
The Right Temporal Variant of Frontotemporal Dementia Is Not Genetically Sporadic: A Case Series
Abstract
Background:
Right temporal variant frontotemporal dementia (rtvFTD) has been generally considered as a right sided variant of semantic variant primary progressive aphasia (svPPA), which is a genetically sporadic disorder. Recently, we have shown that rtvFTD has a unique clinical syndrome compared to svPPA and behavioral variant frontotemporal dementia.
Objective:
We challenge the assumption that rtvFTD is a sporadic, non-familial variant of FTD by identifying potential autosomal dominant inheritance and related genes in rtvFTD.
Methods:
We collected all subjects with a diagnosis of FTD or primary progressive aphasia who had undergone genetic screening (n = 284) and subsequently who had a genetic variant (n = 48) with a diagnosis of rtvFTD (n = 6) in 2 specialized memory clinics.
Results:
Genetic variants in FTD related genes were found in 33% of genetically screened rtvFTD cases; including MAPT (n = 4), GRN (n = 1), and TARDBP (n = 1) genes, whereas only one svPPA case had a genetic variant in our combined cohorts. Additionally, 4 out of 6 rtvFTD subjects had a strong family history for dementia.
Conclusion:
Our results demonstrate that rtvFTD, unlike svPPA, is not a pure sporadic, but a heterogeneous potential genetic variant of FTD, and screening for genetic causes for FTD should be performed in patients with rtvFTD.
INTRODUCTION
Frontotemporal dementia (FTD) is a syndrome caused by degeneration of the frontal and/or temporal lobes [1]. Patients with predominant behavioral disturbances and frontotemporal atrophy on neuro-imaging are classified as behavioral variant FTD (bvFTD) [2], whereas the language predominant subtypes of FTD are classified under the umbrella of primary progressive aphasia (PPA) and have been associated with left hemisphere atrophy [3].
Over the years, the genetics of FTD have been broadly explored. The autosomal dominant inherita-nce pattern has been found higher in bvFTD, whereas semantic variant PPA (svPPA) is typically a non-familial sporadic disease [4–7]. Pathogenic variants are most common in the microtubule associated protein tau gene (MAPT), the progranulin gene (GRN), and a hexanucleotide repeat expansion in the chromosome 9 open reading frame 72 gene (C9orf72), whereas a variety of rare pathogenic variants has been described as well [5].
Currently, diagnostic criteria for a variant of FTD presenting with behavioral changes, memory deficit, and prosopagnosia in the presence of right temporal atrophy (rtvFTD) are lacking [8]. Because of the atrophy pattern, theoretically, rtvFTD is considered a right variant of svPPA [3, 9, 10] and the general assumption is that it is also a sporadic disease.
Only one study focusing on the underlying genetic and pathological features in rtvFTD, showed a positive family history in 45% of the patients with pos-tmortem diagnostic confirmation [11]. Thus, we set out to investigate whether rtvFTD could be potentially a genetic disorder.
METHODS
In this report, out of 636 patients from the Amsterdam dementia cohort (ADC) with a clinical diagnosis of bvFTD (n = 450), non-fluent variant PPA (n = 32), logopenic variant PPA (n = 18), svPPA (n = 65), and rtvFTD (n = 71) (January 2000–November 2019) [12], we included 148 cases who had undergone gen-etic screening. Additionally, 136 FTD/PPA patients with genetic screening from the Istanbul University dementia cohort (IUDC) (November 1999-January 2020) [13] were included (total genetically screened patients, n = 284). Genetic screening was offered in case of a positive family history or when this was requested by the patient/caregiver. All included pat-ients were screened for a variant in the GRN and MAPT genes. Additionally, a subset of patients was screened for the hexanucleotide repeat expansion in the C9orf72 gene (n = 189) and/or the variants in other dementia genes with whole-exome sequencing (WES) (n = 77) (Supplementary Material 1). In 48 patients, pathogenic variants or variants of unknown significance (VUS) [14] in the FTD related genes were identified and six out of them met the clinical and the radiological characteristics of rtvFTD [8] (Supplementary Figure 1). Of note, in all subjects, the atrophy scores of the right temporal lobe [15–17] were higher (at least 1 grade) than the left temporal lobe and the frontal lobes that were assessed by a well experienced neuroradiologist who was blind to the clinical diagnosis (FB). Additionally, in our sample, the frontal atrophy scores were less than grade-1 [16] and none of the subjects met the diagnostic criteria of svPPA [3], while all of the fulfilled at least two symptoms out of prosopagnosia, episodic memory impairment, and behavioral change [8], even if they had an accompanying left temporal atrophy on the initial MRI. All subjects gave their written informed consent for the use of their clinical and genetic data for research purposes. Details of the genetic and pathological assessment are reported in Supplementary Material 1.
Fig. 1
Patient selection.
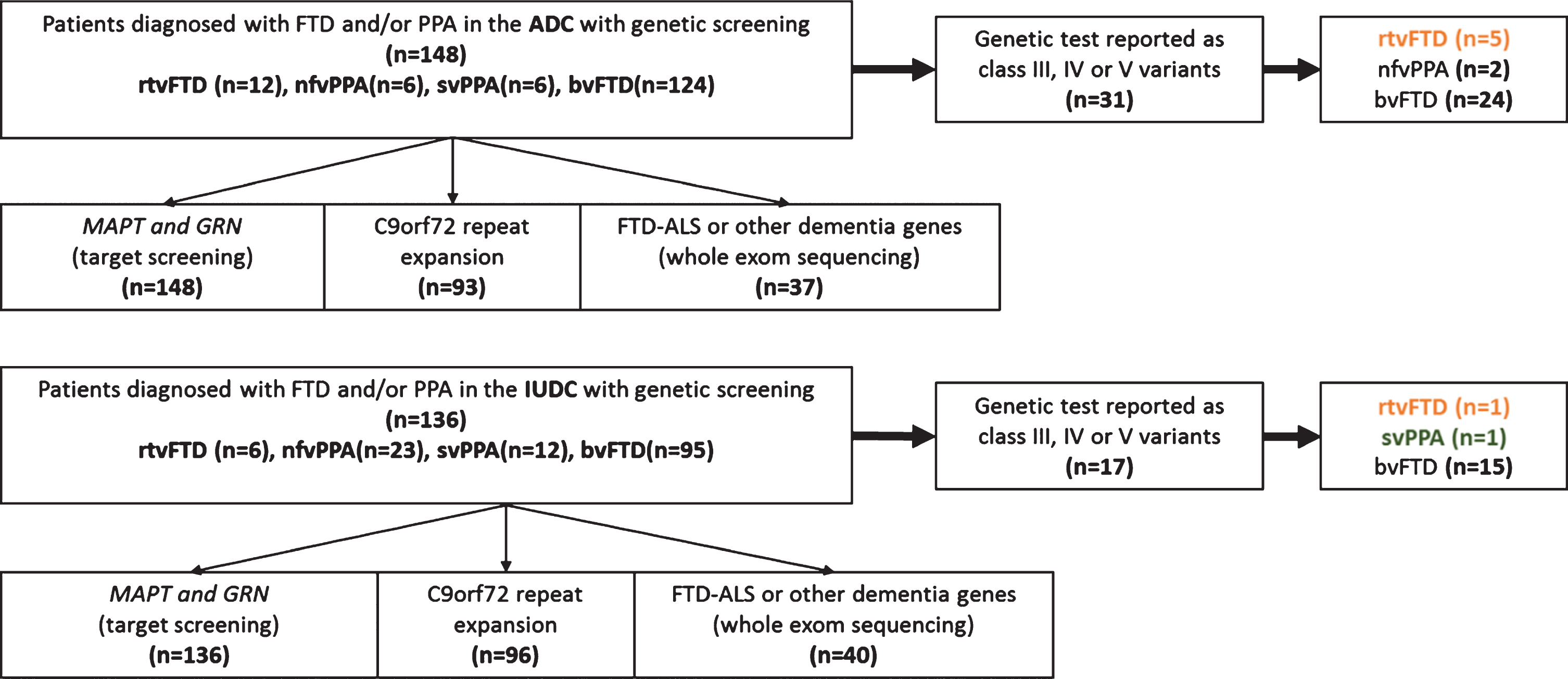
RESULTS
Demographic, clinical features are displayed in Table 1, and detailed case histories are reported in Supplementary Material 2.
Table 1
Demographic and clinical data
| Case 1 | Case 2 | Case 3 | Case 4 | Case 5 | Case 6 | |
| Institution | ADC | ADC | ADC | ADC | IUDC | ADC |
| Age (y) | 59 | 64 | 58 | 53 | 63 | 58 |
| Sex | Male | Female | Male | Female | Male | Female |
| Handedness | Right | Right | Right | Right | Right | Right |
| Symptom duration (y) | 2 | 8 | 4 | 1 | 1 | 11 |
| MTA (Right/left) | 4/1 | 4/2 | 2/1 | 3/2 | 4/2 | 3/1 |
| PET | N.A. | N.A. | Right temporal | N.A. | N.A. | N.A. |
| hypo-perfusion | ||||||
| Gene | GRN | MAPT | MAPT | MAPT | MAPT | TARDBP |
| Variant | Gln130 Serfs*125 | Ser305Thr | Ser352Leu | Arg406Trp | Pro301Leu | Ile383Val |
| Pathogenicity | Pathogenic [37] | Likely pathogenic [38]. Other variants in this codon reported as pathogenic | Unknown significance [43]. Heterozygous in our patient, homozygous in the reported patient | Pathogenic [44]. | Pathogenic [45]. | Unknown significance [28–30]. No data about pathogenicity in the reported patients |
| Pathological confirmation | N.A. | N.A. | Suggestive for primary tau mutation | N.A. | N.A. | N.A. |
| Modified Goldman score | 2 | 1 | 4 | 1 | 1 | 3 |
| APOE | E3E3 | E3E3 | E3E4 | E3E4 | N.A. | N.A. |
| CSF, pg/mL* | ||||||
| Aβ42 | 1073 | 1101 | 716 | 1270 | N.A. | 1574 |
| Tau | 326 | 353 | 717 | 512 | N.A. | 311 |
| P-tau | 38 | 54 | 70 | 80 | N.A. | 37 |
| Cognitive Tests | ||||||
| MMSE | 26/30 | 28/30 | 23/30 | 28/30 | 29/30 | 29/30 |
| FAB | 16/18 | - | 14/18 | 18/18 | N.A. | 18/18 |
| VAT-A | 4/12 | N.A. | 10/12 | |||
| RAVLT | - | - | - | 8/30 | N.A. | 22/30 |
| delayed recall | ||||||
| VAT naming | 12/12 | 12/12 | 10/12 | 12/12 | N.A. | 10/12 |
| TMT A | 41” (A) | 77,6” (LA) | - | 52” (A) | N.A. | 32” (A) |
| TMT B | 88” (A) | 192,7” (LA) | - | 71” (A) | N.A. | 70” (A) |
| VOSP-Dot | 10/10 | 10/10 | 10/10 | 10/10 | N.A. | 10/10 |
| Counting | ||||||
| VOSP-FL | 20/20 | 20/20 | - | 19/20 | N.A. | - |
ADC, Amsterdam Dementia Cohort; IUDC, Istanbul University Dementia Cohort; MTA, mesial temporal atrophy; PET, positron emission tomography; APOE, Apolipoprotein E; CSF, cerebrospinal fluid; Aβ42, amyloid-β 42; P-tau, phospho tau; MMSE: Mini-Mental State Examination; FAB, Frontal assessment battery; TMT, Trail making test; VAT, Visual association test; RAVLT, Dutch version of the Rey Auditory Verbal Learning Test; VOSP, Visual objective and space perception; FL, Fragmented letters; L, Low; VL, Very low; HA, High average; LA, Low average; A, Average. *Cutoff value for CSF Aβ42 indicating Alzheimer’s disease pathology is <550 pg/mL, Tau >375 pg/mL, P-tau>52 pg/mL.
In our combined cohorts, genetic variants in FTD related genes were found in 33% of genetically sc-reened rtvFTD subjects (6 out of 18 genetically scr-eened rtvFTD), whereas only one svPPA (1 out of 18 genetically screened svPPA) subject had a genetic variant.
Summary of the cases
Case 1: A 59-year-old male presented with be-havioral problems, memory deficit, depression, topo-graphagnosia, and developed swallowing problems and mutism. The modified Goldman score [4] for fa-mily history was 2. We identified a heterozygous pat-hogenic variant in the GRN gene (NM 002087.3) c.388_391del, p.(Gln130Serfs*125).
Case 2: A 64-year-old female presented with prosopagnosia, behavioral changes, memory deficit, depression, and developed topographagnosia and motor restless. The modified Goldman score [4] for family history was 1. We identified a heterozygous likely pathogenic variant in the MAPT gene (NM 005910.5) c.914G>C, p.(Ser305Thr).
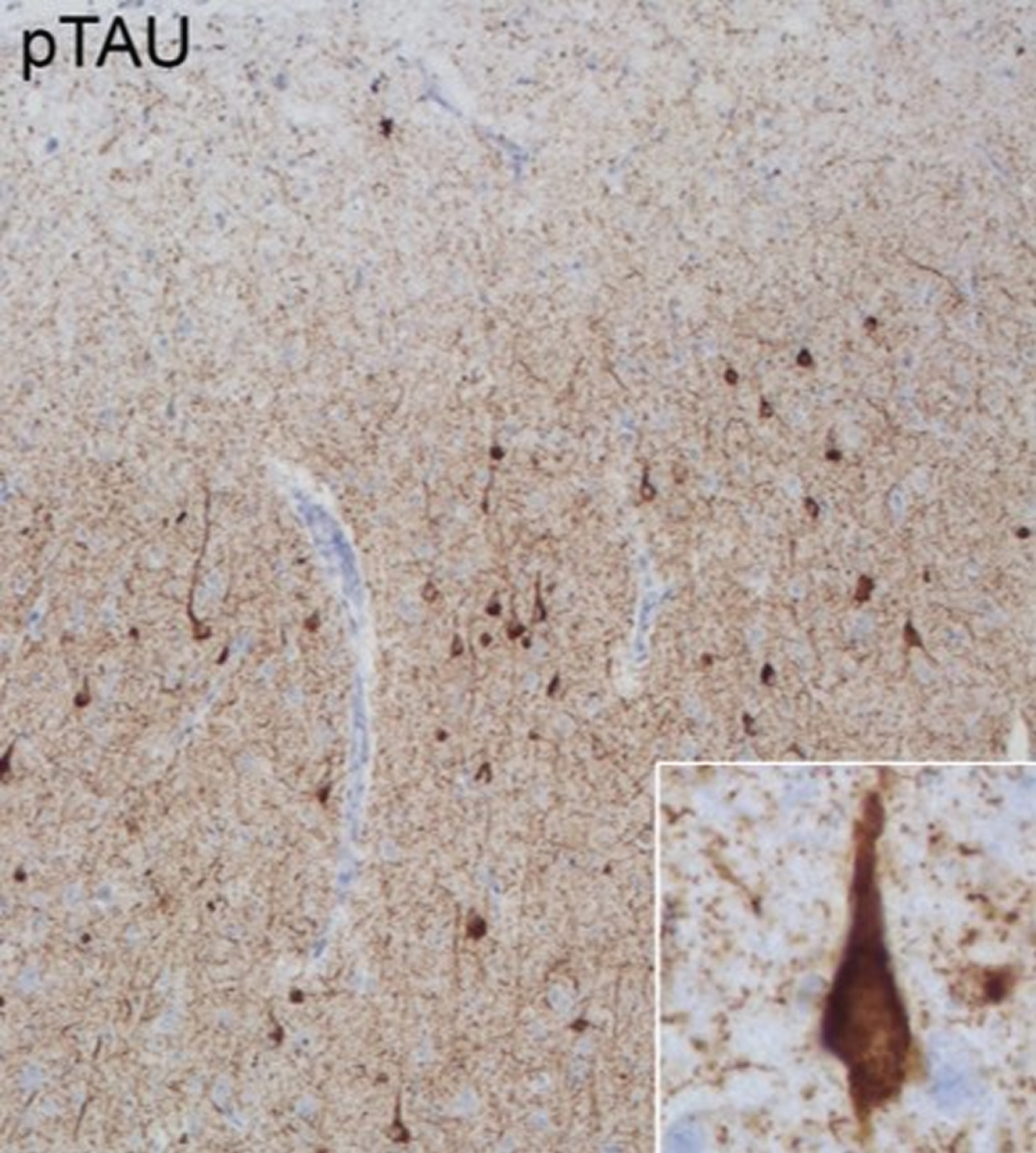
Case 3: A 58-year-old male presented with behavioral changes, depression, memory deficits, and dev-eloped prosopagnosia and atypical parkinsonism. The modified Goldman score [4] for family history was 4. We identified a heterozygous VUS in the MAPT gene (NM 005910.5) c.1055C>T, p.(Ser352Leu). In addition, extensive 3R and 4R tauopa-thy was reported in his autopsy which is suggestive for a pathogenic mutation in the MAPT gene [18] (Fig. 2).
Case 4: A 53-year-old female presented with memory deficits, depression, apathy, and developed anomia and several behavioral problems. The modified Goldman score [4] for family history was 1. We identified a heterozygous pathogenic variant in the MAPT gene, (NM 005910.5) c.1216C>T, p.(Arg406Trp).
Fig. 2
Pathological features of Case 3. Anterior cingulate cortex stained with phospho-tau (p-tau) monoclonal antibody (AT8: Pierce Biotechnology, Rockford, IL, USA). Extensive 3R and 4R tauopathy which is characteristic for MAPT related frontotemporal lobar degeneration is observed in neurons across all layers.
Case 5: A 63-year-old male presented with beh-avioral changes, prosopagnosia, anomia, and single word comprehension deficit, and developed topogra-phagnosia. The modified Goldman score [4] for family history was 1. We identified a heterozygous pathogenic variant in the MAPT gene (NM 005910.5) c.902C>T p.(Pro301Leu).
Case 6: A 58-year-old female presented with somatic and behavioral problems, memory deficit, and motor restless. The modified Goldman score [4] for family history was 3. We identified a heterozygous VUS in the TARDBP gene, (NM007375.3) c.1147A>G, p.(Ile383Val).
DISCUSSION
RtvFTD and svPPA are generally considered sporadic, non familial variants of FTD. In our combined cohorts, we can confirm that in svPPA rarely (∼5%) class III-V genetic variants in FTD related genes are found. However, 33% of rtvFTD patients that were screened for genetic mutations in FTD genes had a genetic variant. Moreover, these variants were in three different genes (MAPT, GRN, and TARDBP). This demonstrates that rtvFTD patients, unlike svPPA, are a heterogenous group that should be screened for genetic mutations.
The genetic diagnosis of four out of six rtvFTD cases was FTLD-MAPT. Previous clinico-rad-iological studies have shown that FTLD-MAPT links to bilateral anterior temporal atrophy [19], which might include rtvFTD. Moreover, the relationship between rtvFTD and MAPT mutations has been previously reported [11].
Besides the MAPT gene, the association between rtvFTD with variants in the GRN gene has been confirmed in separate case reports [20–23]. In many cases with a variant in the GRN gene, the asymmetric atrophy extends to the parietal lobe, which was not the case in our patient. Our finding underscores the observation that a pathogenic variant status in the GRN gene may be associated with an asymmetric atrophy pattern [24, 25], which can also involve uniquely the temporal lobe.
Although TARDBP gene mutations have been described in sporadic and familial amyotrophic lateral sclerosis (ALS) in early studies [26, 27], it has subsequently been associated with FTD without ALS [28–33]. Additionally, the heterozygous variant of Case 6 has been reported in subjects with temporal variant FTD without ALS [28–30].
In our study, four out of six patients had a strong family history for dementia. In the literature, a positive family history was reported in 37.5% (15 out of 40) of patients with rtvFTD [combined Chan et al. [34] and Josephs et al. [11]]. This percentage is quite high compared to svPPA in which a suggestive family history is identified in less than 5% of patients [6, 35]
Nonetheless, it is still unknown whether rtvFTD and svPPA share the same pathophysiology. A recent GWAS metadata analysis [36] has revealed that the svPPA gene network is uniquely associated with TAR DNA binding protein 43 metabolism. From this perspective, accompanying tauopathy in rtv-FTD resembles the heterogeneous pathophysiology of bvFTD, rather than svPPA. On the other hand, although C9orf72 is the most common worldwide cause of genetic FTD [5], it should be noted that this variant was not found either in our study or other rtvFTD cohorts [11, 34]. Therefore, further research into the pathophysiological background of rtvFTD and how this relates to the other FTD subtypes is warranted.
In conclusion, currently, there is no consensus on whether rtvFTD is a mirror variant of svPPA or should be lumped with svPPA. Although reminiscent of svPPA, our findings show that rtvFTD, unlike svPPA, often has a genetic basis and the genetic variants are found in multiple genes. Therefore, genetic screening is essential in patients with rtvFTD.
ACKNOWLEDGMENTS
We are very grateful for the generous contribution of the patients and their relatives. Research of the Alzheimer Center Amsterdam is part of the neu-rodegeneration research program of Amsterdam Neuroscience. The Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. WF holds the Pasman chair. Dr. HUE has received research support from the Turkish Neurological Society. FB is supported by the NIHR biomedical research center at UCLH.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1191r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-201191.
REFERENCES
[1] | Neary D , Snowden JS , Gustafson L , Passant U , Stuss D , Black S , Freedman M , Kertesz A , Robert PH , Albert M , Boone K , Miller BL , Cummings J , Benson DF ((1998) ) Frontotemporal lobar degeneration: A consensus on clinical diagnostic criteria. Neurology 51: , 1546–1554. |
[2] | Rascovsky K , Hodges JR , Knopman D , Mendez MF , Kramer JH , Neuhaus J , van Swieten JC , Seelaar H , Dopper EG , Onyike CU , Hillis AE , Josephs KA , Boeve BF , Kertesz A , Seeley WW , Rankin KP , Johnson JK , Gorno-Tempini ML , Rosen H , Prioleau-Latham CE , Lee A , Kipps CM , Lillo P , Piguet O , Rohrer JD , Rossor MN , Warren JD , Fox NC , Galasko D , Salmon DP , Black SE , Mesulam M , Weintraub S , Dickerson BC , Diehl-Schmid J , Pasquier F , Deramecourt V , Lebert F , Pijnenburg Y , Chow TW , Manes F , Grafman J , Cappa SF , Freedman M , Grossman M , Miller BL ((2011) ) Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 134: , 2456–2477. |
[3] | Gorno-Tempini ML , Hillis AE , Weintraub S , Kertesz A , Mendez M , Cappa SF , Ogar JM , Rohrer JD , Black S , Boeve BF , Manes F , Dronkers NF , Vandenberghe R , Rascovsky K , Patterson K , Miller BL , Knopman DS , Hodges JR , Mesulam MM , Grossman M ((2011) ) Classification of primary progressive aphasia and its variants. Neurology 76: , 1006–1014. |
[4] | Rohrer JD , Guerreiro R , Vandrovcova J , Uphill J , Reiman D , Beck J , Isaacs AM , Authier A , Ferrari R , Fox NC , Mackenzie IR , Warren JD , de Silva R , Holton J , Revesz T , Hardy J , Mead S , Rossor MN ((2009) ) The heritability and genetics of frontotemporal lobar degeneration. Neurology 73: , 1451–1456. |
[5] | Greaves CV , Rohrer JD ((2019) ) An update on genetic frontotemporal dementia. J Neurol 266: , 2075–2086. |
[6] | Landin-Romero R , Tan R , Hodges JR , Kumfor F ((2016) ) An update on semantic dementia: Genetics, imaging, and pathology. Alzheimers Res Ther 8: , 52. |
[7] | Rohrer JD ((2014) ) The genetics of primary progressive aphasia. Aphasiology 28: , 941–947. |
[8] | Ulugut Erkoyun H , Groot C , Heilbron R , Nelissen A , van Rossum J , Jutten R , Koene T , van der Flier WM , Wattjes MP , Scheltens P , Ossenkoppele R , Barkhof F , Pijnenburg Y ((2020) ) A clinical-radiological framework of the right temporal variant of frontotemporal dementia. Brain 143: , 2831–2843. |
[9] | Seeley WW , Bauer AM , Miller BL , Gorno-Tempini ML , Kramer JH , Weiner M , Rosen HJ ((2005) ) The natural history of temporal variant frontotemporal dementia. Neurology 64: , 1384–1390. |
[10] | Brambati SM , Rankin KP , Narvid J , Seeley WW , Dean D , Rosen HJ , Miller BL , Ashburner J , Gorno-Tempini ML ((2009) ) Atrophy progression in semantic dementia with asymmetric temporal involvement: A tensor-based morphometry study. Neurobiol Aging 30: , 103–111. |
[11] | Josephs KA , Whitwell JL , Knopman DS , Boeve BF , Vemuri P , Senjem ML , Parisi JE , Ivnik RJ , Dickson DW , Petersen RC , Jack CR Jr. ((2009) ) Two distinct subtypes of right temporal variant frontotemporal dementia. Neurology 73: , 1443–1450. |
[12] | van der Flier WM , Scheltens P ((2018) ) Amsterdam Dementia Cohort: Performing research to optimize care. J Alzheimers Dis 62: , 1091–1111. |
[13] | Guven G , Lohmann E , Bras J , Gibbs JR , Gurvit H , Bilgic B , Hanagasi H , Rizzu P , Heutink P , Emre M , Erginel-Unaltuna N , Just W , Hardy J , Singleton A , Guerreiro R ((2016) ) Mutation frequency of the major frontotemporal dementia genes, MAPT, GRN and C9ORF72 in a Turkish cohort of dementia patients. PLoS One 11: , e0162592. |
[14] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , Grody WW , Hegde M , Lyon E , Spector E , Voelkerding K , Rehm HL , ACMG Laboratory Quality Assurance Committee ((2015) ) Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: , 405–424. |
[15] | Scheltens P , Leys D , Barkhof F , Huglo D , Weinstein HC , Vermersch P , Kuiper M , Steinling M , Wolters EC , Valk J ((1992) ) Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: Diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry 55: , 967–972. |
[16] | Kipps CM , Davies RR , Mitchell J , Kril JJ , Halliday GM , Hodges JR ((2007) ) Clinical significance of lobar atrophy in frontotemporal dementia: Application of an MRI visual rating scale. Dement Geriatr Cogn Disord 23: , 334–342. |
[17] | Harper L , Barkhof F , Fox NC , Schott JM ((2015) ) Using visual rating to diagnose dementia: A critical evaluation of MRI atrophy scales. J Neurol Neurosurg Psychiatry 86: , 1225–1233. |
[18] | Kovacs GG ((2015) ) Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol Appl Neurobiol 41: , 3–23. |
[19] | Whitwell JL , Jack CR , Jr. , Boeve BF , Senjem ML , Baker M , Rademakers R , Ivnik RJ , Knopman DS , Wszolek ZK , Petersen RC , Josephs KA ((2009) ) Voxel-based morphometry patterns of atrophy in FTLD with mutations in MAPT or PGRN. Neurology 72: , 813–820. |
[20] | Huey ED , Grafman J , Wassermann EM , Pietrini P , Tierney MC , Ghetti B , Spina S , Baker M , Hutton M , Elder JW , Berger SL , Heflin KA , Hardy J , Momeni P ((2006) ) Characteristics of frontotemporal dementia patients with a Progranulin mutation. Ann Neurol 60: , 374–380. |
[21] | Beck J , Rohrer JD , Campbell T , Isaacs A , Morrison KE , Goodall EF , Warrington EK , Stevens J , Revesz T , Holton J , Al-Sarraj S , King A , Scahill R , Warren JD , Fox NC , Rossor MN , Collinge J , Mead S ((2008) ) A distinct clinical, neuropsychological and radiological phenotype is associated with progranulin gene mutations in a large UK series. Brain 131: , 706–720. |
[22] | Pietroboni AM , Fumagalli GG , Ghezzi L , Fenoglio C , Cortini F , Serpente M , Cantoni C , Rotondo E , Corti P , Carecchio M , Bassi M , Bresolin N , Galbiati D , Galimberti D , Scarpini E ((2011) ) Phenotypic heterogeneity of the GRN Asp22fs mutation in a large Italian kindred. J Alzheimers Dis 24: , 253–259. |
[23] | Cannon A , Fujioka S , Rutherford NJ , Ferman TJ , Broderick DF , Boylan KB , Graff-Radford NR , Uitti RJ , Rademakers R , Wszolek ZK , Dickson DW ((2013) ) Clinicopathologic variability of the GRN A9D mutation, including amyotrophic lateral sclerosis. Neurology 80: , 1771–1777. |
[24] | Whitwell JL , Weigand SD , Boeve BF , Senjem ML , Gunter JL , DeJesus-Hernandez M , Rutherford NJ , Baker M , Knopman DS , Wszolek ZK , Parisi JE , Dickson DW , Petersen RC , Rademakers R , Jack CR Jr. , Josephs KA ((2012) ) Neuroimaging signatures of frontotemporal dementia genetics: C9ORF72, tau, progranulin and sporadics. Brain 135: , 794–806. |
[25] | Rohrer JD , Paviour D , Bronstein AM , O’Sullivan SS , Lees A , Warren JD ((2010) ) Progressive supranuclear palsy syndrome presenting as progressive nonfluent aphasia: A neuropsychological and neuroimaging analysis. Mov Disord 25: , 179–188. |
[26] | Gitcho MA , Baloh RH , Chakraverty S , Mayo K , Norton JB , Levitch D , Hatanpaa KJ , White III CL , Bigio EH , Caselli R , Baker M , Al-Lozi MT , Morris JC , Pestronk A , Rademakers R , Goate AM , Cairns NJ ((2008) ) TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol 63: , 535–538. |
[27] | Yokoseki A , Shiga A , Tan C-F , Tagawa A , Kaneko H , Koyama A , Eguchi H , Tsujino A , Ikeuchi T , Kakita A , Okamoto K , Nishizawa M , Takahashi H , Onodera O ((2008) ) TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol 63: , 538–542. |
[28] | Gelpi E , van der Zee J , Turon Estrada A , Van Broeckhoven C , Sanchez-Valle R ((2014) ) TARDBP mutation p.Ile383Val associated with semantic dementia and complex proteinopathy. Neuropathol Appl Neurobiol 40: , 225–230. |
[29] | Gonzalez-Sanchez M , Puertas-Martin V , Esteban-Perez J , Garcia-Redondo A , Borrego-Hernandez D , Mendez-Guerrero A , Llamas-Velasco S , Herrero-San Martin A , Cordero-Vazquez P , Herrero-Manso MC , Perez-Martinez DA , Villarejo-Galende A ((2018) ) TARDBP mutation associated with semantic variant primary progressive aphasia, case report and review of the literature. Neurocase 24: , 301–305. |
[30] | Caroppo P , Camuzat A , Guillot-Noel L , Thomas-Antérion C , Couratier P , Wong TH , Teichmann M , Golfier V , Auriacombe S , Belliard S , Laurent B , Lattante S , Millecamps S , Clot F , Dubois B , van Swieten JC , Brice A , Le Ber I ((2016) ) Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol Genet 2: , e80. |
[31] | Cheng YW , Lee MJ , Chen TF , Cheng TW , Lai YM , Hua MS , Chiu MJ ((2016) ) A single nucleotide TDP-43 mutation within a Taiwanese family: A multifaceted demon. Amyotroph Lateral Scher Frontotemp Degenerat 17: , 292–294. |
[32] | Floris G , Borghero G , Cannas A , Di Stefano F , Murru MR , Corongiu D , Cuccu S , Tranquilli S , Cherchi MV , Serra A , Loi G , Marrosu MG , Chiò A , Marrosu F ((2015) ) Clinical phenotypes and radiological findings in frontotemporal dementia related to TARDBP mutations. J Neurol 262: , 375–384. |
[33] | Moreno F , Rabinovici GD , Karydas A , Miller Z , Hsu SC , Legati A , Fong J , Schonhaut D , Esselmann H , Watson C , Stephens ML , Kramer J , Wiltfang J , Seeley WW , Miller BL , Coppola G , Grinberg LT ((2015) ) A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques. Acta Neuropathol Commun 3: , 19. |
[34] | Chan D , Anderson V , Pijnenburg Y , Whitwell J , Barnes J , Scahill R , Stevens JM , Barkhof F , Scheltens P , Rossor MN , Fox NC ((2009) ) The clinical profile of right temporal lobe atrophy. Brain 132: , 1287–1298. |
[35] | Godbolt AK , Josephs KA , Revesz T , Warrington EK , Lantos P , King A , Fox NC , Al Sarraj S , Holton J , Cipolotti L , Khan MN , Rossor MN ((2005) ) Sporadic and familial dementia with ubiquitin-positive tau-negative inclusions: Clinical features of one histopathological abnormality underlying frontotemporal lobar degeneration. Arch Neurol 62: , 1097–1101. |
[36] | Bonham LW , Steele NZR , Karch CM , Broce I , Geier EG , Wen NL , Momeni P , Hardy J , Miller ZA , Gorno-Tempini ML , Hess CP , Lewis P , Miller BL , Seeley WW , Manzoni C , Desikan RS , Baranzini SE , Ferrari R , Yokoyama JS , International FTDGC ((2019) ) Genetic variation across RNA metabolism and cell death gene networks is implicated in the semantic variant of primary progressive aphasia. Sci Rep 9: , 10854–10854. |
[37] | Baker M , Mackenzie IR , Pickering-Brown SM , Gass J , Rademakers R , Lindholm C , Snowden J , Adamson J , Sadovnick AD , Rollinson S , Cannon A , Dwosh E , Neary D , Melquist S , Richardson A , Dickson D , Berger Z , Eriksen J , Robinson T , Zehr C , Dickey CA , Crook R , McGowan E , Mann D , Boeve B , Feldman H , Hutton M ((2006) ) Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 442: , 916–919. |
[38] | Meeter LH , Dopper EG , Jiskoot LC , Sanchez-Valle R , Graff C , Benussi L , Ghidoni R , Pijnenburg YA , Borroni B , Galimberti D , Laforce RJ , Masellis M , Vandenberghe R , Ber IL , Otto M , van Minkelen R , Papma JM , Rombouts SA , Balasa M , Oijerstedt L , Jelic V , Dick KM , Cash DM , Harding SR , Jorge Cardoso M , Ourselin S , Rossor MN , Padovani A , Scarpini E , Fenoglio C , Tartaglia MC , Lamari F , Barro C , Kuhle J , Rohrer JD , Teunissen CE , van Swieten JC ((2016) ) Neurofilament light chain: A biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol 3: , 623–636. |
[39] | Boeve BF , Tremont-Lukats IW , Waclawik AJ , Murrell JR , Hermann B , Jack CR Jr. , Shiung MM , Smith GE , Nair AR , Lindor N , Koppikar V , Ghetti B ((2005) ) Longitudinal characterization of two siblings with frontotemporal dementia and parkinsonism linked to chromosome 17 associated with the S305N tau mutation. Brain 128: , 752–772. |
[40] | Iijima M , Tabira T , Poorkaj P , Schellenberg GD , Trojanowski JQ , Lee VM , Schmidt ML , Takahashi K , Nabika T , Matsumoto T , Yamashita Y , Yoshioka S , Ishino H ((1999) ) A distinct familial presenile dementia with a novel missense mutation in the tau gene. Neuroreport 10: , 497–501. |
[41] | Skoglund L , Viitanen M , Kalimo H , Lannfelt L , Jonhagen ME , Ingelsson M , Glaser A , Herva R ((2008) ) The tau S305S mutation causes frontotemporal dementia with parkinsonism. Eur J Neurol 15: , 156–161. |
[42] | Stanford PM , Halliday GM , Brooks WS , Kwok JB , Storey CE , Creasey H , Morris JG , Fulham MJ , Schofield PR ((2000) ) Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: Expansion of the disease phenotype caused by tau gene mutations. Brain 123: (Pt 5), 880–893. |
[43] | Nicholl DJ , Greenstone MA , Clarke CE , Rizzu P , Crooks D , Crowe A , Trojanowski JQ , Lee VM , Heutink P ((2003) ) An English kindred with a novel recessive tauopathy and respiratory failure. Ann Neurol 54: , 682–686. |
[44] | Ygland E , van Westen D , Englund E , Rademakers R , Wszolek ZK , Nilsson K , Nilsson C , Landqvist Waldo M , Alafuzoff I , Hansson O , Gustafson L , Puschmann A ((2018) ) Slowly progressive dementia caused by MAPT R406W mutations: Longitudinal report on a new kindred and systematic review. Alzheimers Res Ther 10: , 2. |
[45] | Shi Z , Liu S , Xiang L , Wang Y , Liu M , Liu S , Han T , Zhou Y , Wang J , Cai L , Gao S , Ji Y ((2016) ) Frontotemporal dementia-related gene mutations in clinical dementia patients from a Chinese population. J Hum Genet 61: , 1003–1008. |

