
Safety, Tolerability, and Pharmacokinetics of Crenezumab in Patients with Mild-to-Moderate Alzheimer’s Disease Treated with Escalating Doses for up to 133 Weeks
Abstract
Background:
Crenezumab is a fully humanized, monoclonal anti-amyloid-β immunoglobulin G4 antibody.
Objective:
This Phase Ib study (NCT02353598) evaluated the safety, tolerability, and pharmacokinetics of crenezumabat doses of ≤120 mg/kg administered intravenously every 4 weeks (q4w). Immunogenicity and exploratory biomarkers were also evaluated.
Methods:
In this multicenter, double-blind study, participants (aged 50–90 years) with mild-to-moderate Alzheimer’s disease (AD) and amyloid-positive positron emission tomography (PET) scan were randomized to receive crenezumab 30 or 45 mg/kg (Cohort 1, n = 21), 60 mg/kg (Cohort 2, n = 21), or 120 mg/kg (Cohort 3, n = 19) or corresponding placebo (n = 14) intravenously q4w for 13 weeks. Seventy-one participants were subsequently enrolled in an optional open-label extension (OLE) and received crenezumab at the originally assigned dose level, except for Cohort 3 (crenezumab 60 mg/kg during OLE). Participants received regular brain MRIs to assess amyloid-related imaging abnormalities (ARIA). Results up to Week 133 are reported.
Results:
Approximately 94% of participants experienced ≥1 adverse event (AE). Most AEs were mild or moderate; 15.5% experienced a Grade ≥3 AE. No ARIA-edema/effusion (ARIA-E) events were observed. New ARIA-micro hemorrhages and hemosiderosis (ARIA-H) were reported in 4.9% (double-blind treatment period) and 9.9% (combined double-blind treatment and OLE periods) of participants. Steady-state trough concentrations of crenezumab were dose-proportional and maintained for each dose level.
Conclusion:
Crenezumab doses of ≤120 mg/kg intravenously q4w were well tolerated. The observed safety profile for ≤133 weeks of treatment in a mild-to-moderate AD population was similar to that seen in previous trials.
INTRODUCTION
Approximately 47 million people globally are living with dementia [1], of whom 60–80% have Alzheimer’s disease (AD) [2]. Neuropathologically, AD is characterized by amyloid plaques and tau tangles in the brain [3], and evidence suggests that accumulation of disease-defining pathological proteins precedes clinical symptoms by many years [3, 4]. In AD, amyloid burden results from the accumulation of insoluble amyloid-β (Aβ) fibrils and plaques as well as soluble Aβ peptide monomers and oligomers [3, 5]. In vitro and in vivo data have demonstrated that soluble Aβ oligomers are neurotoxic [6–9]; therefore, reducing Aβ oligomers in the brain may alleviate cognitive decline and reduce neurodegeneration [10, 11]. Although currently approved therapies manage some symptoms of AD, their benefit is modest, and firm evidence of modification of disease progression is lacking [12].
Crenezumab (RO5490245) is a fully humanized, anti-Aβ monoclonal immunoglobulin G4 (IgG4) antibody that binds to monomeric and aggregated forms of Aβ and has a higher preferential binding affinity for oligomeric Aβ species [13–15]. In vitro, crenezumab prevents Aβ aggregation, promotes disaggregation, and provides a neuroprotective effect by blocking the interaction between Aβ oligomers and neurons [13, 14]. Following in vivo dosing in AD transgenic mice, crenezumab localized to brain areas with putative high concentrations of Aβ oligomers (i.e., hippocampal mossy fiber tract and the periphery of amyloid plaques) but not to the dense core of plaques or vascular amyloid [16]. Additionally, the low effector function of the IgG4 backbone and lack of crenezumab binding to vascular amyloid are hypothesized to minimize inflammation in brain vasculature and result in a reduced risk of amyloid-related imaging abnormalities (ARIA) and localized microvascular damage [13]; this may allow for high doses of crenezumab to be administered without compromising safety.
The completed Phase II ABBY (NCT01343966) [17] and BLAZE (NCT01397578) [18] studies evaluated the safety and efficacy of crenezumab. Crenezumab was administered at two doses (300 mg subcutaneously [SC] every 2 weeks and 15 mg/kg intravenously [IV] every 4 weeks [q4w]) for 68 weeks in individuals with mild-to-moderate AD, with the option to enroll in an open-label extension (OLE) phase (NCT01723826). Although the ABBY and BLAZE trials did not meet their primary endpoints, post hoc exploratory analyses of ABBY suggested a trend favoring reduced cognitive decline in progressively milder subgroups in the crenezumab-treated patients who received the higher dose of the two doses tested (i.e., crenezumab 15 mg/kg IV q4w). Data from the BLAZE study suggested a trend toward reduced amyloid accumulation as measured by amyloid positron emission tomography (PET) in the group receiving the higher dose [18].
Results presented here are from a multicenter, randomized, Phase Ib study (GN29632 [NCT02353598]) that consisted of a double-blind, placebo-controlled, dose-escalation treatment period followed by an OLE period. This study was designed to investigate the safety, tolerability, and pharmacokinetics (PK) of crenezumab delivered at higher doses than those used in previous Phase II studies [17, 18], and the results reported here not only provide a better understanding of the safety associated with longer use (up to Week 133) of doses of crenezumab that are higher than used in Phase II, but also include safety and PK data of a dose higher than the one tested in Phase III (i.e., 120 mg/kg IV q4w). Based on interim safety data from this Phase Ib study, a fourfold higher dose (60 mg/kg IV q4w) of crenezumab than the high dose used in Phase II was selected for the Phase III CREAD (NCT02670083) and CREAD2 (NCT03114657) trials that investigated the efficacy and safety of crenezumab compared with placebo in patients with early (prodromal-to-mild) AD [19, 20]. The CREAD and CREAD2 trials were discontinued following a preplanned interim analysis of CREAD which demonstrated that the study was unlikely to meet the primary endpoint. No adverse safety signals for crenezumab were observed in this interim analysis, and the overall safety profile of crenezumab was similar to that seen in previous trials. Based on the results from this analysis, the crenezumab clinical development program in sporadic AD was terminated. In addition to the CREAD Phase III trials, the CREAD OLE study (BN40031 [NCT03491150]) and the Phase Ib study reported here were stopped early. Currently, the efficacy and safety of crenezumab continue to be studied in participants at risk for autosomal dominant AD in the Phase II Alzheimer’s Prevention Initiative trial (NCT01998841). Trial participants are clinically asymptomatic at study entry and carry the presenilin 1 E280A autosomal dominant mutation [21].
Here we report the safety, PK, pharmacodynamics (PD), immunogenicity, and imaging biomarker data from this Phase Ib study in patients treated with crenezumab for up to 133 weeks. Safety data are presented in two parts: the ascending-dose, 13-week, double-blind, and placebo-controlled treatment period; and, for participants who entered the OLE, the combined 13-week double-blind treatment and OLE periods for up to 133 weeks.
MATERIALS AND METHODS
Objectives
The primary objective of this Phase Ib study was to evaluate the safety and tolerability of multiple doses of crenezumab in individuals with mild-to-moderate AD. A secondary objective was to further characterize the PK characteristics of crenezumab in the serum of individuals with mild-to-moderate AD. Exploratory objectives evaluated the effect of crenezumab on imaging biomarkers from magnetic resonance imaging (MRI) and PET scans and on plasma Aβ concentrations. Exploratory clinical efficacy assessments were also evaluated in this study; however, formal analyses of clinical efficacy measures were not performed due to the small sample size.
Study design
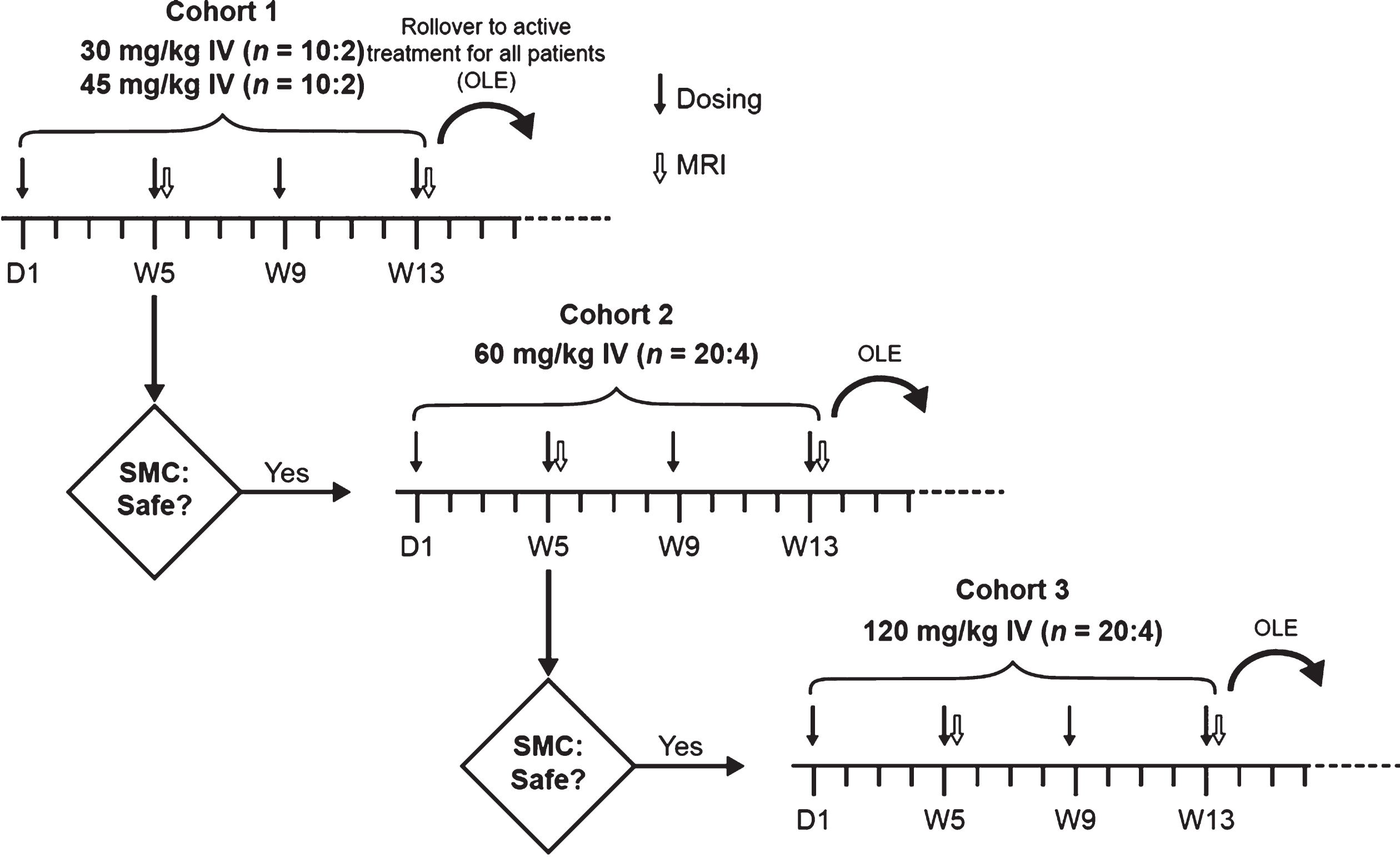
This was a 13-week, multicenter, randomized, double-blind, parallel-group, multiple ascending-dose study with three cohorts, followed by an OLE period. Participants with mild-to-moderate AD from 11 trial sites in the United States were randomized 5:1 (active drug to placebo) to receive infusion of crenezumab or placebo IV q4w as follows: Cohort 1 received 30 or 45 mg/kg of crenezumab or placebo (5:1:5:1); Cohort 2 received 60 mg/kg of crenezumab or placebo; and Cohort 3 received 120 mg/kg of crenezumab or placebo. Infusions were administered on Day 1 and during Weeks 5, 9, and 13 (Fig. 1). After completing the placebo-controlled phase at Week 13, participants could enter the OLE phase of the study, during which they received crenezumab at the originally assigned dose level for their cohort, except for participants in Cohort 3, who, per the study design, switched from crenezumab 120 to 60 mg/kg IV q4w. Following a protocol amendment, participants in Cohort 1 had the option to increase to 60 mg/kg q4w at or after Week 133. However, to allow for comparison between the dose levels, only data up to Week 133 are reported.
Fig. 1
Study design. Participants were enrolled sequentially. The next ascending dose cohort (60 or 120 mg/kg) was initiated if all previous dosing cohorts were supported by adequate safety and tolerability and provided that no previous dosing cohort was closed to enrollment due to safety concerns by the time the last participant enrolled in the study had completed the Week 5 MRI scan and the second study drug administration. Participation in the OLE phase was optional. The OLE dose was equivalent to the dose assigned during the double-blind randomization phase for all participants, except for those in the 120 mg/kg cohort who received 60 mg/kg IV every 4 weeks upon starting the OLE period. Additionally, at or after Week 133, participants in the 30 or 45 mg/kg dosing cohort were given the option to increase the dose of active drug to 60 mg/kg IV every 4 weeks. D, Day; IV, intravenously; MRI, magnetic resonance imaging; OLE, open-label extension; SMC, safety monitoring committee; W, week.
This study was conducted in accordance with the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use E6 Guideline for Good Clinical Practice and the principles of the Declaration of Helsinki. A central investigational review board and individual site institutional review boards reviewed and approved the protocols as well as informed consent forms. All participants provided informed consent. The first participant was enrolled on February 26, 2015, and the last participant’s last visit occurred on March 25, 2019. The end of this study was identified as the date when the crenezumab clinical development program in sporadic AD was terminated (January 30, 2019) based on results from the preplanned interim analysis of the Phase III CREAD study described earlier.
Participants
Eligible patients were 50–90 years of age with a clinical diagnosis of probable mild-to-moderate AD based on the National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorders Association criteria, or probable major neurocognitive disorder due to AD of mild-to-moderate severity based on the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria. Additionally, all participants were required to have a Clinical Dementia Rating global score of 0.5 or 1, a Mini-Mental State Examination score of 18–28 inclusive at screening, a Geriatric Depression Scale-15 score of <6, adequate visual and auditory acuity sufficient for performing neuropsychological testing, and an [18F]-florbetapir PET scan positive for cerebral amyloid by a qualitative read conducted by a central PET laboratory. All participants were required to have availability of a study partner/caregiver who had cognitive capacity to provide accurate information regarding the participant’s cognitive and functional abilities and ≥10 h per week of contact with the participant.
Individuals were eligible for study participation regardless of whether they were receiving approved standard-of-care drug treatments for AD, provided that the dose of the approved treatment had been stable for ≥3 months prior to screening. Participants were excluded if they had received prior administration of crenezumab or any other therapeutic agent targeting Aβ, any investigational active or passive immunotherapies, or other long-acting biologic agent evaluated to prevent or postpone cognitive decline within 1 year of screening.
Assessments
Safety
Safety was monitored throughout the trial by characterization of adverse events (AEs), electrocardiogram parameters, MRI brain scans, chemistry and hematology laboratory values, vital signs, and physical and neurological examinations. The incidence, nature, and severity of AEs and serious AEs (SAEs) were reported. AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0 (NCI CTCAE v4.0). AEs that occurred during or within 24 h after study drug administration and were determined by the investigator to be related to study drug infusion were designated as infusion-related reactions. AEs of special interest were also monitored and included cases of potential drug-induced liver injury with elevated alanine aminotransferase or aspartate aminotransferase combined with either elevated bilirubin or clinical jaundice, suspected transmission of an infectious agent by the study drug, and pneumonia based on a potential signal observed in the Phase II studies [17, 18]. The incidence of suicidal ideation, suicidal behavior, and self-injurious behavior without suicidal intent using the Columbia-Suicide Severity Rating Scale was also assessed. Safety data per dose group are presented up to Week 133.
Participants underwent MRI scans at screening and following study drug administration at Weeks 5 and 13 of the double-blind treatment period and every 12 weeks up to Week 61 and every 24 weeks up to Week 133 during the OLE period. Monitoring by brain MRI included the following sequences: high-resolution T1-weighted, T2*-weighted, and T2-weighted fluid-attenuated inversion recovery. Participants who did not enroll in the OLE period received one post-treatment safety assessment follow-up visit, including an MRI scan, 8 weeks after their final dose (Week 21). Scans were centrally read.
Dose-limiting criteria
During the double-blind, placebo-controlled treatment period, additional doses of the study drug were not to be administered if a participant experienced a dose-limiting toxicity assessed by the investigator as related to the study drug. This included NCI CTCAE v4.0 Grade ≥3 AEs; NCI CTCAE v4.0 clinically significant Grade ≥2 neurology AEs, such as ataxia, central nervous system cerebrovascular ischemia, encephalopathy, pyramidal tract dysfunction, neuropathy cranial/sensory/motor, or seizure; or any severe or serious infusion-related reaction occurring within 24 h after study drug administration that did not promptly resolve with a reduced infusion rate and/or supportive care.
Study drug was to be discontinued during the double-blind, placebo-controlled treatment period or OLE phase if participants presented with >10 microhemorrhages and/or hemosiderosis (ARIA-H) or were diagnosed with three recurrent, symptomatic ARIA-edema/effusion (ARIA-E) events. In addition, the study drug dose was reduced if participants experienced >8 ARIA-hemosiderin deposits cumulatively. If participants presented with asymptomatic ARIA-E with a Barkhof grand total score of ≥4 [22] or symptomatic ARIA-E (any size), the study drug was to be interrupted and monthly MRI monitoring conducted until abnormalities resolved. Upon resolution, the study drug was reintroduced and the participant received an MRI scan approximately 4 weeks later.
Pharmacokinetics and pharmacodynamics
Blood samples were collected from all participants through Week 133 to determine crenezumab concentration and plasma Aβ1-40 and Aβ1-42. Serum crenezumab concentrations were analyzed using a validated enzyme-linked immunosorbent assay with a lower limit of quantification of 0.05 μg/mL. Total monomeric plasma Aβ1-40 and Aβ1-42 levels corresponding to free and crenezumab-bound Aβ levels were measured using a robust Elecsys® drug-tolerant, noncommercial prototype assay on the cobas e 411 analyzer (Roche Diagnostics International Ltd, Rotkreuz, Switzerland), with a lower limit of detection of <2 pg/mL (for both assays).
Immunogenicity sampling
Blood samples were collected from all participants through Week 133 to evaluate antidrug antibodies (ADAs). ADAs were determined using a validated in-solution bridging immunoassay able to detect 500 ng/mL of an ADA surrogate positive control in the presence of 75 μg/mL of crenezumab.
Imaging biomarkers
Measurements were taken of whole brain, ventricular, and hippocampal volumes from the T1-weighted MRI scans at screening and Weeks 25, 49, 61, 85, 109, and 133. Measurements of fibrillar amyloid burden were taken from PET imaging using [18F]-florbetapir at screening, Week 53 and 105 by calculating the standard uptake value ratio (SUVR) using a composite cortical region (anterior/posterior cingulate and frontal, temporal, and parietal lobes) and a subcortical white matter reference region, which is preferred for longitudinal analysis [18].
Statistical analyses
Target enrollment for this study was 72 participants in total. Safety outcomes were assessed in all randomized participants who received at least one dose of the study drug, and participants were grouped according to the treatment actually received. AEs, changes in laboratory test results, and changes in vital signs were summarized.
PK and plasma PD analyses included samples during the double-blind portion of the study up to Week 13 from all randomized participants who received at least one dose of the study drug (placebo or crenezumab) and had at least one evaluable postdose PK sample and plasma PD sample, respectively. To further characterize the PK and PD characteristics of crenezumab in individuals with mild-to-moderate AD, serum PK and plasma PD data collected during the double-blind treatment period in the Phase Ib study and during the Phase II ABBY study [17] were pooled to assess the dose proportionality over a wider dose range.
The prevalence of ADAs was determined for the baseline visit by calculating the proportion of participants with confirmed positive ADA levels at baseline relative to the total number of participants with available baseline samples. The incidence of treatment-emergent ADAs was determined for the proportion of participants with confirmed post-baseline positive ADAs relative to the total number of participants receiving at least one dose of crenezumab and who had at least one post-baseline sample available for the ADA analysis. For the immunogenicity assessments, the number and percentage of participants with confirmed positive anti-crenezumab antibody levels in each treatment arm were summarized.
The MRI and PET biomarker analyses across the double-blind and OLE phases of the study were exploratory; this study was not powered to assess differences in imaging measurements. Since all change-from-baseline values were calculated for imaging visits that occurred after the double-blind dosing was complete (starting at Week 25 for MRI and Week 53 for PET), participants were grouped into three cohorts based on the dose received during the OLE phase: 30, 45, and 60 mg/kg for the imaging biomarker analyses.
The safety data presented herein are for the 13-week, double-blind, treatment phase and, for participants who entered the OLE, the combined 13-week, double-blind treatment and OLE periods for up to Week 133.
All statistical analyses presented are descriptive.
RESULTS
Participant population
Between February 26, 2015, and August 31, 2016, 77 participants were enrolled in the double-blind, placebo-controlled phase of this study; 75 received at least one dose of study treatment (crenezumab 30 or 45 mg/kg or placebo [Cohort 1], n = 26; crenezumab 60 mg/kg or placebo [Cohort 2], n = 26; and crenezumab 120 mg/kg or placebo [Cohort 3], n = 23). Of those, 61 participants received crenezumab according to the assigned cohort dosing level and 14 received placebo (Supplementary Figure 1). Baseline characteristics and treatment exposure for all participants receiving at least one dose of study treatment (N = 75) are presented in Table 1. Overall, 75% of dosed participants (56/75) were carriers of at least one E4 allele of apolipoprotein E. There were no notable imbalances in carrier frequency or baseline characteristics between cohorts or treatment groups (Table 1).
Table 1
Baseline characteristics and treatment exposure in all participants enrolled and receiving at least one dose of treatment (N = 75) in the study
| Placebo | Crenezumab | ||||
| (n = 14) | 30 mg/kg | 45 mg/kg | 60 mg/kg | 120 ⟶ 60 mg/kg | |
| (n = 10) | (n = 11) | (n = 21) | (n = 19) | ||
| Mean age, years (range) | 71.4 (57–84) | 73.4 (54–82) | 73.3 (57–82) | 72.9 (51–87) | 69.4 (54–88) |
| Male, n (%) | 6 (42.9) | 6 (60.0) | 4 (36.4) | 13 (61.9) | 10 (52.6) |
| ApoE status, n (%) | |||||
| E2/E3 | 1 (7.1) | 0 | 0 | 0 | 1 (5.3) |
| E2/E4 | 0 | 0 | 0 | 0 | 1 (5.3) |
| E3/E3 | 3 (21.4) | 4 (40.0) | 1 (9.1) | 4 (19.0) | 5 (26.3) |
| E3/E4 | 8 (57.1) | 6 (60.0) | 7 (63.6) | 14 (66.7) | 7 (36.8) |
| E4/E4 | 2 (14.3) | 0 | 3 (27.3) | 3 (14.3) | 5 (26.3) |
| MMSE score, mean (SD), range | 20.9 (2.8), 18–25 | 23.3 (3.8), 18–28 | 22.2 (2.7), 18–26 | 23.2 (3.3), 18–29 | 22.9 (3.1), 18–28 |
| Median duration of crenezumab exposure, | 132.43 (12.1–196.6) | ||||
| weeks (range)a | |||||
aMedian duration of exposure is reported for participants who enrolled in the open-label extension period (crenezumab 30/45 mg/kg, n = 23; crenezumab 60 mg/kg, n = 48). ApoE, apolipoprotein E; MMSE, Mini-Mental State Examination.
A total of 71 participants entered the OLE phase of the study (OLE crenezumab 30 or 45 mg/kg, n = 23; OLE crenezumab 60 mg/kg, n = 48; Supplementary Figure 1), most (n = 58 [81.7%]) were older than 65 years of age (medianage [range], 73 years [51–88 years]), male (52.1%), white (100%), and not Hispanic or Latino (94.4%). Approximately 74.6% of the OLE population (n = 53/71) were apolipoprotein E4 carriers. All patients enrolled in the OLE phase discontinued the study early, the majority of them due to premature study termination (see Supplementary Figure 1 for details).
Safety overview
Ascending-dose, 13-week, double-blind, placebo-controlled treatment period
Safety results for the randomized, 13-week, double-blind, placebo-controlled treatment phase of the study are presented in Table 2. Overall, most AEs were low grade and no protocol-defined dose-limiting toxicities or deaths occurred. The most common AEs were headache (9.8%), fatigue (4.9%), cerebral micro hemorrhage (4.9%), and dizziness (4.9%). All four SAEs that occurred were assessed by the investigators as not related to the study drug. In the two participants who experienced an AE during the 13-week double-blind, placebo-controlled treatment period and were discontinued from the study drug due to AEs (discontinuation occurred after completion of the double-blind, placebo-controlled treatment period during the safety follow-up [n = 1] or during the OLE phase prior to receiving any doses during the OLE [n = 1]), only one AE (Cohort 2, crenezumab 60 mg/kg: Grade 2 confusional state that started on Day 59) was assessed as related to the study treatment (Table 2). No clear dose relationship was observed with respect to the nature, severity, and frequency of AEs.
Table 2
Overview of safety during the 13-week, double-blind, randomized treatment phase
| Adverse events, n (%) | Crenezumab | ||||
| Placebo | 30 mg/kg | 45 mg/kg | 60 mg/kg | 120 mg/kg | |
| (n = 14) | (n = 10) | (n = 11) | (n = 21) | (n = 19) | |
| Participants with≥1 AE | 6 (42.9) | 8 (80.0) | 7 (63.6) | 14 (66.7) | 5 (26.3) |
| AE related to study drug per investigatora | 1 (7.1) | 1 (10.0) | 4 (36.4) | 3 (14.3) | 1 (5.3) |
| AE Grade≥3 (severe, life-threatening, or resulting in death)b | 0 | 1 (10.0) | 0 | 0 | 0 |
| Serious AEc | 0 | 1 (10.0) | 0 | 2 (9.5) | 0 |
| Treatment withdrawal due to AEd | 0 | 1 (10.0) | 0 | 1 (4.8) | 0 |
aPlacebo: Grade 1 ventricular extra systoles; 30 mg/kg: Grade 1 dysgeusia and Grade 1 oral disorder (n = 1); 45 mg/kg: Grade 1 headache (n = 1), Grade 1 cerebral microhemorrhage (n = 2), Grade 1 dizziness and Grade 1 headache (n = 1); 60 mg/kg: Grade 1 headache (n = 1), Grade 1 fatigue and Grade 1 hallucination (n = 1), Grade 2 agitation, Grade 2 confusional state, Grade 2 diarrhea, and Grade 2 fatigue (n = 1); 120 mg/kg: Grade 1 vision blurred and Grade 1 visual field defect (n = 1). bPer National Cancer Institute Common Terminology Criteria for Adverse Events version 4.0. 30 mg/kg: Grade 3 malignant melanoma. c30 mg/kg: Grade 3 malignant melanoma; 60 mg/kg: Grade 1 accidental overdose and Grade 2 pneumonia (n = 1); Grade 1 atypical chest pain (n = 1); all events assessed by investigator as unrelated to study drug. d30 mg/kg: Grade 3 malignant melanoma assessed by investigator as unrelated to study drug (participant completed all four treatments in the double-blind period and discontinued the study during the safety follow-up); 60 mg/kg: Grade 2 confusional state assessed as related to study drug by investigator. AE, adverse event.
No ARIA-E events or macro hemorrhages were observed. New ARIA-H events occurred in 4.9% of participants treated with crenezumab (Cohort 1, crenezumab 45 mg/kg, n = 2; Cohort 2, crenezumab 60 mg/kg, n = 1). These events were asymptomatic and did not lead to study drug modifications. No new ARIA-H events were reported in participants in Cohort 3 (120 mg/kg) or in participants receiving placebo.
Combined 13-week double-blind treatment and OLE periods for up to 133 weeks
Safety results from both the double-blind treatment and OLE periods for participants who enrolled in the OLE phase of the study up to Week 133 are presented in Table 3. Overall, the majority of AEs were classified as mild (Grade 1, 31.0%) or moderate (Grade 2, 47.9%); no deaths occurred. The most commonly reported AEs in ≥10% of participants treated with crenezumab were fall (22.5%), upper respiratory tract infection (18.3%), headache (16.9%), anxiety (15.5%), urinary tract infection (12.7%), and depression (11.3%). None of the SAEs (n = 11) reported were considered related to the study drug. AEs that led to study drug discontinuation occurred in five participants; three AEs were considered related to the study drug (OLE crenezumab 45 mg/kg: Grade 1 cerebral hemosiderin deposition and Grade 1 cerebral microhemorrhage on Day 758 during the OLE period, n = 1; OLE crenezumab 60 mg/kg: Grade 2confusional state that started on Day 59 of the double-blind treatment phase, n = 1).
Table 3
Overview of safety during the randomized treatment period and up to Week 133 in the OLE phase in participants enrolled in the OLE phase (N = 71)
| Adverse events, n (%) | OLE crenezumab 30/45 mg/kg | OLE crenezumab 60 mg/kga | Total |
| (n = 23) | (n = 48) | (N = 71) | |
| Participants with≥1 AE | 23 (100) | 44 (91.7) | 67 (94.4) |
| AE related to study drug per investigator | 7 (30.4) | 8 (16.7) | 15 (21.1) |
| Total number of deaths | 0 | 0 | 0 |
| AE grade≥3 (severe, life-threatening, or resulting in death)b | 2 (8.7) | 9 (18.8) | 11 (15.5) |
| Serious AEc | 3 (13.0) | 8 (16.7) | 11 (15.5) |
| Treatment withdrawal due to AEd | 1 (4.3) | 4 (8.3) | 5 (7.0) |
aIncludes participants switching from crenezumab 120 mg/kg during the randomized phase to 60 mg/kg during the OLE phase. bAll events assessed by investigator as unrelated to study drug. cAll events assessed by investigator as unrelated to study drug. OLE crenezumab 30/45 mg/kg: Grade 3 malignant melanoma (n = 1), Grade 2 chest pain (n = 1), Grade 3 fall (n = 1); OLE crenezumab 60 mg/kg: nephrolithiasis (Grade 2, n = 1; Grade 3, n = 1), Grade 1 accidental overdose, Grade 2 pneumonia, and Grade 3 subdural hematoma (n = 1), Grade 1 contusion (n = 2), Grade 1 dyspnea (n = 1), Grade 1 noncardiac chest pain and Grade 2 pulmonary embolism (n = 1), Grade 3 small intestine obstruction (n = 1). dOLE crenezumab 30/45 mg/kg: Grade 1 cerebral hemosiderin deposition and Grade 1 cerebral microhemorrhage (n = 1) assessed by investigator as related to study drug; OLE crenezumab 60 mg/kg: Grade 2 confusional state (n = 1) assessed by investigator as related to study drug (participant first experienced confusional state during the double-blind treatment period and did not receive any dose of crenezumab in the OLE period prior to discontinuation of treatment), Grade 3 atrial fibrillation (n = 1) assessed by investigator as unrelated to study drug, Grade 3 subdural hematoma (n = 1) assessed by investigator as unrelated to study drug, Grade 2 pulmonary embolism (n = 1) assessed by investigator as unrelated to study drug. AE, adverse event; OLE, open-label extension.
In the combined double-blind treatment and OLE periods, no ARIA-E events were observed. New ARIA-H events were reported in 9.9% of participants (n = 7): four in the OLE crenezumab 30 or 45 mg/kg group and three in the OLE 60 mg/kg group. In the OLE 30 or 45 mg/kg group, one participant (crenezumab 45 mg/kg dose) who developed 7 micro hemorrhages cumulatively also experienced Grade 1 siderosis and discontinued the study drug early because of the AE. Furthermore, one participant with micro hemorrhages in the OLE 60 mg/kg group discontinued study treatment because of a traumatic subdural hematoma (e.g., macrohemorrhage) caused by a head injury; this event was assessed by the investigator as unrelated to the study drug.
Infusion-related reactions
As described previously, infusion-related reactions were AEs that occurred during or within 24 h after study drug administration and were deemed related to the infusion by the investigator. During the combined double-blind treatment and OLE periods, six participants experienced eight infusion-related reactions: six occurred in four participants during the 13-week double-blind treatment period and resolved without treatment (Cohort 1, crenezumab 30 mg/kg: Grade 1 dysgeusia and Grade 1 oral disorder [n = 1] on Day 1; Cohort 1, placebo 45 mg/kg: Grade 1 ventricular extrasystoles [n = 1] on Day 88; Cohort 2, crenezumab 60 mg/kg: Grade 1 headache [n = 1] on Day 2; Cohort 3, crenezumab 120 mg/kg: Grade 1 vision blurred and Grade 1 vision field defect on Day 88 [n = 1]), and two were reported during the OLE phase (OLE crenezumab 60 mg/kg: Grade 2 hypertension [n = 1] on Day 118 that resolved without treatment and Grade 1 infusion site rash [n = 1] on Day 139 that resolved with treatment). No changes were made to the study drug dose.
Adverse events of special interest
Ascending-dose, 13-week, double-blind, placebo-controlled treatment period
Among participants who received crenezumab during the double-blind treatment period, two events of pneumonia occurred (crenezumab 60 mg/kg, Grade 2 SAE [n = 1]; crenezumab 120 mg/kg, Grade 1 AE [n = 1]). No changes were made to the study drug dose, and both participants recovered. There were no reported pneumonia events in participants receiving placebo.
Combined 13-week double-blind treatment and OLE periods up to 133 weeks
During the combined double-blind treatment and OLE periods, approximately 5.6% of participants (4/71), all in the OLE crenezumab 60 mg/kg group, developed pneumonia; two events were reported during the double-blind treatment phase and two events occurred during the OLE period, including one participant diagnosed with “pneumonia influenza.” All patients recovered, no changes were made to the study drug dose, and none of the events were considered by the investigator to be related to the study drug.
Pharmacokinetics and pharmacodynamics
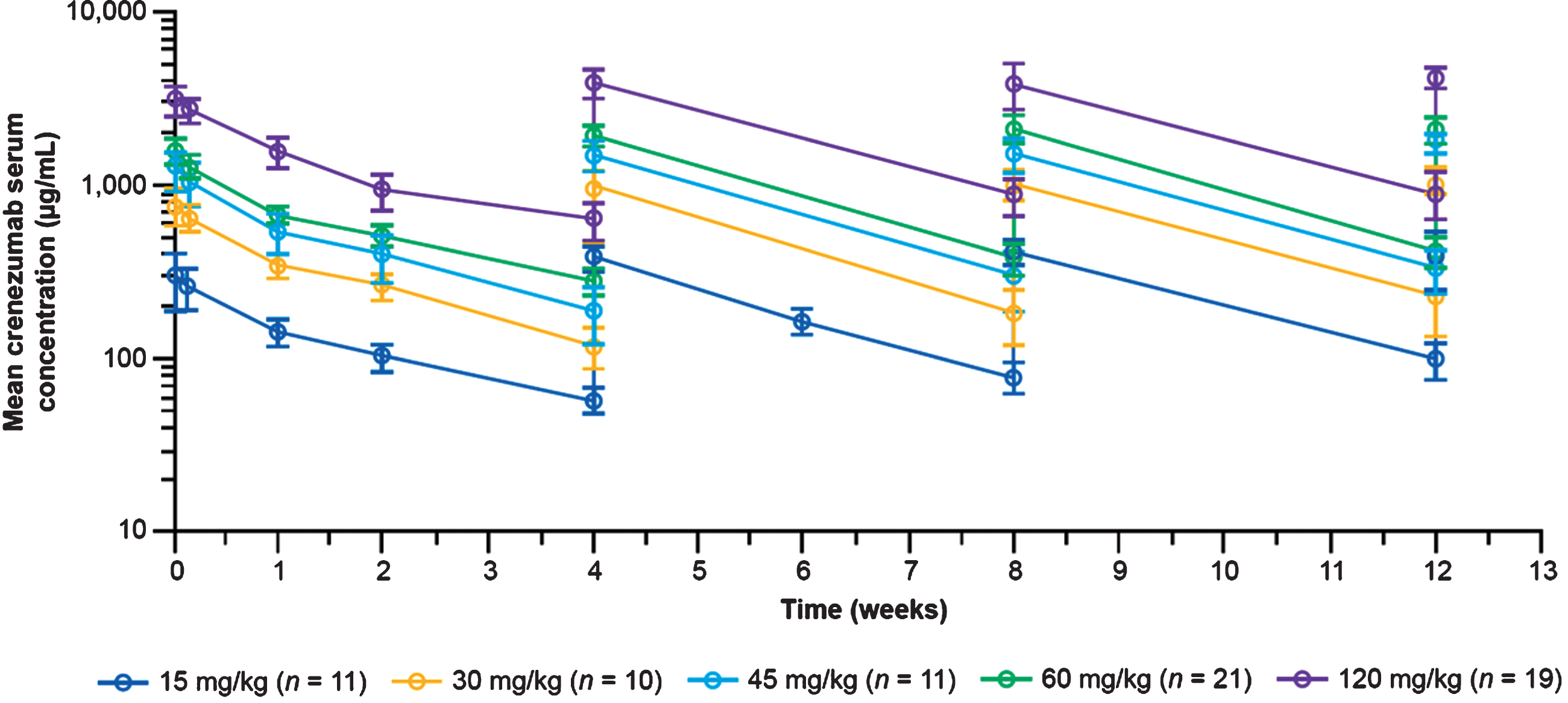
Crenezumab exhibited biphasic disposition over the 28-day period following the initial dose, and steady-state concentrations were achieved within 13 weeks (Fig. 2), with a modest accumulation ratio of 1.4 (range, 0.69–1.50) for post-infusion concentrations (obtained 60–90 minutes post end of infusion) and 1.5 (range, 0.52–4.31) for trough concentrations (obtained prior to the next crenezumab dose); accumulation ratios were calculated for each participant separately. Crenezumab serum concentrations increased proportionally with doses between 15 and 120 mg/kg q4w. At 60 mg/kg q4w, the mean steady-state crenezumab concentration after infusion (Cpeak at Week 13) was 2,090 μg/mL (SD±383 μg/mL; n = 21) and the mean steady-state trough concentration (Cmin at Week 13) was 416 μg/mL (SD±84.1 μg/mL; n = 21). For reference, the corresponding values in the Phase II ABBY trial for 15 mg/kg q4w were 454 μg/mL (SD±114 μg/mL) and 118 μg/mL (SD±71 μg/mL), respectively [17]. Of note, this shows that there was minimal overlap in exposure between the 60 mg/kg dose and the 15 mg/kg dose tested in Phase II, given that the trough concentration after the 60 mg/kg dose was similar to the post-infusion concentration after the 15 mg/kg dose.
Fig. 2
Mean (SD) serum crenezumab concentrations. Data shown are from the Phase II ABBY study (SRI cohort) for the 15 mg/kg dose and from the Phase Ib (GN29632) study reported here for the 30–120 mg/kg doses during the 13-week, randomized, placebo-controlled period. SD, standard deviation; SRI, safety run-in.
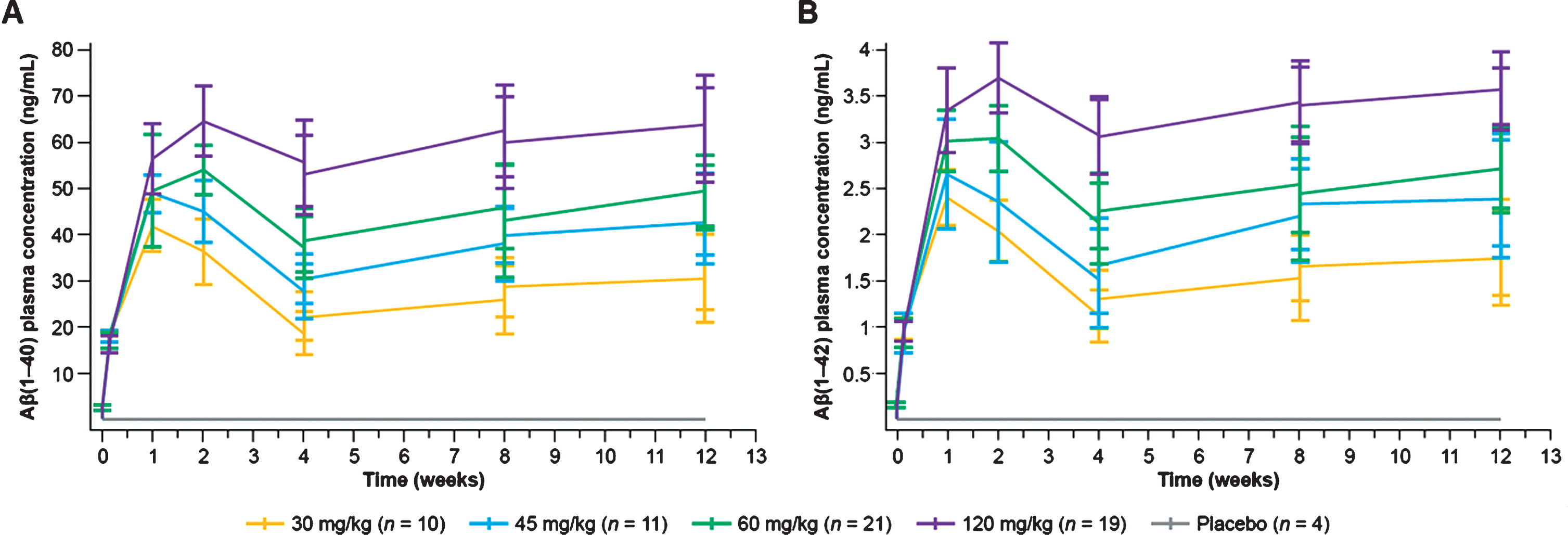
Total plasma monomeric Aβ1–40 and Aβ1–42 levels increased after crenezumab administration in a dose-dependent but not dose-proportional manner (Fig. 3), demonstrating peripheral target engagement [23]. At 60 mg/kg q4w, the plasma Aβ1–40 and Aβ1–42 mean trough concentrations at Week 13 were 48.1 ng/mL (SD±7.25 ng/mL; n = 21) and 2.72 ng/mL (SD±0.45 ng/mL; n = 21), respectively (Fig. 3). For reference, the corresponding total plasma Aβ1–40 and Aβ1–42 values for 15 mg/kg q4w after 68 weeks of treatment were 17.6 and 1.12 ng/mL, respectively [18]. The increase in total plasma Aβ1–40 and Aβ1–42 levels was delayed compared with crenezumab concentrations and reached maximum levels 7–14 days after the initial crenezumab dose [24].
Fig. 3
Plasma Aβ1–40(A) and Aβ1–42 (B) concentrations following administration of crenezumab during the 13-week, double-blind, randomized treatment phase. Aβ, amyloid-β.
Immunogenicity
The baseline prevalence of ADAs was 17.6% (13/74 participants), and all ADA-positive samples had only minimal detectable titer values in the assay. The incidence of treatment-emergent ADAs was 0% in all crenezumab treatment arms.
Imaging biomarkers
Participants who received crenezumab 30 mg/kg (n = 11), those who received 45 mg/kg (n = 12), and those who received 60 mg/kg (n = 48) during the OLE phase had similar rates of brain atrophy: an increase in ventricular volume (12% per year) and decreases in both hippocampal (3% per year) and whole brain (1.5% per year) volumes (Supplementary Figure 2). Amyloid PET SUVR using the subcortical white matter as a reference region suggested that participants who received the higher doses (45 and 60 mg/kg IV q4w) may have stabilized SUVR over time, whereas mean SUVR in participants in the lowest dose group (30 mg/kg IV q4w) increased (Supplementary Figure 3).
DISCUSSION
Safety
Overall, crenezumab was generally well tolerated at the doses evaluated in this study, with a safety profile consistent with the known safety profile of crenezumab from previous trials [17, 18]. The highest crenezumab dose administered in this study was 120 mg/kg IV q4w during the double-blind treatment period, and no dose-limiting toxicities were reported. Safety analyses of crenezumab in participants treated during the double-blind dose-escalation treatment phase showed that AEs were balanced across system organ classes, and the majority of AEs were low grade and non-serious. Similarly, in participants treated for up to Week 133 (combination of the double-blind treatment and OLE data), the majority of AEs were classified as mild or moderate and there were no new safety signals identified.
A low number of ARIA events were observed in the previous Phase II ABBY and BLAZE studies [17, 18]. Similarly, in both the double-blind treatment phase and up to Week 133 in the OLE phase in this study, no ARIA-E cases were observed and the number of ARIA-H events did not increase with higher doses of crenezumab. The low number of ARIA events in this study further support the hypothesis that the low effector function of the IgG4 backbone and/or the lack of vascular amyloid binding of crenezumab lower the risk of inflammation or microvascular damage [13, 25].
In the previous Phase II studies of crenezumab, a numerical imbalance of pneumonia cases was observed in the crenezumab group versus the placebo group [17, 18]. In the current study, the majority of pneumonia events were considered non-serious, were deemed not related to the study drug, and resolved without interrupting the dose of crenezumab. Moreover, the incidence of pneumonia observed in this study is consistent with the previously reported pneumonia incidence in this age group [26].
Pharmacokinetics and plasma pharmacodynamics
Crenezumab serum concentrations between 15 and 120 mg/kg q4w increased proportionally with the dose, with a half-life consistent with that of IgG monoclonal antibodies [23]. These results agree with projected exposures based on prior studies. Dose-dependent increases in total plasma monomeric Aβ1–40 and Aβ1–42 levels demonstrate that a higher degree of peripheral target engagement than observed in phase II studies is achievable with the 60 mg/kg IV q4w dose.
Immunogenicity
Although no positive post-baseline ADAs were detected following treatment with crenezumab, the high amount of circulating crenezumab could have interfered with detection of ADAs in some samples; thus, negative results do not completely rule out the presence of ADAs.
Imaging biomarkers
Because this study was not designed to detect differences in imaging measurements, no substantial conclusions can be made from comparisons between treatment arms due to the small sample size, short double-blind treatment phase followed by an active OLE period with no control group, and non-concurrent randomization of dose groups. However, the lack of observable differences in atrophy measures between the treatment groups is consistent with findings from the Phase II ABBY and BLAZE studies [17, 18]. Similarly, slower amyloid accumulation with the higher doses of crenezumab in this study appears consistent with results from the BLAZE study, in which the higher of two doses tested (15 mg/kg IV q4w) exhibited a reduced amyloid accumulation versus placebo [18].
Conclusions
In this Phase Ib trial, crenezumab was generally well tolerated at doses up to 120 mg/kg IV q4w, with a safety profile consistent with that observed in prior trials. Crenezumab serum concentrations increased dose-proportionally across the dose range evaluated, and steady-state concentrations were maintained. Total plasma Aβ1–40 and Aβ1–42levels increased following exposure to crenezumab in a dose-dependent manner, indicating peripheral target engagement.
ACKNOWLEDGMENTS
We would like to thank all the investigators, clinicians, and research staff who supported this trial, the individuals who participated in the trial, and their families. The following investigators, coordinators, and sites were involved with this study: Dr Marc Agronin (principal investigator), Dr Roberto Gil (sub-investigator), Niurka Colina (study coordinator), and Leo Then (study coordinator), Miami Jewish Health, Miami, FL; Dr Adam Boxer (principal investigator), Dr Cynthia Barton (sub-investigator), Dr Phi Dan Luong (sub-investigator), Dr Zachary Miller (sub-investigator), Dr Raeloni Porlaris (sub-investigator), Vivian Cheng (study coordinator), Noelle Ohanesian (study coordinator), and Ryan Powers (study coordinator), UCSF Memory and Aging Center, San Francisco, CA; Dr Mark Brody (principal investigator), Dr Janice Miller (sub-investigator), Dr Paayal Patel (sub-investigator), Dr Cynthia Stimeck (sub-investigator), Daisy Acevedo (study coordinator), Jessica Espinoza (study coordinator), Polina Kaplun (study coordinator), and Ana Scolari-Fuquay (study coordinator), Brain Matters Research, Delray Beach, FL; Dr Christopher Galloway (principal investigator), Dr Craig Curtis (principal investigator), Dr Judith A. Chalykoff (sub-investigator), Dr Von Garcia (sub-investigator), Dr Christine Murphy (sub-investigator), Dr Esteban Olivera (sub-investigator), Dr Gretchen Pohaski (sub-investigator), Dr Katy Smith (sub-investigator), Dr George Stoica (sub-investigator), Dr Lauren Trottier (sub-investigator), Dr Jennifer West (sub-investigator), Nadia Afghani (study coordinator), Stephanie Colina (study coordinator), Mark Daley (study coordinator), and Heather King (study coordinator), Bioclinica Research, Orlando, FL; Dr Martin Farlow (principal investigator), Dr Jared R. Brosch (sub-investigator), Jill Buck (study coordinator), Lyla Christner (study coordinator), and Madeline Naylor (study coordinator), Indiana University School of Medicine, Indianapolis, IN; Dr Lawrence Honig (principal investigator), Katrina Cuasay (study coordinator), and Evelyn Dominguez (study coordinator), Columbia University Irving Medical Center, New York, NY; Dr Ronald Schwartz (principal investigator), Sarah Crimmins (study coordinator), and Kayleigh Moring (study coordinator), Hattiesburg Clinic, Hattiesburg, MS; Dr Franco Sicuro (principal investigator), Dr Angela Clemente (sub-investigator), Dr Ronni Kahn (sub-investigator), Dr Kevin Mayse (sub-investigator), Becky Gedney (pharmacist), Kim Bichsel (study coordinator), and Sarah van Winkle (study coordinator), Millennium Psychiatric Associates, LLC, Creve Coeur, MO; Dr John Stoukides (principal investigator) and Laurie Given (study coordinator), Rhode Island Mood and Memory Research Institute, East Providence, RI; Dr Jewel White (principal investigator), Dr James McDonough (principal investigator), Dr David Subich (principal investigator), Christian Beierschmitt (study coordinator), Madison Bennett (study coordinator), Matthew Bryant (study coordinator), Amy Pucek (study coordinator), and Austin Wing (study coordinator), Bioclinica Research, The Villages, FL; and Dr Anton Porsteinsson (principal investigator), Dr Michelle Cervello (sub-investigator), Dr Audrey Rice (sub-investigator), and Susan Salem-Spencer (study coordinator), University of Rochester, Rochester, NY. We would also like to thank William Cho, an employee of Genentech, Inc. at the time of this study, for his scientific contributions.
This study was funded by F. Hoffmann-La Roche Ltd, Basel, Switzerland. Medical writing support was provided by Angela Morris, PhD, and Rachel Johnson, PhD, of Health Interactions, Inc, and funded by F. Hoffmann-La Roche Ltd.
ELECSYS, COBAS, and COBAS E are registered trademarks of Roche.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/20-0134r1).
DATA SHARING STATMENT
Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-200134.
REFERENCES
[1] | Prince M , Comas-Herrera A , Knapp M , Guerchet M , Karagiannidou M ((2016) ) World Alzheimer Report 2016. Improving healthcare for peopleliving with dementia coverage: Quality and costs now and in the future. Alzheimer’s Disease International, London, UK. |
[2] | Alzheimer’s Association, What is Alzheimer’s? Alzheimer’s and Dementia basics, https://www.alz.org/alzheimers-dementia/what-is-alzheimers |
[3] | Masters CL , Bateman R , Blennow K , Rowe CC , Sperling RA , Cummings JL ((2015) ) Alzheimer’s disease. Nat Rev Dis Primers 1: , 15056. |
[4] | Cummings J , Gould H , Zhong K ((2012) ) Advances in designs for Alzheimer’s disease clinical trials. Am J Neurodegener Dis 1: , 205–216. |
[5] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[6] | Hyman B , West H , Rebeck G , Buldyrev S , Mantegna R , Ukleja M , Havlin S , Stanley H ((1995) ) Quantitative analysis of senile plaques in Alzheimer’s disease: Observation of log-normal size distribution and molecular epidemiology of differences associated with apolipoprotein E genotype and trisomy 21 (Down syndrome). Proc Natl Acad Sci U S A 92: , 3586–3590. |
[7] | Spires-Jones T , Meyer-Luehmann M , Osetek J , Jones P , Stern E , Bacskai B , Hyman B ((2007) ) Impaired spine stability underlies plaque-related spine loss in an Alzheimer’s disease mouse model. Am J Pathol 171: , 1304–1311. |
[8] | Koffie R , Meyer-Luehmann M , Hashimoto T , Adams K , Mielke M , Garcia-Alloza M , Micheva K , Smith S , Kim M , Lee V , Hyman B , Spires-Jones T ((2009) ) Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106: , 4012–4017. |
[9] | Serrano-Pozo A , William C , Ferrer I , Uro-Coste E , Delisle M , Maurage C , Hock C , Nitsch R , Masliah E , Growdon J , Frosch M , Hyman B ((2010) ) Benefical effect of human anti-amyloid-β active immunization on neurite morphology and tau pathology. Brain 133: , 1312–1327. |
[10] | Palop J , Mucke L ((2010) ) Amyloid-β-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci 13: , 812–818. |
[11] | Mucke L , Selkoe D ((2012) ) Neurotoxicity of amyloid β-protein: Synaptic and network dysfunction. Cold Spring Harb Perspect Med 2: , a006338. |
[12] | Schneider LS , Mangialasche F , Andreasen N , Feldman H , Giacobini E , Jones R , Manua V , Mecocci P , Winblad B , Kivipelto M ((2014) ) Clinical trials and late-stage drug development for Alzheimer’s disease: An appraisal from 1984 to 2014. J Intern Med 275: , 251–283. |
[13] | Adolfsson O , Pihlgren M , Toni N , Varisco Y , Buccarello AL , Antoniello K , Lohmann S , Piorkowska K , Gafner V , Atwal JK , Maloney J , Chen M , Gogineni A , Weimer RM , Mortensen DL , Friesenhahn M , Ho C , Paul R , Pfeifer A , Muhs A , Watts RJ ((2012) ) An effector-reduced anti-β-amyloid (Aβ) antibody with unique aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J Neurosci 32: , 9677–9689. |
[14] | Ultsch M , Li B , Maurer T , Mathieu M , Adolfsson O , Muhs A , Pfeifer A , Pihlgren M , Bainbridge TW , Reichelt M , Ernst JA , Eigenbrot C , Fuh G , Atwal JK , Watts RJ , Wang W ((2016) ) Structure of crenezumab complex with Aβ shows loss of β-hairpin. Sci Rep 6: , 39374. |
[15] | Yang T , Dang Y , Ostaszewski B , Mengel D , Steffen V , Rabe C , Bittner T , Walsh DM , Selkoe DJ ((2018) ) Target engagement in an AD trial: Crenezumab lowers Aβ oligomer levels in CSF. Alzheimers Dement 14 (Suppl): , P1669–P1670. |
[16] | Meilandt WJ , Maloney JA , Imperio J , Lalehzadeh G , Earr T , Crowell S , Bainbridge TW , Lu Y , Ernst JA , Fuji RN , Atwal JK ((2019) ) Characterization of the selective in vitro and in vivo binding properties of crenezumab to oligomeric Aβ. Alzheimers Res Ther 11: , 97. |
[17] | Cummings J , Cohen S , Van Dyck CH , Brody M , Curtis C , Cho W , Ward M , Friesenhahn M , Rabe C , Brunstein F , Quartino A , Honigberg LA , Fuji R , Clayton D , Mortensen D , Ho C , Paul R ((2018) ) ABBY: A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology 90: , e1889–e1897. |
[18] | Salloway S , Honigberg LA , Cho W , Ward M , Friesenhahn M , Brunstein F , Quartino A , Clayton D , Mortensen D , Bittner T , Ho C , Rabe C , Schauer SP , Wildsmith KR , Fuji RN , Suliman S , Reiman EM , Chen K , Paul R ((2018) ) Amyloid positron emission tomography and cerebrospinal fluid results from a crenezumab anti-amyloid-beta antibody double-blind, placebo-controlled, randomized phase II study in mild-to-moderate Alzheimer’s disease (BLAZE). Alzheimers Res Ther 10: , 96. |
[19] | ClinicalTrials.gov, CREAD Study: A Study of Crenezumab Versus Placebo to Evaluate the Efficacy and Safety in Participants With Prodromal to Mild Alzheimer’s Disease (AD). ClinicalTrials.gov Identifier: NCT02670083, https://clinicaltrials.gov/ct2/show/NCT02670083. |
[20] | ClinicalTrials.gov, A Study of Crenezumab Versus Placebo to Evaluate the Efficacy and Safety in Participants With Prodromal to Mild Alzheimer’s Disease (AD) (CREAD 2). ClinicalTrials.gov Identifier: NCT03114657, https://clinicaltrials.gov/ct2/show/NCT03114657 |
[21] | Tariot PN , Lopera F , Langbaum JB , Thomas RG , Hendrix S , Schneider LS , Rios-Romenets S , Giraldo M , Acosta N , Tobon C , Ramos C , Espinosa A , Cho W , Ward M , Clayton D , Friesenhahn M , Mackey H , Honigberg L , Sanabria Bohorquez S , Chen K , Walsh T , Langlois C , Reiman EM ((2018) ) The Alzheimer’s prevention initiative autosomal-dominant alzheimer’s disease trial: A study of crenezumab versus placebo in preclinical PSEN1 E280A mutation carriers to evaluate efficacy and safety in the treatment of autosomal-dominant Alzheimer’s disease, including a placebo-treated noncarrier cohort. Alzheimers Dement 4: , 150–160. |
[22] | Barkhof F , Daams M , Scheltens P , Brashear HR , Arrighi HM , Bechten A , Morris K , McGovern M , Wattjes MP ((2013) ) An MRI rating scale for amyloid-related imaging abnormalities with edema or effusion. AJNR Am J Neuroradiol 34: , 1550–1555. |
[23] | Yoshida K , Moein A , Bittner T , Ostrowitzki S , Lin H , Honigberg L , Jin JY , Quartino A , ((2020) ) Pharmacokinetics and pharmacodynamic effect of crenezumab on plasma and cerebrospinal fluid beta-amyloid in patients with mild-to-moderate Alzheimer’s disease. Alzheimers Res Ther 12: , 16. |
[24] | Lin H , Schneider A , Quartino A , Bittner T , Hu N , Smith J , Cho W , Ostrowitzki S ((2018) ) Safety, tolerability and pharmacokinetics of crenezumab in mild-to-moderate AD patients treated with escalating doses for up to 32 months. Poster presentation at the 70th Annual American Academy of Neurology (AAN) Annual Meeting, Los Angeles, CA, USA. |
[25] | Atwal JK , Ferl G , Meilandt WJ , Wetzel-Smith M , Li B , Bainbridge TW , Reichelt M , Ultsch M , Ernst JA , Wang W , Crowell S , Quartino AL , Fuji RN ((2017) ) Crenezumab’s preferential binding of oligomeric amyloid-beta species underlies its unique mechanism of action. Neurology 88: , S7.005. |
[26] | Kolditz M , Tesch F , Mocke L , Hoffken G , Ewig S , Schmitt J ((2016) ) Burden and risk factors of ambulatory or hospitalized CAP: A population based cohort study. Respir Med 121: , 32–38. |


