
Reduction of Dendritic Inhibition in CA1 Pyramidal Neurons in Amyloidosis Models of Early Alzheimer’s Disease
Abstract
Background:
In an early stage of Alzheimer’s disease (AD), before the formation of amyloid plaques, neuronal network hyperactivity has been reported in both patients and animal models. This suggests an underlying disturbance of the balance between excitation and inhibition. Several studies have highlighted the role of somatic inhibition in early AD, while less is known about dendritic inhibition.
Objective:
In this study we investigated how inhibitory synaptic currents are affected by elevated Aβ levels.
Methods:
We performed whole-cell patch clamp recordings of CA1 pyramidal neurons in organotypic hippocampal slice cultures after treatment with Aβ-oligomers and in hippocampal brain slices from AppNL-F-G mice (APP-KI).
Results:
We found a reduction of spontaneous inhibitory postsynaptic currents (sIPSCs) in CA1 pyramidal neurons in organotypic slices after 24 h Aβ treatment. sIPSCs with slow rise times were reduced, suggesting a specific loss of dendritic inhibitory inputs. As miniature IPSCs and synaptic density were unaffected, these results suggest a decrease in activity-dependent transmission after Aβ treatment. We observed a similar, although weaker, reduction in sIPSCs in CA1 pyramidal neurons from APP-KI mice compared to control. When separated by sex, the strongest reduction in sIPSC frequency was found in slices from male APP-KI mice. Consistent with hyperexcitability in pyramidal cells, dendritically targeting interneurons received slightly more excitatory input. GABAergic action potentials had faster kinetics in APP-KI slices.
Conclusion:
Our results show that Aβ affects dendritic inhibition via impaired action potential driven release, possibly due to altered kinetics of GABAergic action potentials. Reduced dendritic inhibition may contribute to neuronal hyperactivity in early AD.
INTRODUCTION
In the early phase of Alzheimer’s disease (AD), before patients experience behavioral problems, hyperactive neuronal networks in the brain have been reported [1–3]. It has even been suggested that neuronal hyperactivity is the primary neuronal dysfunction in early AD [4]. The hyperactivity increases the prevalence of epilepsy in the early phases of the disease, both in transgenic mouse models [5–8] and in human patients [1–3]. Several studies have linked hyperactivity to increased amyloid-β (Aβ) levels [5, 7], which are already elevated in the early stages of the disease [9, 10].
A hyperactive network indicates a disturbed balance of excitation and inhibition, via increased excitation, decreased inhibition, or both. Increased activity of excitatory neurons in early AD has been reported by several laboratories [7, 8, 11, 12] and various mechanism have been proposed, including dendritic hyperexcitability [11], dendritic instability [12], and impaired glutamate reuptake [13]. Recently, altered inhibition is getting more attention as a possible contributor to network hyperexcitability in AD [14, 15]. Parvalbumin (PV) interneurons provide strong perisomatic inhibition to principal cells and changes in PV neurons will have a direct impact on network activity. Cortical PV neurons show reduced activity [6, 16], whereas hippocampal PV neurons were actually found hyperactive in early AD [17]. In both brain regions, restoring PV interneuron activity alleviates disease pathology [6, 7, 16, 17].
In many AD mouse models, amyloid-β protein precursor (AβPP) is overexpressed to elevate Aβ levels. In wildtype mice, AβPP is expressed mostly in neurons and shows region- and cell-specific differences [18], which are lost in these mouse models [19]. Interestingly, dendritic innervating GABAergic interneurons in the hippocampal CA1 region express AβPP at a high level [18, 20]. Dendritic innervating interneurons may therefore be sensitive to local changes in Aβ, but their role in early AD has been largely unexplored. Given the evidence for enhanced dendritic excitability in the CA1 region early in AD [11, 12], we assessed how dendritic inhibition is affected by elevated Aβ levels.
Here we use two amyloidosis models to examine how elevated Aβ levels affect inhibitory currents in CA1 pyramidal neurons. We applied soluble Aβ oligomers in organotypic hippocampal slice cultures to study acute effects of elevated Aβ on inhibitory neurotransmission. We compared our results with hippocampal slices of young adult AppNL-G-F mice, a second generation amyloidosis mouse model with three knock-in mutations in the humanized App-gene [21]. AppNL-G-F mice chronically overproduce Aβ without potential side-effects of AβPP overexpression [19].
MATERIALS AND METHODS
Mice
All animal experiments were performed in compliance with the guidelines for the welfare of experi-mental animals issued by the Federal Government of The Netherlands. All animal experiments were approved by the Animal Ethical Review Committee (DEC) of Utrecht University. GAD65-GFP mice [22] express GFP in ∼20% of GABAergic cells, which mainly innervate the dendrites of CA1 pyramidal neurons in the hippocampus [23]. AppNL-G-F mice express three mutations in the humanized APP gene, leading to an increased production of Aβ42. AppNL-G-F mice were crossed with the GAD65-GFP line (AppNL-F-G/GAD65-GFP) to identify dendritically targeting interneurons in this amyloidosis mouse model. The AppNL-G-F line was kept homozygous and GAD65-GFP line was heterozygous. We refer to this crossing as APP-KI mice throughout the manuscript. GAD65-GFP mice were used as control mice.
Brain slices
Organotypic hippocampal slice cultures were prepared from postnatal day 6–8 old GAD65-GFP mice as previously described [24, 25]. In short, mice of both sexes were decapitated and their brains were placed in ice cold Gey’s Balanced Salt Solution (GBSS, in mM: 137 NaCl, 5 KCl, 1.5 CaCl2, 1 MgCl2, 0.3 MgSO4, 0.2 KH2PO4, 0.85 Na2HPO4). The hippocampus was extracted and transverse hippocampal slices of 400μm thickness were cut with a McIlwain tissue chopper (Brinkmann Instruments). Slices were kept in an incubator at 35°C with 5% CO2. Slices were used after 10–20 days in vitro (DIV). Slices were treated for 24 h with 0.4μg/ml Aβ-oligomers (Crossbeta Biosciences, Utrecht) or control vehicle before the experiments.
Acute hippocampal slices were made from GAD65-GFP (control) and APP-KI mice. Male and female mice at the age of 8 to 14 weeks were sedated using isoflurane and decapitated. The brain was quickly removed and placed in ice-cold slicing medium (in mM: 92 CholineCl, 2.5 KCL, 1.2 NaH2PO4, 30 NaHCO3, 20 HEPES, 25 glucose, 5 Na-ascorbate, 3 Na-pyruvate, 10 MgSO4·7H2O, 0.5 CaCl2·2H2O, pH ∼7.3, ∼315 mOsm) for 2 min. Subsequently, the cerebellum and the prefrontal cortex were removed and the brain was placed in the vibratome chamber (LEICA VT 1000S) in ice-cold carbogenated artificial cerebrospinal fluid (ACSF; in mM: 126 NaCl, 3 KCl, 2.5 CaCl2·2H2O, 1.3 MgCl2·7H2O, 26 NaHCO3, 1.25 Na2H2Po4, 20 glucose, pH ∼7.3, ∼315 mOsm). Coronal hippocampal slices of 300μm thickness were made. Afterwards, slices were transferred to a slice chamber with ACSF at 35°C for 10 min. The slices were kept in ACSF at room temperature for at least 1 h before the experiment. All dissections were performed in the morning and the experiments after lunch.
Treatment
Organotypic slices were treated for 24 h with 0.4μg/ml Aβ-oligomers (Crossbeta Biosciences, Utrecht). At this concentration, these Aβ oligomers are able to block LTP in vivo [26]. The vehicle solution was used as control (20 mM HEPES, 150 mM NaCl, 200 mM sucrose). The oligomers used in this study are chemically stabilized and uniform in size, no monomers and fibrils were present in the solution. The oligomers have been obtained using recombinant full length amyloid-β 1–42 peptide as starting material and based on a protocol that is described before [27].
Electrophysiology
Slices were transferred to a recording chamber which was perfused with carbonated, heated ASCF (32°C). Whole-cell patch clamp measurements were recorded with a MultiClamp 700B amplifier (Molecular Devices) and stored using pClamp 10 software. Recordings were filtered with a 3 kHz Bessel filter. Thick-walled borosilicate pipettes of 4–6 MΩ were filled with two different internal solutions. For the recording of sIPSCs (in mM): 70 K-gluconate, 70 KCl, 0.5 EGTA, 10 HEPES, 4 MgATP, 0.4 NaGTP, and 4 Na2 phosphocreatine. For the recording of sEPSCs (in mM): 140 K-gluconate, 4 KCl, 0.5 EGTA, 10 HEPES, 4 MgATP, 0.4 NaGTP, and 4 Na2 phosphocreatine. Cells were discarded if series resistance was above 35 MΩ or if the resting membrane potential exceeded –50 mV. Recordings were excluded when the series resistance after the recording deviated more than 30% from its original value.
Inhibitory currents (sIPSCs) were isolated by adding APV (1μM) and DNQX (1μM) to the ASCF, which block AMPA and NMDA currents respectively. No blockers were added for recordings of sEPSCs and action potential. sIPSCs and sEPSCs were recorded at –70 mV holding potential.
Immunohistochemistry
Organotypic slices were fixed after 24 h Aβ or vehicle treatment with 4% PFA for 30 min. After washing, slices were permeabilized with 0.5% TitronX-100 in PBS for 15 min. Slices were subsequently blocked with 10% goat serum and 0.2% TitronX-100 in PBS for 1 h, before primary antibodies were added in blocking buffer for overnight incubation at 4°C (rabbit VGAT 1/1000, SySy: 131003; mouse Gephyrin 1/1000, SySy: 147011). Secondary antibodies were added the next day for 3.5 h at room temperature (α-rabbit 405 1/500, Life Technologies A31556; α-mouse 647 1/500, Life Technologies A21236).
Confocal imaging
Fixed slices were imaged under a Zeiss LSM700 inverted confocal laser scanning microscope using Zen 2011 software (Zeiss). The 405 nm and 647 nm antibodies were excited with a 405 nm and 633 nm laser. Stained organotypic hippocampal slices were viewed with a 63×/1.40 NA oil immersion objective (Plan-Apochromat, Zeiss) at 1.3× digital zoom. Image stacks (123μm×123μm, 1024×1024 pixels) with a 0.3μm step size in z were acquired (range: 18–22 images). Per slice, 5 different Z-stacks were obtained from the stratum radiatum and the stratum lacunosum-moleculare border in the CA1 area.
Analysis
Spontaneous and miniature inhibitory postsynaptic currents (sIPSCs and mIPSCs) were analyzed with MiniAnalysis (synaptosoft). Only events with a rise time <7 ms were included in our analysis. This excludes slow rise time events resulting from spillover [28, 29]. Action potential (AP) parameters were extracted in Matlab (MathWorks) using a custom script. AP threshold was manually determined from the change in slope at the AP rising phase. AP amplitude was calculated from threshold to peak. AP rise and decay times were determined between 10% and 90% of amplitude; halfwidth at 50% of amplitude. Afterhyperpolarization was calculated from the threshold to minimum. The rebound potential after a current injection was calculated from pre-train baseline to minimum right after the train. Cumulative distributions were generated from 150 events per cell.
Immunofluorescent images were analyzed in Fiji using a custom macro. Three to six Z-stacks were averaged and filtered. Puncta from thresholded images were counted. Synapses were counted when the VGAT and gephyrin channel overlapped for at least 33%.
Statistical analysis was performed in Prism (Graphpad software). For the comparison of two groups we either used the unpaired two-tailed t-test (t; parametric) or a Mann-Whitney (MW; nonparametric). Determining a normal distribution was done with the D’Agostino & Pearson test. Welch’s t-test was used for two groups with unequal variance (Fig. 4B). The IEI of slow and fast rise time events were tested with 2-way ANOVA and a Sidak’s post-hoc test. The p-values are indicated in the figure legends. Error bars indicate standard error of the mean. Significance is reported as *p≤0.05; **p≤0.01; ***p≤0.001
Splitting IPSCs based on their rise time
The rise times of individual sIPSCs and mIPSCs were plotted in a histogram and fitted with a double Gaussian distribution [30] (Equation 1).
(1)
The coefficients of the fit were used to draw the two single exponentials (Equation 2 & 3).
(2)
(3)
The intersection between Y1 and Y2 was used as the cutoff rise time. Events with a rise time smaller than the cutoff were categorized as fast events and events with larger rise time were labeled slow rise time events (Supplementary Figure 1). We determined the cutoff value in the control group and used this value to separate the control and the experimental group. The cutoff value for the experimental group did not deviate much from the control, yet to have equal comparison between groups, we only utilized the cutoff value of the control group.
RESULTS
Reduced dendritic inhibition after 24 h Aβ treatment
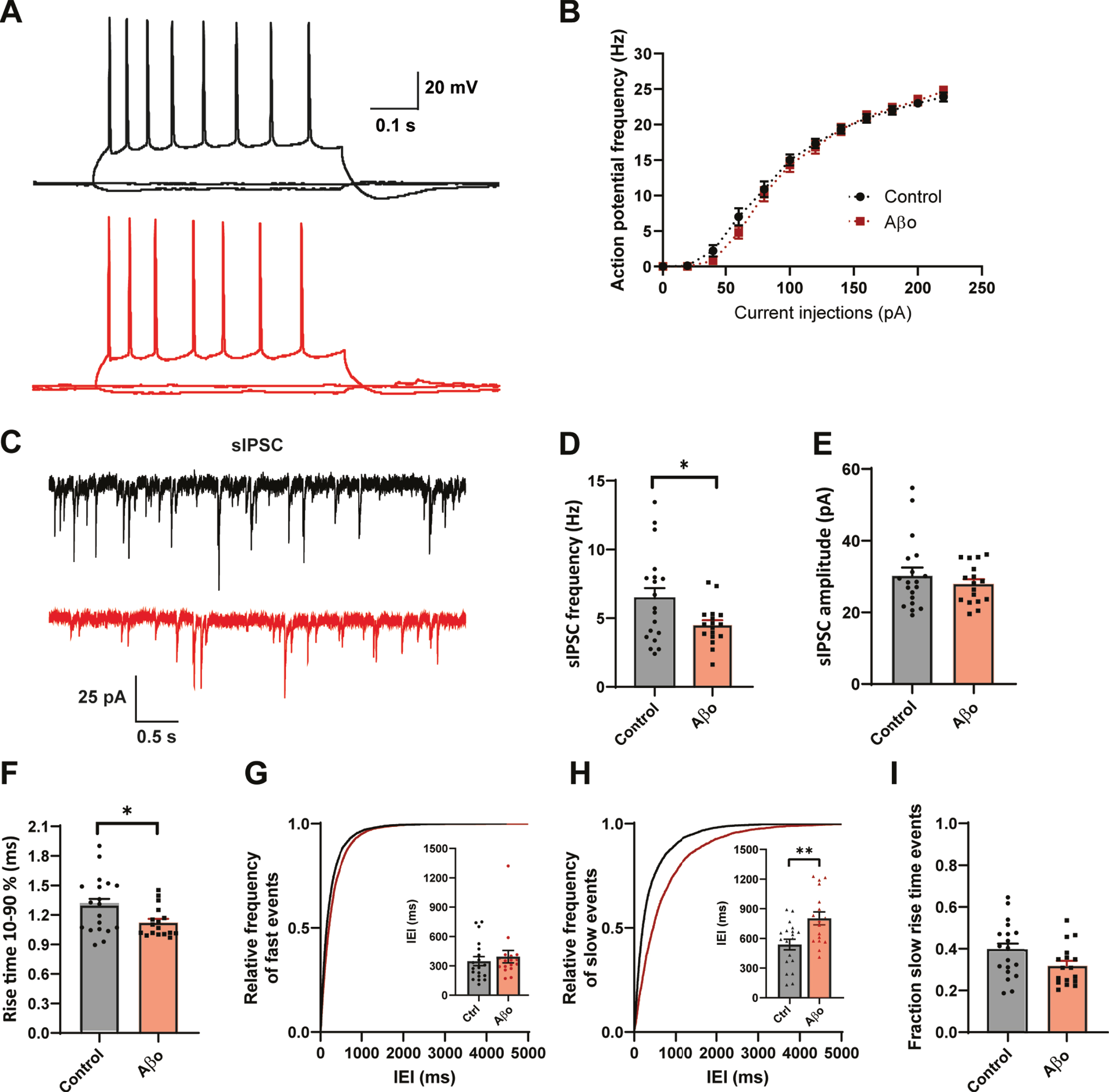
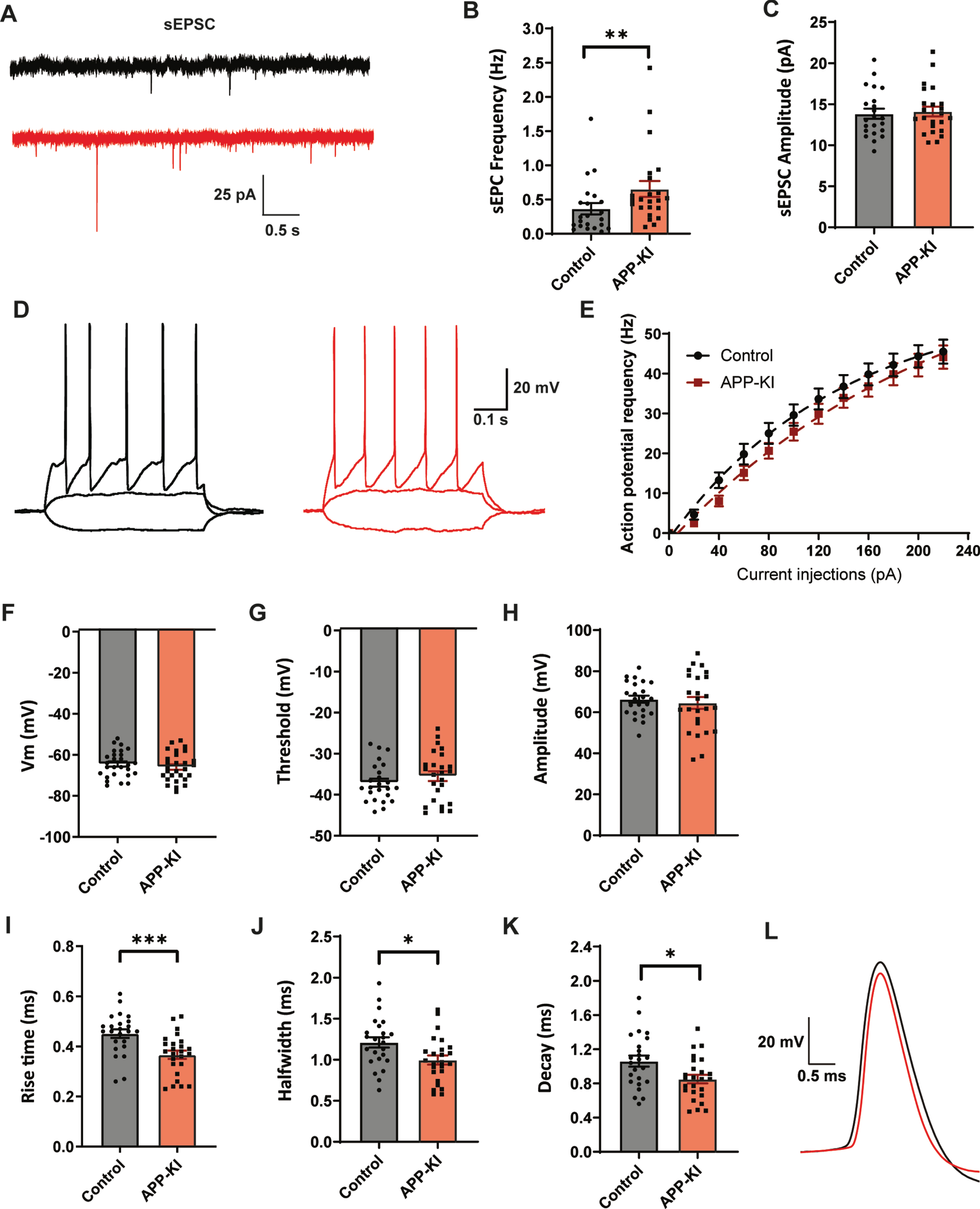
To address acute effects of Aβ on inhibitory synaptic transmission we treated organotypic hippocampal slice cultures from GAD65-GFP mice for 24 h with 0.4μg/ml Aβ-oligomers or control vehicle. Whole-cell patch clamp recordings were made from CA1 pyramidal neurons. Aβ treatment did not affect AP firing rates with increasing current injections in these cells (Fig. 1A, B). Beside a small change in AP decay time, we found no other changes in AP parameters (Supplementary Figure 1). We investigated the inhibitory synaptic input in the CA1 neurons by recording spontaneous inhibitory postsynaptic currents (sIPSCs) (Fig. 1C). We observed that sIPSC frequency was significantly decreased in CA1 pyramidal neurons after Aβ treatment (Fig. 1D), whereas sIPSC amplitude was unaffected (Fig. 1E). Remarkably, the average rise time of the sIPSCs was faster in the Aβ treated slices (Fig. 1F). The sIPSC rise time depends on the cellular location of the inhibitory synapse, with somatic synapses producing sIPSCs with faster rise time and synapses on the dendrites generate slower rise time events due to dendritic filtering [30–33]. To examine if somatic and dendritic synaptic inputs were differentially affected, we separated the sIPSCs in two groups (see Methods and Supplementary Figure 2) and analyzed interevent intervals (IEIs) of sIPSCs with fast and slow rise times for each recording. The mean IEI of fast sIPSCs was similar in treated and control neurons (Fig. 1G), whereas the mean IEI of slow events in Aβ-treated slices was increased (Fig. 1H). This suggests a specific reduction in sIPSCs with slow rise times, presumably originating from dendritic synapses. Accordingly, we observed a trend towards reduction in the fraction of sIPSCs with a slow rise time in Aβ-treated slices (Fig. 1I). We verified that the reduction in slow sIPSCs after Aβ treatment was not due to differences in series resistance (Supplementary Table 1). Our results suggest that 24 h Aβ treatment specifically reduces inhibitory transmission at dendritic synapses.
Fig. 1
24 h Aβ oligomer treatment reduces dendritic inhibition in organotypic hippocampal slices. A) Example traces of action potentials after current injections. B) No change in action potential firing rates in control slices (black, n = 20, N = 6) and Aβ-treated slice (red, n = 18, N = 6) with increasing current injections. C) Representative sIPSC recording from control (black) and Aβ-oligomer treated (red) slices. D, E) Aβ reduces sIPSC frequency (D; p = 0.028, t), but leaves sIPSC amplitude (E; p = 0.40, t) unchanged. F) Rise time of sIPSCs was faster in the Aβ-treated slices (p = 0.029, t). G, H) Cumulative distribution of inter-event intervals (IEIs) of fast rise time (G) and slow rise time (H) events (p = 0.82 and p = 0.003, Sidak). The inserts show the mean IEI (I) Fraction of sIPSCs with slow rise times in Aβ-treated and control slices (p = 0.08, t). (Data in C–I: control n = 21, N = 6 Aβ n = 20, N = 6).
No change in synapse numbers after 24 h Aβ treatment
A reduction in the frequency of sIPSCs may reflect decreased presynaptic activity and/or a reduction of the number of (dendritic) synapses. To elucidate the role of activity, we blocked neuronal activity with TTX and recorded miniature inhibitory postsynaptic currents (mIPSCs) in organotypic slices (Fig. 2A). We found that mIPSC frequency (Fig. 2B), amplitude (Fig. 2C), and rise time (Fig. 2D) were similar in treated and control slices. The distribution of fast and slow mIPSCs was also unaffected by Aβ treatment (Fig. 2E–G). A lack of effect on mIPSCs suggests that altered activity is responsible for the reduced dendritic inhibition.
Fig. 2
No effect on synapse number after 24 h Aβ oligomer application. A) Representative mIPSC recordings from control (black) and Aβ-oligomer treated (red) slices. B–D) The frequency (B; p = 0.9, MW), amplitude (C; p = 0.68, t), and rise time (D; p = 0.23, MW) of mIPSCs were not changed. E, F) Cumulative distribution of inter-event intervals (IEIs) of fast rise time (E) and slow rise time (F) events (p = 0.42 and p = 0.66, Sidak). The inserts show the mean IEI. G) Fraction of sIPSCs with slow rise times in Aβ-treated and control slices. H) Representative images of immunofluorescent staining against postsynaptic gephyrin and presynaptic VGAT. Scale bar denotes 3μm. I, J) The density of VGAT (I; p = 0.9, t) and gephyrin (J; p = 0.5, t) puncta was similar in control and Aβ-treated slices. K) Synapse density, determined by overlap of the VGAT and gephyrin puncta, was not altered by Aβ treatment (p = 0.9, MW). (Data in B–G: control n = 12, N = 4; Aβ n = 12, N = 4. Data in H–K: control n = 15, N = 3; Aβ n = 15, N = 3).
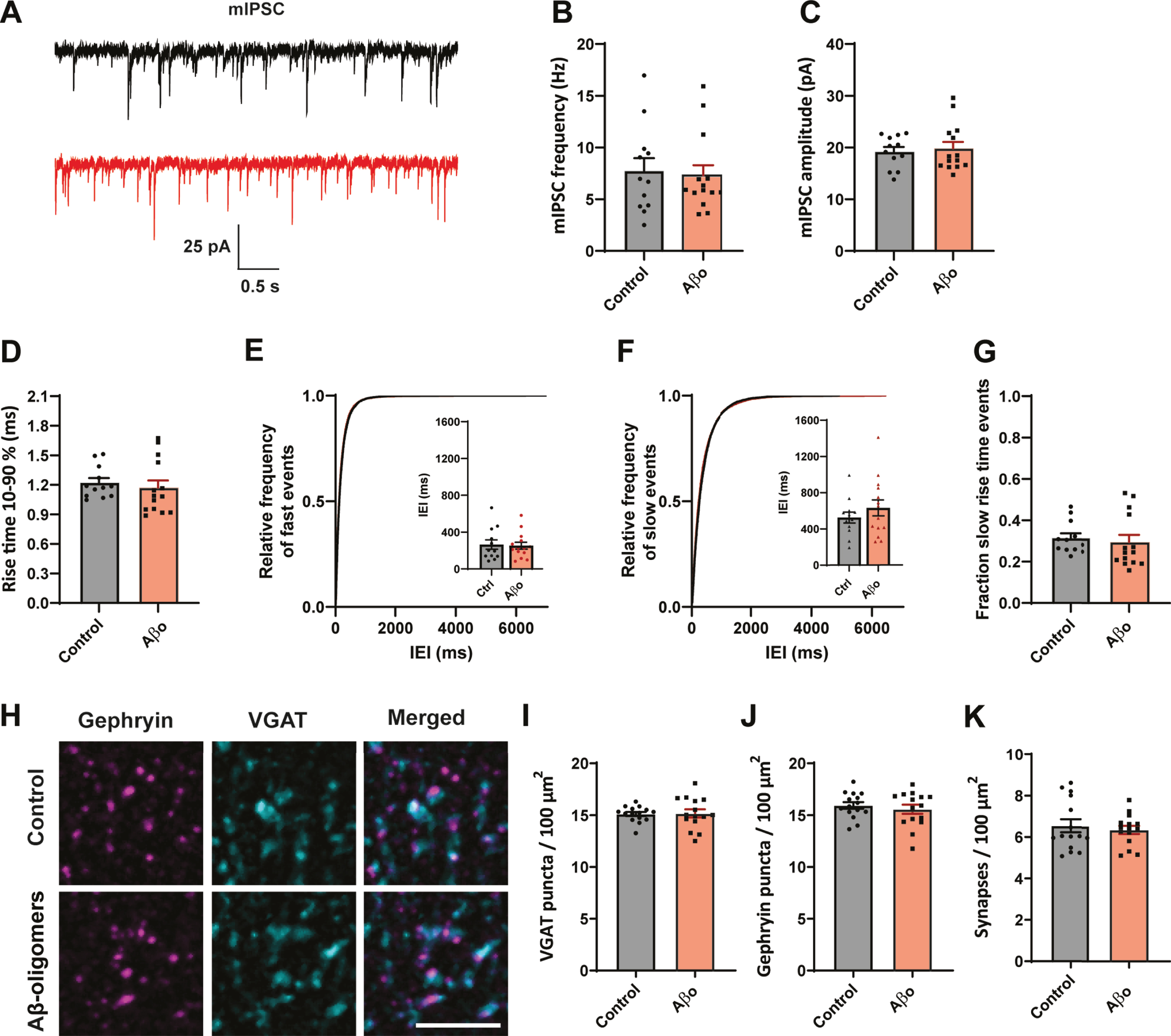
To further assess a possible change in synapse numbers, we performed immunohistochemistry to visualize inhibitory synapses in the dendritic layers of the hippocampus using presynaptic VGAT and postsynaptic gephyrin (Fig. 2H). The density of VGAT (Fig. 2I) and gephyrin puncta (Fig. 2J) was similar in organotypic slices that were treated with Aβ or control vehicle for 24 h. The density of inhibitory synapses, determined by overlap of VGAT and gephyrin puncta, did not change (Fig. 2K).
These results suggest that inhibitory synapse numbers were not affected by Aβ-oligomer treatment and that the observed reduction in dendritic inhibitory currents in CA1 neurons was due to altered activity-dependent GABA release.
Similar changes in inhibition in a chronic model of amyloidosis
To address if our findings with acute Aβ treatment are representative for early AD, in which Aβ levels are chronically elevated, we made use of an APP-KI mouse model [21]. In APP-KI mice, plaques appear around two months [21] and cognitive impairments are reported at 6 months of age [34, 35]. We performed whole-cell patch clamp recordings of CA1 pyramidal cells in acute hippocampal slices of 8–14 week old APP-KI and control mice. This is the age at which the first plaques start to appear in APP-KI mice [18, 21]. In contrast to Aβ-treated organotypic slices, CA1 pyramidal neurons in APP-KI slices displayed an elevated AP firing rate with increasing current injections (Fig. 3A, B), indicating that cellular excitability was increased when Aβ levels were chronically elevated. We did not observe any difference in resting membrane potential, AP amplitude or AP halfwidth compared to control (Fig. 3C–E). A small, but significant, reduction in AP threshold was observed (Fig. 3F). Other AP parameters were not changed (Supplementary Figure 3). Together this suggests an increased excitability of hippocampal pyramidal neurons in young APP-KI mice, which is in line with the hyperactivity reported in other mouse models at comparable age [5–8].
Fig. 3
Increased excitability and mild changes IPSCs in APP-KI mice. A) Representative traces of current injections in CA1 pyramidal neurons of control (black) and APP-KI (red) slices. B) APP-KI cells were able to fire action potentials at a higher frequency than control cells at increasing current injections (p < 0.0001 2w ANOVA). C) Resting membrane voltage of CA1 pyramidal neurons (p = 0.73, t). D–F) Action potential amplitude (D, measured from threshold; p = 0.88, t), AP halfwidth (E; p = 0.71, MW), and AP threshold (F; p = 0.027, t) in APP-KI and control cells. G) Representative sIPSC recording from control (black) and APP-KI (red) slice. H–J) Frequency (H; p = 0.12, t), amplitude (I; p = 0.79, t), and rise time (J; p = 0.18, MW) of sIPSCs in APP-KI and control cells. K-L) Cumulative distribution of inter-event intervals (IEIs) of fast rise time (K) and slow rise time (L) events (p = 0.96 and p = 0.96, Sidak). The inserts show the mean IEI. M) Fraction of sIPSCs with slow rise times in Aβ-treated and control slices (p = 0.21, t). (Data in B–F: control n = 20, N = 9; APP-KI n = 18, N = 7. Data in H–K: control n = 20, N = 9; Aβ n = 20, N = 7).
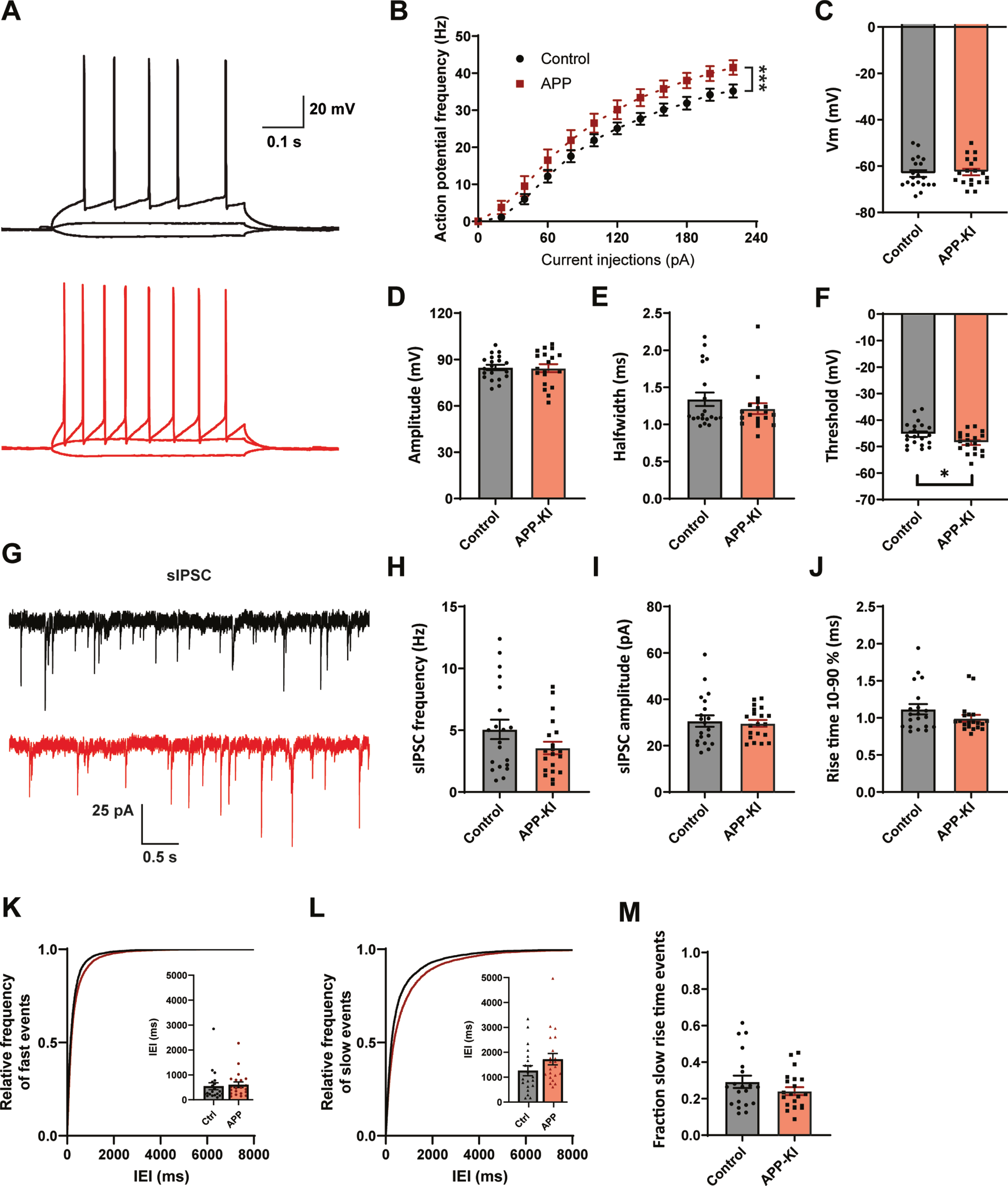
We investigated the inhibitory synaptic input by recording sIPSCs in these CA1 pyramidal neurons (Fig. 3G). The frequency of sIPSCs appeared slightly reduced in APP-KI cells, but the difference did not reach significance (Fig. 3H). There was no difference in sIPSC amplitude or rise time (Fig. 3I-J). When we separated sIPSCs based on rise time, we observed a small increase in IEI only for slow sIPSCs (Fig. 3K-L). The fraction of slow sIPSCs was only mildly reduced (Fig. 3M). Although the pattern was similar to the organotypic slices, the reduction in sIPSCs was only mild in APP-KI slices and did not reach significance.
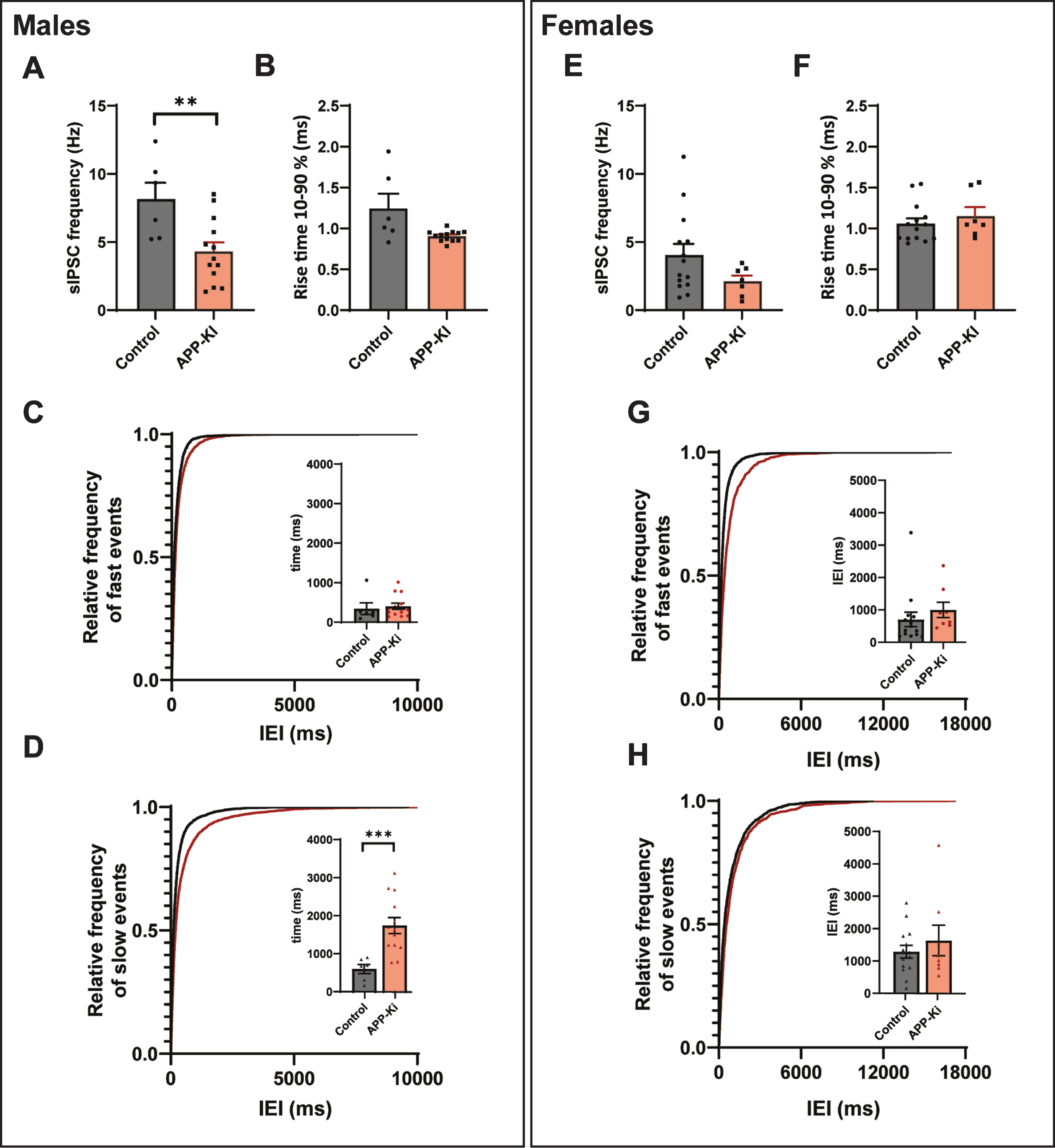
APP-KI males have a stronger reduction in inhibition
It is well documented that gender has a significant influence on the progression of AD, with women affected more severely at the late stage of the disease, which may be related to the loss of estrogen [36–38]. As the mice in our experiments were sexually mature, we explored whether differences between the sexes played a role in our results. In line with previous reports [39, 40], the frequency of sIPSCs was lower in slices from females compared to male mice (Fig. 4A, E). We found that sIPSC frequency was significantly decreased in neurons recorded in slices from male APP-KI mice (Fig. 4A) and rise times appeared reduced (Fig. 4B), while there was no significant change in sIPSCs frequency or rise time recorded in slices from females (Fig. 4E, F). Separation by rise time showed that the reduction of inhibitory transmission in the males was due to a loss of slow rise time events, suggesting a specific decrease in dendritic inhibition (Fig. 4C, D). There was no difference in dendritic inhibition in the females (Fig. 4G, H).
Fig. 4
Sex differences in APP-KI mice. A, B) The frequency (p = 0.008, t) and rise time (p = 0.11, t) of sIPSCs, recorded in slices from male APP-KI and control mice. C, D) Cumulative distribution of inter-event intervals (IEIs) of fast rise time (C) and slow rise time (D) events recorded in slices from male APP-KI and control mice (p = 0.96 and p = 0.0001, Sidak). The inserts show the mean IEI. E, F) The frequency (Mann-Whitney p = 0.13) and rise time (t-test p = 0.87) of sIPSCs, recorded in slices from female APP-KI and control mice. G, H) Same as is C and D, but for sIPSCs recorded in female mice (p = 0.70 and p = 0.62, Sidak). Same data as in Fig. 3H–M, but separated by sex. (Data in A–D: control n = 6, N = 3; APP-KI n = 13, N = 4. Data in D–H: control n = 11, N = 2; Aβ n = 7, N = 5).
Changed action potential kinetics of GABAergic neurons
Dendritic inhibition onto CA1 pyramidal cells is provided by different types of GABAergic interneurons. Some of these GABAergic cells, located on the border of the stratum radiatum and stratum lacunosum-moleculare, reportedly express AβPP at high levels [18]. We wondered if these GABAergic cells were responsible for the reduced dendritic inhibition observed in APP-KI slices. To visualize these cells we used GAD65-GFP mice, in which a subset of dendritically targeting GABAergic cells, partially overlapping with AβPP-expressing cells [18], are labeled with GFP [23].
We performed whole-cell patch clamp recordings of GFP-expressing GABAergic interneurons in the dendritic layers of the hippocampal CA1. We observed an increase in the frequency of excitatory postsynaptic currents (sEPSCs) in GABAergic cells in APβP-KI slices compared to control (Fig. 5A, B), consistent with enhanced excitability of pyramidal cells (Fig. 1A, B). The amplitudes of sEPSCs were not different (Fig. 5C). AP firing rates of GABAergic neurons, recorded with increasing current injections, were not different in slices from APP-KI and control mice (Fig. 5D, E) and there was also no difference in resting membrane potential (Fig. 5F), AP threshold (Fig. 5G), and AP amplitude (Fig. 5H). However, AP rise time (Fig. 5I), halfwidth (Fig. 5J), and decay time (Fig. 5K) were all strongly reduced in APP-KI slices. This indicates that the APs of GFP-expressing GABAergic neurons are narrower and faster in APP-KI slices (Fig. 5L). These results indicate that dendritically innervating inhibitory neurons on the border of the stratum radiatum and stratum lacunosum-moleculare have altered AP kinetics in APP-KI slices.
Fig. 5
Dendritically targeting interneurons in APP-KI mice have faster action potentials. A) Representative sEPSC recordings in GFP-expressing interneurons in hippocampal slice from control and APP-KI mice. B, C) Frequency (B; p = 0.009, MW), and amplitude (C; p = 0.74, t) of sEPSCs. D) Representative traces of current injections in GFP-expressing interneurons from control (black) and APP-KI mice (red). E) No change in action potential firing rate with increasing current injections (control: n = 24, APP-KI: n = 25). F–H) Resting membrane potential (F; p = 0.41, t), action potential threshold (G; p = 0.32, t), and action potential amplitude (H; p = 0.59, t) in APP-KI and control GFP-expressing interneurons. I–K) Action potential rise time (I; p = 0.0008, t), halfwidth (J; p = 0.014, t), and decay (K; p = 0.13, t) in APP-KI and control GFP-expressing interneurons. L) Representative action potentials of control (black) and APP-KI (red), highlighting the faster AP kinetics in APP-KI interneurons. (Data in B-C: control n = 22, N = 8; APP-KI n = 23, N = 8. Data in D–H: control n = 24, N = 8; Aβ n = 25, N = 8).
DISCUSSION
In this study we examined synaptic inhibition in two amyloidosis models of early AD. A 24 h treatment with Aβ oligomers significantly reduced inhibitory transmission in organotypic hippocampal slices, with a specific loss of slow rise time events presumably originating from dendritic inhibitory synapses. Analysis of synapse numbers and mIPSCs indicated that the sIPSC reduction is due to a decrease in action-potential driven GABA release. A similar, but weaker, pattern could be identified in slices of APP-KI mice, which have chronically elevated Aβ levels. The weaker effect in the APP-KI mice was due to a sex-specific effect. A specific loss of slow rise time inhibitory currents, i.e., the same pattern as in the organotypic slices, was observed only in male APP-KI mice, whereas inhibitory currents were similar in slices from female APP-KI and control mice. Furthermore, dendritically innervating GABAergic interneurons in slices from APP-KI mice displayed altered AP kinetics.
Inhibition has been reported altered in several mouse models of early AD [6, 7, 16, 35]. In particular, the somatically innervating PV interneurons have been implicated in AD, with cortical PV cells reported to be hypoactive [6, 16], while hippocampal PV neurons are reported hyperactive [7, 17]. In our study we did not find indications for altered activity of PV cells. Hippocampal PV interneurons project mainly to the somata of the pyramidal cells [42, 43]. If their activity was affected, we would have observed a change in fast rise time IPSCs. However, we only observed changes in slow rise time synaptic events. In our recordings we isolated inhibitory currents by blocking excitation with APV and DNQX, which also blocks excitatory input to GABAergic neurons, including PV cells. In the studies reporting increased PV activity excitatory transmission was not blocked [7, 17]. The apparent discrepancy between our current results and these earlier reports may therefore be resolved if the reported hyperactivity of PV cells requires excitatory input. In addition, cell-specific and age-dependent differences between model systems cannot be excluded [19].
Blocking Aβ production in interneurons has recently been shown to alleviate plaque load in young AD mice, indicating that GABAergic neurons contribute to early AD pathology [18], but it is unclear if a specific group of interneurons is involved. Our data suggests that Aβ does not equally affect all inhibitory synapses. We observed a specific reduction in inhibitory currents with slow rise time in APP-KI slices, as well as in Aβ-treated organotypic slices. Our analysis excludes that a difference in spillover or series resistance is responsible, and we conclude that Aβ specifically impairs dendritic inhibitory synapses. CA1 pyramidal neurons receive a wide variety of GABAergic input, with the vast majority of all inhibitory input originating from the dendrites [44]. The observed reduction in dendritic inhibition may originate from several interneuron types. Somatostatin levels and somatostatin containing O-LM interneurons are reported to gradually decrease in a mouse model of AD [45]. A later study showed that this is due to a reduced cholinergic drive from the septum [46]. In addition, an early loss of calretinin expressing interneurons was reported in APP/PS1 mice [47]. In our experiments we investigated a subset of interneurons at the border between stratum radiatum and stratum lacunosum-moleculare (sRad/sLM border), which partially overlap with interneurons with high expression of AβPP [18, 20]. These regular firing interneurons do not contain somatostatin and only a fraction expresses calretinin [23] and they primarily innervate the dendrites of pyramidal neurons [23, 48]. We found that excitability of these GABAergic neurons was not affected, while their excitatory drive was slightly enhanced, making it unlikely that the observed reduction in sIPSC frequency is due to less AP firing by these cells. Interestingly, we observed that APs recorded in dendritically targeting GABAergic neurons from APP-KI slices were narrower and faster compared to control slices. A large fraction of GFP-labeled GABAergic neurons at the sRad/sLM border are cholecystokinin (CCK)-positive interneurons [23], which all express AβPP [18]. CCK-positive interneurons are known to display prominent asynchronous neurotransmitter release [49–51], and a narrower AP would render AP-dependent GABA release inefficient at their terminals due to reduced opening of voltage gated calcium channels [52]. Furthermore, asynchronous release depends on N-type calcium channels [50, 53], which are susceptible to Aβ42 exposure [54]. In addition, Aβ may interfere with the physiological function of AβPP at GABAergic synapses [20] via interactions with reelin [55] or presynaptic GABAB receptors [56, 57].
We observed a strong effect of sex in our data. Sex is known to be a major risk factor in AD, with older women being more susceptible to AD and showing a faster disease progress [36–38]. This pattern has also been described in transgenic mouse models [58–61]. Specifically in AppNL-F-G mice, females have been reported to have heavier plaque load than males in late stages of the disease, although this was not associated with any apparent difference in behavior [34]. Surprisingly, we found that sIPSCs were stronger affected in slices from male APP-KI mice than from females (Fig. 4). One possible explanation is that in young females, the brain is protected by estrogen. The drop in estrogen levels in older females may render their brains vulnerable to Aβ toxicity [62–64]. Our APP-KI females are young fertile animals, high in estrogen, which may protect them against Aβ toxicity [63, 64].
Together our results indicate that an Aβ-dependent reduction in inhibition occurs in an early phase of AD. Our findings suggest that this is due to a specific impairment of AP-driven GABA release at dendritic inhibitory synapses, possibly as a result of altered AP kinetics. A specific impairment in dendritic inhibition may be an important contribution to enhanced dendritic excitability [11, 12] and hyperactive networks [4, 5, 7, 8] that form the earliest neuronal dysfunction in early AD.
ACKNOWLEDGMENTS
The authors thank Joris de Wit and Bart de Strooper for their generous donation of the AppNL-F-G/Gad65-GFP mouse line, Guus Scheefhals and Crossbeta Biosciences for the generous donation of the Aβ-oligomers, Christine Lützkendorf for her assistance with the immunohistochemistry and preparation of brain slices, and René van Dorland for excellent technical support. This research was supported by Alzheimer Nederland (WE.03-2016-04 and WE.03-2018-11).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0527r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200527.
REFERENCES
[1] | Vossel KA , Beagle AJ , Rabinovici GD , Shu H , Lee SE , Naasan G , Hegde M , Cornes SB , Henry ML , Nelson AB , Seeley WW , Geschwind MD , Gorno-Tempini ML , Shih T , Kirsch HE , Garcia PA , Miller BL , Mucke L ((2013) ) Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol 70: , 1158–1166. |
[2] | Vossel KA , Ranasinghe KG , Beagle AJ , Mizuiri D , Honma SM , Dowling AF , Darwish SM , Van Berlo V , Barnes DE , Mantle M , Karydas AM , Coppola G , Roberson ED , Miller BL , Garcia PA , Kirsch HE , Mucke L , Nagarajan SS ((2016) ) Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann Neurol 80: , 858–870. |
[3] | Irizarry MC , Jin S , He F , Emond JA , Raman R , Thomas RG , Sano M , Quinn JF , Tariot PN , Galasko DR , Ishihara LS , Weil JG , Aisen PS ((2012) ) Incidence of new-onset seizures in mild to moderate Alzheimer disease. Arch Neurol 69: , 368–372. |
[4] | Busche MA , Konnerth A ((2015) ) Neuronal hyperactivity - A key defect in Alzheimer’s disease? Bioessays 37: , 624–632. |
[5] | Busche MA , Chen X , Henning HA , Reichwald J , Staufenbiel M , Sakmann B , Konnerth A ((2012) ) Critical role of soluble amyloid-β for early hippocampal hyperactivity in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 109: , 8740–8745. |
[6] | Verret L , Mann EO , Hang GB , Barth AMI , Cobos I , Ho K , Devidze N , Masliah E , Kreitzer AC , Mody I , Mucke L , Palop JJ ((2012) ) Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149: , 708–721. |
[7] | Palop JJ , Chin J , Roberson ED , Wang J , Thwin MT , Bien-Ly N , Yoo J , Ho KO , Yu G-Q , Kreitzer A , Finkbeiner S , Noebels JL , Mucke L ((2007) ) Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55: , 697–711. |
[8] | Minkeviciene R , Rheims S , Dobszay MB , Zilberter M , Hartikainen J , Fülöp L , Penke B , Zilberter Y , Harkany T , Pitkänen A , Tanila H ((2009) ) Amyloid β-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29: , 3453–3462. |
[9] | Jack CR , Knopman DS , Jagust WJ , Shaw LM , Aisen PS , Weiner MW , Petersen RC , Trojanowski JQ ((2010) ) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9: , 119–128. |
[10] | Jack CR , Knopman DS , Jagust WJ , Petersen RC , Weiner MW , Aisen PS , Shaw LM , Vemuri P , Wiste HJ , Weigand SD , Lesnick TG , Pankratz VS , Donohue MC , Trojanowski JQ ((2013) ) Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol 12: , 207–216. |
[11] | Hall AM , Throesch BT , Buckingham SC , Markwardt SJ , Peng Y , Wang Q , Hoffman DA , Roberson ED ((2015) ) Tau-dependent Kv4.2 depletion and dendritic hyperexcitability in a mouse model of Alzheimer’s disease. J Neurosci 35: , 6221–6230. |
[12] | Šišková Z , Justus D , Kaneko H , Friedrichs D , Henneberg N , Beutel T , Pitsch J , Schoch S , Becker A , von der Kammer H , Remy S ((2014) ) Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer’s disease. Neuron 84: , 1023–1033. |
[13] | Zott B , Simon MM , Hong W , Unger F , Chen-Engerer H-J , Frosch MP , Sakmann B , Walsh DM , Konnerth A ((2019) ) A vicious cycle of β amyloid–dependent neuronal hyperactivation. Science 365: , 559–565. |
[14] | Palop JJ , Mucke L ((2016) ) Network abnormalities and interneuron dysfunction in Alzheimer disease. Nat Rev Neurosci 17: , 777–792. |
[15] | Ambrad Giovannetti E , Fuhrmann M ((2019) ) Unsupervised excitation: GABAergic dysfunctions in Alzheimer’s disease. Brain Res 1707: , 216–226. |
[16] | Martinez-Losa M , Tracy TE , Ma K , Verret L , Clemente-Perez A , Khan AS , Cobos I , Ho K , Gan L , Mucke L , Alvarez-Dolado M , Palop JJ ((2018) ) Nav1.1-overexpressing interneuron transplants restore brain rhythms and cognition in a mouse model of Alzheimer’s disease. Neuron 98: , 75–89.e5. |
[17] | Hijazi S , Heistek TS , Scheltens P , Neumann U , Shimshek DR , Mansvelder HD , Smit AB , van Kesteren RE ((2019) ) Early restoration of parvalbumin interneuron activity prevents memory loss and network hyperexcitability in a mouse model of Alzheimer’s disease. Mol Psychiatry, doi: 10.1038/s41380-019-0483-4 |
[18] | Rice HC , Marcassa G , Chrysidou I , Horré K , Young-Pearse TL , Müller UC , Saito T , Saido TC , Vassar R , De Wit J , De Strooper B ((2020) ) Contribution of GABAergic interneurons to amyloid-β plaque pathology in an APP knock-in mouse model. Mol Neurodegener 15: , 3. |
[19] | Sasaguri H , Nilsson P , Hashimoto S , Nagata K , Saito T , De Strooper B , Hardy J , Vassar R , Winblad B , Saido TC ((2017) ) APP mouse models for Alzheimer’s disease preclinical studies. EMBO J 36: , e201797397. |
[20] | Wang B , Wang Z , Sun L , Yang XL , Li H , Cole AL , Rodriguez-rivera J , Lu H , Zheng H ((2014) ) The amyloid precursor protein controls adult hippocampal neurogenesis through GABAergic interneurons. J Neurosci 34: , 13314–13325. |
[21] | Saito T , Matsuba Y , Mihira N , Takano J , Nilsson P , Itohara S , Iwata N , Saido TC ((2014) ) Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci 17: , 661–663. |
[22] | López-Bendito G , Sturgess K , Erdélyi F , Szabó G , Molnár Z , Paulsen O ((2004) ) Preferential origin and layer destination of GAD65-GFP cortical interneurons. Cereb Cortex 14: , 1122–1133. |
[23] | Wierenga CJ , Müllner FE , Rinke I , Keck T , Stein V , Bonhoeffer T ((2010) ) Molecular and electrophysiological characterization of GFP-expressing CA1 interneurons in GAD65-GFP mice. PLoS One 5: , e15915. |
[24] | Frias CP , Liang J , Bresser T , Scheefhals L , Kesteren M van , Dorland R van , Hu HY , Bodzeta A , Van Bergen en Henegouwen PMP , Hoogenraad CC , Wierenga CJ ((2019) ) Semaphorin4D induces inhibitory synapse formation by rapid stabilization of presynaptic boutons via MET co-activation. J Neurosci 39: , 4221–4237. |
[25] | Hu HY , Kruijssen DL , Frias CP , Rózsa B , Hoogenraad CC , Wierenga CJ ((2019) ) Endocannabinoid signaling mediates local dendritic coordination between excitatory and inhibitory synapses. Cell Rep 27: , 666–675. |
[26] | Tepper AW , de Boer EC , Hoogveld E , Vis JD , Schut IC , Diggelen F Van , Vereyken IJ , Hoffmann M , Scheefhals GA ((2015) ) Stable amyloid oligomers as tractable drug targets and versatile research tools in AD and PD. In AD/PD 2015: International Conference on Alzheimer’s and Parkinson’s Diseases, Nice, http://www.crossbeta.com/wordpress/wp-content/uploads/2017/03/150305AT-Crossbeta-Poster-ADPD.pdf |
[27] | Lambert MP , Barlow AK , Chromy BA , Edwards C , Freed R , Liosatos M , Morgan TE , Rozovsky I , Trommer B , Viola KL , Wals P , Zhang C , Finch CE , Krafft GA , Klein WL ((1998) ) Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 95: , 6448–6453. |
[28] | Clements JD , Westbrook GL ((1991) ) Activation kinetics reveal the number of glutamate and glycine binding sites on the N-methyl-d-aspartate receptor. Neuron 7: , 605–613. |
[29] | Capogna M , Pearce RA ((2011) ) GABAA,slow: Causes and consequences. Trends Neurosci 34: , 101–112. |
[30] | Wierenga CJ , Wadman WJ ((1999) ) Miniature inhibitory postsynaptic currents in CA1 pyramidal neurons after kindling epileptogenesis. J Neurophysiol 82: , 1352–1362. |
[31] | Rall W ((1967) ) Distinguishing theoretical synaptic potentials computed for different soma-dendritic distributions of synaptic input. J Neurophysiol 30: , 1138–1168. |
[32] | Ledri M , Nikitidou L , Erdelyi F , Szabo G , Kirik D , Deisseroth K , Kokaia M ((2012) ) Altered profile of basket cell afferent synapses in hyper-excitable dentate gyrus revealed by optogenetic and two-pathway stimulations. Eur J Neurosci 36: , 1971–1983. |
[33] | Bekkers JM , Clements JD ((1999) ) Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol 516: , 227–248. |
[34] | Masuda A , Kobayashi Y , Kogo N , Saito T , Saido TC , Itohara S ((2016) ) Cognitive deficits in single App knock-in mouse models. Neurobiol Learn Mem 135: , 73–82. |
[35] | Sakakibara Y , Sekiya M , Saito T , Saido TC , Iijima KM ((2018) ) Cognitive and emotional alterations in App knock-in mouse models of Aβ amyloidosis. BMC Neurosci 19: , 46. |
[36] | Laws KR , Irvine K , Gale TM ((2018) ) Sex differences in Alzheimer’s disease. Curr Opin Psychiatry 31: , 133–139. |
[37] | Ferretti MT , Iulita MF , Cavedo E , Chiesa PA , Dimech AS , Chadha AS , Baracchi F , Girouard H , Misoch S , Giacobini E , Depypere H , Hampel H ((2018) ) Sex differences in Alzheimer disease —The gateway to precision medicine. Nat Rev Neurol 14: , 457–469. |
[38] | Van Der Flier WM , Scheltens P ((2005) ) Epidemiology and risk factors of dementia.v. Neurol Pract 76: (Suppl 5), 2–7. |
[39] | Murphy DD , Cole NB , Greenberger V , Segal M ((1998) ) Estradiol increases dendritic spine density by reducing GABA neurotransmission in hippocampal neurons. J Neurosci 18: , 2550–2559. |
[40] | Chudomel O , Herman H , Nair K , Moshe SL , Galanopoulou AS ((2009) ) Age- and gender-related differences in GABAA receptor-mediated postsynaptic currents in GABAergic neurons of the Substantia Nigra reticulata in the rat. Neuroscience 163: , 155–167. |
[41] | Kiss E , Gorgas K , Schlicksupp A , Groß D , Kins S , Kirsch J , Kuhse J ((2016) ) Biphasic alteration of the inhibitory synapse scaffold protein gephyrin in early and late stages of an Alzheimer disease model. Am J Pathol 186: , 2279–2291. |
[42] | Cobb SR , Halasy K , Vida I , Nyíri G , Tamás G , Buhl EH , Somogyi P ((1997) ) Synaptic effects of identified interneurons innervating both interneurons and pyramidal cells in the rat hippocampus. Neuroscience 79: , 629–648. |
[43] | Sik A , Penttonen M , Ylinen A , Buzsaki G ((1995) ) Hippocampal CA1 interneurons: An in vivo intracellular labeling study. J Neurosci 15: , 6651–6665. |
[44] | Megías M , Emri Z , Freund TF , Gulyás AI , Megias M ((2001) ) Total number and distribution of inhibitory and excitatory synapses on hippocampal CA1 pyramidal cells. Neuroscience 102: , 527–540. |
[45] | Ramos B , Baglietto-Vargas D , Rio JC del , Moreno-Gonzalez I , Santa-Maria C , Jimenez S , Caballero C , Lopez-Tellez JF , Khan ZU , Ruano D , Gutierrez A , Vitorica J ((2006) ) Early neuropathology of somatostatin/NPY GABAergic cells in the hippocampus of a PS1×APP transgenic model of Alzheimer’s disease. Neurobiol Aging 27: , 1658–1672. |
[46] | Schmid LC , Mittag M , Poll S , Steffen J , Wagner J , Geis H-R , Schwarz I , Schmidt B , Schwarz MK , Remy S , Fuhrmann M ((2016) ) Dysfunction of somatostatin-positive interneurons associated with memory deficits in an Alzheimer’s disease model. Neuron 92: , 1–12. |
[47] | Baglietto-Vargas D , Moreno-Gonzalez I , Sanchez-Varo R , Jimenez S , Trujillo-Estrada L , Sanchez-Mejias E , Torres M , Romero-Acebal M , Ruano D , Vizuete M , Vitorica J , Gutierrez A ((2010) ) Calretinin interneurons are early targets of extracellular amyloid-β pathology in PS1/AβPP Alzheimer mice hippocampus. J Alzheimers Dis 21: , 119–132. |
[48] | Vida I , Halasy K , Szinyei C , Somogyi P , Buhl EH ((1998) ) Unitary IPSPs evoked by interneurons at the stratum radiatum-stratum lacunosum-moleculare border in the CA1 area of the rat hippocampus in vitro. J Physiol 506: , 755–773. |
[49] | Daw MI , Tricoire L , Erdélyi F , Szabó G , McBain CJ ((2009) ) Asynchronous transmitter release from cholecystokinin-containing inhibitory interneurons is widespread and target-cell independent. J Neurosci 29: , 11112–11122. |
[50] | Hefft S , Jonas P ((2005) ) Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8: , 1319–1328. |
[51] | Daw MI , Pelkey KA , Chittajallu R , McBain CJ ((2010) ) Presynaptic kainate receptor activation preserves asynchronous GABA release despite the reduction in synchronous release from hippocampal cholecystokinin interneurons. J Neurosci 30: , 11202–11209. |
[52] | Yang YM , Wang LY ((2006) ) Amplitude and kinetics of action potential-evoked Ca2+current and its efficacy in triggering transmitter release at the developing calyx of Held synapse. J Neurosci 26: , 5698–5708. |
[53] | Medrihan L , Cesca F , Raimondi A , Lignani G , Baldelli P , Benfenati F ((2013) ) Synapsin II desynchronizes neurotransmitter release at inhibitory synapses by interacting with presynaptic calcium channels. Nat Commun 4: , 1512. |
[54] | Kašparová J , Lisá V , Tuček S , Doležal V ((2001) ) Chronic exposure of NG108-15 cells to amyloid β peptide (Aβ1-42) abolishes calcium influx via N-type calcium channels. Neurochem Res 26: , 1079–1084. |
[55] | Hoe H , Lee KJ , Carney RSE , Lee J , Markova A , Lee J , Howell BW , Hyman BT , Pak DTS , Bu G , Rebeck GW ((2009) ) Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J Neurosci 29: , 7459–7473. |
[56] | Rice HC , De Malmazet D , Schreurs A , Frere S , Van Molle I , Volkov AN , Creemers E , Vertkin I , Nys J , Ranaivoson FM , Comoletti D , Savas JN , Remaut H , Balschun D , Wierda KD , Slutsky I , Farrow K , De Strooper B , De Wit J ((2019) ) Secreted amyloid-β precursor protein functions as a GABA BR1a ligand to modulate synaptic transmission. Science 363: , eaao4827. |
[57] | Dinamarca MC , Raveh A , Schneider A , Fritzius T , Früh S , Rem PD , Stawarski M , Lalanne T , Turecek R , Choo M , Besseyrias V , Bildl W , Bentrop D , Staufenbiel M , Gassmann M , Fakler B , Schwenk J , Bettler B ((2019) ) Complex formation of APP with GABA B receptors links axonal trafficking to amyloidogenic processing. Nat Commun 10: , 1331. |
[58] | King DL , Arendash GW , Crawford F , Sterk T , Menendez J , Mullan MJ ((1999) ) Progressive and gender-dependent cognitive impairment in the APP(SW) transgenic mouse model for Alzheimer’s disease. Behav Brain Res 103: , 145–162. |
[59] | Richetin K , Petsophonsakul P , Roybon L , Guiard BP , Rampon C ((2017) ) Differential alteration of hippocampal function and plasticity in females and males of the APPxPS1 mouse model of Alzheimer’s disease. Neurobiol Aging 57: , 220–231. |
[60] | Van Duijn S , Nabuurs RJA , Van Duinen SG , Natté R , Van Buchem MA , Alia A ((2013) ) Longitudinal monitoring of sex-related in vivo metabolic changes in the brain of Alzheimer’s disease transgenic mouse using magnetic resonance spectroscopy. J Alzheimers Dis 34: , 1051–1059. |
[61] | Sala Frigerio C , Wolfs L , Fattorelli N , Thrupp N , Voytyuk I , Schmidt I , Mancuso R , Chen WT , Woodbury ME , Srivastava G , Möller T , Hudry E , Das S , Saido T , Karran E , Hyman B , Perry VH , Fiers M , De Strooper B ((2019) ) The major risk factors for Alzheimer’s disease: Age, sex, and genes modulate the microglia response to Aβ plaques. Cell Rep 27: , 1293–1306.e6. |
[62] | Viña J , Lloret A ((2010) ) Why women have more Alzheimer’s disease than men: Gender and mitochondrial toxicity of amyloid-β peptide. J Alzheimers Dis 20: , S527–S533. |
[63] | Pike CJ ((2017) ) Sex and the development of Alzheimer’s disease. J Neurosci Res 95: , 671–680. |
[64] | Daniel JM ((2013) ) Estrogens, estrogen receptors, and female cognitive aging: The impact of timing. Horm Behav 63: , 231–237. |


