
Serum Hepcidin Levels in Cognitively Normal Older Adults with High Neocortical Amyloid-β Load
Abstract
Background/Objective:
Hepcidin, an iron-regulating hormone, suppresses the release of iron by binding to the iron exporter protein, ferroportin, resulting in intracellular iron accumulation. Given that iron dyshomeostasis has been observed in Alzheimer’s disease (AD) together with elevated serum hepcidin levels, the current study examined whether elevated serum hepcidin levels are an early event in AD pathogenesis by measuring the hormone in cognitively normal older adults at risk of AD, based on high neocortical amyloid-β load (NAL).
Methods:
Serum hepcidin levels in cognitively normal participants (n = 100) aged between 65–90 years were measured using ELISA. To evaluate NAL, all participants underwent 18F-florbetaben positron emission tomography. A standard uptake value ratio (SUVR)<1.35 was classified as low NAL (n = 65) and ≥1.35 (n = 35) was classified as high NAL.
Results:
Serum hepcidin was significantly higher in participants with high NAL compared to those with low NAL before and after adjusting for covariates: age, gender, and APOE ɛ4 carriage (p < 0.05). A receiver operating characteristic curve based on a logistic regression of the same covariates, the base model, distinguished high from low NAL (area under the curve, AUC = 0.766), but was outperformed when serum hepcidin was added to the base model (AUC = 0.794) and further improved with plasma Aβ42/40 ratio (AUC = 0.829).
Conclusion:
The present findings indicate that serum hepcidin is increased in individuals at risk for AD and contribute to the body of evidence supporting iron dyshomeostasis as an early event of AD. Further, hepcidin may add value to a panel of markers that contribute toward identifying individuals at risk of AD; however, further validation studies are required.
INTRODUCTION
There is increasing evidence of a disruption in iron homeostasis in the brain in Alzheimer’s disease (AD) pathogenesis [1–3]. This iron dysregulation is also reflected in the blood wherein altered levels of the iron storage and transfer protein (ferritin and transferrin) have been observed in AD patients compared with healthy controls [4]. Interestingly, higher serum ferritin has also been reported in cognitively normal individuals at risk of AD, based on high neocortical amyloid-β load (NAL) [5–7].
Hepcidin, a key protein involved in iron homeostasis, is an iron-regulating hormone. Hepcidin suppresses the release of iron by binding and internalizing the iron exporter protein, ferroportin, resulting in intracellular iron accumulation. Hepcidin levels have been reported to be lower in AD brain tissue compared to age-matched healthy adults [8]. Further, lower hepcidin levels have also been reported in AD transgenic mouse (Tg2576) brains compared to wild type brains [8]. In contrast, in the blood, hepcidin levels are significantly higher in AD patients compared to control participants [9–11]. Furthermore, blood hepcidin levels have also been reported to have a positive association with AD severity [9, 10].
The current study investigated whether elevated hepcidin levels in the blood precede the onset of the clinical symptoms of AD by measuring this iron-regulating hormone in cognitively normal older adults at risk of AD. Given that the onset of abnormal NAL build-up, assessed using positron emission tomography (PET) occurs approximately two decades prior to the clinical manifestation of AD, is a prodromal feature and biomarker of AD [12], serum hepcidin levels were compared between cognitively normal older adults with high NAL and low NAL. Further, the current study also evaluated if hepcidin has potential to distinguish between individuals with high NAL and low NAL.
MATERIALS AND METHODS
Participants
All study participants were from the Kerr Anglican Retirement Village Initiative in Ageing Health (KARVIAH) cohort. All participants met the screening (inclusion and exclusion) criteria. Briefly, the inclusion criteria required volunteers to be within an age range of 65–90 years, have good general health and have no known significant cerebral vascular disease based on their medical history, be fluent in English, have adequate/corrected vision and hearing to enable testing, and have no objective memory impairment as determined by a Montreal Cognitive Assessment (MoCA) score ≥26. MoCA scores between 18–25 were assessed on a case by case basis by the study neuropsychologist following score stratification according to age and education [13]. The exclusion criteria included diagnosis of dementia based on the revised criteria from the National Institute on Aging - Alzheimer’s Association [14], presence of acute functional psychiatric disorder, history of stroke, depression (based on the Depression, Anxiety, Stress Scales) and uncontrolled hypertension (systolic BP > 170 or diastolic BP > 100). One hundred and five participants out of the 134 volunteers meeting the inclusion/exclusion criteria underwent blood collection, neuroimaging, and neuropsychometric evaluation, while the remaining volunteers either declined neuroimaging or withdrew from the study. One hundred participants of the above 105 participants were considered as cognitively normal based on their Mini-Mental State Examination score (MMSE≥26) for the current study. All volunteers provided written informed consent prior to participation and the Bellberry Human Research Ethics Committee, Australia, and Macquarie University Human Research Ethics Committee provided approval for the study.
Assessment of neocortical amyloid-β load via PET
Study participants underwent PET imaging using ligand 18F-florbetaben (FBB) and magnetic resonance imaging (MRI) at Macquarie Medical Imaging in Sydney within a time frame of three months from blood collection. For PET, an intravenous bolus of FBB was slowly administered over 30 s, while participants were in a rested position and images were acquired over a 20 min scan, in 5 min acquisitions, beginning 50 min post injection. Neocortical amyloid-β load was calculated as the mean SUVR of the frontal, superior parietal, lateral temporal, lateral occipital, and anterior and posterior cingulate regions using image processing software, CapAIBL [15, 16]. Using a SUVR cut-off score of 1.35 (derived from a hierarchical cluster analysis), participants were categorized into low NAL (SUVR < 1.35, n = 65) and high NAL (SUVR≥1.35, n = 35) [17]. Ninety-six of the 100 participants within the current study passed all standard MRI inclusion/exclusion criteria and underwent MRI as described previously using a General Electric 3Tesla scanner (Model 750W) [18]. Hippocampal volume was calculated from the images acquired and was normalized to the total intracranial volume comprising the cerebrospinal fluid, grey matter, and white matter.
Blood collection and APOE genotyping
All participants fasted for a minimum of 10 h overnight prior to blood withdrawal. The blood drawn was fractionated as described previously by Ellis et al. [19], and stored at –80°C. Apolipoprotein E (APOE) genotype was determined from purified genomic DNA extracted from 0.5 ml whole blood as previously described [5].
Measurement of serum hepcidin
Serum samples stored at –80°C were thawed on ice prior to using the Hepcidin-25 (bioactive) DRG ELISA kits (DRG International, Springfield, NJ, USA) to measure serum hepcidin levels. All reagents were prepared according to the manufacturer’s instructions and the assay was run according to the manufacturer’s instructions. Briefly, 20 μL of each standard, control and test sample were dispensed into appropriate wells of a pre-coated antibody plate (in duplicates) following which 50 μL of the enzyme conjugate was dispensed into each well and mixed thoroughly for 10 s. The plate was then incubated at room temperature for 60 min, following which the contents of the wells were briskly shaken out and the wells were washed with wash solution. Subsequently, 100 μL of enzyme complex was dispensed into each well and incubated for 30 min at room temperature. Following incubation, the contents of the wells were emptied and washed with wash solution. The substrate solution was added to each well and incubated for 20 min at room temperature following which the stop solution was added to stop the enzymatic reaction and the plate was read at 450±10 nm. The intensity of the color developed after incubation was inversely proportional to the concentration of hepcidin-25 in the patient sample. The detection range of the assay was 0.153 ng/mL–81 ng/mL. The limit of quantification (LoQ) was 1.149 ng/mL and coefficients of variation (CVs) obtained were < 10%.
Measurement of serum C-reactive protein, ferritin, iron, transferrin, and ceruloplasmin levels and plasma Aβ40 and Aβ42
Serum C-reactive protein (CRP) levels were measured employing the Siemens ADVIA® wide range C-reactive protein assay (wr-CRP) at Laverty Pathology, New South Wales (NSW), Australia. Serum ferritin levels were measured using a sandwich immunoassay utilizing direct chemiluminometric technology (ADVIA Centaur® Ferritin assay, Siemens Healthcare and Diagnostics) at Laverty Pathology, NSW, Australia. Serum iron and transferrin levels were measured using colorimetric and immunoturbidimetric methods, respectively, using the ADVIA® Chemistry systems, Siemens Healthcare Diagnostics at Laverty Pathology, NSW, Australia. Ceruloplasmin levels were measured immunoturbidimetrically on the Roche cobas® c system at Laverty Pathology, NSW, Australia. The ultra-sensitive single molecule array (Simoa, Quanterix) platform was employed to measure plasma Aβ40 and Aβ42 concentrations as described previously [20]. Ninety-five of the 100 participants from the current study had reliable data for both plasma Aβ40 and Aβ42 and were employed within the current study.
Dietary iron intake
Dietary iron intake was investigated to establish whether diet attributed to the comparisons of the iron-related proteins being made between the low and high NAL groups, employing data from the Cancer Council of Victoria Food Frequency Questionnaire (CCVFFQ) [21]. The CCVFFQ is a paper-based semi-quantitative seventy-four item questionnaire. The CCVFFQ was optically scanned to obtain nutrient intakes in gram/day. Participants filled out the CCVFFQ when they visited the research center for their assessment.
Neuropsychological testing
Study participants underwent a comprehensive battery of neuropsychological tests with MMSE scores utilized as the primary measure for global cognitive ability assessment within the current study [22]. The scores obtained from a MMSE can fall within a range of 0–30, indicating severe impairment (0) to no impairment (30).
Statistical analyses
Descriptive statistics including means and standard deviations were calculated for high and low NAL groups. Chi-square tests were employed to compare gender and APOE ɛ4 carrier status between high and low NAL groups. Response variables were log transformed as necessary to better approximate normality and variance homogeneity. Linear models were employed to compare serum hepcidin levels between high and low NAL groups, with and without adjusting for covariates age, gender, APOE ɛ4 carrier status, and CRP. Logistic regression with high/low NAL as response was used to evaluate predictive models. Spearman’s (rs) correlation coefficient was employed for all continuous variable correlations. Within the current study, p < 0.05 was considered significant for all analyses carried out. All analyses were carried out using IBM® SPSS® Version 23, while receiver operating characteristic (ROC) curves were generated using the package Deducer on R (version 3.2.5).
RESULTS
The study cohort characteristics including participant demographics, APOE ɛ4 carriage, MMSE scores, hippocampal volume, and NAL, are presented in Table 1. The frequency of APOE ɛ4 carriers was significantly higher in participants with high NAL compared to those with low NAL (p < 0.0001), as expected [23]. No statistically significant differences were observed in age, gender, MMSE scores, and hippocampal volume between the high NAL and low NAL groups.
Table 1
Characteristics of study participants
| Low NAL (n = 65) | High NAL (n = 35) | p | |
| Gender (M/F) | 19/46 | 13/22 | 0.419 |
| Age (y, mean±SD) | 77.62±5.56 | 79.23±5.38 | 0.165 |
| APOE ɛ4 carriers % (n) | 7.69 (5) | 45.71 (16) | <0.0001 |
| APOE ɛ2 carriers % (n) | 21.54 (14) | 14.29 (5) | 0.378 |
| MMSE (mean±SD) | 28.51±1.16 | 28.80±1.11 | 0.225 |
| MoCA (mean±SD) | 27.43±1.67 | 27.03±1.92 | 0.278 |
| HV% (left; right lobes, mean±SD, n Low | 0.195±0.0201; | 0.194±0.0197; | 0.805; |
| NAL = 64, n high NAL = 32) | 0.199±0.0214 | 0.200±0.0186 | 0.891 |
| NAL (mean±SD) | 1.16±0.09 | 1.71±0.26 | – |
| Plasma Aβ42/40 (mean±SD, n Low | 0.052±0.008 | 0.047±0.005 | 0.004† |
| NAL = 63, n high NAL = 32) |
Baseline characteristics including gender, age, APOE ɛ4/ɛ2 carrier status, Mini-Mental State Examination (MMSE) scores, Montreal Cognitive Assessment (MoCA) scores (adjusted for education), hippocampal volume (HV; normalized by intracranial volume), and neocortical amyloid-β load (NAL; represented by the standard uptake value ratio (SUVR) of the ligand 18F-florbetaben in the neocortical region normalized with that in the cerebellum) compared between participants with low NAL (SUVR < 1.35) and high NAL (SUVR≥1.35) are presented. Additionally, plasma Aβ42/40 ratios are also presented. †indicates p-values obtained from variables transformed to the logarithmic scale for analyses. Chi-square tests or linear models were employed, as appropriate. p < 0.05 was considered as significant.
Hepcidin levels were significantly higher (∼33%, p = 0.036) in participants with high NAL (mean±SD:30.49±19.34 μg/L) compared to those with low NAL (22.88±15.72 μg/L) (Fig. 1). This observation remained significant after adjusting for the AD risk factors, age, gender, and APOE ɛ4 carrier status, as covariates (p = 0.018) and continued to remain significant after adjusting for the inflammation marker, CRP (p = 0.029). Additionally, serum ferritin concentrations were significantly higher in individuals with high NAL compared to low NAL, while no significant differences were observed in serum iron and other iron related measures (transferrin, transferrin saturation, and ceruloplasmin) in individuals with high NAL compared to those with low NAL (Table 2) as observed previously [5].
Fig.1
Comparison of serum hepcidin and other iron related proteins between participants with low and high NAL. A) Serum hepcidin (n low NAL = 65, n high NAL = 35), B) serum ferritin (n low NAL = 64, n high NAL = 34), C) serum iron (n low NAL = 64, n high NAL = 35), D) transferrin (n low NAL = 64, n high NAL = 35), E) transferrin saturation% (n low NAL = 64, n high NAL = 35), and F) ceruloplasmin (n low NAL = 63, n high NAL = 35) were compared between participants with high NAL and low NAL. Serum hepcidin (p = 0.036) and ferritin (p = 0.013) were significantly higher in high NAL compared to low NAL, while the other measures were non-significant (p > 0.05). Error bars represent standard deviation and line segments represent the mean. *represents p < 0.05. NAL, neocortical amyloid-β load.
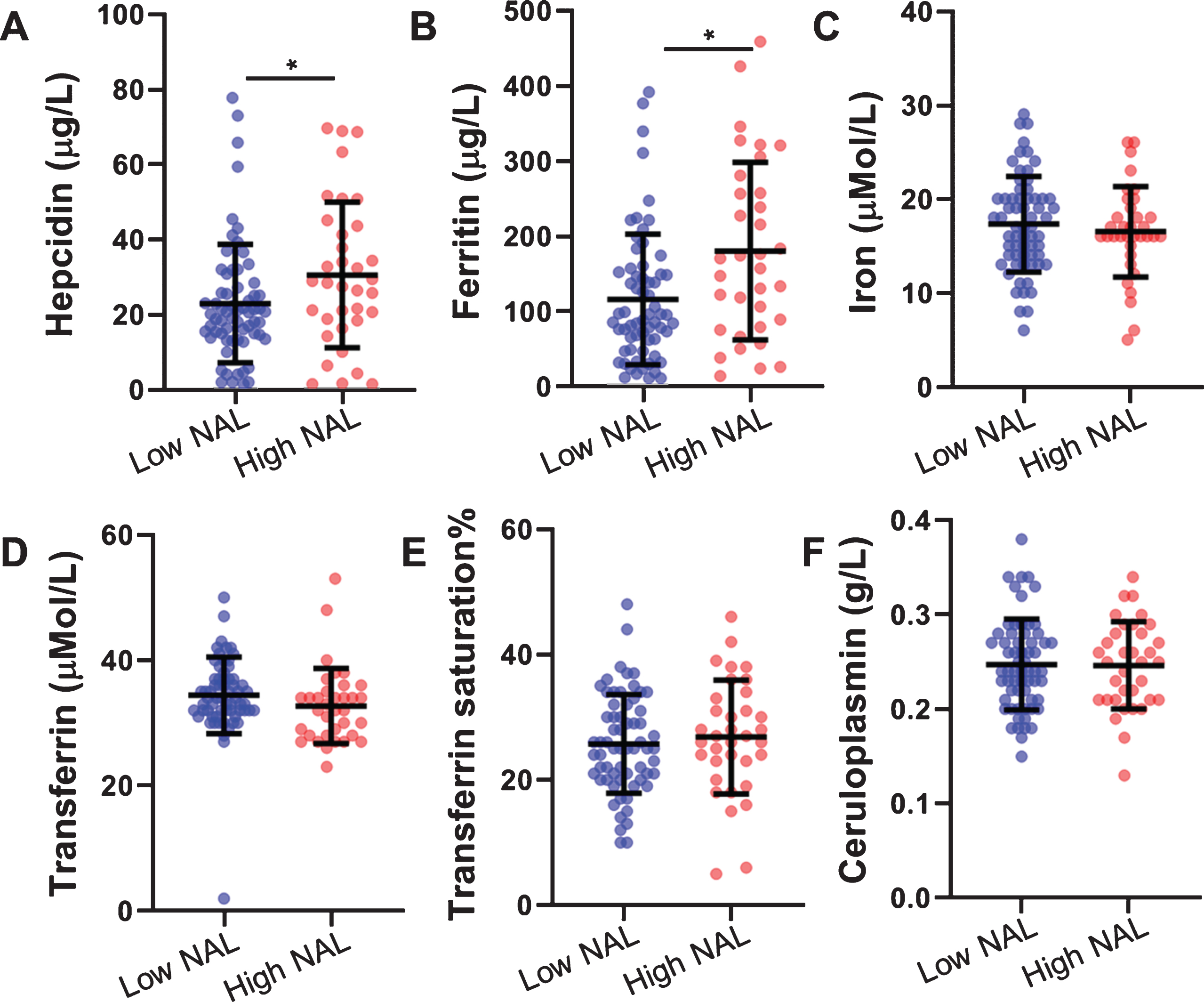
Table 2
Comparison of serum iron related proteins between participants with low and high NAL
| Low NAL | High NAL | p | pa | pb | |
| Serum hepcidin (g/L) | 22.88±15.72 | 30.49±19.34 | 0.036 | 0.018 | 0.029 |
| Serum ferritin (μg/L, n Low NAL = 64, n high NAL = 34) | 115.53±86.73 | 179.65±117.97 | 0.013† | 0.023† | 0.032† |
| Serum iron (μmol/L, n Low NAL = 64, n high NAL = 35) | 17.31±5.09 | 16.51±4.82 | 0.453 | 0.868 | 0.777 |
| Serum transferrin (μmol/L, n Low NAL = 64, n high NAL = 35) | 34.43±6.06 | 32.71±6.01 | 0.181 | 0.128 | 0.155 |
| Transferrin saturation (%, n Low NAL = 64, n high NAL = 35) | 25.73±7.88 | 26.86±9.08 | 0.522 | 0.173 | 0.156 |
| Ceruloplasmin (g/L, n Low NAL = 63, n high NAL = 35) | 0.247±0.048 | 0.246±.046 | 0.932 | 0.769 | 0.973 |
Concentrations were compared between study participants with low and high neocortical amyloid-β load (NAL) represented by the standard uptake value ratio (SUVR) of the ligand 18F-Florbetaben in the neocortical region normalized with that in the cerebellum. Low NAL (n = 65, unless mentioned otherwise) was defined as SUVR < 1.35 while high NAL (n = 35, unless mentioned otherwise) was defined as SUVR≥1.35. †indicates p-values obtained from variables transformed to the logarithmic scale for analyses. pa values have been adjusted for age, gender, and APOE ɛ4 status, pb values have been adjusted for age, gender, APOE ɛ4 status, and C-reactive protein (CRP). p < 0.05 was considered as significant. Data are presented in mean±SD.
Further, to explore whether dietary iron intake directly attributed to the observations in the present study, we assessed the dietary intake of iron between low NAL and high NAL participants, employing data from the CCVFFQ. However, no significant difference was observed in the dietary intake of iron between the two groups, before (mean±SD: low NAL = 11.8±3.9 mg/day; high NAL = 12.3±4.3 mg/day; p = 0.536) and after adjusting for covariates age, gender, and APOE ɛ4 allele status (p = 0.573).
On investigating the association between hepcidin and AD risk factors, no significant association was observed between serum hepcidin levels and age (rs = 0.022, p = 0.827), APOE ɛ4 carriage (carriers (mean±SD):26.25±17.19; non-carriers: 25.36±17.52, p = 0.836), or gender (males (mean±SD):22.54±17.36; females: 26.95±17.32, p = 0.238).
Further, serum hepcidin levels correlated with serum iron levels (rs = 0.228, p = 0.023). Serum hepcidin levels also correlated with serum ferritin (rs = 0.731, p < 0.0001), serum transferrin (rs = –0.425, p < 0.0001), saturated transferrin (rs = 0.355, p < 0.0005), and inflammatory protein, CRP (rs = 0.2, p = 0.046) (Fig. 2). Further, a trend of association was observed between hepcidin and ceruloplasmin (rs = 0.191, p = 0.059). After stratifying participants by NAL status, ferritin, transferrin, and saturated transferrin continued to significantly correlate with hepcidin in both low and high NAL participants. However, serum iron, ceruloplasmin, and CRP only correlated significantly with hepcidin in the low NAL participants (Table 3).
Fig.2
Association of serum hepcidin with iron, iron related proteins, and CRP. Serum hepcidin levels correlated with levels of A) serum iron (n = 99), B) serum ferritin (n = 98), C) serum transferrin (n = 99), D) transferrin saturation% (n = 99), and E) C-reactive protein (n = 100), as determined using Spearman’s correlation coefficient (rs).
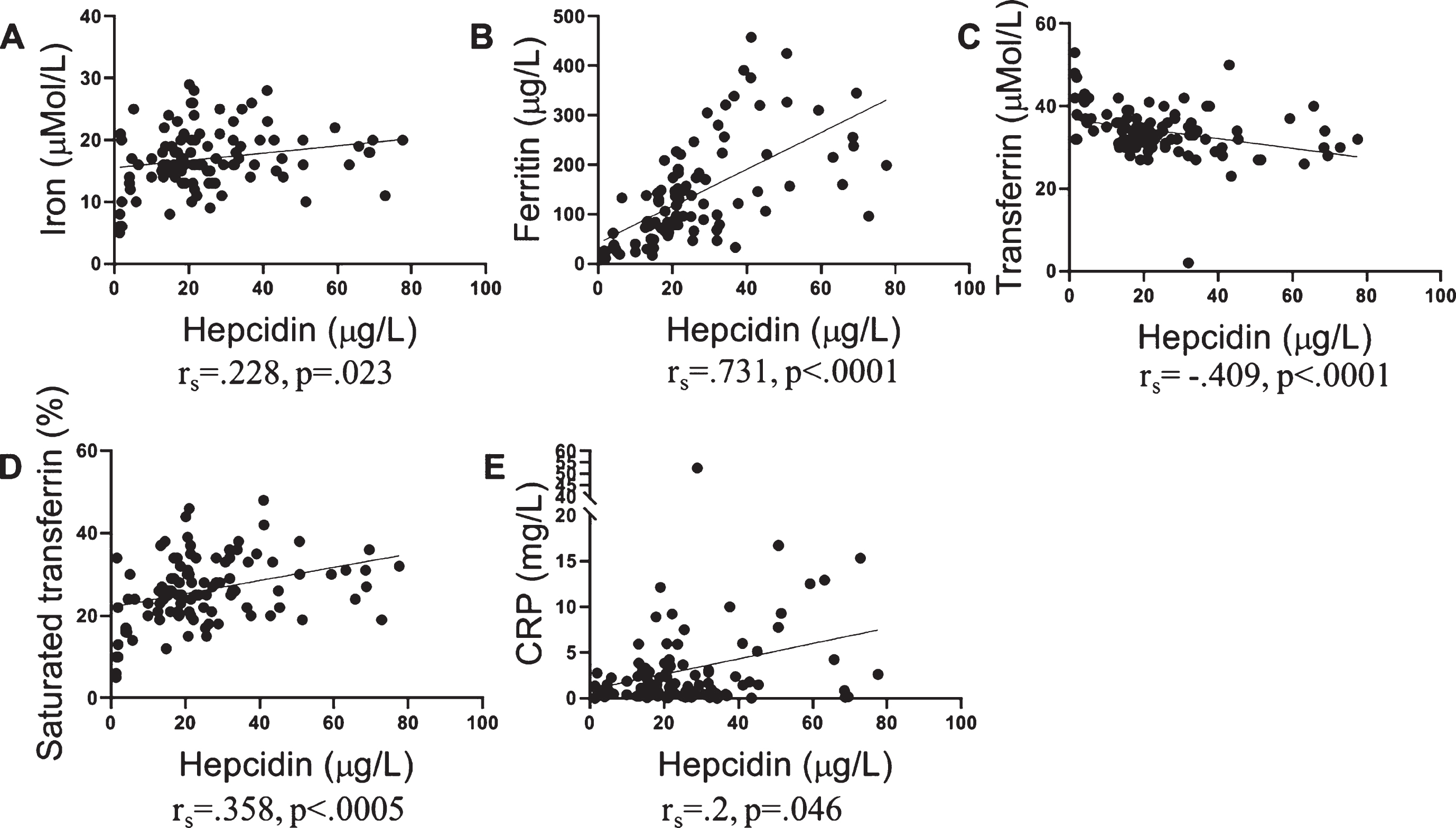
Table 3
Association of serum hepcidin with serum iron, iron related proteins, and C-reactive protein
| All participants | Low NAL | High NAL | ||||
| rs | p | rs | p | rs | p | |
| Iron | 0.228 | 0.023 | 0.252 | 0.045 | 0.271 | 0.115 |
| Ferritin | 0.731 | <0.0001 | 0.686 | <0.0001 | 0.731 | <0.0001 |
| Transferrin | –0.425 | <0.0001 | –0.331 | 0.008 | –0.507 | 0.002 |
| Transferrin saturation | 0.355 | <0.0005 | 0.326 | 0.009 | 0.415 | 0.013 |
| Ceruloplasmin | 0.191 | 0.059 | 0.333 | 0.008 | –0.033 | 0.851 |
| C-reactive protein | 0.200 | 0.046 | 0.247 | 0.047 | 0.258 | 0.134 |
Correlations between serum hepcidin levels and iron, ferritin, transferrin, saturated transferrin, and ceruloplasmin levels were examined in all participants and after stratification based on the neocortical amyloid-β load (NAL) status using Spearman’s correlation coefficient (rs). p < 0.05 was considered as significant.
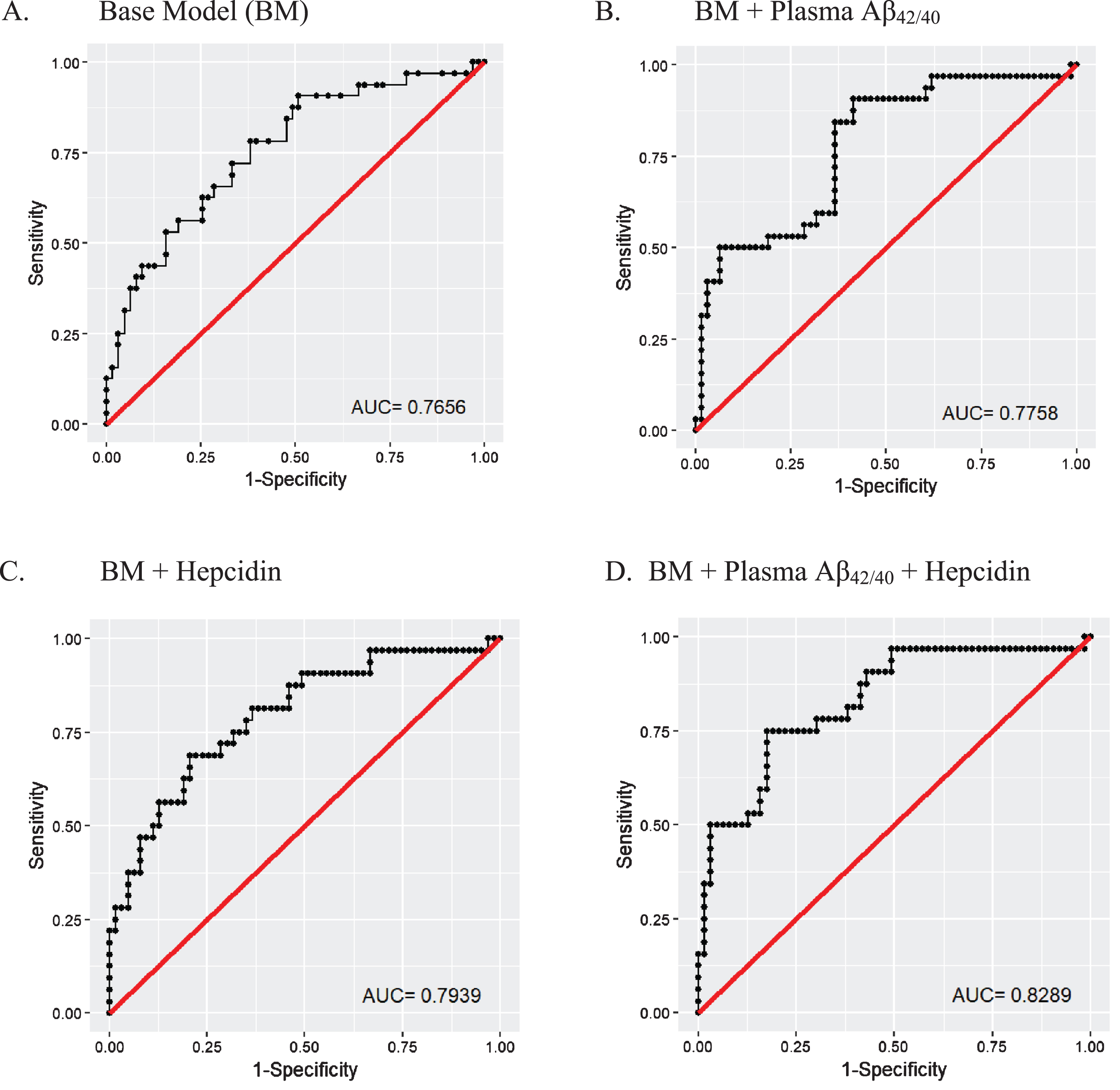
To evaluate the potential of serum hepcidin as a biomarker to distinguish between low and high NAL, receiver operating characteristic (ROC) curves were generated using logistic regressions. A ‘base’ model incorporating the major risk factors for AD (age, APOE ɛ4 allele status, and gender) was generated, to which another promising blood marker, plasma Aβ42/40 ratio [20, 24] or (/and) serum hepcidin was (were) added such that a ‘base + plasma Aβ42/40’ model, a ‘base + serum hepcidin’ model and a ‘base + plasma Aβ42/40 + serum hepcidin’ model were generated. The area under the curves (AUCs) of the ‘base + plasma Aβ42/40’ model (AUC = .776, 78% sensitivity, 63.5% specificity), the ‘base + serum hepcidin’ model (AUC = .794, 78% sensitivity, 65.1% specificity) and the ‘base + plasma Aβ42/40 + serum hepcidin’ model (AUC = .829, 78% sensitivity, 69.8% specificity) were observed to outperform the ‘base’ model (AUC = .766, 78% sensitivity, 61.9% specificity) in distinguishing between high NAL and low NAL (Fig. 3).
Fig.3
Receiver operating characteristic (ROC) curves for the prediction of high neocortical amyloid-β load. ROCs were created for (A) a ‘base model’ comprising risk factors for AD, namely age, gender, and APOE ɛ4 allele status (BM), (B) BM + plasma Aβ42/40, (C) BM + hepcidin, and (D) BM + plasma Aβ42/40 ratio + hepcidin. The BM was outperformed by models B, C, and D. Logistic regression models were employed to perform the analyses. All models were generated using data from the 95 participants that had data for plasma Aβ42/40 (Low NAL = 63, High NAL = 32).
DISCUSSION
Observations from the current study indicate that serum hepcidin concentration is increased in cognitively normal older adults at risk for AD, characterized by high NAL, prior to hippocampal atrophy. Our findings of increased serum hepcidin in individuals with high NAL build on the earlier work of Sternberg and colleagues and Kweon and colleagues who observed increased serum hepcidin levels in clinically diagnosed AD patients, by demonstrating that changes in this hormone is an early event in the pathogenesis of AD [9, 10]. Additionally, the observations in the present study did not appear to be traceable to the participants’ dietary intake of iron as assessed from the CCVFFQ, suggesting the observations were associated with iron dyshomeostasis within the asymptomatic at-risk state of the disease, rather than dietary iron intake. However, it is acknowledged that data from food frequency questionnaires do not necessarily reflect an accurate evaluation of the dietary intake of iron but rather serve as a suitable reflection of exposure to certain food and nutrient types over a period of time. Further, within the current study we also observed that serum hepcidin along with the AD risk factors age, APOE ɛ4 status, gender, and plasma Aβ42/40 ratios (a blood biomarker reflecting brain amyloid status), distinguished between low NAL and high NAL (AUC = .83); however, it is acknowledged that hepcidin’s contribution alone is relatively modest.
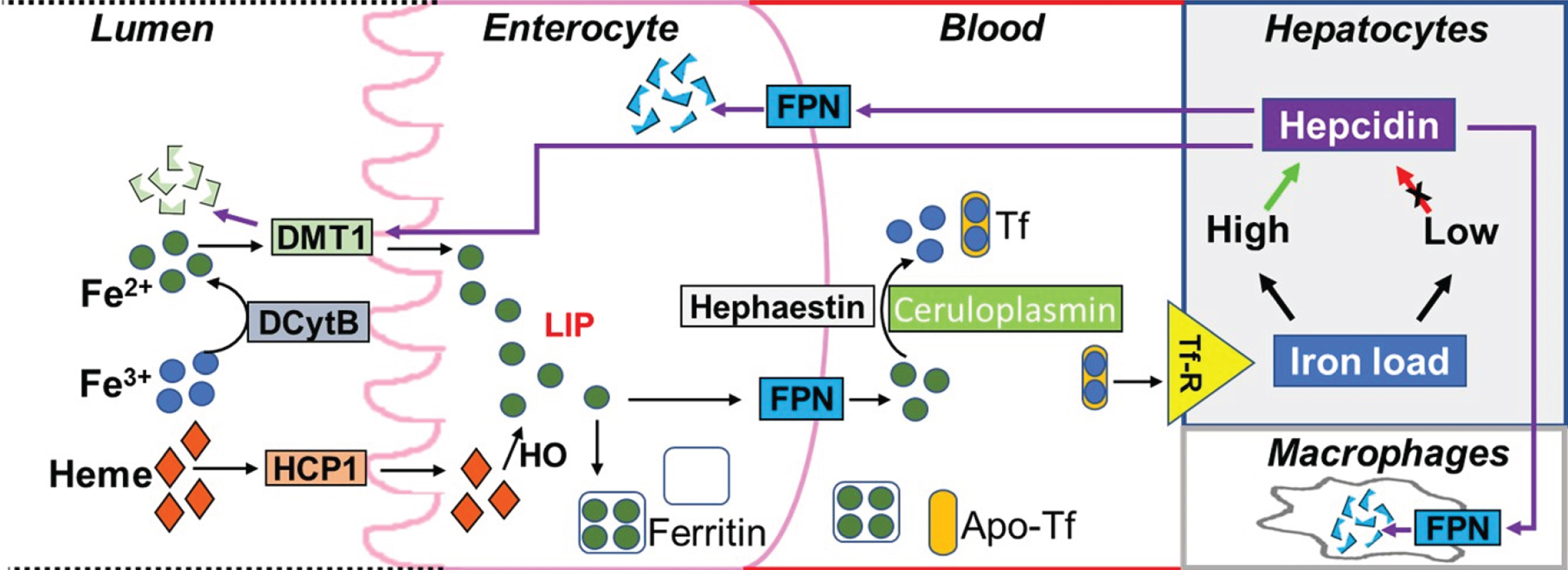
Hepcidin regulates blood iron levels by inhibiting the uptake of dietary iron from the intestine. It also has roles in preventing the release of iron from recycled red blood cells in macrophages as well as preventing the release into the circulation of stored iron from hepatocytes. The mechanisms by which hepcidin inhibits iron entry into circulation, from macrophages and other iron exporting cells, involves binding to the iron exporter protein, ferroportin, followed by internalization and degradation of the hepcidin-ferroportin complex [3, 25, 26]. Studies have also suggested that the primary mechanism by which hepcidin inhibits iron entry from enterocytes into circulation, involves decreasing iron absorption into the enterocytes by promoting degradation and internalization of the apical divalent metal transporter-1 (DMT1), the protein carrying Fe2+ from the intestinal lumen into the enterocytes prior to release into circulation by ferroportin [27–29] (Fig. 4).
Fig.4
Regulation of blood iron levels by hepcidin. Dietary iron in the lumen of duodenum is present in the form of heme and ferric ions (Fe3+). While heme is absorbed into enterocytes via the heme carrier protein 1 (HCP1), ferric ions are first converted to ferrous ions (Fe2+) by duodenal Cytochrome B (DCytB) to enable binding to the divalent metal transporter 1 (DMT1) thereby facilitating transport into enterocytes. Ferrous ions transported by DMT1 and the ferrous ions generated from heme (via heme oxygenase 1, HO) contribute to the labile iron pool (LIP). Ferrous ions in the LIP are then either stored in the iron storage protein, ferritin, or released into the blood via ferroportin (FPN) present on the basolateral membrane of the enterocyte with subsequent conversion to ferric ions via hephaestin (on the membrane) or ceruloplasmin (in the blood). These ferric ions can then be loaded into the iron carrier protein, transferrin (Tf), which bind to transferrin receptors (Tf-R) on tissue cell membranes resulting in iron transport to different tissues. When iron load in the tissue is high, hepcidin is released resulting in the internalization and degradation of the hepcidin-ferroportin complex (or hepcidin-DMT1 complex). This prevents further iron release by FPN into the blood or dietary iron absorption by enterocytes via DMT1, respectively. Hepcidin also prevents the release of iron from recycled red blood cells in macrophages. Alternatively, when iron load is low, the secretion of hepcidin is inhibited so that FPN can release iron into the blood.
We posit that the higher circulating hepcidin levels observed in individuals with high NAL could be attributed to the iron overload observed in AD pathogenesis [5, 30]. In response to iron loading in AD pathogenesis, we suggest that the liver secretes hepcidin into circulation and this circulating hepcidin may cross the blood-brain barrier to suppress ferroportin function. This may also explain the observation of lower brain hepcidin and ferroportin levels in AD observed by Raha and colleagues [8], as hepcidin binds to ferroportin (located on the cell membrane) following degradation of the hepcidin-ferroportin complex to downregulate ferroportin expression. Further, in support of our hypothesis, there is evidence suggesting that at least a proportion of the hepcidin in the brain may be originating outside the brain [31]. Additionally, our observations within the current study also fall in line with the “hepcidin-ferroportin neuronal iron overload” hypothesis suggested by Hofer and Perry [32].
Within the current study, a positive association between serum hepcidin and ferritin, and an inverse association between serum hepcidin and transferrin was observed in both low NAL and high NAL participants. However, significant associations of hepcidin with iron and ceruloplasmin (a ferroxidase that promotes iron loading into transferrin [33]), were only observed in individuals with low NAL. These associations in low NAL participants or healthy control individuals, as illustrated in Figure 4, could be attributed to the physiological role of hepcidin in maintaining serum iron levels, and the role of ceruloplasmin in promoting iron loading into transferrin [33], thereby influencing tissue iron load, and in turn, triggering optimal hepcidin secretion via this feedback mechanism to maintain iron homeostasis. The absence of a significant association of hepcidin with serum iron and ceruloplasmin in the high NAL group may therefore reflect a disruption in iron homeostasis.
Additionally, a significant association of hepcidin with the inflammation marker, CRP, was only observed in individuals at no apparent risk of AD or with low NAL. These observations are consistent with a study showing increasing serum hepcidin concentrations with increasing CRP in a general population [34]. While serum hepcidin levels were observed to be associated with inflammation in individuals with low NAL, this was not the case in individuals with high NAL (or at risk for AD), wherein no significant association was observed between serum hepcidin and the inflammation marker, CRP. Moreover, hepcidin levels remained significantly higher in high NAL after adjusting for CRP, indicating that the increased hepcidin levels in high NAL observed in the current study are potentially independent of systemic inflammation.
It is acknowledged that the current study has its limitations given its modest sample size and cross-sectional design. Longitudinal studies will provide more insight into the role of hepcidin across the disease pathogenesis trajectory. Further, within the current study, CRP was employed as a marker of inflammation, as this data was available to us. While interleukin-6 (IL-6) induces hepcidin through the induction of the signal transducer and activator of transcription 3 (STAT3) pathway [35], IL-6 has been reported to control CRP expression [36] encouraging the use of CRP as a suitable proxy for IL-6 within the present study. However, further studies using IL-6 are required to confirm whether hepcidin alterations in AD pathogenesis are independent of inflammation.
To conclude, serum hepcidin is higher in older adults with high NAL, prior to cognitive decline and hippocampal atrophy. The observation of increased hepcidin concentrations in individuals at risk for AD contributes to the body of evidence supporting iron dyshomeostasis as an early event of AD. Dysregulation of iron homeostasis point to the importance of investigating related biological pathways to better understand the molecular pathology of AD. Additionally, our findings indicate that hepcidin may add value to a panel of markers that contribute toward differentiating individuals at risk of AD. However, it is acknowledged that further validation studies are required. Our findings clearly highlight that a disruption in iron homeostasis in the blood is an early feature in the pathogenesis of AD.
ACKNOWLEDGMENTS
We thank the participants and their families for their participation and cooperation, and the Anglicare, KaRa Minds and Australian Alzheimer’s Research Foundation (AARF) research and support staff for their contributions to this study. We acknowledge the KaRa Institute Benefaction grant awarded to PC for this study. We specially thank Ms. Candice ManYan, Ms. Bethany Ball, Ms. Emma Toovey, Ms. Kate Fredericks, and Ms. Catherine Brown for their contributions to this study. We thank Dr. Abhay Singh for his help with using R. We also thank the staff of the Macquarie Medical Imaging centre in Macquarie University Hospital, Sydney, for their contributions. Florbetaben is a proprietary PET radiopharmaceutical owned by Piramal Imaging. For this study, Florbetaben was manufactured and supplied under GMP conditions by Cyclotek (Australia) Pty Ltd. HZ is a Wallenberg Scholar supported by grants from the Swedish Research Council (#2018-02532), the European Research Council (#681712), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-720931), the Alzheimer Drug Discovery Foundation (ADDF), USA (#201809-2016862), and the UK Dementia Research Institute at UCL. KB is supported by the Swedish Research Council (#2017-00915), the Alzheimer Drug Discovery Foundation (ADDF), USA (#RDAPB-201809-2016615), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement (#ALFGBG-715986), and European Union Joint Program for Neurodegenerative Disorders (JPND2019-466-236).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0162r2).
REFERENCES
[1] | Zecca L , Youdim MB , Riederer P , Connor JR , Crichton RR ((2004) ) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5: , 863–873. |
[2] | Benarroch EE ((2009) ) Brain iron homeostasis and neurodegenerative disease. Neurology 72: , 1436–1440. |
[3] | Belaidi AA , Bush AI ((2016) ) Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J Neurochem 139 Suppl 1: , 179–197. |
[4] | Giambattistelli F , Bucossi S , Salustri C , Panetta V , Mariani S , Siotto M , Ventriglia M , Vernieri F , Dell’acqua ML , Cassetta E , Rossini PM , Squitti R ((2012) ) Effects of hemochromatosis and transferrin gene mutations on iron dyshomeostasis, liver dysfunction and on the risk of Alzheimer’s disease. Neurobiol Aging 33: , 1633–1641. |
[5] | Goozee K , Chatterjee P , James I , Shen K , Sohrabi HR , Asih PR , Dave P , ManYan C , Taddei K , Ayton SJ , Garg ML , Kwok JB , Bush AI , Chung R , Magnussen JS , Martins RN ((2018) ) Elevated plasma ferritin in elderly individuals with high neocortical amyloid-beta load. Mol Psychiatry 23: , 1807–1812. |
[6] | Dubois B , Hampel H , Feldman HH , Scheltens P , Aisen P , Andrieu S , Bakardjian H , Benali H , Bertram L , Blennow K , Broich K , Cavedo E , Crutch S , Dartigues JF , Duyckaerts C , Epelbaum S , Frisoni GB , Gauthier S , Genthon R , Gouw AA , Habert MO , Holtzman DM , Kivipelto M , Lista S , Molinuevo JL , O’Bryant SE , Rabinovici GD , Rowe C , Salloway S , Schneider LS , Sperling R , Teichmann M , Carrillo MC , Cummings J , Jack CR Jr. , Proceedings of the Meeting of the International Working G, the American Alzheimer’s Association on “The Preclinical State of AD, July, Washington, DC, USA ((2016) ) Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement 12: , 292–323. |
[7] | Dubois B , Feldman HH , Jacova C , Hampel H , Molinuevo JL , Blennow K , DeKosky ST , Gauthier S , Selkoe D , Bateman R , Cappa S , Crutch S , Engelborghs S , Frisoni GB , Fox NC , Galasko D , Habert MO , Jicha GA , Nordberg A , Pasquier F , Rabinovici G , Robert P , Rowe C , Salloway S , Sarazin M , Epelbaum S , de Souza LC , Vellas B , Visser PJ , Schneider L , Stern Y , Scheltens P , Cummings JL ((2014) ) Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol 13: , 614–629. |
[8] | Raha AA , Vaishnav RA , Friedland RP , Bomford A , Raha-Chowdhury R ((2013) ) The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol Commun 1: , 55. |
[9] | Sternberg Z , Hu Z , Sternberg D , Waseh S , Quinn JF , Wild K , Jeffrey K , Zhao L , Garrick M ((2017) ) Serum hepcidin levels, iron dyshomeostasis and cognitive loss in Alzheimer’s disease. Aging Dis 8: , 215–227. |
[10] | Kweon OJ , Youn YC , Lim YK , Lee MK , Kim HR ((2019) ) Clinical utility of serum hepcidin and iron profile measurements in Alzheimer’s disease. J Neurol Sci 403: , 85–91. |
[11] | Victor M , Savina H , Julia P , Vasil V , Maria P , Latchezar T , Kamen T , Borislav M , Radoslava E , Ivo B ((2017) ) The role of iron homeostasis in Alzheimer’s disease. J Alzheimers Neurodegener Dis 3: , 011. |
[12] | Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , Szoeke C , Macaulay SL , Martins R , Maruff P , Ames D , Rowe CC , Masters CL , Australian Imaging Biomarkers and Lifestyle (AIBL) Research Group ((2013) ) Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol 12: , 357–367. |
[13] | Rossetti HC , Lacritz LH , Cullum CM , Weiner MF ((2011) ) Normative data for the Montreal Cognitive Assessment (MoCA) in a population-based sample. Neurology 77: , 1272–1275. |
[14] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr. , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[15] | Zhou L , Salvado O , Dore V , Bourgeat P , Raniga P , Macaulay SL , Ames D , Masters CL , Ellis KA , Villemagne VL , Rowe CC , Fripp J , AIBL Research Group ((2014) ) MR-less surface-based amyloid assessment based on 11C PiB PET. PLoS One 9: , e84777. |
[16] | Bourgeat P , Villemagne VL , Dore V , Brown B , Macaulay SL , Martins R , Masters CL , Ames D , Ellis K , Rowe CC , Salvado O , Fripp J , AIBL Research Group ((2015) ) Comparison of MR-less PiB SUVR quantification methods.S. Neurobiol Aging 36 Suppl 1: , 159–166. |
[17] | Burnham SC , Faux NG , Wilson W , Laws SM , Ames D , Bedo J , Bush AI , Doecke JD , Ellis KA , Head R , Jones G , Kiiveri H , Martins RN , Rembach A , Rowe CC , Salvado O , Macaulay SL , Masters CL , Villemagne VL , Alzheimer’s Disease Neuroimaging Initiative, Australian Imaging, Biomarkers and Lifestyle Study Research Group ((2014) ) A blood-based predictor for neocortical Abeta burden in Alzheimer’s disease: Results from the AIBL study. Mol Psychiatry 19: , 519–526. |
[18] | Goozee K , Chatterjee P , James I , Shen K , Sohrabi HR , Asih PR , Dave P , Ball B , ManYan C , Taddei K , Chung R , Garg ML , Martins RN ((2017) ) Alterations in erythrocyte fatty acid composition in preclinical Alzheimer’s disease. Sci Rep 7: , 676. |
[19] | Ellis KA , Bush AI , Darby D , De Fazio D , Foster J , Hudson P , Lautenschlager NT , Lenzo N , Martins RN , Maruff P , Masters C , Milner A , Pike K , Rowe C , Savage G , Szoeke C , Taddei K , Villemagne V , Woodward M , Ames D , AIBL Research Group ((2009) ) The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: Methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease. Int Psychogeriatr 21: , 672–687. |
[20] | Chatterjee P , Elmi M , Goozee K , Shah T , Sohrabi HR , Dias CB , Pedrini S , Shen K , Asih PR , Dave P , Taddei K , Vanderstichele H , Zetterberg H , Blennow K , Martins RN ((2019) ) Ultrasensitive detection of plasma amyloid-beta as a biomarker for cognitively normal elderly individuals at risk of Alzheimer’s disease. J Alzheimers Dis 71: , 775–783. |
[21] | Ireland P , Jolley D , Giles G , O’Dea K , Powles J , Rutishauser I , Wahlqvist ML , Williams J ((1994) ) Development of the Melbourne FFQ: A food frequency questionnaire for use in an Australian prospective study involving an ethnically diverse cohort. Asia Pac J Clin Nutr 3: , 19–31. |
[22] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[23] | Polvikoski T , Sulkava R , Haltia M , Kainulainen K , Vuorio A , Verkkoniemi A , Niinisto L , Halonen P , Kontula K ((1995) ) Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N Engl J Med 333: , 1242–1247. |
[24] | Nakamura A , Kaneko N , Villemagne VL , Kato T , Doecke J , Dore V , Fowler C , Li QX , Martins R , Rowe C , Tomita T , Matsuzaki K , Ishii K , Ishii K , Arahata Y , Iwamoto S , Ito K , Tanaka K , Masters CL , Yanagisawa K ((2018) ) High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 554: , 249–254. |
[25] | Nemeth E , Tuttle MS , Powelson J , Vaughn MB , Donovan A , Ward DM , Ganz T , Kaplan J ((2004) ) Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 306: , 2090–2093. |
[26] | Delaby C , Pilard N , Goncalves AS , Beaumont C , Canonne-Hergaux F ((2005) ) Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 106: , 3979–3984. |
[27] | Mena NP , Esparza A , Tapia V , Valdes P , Nunez MT ((2008) ) Hepcidin inhibits apical iron uptake in intestinal cells. Am J Physiol Gastrointest Liver Physiol 294: , G192–198. |
[28] | Chaston T , Chung B , Mascarenhas M , Marks J , Patel B , Srai SK , Sharp P ((2008) ) Evidence for differential effects of hepcidin in macrophages and intestinal epithelial cells. Gut 57: , 374–382. |
[29] | Brasse-Lagnel C , Karim Z , Letteron P , Bekri S , Bado A , Beaumont C ((2011) ) Intestinal DMT1 cotransporter is down-regulated by hepcidin via proteasome internalization and degradation. Gastroenterology 140: , 1261–1271 e1261. |
[30] | Smith MA , Zhu X , Tabaton M , Liu G , McKeel DW Jr. , Cohen ML , Wang X , Siedlak SL , Dwyer BE , Hayashi T , Nakamura M , Nunomura A , Perry G ((2010) ) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 19: , 363–372. |
[31] | Raha-Chowdhury R , Raha AA , Forostyak S , Zhao J-W , Stott SRW , Bomford A ((2015) ) Expression and cellular localization of hepcidin mRNA and protein in normal rat brain. BMC Neurosci 16: , 24. |
[32] | Hofer T , Perry G ((2016) ) Nucleic acid oxidative damage in Alzheimer’s disease-explained by the hepcidin-ferroportin neuronal iron overload hypothesis? J Trace Elem Med Biol 38: , 1–9. |
[33] | Osaki S , Johnson DA , Frieden E ((1966) ) The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J Biol Chem 241: , 2746–2751. |
[34] | Galesloot TE , Vermeulen SH , Geurts-Moespot AJ , Klaver SM , Kroot JJ , van Tienoven D , Wetzels JFM , Kiemeney LALM , Sweep FC , den Heijer M , Swinkels DW ((2011) ) Serum hepcidin: Reference ranges and biochemical correlates in the generalopulation. Blood 117: , e218–e225. |
[35] | Pietrangelo A , Dierssen U , Valli L , Garuti C , Rump A , Corradini E , Ernst M , Klein C , Trautwein C ((2007) ) STAT3 is required for IL-6-gp130–dependent activation of hepcidin in vivo. Gastroenterology 132: , 294–300. |
[36] | Pepys MB , Hirschfield GM ((2003) ) C-reactive protein: A critical update. J Clin Invest 111: , 1805–1812. |


