
TAR DNA-Binding Protein 43 Is Associated with Rate of Memory, Functional and Global Cognitive Decline in the Decade Prior to Death
Abstract
Background:
Transactive response DNA-binding protein of 43 kDa (TDP-43) is associated with memory impairment and overall cognitive decline. It is unclear how TDP-43 contributes to the rate of clinical decline.
Objective:
To determine whether cross-sectional and longitudinal cognitive and functional decline are associated with anatomical distribution of TDP-43 in the brain.
Methods:
Longitudinal clinical-neuropathologic autopsy cohort study of 385 initially cognitively normal/mildly impaired older adults prospectively followed until death. We investigated how TDP-43, amyloid-β (Aβ), tau neurofibrillary tangles (NFT), Lewy body disease (LBD), age, sex, and genetics are associated with clinical scores and rates of their longitudinal decline.
Results:
Of 385 participants, 260 (68%) had no TDP-43, 32 (8%) had TDP-43 limited to amygdala, and 93 (24%) had TDP-43 in the hippocampus and beyond. Higher TDP-43 and Braak NFT stages independently were associated with faster decline in global cognition, functional performance measured by Clinical Dementia Rating scale, and naming and episodic memory, whereas older age was associated with slower rate of cognitive, psychiatric, and functional decline. Cross-sectionally the following associations were found: higher TDP-43 and Braak NFT - worse performance; higher Aβ burden - worse global cognition, more behavioral changes, the latter also with higher LBD; older age - worse naming, lower frequency of behavioral changes; female sex - more impaired naming and better preserved episodic memory. There were no genetic associations.
Conclusion:
The association of TDP-43 distribution with decline in cognitive and functional performance suggests that TDP-43 is playing a role in the clinical progression to dementia. Further characterization of clinical features associated with TDP-43 can facilitate establishment of antemortem diagnosis.
INTRODUCTION
Phosphorylated transactive response DNA-bind-ing protein ∼43 kDa (TDP-43) is a recognized independent driver of neurodegeneration in individuals with and without Alzheimer’s disease neuropathological changes (ADNC) [1–3]. In individuals with ADNC (presence of amyloid-β [Aβ] and tau on brain autopsy), the hippocampus is particularly vulnerable to abnormal TDP-43 deposition, with hippocampal volumes being smaller at baseline and atrophying faster over time, independent of concomitant ADNC [2, 4, 5]. The hippocampus is critical for episodic memory encoding and recall [6], as well as other cognitive functions; therefore, TDP-43-mediated hippocampal atrophy, as well as neurodegeneration of other regions (e.g., amygdala) [4] could clinically translate into more advanced memory impairment and overall cognitive and functional decline in individuals who had been found to be TDP-43 positive on brain autopsy compared to their TDP-43 negative counterparts, regardless of ADNC [7–11].
Detailed cognitive, functional, and neuropsychological data comparing TDP-43 positive and TDP-43 negative individuals are limited and mostly cross-sectional in nature [7, 8, 12, 13]. Hence, it is unclear to what extent TDP-43 contributes to an individual’s rate of clinical decline, whether TDP-43 stage-dependent rate of hippocampal neurodegeneration [2] translates into faster cognitive decline, and if so whether this rate of decline is likewise TDP-43 distribution (stage)-dependent.
In this study we sought to determine whether baseline and longitudinal change in cognition, particularly those related to episodic memory and functional performance are associated with TDP-43 stage: TDP-43 negative (T0), TDP-43 present and limited to being deposited in the amygdala (T1), and TDP-43 positive with extension into the hippocampus and beyond (T2). To address our aim, we analyzed a large cohort of participants who had been recruited prospectively, followed by behavioral neurologists and neuropsychologists until death, and had brain autopsy. We hypothesized that higher TDP-43 stage would be associated with greater cross-sectional episodic memory loss and faster rates of memory loss over time.
MATERIALS AND METHODS
Study design and participants
We conducted a longitudinal clinical-pathologic study using an autopsy cohort of participants who were enrolled in the Mayo Clinic Alzheimer’s Disease Research Center (ADRC), the Mayo Clinic Alzheimer’s Disease Participant Registry (ADPR), or the Mayo Clinic Study of Aging (MCSA) [14], were prospectively followed to the time of their death and had a brain autopsy. All participants had died between May 12, 1999 and December 31, 2015. From this cohort we identified all participants who met the following inclusion criteria: 1) the participant had TDP-43 status and TDP-43 stage determined (n = 746); 2) the participant had Braak neurofibrillary tangle (NFT) stage and Aβ neuritic plaque score determined in accordance with the National Institute on Aging - Alzheimer’s Association (NIA-AA) neuropathologic diagnostic criteria [15] (n = 746); 3) the participant had at least one follow-up clinical exam data available (n = 657); 4) at initial evaluation the participant had Clinical Dementia Rating Sum of Boxes (CDRSB)≤1 (n = 385). A total of 385 of participants met our inclusion criteria. Participants with initial CDRSB > 1 were excluded in order to estimate the earliest time of onset of potential effect of TDP-43 on cognitive and functional decline and to limit the missing data due to untestable state of functioning early in follow-up. All participants had been clinically evaluated by a board-certified behavioral neurologist and neuropsychologist and had completed clinical and neuropsychological testing on a yearly basis up to the time of death; cognitive status was determined by consensus after each research visit.
At the time of enrollment, participants’ demographics (age, sex, etc.) were collected and apoli-poprotein E epsilon 4 (APOE ɛ4) genotyping was performed as previously described [16].
Standard protocol approvals, registrations, and participant consents
This study has been approved by the Mayo Clinic Institutional Review Board, and ADPR and MCSA participants were also approved by the Olmsted Medical center Institutional Review Board. All participants and/or their proxies signed a written informed consent form before taking part in any research activities in accordance with the Declaration of Helsinki.
Clinical evaluation
The following battery of tests was administered at each visit: Mini-Mental Status Examination (MMSE) [17], a test of general cognitive function; modified Clinical Dementia Rating sum of boxes (CDRSB) [18], a measure of functional performance; and the Neuropsychiatric Inventory Questionnaire (NPI) [19] completed by a partner/caregiver assessing the presence of behavioral/psychiatric changes in the participants.
The neuropsychological evaluation of participants was overseen by an experienced neuropsychologist independent of the neurological assessment. The following battery of tests was completed: 60-item Boston Naming Test (BNT) [20] assessing confrontational naming and, hence, language; Wechsler Memory Scale-Revised Logical Memory I (WMSR Log Mem I) [21], an episodic memory test which assesses immediate recall of paragraph-length stories; the Rey Auditory Verbal Learning Test (AVLT) [22], a test of episodic verbal memory, is a list learning test that includes five learning trials, an interference trial, immediate recall and delayed recall trials, and recognition. Briefly, a list of 15 words is read aloud to the participant after which the participant is asked to immediately recall as many words as he/she remembered; this procedure is repeated for 5 consecutive trials. The Sum AVLT 1–5 score represents the total number of words the participant could cumulatively recall from all 5 learning trials. After the 5th trial, a second list of 15 new words is read to the participant after which the participant is asked to recall them. Immediately after, the participant is asked to recall the words from the 1st list (trial 6). Thirty minutes after the introduction of the 2nd word list and the completion of other neuropsychological testing, the participant is asked again to recall the words from the 1st list (trial 7). The Sum AVLT 1–7 score represents the total number of words from the 1st word list the participant could cumulatively recall from all 7 trials. The AVLT Delayed Recall represents the number of words from the 1st list that the participant could recall on trial 7.
Pathologic evaluation
All cases underwent pathological examination according to the recommendations of the NIA-AA [15]. Each participant was assigned a Braak NFT stage 0-VI [23] and neocortical Aβ neuritic plaque density score 0–3 according to the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) [24].
Cases were designated as TDP-43-positive if TDP-43 immunoreactive neuronal cytoplasmic inclusions, dystrophic neurites, neuronal intranuclear or NFT-associated inclusions were identified in the amygdala [25, 26]. For all cases in which TDP-43 was observed in the amygdala, we subsequently assessed for the presence of TDP-43 in the hippocampus (subiculum, CA1 and dentate fascia) and beyond (entorhinal cortex, occipitotemporal cortex, insula, basal nucleus, inferior temporal cortex, inferior olive, substantia nigra, midbrain tegmentum, basal ganglia, middle frontal gyrus) [25, 26]. All cases were then classified as: TDP-43 stage 0 (T0) = those without any TDP-43 immunoreactivity; TDP-43 stage 1 (T1) = those with TDP-43 restricted to the amygdala; and TDP-43 stage≥2 (T2) = those with TDP-43 in hippocampus with or without lesions beyond the hippocampus [2, 5]. This categorization was used to allow the TDP-43 effect to vary by stage 0 versus stage 1 versus stage≥2 [25, 26]. The grouping of those in stages 2 and above was based on the commonality of hippocampal TDP-43 and to keep the analyses tractable [2].
The presence of Lewy bodies in the brainstem, limbic system or amygdala, or neocortex was documented and staged according to NACC (NACCLEWY) [27]: 0 = no Lewy bodies; 1 = brainstem-predominant Lewy bodies; 2 = amygdala-predominant or limbic/transitional Lewy body disease (LBD); 3 = diffuse/neocortical LBD. For vascular pathology we used the following staging scheme: 0 = no vascular infarcts; 1 = microinfarcts or lacunar/large infarcts; 2 = both microinfarcts and lacunar/larger infarcts present [28].
Statistical analysis
For descriptive summaries, we defined a participant’s baseline measure as their earliest clinical evaluation and defined their rate of change based on the slope of a person-specific least squares regression line fit to their clinical score versus age. For those with only two time points, the least squares fit coincides with a line drawn through their two data points. We then compared TDP-43 groups on these numeric baseline and rate measures using ANOVA and pairwise Wilcoxon rank-sum tests.
We used linear mixed effects models to assess the changes in clinical scores with each clinical score as an outcome, and time from clinical exam to death (centered at 5 years before death with 1 year per unit increase), sex, APOE ɛ4 status, age at death (centered at 80 years old with 5 years per unit increase), Braak NFT stage, Aβ neuritic plaque score, LBD stage, and TDP-43 stage as explanatory variables. Braak NFT stage, Aβ neuritic plaque score, LBD stage, and TDP-43 stage are treated as continuous in order to increase power of analysis.
Each explanatory variable was included so that the covariate could have an effect on mean clinical score at a point in time and on mean annual change in clinical scores. In this way one mixed effect model can address both cross-sectional and longitudinal effects. The associations between each explanatory variable and outcome variable are shown after adding the effects of all the other explanatory variables in the model. The estimates are expressed as points of the clinical score of a certain test; however, due to the different scales used for each test the effect sizes might differ. The results of cross-sectional and longitudinal models can be interpreted as following: at a given point in time a patient with one level increase in TDP-43 stage will score 2 points lower on Boston naming test (1 point = 1 word; maximum score = 60 points) than a patient with TDP-43 stage one level lower. In terms of rate, a patient with one level increase in TDP-43 stage will be scoring approximately 0.2 points lower per year on BNT compared to a patient with TDP-43 stage one level lower which results in naming 1 word less in 5 years. We modeled time as age at death minus age at clinical evaluation. The mixed models included participant-specific random intercepts and random slopes. All analyses were performed using R version 3.6.2. Mixed models were fit using the lme4 package version 1.1-21.
We also performed a subanalysis excluding all patients with high ADNC, i.e., Braak NFT stage VI and frequent neuritic plaques (C3), n = 45, in order to see whether associations and direction of the rate of decline are maintained when high likelihood ADNC cases are removed.
RESULTS
Demographic, genetic, pathologic, and clinical characteristics of the participants are summarized in Table 1. Of 385 participants included in this study, 125 (32%) had TDP-43 immunoreactive inclusions. TDP-43 positive participants were more likely to be APOE ɛ4 carriers, have more advanced ADNC and LBD, be older and more cognitively impaired at the last evaluation prior to death; these differences were not statistically significant between the TDP-43 stages (T1 versus T2) and in case of LBD also T0 and T1 except for age at last clinical evaluation. All participants had at least two clinical evaluations with the majority in all groups having more than four evaluations prior to death, and those with T2 having the highest number of visits. For each participant we calculated a rate of change on each clinical score and summarized these rates along with baseline values in Table 2; the difference shown between TDP-43 groups in baseline scores and rate of decline in performance are from univariate analysis unadjusted for potential confounders.
Table 1
Demographic, genetic, pathologic, and clinical characteristics of participants grouped by TDP-43 stage
| Characteristic | T0 | T1 | T2 | Overalla | T0/T1 | T0/T2 | T1/T2 |
| n = 260 | n = 32 | n = 93 | p | p | p | p | |
| Female, n (%) | 131 (50%) | 17 (53%) | 58 (62%) | 0.13 | |||
| APOE ɛ4 carrier, n (%) | 71 (27%) | 16 (50%) | 36 (39%) | 0.01 | 0.01 | 0.048 | 0.30 |
| Amyloid- β, CERAD, n (%) | < 0.001 | 0.005 | < 0.001 | 0.61 | |||
| C0 | 81 (31%) | 7 (22%) | 15 (16%) | ||||
| C1 | 65 (25%) | 3 (9%) | 16 (17%) | ||||
| C2 | 66 (25%) | 7 (22%) | 25 (27%) | ||||
| C3 | 48 (18%) | 15 (47%) | 37 (40%) | ||||
| Tau, Braak NFT stage, n (%) | < 0.001 | < 0.001 | < 0.001 | 0.38 | |||
| 0 | 5 (2%) | 0 (0%) | 0 (0%) | ||||
| I | 22 (8%) | 1 (3%) | 3 (3%) | ||||
| II | 60 (23%) | 6 (19%) | 8 (9%) | ||||
| III | 49 (19%) | 2 (6%) | 11 (12%) | ||||
| IV | 59 (23%) | 4 (12%) | 18 (19%) | ||||
| V | 49 (19%) | 6 (19%) | 26 (28%) | ||||
| VI | 16 (6%) | 13 (41%) | 27 (29%) | ||||
| Lewy body disease stage, n (%) | 0.003 | 0.17 | 0.001 | 0.30 | |||
| 0 | 207 (80%) | 22 (71%) | 55 (60%) | ||||
| 1 | 10 (4%) | 0 (0%) | 4 (4%) | ||||
| 2 | 27 (10%) | 6 (19%) | 17 (18%) | ||||
| 3 | 16 (6%) | 3 (10%) | 16 (17%) | ||||
| Vascular score, n (%) | 0.58 | ||||||
| 0 | 133 (51%) | 18 (56%) | 48 (52%) | ||||
| 1 | 97 (37%) | 10 (31%) | 39 (42%) | ||||
| 2 | 30 (12%) | 4 (12%) | 6 (6%) | ||||
| Diagnosis at last exam, n (%) | < 0.001 | 0.003 | < 0.001 | 0.68 | |||
| CU | 148 (57%) | 10 (31%) | 20 (22%) | ||||
| MCI | 49 (19%) | 5 (16%) | 14 (15%) | ||||
| AD | 41 (16%) | 14 (44%) | 45 (48%) | ||||
| Other | 22 (8%) | 3 (9%) | 14 (15%) | ||||
| Age at clinical exam, y | |||||||
| Baseline | 81 (44, 98) | 76 (59, 97) | 81 (54, 95) | 0.054 | |||
| Last | 87 (53, 102) | 83 (69, 105) | 88 (60, 101) | 0.02 | 0.10 | 0.04 | 0.01 |
| Years from baseline to death | 7.1 (0.3, 22.4) | 8.0 (2.6, 16.6) | 9.9 (1.3, 18.2) | < 0.001 | 0.35 | < 0.001 | 0.03 |
| No. of visits | 4.5 (1.0, 15.0) | 5.5 (2.0, 14.0) | 6.0 (1.0, 14.0) | < 0.001 | 0.10 | < 0.001 | 0.36 |
| 2 | 30 (12%) | 4 (12%) | 6 (6%) | ||||
| 3 | 42 (16%) | 4 (12%) | 7 (8%) | ||||
| 4 | 37 (14%) | 4 (12%) | 7 (8%) | ||||
| > 4 | 130 (50%) | 20 (62%) | 69 (74%) |
Data shown are n (%) or median (range). ap-values for continuous variables are from Wilcoxon rank sum test or ANOVA; p-values for categorical variables are from Fisher’s Exact test. CU, cognitively unimpaired; MCI, mild cognitive impairment; AD, Alzheimer’s disease.
Table 2
Summary of clinical scores of participants grouped by TDP-43 stage
| Clinical test/max score | T0 | T1 | T2 | Overalla | T0/T1 | T0/T2 | T1/T2 |
| n = 260 | n = 32 | n = 93 | p | p | p | p | |
| MMSE/30 | |||||||
| Baseline | 28 (21, 30) | 28 (23, 30) | 27 (20, 30) | 0.41 | |||
| Rate | –0.23 (–5.0, 2.82) | –0.82 (–4.1, 0.62) | –0.83 (–8.0, 0.86) | < 0.001 | 0.003 | < 0.001 | 0.60 |
| CDRSB/18b | |||||||
| Baseline | 0.0 (0.0, 1.0) | 0.0 (0.0, 1.0) | 0.0 (0.0, 1.0) | < 0.001 | 0.01 | < 0.001 | 0.92 |
| Rate | 0.00 (–0.35, 3.58) | 0.76 (0.00, 5.73) | 0.70 (–0.82, 4.17) | < 0.001 | 0.001 | < 0.001 | 0.77 |
| BNT/60 | |||||||
| Baseline | 54 (25, 60) | 52 (25, 60) | 53 (9, 60) | 0.053 | |||
| Rate | –0.24 (–9.4, 3.91) | –0.92 (–7.9, 1.53) | –1.41 (–10.7, 1.11) | < 0.001 | 0.02 | < 0.001 | 0.53 |
| WMSR Log Mem I/50 | |||||||
| Baseline | 20 (0, 39) | 15 (4, 39) | 17 (2, 34) | < 0.001 | 0.02 | < 0.001 | 0.76 |
| Rate | –0.40 (–9.7, 10.97) | –0.92 (–5.93, 4.54) | –1.29 (–4.1, 5.64) | < 0.001 | 0.051 | < 0.001 | 0.64 |
| Total NPI severity/36b | |||||||
| Baseline | 0 (0, 9) | 1 (0, 9) | 0 (0, 10) | 0.008 | 0.002 | 0.18 | 0.10 |
| Rate | 0.02 (–3.85, 3.76) | 0.32 (–1.69, 2.55) | 0.17 (–1.44, 6.43) | 0.35 | |||
| AVLT Delayed Recall/15 | |||||||
| Baseline | 5 (0, 15) | 4 (0, 15) | 3 (0, 12) | < 0.001 | 0.02 | < 0.001 | 0.64 |
| Rate | –0.12 (–9.69, 6.75) | –0.15 (–3.94, 3.03) | –0.30 (–2.58, 1.71) | 0.17 | |||
| Sum AVLT 1–5/75 | |||||||
| Baseline | 34 (13, 60) | 33 (19, 62) | 31 (13, 48) | 0.06 | |||
| Rate | –0.60 (–17.44, 10.97) | –0.85 (–10.10, 6.06) | –1.44 (–13.89, 4.29) | < 0.001 | 0.28 | < 0.001 | 0.27 |
| Sum AVLT 1–7/105 | |||||||
| Baseline | 46 (15, 89) | 42 (21, 92) | 40 (13, 73) | 0.007 | 0.15 | 0.003 | 0.68 |
| Rate | –0.93 (–31.97, 22.78) | –1.33 (–13.22, 10.61) | –2.41 (–20.43, 7.72) | < 0.001 | 0.35 | < 0.001 | 0.17 |
Data shown are n (%) or median (range). ap-values are from Wilcoxon rank sum test. bHigher score indicates greater cognitive impairment.
Linear mixed effects model results are summarized in Fig. 1 and Tables 3 and 4. For the cross-sectional component of these model-based analyses, higher TDP-43 stage was independently associated with significantly worse scores on all tests except for NPI-Q. Higher Braak NFT stage was associated with worse performance on all clinical tests. Higher Aβ neuritic plaque score was independently associated with worse scores on MMSE, CDRSB, and NPI-Q. Higher LBD stage was associated with worse scores on NPI-Q only. Other important associations identified were: older age was associated with lower scores on BNT and NPI-Q; female sex was associated with worse performance on BNT and better performance on all subscores of AVLT. While education years differed significantly between men and women (median 15 years [range 6–20] in men versus median 14 years [range 6–20], p = 0.03), when models were adjusted for education, sex differences were still present for BNT and AVLT in cross-sectional model (p < 0.001).
Fig. 1
Forest plot showing estimates and 95% confidence intervals for cross-sectional and longitudinal models Forest plots that do not cross red dotted line represent statistically significant associations (p < 0.05). In our model, age at death was centered at 80 years old with 5 years per unit increase, and time to death was centered at 5 years before death with one year per unit increase.
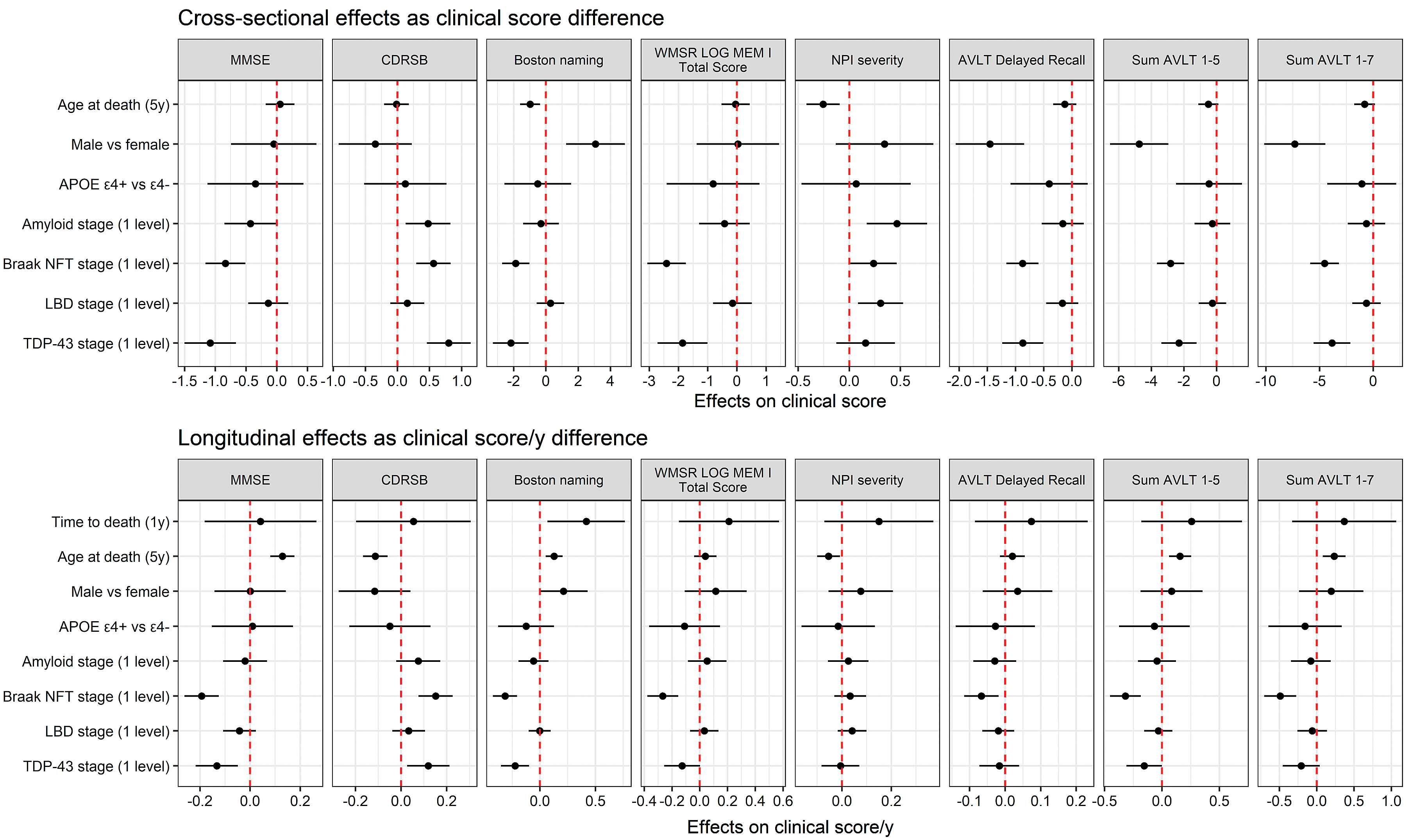
Table 3
Estimates and 95% confidence intervals from linear mixed-effects models for clinical battery of tests
| Clinical test | MMSE model | CDRSB model | BNT model | NPI model | ||||
| Estimatea | p | Estimate | p | Estimate | p | Estimate | p | |
| Cross-sectional | ||||||||
| Age at death (5 y) | 0.05 (–0.18, 0.29) | 0.66 | –0.02 (–0.21, 0.18) | 0.87 | –0.98 (–1.60, –0.36) | 0.002 | –0.26 (–0.42, –0.09) | 0.002 |
| Male versus female | –0.05 (–0.74, 0.64) | 0.89 | –0.35 (–0.92, 0.22) | 0.23 | 3.07 (1.25, 4.89) | < 0.001 | 0.35 (–0.13, 0.82) | 0.16 |
| APOE ɛ4 + versus ɛ4- | –0.35 (–1.13, 0.43) | 0.38 | 0.12 (–0.52, 0.76) | 0.71 | –0.51 (–2.57, 1.56) | 0.63 | 0.07 (–0.47, 0.60) | 0.81 |
| Aβ stage (1 level)b | –0.43 (–0.85, –0.004) | 0.048 | 0.48 (0.13, 0.83) | 0.007 | –0.31 (–1.42, 0.81) | 0.59 | 0.47 (0.17, 0.76) | 0.002 |
| Braak NFT stage (1 level) | –0.84 (–1.16, –0.51) | < 0.001 | 0.56 (0.29, 0.83) | < 0.001 | –1.87 (–2.72, –1.02) | < 0.001 | 0.24 (0.01, 0.46) | 0.04 |
| LBD stage (1 level) | –0.14 (–0.46, 0.19) | 0.41 | 0.15 (–0.11, 0.42) | 0.26 | 0.28 (–0.57, 1.14) | 0.52 | 0.31 (0.09, 0.53) | 0.007 |
| TDP-43 stage (1 level) | –1.08 (–1.50, –0.66) | < 0.001 | 0.80 (0.46, 1.14) | < 0.001 | –2.17 (–3.28, –1.07) | < 0.001 | 0.16 (–0.13, 0.45) | 0.28 |
| Longitudinal | ||||||||
| Time to death (1 y) | 0.04 (–0.18, 0.26) | 0.71 | 0.05 (–0.20, 0.30) | 0.67 | 0.42 (0.07, 0.77) | 0.02 | 0.15 (–0.07, 0.37) | 0.18 |
| Age at death (5 y) | 0.13 (0.08, 0.18) | < 0.001 | –0.11 (–0.17, –0.06) | < 0.001 | 0.13 (0.05, 0.20) | 0.001 | –0.05 (–0.10, –0.01) | 0.02 |
| Male versus female | 0.0004 (–0.14, 0.14) | > 0.99 | –0.12 (–0.27, 0.04) | 0.15 | 0.21 (–0.01, 0.43) | 0.06 | 0.08 (–0.05, 0.21) | 0.25 |
| APOE ɛ4 + versus ɛ4- | 0.01 (–0.15, 0.17) | 0.91 | –0.05 (–0.23, 0.13) | 0.59 | –0.13 (–0.38, 0.13) | 0.33 | –0.01 (–0.16, 0.13) | 0.85 |
| Aβ stage (1 level) | –0.02 (–0.11, 0.07) | 0.66 | 0.08 (–0.02, 0.17) | 0.12 | –0.06 (–0.19, 0.08) | 0.40 | 0.03 (–0.06, 0.11) | 0.54 |
| Braak NFT stage (1 level) | –0.19 (–0.26, –0.12) | < 0.001 | 0.15 (0.08, 0.23) | < 0.001 | –0.31 (–0.42, –0.21) | < 0.001 | 0.03 (–0.03, 0.10) | 0.30 |
| LBD stage (1 level) | –0.04 (–0.11, 0.02) | 0.21 | 0.03 (–0.04, 0.11) | 0.36 | –0.00 (–0.10, 0.10) | 0.95 | 0.04 (–0.02, 0.10) | 0.16 |
| TDP-43 stage (1 level) | –0.13 (–0.22, –0.05) | 0.002 | 0.12 (0.03, 0.21) | 0.01 | –0.22 (–0.35, –0.10) | < 0.001 | –0.01 (–0.08, 0.07) | 0.89 |
aEstimates are expressed in terms of 1 point clinical score change. bAmyloid-β neuritic plaque score, CERAD C0-3.
Table 4
Estimates and 95% confidence intervals from linear mixed-effects models for memory measures
| Clinical test | WMSR Log | AVLT Delayed | Sum AVLT | Sum AVLT | ||||
| Mem I model | Recall model | 1–5 model | 1–7 model | |||||
| Estimatea | p | Estimate | p | Estimate | p | Estimate | p | |
| Cross-sectional | ||||||||
| Age at death (5 y) | –0.04 (–0.53, 0.44) | 0.86 | –0.13 (–0.33, 0.08) | 0.23 | –0.51 (–1.11, 0.10) | 0.10 | –0.81 (–1.78, 0.16) | 0.10 |
| Male versus female | 0.03 (–1.38, 1.45) | 0.96 | –1.45 (–2.06, –0.85) | < 0.001 | –4.74 (–6.53, –2.95) | < 0.001 | –7.32 (–10.2, –4.47) | < 0.001 |
| APOE ɛ4 + versus ɛ4- | –0.82 (–2.41, 0.77) | 0.31 | –0.41 (–1.09, 0.28) | 0.24 | –0.47 (–2.49, 1.55) | 0.65 | –1.08 (–4.29, 2.14) | 0.51 |
| Aβ stage (1 level)b | –0.43 (–1.29, 0.44) | 0.33 | –0.16 (–0.54, 0.21) | 0.38 | –0.26 (–1.36, 0.84) | 0.64 | –0.63 (–2.38, 1.11) | 0.48 |
| Braak NFT stage (1 level) | –2.41 (–3.07, –1.75) | < 0.001 | –0.88 (–1.16, –0.59) | < 0.001 | –2.82 (–3.66, –1.98) | < 0.001 | –4.54 (–5.88, –3.20) | < 0.001 |
| LBD stage (1 level) | –0.15 (–0.82, 0.51) | 0.65 | –0.17 (–0.46, 0.11) | 0.23 | –0.26 (–1.10, 0.59) | 0.55 | –0.64 (–1.97, 0.70) | 0.35 |
| TDP-43 stage (1 level) | –1.86 (–2.72, –1.01) | < 0.001 | –0.87 (–1.24, –0.51) | < 0.001 | –2.31 (–3.39, –1.23) | < 0.001 | –3.85 (–5.57, –2.13) | < 0.001 |
| Longitudinal | ||||||||
| Time to death (1 y) | 0.21 (–0.15, 0.57) | 0.25 | 0.07 (–0.08, 0.23) | 0.36 | 0.26 (–0.18, 0.70) | 0.25 | 0.37 (–0.33, 1.06) | 0.30 |
| Age at death (5 y) | 0.04 (–0.04, 0.12) | 0.34 | 0.02 (–0.01, 0.06) | 0.25 | 0.16 (0.06, 0.25) | 0.001 | 0.23 (0.08, 0.39) | 0.003 |
| Male versus female | 0.11 (–0.11, 0.34) | 0.31 | 0.03 (–0.06, 0.13) | 0.49 | 0.08 (–0.19, 0.35) | 0.55 | 0.19 (–0.24, 0.63) | 0.38 |
| APOE ɛ4 + versus ɛ4- | –0.11 (–0.37, 0.15) | 0.40 | –0.03 (–0.14, 0.08) | 0.63 | –0.07 (–0.37, 0.24) | 0.67 | –0.16 (–0.65, 0.33) | 0.53 |
| Aβ stage (1 level) | 0.05 (–0.09, 0.19) | 0.45 | –0.03 (–0.09, 0.03) | 0.34 | –0.04 (–0.21, 0.12) | 0.60 | –0.08 (–0.34, 0.19) | 0.57 |
| Braak NFT stage (1 level) | –0.27 (–0.38, –0.15) | < 0.001 | –0.07 (–0.12, –0.02) | 0.007 | –0.32 (–0.45, –0.18) | < 0.001 | –0.49 (–0.70, –0.28) | < 0.001 |
| LBD stage (1 level) | 0.03 (–0.07, 0.13) | 0.53 | –0.02 (–0.06, 0.03) | 0.39 | –0.03 (–0.16, 0.09) | 0.60 | –0.06 (–0.26, 0.14) | 0.55 |
| TDP-43 stage (1 level) | –0.13 (–0.26, 0.00) | 0.054 | –0.02 (–0.07, 0.04) | 0.56 | –0.16 (–0.31, –0.0005) | 0.049 | –0.21 (–0.46, 0.04) | 0.10 |
aEstimates are expressed in terms of 1 point clinical score change. bAmyloid-β neuritic plaque score, CERAD C0-3.
For the longitudinal component of our model-based analysis, higher TDP-43 stage was independently associated with faster rates of decline in performance on MMSE, CDRSB, BNT, sum AVLT 1–5, and a trend of faster rates of decline in performance on WMSR Logical Memory I and sum AVLT 1–7. Higher Braak NFT stage was independently associated with faster rate of decline in performance on all tests except for NPI. In contrast, when controlling for all other explanatory variables (sex, APOE ɛ4, Aβ neuritic plaque score, LBD, TDP-43, and Braak NFT stages), we found that older age was associated with a slower rate of decline in performance on MMSE, CDRSB, BNT, NPI, sum AVLT 1–5, and sum AVLT 1–7 (Fig. 1, Tables 3 and 4).
In the subanalysis performed with high likelihood ADNC cases excluded, all of the above reported associations persisted.
DISCUSSION
In this clinical-pathologic study of 385 autopsied older adults with ample longitudinal clinical evaluation, we found that after controlling for sex, age, APOE ɛ4 carriership status, Braak NFT stage, Aβ neuritic plaque score, and LBD stage, an increase in TDP-43 stage is independently associated with faster rate of decline in global cognition (MMSE), functional performance (CDRSB), naming (BNT) and episodic memory (sum AVLT 1–5). These longitudinal associations including all measures of episodic memory (WMSR Log Mem I, all AVLT subscores) were also observed with increasing Braak NFT stage. Older age, on the other hand, was associated with slower rate of decline in global cognition, functional performance, naming, episodic memory, and accumulation of psychiatric symptoms (NPI).
Our finding of TDP-43 being associated with greater impairment in global cognition, functional performance, naming, and episodic memory at a single point in time is in keeping with previous reports from our group [7, 8], as well as other centers [11, 12, 29, 30]; as in our previous studies, these association are stage-dependent with higher TDP-43 stage relating to greater impairment [8, 25, 26]. The novelty of this study is that higher TDP-43 stage is also associated with faster decline in global cognition, functional performance, and poorer naming and memory performances which were also stage-dependent.
Faster rates of global cognitive decline and episodic memory deterioration have previously been reported by colleagues at Rush University Medical Center which we have now validated in an independent cohort [31–33]. In addition, we also link TDP-43 stage to faster decline in naming performance and to functional decline. Given that entorhinal cortex is a functional hub for naming [34] and an increase of TDP-43 stage is associated with progressive atrophy affecting the entorhinal cortex [4], this association is not surprising.
Episodic memory is functionally attributed to the hippocampal formation [6, 34] and, hence, might be the most vulnerable to TDP-43 deposition and progression given that faster rates of hippocampal atrophy are linked to TDP-43 and that atrophy accelerates with increase in TDP-43 stage [2, 5]. Therefore, our clinical findings related to the rate of episodic memory decline might mirror TDP-43-mediated accelerated rate of hippocampal atrophy. An interesting question is whether an increase in TDP-43 stages has a greater effect on encoding (sum AVLT 1–5) compared to retrieval (AVLT Delayed Recall) as faster rate of decline in performance only on certain AVLT subscores has been found in this study. This might be the case with varying susceptibility of hippocampal subfields to TDP-43 aggregation and TDP-43-associated rate of atrophy which clinically could translate into different degrees of impairment of particular components of episodic memory. A clinical-neuropathologic study assessing the associations between TDP-43 burden and/or atrophy in hippocampal subfields (e.g., CA2/3 versus subiculum) and the components of episodic memory is needed to test this hypothesis.
Wilson et al. [33] found an association between higher TDP-43 burden and faster rates of decline in working memory which functionally is connected to frontal regions [35]. We did not test for the association with working memory in this study, although we have shown that hypometabolism on [18F]-flurodeoxyglucose PET in frontal regions is a feature of TDP-43 deposition even when TDP-43 deposition is limited to amygdala, and in the absence of significant atrophy on magnetic resonance imaging [36]. Therefore, impairment in working memory may be related to TDP-43-mediated functional network disruption between frontal and limbic regions rather than direct TDP-43 aggregation/atrophy of frontal neocortex.
A recent study reported that neuropsychiatric symptoms were less frequent in patients with concomitant TDP-43 and AD pathologies compared to pure AD suggesting a neuroprotective effect of TDP-43 in regards to emergence of psychiatric features in patients with AD and, hence, warranting longitudinal studies [37]. We, however, did not observe a positive or negative association between the severity of psychiatric symptoms and TDP-43 stage in our longitudinal or cross-sectional analyses. Hence, we cannot confirm or refute this observation. Given our findings and evidence from colleagues [33], increase in TDP-43 distribution may lead to acceleration in decline in multiple cognitive domains resulting in overall functional decline and potentially faster progression to dementia.
Increasing Braak NFT stage was also associated with faster rates of decline in global cognition, functional performance, naming and episodic memory. The effect of tau on accelerated decline in various cognitive domains has also been reported by another group of researchers [31–33]. In addition to that, here we show in an autopsy-confirmed cohort that an increase in Braak NFT stage is associated with faster overall functional decline measured by CDRSB. Overall, NFT-tau related to an accelerated decline in performance on all administered tests with the exception of NPI. Taken together, this finding from our study and the others is evidence that increases in NFT-tau may have a more widespread effect affecting even more cognitive domains, both cross-sectionally and longitudinally, which can be related to its more widespread distribution in neocortex compared to TDP-43 [23, 25, 26]. It is unclear whether there is an interaction between NFT-tau and TDP-43 that might affect the rate of decline on clinical tests and further research is warranted; however, we did not find the modification by NFT-tau of the TDP-43 effect on rates of hippocampal atrophy in our earlier study [5]. Another recent study also reported finding lower MMSE final scores in patients with higher TDP-43 stage independent of both early and late tau pathology; however, no difference in rate of decline was found possibly due to the smaller sample size and reduced power [30].
Age is an important variable associated with rate of decline in cognitive performance and had an opposite association to TDP-43 and tau—older age was associated with slower decline in performance and function. Interestingly, we found a similar relationship between age and rate of hippocampal atrophy [5] which perhaps is the product of acceleration resulting in the most drastic change in hippocampal volume over time happens earlier in life and once a certain “volume threshold” has been reached, that further decline is relatively slower compared to early rates [38]. Given that older age was associated with poorer performance cross-sectionally, the “threshold” theory might be appropriate. Another possibility is that older participants in this group, particularly those with hippocampal TDP-43, had more research visits compared to TDP-43 negative participants. Each research visit included administration of the same battery of tests; therefore, the age-related improvement in performance could be a potential result of practice effects [39]. Lastly, slower rates of decline at older age can be the effect of more severity tau deposition in younger patients [40].
Unlike with TDP-43, NFT-tau, and age, no other associations with rate of decline were found. Cross-sectionally, we found evidence suggesting that an increase in Aβ neuritic plaque score was related to poorer functional performance and a higher fre-quency of behavioral/psychiatric symptoms as measured by the NPI-Q the latter finding being discordant with a study from another group on neuropsychiatric symptoms and ADNC where only associations with Braak NFT stage were found [41]. A higher frequency of behavioral/psychiatric symptoms was also found to be associated with higher LBD stage which is consistent with findings from another center where psychosis, hallucinations and delusions in patients with pathologically confirmed AD were also associated with higher LBD stage [42]. Our study also further suggests that that association is not being driven by tau NFT, Aβ, or TDP-43. Our findings of female advantage on verbal memory tasks are consistent with previous reports and might reflect a sex-specific form of cognitive reserve and, therefore, emphasize the need for sex-adjustments for AVLT [43–45]. Men, on the other hand, historically outperform women on naming tests with a possible explanation of greater left hemispheric lateralization for language in men compared to women and, therefore, naming advantage [46].
Strengths of this study include the longitudinal design, large sample size, ample clinical and pathologic data, and robust statistical analyses. There are also limitations. While we concentrated on the association of TDP-43 with global cognition, functional performance, behavior and particularly episodic memory, there are other associations worth exploring. We also acknowledge that our cohort is not racially diverse with almost all participants being Caucasian of European descent and of non-Hispanic ethnicity which may limit generalizability to other populations.
The present study provides further evidence that TDP-43 has an impact on clinical signs and symptoms and furthermore contributes to faster rates of cognitive, behavioral, and functional decline along with NFT-tau. Hence, unraveling peculiarities of the clinical phenotype of participants with TDP-43 in combination with reported potential imaging associations [7, 36] can aid in antemortem diagnosis and prognosis, and will be important for clinical trial enrollment and surveillance.
ACKNOWLEDGMENTS
We thank the families of the participants who donated their brains to science allowing for the completion of this study. This study was funded by National Institutes of Health grants R01 AG37491-11 (KAJ), P30 AG062677 (RCP) and U01 AG006786 (RCP). These grants served for the design and conduct of the study, collection, management and analysis of the collected data. The sponsor had no role in study design; collection, analysis and interpretation of data; writing the report; or in the decision to submit the article for publication.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-1166r1).
REFERENCES
[1] | Neumann M , Sampathu DM , Kwong LK , Truax AC , Micsenyi MC , Chou TT , Bruce J , Schuck T , Grossman M , Clark CM ((2006) ) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314: , 130–133. |
[2] | Josephs KA , Dickson DW , Tosakulwong N , Weigand SD , Murray ME , Petrucelli L , Liesinger AM , Senjem ML , Spychalla AJ , Knopman DS ((2017) ) Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer’s disease: A longitudinal retrospective study. Lancet Neurol 16: , 917–924. |
[3] | Nelson PT , Dickson DW , Trojanowski JQ , Jack CR , Boyle PA , Arfanakis K , Rademakers R , Alafuzoff I , Attems J , Brayne C ((2019) ) Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 142: , 1503–1527. |
[4] | Bejanin A , Murray ME , Martin P , Botha H , Tosakulwong N , Schwarz CG , Senjem ML , Chételat G , Kantarci K , Jack CR Jr ((2019) ) Antemortem volume loss mirrors TDP-43 staging in older adults with non-frontotemporal lobar degeneration. Brain 142: , 3621–3635. |
[5] | Buciuc M , Wennberg AM , Weigand SD , Murray ME , Senjem ML , Spychalla AJ , Boeve BF , Knopman DS , Jack CR , Kantarci K ((2020) ) Effect modifiers of TDP-43-associated hippocampal atrophy rates in patients with Alzheimer’s disease neuropathological changes. J Alzheimers Dis 73: , 1511–1523. |
[6] | Eichenbaum H ((1993) ) Memory, amnesia, and the hippocampal system, MIT Press. |
[7] | Josephs KA , Whitwell JL , Knopman DS , Hu WT , Stroh DA , Baker M , Rademakers R , Boeve BF , Parisi JE , Smith GE ((2008) ) Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 70: , 1850–1857. |
[8] | Josephs KA , Whitwell JL , Weigand SD , Murray ME , Tosakulwong N , Liesinger AM , Petrucelli L , Senjem ML , Knopman DS , Boeve BF ((2014) ) TDP-43 is a key player in the clinical features associated with Alzheimer’s disease. Acta Neuropathol 127: , 811–824. |
[9] | Buciuc M , Whitwell JL , Tosakulwong N , Weigand SD , Murray ME , Boeve BF , Knopman DS , Parisi JE , Petersen RC , Dickson DW ((2020) ) Association between transactive response DNA-binding protein of 43 kDa type and cognitive resilience to Alzheimer’s disease: A case-control study. Neurobiol Aging 92: , 92–97. |
[10] | Latimer CS , Burke BT , Liachko NF , Currey HN , Kilgore MD , Gibbons LE , Henriksen J , Darvas M , Domoto-Reilly K , Jayadev S ((2019) ) Resistance and resilience to Alzheimer’s disease pathology are associated with reduced cortical pTau and absence of limbic-predominant age-related TDP-43 encephalopathy in a community-based cohort. Acta Neuropathol Commun 7: , 9. |
[11] | Robinson JL , Corrada MM , Kovacs GG , Dominique M , Caswell C , Xie SX , Lee VM-Y , Kawas CH , Trojanowski JQ ((2018) ) Non-Alzheimer’s contributions to dementia and cognitive resilience in The 90+Study. Acta Neuropathol 136: , 377–388. |
[12] | Nelson PT , Abner EL , Schmitt FA , Kryscio RJ , Jicha GA , Smith CD , Davis DG , Poduska JW , Patel E , Mendiondo MS ((2010) ) Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol 20: , 66–79. |
[13] | Robinson JL , Geser F , Corrada MM , Berlau DJ , Arnold SE , Lee VM-Y , Kawas CH , Trojanowski JQ ((2011) ) Neocortical and hippocampal amyloid-β and tau measures associate with dementia in the oldest-old. Brain 134: , 3708–3715. |
[14] | Roberts RO , Geda YE , Knopman DS , Cha RH , Pankratz VS , Boeve BF , Ivnik RJ , Tangalos EG , Petersen RC , Rocca WA ((2008) ) The Mayo Clinic Study of Aging: Design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 30: , 58–69. |
[15] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E ((2012) ) National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[16] | Crook R , Hardy J , Duff K ((1994) ) Single-day apolipoprotein E genotyping. J Neurosci Methods 53: , 125–127. |
[17] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[18] | Hughes CP , Berg L , Danziger W , Coben LA , Martin RL ((1982) ) A new clinical scale for the staging of dementia. Br J Psychiatry 140: , 566–572. |
[19] | Kaufer DI , Cummings JL , Ketchel P , Smith V , MacMillan A , Shelley T , Lopez OL , DeKosky ST ((2000) ) Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 12: , 233–239. |
[20] | Kaplan E , Goodglass H , Weintraub S ((2001) ) Boston Naming Test, Pro-ed. |
[21] | ((2011) ) WMS-R. In Encyclopedia of Clinical Neuropsychology, Kreutzer JS, DeLuca J, Caplan B, eds. Springer, New York. |
[22] | Rey A ((1958) ) L’examen clinique en psychologie. |
[23] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[24] | Mirra SS , Heyman A , McKeel D , Sumi S , Crain BJ , Brownlee L , Vogel F , Hughes J , Van Belle G , Berg L ((1991) ) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41: , 479–479. |
[25] | Josephs KA , Murray ME , Whitwell JL , Parisi JE , Petrucelli L , Jack CR , Petersen RC , Dickson DW ((2014) ) Staging TDP-43 pathology in Alzheimer’s disease. Acta Neuropathol 127: , 441–450. |
[26] | Josephs KA , Murray ME , Whitwell JL , Tosakulwong N , Weigand SD , Petrucelli L , Liesinger AM , Petersen RC , Parisi JE , Dickson DW ((2016) ) Updated TDP-43 in Alzheimer’s disease staging scheme. Acta Neuropathol 131: , 571–585. |
[27] | Beekly DL , Ramos EM , Lee WW , Deitrich WD , Jacka ME , Wu J , Hubbard JL , Koepsell TD , Morris JC , Kukull WA ((2007) ) The National Alzheimer’s Coordinating Center (NACC) database: The uniform data set. Alzheimer Dis Assoc Disord 21: , 249–258. |
[28] | Wennberg AM , Whitwell JL , Tosakulwong N , Weigand SD , Murray ME , Machulda MM , Petrucelli L , Mielke MM , Jack CR Jr , Knopman DS ((2019) ) The influence of tau, amyloid, alpha-synuclein, TDP-43, and vascular pathology in clinically normal elderly individuals. Neurobiol Aging 77: , 26–36. |
[29] | Nag S , Yu L , Wilson RS , Chen E-Y , Bennett DA , Schneider JA ((2017) ) TDP-43 pathology and memory impairment in elders without pathologic diagnoses of AD or FTLD. Neurology 88: , 653–660. |
[30] | McAleese KE , Walker L , Erskine D , Johnson M , Koss D , Thomas AJ , Attems J ((2020) ) Concomitant LATE-NC in Alzheimer’s disease is not associated with increased tau or amyloid-β pathological burden. Neuropathol Appl Neurobiol 46: , 722–734. |
[31] | Boyle PA , Yang J , Yu L , Leurgans SE , Capuano AW , Schneider JA , Wilson RS , Bennett DA ((2017) ) Varied effects of age-related neuropathologies on the trajectory of late life cognitive decline. Brain 140: , 804–812. |
[32] | Wilson RS , Capuano AW , Bennett DA , Schneider JA , Boyle PA ((2016) ) Temporal course of neurodegenerative effects on cognition in old age. Neuropsychology 30: , 591. |
[33] | Wilson RS , Yu L , Trojanowski JQ , Chen E-Y , Boyle PA , Bennett DA , Schneider JA ((2013) ) TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 70: , 1418–1424. |
[34] | Van Strien N , Cappaert N , Witter M ((2009) ) The anatomy of memory: An interactive overview of the parahippocampal–hippocampal network. Nat Rev Neurosci 10: , 272–282. |
[35] | Squire LR ((2009) ) Memory and brain systems: 1969–2009. J Neurosci 29: , 12711–12716. |
[36] | Buciuc M , Botha H , Murray ME , Schwarz CG , Senjem ML , Jones DT , Knopman DS , Boeve BF , Petersen RC , Jack CR ((2020) ) Utility of FDG-PET in diagnosis of Alzheimer-related TDP-43 proteinopathy. Neurology 95: , e23–e34. |
[37] | Liu KY , Reeves S , McAleese KE , Attems J , Francis P , Thomas A , Howard R ((2020) ) Neuropsychiatric symptoms in limbic-predominant age-related TDP-43 encephalopathy and Alzheimer’s disease. Brain 143: , 3842–3849. |
[38] | Josephs KA , Martin PR , Weigand SD , Tosakulwong N , Buciuc M , Murray ME , Petrucelli L , Senjem ML , Spychalla AJ , Knopman DS ((2020) ) Protein contributions to brain atrophy acceleration in Alzheimer’s disease and primary age-related tauopathy. Brain 143: , 3463–3476. |
[39] | Rabbitt P , Lunn M , Wong D , Cobain M ((2008) ) Age and ability affect practice gains in longitudinal studies of cognitive change. J Gerontol B Psychol Sci Soc Sci 63: , P235–P240. |
[40] | Whitwell JL , Martin P , Graff-Radford J , Machulda MM , Senjem ML , Schwarz CG , Weigand SD , Spychalla AJ , Drubach DA , Jack CR Jr ((2019) ) The role of age on tau PET uptake and gray matter atrophy in atypical Alzheimer’s disease. Alzheimers Dement 15: , 675–685. |
[41] | Ehrenberg AJ , Suemoto CK , França Resende EdP , Petersen C , Leite REP , Rodriguez RD , Ferretti-Rebustini REdL , You M , Oh J , Nitrini R ((2018) ) Neuropathologic correlates of psychiatric symptoms in Alzheimer’s disease. J Alzheimers Dis 66: , 115–126. |
[42] | Fischer CE , Qian W , Schweizer TA , Millikin CP , Ismail Z , Smith EE , Lix LM , Shelton P , Munoz DG ((2016) ) Lewy bodies, vascular risk factors, and subcortical arteriosclerotic leukoencephalopathy, but not Alzheimer pathology, are associated with development of psychosis in Alzheimer’s disease. J Alzheimers Dis 50: , 283–295. |
[43] | Caselli RJ , Dueck AC , Locke DE , Baxter LC , Woodruff BK , Geda YE ((2015) ) Sex-based memory advantages and cognitive aging: A challenge to the cognitive reserve construct?. J Int Neuropsychol Soc 21: , 95. |
[44] | Sundermann EE , Maki PM , Rubin LH , Lipton RB , Landau S , Biegon A , Initiative AsDN ((2016) ) Female advantage in verbal memory: Evidence of sex-specific cognitive reserve. Neurology 87: , 1916–1924. |
[45] | Stricker NH , Christianson TJ , Lundt ES , Alden EC , Machulda MM , Fields JA , Kremers WK , Jack CR Jr , Knopman DS , Mielke MM , Petersen RC ((2020) ) Mayo Normative Studies: Regression-based normative data for the Auditory Verbal Learning Test for ages 30-91 years and the importance of adjusting for sex. J Int Neuropsychol Soc, doi: 10.1017/S1355617720000752 |
[46] | Gordon HW ((1980) ) Cerebral organization in bilinguals: I. Lateralization. Brain Lang 9: , 255–268. |


