
Plasma High Density Lipoprotein Small Subclass is Reduced in Alzheimer’s Disease Patients and Correlates with Cognitive Performance
Abstract
Background:
The link between cholesterol and Alzheimer’s disease (AD) has received much attention, as evidence suggests high levels of cholesterol might be an AD risk factor. The carriage of cholesterol and lipids through the body is mediated via lipoproteins, some of which, particularly apolipoprotein E (ApoE), are intimately linked with AD. In humans, high density lipoprotein (HDL) is regarded as a “good” lipid complex due to its ability to enable clearance of excess cholesterol via ‘cholesterol reverse transport’, although its activities in the pathogenesis of AD are poorly understood. There are several subclasses of HDL; these range from the newly formed small HDL, to much larger HDL.
Objective:
We examined the major subclasses of HDL in healthy controls, mild cognitively impaired, and AD patients who were not taking statins to determine whether there were HDL profile differences between the groups, and whether HDL subclass levels correlated with plasma amyloid-β (Aβ) levels or brain Aβ deposition.
Methods:
Samples from AIBL cohort were used in this study. HDL subclass levels were assessed by Lipoprint while Aβ1–42 levels were assessed by ELISA. Brain Aβ deposition was assessed by PET scan. Statistical analysis was performed using parametric and non-parametric tests.
Results:
We found that small HDL subclass is reduced in AD patients and it correlates with cognitive performance while plasma Aβ concentrations do not correlate with lipid profile or HDL subfraction levels.
Conclusion:
Our data indicate that AD patients exhibit altered plasma HDL profile and that HDL subclasses correlate with cognitive performances.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease, pathologically characterized by the extracellular deposition of amyloid-β (Aβ) in the brain and the accumulation of hyperphosphorylated tau filaments in neurons. While a small portion of AD cases can be attributed to a genetic predisposition, sporadic or late-onset AD (LOAD) accounts for the majority of cases.
In the periphery, cholesterol is transported to the cells where it accumulates through the action of very low-density lipoproteins (VLDL) and low-density lipoproteins (LDL), which are referred to as ‘bad cholesterol’ as their action increases the cholesterol levels and the risk for several diseases associated with it. The cholesterol balance is maintained by the action of high-density lipoproteins (HDL), which are referred to as ‘good cholesterol’, as their duty is to remove excess cholesterol by transporting it to the liver for its elimination. However, the lipid environment in the brain is very different compared to the lipid environment in the periphery, and these differences are maintained by the protective features of the blood-brain barrier (BBB), which isolates the brain from the periphery and forces the brain to generate cholesterol in situ. In the brain, cholesterol is carried by HDL-like particles, in which Apolipoprotein E (ApoE), produced by astrocytes and microglia [1–4], is the main apolipoprotein (whereas in plasma it is ApoA-I) and cholesterol-carrier in the brain, followed by ApoA-I, which is not produced in the brain but it can be transported across in the choroid plexus [5]. These differences are at the basis for four different CSF lipoprotein classes, CSF-Lp ApoA-I, CSF-Lp ApoE, CSF-Lp ApoE/ApoA-I, and CSF-Lp (without ApoE and ApoA-I) [6]. Additionally, other lipoproteins that are common in the peripheral circulation, such as LDL and VLDL, are not present in the brain environment, leaving the whole load of cholesterol transport duties to CSF-Lp particles. Finally, the elimination of excess of cholesterol requires the conversion of cholesterol to a more soluble 24S-hydroxycholesterol, which allows it to cross the BBB into the peripheral circulation, where it is picked up by plasma lipoprotein and redirected to the liver for its elimination [7].
Overall, lipid metabolism has been linked to AD pathogenesis in several ways. Several reports suggest that high levels of intracellular cholesterol cause an increase in Aβ deposition in the brain [8–10], whereas low cholesterol levels have been shown to increase processing of the amyloid-β protein precursor (AβPP) via the non-amyloidogenic pathway [11–14]. In addition, longitudinal studies have shown that mid-life obesity and cardiovascular disease are also risk factors for AD [15, 16] and peripheral biomarker studies have found preclinical AD-related biomarkers are associated with lipid metabolism [17–19]. To date, studies of cholesterol levels in AD have produced conflicting results. Some studies have found increased levels of cholesterol in AD [20–22], while other reports have not [23–25]. It has also been reported that individuals who subsequently develop AD exhibit decreased cholesterol levels before clinical manifestation of the disease [26]. Despite such reports, hypercholesterolemia is still considered an early risk factor for developing AD [27] and has been associated with impaired memory recall in the elderly [28]. In support of these studies, mice fed with high-fat diets exhibit altered brain mass and amyloid levels in the brain [9, 29, 30], while elevated plasma triglyceride (TG) levels have been shown to precede amyloid deposition [31]. Consistent with the notion that high cholesterol levels increase disease risk, statins have shown the potential to reduce the risk for AD, though their effectiveness appears to require administration well before clinical manifestation of symptoms [32–35]. More recently, statins have also been shown to be capable of raising HDL levels, most likely through a mechanism independent of lowering LDL levels [36, 37]. There are studies which have reported altered HDL levels in AD compared to controls [21, 22], and some that have reported no such association [23, 24, 38]. It is, however, important to note that low plasma HDL levels have recently been associated with higher cerebral Aβ deposition [39] and that ApoA-I, the major constituent of plasma HDL, has been reported to be lower in AD compared to controls [40–43]. ApoA-I binds to the Aβ peptide as well as its precursor protein AβPP, and has been shown to reduce Aβ-induced toxicity and Aβ aggregation into β-pleated sheets [44, 45]. In AD mouse models, ApoA-I overexpression reduces cognitive deterioration and neuroinflammation, whereas its deficiency increases cognitive defects and cerebral amyloid angiopathy [46, 47].
It is most likely the first clear link between AD and lipid metabolism was reported when APOE polymorphisms were shown to influence AD [48]. The APOE gene is located on chromosome 19, and there are three allelic variations of APOE (ɛ2, ɛ3, ɛ4), with the possession of the ɛ4 allele (APOE ɛ4) to be the predominant genetic risk factor for the late-onset form of the disease (carried by ∼50% of all sporadic AD patients, yet is only found at a frequency of around 14% in the general population), whereas possession of the APOE ɛ2 allele appears to be protective [49, 50]. It is still not clear how possession of the APOE ɛ4 allele increases AD risk, but ApoE appears to be required for Aβ aggregation, and ApoE ɛ4 has been shown to be the most efficient at promoting such oligomerization; ApoE also helps remove Aβ from the brain to the periphery, yet again, ApoE ɛ4 is the least efficient at this task [51, 52]. The ɛ4 allele is also the least efficient in promoting cholesterol efflux from neuronal cells [53], and cholesterol bound to ApoE ɛ4 displays a lower rate of cellular uptake [54].
Clearly, there is still a need for further studies to increase our understanding of changes to plasma cholesterol levels in AD. Therefore, we evaluated plasma cholesterol and lipoproteins further, by investigating plasma lipoprotein subgroups, to provide more insight into the early pathogenic events involved in the development of AD. An aspect overlooked by most previous studies is that cholesterol is transported by at least four different subclasses of lipoproteins in the blood stream: chylomicrons, VLDL, LDL, and HDL. HDL also exists in different forms, from the newly generated HDL small (which have been reported to display anti-oxidant features), to the more mature HDL large. Changes in the levels of these subclasses may be missed if not measured individually. In this study, we evaluated the plasma levels of HDL subclasses, as well as levels of cholesterol, LDL, total HDL, and TG, to determine whether more detailed lipid profiles would reveal differences between healthy controls, MCI, and AD patients. In addition, we evaluated the cerebral Aβ deposition in this cohort, measured as neocortical standardized uptake value ratio (SUVR) score from 11C-PiB positron emission tomography (PET) analysis and investigated correlations with regards to HDL levels to determine whether HDL subclasses levels could reflect brain amyloid deposition.
MATERIALS AND METHODS
The AIBL study was approved by the ethics committees of St. Vincent’s Health and Austin Health in Melbourne, Hollywood Private Hospital and Edith Cowan University in Perth (Australia). All volunteers gave written and informed consent before participating in our study. A total of 486 participants, divided into healthy controls (HC), mild cognitive impaired (MCI), and Alzheimer’s patients (AD) from the AIBL cohort were used, after ensuring that this subset was not undergoing statin treatment, and that conversion to a different status did not occur in the following 18 months. A full description of the recruitment process has already been published. Exclusion criteria included a history of non-AD dementia, schizophrenia, bipolar disorder, current depression (GDS score above 5/15), Parkinson’s disease, uncontrolled hypertension (systolic BP > 170 or diastolic BP > 100), cancer (other than basal cell skin carcinoma) within the last two years, symptomatic stroke, uncontrolled diabetes, or current regular alcohol use exceeding two standard drinks per day for women or four per day for men [55]. The AIBL Study clinical panel meets on a monthly basis to discuss baseline classification for each set of patients recently tested and ensures diagnoses were made in accordance with the NINCDS-ARDA criteria [56, 57]. Various body parameters were evaluated at the examinations, such as weight, height, blood pressure, and pulse rate. Blood was drawn from overnight fasting participants and collected to obtain plasma or serum for our analysis. APOE status was determined by genotyping cells from whole blood as previously described [17]. Total cholesterol, LDL, HDL, and TG levels were assessed in plasma.
The Lipoprint system (Quantimetrix, Redondo Beach, CA, USA) was used to examine the serum HDL profiles of study participants following the kit instructions. Briefly, 25μl samples of serum were combined with 200μL of the supplied loading buffer in glass gel tubes and allowed to polymerize for 30 min. Samples were then separated in an electrophoretic chamber for 1–1.5 h at 3 mA per tube. Finally, gels were scanned and the band intensity of each subfraction was obtained using the supplied software. The HDL analysis provided values for 10 subclasses of HDL which we divided into three major groups named HDL Large (subclasses 1 through 3), HDL Intermediate (subclasses 4–6), and HDL Small (subclasses 7–10). Initial analysis indicated a good correlation between HDL Large and HDL-2 (1.063–1.125 g/ml) and between HDL Intermediate + Small and HDL-3 (1.125–1.21 g/ml) (personal communication). Upon assessing the subclass percentage in serum, absolute levels (mg/dl) in each subclass (L, I, and S) were determined using the plasma HDL concentration measured in the initial blood drawn analysis which was routinely performed in all participants. Although plasma and serum are different biological fluids, it has been reported that HDL concentration is almost identical [58, 59].
Plasma Aβ1–42 concentrations were measured using a commercially available ELISA kit (INNO-BIA, Innogenetics, Gent, Belgium) following manufacturer’s instructions.
PET scans consisting of 30 min acquisitions were performed 40 min after injection of 370 MBq 11C-PiB. PET images were processed using a semi-automatic region-of-interested method as previously described [60]. Standardized uptake values (SUV) for 11C-PiB were calculated for all brain regions examined. The SUV ratio (SUVR) was calculated dividing all regional SUV by the cerebellar cortex SUV. However, the centiloid scale was recently proposed to provide a standard quantification of Aβ-PET images. In the centiloid scale, the Aβ burden can be expressed with values ranging from 0 (the typical Aβ burden in young controls) to 100 (the typical Aβ burden in mild AD patients) [61]. Centiloid values were generated using CapAIBL as described elsewhere [62].
Statistical comparison of means in different groups was based on ANCOVA (Analysis of Covariance) where adjustment was made for covariates such as age, gender, site, and APOE status. For non-normal distributions, a non-parametric ANOVA (Kruskal-Wallis) and non-parametric U Test (Mann-Whitney) were used. Associations between continuous variates were assessed using Spearman’s correlation. Due to the non-normal distribution of our variables, most of our analysis were performed using non-parametric tests, which limits our power to adjust for several covariates. We acknowledge this is a limitation of our study. A p-value less than 0.05 was regarded as significant. Analyses were carried out using TIBCO Spotfire S+ version 8.2 (Boston, MA) and SPSS version 25 (Chicago, IL, USA).
RESULTS
The basic demographics of the study participants are summarized in Table 1. In total, 347 HC, 55 MCI, and 84 AD patients were studied (all participants were 65-years old and older). All samples were evaluated for total cholesterol, LDL, HDL, and TG levels. Using Kruskal-Wallis analysis, we compared the levels of cholesterol, LDL, HDL, and triglycerides among HC, MCI, and AD patients and we did not observe any significant difference (Table 1). Additionally, the effect of APOE genotype was assessed in each clinical group by Mann-Whitney U Test. The levels of cholesterol and LDL were significantly higher APOE ɛ4-carrier in HC only, but not in MCI or AD. Conversely, HDL and TG levels were not affected by the presence of APOE ɛ4 allele in any clinical group (Supplementary Table 1). To determine whether the number of APOE ɛ4 alleles affected the lipid profile, we performed the Kruskal-Wallis analysis in each clinical group. Again, the cholesterol and LDL levels were significantly affected by APOE genotype in HC only with APOE ɛ4 homozygous individuals displaying highest levels of cholesterol and LDL. HDL and TG levels were not affected by the number of APOE ɛ4 alleles in any clinical groups (data not shown).
Table 1
Comparison of demographic characteristics and cholesterol, LDL, HDL, and triglyceride levels among HC, MCI, and AD participants
| HC | MCI | AD | ANOVA (p) | |
| N | 347 | 55 | 84 | |
| Age (y) | 72±6 | 79±6 | 81±7 | |
| M/F | 148/199 | 25/30 | 34/50 | |
| Melbourne/Perth | 196/151 | 27/28 | 53/31 | |
| APOE ɛ4 (no/yes) | 2871/76 | 23/32 | 36/48 | |
| Cholesterol (mg/dl) | 222±37 | 215±38 | 226±44 | 0.19 |
| LDL (mg/dl) | 134±33 | 128±34 | 135±39 | 0.46 |
| HDL (mg/dl) | 66±18 | 63±14 | 66±18 | 0.73 |
| TG (mg/dl) | 113±50 | 119±56 | 129±85 | 0.14 |
Values are presented as mean±S.D or as frequency. Non-parametric ANOVA (Kruskal-Wallis analysis) was performed.
The levels of HDL subgroups, expressed as a percentage of the total HDL or in terms of concentration, are listed in Table 2 (top). These analyses were performed using ANCOVA for normal distributions (HDL L% and HDL I%) or Kruskal-Wallis analysis for non-normal distributions (HDL S%, HDL L mg/dl, HDL I mg/dl, and HDL S mg/dl). Using the data reported in percentage (Table 2, bottom), analyses found that differences between the HC, MCI, and AD patients are associated with the HDL Small group only (p < 0.001). A pairwise comparison with Mann-Whitney U Test within the HDL Small group shows that the AD participants exhibit significantly less HDL Small particles when compared to the HC (p < 0.001) (–26%) and MCI (p = 0.004) (–25%) participants. In contrast, comparison of HC with MCI participants showed no differences (p = 0.77). Conversely, there were no significant differences associated with HDL Large (p = 0.10) or HDL Intermediate (p = 0.21). Analysis of the absolute values (Table 2, bottom) resulted in the same conclusion; significant differences associated with the HDL Small values (p = <0.001), while no differences are associated with HDL Large (p = 0.12) or HDL Intermediate (p = 0.56). A further post hoc analysis of the HDL Small group demonstrated the same decrease in HDL Small particles in AD versus HC (p < 0.001) (–24%) and in AD versus MCI (p = 0.006) (–21%), while HC versus MCI displayed no significant difference (p = 0.58). The significance of our results was not affected by performing the same analysis in non-smokers only, which removed an important factor known to modulate HDL [63] (HDL S% : Kruskal-Wallis p < 0.001; Mann-Whitney U Test, HC versus AD p < 0.001, MCI versus AD p = 0.009; HDL S mg/dl: Kruskal-Wallis p < 0.001; Mann-Whitney U Test, HC versus AD p < 0.001, MCI versus AD p = 0.026). We have also performed the same analysis in APOE ɛ4-carriers and APOE ɛ4-non-carriers but we did not find any difference between those groups (Supplementary Table 2). A more detailed analysis assessing the number of APOE ɛ4 alleles (ANCOVA for HDL L% and I% and Kruskal-Wallis for HDL S% and HDL L, I, S mg/dl) did not alter our previous findings in HC and AD, suggesting that HDL subclasses distributions are not affected by APOE genotype in these clinical groups. In MCI, the distribution of HDL L or I (expressed as % or mg/dl) was not affected by the number of APOE ɛ4 alleles. However, Kruskal-Wallis analysis unexpectedly indicated that in MCI, HDL S% subclasses are affected by APOE genotype (p = 0.03) (with APOE ɛ4 homozygous displaying the highest levels), while HDL S mg/dl distribution are not (p = 0.107) (data not shown). However, the low number of APOE ɛ4 homozygous (n = 6) in the MCI group may have affected the results and further analysis is needed.
Table 2
Comparison of HDL sub-distribution in the HC, MCI, and AD groups, expressed as % or mg/dl
| Lipoprotein fractions (% of total HDL) | Lipoprotein fractions (mg/dl) | |||||
| HC | MCI | AD | HC | MCI | AD | |
| HDL L | 34.5±10.2 | 34.5±10.0 | 38.3±10.4 | 24.0±12.4 | 22.7±10.7 | 25.9±11.2 |
| HDL I | 53.9±6.8 | 54.0±6.1 | 53.1±7.4 | 34.9±7.6 | 33.5±6.0 | 34.5±8.4 |
| HDL S | 11.5±5.6 | 11.3±6.0 | 8.5±5.4 | 7.1±3.1 | 6.8±3.2 | 5.4±3.5 |
| General linear model | ||||||
| HDL Subdistribution expressed as % | HDL Subdistribution expressed as mg/dl | |||||
| Overall, p | Individual comparisons, p | Overall, p | Individual comparisons, p | |||
| HDL L | 0.10 | 0.12 | ||||
| HDL I | 0.21 | 0.56 | ||||
| HDL S | <0.001 | HC-o versus MCI 0.77 | <0.001 | HC-o versus MCI 0.58 | ||
| HC-o versus AD < 0.001 | HC-o versus AD < 0.001 | |||||
| MCI versus AD 0.004 | MCI versus AD 0.006 | |||||
ANCOVA (analyses were adjusted for sex, age, site, and APOE ɛ4-carrier status) or non-parametric ANOVA (Kruskal-Wallis analysis) were used and regarded as significant when p < 0.05 (bold). When Kruskal-Wallis analysis was significant, individual comparison was performed using Mann-Whitney U Test and considered significant when p < 0.05 (bold). For HDL S (% and mg/dl), HDL L (mg/dl) and HDL I (mg/dl) non-parametric tests were used. Values are presented as mean±S.D.
Based on the evidence that HDL Small levels were affected by clinical classifications, with levels lower in AD versus HC, we also evaluated if HDL subclass levels were affected by brain amyloid deposition in HC (HC Aβ–versus HC Aβ+). These analyses were performed using ANCOVA for normal distributions (HDL L% and HDL I%) or Mann-Whitney U Test analysis for non-normal distributions (HDL S%, HDL L mg/dl, HDL I mg/dl, and HDL S mg/dl). As shown in Table 3 (top), there was no different distribution of any HDL subclass with regards to brain amyloid deposition. ANCOVA analysis (performed adjusted for age, gender, site, and APOE status) or Mann-Whitney U Test analysis did not reveal any significant difference (Table 3, bottom).
Table 3
Comparison of HDL sub-distribution in HC with low (Aβ–) and high (Aβ+) Aβ deposition, expressed as % or mg/dl
| Lipoprotein fractions (% of total HDL) | Lipoprotein fractions (mg/dl) | |||
| HC Aβ–(n = 49) | HC Aβ+ (n = 21) | HC Aβ–(n = 49) | HC Aβ+ (n = 21) | |
| HDL L | 34.3±11.2 | 34.5±9.5 | 24.4±12.6 | 24.6±12.5 |
| HDL I | 53.9±7.4 | 54.0±5.5 | 35.5±6.9 | 35.6±8.6 |
| HDL S | 11.7±6.8 | 11.5±5.4 | 7.2±3.5 | 7.1±2.4 |
| General linear model | ||||
| HDL Subdistribution (% of total HDL) | HDL Subdistribution (mg/dl) | |||
| p | p | |||
| HDL L | 0.72 | 0.91 | ||
| HDL I | 0.40 | 0.88 | ||
| HDL S | 0.94 | 0.99 | ||
ANCOVA (analyses were adjusted for sex, age, site, and APOE ɛ4-carrier status) or non-parametric Mann-Whitney U Test were used and regarded as significant when p < 0.05. For HDL S (% and mg/dl), HDL L (mg/dl) and HDL I (mg/dl) non-parametric tests were used. Values are presented as mean±S.D.
Using Spearman’s correlation, SUVR-Centiloid values were correlated with HDL or HDL subfractions (HDL Large, Intermediate, and Small) to determine whether alteration in the HDL profile was associated with a different SUVR score in HC and in combined MCI/AD groups. No significant association with HDL, HDL Intermediate, or HDL Small in any of the groups, nor with HDL Large in HC was observed (Table 4, top). However, there was a significant negative association between SUVR and HDL Large in the MCI/AD group only (p = 0.037). We also evaluated if plasma levels of Aβ1–42 correlated with HDL or HDL subclasses in HC or MCI/AD. Again, we did not find any significant correlation between plasma levels of Aβ1–42 and HDL (or any HDL subclasses) in any clinical classification (Table 4, bottom).
Table 4
Correlations between brain Aβ deposition, plasma Aβ1–42 levels, and HDL subclasses
| SUVR-Centiloid | |||||
| HC (n = 70) | MCI/AD (n = 32) | ||||
| mg/dl | ρ | p | ρ | p | |
| HDL | 0.010 | 0.931 | –0.325 | 0.070 | |
| HDL L | 0.083 | 0.493 | –0.371 | 0.037 | |
| HDL I | –0.054 | 0.660 | –0.160 | 0.382 | |
| HDL S | 0.008 | 0.950 | 0.225 | 0.216 | |
| Plasma Aβ1–42 (pg/ml) | |||||
| HC (n = 346) | MCI/AD (n = 135) | ||||
| mg/dl | ρ | p | ρ | P | |
| HDL | 0.013 | 0.815 | 0.118 | 0.174 | |
| HDL L | 0.015 | 0.774 | 0.089 | 0.304 | |
| HDL I | –0.025 | 0.637 | 0.080 | 0.357 | |
| HDL S | 0.041 | 0.443 | 0.088 | 0.310 | |
Spearman’s correlation evaluating brain amyloidosis or plasma Aβ1–42 levels with HDL (or HDL subgroups) was performed in HC, MCI, and AD and considered significant when p < 0.05 (bold).
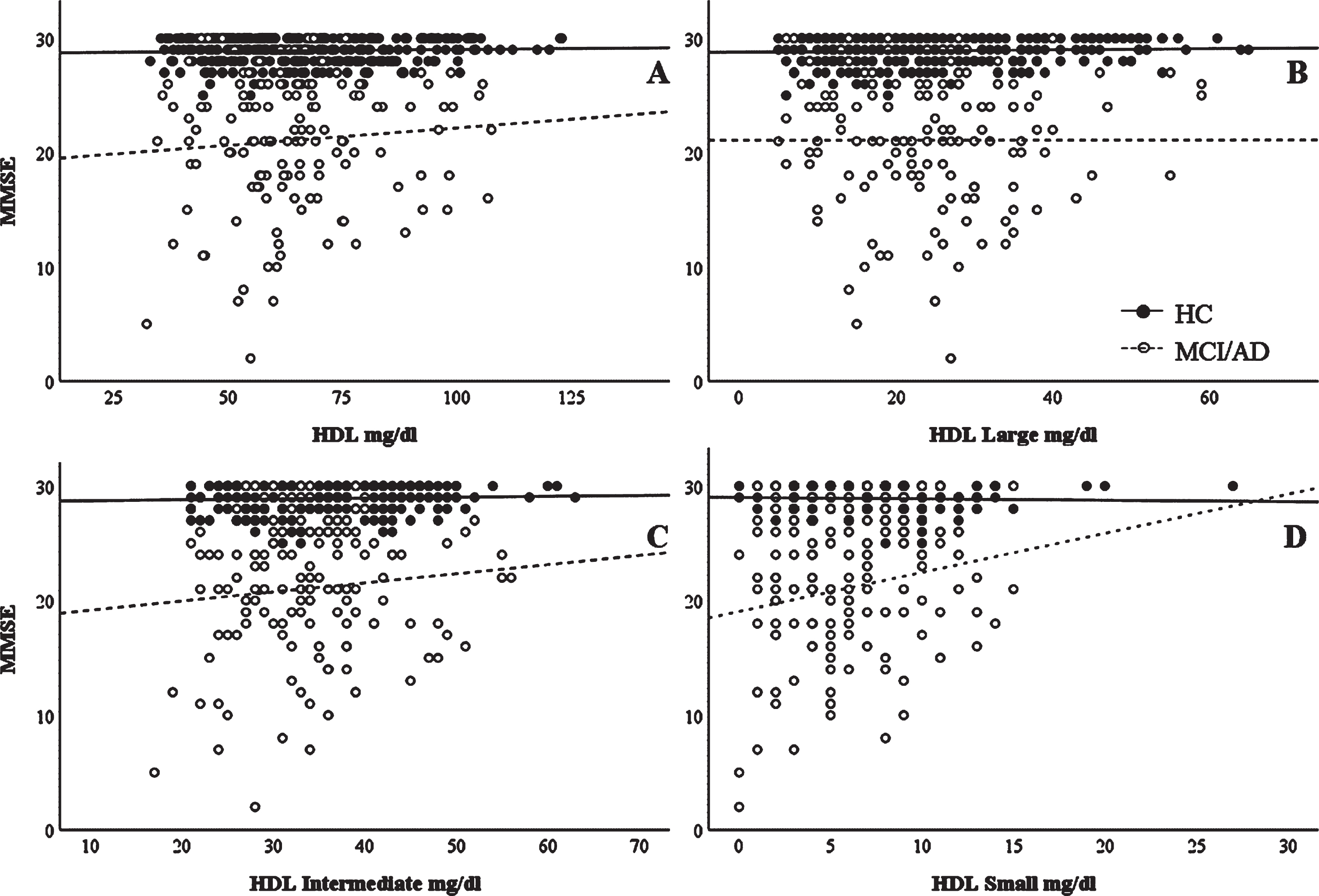
Cognitive performance scores (MMSE) were correlated with the HDL or HDL subfractions in HC and MCI/AD groups to determine whether cognitive performances were associated with HDL profile. HDL Small levels significantly positively correlated with MMSE in the MCI/AD group (ρ= 0.171, p = 0.044), but not in the HC group (ρ= –0.052, p = 0.334). However, we did not find any significant association between HDL, HDL Large and HDL Intermediate with MMSE in any clinical group (in HC: ρ= –0.001, p = 0.989; ρ= –0.004, p = 0.934; and ρ= 0.001, p = 0.986 for HDL, HDL L, and HDL I, respectively; in MCI/AD: ρ= 0.070, p = 0.416; ρ= –0.038, p = 0.653; and ρ= 0.051, p = 0.551 for HDL, HDL L, and HDL I, respectively (Fig. 1).
Fig. 1
Correlations between MMSE scores and HDL subclasses. Unadjusted correlation between MMSE scores and (A) HDL, (B) HDL Large, (C) HDL Intermediate, and (D) HDL Small subclasses. Spearman’s correlation data are reported in the text.
DISCUSSION
The finding over two decades ago that APOE polymorphisms can influence the risk of AD strongly implied a link between lipid metabolism, particularly cholesterol metabolism, and AD. The later findings that brain Aβ plaque deposits can be found commonly in coronary artery disease (CAD) as well as in hypertension patients strengthened this concept, suggesting a neuropathologic link between CAD, hypertension, and AD. It is now known that many related conditions, including CAD, hypertension, mid-life obesity, type 2 diabetes, and insulin resistance all increase AD risk to some extent [64].
However, cholesterol metabolism is substantially different in the periphery and in the brain. While in the periphery, chylomicrons, VLDL, LDL, and HDL are all responsible for cholesterol transport, in the brain the whole burden is carried by HDL-like particles. Additionally, these HDL-like particles have ApoE generated in the brain as the major apolipoprotein, while ApoA-I relies upon transport across the choroid plexus to enter the brain as it is not generated in situ. Furthermore, cholesterol in the brain can be eliminated only after conversion to 24S-hydroxycholesterol, which allows it to cross the BBB and reach the periphery where it is directed to the liver for its excretion [7]. These differences altogether indicate that with regards to cholesterol and its metabolism, the periphery and the brain may display different behaviors that can modulate the risk for AD in different ways.
The notion of a linkage between cholesterol and AD is reported in studies indicating that high-fat diets in animal models induce the deposition of Aβ [9, 29, 30, 65, 66]. Other studies looking for a mechanistic link found that cholesterol can modulate AβPP processing toward the amyloidogenic pathway [27, 67–69], and the conclusions from all these studies were that high levels of cholesterol in the circulation are most likely a risk factor for AD.
Many studies have tested the effects of lowering cholesterol levels on AD or AD risk through the use of statins, either in clinical trials or in vitro studies; however, results have been inconsistent [11–13, 32, 35, 70–74]. A possible reason for the conflicting data may be due to the way in which statins function. In clinical studies, many reports linked the use of statins to a decreased risk for AD, whereas other reports did not observe changes in symptoms, or any altered AβPP processing in AD patients. Overall, the results suggest that statins may be protective before the onset of the disease but become less efficient or ineffective once the disease has started [32–35]. Interestingly, while statins may have beneficial effects in preventing AD due to cholesterol-lowering properties, this may have little to do with cholesterol effects on Aβ production in the brain, as positive results were seen in some studies despite the fact that the statins being tested were not able to cross the BBB, hence effects must have been due to peripheral changes. Alternatively, statins may be able to reduce AD risk due to influences on other biochemical pathways, for example, statins also alter the isoprenylation of small GTPases, another pathway that has been linked to AD [75–78]. In order to eliminate this confounding factor, our sample population excluded all participants who were undergoing statin treatment. In our cohort, we did not observe any marked alteration in total cholesterol, total LDL, total HDL, or TG levels (Table 1) between the different groups. Other studies have reported mostly unchanged levels of lipoproteins, with one or two exceptions. For example, one study found that the charge-based major LDL subfraction, as characterized by capillary isotachophoresis, was associated with both MCI and AD, yet did not find differences in other lipoprotein classes [22]. Another study detected mild hypercholesterolemia in AD patients, with no changes in triglycerides [20–22]. One study demonstrated that low levels of HDL are associated with lower grey matter volume in cognitively healthy adults [79].
It is important to note that comparisons of apparently similar studies are complicated to an extent by the presence of other conditions, and also by the type of lipoprotein measure used, such as HDL-cholesterol levels versus HDL particle levels. For example, in one study of a cohort enriched for cerebrovascular disease and elevated vascular risk, statistical models that controlled for age and APOE ɛ4 alleles revealed independent associations among the levels of LDL-cholesterol, HDL-cholesterol, and the level of Aβ deposition as measured by PiB-PET: in this study, higher LDL-cholesterol and lower HDL-cholesterol levels were both associated with a higher Aβ deposition [39].
Intrigued by the apparent lack of change in total HDL levels, we then compared levels of the respective HDL subclasses between AD, MCI, and controls. As shown in Table 2, HDL exhibited a different profile in the plasma of AD participants. Examination of the HDL profiles revealed lower levels of the smaller HDL particles (HDL Small) in AD participants compared with the MCI or healthy controls. Other studies of plasma HDL have reported, for example: lower overall levels of HDL in vascular dementia, but not AD [21, 80], lower levels of HDL in AD with cardiovascular comorbidities but not in AD alone [81], or lower levels of HDL-cholesterol and ApoA-I in AD [82], showing that results have not been conclusive. These studies did not investigate HDL subclasses, however, and further lipoprotein subclass studies such as ours may reveal more information concerning plasma lipoprotein changes in various conditions. It is interesting to note that these small, dense HDL particles have been shown to be beneficial against atherosclerosis and vascular related oxidative stress [83], which fits with the hypothesis that AD is intimately linked to metabolic syndrome [84]. As metabolic syndrome (as well as type 2 diabetes) linked oxidative stress and vascular damage (leading to vascular insufficiency) are believed to be risk factors for AD, more detailed studies might reveal other changes in small HDL particles which occur at earlier stages of AD pathogenesis. In accordance with the notion that small HDL display protective features, our data indicated that higher levels of HDL small particles were significantly associated with higher MMSE scores in MCI/AD, suggesting a beneficial role of these small dense HDL in the disease.
In other studies, aging has been shown to affect the composition and function of HDL [85], and HDL levels in general have been shown to decrease with aging [86]. In people with exceptional longevity (age > 95), lower HDL levels do appear to be associated with lower cognitive function [85–89]. Furthermore, phospholipid transfer protein (PLTP) is responsible for HDL remodeling and HDL enlargement, and it is also possible that lower levels of small HDL in AD are a consequence of higher PLTP activity, which has already been reported in AD [90]. Additionally, cholesterol efflux has been shown to be abnormal in aging and has been associated with decreased levels of HDL in aged individuals [86].
It has been reported that increased cerebral Aβ deposition is associated with high levels of LDL and low levels of HDL [39]; however, our analysis did not find any association between Aβ deposition levels and the levels of cholesterol, LDL, HDL, or TG in any clinical group (data not shown). We then evaluated whether levels of the HDL subgroups correlated with levels of Aβ deposition in the brain. From our results, cerebral Aβ deposition level appears to be independent of levels of HDL sub-fractions in almost every clinical group, with the sole exception of a significant negative association between SUVR and HDL Large in the MCI/AD group (p = 0.037). Additionally, we did not observe any correlation between Aβ1–42 plasma levels and HDL or HDL subclasses. We have also considered HDL subclasses as potential biomarkers for HC with high amyloidosis for whom the conversion to AD is more likely. However, we did not observe any different HDL subclasses distribution with regards to brain amyloid deposition. These data indicated that, at least in our cohort, in spite of an altered HDL metabolism, there is no correlation between HDL subclasses levels and brain amyloid deposition or plasma levels of Aβ1–42. However, we cannot exclude that a more detailed HDL analysis that includes additional factor would unveil a link between HDL and amyloid deposition. The fact that HDL subclasses are altered in AD but not in HC Aβ+ suggests that an altered HDL metabolism may be a consequence of the disease progression.
Taken together, our data support previous studies which have shown that patients with AD exhibit alterations in their plasma lipoprotein profile, and we suggest that HDL changes can be attributed to lower levels of small HDL particles. Further studies will be necessary to determine at what stage of AD pathogenesis these alterations to small HDL occur, and how they are involved in the progression of the disease.
ACKNOWLEDGMENTS
We thank all the participants who took part in this study and the clinicians who referred participants. The AIBL study (http://www.AIBL.csiro.au) is a collaboration between CSIRO, Edith Cowan University (ECU), The Florey Institute of Neuroscience and Mental Health (FINMH), National Ageing Research Institute (NARI) and Austin Health. The authors acknowledge the financial support of the Cooperative Research Centre for Mental Health (CRCMH), the CRCMH program is an Australian Government Initiative, McCusker Alzheimer’s Research Foundation, Alzheimer’s Australia Research Foundation (AARF), the Science and Industry Endowment Fund, CSIRO, Brightfocus, USA and the WA Dept. of Health. FINMH acknowledges the funding support from the Victorian Government’s Operational Infrastructure Support program. Pfizer International has contributed financial support to assist with analysis of blood samples and to further the AIBL research program. Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0291r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200291.
REFERENCES
[1] | Boyles JK , Pitas RE , Wilson E , Mahley RW , Taylor JM ((1985) ) Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest 76: , 1501–1513. |
[2] | Diedrich JF , Minnigan H , Carp RI , Whitaker JN , Race R , Frey W 2nd , Haase AT ((1991) ) Neuropathological changes in scrapie and Alzheimer’s disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol 65: , 4759–4768. |
[3] | Nakai M , Kawamata T , Taniguchi T , Maeda K , Tanaka C ((1996) ) Expression of apolipoprotein E mRNA in rat microglia. Neurosci Lett 211: , 41–44. |
[4] | Stone DJ , Rozovsky I , Morgan TE , Anderson CP , Hajian H , Finch CE ((1997) ) Astrocytes and microglia respond to estrogen with increased apoE mRNA in vivo and in vitro. Exp Neurol 143: , 313–318. |
[5] | Stukas S , Robert J , Lee M , Kulic I , Carr M , Tourigny K , Fan J , Namjoshi D , Lemke K , DeValle N , Chan J , Wilson T , Wilkinson A , Chapanian R , Kizhakkedathu JN , Cirrito JR , Oda MN , Wellington CL ((2014) ) Intravenously injected human apolipoprotein A-I rapidly enters the central nervous system via the choroid plexus. J Am Heart Assoc 3: , e001156. |
[6] | Koch S , Donarski N , Goetze K , Kreckel M , Stuerenburg HJ , Buhmann C , Beisiegel U ((2001) ) Characterization of four lipoprotein classes in human cerebrospinal fluid. J Lipid Res 42: , 1143–1151. |
[7] | Lütjohann D , Breuer O , Ahlborg G , Nennesmo I , Sidén A , Diczfalusy U , Björkhem I ((1996) ) Cholesterol homeostasis in human brain: Evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci U S A 93: , 9799–9804. |
[8] | Burns MP , Noble WJ , Olm V , Gaynor K , Casey E , LaFrancois J , Wang L , Duff K ((2003) ) Co-localization of cholesterol, apolipoprotein E and fibrillar Abeta in amyloid plaques. Brain Res Mol Brain Res 110: , 119–125. |
[9] | Refolo LM , Malester B , LaFrancois J , Bryant-Thomas T , Wang R , Tint GS , Sambamurti K , Duff K , Pappolla MA ((2000) ) Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis 7: , 321–331. |
[10] | Wahrle S , Das P , Nyborg AC , McLendon C , Shoji M , Kawarabayashi T , Younkin LH , Younkin SG , Golde TE ((2002) ) Cholesterol-dependent gamma-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis 9: , 11–23. |
[11] | Buxbaum JD , Geoghagen NS , Friedhoff LT ((2001) ) Cholesterol depletion with physiological concentrations of a statin decreases the formation of the Alzheimer amyloid Abeta peptide. J Alzheimers Dis 3: , 221–229. |
[12] | Fassbender K , Simons M , Bergmann C , Stroick M , Lutjohann D , Keller P , Runz H , Kuhl S , Bertsch T , von Bergmann K , Hennerici M , Beyreuther K , Hartmann T ((2001) ) Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc Natl Acad Sci U S A 98: , 5856–5861. |
[13] | Kojro E , Gimpl G , Lammich S , Marz W , Fahrenholz F ((2001) ) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci U S A 98: , 5815–5820. |
[14] | Simons M , Keller P , De Strooper B , Beyreuther K , Dotti CG , Simons K ((1998) ) Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc Natl Acad Sci U S A 95: , 6460–6464. |
[15] | Norton S , Matthews FE , Barnes DE , Yaffe K , Brayne C ((2014) ) Potential for primary prevention of Alzheimer’s disease: An analysis of population-based data. Lancet Neurol 13: , 788–794. |
[16] | Polidori MC , Pientka L , Mecocci P ((2012) ) A review of the major vascular risk factors related to Alzheimer’s disease. J Alzheimers Dis 32: , 521–530. |
[17] | Gupta VB , Laws SM , Villemagne VL , Ames D , Bush AI , Ellis KA , Lui JK , Masters C , Rowe CC , Szoeke C , Taddei K , Martins RN ((2011) ) Plasma apolipoprotein E and Alzheimer disease risk: The AIBL study of aging. Neurology 76: , 1091–1098. |
[18] | Mapstone M , Cheema AK , Fiandaca MS ((2014) ) Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med 20: , 415–418. |
[19] | Song F , Poljak A , Crawford J , Kochan NA , Wen W , Cameron B , Lux O , Brodaty H , Mather K , Smythe GA , Sachdev PS ((2012) ) Plasma apolipoprotein levels are associated with cognitive status and decline in a community cohort of older individuals. PLoS One 7: , e34078. |
[20] | Ramdane S , Daoudi-Gueddah D ((2011) ) Mild hypercholesterolemia, normal plasma triglycerides, and normal glucose levels across dementia staging in Alzheimer’s disease: A clinical setting-based retrospective study. Am J Alzheimers Dis Other Demen 26: , 399–405. |
[21] | Xiao Z , Wang J , Chen W , Wang P , Zeng H ((2012) ) Association studies of several cholesterol-related genes (ABCA1, CETP and LIPC) with serum lipids and risk of Alzheimer’s disease. Lipids Health Dis 11: , 163. |
[22] | Zhang B , Matsunaga A , Saku K , Nakano S , Yamada T ((2004) ) Associations among plasma lipoprotein subfractions as characterized by analytical capillary isotachophoresis, apolipoprotein E phenotype, Alzheimer disease, and mild cognitive impairment. Arterioscler Thromb Vasc Biol 24: , e144–146. |
[23] | Isbir T , Agachan B , Yilmaz H , Aydin M , Kara I , Eker E , Eker D ((2001) ) Apolipoprotein-E gene polymorphism and lipid profiles in Alzheimer’s disease. Am J Alzheimers Dis Other Demen 16: , 77–81. |
[24] | Reitz C , Tang MX , Luchsinger J , Mayeux R ((2004) ) Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch Neurol 61: , 705–714. |
[25] | Tan ZS , Seshadri S , Beiser A , Wilson PW , Kiel DP , Tocco M , D’Agostino RB , Wolf PA ((2003) ) Plasma total cholesterol level as a risk factor for Alzheimer disease: The Framingham Study. Arch Intern Med 163: , 1053–1057. |
[26] | Notkola IL , Sulkava R , Pekkanen J , Erkinjuntti T , Ehnholm C , Kivinen P , Tuomilehto J , Nissinen A ((1998) ) Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer’s disease. Neuroepidemiology 17: , 14–20. |
[27] | Pappolla MA , Bryant-Thomas TK , Herbert D , Pacheco J , Fabra Garcia M , Manjon M , Girones X , Henry TL , Matsubara E , Zambon D , Wolozin B , Sano M , Cruz-Sanchez FF , Thal LJ , Petanceska SS , Refolo LM ((2003) ) Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 61: , 199–205. |
[28] | Zhang J , McKeown RE , Hajjar I ((2005) ) Serum cholesterol levels are associated with impaired recall memory among older people. Age Ageing 34: , 178–182. |
[29] | Levin-Allerhand JA , Lominska CE , Smith JD ((2002) ) Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J Nutr Health Aging 6: , 315–319. |
[30] | Pedrini S , Thomas C , Brautigam H , Schmeidler J , Ho L , Fraser P , Westaway D , Hyslop PS , Martins RN , Buxbaum JD , Pasinetti GM , Dickstein DL , Hof PR , Ehrlich ME , Gandy S ((2009) ) Dietary composition modulates brain mass and solubilizable Abeta levels in a mouse model of aggressive Alzheimer’s amyloid pathology. Mol Neurodegener 4: , 40. |
[31] | Burgess BL , McIsaac SA , Naus KE , Chan JY , Tansley GH , Yang J , Miao F , Ross CJ , van Eck M , Hayden MR , van Nostrand W , St George-Hyslop P , Westaway D , Wellington CL ((2006) ) Elevated plasma triglyceride levels precede amyloid deposition in Alzheimer’s disease mouse models with abundant A beta in plasma. Neurobiol Dis 24: , 114–127. |
[32] | Hoglund K , Wiklund O , Vanderstichele H , Eikenberg O , Vanmechelen E , Blennow K ((2004) ) Plasma levels of beta-amyloid(1–40), beta-amyloid(1–42), and total beta-amyloid remain unaffected in adult patients with hypercholesterolemia after treatment with statins. Arch Neurol 61: , 333–337. |
[33] | Jick H , Zornberg GL , Jick SS , Seshadri S , Drachman DA ((2000) ) Statins and the risk of dementia. Lancet 356: , 1627–1631. |
[34] | Riekse RG , Li G , Petrie EC , Leverenz JB , Vavrek D , Vuletic S , Albers JJ , Montine TJ , Lee VM , Lee M , Seubert P , Galasko D , Schellenberg GD , Hazzard WR , Peskind ER ((2006) ) Effect of statins on Alzheimer’s disease biomarkers in cerebrospinal fluid. J Alzheimers Dis 10: , 399–406. |
[35] | Wolozin B , Kellman W , Ruosseau P , Celesia GG , Siegel G ((2000) ) Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 57: , 1439–1443. |
[36] | Barter PJ , Brandrup-Wognsen G , Palmer MK , Nicholls SJ ((2010) ) Effect of statins on HDL-C: A complex process unrelated to changes in LDL-C: Analysis of the VOYAGER Database. J Lipid Res 51: , 1546–1553. |
[37] | McTaggart F , Jones P ((2008) ) Effects of statins on high-density lipoproteins: A potential contribution to cardiovascular benefit. Cardiovasc Drugs Ther 22: , 321–338. |
[38] | den Heijer T , Hofman A , Koudstaal PJ , Breteler MM ((2005) ) Serum lipids and hippocampal volume: The link to Alzheimer’s disease? Ann Neurol 57: , 779–780; author reply 7780. |
[39] | Reed B , Villeneuve S , Mack W , DeCarli C , Chui HC , Jagust W ((2014) ) Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol 71: , 195–200. |
[40] | Harr SD , Uint L , Hollister R , Hyman BT , Mendez AJ ((1996) ) Brain expression of apolipoproteins E, J, and A-I in Alzheimer’s disease. J Neurochem 66: , 2429–2435. |
[41] | Kawano M , Kawakami M , Otsuka M , Yashima H , Yaginuma T , Ueki A ((1995) ) Marked decrease of plasma apolipoprotein AI and AII in Japanese patients with late-onset non-familial Alzheimer’s disease. Clin Chim Acta 239: , 209–211. |
[42] | Liu HC , Hu CJ , Chang JG , Sung SM , Lee LS , Yuan RY , Leu SJ ((2006) ) Proteomic identification of lower apolipoprotein A-I in Alzheimer’s disease. Dement Geriatr Cogn Disord 21: , 155–161. |
[43] | Merched A , Xia Y , Visvikis S , Serot JM , Siest G ((2000) ) Decreased high-density lipoprotein cholesterol and serum apolipoprotein AI concentrations are highly correlated with the severity of Alzheimer’s disease. Neurobiol Aging 21: , 27–30. |
[44] | Koldamova RP , Lefterov IM , Lefterova MI , Lazo JS ((2001) ) Apolipoprotein A-I directly interacts with amyloid precursor protein and inhibits A beta aggregation and toxicity. Biochemistry 40: , 3553–3560. |
[45] | Paula-Lima AC , Tricerri MA , Brito-Moreira J , Bomfim TR , Oliveira FF , Magdesian MH , Grinberg LT , Panizzutti R , Ferreira ST ((2009) ) Human apolipoprotein A-I binds amyloid-beta and prevents Abeta-induced neurotoxicity. Int J Biochem Cell Biol 41: , 1361–1370. |
[46] | Lefterov I , Fitz NF , Cronican AA , Fogg A , Lefterov P , Kodali R , Wetzel R , Koldamova R ((2010) ) Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem 285: , 36945–36957. |
[47] | Lewis TL , Cao D , Lu H , Mans RA , Su YR , Jungbauer L , Linton MF , Fazio S , LaDu MJ , Li L ((2010) ) Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem 285: , 36958–36968. |
[48] | Pericak-Vance MA , Bebout JL , Gaskell PC Jr. , Yamaoka LH , Hung WY , Alberts MJ , Walker AP , Bartlett RJ , Haynes CA , Welsh KA , Earl NL , Heyman A , Clark CM , Roses AD ((1991) ) Linkage studies in familial Alzheimer disease: Evidence for chromosome 19 linkage. Am J Hum Genet 48: , 1034–1050. |
[49] | Benjamin R , Leake A , McArthur FK , Ince PG , Candy JM , Edwardson JA , Morris CM , Bjertness E ((1994) ) Protective effect of apoE epsilon 2 in Alzheimer’s disease. Lancet 344: , 473. |
[50] | Talbot C , Lendon C , Craddock N , Shears S , Morris JC , Goate A ((1994) ) Protection against Alzheimer’s disease with apoE epsilon 2. Lancet 343: , 1432–1433. |
[51] | Sharman MJ , Morici M , Hone E , Berger T , Taddei K , Martins IJ , Lim WL , Singh S , Wenk MR , Ghiso J , Buxbaum JD , Gandy S , Martins RN ((2010) ) APOE genotype results in differential effects on the peripheral clearance of amyloid-beta42 in APOE knock-in and knock-out mice. J Alzheimers Dis 21: , 403–409. |
[52] | Verghese PB , Castellano JM , Holtzman DM ((2011) ) Apolipoprotein E in Alzheimer’s disease and other neurological disorders. Lancet Neurol 10: , 241–252. |
[53] | Michikawa M , Fan QW , Isobe I , Yanagisawa K ((2000) ) Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J Neurochem 74: , 1008–1016. |
[54] | Rapp A , Gmeiner B , Huttinger M ((2006) ) Implication of apoE isoforms in cholesterol metabolism by primary rat hippocampal neurons and astrocytes. Biochimie 88: , 473–483. |
[55] | Ellis KA , Bush AI , Darby D , De Fazio D , Foster J , Hudson P , Lautenschlager NT , Lenzo N , Martins RN , Maruff P , Masters C , Milner A , Pike K , Rowe C , Savage G , Szoeke C , Taddei K , Villemagne V , Woodward M , Ames D ((2009) ) The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: Methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr 21: , 672–687. |
[56] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[57] | Winblad B , Palmer K , Kivipelto M , Jelic V , Fratiglioni L , Wahlund LO , Nordberg A , Backman L , Albert M , Almkvist O , Arai H , Basun H , Blennow K , de Leon M , DeCarli C , Erkinjuntti T , Giacobini E , Graff C , Hardy J , Jack C , Jorm A , Ritchie K , van Duijn C , Visser P , Petersen RC ((2004) ) Mild cognitive impairment–beyond controversies, towards a consensus: Report of the International Working Group on Mild Cognitive Impairment. J Intern Med 256: , 240–246. |
[58] | Beheshti I , Wessels LM , Eckfeldt JH ((1994) ) EDTA-plasma vs serum differences in cholesterol, high-density-lipoprotein cholesterol, and triglyceride as measured by several methods. Clin Chem 40: , 2088–2092. |
[59] | Folsom AR , Kuba K , Leupker RV , Jacobs DR , Frantz ID Jr. ((1983) ) Lipid concentrations in serum and EDTA-treated plasma from fasting and nonfasting normal persons, with particular regard to high-density lipoprotein cholesterol. Clin Chem 29: , 505–508. |
[60] | Villemagne VL , Pike KE , Chetelat G , Ellis KA , Mulligan RS , Bourgeat P , Ackermann U , Jones G , Szoeke C , Salvado O , Martins R , O’Keefe G , Mathis CA , Klunk WE , Ames D , Masters CL , Rowe CC ((2011) ) Longitudinal assessment of Abeta and cognition in aging and Alzheimer disease. Ann Neurol 69: , 181–192. |
[61] | Klunk WE , Koeppe RA , Price JC , Benzinger TL , Devous MD Sr. , Jagust WJ , Johnson KA , Mathis CA , Minhas D , Pontecorvo MJ , Rowe CC , Skovronsky DM , Mintun MA ((2015) ) The Centiloid Project: Standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement 11: , 1–15 e11-14. |
[62] | Bourgeat P , Dore V , Fripp J , Ames D , Masters CL , Salvado O , Villemagne VL , Rowe CC ((2018) ) Implementing the centiloid transformation for (11)C-PiB and beta-amyloid (18)F-PET tracers using CapAIBL. Neuroimage 183: , 387–393. |
[63] | Gepner AD , Piper ME , Johnson HM , Fiore MC , Baker TB , Stein JH ((2011) ) Effects of smoking and smoking cessation on lipids and lipoproteins: Outcomes from a randomized clinical trial. Am Heart J 161: , 145–151. |
[64] | Murray IV , Proza JF , Sohrabji F , Lawler JM ((2011) ) Vascular and metabolic dysfunction in Alzheimer’s disease: A review. Exp Biol Med (Maywood) 236: , 772–782. |
[65] | Oksman M , Iivonen H , Hogyes E , Amtul Z , Penke B , Leenders I , Broersen L , Lutjohann D , Hartmann T , Tanila H ((2006) ) Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis 23: , 563–572. |
[66] | Park SH , Kim JH , Choi KH , Jang YJ , Bae SS , Choi BT , Shin HK ((2013) ) Hypercholesterolemia accelerates amyloid beta-induced cognitive deficits. Int J Mol Med 31: , 577–582. |
[67] | Guardia-Laguarta C , Coma M , Pera M , Clarimon J , Sereno L , Agullo JM , Molina-Porcel L , Gallardo E , Deng A , Berezovska O , Hyman BT , Blesa R , Gomez-Isla T , Lleo A ((2009) ) Mild cholesterol depletion reduces amyloid-beta production by impairing APP trafficking to the cell surface. J Neurochem 110: , 220–230. |
[68] | Lesser GT , Beeri MS , Schmeidler J , Purohit DP , Haroutunian V ((2011) ) Cholesterol and LDL relate to neuritic plaques and to APOE4 presence but not to neurofibrillary tangles. Curr Alzheimer Res 8: , 303–312. |
[69] | Xiong H , Callaghan D , Jones A , Walker DG , Lue LF , Beach TG , Sue LI , Woulfe J , Xu H , Stanimirovic DB , Zhang W ((2008) ) Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis 29: , 422–437. |
[70] | Hoglund K , Thelen KM , Syversen S , Sjogren M , von Bergmann K , Wallin A , Vanmechelen E , Vanderstichele H , Lutjohann D , Blennow K ((2005) ) The effect of simvastatin treatment on the amyloid precursor protein and brain cholesterol metabolism in patients with Alzheimer’s disease. Dement Geriatr Cogn Disord 19: , 256–265. |
[71] | Ishii K , Tokuda T , Matsushima T , Miya F , Shoji S , Ikeda S , Tamaoka A ((2003) ) Pravastatin at 10 mg/day does not decrease plasma levels of either amyloid-beta (Abeta) 40 or Abeta 42 in humans. Neurosci Lett 350: , 161–164. |
[72] | Simons M , Schwarzler F , Lutjohann D , von Bergmann K , Beyreuther K , Dichgans J , Wormstall H , Hartmann T , Schulz JB ((2002) ) Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol 52: , 346–350. |
[73] | Shepardson NE , Shankar GM , Selkoe DJ ((2011) ) Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch Neurol 68: , 1385–1392. |
[74] | Shepardson NE , Shankar GM , Selkoe DJ ((2011) ) Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch Neurol 68: , 1239–1244. |
[75] | Eckert GP , Hooff GP , Strandjord DM , Igbavboa U , Volmer DA , Muller WE , Wood WG ((2009) ) Regulation of the brain isoprenoids farnesyl- and geranylgeranylpyrophosphate is altered in male Alzheimer patients. Neurobiol Dis 35: , 251–257. |
[76] | Hooff GP , Peters I , Wood WG , Muller WE , Eckert GP ((2010) ) Modulation of cholesterol, farnesylpyrophosphate, and geranylgeranylpyrophosphate in neuroblastoma SH-SY5Y-APP695 cells: Impact on amyloid beta-protein production. Mol Neurobiol 41: , 341–350. |
[77] | Hooff GP , Wood WG , Muller WE , Eckert GP ((2010) ) Isoprenoids, small GTPases and Alzheimer’s disease. Biochim Biophys Acta 1801: , 896–905. |
[78] | Pedrini S , Carter TL , Prendergast G , Petanceska S , Ehrlich ME , Gandy S ((2005) ) Modulation of statin-activated shedding of Alzheimer APP ectodomain by ROCK. PLoS Med 2: , e18. |
[79] | Ward MA , Bendlin BB , McLaren DG , Hess TM , Gallagher CL , Kastman EK , Rowley HA , Asthana S , Carlsson CM , Sager MA , Johnson SC ((2010) ) Low HDL cholesterol is associated with lower gray matter volume in cognitively healthy adults. Front Aging Neurosci 2: , 29. |
[80] | Reitz C , Tang MX , Schupf N , Manly JJ , Mayeux R , Luchsinger JA ((2010) ) Association of higher levels of high-density lipoprotein cholesterol in elderly individuals and lower risk of late-onset Alzheimer disease. Arch Neurol 67: , 1491–1497. |
[81] | Dias IH , Polidori MC , Li L , Weber D , Stahl W , Nelles G , Grune T , Griffiths HR ((2014) ) Plasma levels of HDL and carotenoids are lower in dementia patients with vascular comorbidities. J Alzheimers Dis 40: , 399–408. |
[82] | Raygani AV , Rahimi Z , Kharazi H , Tavilani H , Pourmotabbed T ((2006) ) Association between apolipoprotein E polymorphism and serum lipid and apolipoprotein levels with Alzheimer’s disease. Neurosci Lett 408: , 68–72. |
[83] | Kontush A , Chantepie S , Chapman MJ ((2003) ) Small, dense HDL particles exert potent protection of atherogenic LDL against oxidative stress. Arterioscler Thromb Vasc Biol 23: , 1881–1888. |
[84] | Barron AM , Rosario ER , Elteriefi R , Pike CJ ((2013) ) Sex-specific effects of high fat diet on indices of metabolic syndrome in 3xTg-AD mice: Implications for Alzheimer’s disease. PLoS One 8: , e78554. |
[85] | Holzer M , Trieb M , Konya V , Wadsack C , Heinemann A , Marsche G ((2013) ) Aging affects high-density lipoprotein composition and function. Biochim Biophys Acta 1831: , 1442–1448. |
[86] | Berrougui H , Khalil A ((2009) ) Age-associated decrease of high-density lipoprotein-mediated reverse cholesterol transport activity. Rejuvenation Res 12: , 117–126. |
[87] | Arai Y , Hirose N ((2004) ) Aging and HDL metabolism in elderly people more than 100 years old. J Atheroscler Thromb 11: , 246–252. |
[88] | Atzmon G , Gabriely I , Greiner W , Davidson D , Schechter C , Barzilai N ((2002) ) Plasma HDL levels highly correlate with cognitive function in exceptional longevity. J Gerontol A Biol Sci Med Sci 57: , M712–715. |
[89] | Walter M ((2009) ) Interrelationships among HDL metabolism, aging, and atherosclerosis. Arterioscler Thromb Vasc Biol 29: , 1244–1250. |
[90] | Vuletic S , Jin LW , Marcovina SM , Peskind ER , Moller T , Albers JJ ((2003) ) Widespread distribution of PLTP in human CNS: Evidence for PLTP synthesis by glia and neurons, and increased levels in Alzheimer’s disease. J Lipid Res 44: , 1113–1123. |


