
Genetics Contributes to Concomitant Pathology and Clinical Presentation in Dementia with Lewy Bodies
Abstract
Background:
Dementia with Lewy bodies (DLB) is a complex, progressive neurodegenerative disease with considerable phenotypic, pathological, and genetic heterogeneity.
Objective:
We tested if genetic variants in part explain the heterogeneity in DLB.
Methods:
We tested the effects of variants previously associated with DLB (near APOE, GBA, and SNCA) and polygenic risk scores for Alzheimer’s disease (AD-PRS) and Parkinson’s disease (PD-PRS). We studied 190 probable DLB patients from the Alzheimer’s dementia cohort and compared them to 2,552 control subjects. The p-tau/Aβ1–42 ratio in cerebrospinal fluid was used as in vivo proxy to separate DLB cases into DLB with concomitant AD pathology (DLB-AD) or DLB without AD (DLB-pure). We studied the clinical measures age, Mini-Mental State Examination (MMSE), and the presence of core symptoms at diagnosis and disease duration.
Results:
We found that all studied genetic factors significantly associated with DLB risk (all-DLB). Second, we stratified the DLB patients by the presence of concomitant AD pathology and found that APOE ɛ4 and the AD-PRS associated specifically with DLB-AD, but less with DLB-pure. In addition, the GBA p.E365K variant showed strong associated with DLB-pure and less with DLB-AD. Last, we studied the clinical measures and found that APOE ɛ4 associated with reduced MMSE, higher odds to have fluctuations and a shorter disease duration. In addition, the GBA p.E365K variant reduced the age at onset by 5.7 years, but the other variants and the PRS did not associate with clinical features.
Conclusion:
These finding increase our understanding of the pathological and clinical heterogeneity in DLB.
INTRODUCTION
Dementia with Lewy bodies (DLB) is a complex, progressive neurodegenerative disease. DLB is a clinical diagnosis based on the presence of dementia in combination with one or more of four core symptoms: Parkinsonian features (i.e., bradykinesia, postural instability, and rigidity), visual hallucinations, fluctuations in alertness and cognition, and rapid eye-movement sleep behavior disorder (RBD) [1, 2]. There is considerable phenotypic heterogeneity in terms of clinical symptoms and disease course in DLB. Next to clinical heterogeneity there is also pathological heterogeneity in DLB. The pathological hallmark of DLB is intraneuronal aggregates of pathological alpha-synuclein protein in Lewy bodies and Lewy neurites [3, 4], but in roughly half of DLB patients it is accompanied by Alzheimer’s disease (AD) related pathology. AD pathology is defined by extracellular amyloid-β accumulation and intracellular tau deposition [5, 6]. In general, concomitant AD in patients with DLB is best studied at autopsy. However, pathological series are inherently biased due to their retrospective character as only a highly selected subset approves autopsy and AD pathology is measured at the end stage of the disease. This could be overcome by studying concomitant AD pathology in vivo in DLB patients [7, 8]. In previous studies it has been shown that the ratio of t-tau/Aβ1–42 measured in cerebrospinal fluid (CSF) is a good proxy measure to distinguish DLB patients with AD co-pathology from those with low/no AD copathology at autopsy [8].
Both clinical and pathological heterogeneity is poorly understood. By identifying the underlying biological contributions to the heterogeneity between DLB patients we may improve individualized prognosis and personalized treatment [9]. A contributor to heterogeneity within DLB might be its underlying genetic background. The heritability of DLB has been estimated to be 60%[10]. Genome-wide association studies (GWAS) have identified individual variants near or in the APOE (Apolipoprotein E), GBA (glucocerebrosidase), and SNCA (Alpha-synuclein) genes to be consistently associated with DLB [11–17]. Therefore, it is conceivable that these variants also explain part of the clinical and pathological heterogeneity within DLB. Indeed, neuropathological series suggest GBA and APOE are associated with a more aggressive disease course [13, 18]. APOE has been associated with more frequent amyloid pathology, while GBA variants were more often observed in DLB without AD pathology (‘pure’ DLB) [6, 19, 20]. Next to these proven DLB variants, we might study the underlying genetic background of DLB by utilizing the findings from large GWAS of AD and Parkinson’s disease (PD), as these have clinical and pathological features that overlap with DLB. The most recent GWAS, showed that 39 genomic loci (next to APOE) associated with AD [21] and over 90 genomic loci associated with PD [22]. The relatively small effects of these genomic loci can be combined in polygenic risk scores (PRS) for AD and PD. Indeed, a PRS for AD has reported to be associated with autopsy confirmed Lewy-body pathology [23]. However, this association has not been replicated and it is unclear whether this is also true for a PD genetic risk score and if the scores associate with clinical and pathological heterogeneity.
Here we studied the effects of DLB-associated genetic risk factors in relation to concomitant AD pathology, clinical features of DLB and disease duration in a clinical cohort of DLB patients. We aimed to study the associations of the three established genetic DLB risk factors (APOE, GBA, SNCA) and PRSs for AD/PD with; 1) the risk of DLB in the presence (and absence) of concomitant AD pathology, and 2) age at diagnosis, cognitive performance, the core clinical features of DLB and disease duration).
METHODS
Study population
We included DLB patients who visited the Alzheimer Center Amsterdam between 2001 and 2018 from the Amsterdam Dementia cohort (ADC), with available genotyping data [24]. The diagnosis of DLB was made in a multidisciplinary consensus meeting according to the clinical diagnostic consensus criteria for probable DLB [1, 2]. As part of the clinical diagnostic work-up, all patients had received an extensive standardized and multidisciplinary work-up, including medical history, physical and neurological examinations, neuropsychological testing, electroencephalography (EEG) or magnetoencephalography (MEG), brain magnetic resonance imaging (MRI) and laboratory tests. We identified 197 patients with probable DLB in the ADC; genome-wide array was available for 190 (96%) and these were included in the study. Of the 190 patients, 154 (81%) had AD biomarker level results in CSF available (Aβ1–42, total tau, and p-tau). A (123)I-FP-CIT-SPECT (DAT-SPECT) was performed at indication of the clinician in 89 (47%) of the DLB patients and 80 (90%) of these scans showed presynaptic dopaminergic deficits and were rated as abnormal. We compared the DLB patients with 2,552 cognitively normal controls of whom genome-wide array data were available. Controls originated from three sources. We included 867 subjects who visited the Alzheimer Center Amsterdam with subjective concerns of cognitive decline, but who showed no abnormalities on clinical or cognitive testing and did not fulfill criteria for mild cognitive impairment, dementia, or other medical conditions potentially causing cognitive decline (i.e., cognitively normal) [24]. Second, population controls from the Longitudinal Aging Study Amsterdam (LASA, n = 1,648) [25, 26]. Third, individuals of a subset of the Netherlands Brain Bank (NBB, http://www.brainbank.nl, n = 37) without diagnosis of neurodegenerative disease at autopsy. Supplementary Table 1 shows the demographics of the three control cohorts. All studies were approved by the local medical ethics committee and all subjects gave written informed consent for the use of their clinical, biochemical, and genetic data for research purposes.
Genotyping
Genetic variants were determined as previously described by standard imputation methods [27]. In brief, all cases and controls were genotyped using the Illumina Global Screening Array and we applied established quality control methods [28]. We used for imputation only high-quality genotyping (variant call rate > 98%) in all individuals (individual call rate > 98%) and variants departing from Hardy–Weinberg equilibrium were removed (p < 1×10–6). We removed individuals with sex mismatches, individuals of non-European ancestry (based on 1000 Genomes) [29] and removed one individual from pairs of individuals that have a family relation (identity-by-descent≥0.3) [30]. Genotypes were prepared for imputation using provided scripts (HRC-1000G-check-bim.pl, https://www.well.ox.ac.uk/∼wrayner/tools/) [31]. This script compares variant ID, strand and allele frequencies to the haplotype reference panel (HRC v1.1, April 2016) [31]. Finally, all autosomal variants were submitted to the Michigan imputation server [28]. The server used SHAPEIT2 (v2.r790) to phase data and imputation to the reference panel (v1.1) was performed with Minimac3 [28] and the variants of interest were extracted.
Genetic variants associated with DLB and polygenic risk scores
We selected three previously described genetic risk factors that were associated with DLB [11–16]: rs429358 (NC_000019.9:g.45411941T > C, determines the APOE ɛ4 allele, imputation quality = 0.99) in APOE, rs2230288 (NC_000001.10:g.155206167C > T or p.E365K, imputation quality = 0.96) in GBA, rs7681440 (NC_000004.11:g.90756550C > G, imputation quality = 0.98) near SNCA. The variant near BCL7C/ STX1B (rs897984) was considered, but not selected as it did not associate with DLB in the replication phase, despite adequate sample size.
Polygenic risk scores (PRS)
We calculated a weighted PRS for AD, based on the 39 genetic variants that showed genome-wide significant (GWS) evidence of association with AD [21] (Supplementary Table 2). APOE variants were excluded from the AD-PRS. The weighted PRS for PD was based on 90 genetic variants that showed GWS evidence of association with PD [22] (Supplementary Table 3). For the PRS for PD we excluded variants in or near SNCA (rs356182, rs5019538) and GBA (rs35749011, rs76763715), as variants near these genes have been associated with DLB. The PRSs were generated by multiplying the genotype dosage of each risk allele for each variant by its respective weight and then summing across all variants. The PRSs were normalized (mean = 0, standard deviation = 1). The results odds ratios (OR) or hazard ratios (HR) can be interpreted as the difference per one standard deviation increase in the PRS. The selected variants were directly genotyped or imputed with high quality (median imputation score R2 = 0.98).
Clinical measures in DLB patients
The age at diagnosis was fixed at the age a person was first diagnosed with probable DLB. Global cognitive functioning was measured by the Mini-Mental State Examination (MMSE, score from 0 to 30) [32]. DLB core features were (re-)assessed according to the McKeith 2017 criteria at the time of diagnosis [2]. The presence of visual hallucinations was assessed with the Neuropsychiatric Inventory [33]. The presence of parkinsonism was assessed by a preformatted checklist of the neurological exam scoring on the presence of bradykinesia, rigidity, and/or tremor. The presence of fluctuations and RBD was assessed by reviewing patient’s medical charts by two independent raters. Fluctuations were rated positively when patient or caregivers reported that the patients’ attention fluctuated during the day and over the weeks. RBD was rated positively when caregivers reported that the patient seem to ‘act out’ their dreams and if the patient moves extensively during sleep.
Concomitant AD pathology
Concomitant AD pathology in DLB patients, hereinafter referred to as DLB-AD, was defined as a ratio of CSF phosphorylated tau (Ptau)/Aβ1–42≥0.054 (Willemse EAJ, in preparation). CSF was obtained by lumbar puncture between the L3/L4, L4/L5, or L5/S1 intervertebral space using a 25-gauge needle and a syringe and collected into 10 mL polypropylene tubes (Sarstedt, Nümbrecht, Germany), following the international biobanking consensus guidelines for CSF [34]. CSF was routinely analyzed for levels of Aβ1–42, total tau, and p-tau with commercial Enzyme-linked immunosorbent assays (ELISA) (Innotest®, Fujirebio, Gent, Belgium). Aβ1–42 measures were adjusted for an upward drift over time as previously described [35]. Of the 190 patients with DLB, 154 (81%) patients had CSF measures of Aβ1–42 and total tau.
Mortality
For each DLB patient all-cause mortality information (died yes/no, and date of death) was collected using the Dutch municipal population register (until May 1, 2020). We defined survival time as the time (in years) between the year of the patient’s diagnosis, and either the date of death, or May 1, 2020 for alive patients.
Statistical analyses
We assumed additive genetic effects for all variants. Variant effects were reported for the allele that increases the risk on DLB according to literature (for SNCA this is not the minor allele). All analyses were adjusted for 5 ancestry components to control for confounding by population stratification [36]. First, we performed a case-control analysis to confirm the association with risk for DLB and associate the AD-PRS and PD-PRS with DLB. We compared all DLB cases with controls, calculated OR and estimated 95%confidence intervals (CI) using logistic regression models. Then, we split our case group in DLB-AD and pure-DLB and re-calculated OR and CIs using logistic regression models for both subgroups separately. Subsequently, we studied the effects of the genetic variants on clinical features within the DLB cases. With linear regression models we tested the association of the genetic variants on age at onset and MMSE. With logistic regression models we tested the association of the genetic variants and PRSs with the four core features of DLB. With Cox proportional hazards models we tested the effect on survival as time (in years) from DLB onset to death, while adjusting for age at diagnosis. Finally, we tested for statistical interaction (multiplicative) between the clinical features (age at onset, MMSE, core symptoms, and survival) and concomitant AD pathology. Associations with a Pfdr < 0.05 after correction for multiple testing using a false discovery rate (fdr) [37] were considered significant. All analyses were performed in R (verion 3.6.0), the ‘survival’ package (version 2.44-1.1) was used for Cox proportional hazard models and the package ‘forestplot’ was used to create the forest plots.
RESULTS
Study population
Demographic, clinical characteristics, and CSF biomarker values/measures of cases and controls are presented in Table 1. The 190 patients with DLB had an average age at diagnosis of 69 years (SD = 7), and 81%of the patients were male; therefore, sex was included as covariate in all analyses. In the 157 cases an AD biomarker profile was present. A profile indicating the presence of AD (DLB-AD) was present in 85 (54%) and 72 (46%) DLB patients did not have an AD biomarker profile and therefore classified as DLB-pure.
Table 1
Characteristics of the study population
| DLB (all, n = 190) | Controls (n = 2,552) | |
| Sex, n (%) | ||
| Male | 155 (81.6%) | 1318 (51.6%) |
| Female | 35 (18.4%) | 1234 (48.4) |
| Agea, mean (SD) | 69.0 (6.8) | 62 (7.9) |
| MMSE, mean (SD) | 22.5 (4.7) | 28 (2) |
| DLB symptoms, n (%) | ||
| Visual hallucinations | 122 (65.2%) | |
| Parkinsonism | 132 (71.7%) | |
| Fluctuations | 134 (80.7%) | |
| RBD | 100 (69.9%) | |
| CSF markers, mean (SD)b | ||
| Aβ42 (pg/ml) | 828 (238) | 1087 (236) |
| tau (pg/ml) | 352 (201) | 282 (159) |
| p-tau (pg/ml) | 52.1 (23.6) | 47.6 (21) |
| With AD biomarker profile (%) | 82 (53%) | 130 (19.8%) |
| Survival (DLB patients only) | ||
| Dead (n, %) | 118 (69.8%) | |
| Follow-up time, years | 4.7 (2.2) |
Data are presented as mean (SD) or n (%). aAge at inclusion for controls and age at diagnosis for cases. bCSF available for 656 controls (26%) and 154 (81%) DLB patients. Aβ42, amyloid β1–42; CSF, cerebrospinal fluid; DLB, dementia with Lewy bodies; MMSE, Mini-Mental State Examination; N/A, not applicable; p-tau, tau phosphorylated at threonine 181; RBD, rapid eye movement (REM) sleep behavior disorder.
Association with risk of DLB, DLB-AD, and DLB-pure
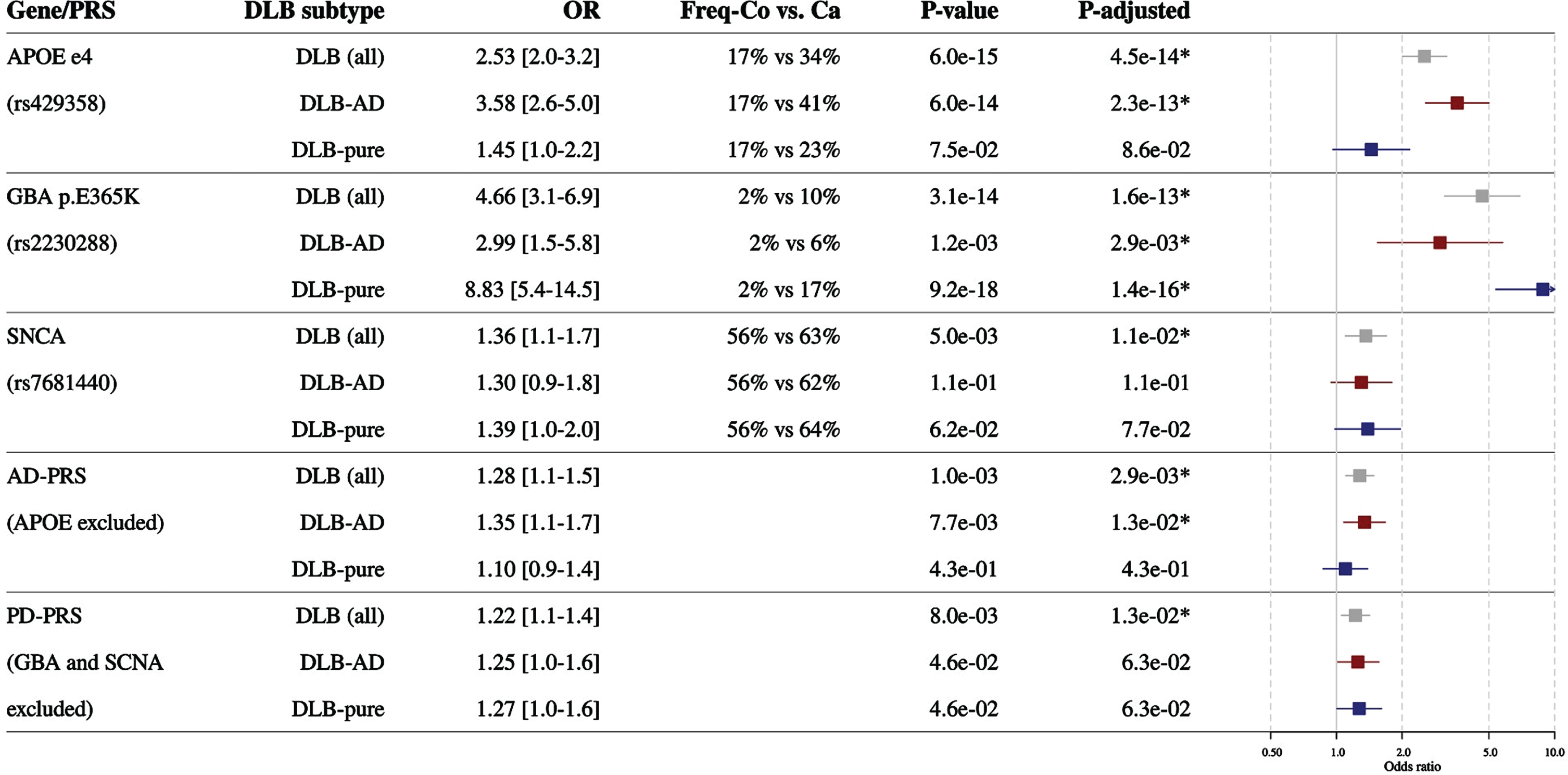
Also in our cohort the three DLB variants were associated with an increased risk of DLB (Fig. 1): APOE ɛ4 (OR = 2.5, pfdr = 4.5×10–14), GBA (OR = 4.7, pfdr = 1.6×10–13), and SNCA (OR = 1.4, pfdr = 1.1×10–2). We show that a both a higher genetic risk for AD (the AD-PRS) as well as a higher genetic risk for PD (the PD-PRS) was associated with an increased risk for DLB. The AD-PRS increased the odds for DLB with 1.3 per 1-SD increase in the PRS (pfdr = 2.9×10–2), PD-PRS increased the odds for DLB with 1.2 per 1-SD increase in the PRS (pfdr = 1.3×10–2). When we subsequently analysed DLB-AD and DLB-pure separately, we found strong differential effects for APOE ɛ4 and GBA on the risk for DLB-AD and DLB-pure (Fig. 1). The risk of APOE ɛ4 allele was associated with a 3.6-fold (pfdr = 2.3×10–13) increased risk of DLB-AD, compared to 1.5-fold increased risk of DLB-pure, which was not significant (pfdr = 8.6×10–2). In contrast, GBA E365K associated with an 8.8-fold (pfdr = 1.4×10–16) increased risk of DLB-pure, compared to a 3-fold (pfdr = 2.9×10–3) increased risk with DLB-AD. The AD-PRS only associated with DLB-AD (OR = 1.4, pfdr = 1.3×10–2), but not with DLB-pure. For SNCA and the PD-PRS the effects were comparable in DLB-AD and DLB-pure (Fig. 1). All allele counts and frequencies in all groups are reported in Supplementary Table 4. Important to note is that there were no major differences in the frequencies of the variants between the different control populations. All single variant associations from the AD-PRS and PD-PRS are in Supplementary Tables 5 and 6. We screened the single variant effects for interesting insights. In the AD-PRS no single variant association stood out. In the PD-PRS the genes associated with the two most significant single variants were involved in the same biological process. These were rs6825004 intronic to Scavenger Receptor Class B Member 2 (SCARB2) and a missense p.Met311Thr in Transmembrane Protein 175 (TMEM175). For both variants, the alleles that increased PD risk showed larger OR for DLB than for PD; for SCARB2 ORDLB = 1.76 versus ORPD = 1.06 and for TMEM175 ORDLB = 1.24 versus ORPD = 1.49. This is interesting as both SCARB2 and TMEM175 are genes of which the protein product is involved in lysosome functioning.
Fig. 1
Association of genetic factors with the risk of dementia with risk variants with all DLB (grey), DLB with concomitant AD pathology (DLB-AD, red), and DLB without concomitant AD pathology (DLB-pure, blue). The allele frequency in cases and controls is shown (note this is not the same as carrier frequency). Effects are calculated by comparing the different DLB case groups to the same group of controls. DLB, dementia with Lewy bodies; DLB-AD, DLB with concomitant AD pathology; DLB-pure, DLB without concomitant AD pathology; APOE, Apolipoprotein E; GBA, Glucocerebrosidase; PRS, polygenic risk score; SNCA, Alpha-synuclein; OR, odds ratio.
Association with clinical features of DLB
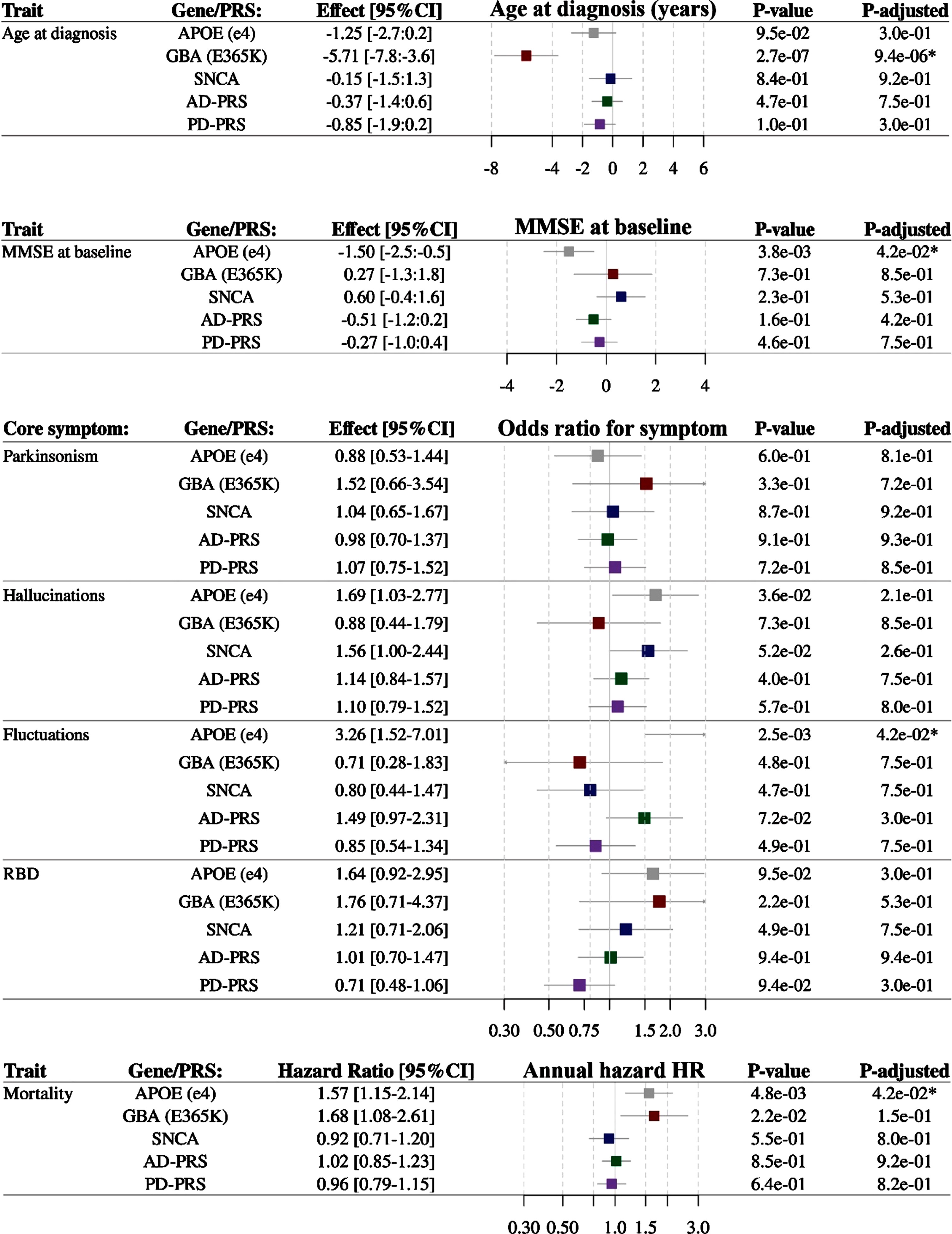
Associations between genetic variants and clinical features of DLB are shown in Fig. 2. There was a strong association between GBA and age at diagnosis of disease. Carriers of the GBA risk variant had a 5.7-year earlier age at diagnosis of DLB per risk allele (95%CI 3.6 to 7.8 years, pfdr = 9.4×10–6). MMSE at time of diagnosis was 1.5 point lower in APOE ɛ4 carriers (pfdr = 4.2×10–2). APOE ɛ4 carriers were more likely to experience fluctuations as a core symptom (OR = 3.3, pfdr 4.2×10–2). None of the genetic variants associated with RBD, hallucinations or parkinsonism (pfdr < 0.05). The APOE ɛ4 allele predisposed for a 1.6-fold increased risk of mortality (pfdr = 4.2×10–2, Fig. 2). Finally, we tested for interaction effect of the genetic variants with concomitant AD pathology (Supplementary Table 7) on the clinical measures, but there were no significant interactions.
Fig. 2
Association of genetic factors with; age at diagnosis (years), Mini-Mental State Examination (MMSE) at baseline (points on a 0–30 scale), presence of core symptoms of DLB (odds to have the symptom at diagnosis) and mortality after diagnosis (annualized hazard ratio). APOE, Apolipoprotein E; GBA, Glucocerebrosidase; SNCA, Alpha-synuclein; MMSE, Mini-Mental State Examination; PRS, polygenic risk score; OR, odds ratio; HR, hazard ratio.
DISCUSSION
Better understanding the genetics underlying DLB is important to understand the clinical presentation and predict the disease course. Here, we showed that genetic factors known to be associated with risk of DLB, are also important drivers of pathological and clinical heterogeneity in a clinical DLB cohort. GBA specifically predisposed for DLB-pure and earlier onset, while APOE predisposed for DLB-AD, lower MMSE, and a more progressive disease course. This suggests that APOE and GBA differentially affect the biological processes that lead to DLB. In addition, we found that the polygenic risk for AD and PD both increased the risk for DLB emphasizing the role of to-be-discovered genetic factors in DLB. The association of the AD-PRS was stronger with DLB-AD, but there was no association with specific clinical features.
We extend previous findings in postmortem series of the association of APOE and GBA with DLB in the presence or absence of AD pathology, by analyzing the CSF and measuring AD pathology in vivo. In this large cohort of DLB patients, the GBA missense mutation (p.E365K) was a strong risk factor for DLB-pure and less so for DLB-AD. The 8.8-fold risk increase for DLB-pure is very high (usually in the range 1.1 to 1.5) [38] for a relatively common variant (2%of the European ancestry populations carries the variant) making this variant a major contributor to pure DLB in the general population. It is likely that the contribution of variants in GBA is even larger because other GBA variants were missed as we did not use sequencing techniques. Previously, APOE was shown a risk factor for AD-DLB as well as pure DLB [6, 39]. We confirm this association but note that APOE ɛ4 was only very weakly associated with DLB-pure (OR = 1.5). The smaller effect for APOE ɛ4 for pure DLB compared to previous estimates might be due to the fact that we assessed AD pathology earlier in the disease course. A stronger effect might be expected in pathological studies if APOE causes AD pathology later in the DLB disease course. The strong differential risks of GBA and APOE variants in relation to the presence of amyloid pathology warrant that future gene–discovery studies should consider stratified analyses for DLB-AD and DLB-pure to discover genetic factors that only effect one of both. Of interest is our observation that the single variants from the PD-PRS most strongly associated with all DLB were variants near two genes (TMEM175 and SCARB2) both involved in lysosomal function/reordering and the larger OR for DLB compared to PD suggests that PD-genes that are involved in lysosome function also play an important role in the development of DLB.
In addition to modifications of disease risk our study shows that the genetic variants influence clinical features of DLB patients. APOE ɛ4 showed most effects; APOE ɛ4 associated with lower MMSE, more fluctuations, and shorter survival after diagnosis. The association with survival after diagnosis is a replication of previous findings [18, 40]. It is conceivable that the observed shorter survival is due to the higher prevalence of concomitant AD pathology conferred by APOE ɛ4, which in turn leads to shorter survival. However, we did not observe an interaction with AD pathology, and therefore these results support that APOE has an independent effect on formation of alpha-synuclein pathology [19, 39], which leads to a more progressive disease course. We have previously reported a lower MMSE at baseline in the presence of amyloid pathology [7, 41], which could explain the effect of APOE ɛ4 on MMSE. However, we found no interaction between APOE ɛ4 and concomitant AD pathology in this study, suggesting the effect of APOE ɛ4 on MMSE is independent of AD pathology [42]. More investigations are necessary to confirm these associations. With respect to the core symptoms of DLB, we only found effects on fluctuations (with APOE ɛ4) which in turn are highly correlated with cognitive function. Only the GBA variant had an effect on age at diagnosis; it reduced the age by almost 6 years, which is in line with an earlier report [13]. Of note, the four DLB patients who carried two copies of GBA p.E365K had an average onset age of 51 years, suggesting the age effect is larger for homozygote carriers. PD carriers of GBA mutations also have an earlier onset, but they are also more likely to have higher UPDRS-III scores, develop dementia faster, and have a shorter time until dopamine wearing-off phenomena [43, 44]. Our findings in DLB are in contrast with these findings in PD, as in our DLB patients, the GBA variant did not associate with other clinical measures.
Limitations
The main strength of this study is the CSF measures and structured assessment of the patients with DLB. A total of 190 patients is low in absolute numbers of patients for genetic association studies, yet we were able to replicate the associations with DLB of all genetic factors, possibly because all underwent standardized work up with comprehensive clinical assessment and extensive diagnostics. A limitation is that our ascertainment of some of the clinical core symptoms was retrospective from patient medical records as not all patients were systematically assessed for the presence/absence of the four core symptoms. For example, the use of standardized rating scales for fluctuations and RBD might have increased the chance of finding associations. A last limitation is that the patients were recruited entirely in a memory clinic, creating a possible selection bias in the type of DLB-patients that were included [45].
ACKNOWLEDGMENTS
Research of Alzheimer center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. Wiesje van der Flier holds the Pasman chair. The clinical database structure was developed with funding from Stichting Dioraphte. Genotyping of the ADC samples was performed in the context of EADB (European Alzheimer DNA biobank) funded by the JPco-fuND FP-829-029 (ZonMW projectnumber 733051061). DLB-specific research was further funded by the Scientific Excellence Program of Amsterdam Neuroscience, the Memorabel grant programme of the Netherlands Organisation for Health Research and Development (ZonMW grant 733050509), and Stichting Alzheimer Nederland. The Longitudinal Aging Study Amsterdam is supported by a grant from the Netherlands Ministry of Health Welfare and Sports, Directorate of Long-Term Care. The work of S.J. van der Lee is supported by ZonMW-Memorabel (grant number 73305012). S.J. van der Lee, Wiesje M. van der Flier, and Henne H. Holstege are recipient of ABOARD, which is a public-private partnership receiving funding from ZonMW (#73305095007) and Health Holland, Topsector Life Sciences & Health (PPP-allowance; #LSHM20106). More than 30 partners participate in ABOARD. ABOARD also receives funding from Edwin Bouw Fonds and Gieskes-Strijbisfonds.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/21-0365r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-210365.
REFERENCES
[1] | McKeith IG , Dickson DW , Lowe J , Emre M , O’Brien JT , Feldman H , Cummings J , Duda JE , Lippa C , Perry EK , Aarsland D , Arai H , Ballard CG , Boeve B , Burn DJ , Costa D , Del Ser T , Dubois B , Galasko D , Gauthier S , Goetz CG , Gomez-Tortosa E , Halliday G , Hansen LA , Hardy J , Iwatsubo T , Kalaria RN , Kaufer D , Kenny RA , Korczyn A , Kosaka K , Lee VM , Lees A , Litvan I , Londos E , Lopez OL , Minoshima S , Mizuno Y , Molina JA , Mukaetova-Ladinska EB , Pasquier F , Perry RH , Schulz JB , Trojanowski JQ , Yamada M , Consortium on DLB ((2005) ) Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 65: , 1863–1872. |
[2] | McKeith IG , Boeve BF , Dickson DW , Halliday G , Taylor JP , Weintraub D , Aarsland D , Galvin J , Attems J , Ballard CG , Bayston A , Beach TG , Blanc F , Bohnen N , Bonanni L , Bras J , Brundin P , Burn D , Chen-Plotkin A , Duda JE , El-Agnaf O , Feldman H , Ferman TJ , Ffytche D , Fujishiro H , Galasko D , Goldman JG , Gomperts SN , Graff-Radford NR , Honig LS , Iranzo A , Kantarci K , Kaufer D , Kukull W , Lee VMY , Leverenz JB , Lewis S , Lippa C , Lunde A , Masellis M , Masliah E , McLean P , Mollenhauer B , Montine TJ , Moreno E , Mori E , Murray M , O’Brien JT , Orimo S , Postuma RB , Ramaswamy S , Ross OA , Salmon DP , Singleton A , Taylor A , Thomas A , Tiraboschi P , Toledo JB , Trojanowski JQ , Tsuang D , Walker Z , Yamada M , Kosaka K ((2017) ) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: , 88–100. |
[3] | Spillantini MG , Crowther RA , Jakes R , Hasegawa M , Goedert M ((1998) ) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95: , 6469–6473. |
[4] | Vekrellis K , Xilouri M , Emmanouilidou E , Rideout HJ , Stefanis L ((2011) ) Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol 10: , 1015–1025. |
[5] | Howlett DR , Whitfield D , Johnson M , Attems J , O’Brien JT , Aarsland D , Lai MK , Lee JH , Chen C , Ballard C , Hortobagyi T , Francis PT ((2015) ) Regional multiple pathology scores are associated with cognitive decline in Lewy body dementias. Brain Pathol 25: , 401–408. |
[6] | Irwin DJ , Grossman M , Weintraub D , Hurtig HI , Duda JE , Xie SX , Lee EB , Van Deerlin VM , Lopez OL , Kofler JK , Nelson PT , Jicha GA , Woltjer R , Quinn JF , Kaye J , Leverenz JB , Tsuang D , Longfellow K , Yearout D , Kukull W , Keene CD , Montine TJ , Zabetian CP , Trojanowski JQ ((2017) ) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol 16: , 55–65. |
[7] | van Steenoven I , Aarsland D , Weintraub D , Londos E , Blanc F , van der Flier WM , Teunissen CE , Mollenhauer B , Fladby T , Kramberger MG , Bonanni L , Lemstra AW , European DLB consortium ((2016) ) Cerebrospinal fluid Alzheimer’s disease biomarkers across the spectrum of Lewy body diseases: Results from a large multicenter cohort. J Alzheimers Dis 54: , 287–295. |
[8] | Irwin DJ , Xie SX , Coughlin D , Nevler N , Akhtar RS , McMillan CT , Lee EB , Wolk DA , Weintraub D , Chen-Plotkin A , Duda JE , Spindler M , Siderowf A , Hurtig HI , Shaw LM , Grossman M , Trojanowski JQ ((2018) ) CSF tau and beta-amyloid predict cerebral synucleinopathy in autopsied Lewy body disorders. Neurology 90: , e1038–e1046. |
[9] | Coughlin DG , Hurtig HI , Irwin DJ ((2020) ) Pathological influences on clinical heterogeneity in Lewy body diseases. Mov Disord 35: , 5–19. |
[10] | Guerreiro R , Escott-Price V , Hernandez DG , Kun-Rodrigues C , Ross OA , Orme T , Neto JL , Carmona S , Dehghani N , Eicher JD , Shepherd C , Parkkinen L , Darwent L , Heckman MG , Scholz SW , Troncoso JC , Pletnikova O , Dawson T , Rosenthal L , Ansorge O , Clarimon J , Lleo A , Morenas-Rodriguez E , Clark L , Honig LS , Marder K , Lemstra A , Rogaeva E , St George-Hyslop P , Londos E , Zetterberg H , Barber I , Braae A , Brown K , Morgan K , Troakes C , Al-Sarraj S , Lashley T , Holton J , Compta Y , Van Deerlin V , Serrano GE , Beach TG , Lesage S , Galasko D , Masliah E , Santana I , Pastor P , Diez-Fairen M , Aguilar M , Tienari PJ , Myllykangas L , Oinas M , Revesz T , Lees A , Boeve BF , Petersen RC , Ferman TJ , Graff-Radford N , Cairns NJ , Morris JC , Pickering-Brown S , Mann D , Halliday GM , Hardy J , Trojanowski JQ , Dickson DW , Singleton A , International Parkinson’s Disease Genomics Consortium, Stone DJ , Bras J ((2019) ) Heritability and genetic variance of dementia with Lewy bodies. Neurobiol Dis 127: , 492–501. |
[11] | Bras J , Guerreiro R , Darwent L , Parkkinen L , Ansorge O , Escott-Price V , Hernandez DG , Nalls MA , Clark LN , Honig LS , Marder K , Van Der Flier WM , Lemstra A , Scheltens P , Rogaeva E , St George-Hyslop P , Londos E , Zetterberg H , Ortega-Cubero S , Pastor P , Ferman TJ , Graff-Radford NR , Ross OA , Barber I , Braae A , Brown K , Morgan K , Maetzler W , Berg D , Troakes C , Al-Sarraj S , Lashley T , Compta Y , Revesz T , Lees A , Cairns N , Halliday GM , Mann D , Pickering-Brown S , Dickson DW , Singleton A , Hardy J ((2014) ) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 23: , 6139–6146. |
[12] | Hardy J , Crook R , Prihar G , Roberts G , Raghavan R , Perry R ((1994) ) Senile dementia of the Lewy body type has an apolipoprotein E epsilon 4 allele frequency intermediate between controls and Alzheimer’s disease. Neurosci Lett 182: , 1–2. |
[13] | Nalls MA , Duran R , Lopez G , Kurzawa-Akanbi M , McKeith IG , Chinnery PF , Morris CM , Theuns J , Crosiers D , Cras P , Engelborghs S , De Deyn PP , Van Broeckhoven C , Mann DM , Snowden J , Pickering-Brown S , Halliwell N , Davidson Y , Gibbons L , Harris J , Sheerin UM , Bras J , Hardy J , Clark L , Marder K , Honig LS , Berg D , Maetzler W , Brockmann K , Gasser T , Novellino F , Quattrone A , Annesi G , De Marco EV , Rogaeva E , Masellis M , Black SE , Bilbao JM , Foroud T , Ghetti B , Nichols WC , Pankratz N , Halliday G , Lesage S , Klebe S , Durr A , Duyckaerts C , Brice A , Giasson BI , Trojanowski JQ , Hurtig HI , Tayebi N , Landazabal C , Knight MA , Keller M , Singleton AB , Wolfsberg TG , Sidransky E ((2013) ) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70: , 727–735. |
[14] | Guerreiro R , Ross OA , Kun-Rodrigues C , Hernandez DG , Orme T , Eicher JD , Shepherd CE , Parkkinen L , Darwent L , Heckman MG , Scholz SW , Troncoso JC , Pletnikova O , Ansorge O , Clarimon J , Lleo A , Morenas-Rodriguez E , Clark L , Honig LS , Marder K , Lemstra A , Rogaeva E , St George-Hyslop P , Londos E , Zetterberg H , Barber I , Braae A , Brown K , Morgan K , Troakes C , Al-Sarraj S , Lashley T , Holton J , Compta Y , Van Deerlin V , Serrano GE , Beach TG , Lesage S , Galasko D , Masliah E , Santana I , Pastor P , Diez-Fairen M , Aguilar M , Tienari PJ , Myllykangas L , Oinas M , Revesz T , Lees A , Boeve BF , Petersen RC , Ferman TJ , Escott-Price V , Graff-Radford N , Cairns NJ , Morris JC , Pickering-Brown S , Mann D , Halliday GM , Hardy J , Trojanowski JQ , Dickson DW , Singleton A , Stone DJ , Bras J ((2018) ) Investigating the genetic architecture of dementia with Lewy bodies: A two-stage genome-wide association study. Lancet Neurol 17: , 64–74. |
[15] | Mata IF , Samii A , Schneer SH , Roberts JW , Griffith A , Leis BC , Schellenberg GD , Sidransky E , Bird TD , Leverenz JB , Tsuang D , Zabetian CP ((2008) ) Glucocerebrosidase gene mutations: A risk factor for Lewy body disorders. Arch Neurol 65: , 379–382. |
[16] | Rongve A , Witoelar A , Ruiz A , Athanasiu L , Abdelnour C , Clarimon J , Heilmann-Heimbach S , Hernandez I , Moreno-Grau S , de Rojas I , Morenas-Rodriguez E , Fladby T , Sando SB , Brathen G , Blanc F , Bousiges O , Lemstra AW , van Steenoven I , Londos E , Almdahl IS , Palhaugen L , Eriksen JA , Djurovic S , Stordal E , Saltvedt I , Ulstein ID , Bettella F , Desikan RS , Idland AV , Toft M , Pihlstrom L , Snaedal J , Tarraga L , Boada M , Lleo A , Stefansson H , Stefansson K , Ramirez A , Aarsland D , Andreassen OA ((2019) ) GBA and APOE epsilon4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Sci Rep 9: , 7013. |
[17] | Orme T , Hernandez D , Ross OA , Kun-Rodrigues C , Darwent L , Shepherd CE , Parkkinen L , Ansorge O , Clark L , Honig LS , Marder K , Lemstra A , Rogaeva E , St George-Hyslop P , Londos E , Zetterberg H , Morgan K , Troakes C , Al-Sarraj S , Lashley T , Holton J , Compta Y , Van Deerlin V , Trojanowski JQ , Serrano GE , Beach TG , Lesage S , Galasko D , Masliah E , Santana I , Pastor P , Tienari PJ , Myllykangas L , Oinas M , Revesz T , Lees A , Boeve BF , Petersen RC , Ferman TJ , Escott-Price V , Graff-Radford N , Cairns NJ , Morris JC , Pickering-Brown S , Mann D , Halliday G , Stone DJ , Dickson DW , Hardy J , Singleton A , Guerreiro R , Bras J ((2020) ) Analysis of neurodegenerative disease-causing genes in dementia with Lewy bodies. Acta Neuropathol Commun 8: , 5. |
[18] | Keogh MJ , Kurzawa-Akanbi M , Griffin H , Douroudis K , Ayers KL , Hussein RI , Hudson G , Pyle A , Cordell HJ , Attems J , McKeith IG , O’Brien JT , Burn DJ , Morris CM , Thomas AJ , Chinnery PF ((2016) ) Exome sequencing in dementia with Lewy bodies. Transl Psychiatry 6: , e728. |
[19] | Dickson DW , Heckman MG , Murray ME , Soto AI , Walton RL , Diehl NN , van Gerpen JA , Uitti RJ , Wszolek ZK , Ertekin-Taner N , Knopman DS , Petersen RC , Graff-Radford NR , Boeve BF , Bu G , Ferman TJ , Ross OA ((2018) ) APOE epsilon4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 91: , e1182–e1195. |
[20] | Tsuang D , Leverenz JB , Lopez OL , Hamilton RL , Bennett DA , Schneider JA , Buchman AS , Larson EB , Crane PK , Kaye JA , Kramer P , Woltjer R , Kukull W , Nelson PT , Jicha GA , Neltner JH , Galasko D , Masliah E , Trojanowski JQ , Schellenberg GD , Yearout D , Huston H , Fritts-Penniman A , Mata IF , Wan JY , Edwards KL , Montine TJ , Zabetian CP ((2012) ) GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 79: , 1944–1950. |
[21] | de Rojas I , Moreno-Grau S , Tesi N , Grenier-Boley B , Andrade V , Jansen IE , Pedersen NL , Stringa N , Zettergren A , Hernández I , et al. ((2021) ) Common variants in Alzheimer’s disease and risk stratification by polygenic risk scores. Nat Commun 12: , 3417. |
[22] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln R , Simon-Sanchez J , Schulte C , Sharma M , Krohn L , Pihlstrom L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB , 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium ((2019) ) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol 18: , 1091–1102. |
[23] | Tan CH , Bonham LW , Fan CC , Mormino EC , Sugrue LP , Broce IJ , Hess CP , Yokoyama JS , Rabinovici GD , Miller BL , Yaffe K , Schellenberg GD , Kauppi K , Holland D , McEvoy LK , Kukull WA , Tosun D , Weiner MW , Sperling RA , Bennett DA , Hyman BT , Andreassen OA , Dale AM , Desikan RS , Alzheimer’s Disease Neuroimaging Initiative ((2019) ) Polygenic hazard score, amyloid deposition and Alzheimer’s neurodegeneration. Brain 142: , 460–470. |
[24] | van der Flier WM , Scheltens P ((2018) ) Amsterdam Dementia Cohort: Performing research to optimize care. J Alzheimers Dis 62: , 1091–1111. |
[25] | Hoogendijk EO , Deeg DJ , Poppelaars J , van der Horst M , Broese van Groenou MI , Comijs HC , Pasman HR , van Schoor NM , Suanet B , Thomese F , van Tilburg TG , Visser M , Huisman M ((2016) ) The Longitudinal Aging Study Amsterdam: Cohort update 2016 and major findings. Eur J Epidemiol 31: , 927–945. |
[26] | Huisman M , Poppelaars J , van der Horst M , Beekman AT , Brug J , van Tilburg TG , Deeg DJ ((2011) ) Cohort profile: The Longitudinal Aging Study Amsterdam. Int J Epidemiol 40: , 868–876. |
[27] | Tesi N , van der Lee SJ , Hulsman M , Jansen IE , Stringa N , van Schoor N , Meijers-Heijboer H , Huisman M , Scheltens P , Reinders MJT , van der Flier WM , Holstege H ((2019) ) Centenarian controls increase variant effect sizes by an average twofold in an extreme case-extreme control analysis of Alzheimer’s disease. Eur J Hum Genet 27: , 244–253. |
[28] | Das S , Forer L , Schonherr S , Sidore C , Locke AE , Kwong A , Vrieze SI , Chew EY , Levy S , McGue M , Schlessinger D , Stambolian D , Loh PR , Iacono WG , Swaroop A , Scott LJ , Cucca F , Kronenberg F , Boehnke M , Abecasis GR , Fuchsberger C ((2016) ) Next-generation genotype imputation service and methods. Nat Genet 48: , 1284–1287. |
[29] | Genomes Project C , Auton A , Brooks LD , Durbin RM , Garrison EP , Kang HM , Korbel JO , Marchini JL , McCarthy S , McVean GA , Abecasis GR ((2015) ) A global reference for human genetic variation. Nature 526: , 68–74. |
[30] | Anderson CA , Pettersson FH , Clarke GM , Cardon LR , Morris AP , Zondervan KT ((2010) ) Data quality control in genetic case-control association studies. Nat Protoc 5: , 1564–1573. |
[31] | McCarthy S , Das S , Kretzschmar W , Delaneau O , Wood AR , Teumer A , Kang HM , Fuchsberger C , Danecek P , Sharp K , Luo Y , Sidore C , Kwong A , Timpson N , Koskinen S , Vrieze S , Scott LJ , Zhang H , Mahajan A , Veldink J , Peters U , Pato C , van Duijn CM , Gillies CE , Gandin I , Mezzavilla M , Gilly A , Cocca M , Traglia M , Angius A , Barrett JC , Boomsma D , Branham K , Breen G , Brummett CM , Busonero F , Campbell H , Chan A , Chen S , Chew E , Collins FS , Corbin LJ , Smith GD , Dedoussis G , Dorr M , Farmaki AE , Ferrucci L , Forer L , Fraser RM , Gabriel S , Levy S , Groop L , Harrison T , Hattersley A , Holmen OL , Hveem K , Kretzler M , Lee JC , McGue M , Meitinger T , Melzer D , Min JL , Mohlke KL , Vincent JB , Nauck M , Nickerson D , Palotie A , Pato M , Pirastu N , McInnis M , Richards JB , Sala C , Salomaa V , Schlessinger D , Schoenherr S , Slagboom PE , Small K , Spector T , Stambolian D , Tuke M , Tuomilehto J , Van den Berg LH , Van Rheenen W , Volker U , Wijmenga C , Toniolo D , Zeggini E , Gasparini P , Sampson MG , Wilson JF , Frayling T , de Bakker PI , Swertz MA , McCarroll S , Kooperberg C , Dekker A , Altshuler D , Willer C , Iacono W , Ripatti S , Soranzo N , Walter K , Swaroop A , Cucca F , Anderson CA , Myers RM , Boehnke M , McCarthy MI , Durbin R , Haplotype Reference Consortium ((2016) ) A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 48: , 1279–1283. |
[32] | Folstein MF , Robins LN , Helzer JE ((1983) ) The Mini-Mental State Examination. Arch Gen Psychiatry 40: , 812. |
[33] | Kaufer DI , Cummings JL , Ketchel P , Smith V , MacMillan A , Shelley T , Lopez OL , DeKosky ST ((2000) ) Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 12: , 233–239. |
[34] | Engelborghs S , Niemantsverdriet E , Struyfs H , Blennow K , Brouns R , Comabella M , Dujmovic I , van der Flier W , Frolich L , Galimberti D , Gnanapavan S , Hemmer B , Hoff E , Hort J , Iacobaeus E , Ingelsson M , Jan de Jong F , Jonsson M , Khalil M , Kuhle J , Lleo A , de Mendonca A , Molinuevo JL , Nagels G , Paquet C , Parnetti L , Roks G , Rosa-Neto P , Scheltens P , Skarsgard C , Stomrud E , Tumani H , Visser PJ , Wallin A , Winblad B , Zetterberg H , Duits F , Teunissen CE ((2017) ) Consensus guidelines for lumbar puncture in patients with neurological diseases. Alzheimers Dement (Amst) 8: , 111–126. |
[35] | Tijms BM , Willemse EAJ , Zwan MD , Mulder SD , Visser PJ , van Berckel BNM , van der Flier WM , Scheltens P , Teunissen CE ((2018) ) Unbiased approach to counteract upward drift in cerebrospinal fluid amyloid-beta 1-42 analysis results. Clin Chem 64: , 576–585. |
[36] | Price AL , Patterson NJ , Plenge RM , Weinblatt ME , Shadick NA , Reich D ((2006) ) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38: , 904–909. |
[37] | Benjamini Y , Hochberg Y ((1995) ) Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc B Methodol 57: , 289–300. |
[38] | Manolio TA , Collins FS , Cox NJ , Goldstein DB , Hindorff LA , Hunter DJ , McCarthy MI , Ramos EM , Cardon LR , Chakravarti A , Cho JH , Guttmacher AE , Kong A , Kruglyak L , Mardis E , Rotimi CN , Slatkin M , Valle D , Whittemore AS , Boehnke M , Clark AG , Eichler EE , Gibson G , Haines JL , Mackay TF , McCarroll SA , Visscher PM ((2009) ) Finding the missing heritability of complex diseases. Nature 461: , 747–753. |
[39] | Tsuang D , Leverenz JB , Lopez OL , Hamilton RL , Bennett DA , Schneider JA , Buchman AS , Larson EB , Crane PK , Kaye JA , Kramer P , Woltjer R , Trojanowski JQ , Weintraub D , Chen-Plotkin AS , Irwin DJ , Rick J , Schellenberg GD , Watson GS , Kukull W , Nelson PT , Jicha GA , Neltner JH , Galasko D , Masliah E , Quinn JF , Chung KA , Yearout D , Mata IF , Wan JY , Edwards KL , Montine TJ , Zabetian CP ((2013) ) APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 70: , 223–228. |
[40] | Larsson V , Torisson G , Londos E ((2018) ) Relative survival in patients with dementia with Lewy bodies and Parkinson’s disease dementia. PLoS One 13: , e0202044. |
[41] | Lemstra AW , de Beer MH , Teunissen CE , Schreuder C , Scheltens P , van der Flier WM , Sikkes SA ((2017) ) Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 88: , 113–118. |
[42] | Vijayaraghavan S , Darreh-Shori T , Rongve A , Berge G , Sando SB , White LR , Auestad BH , Witoelar A , Andreassen OA , Ulstein ID , Aarsland D ((2016) ) Association of butyrylcholinesterase-K allele and apolipoprotein E varepsilon4 allele with cognitive decline in dementia with Lewy bodies and Alzheimer’s disease. J Alzheimers Dis 50: , 567–576. |
[43] | Zhang Y , Shu L , Zhou X , Pan H , Xu Q , Guo J , Tang B , Sun Q ((2018) ) A meta-analysis of GBA-related clinical symptoms in Parkinson’s disease. Parkinsons Dis 2018: , 3136415. |
[44] | Cilia R , Tunesi S , Marotta G , Cereda E , Siri C , Tesei S , Zecchinelli AL , Canesi M , Mariani CB , Meucci N , Sacilotto G , Zini M , Barichella M , Magnani C , Duga S , Asselta R , Solda G , Seresini A , Seia M , Pezzoli G , Goldwurm S ((2016) ) Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann Neurol 80: , 662–673. |
[45] | Morenas-Rodriguez E , Sala I , Subirana A , Pascual-Goni E , Sanchez-Saudinos MB , Alcolea D , Illan-Gala I , Carmona-Iragui M , Ribosa-Nogue R , Camacho V , Blesa R , Fortea J , Lleo A ((2018) ) Clinical subtypes of dementia with Lewy bodies based on the initial clinical presentation. J Alzheimers Dis 64: , 505–513. |


