN2, MAPT, PGRN, CSF1R mutations genotyped and excluded from brain research institute of Niigata University; EOAD, 236 clinically diagnosed early-onset Alzheimer’s disease cases from brain research institute of Niigata University; Num_het, total number of heterozygous mutations from dementia patients; Num_hom, total number of homozygous mutations from dementia patients; MAF, minor allele frequency in the public databases from populations all over the world including GnomAD, TOPMED, jMorp, HGVD, etc.; RR, risk ratio of the probability of dementia in patients with CIAO1 missense variants using the MAF values from public databases as control; OR, odds ratio.
The CIAO1 variant increases the interaction of CIAO1 and immature AβPP
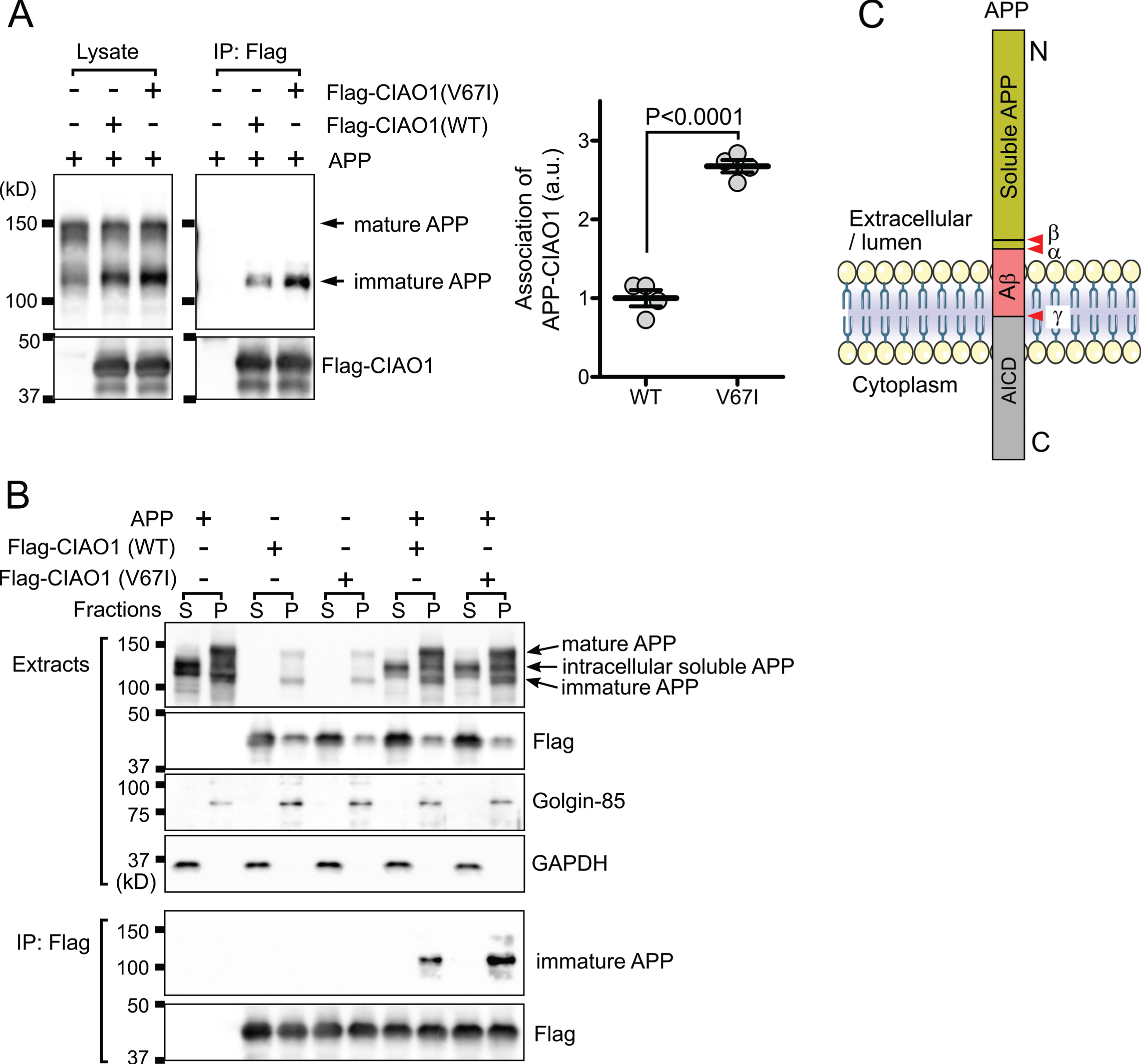
The CIAO1 gene encodes for probable cytosolic iron-sulfur protein assembly protein CIAO1, which is a key component of the cytosolic iron-sulfur protein assembly complex [4]. This molecular complex mediates the incorporation of the iron-sulfur cluster into the iron-sulfur protein and is involved in iron homeostasis [9]. Characteristically, the interaction of CIAO1 and HspB1/Hsp27 has been mapped in a large-scale interactome using immunoprecipitation and subsequent mass spectrometry [10]. HspB1/Hsp27 is a known factor modulating amyloid-β protein precursor (AβPP) expression [11]. Therefore, we examined whether or not CIAO1 and the mutant (V67I) interact with AβPP. After 24 h of transfection, flag-tagged CIAO1 and the mutant were immunoprecipitated, and AβPP in the precipitate was detected by western blotting. Interestingly, CIAO1 and the mutant were specifically associated with the low molecular weight immature AβPP (Fig. 2A). Furthermore, the V67I mutant showed enhanced binding to the immature AβPP (Fig. 2A). Generally, in the secretory pathway, full-length AβPP is roughly distinguished as immature AβPP (N-glycosylated) in the endoplasmic reticulum (ER), and mature AβPP (N- and O-glycosylated, tyrosyl-sulfated) in the Golgi apparatus and plasma membrane [12]. Because immature and soluble AβPP have similar molecular weights, we further separated AβPP molecules by subcellular fractionation. After centrifugation at 100,000×g for 1 h, homogenates were fractioned into cytosol and membrane fractions. Western blotting analysis revealed that intracellular soluble AβPP [13] exhibited different mobility from the immature and mature forms of AβPPs on SDS-PAGE (Fig. 2B, top panel). We unequivocally confirmed the mutant of CIAO1 prominently interacted with the immature AβPP molecules (Fig. 2B, bottom). These data strongly suggest that CIAO1 is involved in AβPP processing in the endoplasmic reticulum and Golgi apparatus. In addition, the V67I mutant may actively participate in and modulate AβPP processing.
Fig. 2
CIAO1 interacts with AβPP. A) Triton-soluble extracts of cells transfected with the indicated plasmids were immunoprecipitated using anti-flag magnetic beads (IP: Flag). Each precipitate was subjected to SDS-PAGE and western blotting. Arrows indicate the mature and immature forms of AβPP. The association of immature AβPP and CIAO1 was quantitatively analyzed in four independent experiments using Image J software. The association of AβPP with CIAO1-WT and CIAO1-V67I was statistically significant according to Student’s t-test. Arbitrary unit (a.u.). B) Cells transfected with the indicated plasmids were fractionated into cytosol (S) and membrane (P) fractions. Each form of AβPP is indicated by an arrow. Golgin-85 and GAPDH were used as marker proteins for the membrane and cytosol fractions, respectively. CIAO1 and the mutant were immunoprecipitated using anti-flag magnetic beads. C) Diagram of the AβPP structure. The cleavage sites for α-, β-, and γ-secretases are indicated (red arrowheads). AβPP intracellular domain (AICD). The anti-AβPP antibody used in this study was raised using a synthetic peptide corresponding to the juxtamembrane region surrounding at proline 620 of the human AβPP protein.
DISCUSSION
The proband and the proband’s younger and older sisters examined by neurologists were diagnosed with Alzheimer’s disease clinically. The proband’s deceased mother and uncle had suffered similar symptoms. Phenotype concordance of 1) an early-onset cognitive decline at around age 60, 2) a relatively long disease duration, and 3) prominent psychiatric symptoms were observed in all family members affected. The proband’s oldest son aged 41 and the second daughter of the proband’s older sister aged 48 need to be followed and watched carefully as they may develop cognitive decline in the future.
To date, variants in CIAO1 have not been found to be related to any human genetic disorders. Although a study showed that CIAO1 bound to oligomeric Aβ in vitro on protein array-based oligomeric Aβ interaction analysis, whether the interaction was specific or not has yet to be demonstrated [14]. We demonstrated CIAO1 specifically interacted with immature AβPP because it interacted with neither soluble nor mature AβPP. Unexpectedly, a more prominent interaction was observed between AβPP and the CIAO1 mutant (V67I) than the wild type. Considering the topology of AβPP (Fig. 2C), CIAO1 may bind to the cytoplasmic domain of AβPP because it does not bind to intracellular soluble AβPP, which lacks a cytoplasmic domain. Although we do not know the exact molecular mechanism for the specific interaction of immature AβPP with CIAO1, one possible explanation would be that ER-to-Golgi transition-associated additional factors might be involved in the interaction between AβPP and CIAO1 (V67I). Thus, the interaction of CIAO1 and AβPP in our preliminary functional confirmation study potentially implies that variants in CIAO1 might influence the Aβ pathway in genetic Alzheimer’s disease.
Unfortunately, the present study was limited to only one pedigree. We did not find definite evidence of deleterious CIAO1 variants in the WGS or WES data from a total of 926 Japanese patients with cognitive decline, indicating that variants in CIAO1 are unlikely to be a common unrecognized cause of cognitive decline. There might be some unknown genetic etiology underlying the cognitive impairment in our family that could not be identified with the present methods. Nonetheless, follow-up studies on larger, prospectively recruited cohorts, as well as on animal models, are needed to further define this understudied condition.
In summary, we report a Japanese family with early-onset Alzheimer’s disease with a heterozygous c.199G > A variant in the CIAO1 gene. Biochemical analysis of the CIAO1 protein suggested that CIAO1 is involved in AβPP processing, and the V67I mutant may actively participate in and modulate AβPP processing. Although the limitation of the present study necessitates caution in drawing conclusions with regard to the genetic association of cognitive decline and CIAO1 variants, we believe that our data call attention to this understudied genotype-phenotype relationship and shed light on the potential molecular mechanism of the involvement of CIAO1 in AβPP processing.
ACKNOWLEDGMENTS
We thank the family members who have participated in this study. We also thank Dr. Yoko Sato for her expert advice on amyloid PET imaging. This work was supported by Grants-in-Aid from the Research Committee for Ataxic Disease (Y.T.), the Ministry of Health, Labor and Welfare, Japan, and JSPS KAKENHI Grant Number JP21K07456 (Y. T.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0706r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-210706.
REFERENCES
[1] | ((2021) ) 2021 Alzheimer’s disease facts and figures. Alzheimers Dement 17: , 327–406. |
[2] | Ferencz B , Gerritsen L ((2015) ) Genetics and underlying pathology of dementia. Neuropsychol Rev 25: , 113–124. |
[3] | Ryan NS , Rossor MN ((2010) ) Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark Med 4: , 99–112. |
[4] | Stehling O , Vashisht AA , Mascarenhas J , Jonsson ZO , Sharma T , Netz DJ , Pierik AJ , Wohlschlegel JA , Lill R ((2012) ) MMS19 assembles iron-sulfur proteins required for DNA metabolism and genomic integrity. Science 337: , 195–199. |
[5] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr. , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[6] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL , Committee ALQA ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[7] | Iwatsubo T , Iwata A , Suzuki K , Ihara R , Arai H , Ishii K , Senda M , Ito K , Ikeuchi T , Kuwano R , Matsuda H , Japanese Alzheimer’s Disease Neuroimaging Initiative, Sun CK , Beckett LA , Petersen RC , Weiner MW , Aisen PS , Donohue MC , Alzheimer’s Disease Neuroimaging Initiative ((2018) ) Japanese and North American Alzheimer’s Disease Neuroimaging Initiative studies: Harmonization for international trials. Alzheimers Dement 14: , 1077–1087. |
[8] | HigasaK, MiyakeN, YoshimuraJ, OkamuraK, NiihoriT, SaitsuH, DoiK, ShimizuM, NakabayashiK, AokiY, TsurusakiY, MorishitaS, KawaguchiT, MigitaO, NakayamaK, NakashimaM, MitsuiJ, NaraharaM, HayashiK, FunayamaR, YamaguchiD, IshiuraH, KoWY, HataK, NagashimaT, YamadaR, MatsubaraY, UmezawaA, TsujiS, MatsumotoN, MatsudaF ((2016) ) Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet 61: , 547–553. |
[9] | Mayank AK , Pandey V , Vashisht AA , Barshop WD , Rayatpisheh S , Sharma T , Haque T , Powers DN , Wohlschlegel JA ((2019) ) An oxygen-dependent interaction between FBXL5 and the CIA-targeting complex regulates iron homeostasis. Mol Cell 75: , 382–393 e385. |
[10] | Ewing RM , Chu P , Elisma F , Li H , Taylor P , Climie S , McBroom-Cerajewski L , Robinson MD , O’Connor L , Li M , Taylor R , Dharsee M , Ho Y , Heilbut A , Moore L , Zhang S , Ornatsky O , Bukhman YV , Ethier M , Sheng Y , Vasilescu J , Abu-Farha M , Lambert JP , Duewel HS , Stewart II , Kuehl B , Hogue K , Colwill K , Gladwish K , Muskat B , Kinach R , Adams SL , Moran MF , Morin GB , Topaloglou T , Figeys D ((2007) ) Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol 3: , 89. |
[11] | Conway M , Nafar F , Straka T , Mearow K ((2014) ) Modulation of amyloid-beta protein precursor expression by HspB1. J Alzheimers Dis 42: , 435–450. |
[12] | Tomita S , Kirino Y , Suzuki T ((1998) ) Cleavage of Alzheimer’s amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J Biol Chem 273: , 6277–6284. |
[13] | Henriques AG , Vieira SI , Crespo-Lopez ME , Guiomar de Oliveira MA , da Cruz e Silva EF , da Cruz e Silva OA ((2009) ) Intracellular sAPP retention in response to Abeta is mapped to cytoskeleton-associated structures. J Neurosci Res 87: , 1449–1461. |
[14] | Olah J , Vincze O , Virok D , Simon D , Bozso Z , Tokesi N , Horvath I , Hlavanda E , Kovacs J , Magyar A , Szucs M , Orosz F , Penke B , Ovadi J ((2011) ) Interactions of pathological hallmark proteins: Tubulin polymerization promoting protein/p25, beta-amyloid, and alpha-synuclein. J Biol Chem 286: , 34088–34100. |

