
Evidence that Brain-Reactive Autoantibodies Contribute to Chronic Neuronal Internalization of Exogenous Amyloid-β1-42 and Key Cell Surface Proteins During Alzheimer’s Disease Pathogenesis
Abstract
Blood-brain barrier (BBB) permeability is a recognized early feature of Alzheimer’s disease (AD). In the present study, we examined consequences of increased BBB permeability on the development of AD-related pathology by tracking selected leaked plasma components and their interactions with neurons in vivo and in vitro. Histological sections of cortical regions of postmortem AD brains were immunostained to determine the distribution of amyloid-β1-42 (Aβ42), cathepsin D, IgG, GluR2/3, and alpha7 nicotinic acetylcholine receptor (α7nAChR). Results revealed that chronic IgG binding to pyramidal neurons coincided with internalization of Aβ42, IgG, GluR2/3, and α7nAChR as well as lysosomal compartment expansion in these cells in regions of AD pathology. To test possible mechanistic interrelationships of these phenomena, we exposed differentiated SH-SY5Y neuroblastoma cells to exogenous, soluble Aβ42 peptide and serum from AD and control subjects. The rate and extent of Aβ42 internalization in these cells was enhanced by serum containing neuron-binding IgG autoantibodies. This was confirmed by treating cells with individual antibodies specific for α7nAChR, purified IgG from AD or non-AD sera, and sera devoid of IgG, in the presence of 100 nM Aβ42. Initial co-localization of IgG, α7nAChR, and Aβ42 was temporally and spatially linked to early endosomes (Rab11) and later to lysosomes (LAMP-1). Aβ42 internalization was attenuated by treatment with monovalent F(ab) antibody fragments generated from purified IgG from AD serum and then rescued by coupling F(ab) fragments with divalent human anti-Fab. Overall, results suggest that cross-linking of neuron-binding autoantibodies targeting cell surface proteins can accelerate intraneuronal Aβ42 deposition in AD.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative disease that currently carries with it a forecast of ever-increasing prevalence and debilitating social and economic costs to those afflicted, caregivers, and their families. It is the most common cause of dementia, now affecting over 5 million people in the United States [1]. Well-recognized hallmarks of AD pathology include excessive amyloid-β deposition intracellularly (within neurons) and extracellularly as amyloid plaques, tau hyperphosphorylation, formation of neurofibrillary tangles, loss of synapses, and progressive neurodegeneration with associated neuroinflammation [2–4]. The evolution of AD pathology can span decades in patients and is thought to progress unnoticed for 10–15 years before telltale symptoms emerge and clinical detection becomes possible using conventional means [5, 6]. Despite extensive research over the past several decades, the factors, conditions, and mechanisms that drive both the macro- and microscopic pathological changes in the brain associated with initiation and progression of this disease remain unresolved.
Much work on postmortem AD brains, the brains of numerous AD-relevant animal models, and cultured cells has been directed toward understanding how amyloid, primarily amyloid-β1-42 (Aβ42), is deposited in the brain within neurons and amyloid plaques throughout AD pathogenesis [3, 7, 8]. Although plaque formation in human AD brains is still generally considered an extracellular event, intracellular Aβ42 deposition has been reported as an early pathological marker in AD brains, in the brains of AD-relevant transgenic animal models, and in various types of cultured neurons and neuron-like cells [9–18]. Aβ42 has been shown to bind with high affinity to the alpha7 subtype of the nicotinic acetylcholine receptor (α7nAChR), which is abundantly present on the surfaces of cholinergic and cholinoceptive neurons (especially on cortical pyramidal cells) [9, 12, 19, 20]. Additionally, in an earlier study, we showed that when α7nAChR-transfected SK-N-MC neuroblastoma cells were exposed to soluble, exogenous human Aβ42 peptide, the resulting α7nAChR-Aβ42 complexes were internalized and accumulated within the lysosomal compartment [12, 20–22]. Also, treatment of adult mouse brain slice cultures with human serum containing brain-reactive autoantibodies or purified antibodies directed against the α7nAChR was found to augment the rate and extent of Aβ42 accumulation selectively in pyramidal neurons [23, 24]. The term “autoantibodies” is used here to reflect the self-reactive property that these IgG species exhibit.
Taken together, the above findings suggest that access of neuron-binding autoantibodies and soluble Aβ42 peptide to the brain parenchyma, presumably mediated by locally increased blood-brain barrier (BBB) permeability, plays a key role in Aβ deposition in the brain during AD pathogenesis. However, details of the mechanisms driving this process are lacking, as is the underlying reason for the apparent requirement for advanced age. In the present study, we examined the consequences of BBB functional compromise on the development of AD-related pathology by examining and tracking the fate of selected leaked plasma components and their interactions with neurons in vivo and in vitro. Results show that, in postmortem AD brain, chronic autoantibody binding was associated with internalization of key neuronal cell surface proteins, GluR2/3 and α7nAChR, and coincident with lysosomal compartment expansion. To determine if these observations in AD brains are mechanistically linked, we investigated the effects of exposure of differentiated SH-SY5Y human neuroblastoma cells to exogenous, soluble Aβ42 and serum from AD and control subjects. The rate and extent of Aβ42 internalization was enhanced by both AD and control sera. In addition, treatment of cells with antibodies specific to α7nAChR or purified IgG obtained from either AD or non-AD controls showed similar enhancement of Aβ42 internalization. We also found that initial co-localization of IgG, α7nAChR, and Aβ42 was temporally and spatially related to the early endosomal marker, Rab11, and to the lysosomal marker, LAMP-1, at later time points. Lastly, Aβ42 and antibody internalization was attenuated by treatment with monovalent F(ab) antibody fragments generated from IgG purified from AD patient sera, and was rescued by crosslinking F(ab) with anti-F(ab) antibodies, suggesting that cross-linking of bivalent neuron-binding autoantibodies contributes to AD pathology by facilitating intracellular Aβ42 deposition.
MATERIALS AND METHODS
Human brain samples
Brain tissue from patients with sporadic AD (n = 23, age-range = 71–88) and age-matched, neurologically normal individuals (n = 14, age-range = 69–83) were obtained from the Harvard Brain Tissue Resource Center (Belmont, MA), the Cooperative Human Tissue Network (Philadelphia, PA), and the UCLA Tissue Resource Center (Los Angeles, CA). Postmortem intervals were <24 h and pathological confirmation of AD was according to criteria defined by the National Institute on Aging and the Reagan Institute Working Group on Diagnostic Criteria from the Neuropathological Assessment of AD [25]. Formalin-fixed tissues were processed for routine paraffin embedding and sectioning according to established protocols (see below). Control tissues exhibited minimal localized microscopic AD-like neuropathology.
Immunohistochemistry (IHC)
IHC was used to investigate the expression and localization of selected proteins in the human brain samples as described previously [23]. Briefly, paraffin-embedded human tissues were deparaffinized using xylene and rehydrated through a graded series of decreasing concentrations of ethanol. Antigenicity was enhanced by microwaving sections in citrate buffer. Endogenous peroxidase was quenched by treating sections with 3% H2O2 for 10 min. Sections were incubated in blocking serum and then treated with primary antibodies at appropriate dilutions for 1 h at room temperature. After a thorough rinse in PBS, corresponding biotin-labeled secondary antibody was applied for 30 min at room temperature. Sections were treated with avidin-peroxidase complex (Vectastain ABC Elite, Vector Laboratories, Inc., Foster City, CA) and visualized with 3-3-diaminobenzidine-4-HCL (DAB)/H2O2 (Imm-Pact-DAB) (Vector). They were then lightly counterstained with hematoxylin, dehydrated through increasing concentrations of ethanol, cleared in xylene, and mounted in Permount. To detect endogenous IgG, anti-human IgG (Vector Laboratories, Inc., Foster City, CA) was used as the primary antibody with no secondary antibody treatment. Controls consisted of brain sections treated with non-immune serum or omission of the primary antibody. Specimens were examined and photographed with a Nikon FXA microscope and digital images were recorded using a Nikon DXM1200F digital camera and analyzed using NIS-Elements Imaging Software (Nikon Instruments Inc. USA) and Cell Profiler image analysis software.
Cell culture
An in vitro model was developed using differentiated human neuroblastoma SH-SY5Y cells (ATCC CRL-2266). Cells were first grown in 10% fetal bovine serum (FBS; Sigma F6178) in Dulbecco’s Minimal Essential Medium (DMEM; Gibco)/F12 (Cellgro; Invitrogen, UK) media until 80% confluence in 35 mm glass-bottom dishes. Cells were then differentiated using 10 μM retinoic acid and 0.2% FBS in DMEM/F12 media for three days. Differentiating cells sprout neuron-like processes that establish connections with cells in the vicinity and were used for experimentation at roughly 70% confluence. Cells were treated with 100 nM HiLyteTM fluor 488 (FITC)-labeled Aβ42 (a physiologically relevant concentration) with or without selected sera for time points ranging from 30 min to 72 h. Selected sera used were obtained from a young non-demented control (YC) subject, an old-aged non-demented control (OC) subject, and an Alzheimer’s disease patient (AD), and were diluted 1:50 in serum-free media [26]. The AD, YC, and OC sera (one from each group) used for these in vitro studies were selected after analyzing and comparing their autoantibody profiles using immunoblotting and Human Protein Microarrays as described previously [26–31]. In these previous studies, we have analyzed the IgG autoantibody signatures of over 100 AD sera and a similar number of control (young and old) sera. Based on these profiles, we selected a single sample from each subject group that best reflected the group. Approval to use these sera was obtained from the University of Medicine and Dentistry-Stratford (now Rowan University) Institutional Review Board. Human sera were heated to 56°C for 25 min to inactivate complement for certain experiments.
Immunocytochemistry (ICC)
Differentiated SH-SY5Y cells were washed in cation-free 1x Hank’s Balanced Salt Solution (HBSS, without calcium or magnesium; Invitrogen, UK) three times prior to experimental protocol, and later were fixed in 4% paraformaldehyde (PFA). If the cell surface membrane was to be stripped so as to not include surface-bound, non-internalized peptide for Aβ42 internalization assays, immediately prior to fixation, three acid washes (0.1 M Glycine, pH 2.5) of 2 min each were performed, followed by two more HBSS washes to remove any residual acid. After fixation, cells were treated with 3% BSA in PBS with Tween (PBS-T) (for removal of cell surface membranes) or PBS alone (for intact cell surface membrane assays) and then probed with primary antibody overnight at 4°C followed by secondary antibodies for 1 h at room temperature. For detection of serum IgG bound to SH-SY5Y cells, anti-human IgG conjugated to an Alexa-fluor 594 (Cy3) fluorophore was used as secondary antibody. Nuclei were counterstained with Hoechst. Confocal microscopy (Nikon) and epifluorescence optics were used to capture images, and NIS-Elements software was used to perform image analysis and quantification of signal intensities.
Peptides and antibodies
For IHC, anti-Aβ42 antibodies were obtained from Millipore International (Temecula, CA) (polyclonal, Cat. No. AB5078 P, dilution 1:50) and Pharmingen (San Diego, CA) (polyclonal Cat. No. 4767, dilution 1:50); biotinylated anti-human IgG antibodies were obtained from Vector Laboratories (Burlingame, CA) (host: goat, Cat. No. PK-6103 and BA-3000, dilution 1:100); mouse anti-GluR2 was obtained from Santa Cruz Biotechnology (Santa Cruz, CA) (polyclonal N19, SC-7611) and Zymed Laboratories (Cat. No. 32-0300); Cathepsin D was obtained from Upstate Biotechnology (Lake Placid, NY) (Cat. No 06-467).
For ICC experiments, exogenous Aβ42 (beta-amyloid 1–42, HiLyteTM fluor 488 (FITC); AnaSpec, Inc., San Jose, CA, USA; AS-60479-01) was used for internalization assays. Aβ42 monomers were prepared by reconstituting 0.1 mg lyophilized powder in 50 μl of 1% NaOH. Samples were diluted to 1 mg/ml in PBS and stored at –20°C in 5 μl aliquots. Cells were treated with 100 nM Aβ42 in serum-free medium prepared immediately prior to use. Medium was added to peptide-treated cells to avoid binding in serum prior to administration. Fresh aliquots were used for each experiment to avoid repeated freeze-thaw cycles that could trigger potential aggregate formation and precipitation/fibrillization. Human serum samples were obtained from Analytical Biological Services Inc. (Wilmington, DE). Samples were numerically coded and included the following information: age, gender, the presence or absence of a detectable neurological disease, an indication of disease severity via a Mini-Mental Status Exam score (MMSE), and postmortem interval. Use of these samples was approved by the UMDNJ-Stratford IRB (now Rowan University).
Primary antibodies for ICC included: anti-synaptic vesicle (SV) 2 (Developmental Studies Hybridoma Bank, dilution 1:1000); anti-Tau6 (Sigma, St. Louis, MO, USA; Cat. No. T8201, dilution 1:500); anti-LAMP1 (Sigma, St. Louis, MO, USA; Cat. No. L1418, dilution 1:200); anti-Rab-11 (Sigma, St. Louis, MO, USA, Cat. No. R5903, diluted 1:100); anti-α7nAChR (Sigma, St. Louis, MO, USA, Cat. No. M220, diluted 1:500). Human serum used as a primary antibody was diluted 1:50. Anti-rabbit secondary IgG Cy3 (1:1500 dilution), anti-mouse IgG-488 (1:500 dilution), and goat anti-human AlexaFluor-488 (1:500 dilution) were used as secondary antibodies.
IgG purification and F(ab) monovalent antibody preparation
The IgG fraction from sera was purified using spin-column chromatography (Pierce Melon Gel IgG Purification Kit) according to manufacturer’s protocol and confirmed using SDS-PAGE and absorption at 280 nm (data not shown). F(ab) fragments were prepared from purified IgG using a Fab preparation kit (Pierce Protein Research Products; ThermoFisher Scientific). Briefly, purified IgG was digested for 4–6 h at 37°C with 1% (w/w) papain at pH 7.0 with 0.01 M cysteine. F(ab) fragments were isolated using filtered chromatography as per the manufacturer’s instructions. The purity of F(ab) fragments was confirmed using spectrophotometry at 280 nm.
Image analysis
Image analysis for ICC experiments was carried out using a Nikon Confocal microscope and NIS-Elements software. The software was used to delineate cells manually as a region of interest (ROI) at 20x by selecting 10 random images on an experimental dish where only entire cells were seen and counted. This roughly accounted for eight cells per image, and ten images per experiment, performed in triplicate, reaching about 240 cells per experiment for analysis. Fluorescence signal intensity emanating from FITC-labeled Aβ42 within the ROI was calculated. After exporting data to an Excel spreadsheet, the summed signal intensity for FITC-Aβ42 was divided by the total area of the ROI to determine the signal intensity (FITC-Aβ42) per unit area of the cells. To measure internalized FITC-Aβ42, surface membranes were first removed using a 0.1 M glycine acid wash prior to image acquisition as described above. To retain surface bound FITC-Aβ42, no acid wash or detergents were included in the protocol.
Statistics
Experiments were run in triplicate and image analysis was blinded to limit bias. Statistical significance of differences between treatment groups was evaluated using the two-tailed Student’s t-test, with levels of significance indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001. Variation within each treatment group was represented by standard deviation from the mean (SEM). Image acquisition parameters were determined by the user and kept identical within a given experiment.
RESULTS
In neurons of AD brains, exogenous Aβ42 and IgG are internalized along with key cell surface proteins and deposited within the lysosomal compartment
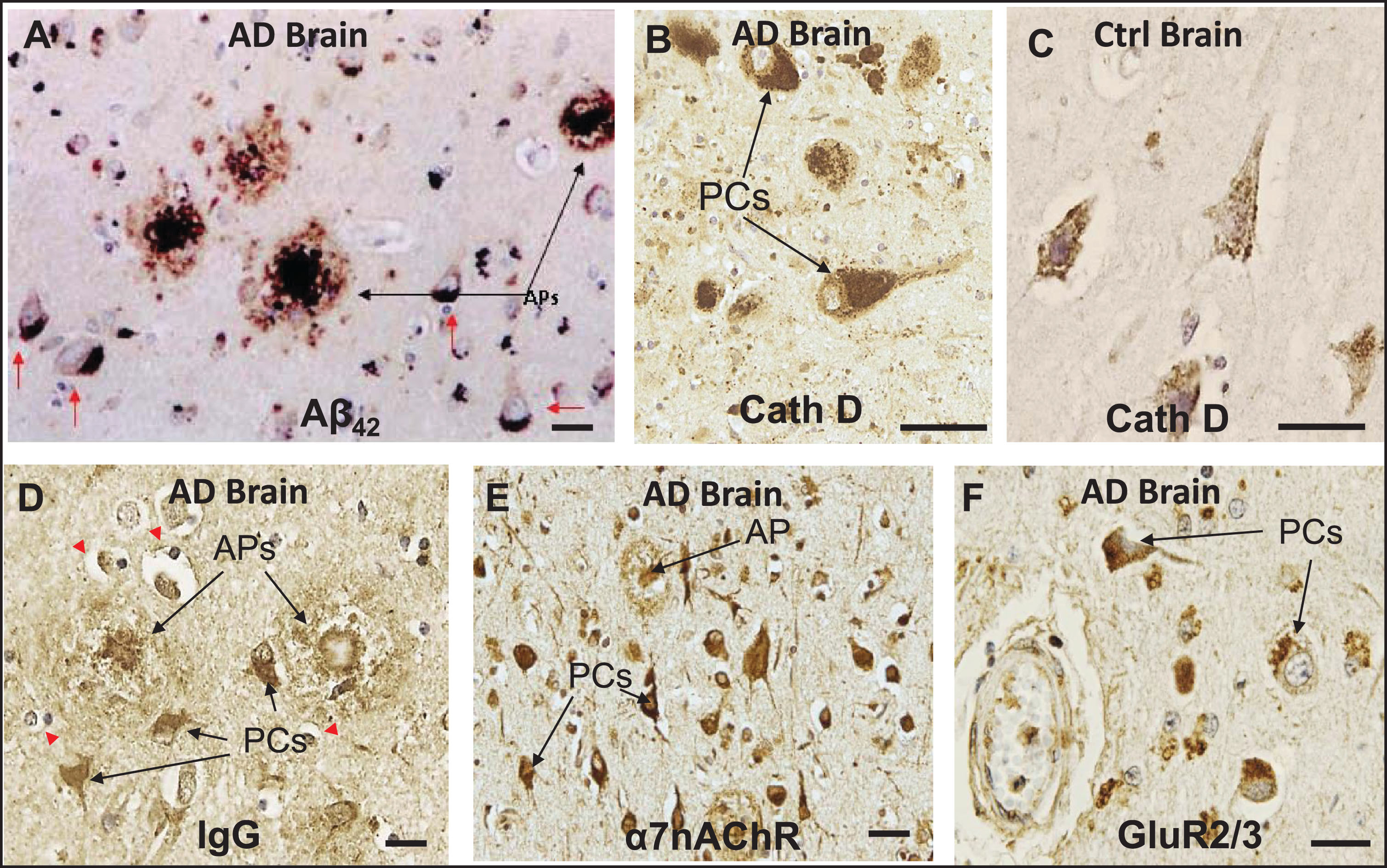
Previous IHC studies of human AD brains and some AD-relevant transgenic mouse models have shown that deposition of Aβ peptides, especially Aβ42, occurs preferentially within the lysosomal compartment of specific neuronal subtypes, especially pyramidal cells, and in amyloid plaques in a number of different brain regions [9, 12, 23, 24, 26, 30, 33] (Fig. 1A–C). Histological sections of AD brains also reveal that, under conditions of BBB compromise, immunoglobulins (Igs) leak into the parenchyma of AD brains and exhibit preferential affinity for the same pyramidal neurons that contain Aβ42 as well as extracellular amyloid plaques (Fig. 1D). Co-localization of IgG and Aβ42 has been demonstrated in cerebral cortex and hippocampus of AD brains in our previous study [23], as well as in the cerebral cortex of diabetic and hypercholesterolemic pigs [33]. IHC also showed intense α7nAChR-positive immunostaining in pyramidal cells and throughout dense-core amyloid plaques, supporting the possibility that at least some plaques are derived from the death and lysis of local Ig- and Aβ42-overburdened neurons (Fig. 1E). A similar localization pattern was seen in pyramidal cells for GluR2/3 (Fig. 1F).
Fig.1
In neurons of AD brains, exogenous Aβ42 and IgG are internalized along with key cell surface proteins and deposited within the lysosomal compartment. A) IHC of a histological section of cerebral cortex of AD brain showing deposition of Aβ42 in amyloid plaques (APs) and in pyramidal cells (PCs). B, C) Cathepsin D (CathD) immunostaining reveals a great expansion of the lysosomal compartment in PCs in AD brains over that of controls. D) Section of AD brain showing preferential affinity of IgG for certain PCs and APs, along with adjacent non-reactive, IgG immuno-negative cells. E) IHC shows intense α7nAChR-positive immunostaining in PCs, and α7nAChR was also detected throughout dense-core APs, supporting the possibility that at least some APs are derived from the death and lysis of local Ig- and Aβ42-overburdened PCs. F) A similar localization pattern was seen in PCs for GluR2/3, a surface receptor found with intracellular distribution, in the vicinity of the cerebrovasculature. Red arrows, Aβ42-burdened neurons. Red arrowheads, IgG immuno-negative cells. Scale bar = 20 μm.
Serum IgG autoantibodies bind to differentiated SH-SY5Y cells
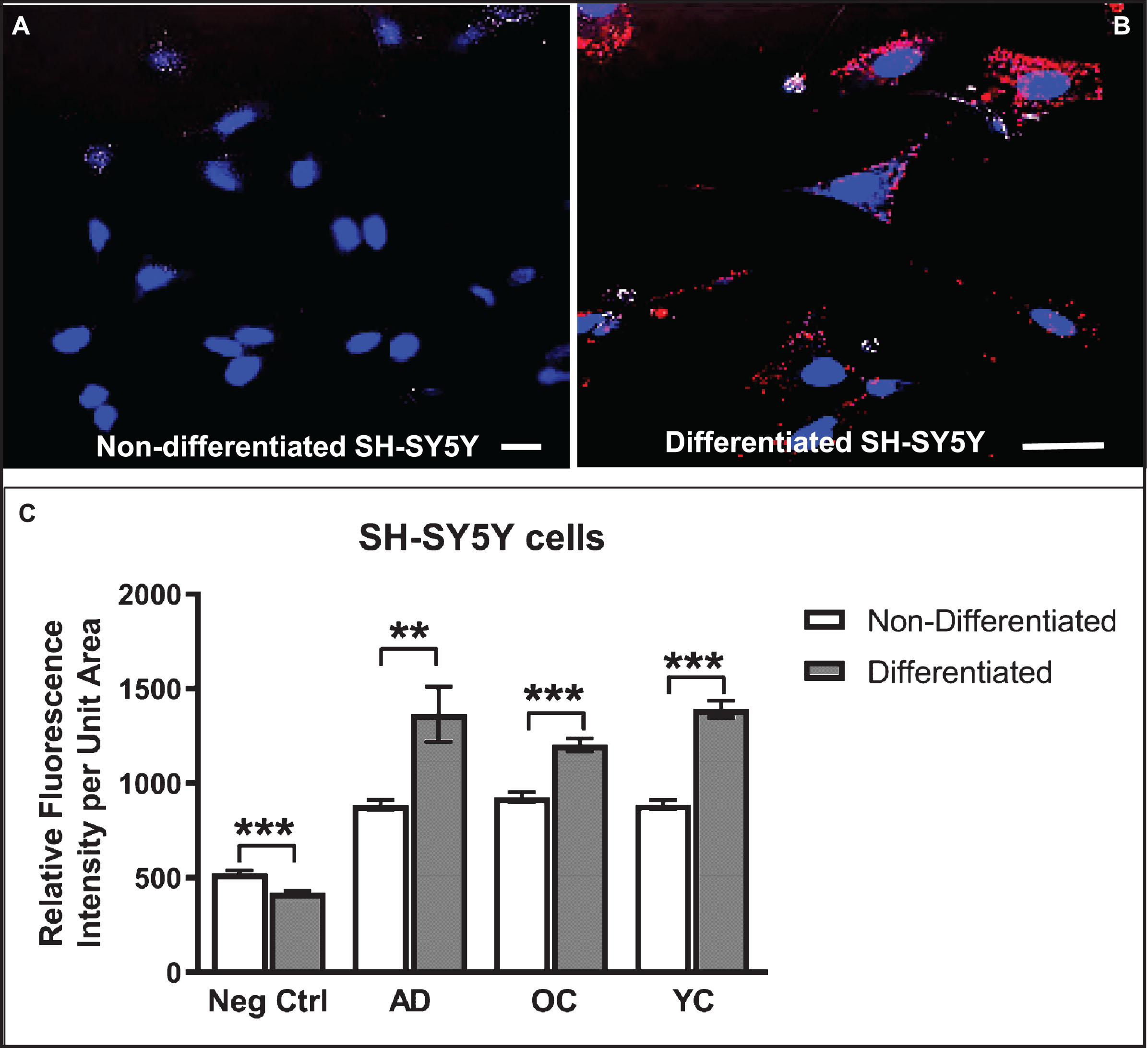
Differentiated SH-SY5Y human neuroblastoma cells were selected as a model system to study the cellular response to conditions thought to approximate those in brain tissue during early stages of AD pathogenesis in the context of increased BBB permeability. SH-SY5Y cells were first induced to differentiate by treatment with 10 μM retinoic acid (RA) for 72 h, a response highlighted by changes in cell shape to one that more resembles neurons, including extension of neurite-like processes (Figs. 2A–D, 3A, B). Immunocytochemistry and confocal microscopy confirmed that differentiation of SH-SY5Y cells was accompanied by increased expression of neuronal proteins consistent with neuronal differentiation, including α7nAChR (Fig. 2A), synaptic vesicle 2a (SV2; Fig. 2B), microtubule-associated protein-2 (Map2; Fig. 2C), and Tau6 (Fig. 2D). These proteins are naturally expressed in this cell type, thus reinforcing their suitability as a neuronal model. Differentiated cells also showed markedly increased reactivity to IgG antibodies present in human serum compared to undifferentiated cells (Fig. 3A–C). Cells not treated with serum as controls (Ctrl) showed lower signals (Fig. 3C). Interestingly, addition of serum from an AD patient did not significantly increase the extent of IgG antibody binding to SH-SY5Y cells over that from a non-demented age-matched older control (OC) individual or sera from a younger non-demented control individual (YC) (Fig. 3C). This suggests that all human sera may contain brain-reactive autoantibodies, regardless of age or the presence or absence of disease, as was shown in our previous study using western blot analysis [31] and immunohistochemistry [23, 31, 33].
Fig.2
Differentiated SH-SY5Y cells are a useful model system to study mechanisms associated with neuronal Aβ42 internalization. SH-SY5Y cells differentiated by treatment with retinoic acid for 72-h. A-D) Differentiation was confirmed by changes in cell shape, extension of neuritic processes, and detection of robust expression of the following neuronal markers of differentiation using ICC: (A) α7nAChR; (B) synaptic vesicle 2a (SV2); (C) microtubule-associated protein 2 (Map-2); and (D) tau6. Scale bar = 5 μm.
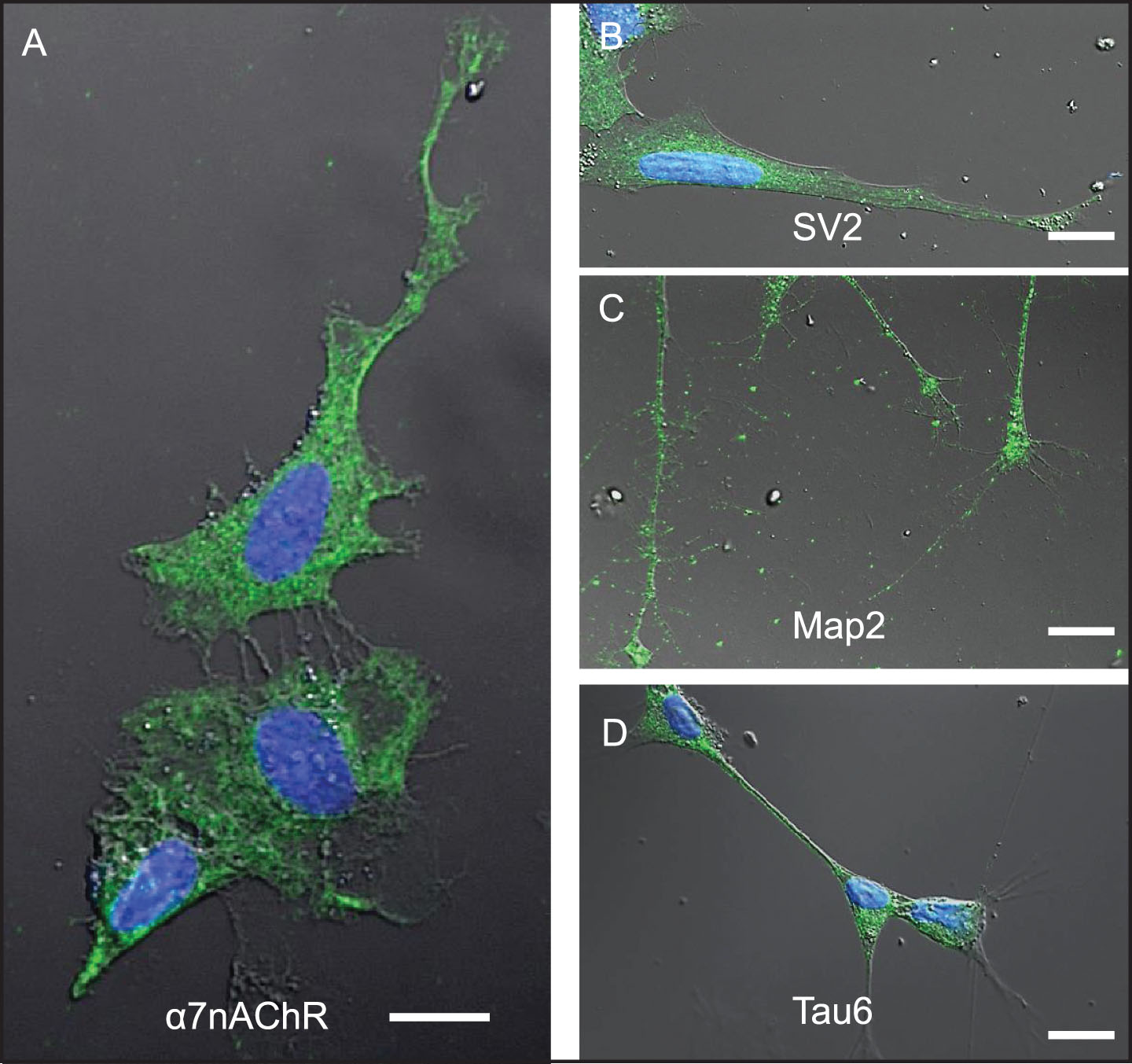
Fig.3
Differentiated SH-SY5Y cells show increased reactivity to IgG autoantibodies present in human serum. A, B) Confocal images of non-differentiated (A) and differentiated (B) SH-SY5Y cells treated with human serum (diluted 1:50) in medium for 24 h and then immunostained with Cy3-labeled (red) anti-human IgG antibodies and counterstained with Hoechst stain (blue) to highlight nuclei. C) Differentiated cells show increased binding of serum IgG over that of non-differentiated cells. Unexpectedly, the extent of IgG binding in cells treated with serum from an AD patient did not differ significantly from an older non-demented subject or younger control. Controls (Ctrl) were performed with omission of serum and addition of secondary antibody. Scale bar = 10 μm. ***p < 0.001. AD, Alzheimer’s disease serum; OC, old-aged matched, non-demented control; YC, younger, non-demented control.
Internalization of exogenous Aβ42 is enhanced by brain-reactive autoantibodies
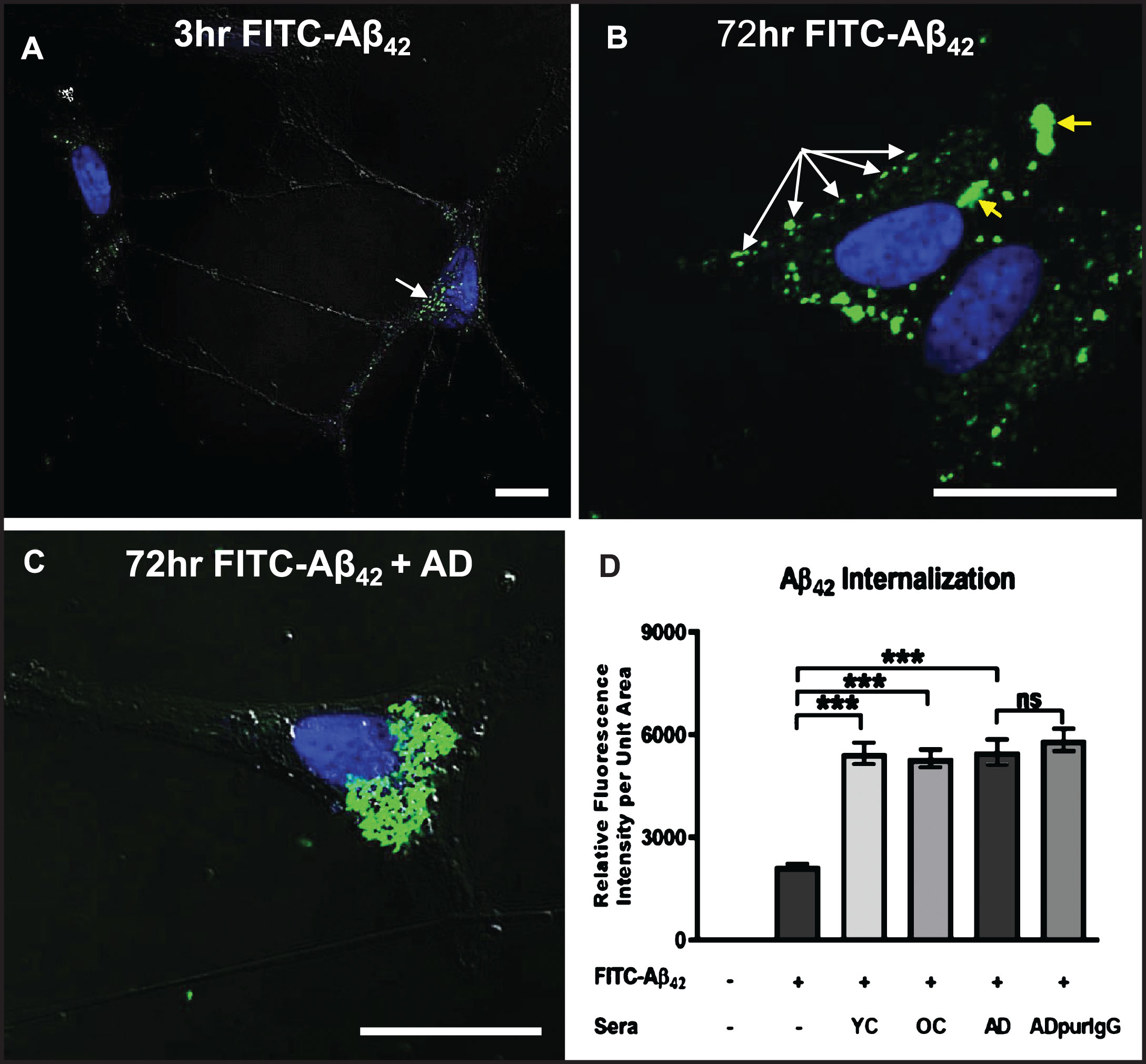
We next investigated the effects of serum autoantibodies on the rate and extent of internalization of exogenous soluble Aβ42 in differentiated SH-SY5Y cells. Exposure of cells to a physiologically relevant dose (100 nM) of FITC-labeled Aβ42 in medium without serum (controls) resulted in detection of some Aβ42 along neurites and within small, uniformly sized granules that tended to cluster preferentially on one side of the nucleus within 3 h (Fig. 4A). At 72 h, many smaller granules containing FITC-labeled Aβ42 were aggregated into larger structures in the cytoplasm, but a significant amount of FITC-labeled Aβ42 remained restricted to smaller granules located primarily along the cell perimeter, suggesting that this Aβ42 may be bound to the outside of the cell surface membrane and aggregated into “patches” that have not yet entered the cell (Fig. 4B). Addition of human AD serum (final dilution = 1 : 50) to the culture medium significantly increased the rate and extent of intracellular FITC-Aβ42 accumulation and aggregation over that of cells treated with FITC-Aβ42 alone for 72 h (Fig. 4C). As in AD brains, there was a clear tendency for internalized Aβ42 to coalesce into large aggregates in close proximity to the nucleus (cf. Fig 1A), with very little FITC-Aβ42 remaining associated with the cell surface membrane (Fig. 4C). Surprisingly, quantitative comparison of the effects of sera from an AD patient, an age-matched non-demented subject (OC), and a younger non-demented control (YC) individual revealed no significant differences in the extent of Aβ42 accumulation at 72 h (Fig. 4D). This increased internalization of FITC-Aβ42 over controls lacking serum was attributed to the IgG fraction of serum since cells treated with IgG purified from AD serum showed levels of intracellular Aβ42 accumulation comparable to cells treated with whole serum (Fig. 4D).
Fig.4
Internalization of exogenous Aβ42 is enhanced by binding of IgG autoantibodies. A, B) Differentiated SH-SY5Y cells treated with a physiologically relevant dose (100 nM) of FITC-labeled Aβ42 for 3 and 72 h in medium without human serum showing that Aβ42 internalization can occur in the absence of serum. A) At 3 h, FITC-Aβ42 was localized to small, dispersed and uniformly sized granules. B) As is typically seen in Aβ42-overburdened neurons in regions with AD pathology, a significant fraction of internalized Aβ42 was concentrated within aggregates at 72 h. C) Addition of AD serum to media containing 100 nM FITC-Aβ42 increased the rate and extent of Aβ42 internalization with very little detected on the cell surface. D) Quantification of the effects of different human sera on the rate and extent of FITC-Aβ42 internalization after 72 h of treatment. Exposure of differentiated SH-SY5Y cells to 100 nM FITC-labeled Aβ42 in media without serum (controls) resulted in detection of a relatively low level of intracellular Aβ42. Addition of human serum to the media greatly increased the rate and extent of FITC-labeled Aβ42 internalization observed at 72 h. Comparison of the effects of serum from an AD patient, an age-matched non-demented subject, and a younger non-demented control individual revealed no significant differences in the amount of Aβ42 accumulation over 72 h, and intracellular Aβ42 levels in these groups were all significantly higher than in cells treated with Aβ42 alone. In addition, IgG purified from AD serum showed comparable Aβ42 internalization rates to that of whole serum, suggesting that, among serum components, IgG is largely responsible for the observed Aβ42 internalization in SH-SY5Y cells. Scale bar = 10 μm. ***p < 0.001. AD, Alzheimer’s disease serum; OC, old-aged matched, non-demented control; YC, young-aged, non-demented control; ADpurIgG, purified IgG from AD sera; white arrow, newly internalized/plasma-membrane bound Aβ42; yellow arrow, aggregates of Aβ42-containing granules.
Internalization of exogenous Aβ42 can be enhanced by autoantibodies directed against a single abundant neuronal surface protein (NSP)
To determine if IgG autoantibodies against a single common and abundant neuronal cell surface protein can influence the rate and extent of exogenous Aβ42 internalization in neurons, we treated differentiated SH-SY5Ycells with 100 nM FITC-labeled Aβ42 for 72 h in medium containing antibody directed against the α7nAChR (diluted 1:500). Fig. 5A shows that, at 72 h, FITC-Aβ42 (green) and cy3-labeled α7nAChR (red) were co-localized as complexes (yellow) that were internalized and trafficked together intracellularly. Treatment of cells with anti-α7nAChR antibodies significantly increased Aβ42 accumulation over that of cells exposed to FITC-Aβ42 only (Fig. 5B). However, the amount of internalized FITC-Aβ42 in anti-α7nAChR-treated cells was less than that in cells treated with purified IgG obtained from AD serum (Fig. 5B). To confirm that binding of IgG autoantibodies is responsible for enhanced FITC-Aβ42 internalization in differentiated SH-SY5Ycells, depletion of IgG from AD serum resulted in a dramatic reduction of the amount of intracellular FITC-Aβ42 to levels below that of cells treated with FITC-Aβ42 only (Fig. 5B). The observation that cells treated with FITC-Aβ42 alone showed higher levels of internalized FITC-Aβ42 than those treated with the same concentration of FITC-Aβ42 in IgG-depleted serum is not unexpected. Human serum comprises of a number of both well-characterized and uncharacterized biomolecules. IgG depleted serum still contains serum components that can compete for the same binding sites on cell surfaces. Also, it has been shown that some serum components have affinity for Aβ42. By removing IgG from the serum, it enhances the ability of other serum components to compete for binding sites on the neuronal cell surface. Secondly, the removal of serum IgG may allow other serum components to form complexes with FITC-Aβ42 with varying (including higher) affinities than in their native states.
Fig.5
Aβ42 internalization is enhanced by anti-α7nAChR autoantibodies. Differentiated cell treated with 100 nM FITC-labeled (green) Aβ42 for 72 h in medium containing antibody directed against the α7nAChR and visualized using Cy3 (red) secondary antibodies. FITC-Aβ42 internalization was enhanced over that in cells treated with FITC-Aβ42 alone, and nearly all was co-localized with α7nAChR (yellow) either in small dispersed granules or uniform clusters of these granules. A) Cell treated for 72 h in medium containing both FITC-Aβ42 and serum from an AD patient showing extensive FITC-Aβ42 internalization and co-localization with α7nAChR. B) Quantification showing increased Aβ42 internalization in cells treated with anti-α7nAChR antibody over those exposed to FITC-Aβ42 alone. Cells treated with IgG purified from AD serum showed a higher level of FITC-Aβ42 internalization than those treated with anti-α7nAChR. Removal of IgG from AD serum caused a dramatic reduction in Aβ42 internalization. Scale bar = 10 μm. *p < 0.05, ***p < 0.001. AD, Alzheimer’s disease serum.
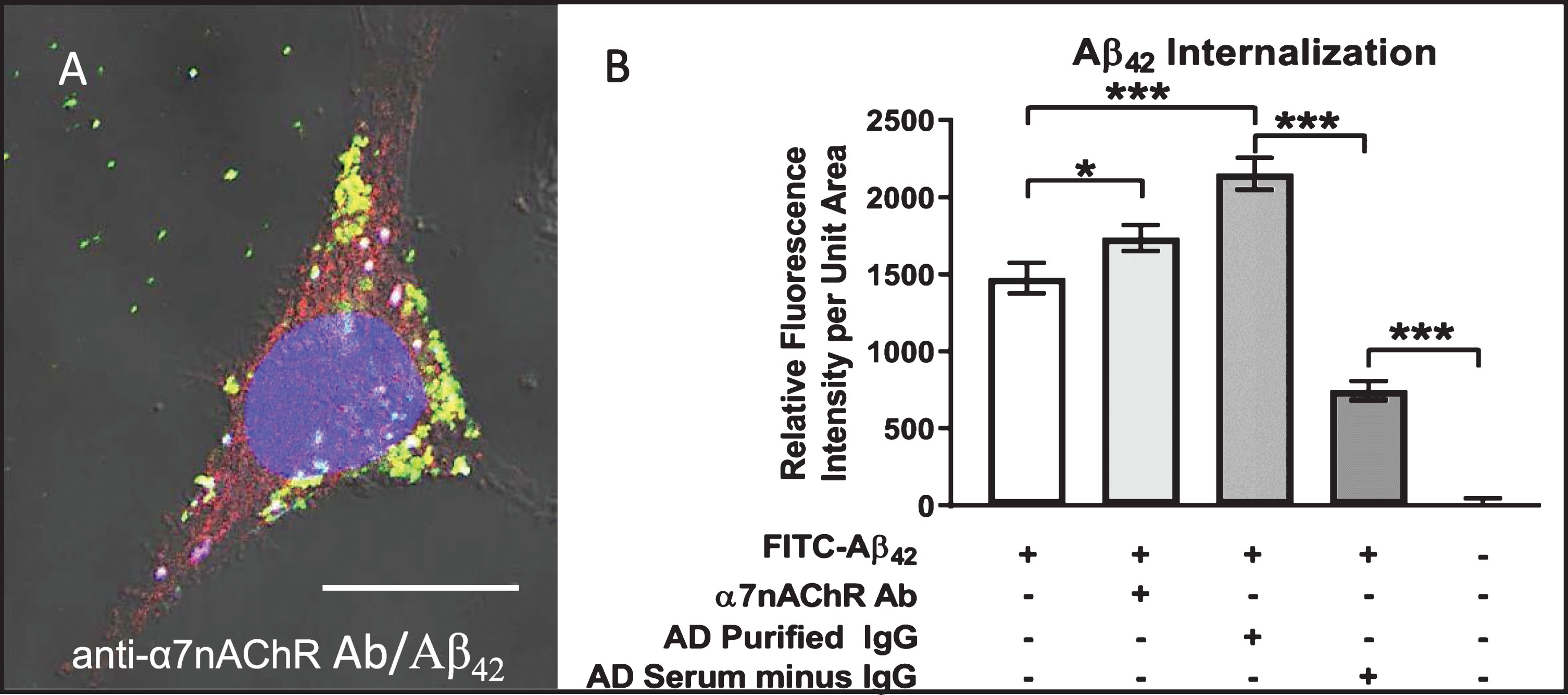
Autoantibody-mediated augmentation of Aβ42 internalization requires an antibody cross-linking step
We have proposed that extensive crosslinking of antigen targets present on neuronal cell surfaces by bivalent autoantibodies triggers endocytosis, a phenomenon that could conceivably provide much of the driving force for intraneuronal accumulation of exogenous Aβ42 as well as chronic depletion of other key proteins and receptors from cell surface membranes in vivo. To test this possibility, we compared the effects of bivalent versus monovalent antibodies on the rate and extent of Aβ42 internalization. Purified monovalent antibody [F(ab)] fragments, capable of binding but not crosslinking antigens on cell surfaces, were prepared by enzymatic cleavage of IgG antibodies isolated from human AD serum. Differentiated SH-SY5Y cells treated with medium containing 100 nM FITC-labeled Aβ42 plus monovalent F(ab) fragments showed reduced Aβ42 internalization and accumulation (Fig. 6). Addition of anti-F(ab) antibodies to medium containing monovalent F(ab) fragments rescued the cross-linking function and increased intracellular Aβ42 to levels comparable to that in cells treated with AD patient serum and IgG purified from AD patient serum (Fig. 6). Lastly, treatment of cells with heat-inactivated (HI) serum caused a modest reduction in Aβ42 internalization, suggesting that complement binding does not make a major contribution to the observed Aβ42 internalization events mediated by serum IgG (Fig. 6). This finding confirms that crosslinking of IgG antibodies bound to target antigens on neuronal cell surfaces enhances Aβ42 internalization via endocytosis.
Fig.6
Aβ42 internalization is driven primarily by IgG cross-linking on neuronal surfaces. Cells were first treated with FITC-Aβ42 to establish a baseline level of Aβ42 internalization in the absence of other factors. Aβ42 was then co-administered with AD serum, purified IgG from AD sera, heat-inactivated (HI) AD sera (to disable binding of complement protein), monovalent F(ab) IgG fragments or monovalent F(ab) fragments co-treated with anti-F(ab) fragment secondary antibody (aFab). The latter was used to restore bivalency and thereby autoantibody cross-linking. In cells treated with monovalent F(ab) fragments (incapable of cross-linking) in conjunction with FITC-Aβ42, Aβ42 internalization was reduced compared to those treated with AD serum, purified IgG from AD serum or heat-inactivated serum. However, when media containing monovalent F(ab) fragments was supplemented with antibodies directed against F(ab) fragments to restore cross-linking abilities, the Aβ42 internalization rate was increased to a level comparable to that of purified IgG from AD serum. Fab, monovalent F(ab) fragment; aFab, anti-F(ab) fragment secondary antibody; AD, Alzheimer’s disease sera; AD purIgG, IgG purified from AD sera; HI, heat-inactivated; ns, non-significant; **p < 0.01; ***p < 0.001.
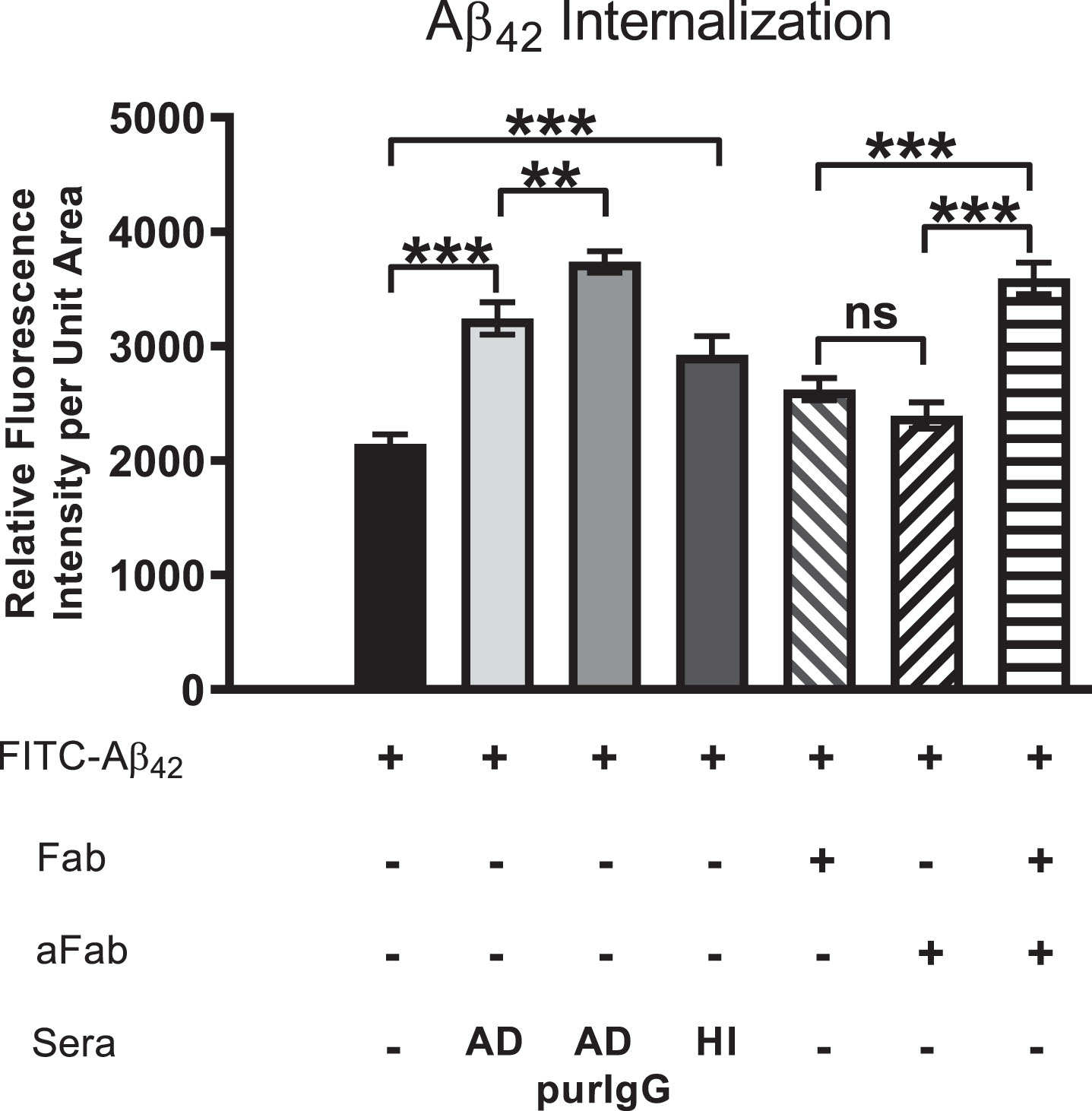
Serum-derived IgG and Aβ42 are internalized via endocytosis and target the lysosomal compartment
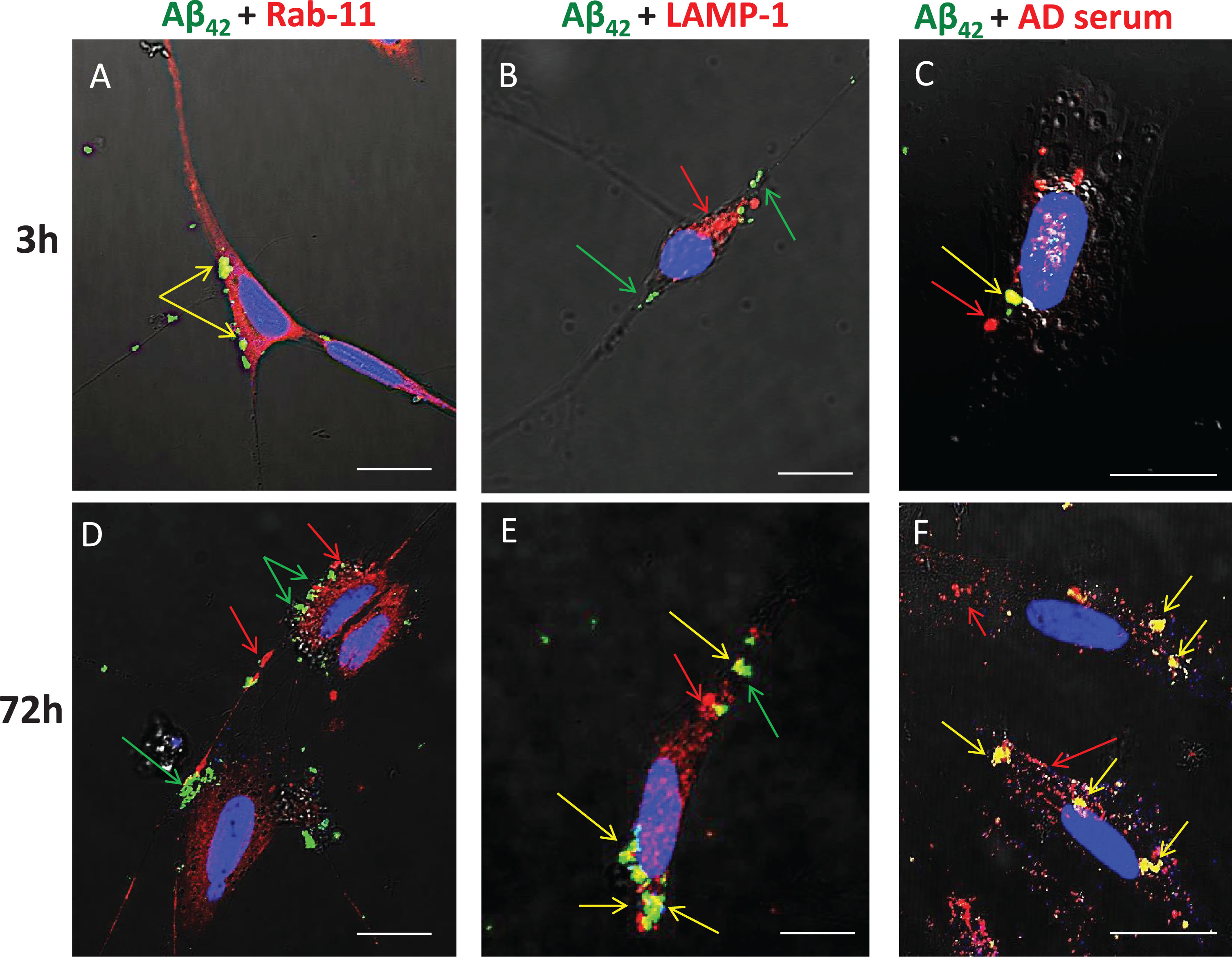
Previous studies have shown that, in human AD brains, chronic endocytosis leads to targeting and expansion of the lysosomal compartment of neurons to the point where it eventually occupies a substantial fraction of total cytoplasmic volume (cf. Fig. 1B). To examine and confirm the key steps of this process using our in vitro model system, SH-SY5Y cells were treated from 3 to 72 h with FITC-Aβ42 with or without AD serum. Immunocytochemistry was used to detect Rab-11 (an early endosome marker) and LAMP-1 (a late endosome/early lysosome marker), and co-localization of these with FITC-Aβ42 was detected by superposition of red and green signals to yield yellow fluorescence (Fig. 7). At 3-h time points, essentially all internalized FITC-Aβ42 was contained within early endosomes, as shown by its co-localization with Rab11 (Fig. 7A); none was associated with lysosomes at this early time point (Fig. 7B). In cells treated for 3 h and 72 h with FITC-Aβ42 in the presence of serum from an AD patient, co-localization of Aβ42 and IgG was evident (Fig. 7C, F), although, as expected, internalization of IgG also occurred independently of Aβ42. At 72 h, little or no co-localization of Aβ42 with Rab-11 was detected (Fig. 7D), but significant accumulations of co-localized Aβ42 and LAMP-1 were observed (Fig. 7E).
Fig.7
Aβ42 is trafficked through endosomes (Rab11) to the lysosome compartment (LAMP-1) following internalization. SH-SY5Y treated from 3 to 72 h with FITC-Aβ42 with or without AD serum. ICC was used to detect Rab-11 (early endosomes) and LAMP-1 (late endosomes/early lysosome marker). Co-localization was detected by superposition of red and green signals to yield yellow fluorescence. At 3 h, nearly all internalized FITC Aβ42 was associated with early endosomes (Rab-11). In cells treated for 3 h and 72 h with FITC Aβ42 in the presence of serum from an AD patient, co-localization of Aβ42 and IgG was evident, although some internalization of Aβ42 also occurred independently of IgG. At 72 h, little or no co-localization of Aβ42 with Rab11 was detected, but significant accumulations of co-localized Aβ42 and LAMP-1 (lysosomes) were common. Rab-11, early endosome; LAMP-1, lysosomal marker; AD, Alzheimer’s disease human serum; green arrow, Aβ42; red arrow, secondary protein of interest; yellow arrow, co-localization; **p < 0.01; ***p < 0.001; Scale bar = 10 μm.
DISCUSSION
In AD patients, Aβ42 peptide progressively accumulates in the brain and is localized preferentially in the walls of blood vessels, amyloid plaques (APs), and the perikaryon of pyramidal neurons, especially cholinergic or cholinoceptive neurons [3, 34]. The source of the Aβ42 that deposits in AD brains remains unknown and controversial. Studies on postmortem AD brain tissue and the brains of transgenic mouse models exhibiting neuronal overexpression of the amyloid-β protein precursor and other AD-relevant proteins have failed to provide a clear answer to the fundamental question of how Aβ peptides are deposited within neurons and amyloid plaques during AD pathogenesis. In the context of increased blood-brain barrier (BBB) permeability, a continual supply of exogenous, soluble, blood-borne Aβ peptides, including Aβ42, can enter into the brain tissue along with other plasma components. This Aβ42 has been shown to be capable of binding to α7nAChRs on the surfaces of cholinoceptive neurons with high affinity [12, 22]. It also binds to many other proteins (albeit with lower affinities) found elsewhere in the blood and brain. Under conditions of BBB leak, the influx of vascular Aβ42 into the brain leads to the formation of Aβ42-α7nAChR complexes on neuronal cell surfaces, which are subsequently internalized, presumably via endocytosis, and accumulate readily within the lysosomal compartment of these cells [35, 36]. Mechanisms driving internalization of Aβ42 and intraneuronal accumulation remain poorly understood.
Our previous studies on the brains of AD and age-matched, non-demented control subjects have shown that a compromised BBB also allows serum immunoglobulins (Igs), especially IgG, to enter the brain parenchyma and interact with the surfaces of neurons. Some of these IgGs can react with brain proteins expressed on the neuronal surfaces (neuron-binding or brain-reactive autoantibodies). The pathophysiological consequences of this binding are largely unknown [3, 9, 21, 22]. In the present study, we investigated this process using differentiated SH-SY5Y human neuroblastoma cells which, like many neurons in the brain, express α7nAChRs on their surfaces. These cells were used to test the hypothesis that, under conditions of BBB compromise where plasma components including antibodies and soluble Aβ42 chronically leak into the brain parenchyma, brain-reactive autoantibodies common in the blood can bind selectively to exposed cell surfaces of neurons that express their cognate antigen targets. Further, we sought to test whether autoantibody-mediated cross-linking of surface antigens, via induced endocytosis, drives internalization of cell surface-bound Aβ42-α7nAChR complexes and thereby facilitates gradual accumulation of Aβ42 in the neuron lysosomal compartment.
The results of the present study highlight the potentially important role of blood-borne, brain-reactive autoantibodies in the pathogenesis of AD. This study reports the following major findings. First, exposure of differentiated SH-SY5Y neuroblastoma cells, which express the α7nAChR, to exogenous, soluble Aβ42 and human serum revealed that the rate and extent of Aβ42 internalization and accumulation in these cells were increased in the presence of serum, regardless of age or whether the serum was from an AD or non-demented (control) subject. Second, this effect was mediated by IgG autoantibodies present in serum, since depletion of IgG from serum reduced the rate and extent of intraneuronal Aβ42 accumulation, whereas treatment with purified IgG enhanced it to levels comparable to that of whole serum. Third, we show that treatment of cells with autoantibodies directed against a single, well-known, abundant cell surface receptor (e.g., the α7nAChR, which is also capable of high-affinity binding to Aβ42) increases the rate and extent of neuronal internalization of exogenous soluble Aβ42. Fourth, both Aβ42 and autoantibody internalization were reduced by treatment of cells with monovalent F(ab) antibody fragments generated from IgG purified from AD patient serum, and was rescued by crosslinking these F(ab) fragments with anti-F(ab) antibodies. These observations suggest that cross-linking of bivalent, neuron-binding autoantibodies accelerates intraneuronal Aβ42 deposition, presumably via endocytosis. Finally, we show that trafficking of internalized Aβ42 appears to be similar to that in AD brains, with initial co-localization of IgG, α7nAChR, and Aβ42 in differentiated SH-SY5Y cells temporally and spatially related to the early endosome marker protein, Rab11, and at later time points to the lysosomal marker, LAMP-1.
Internalization and accumulation of exogenous Aβ42 is enhanced by brain-reactive autoantibodies commonly found in the blood
Recent studies using human protein microarrays have revealed the presence of thousands of different autoantibodies in human sera, irrespective of disease status, suggesting a homeostatic role [26, 37]. Not surprisingly, these studies have revealed that many of these autoantibodies show cross-species reactivity, suggesting affinity for similar, conserved epitopes as we have seen in immunohistochemical preparations of human, rodent and pig brain [23, 31, 33]. The fact that certain specific autoantibodies show increased production in response to the presence of disease, however, favors the idea that one important function of these autoantibodies is the removal of the debris that has entered into the general circulation. This property has led us to explore the potential of some autoantibodies as blood-based biomarkers for use in detecting specific diseases [26, 28–30, 38]. A limitation of the present study is that only a single serum sample was selected for use from each group. In our previous studies, over 100 AD and healthy controls from various ages were analyzed for autoantibody profiles using protein microarrays. This work revealed that the presence of many thousands of IgG autoantibodies in the blood is ubiquitous. Here, as a proof of concept, we chose a representative serum sample from each demographic group to test the potential pathogenic role of autoantibodies in intracellular deposition of Aβ42 peptide. It is well known that, in brain regions exhibiting AD-related pathological changes, loss of synapses and death of the associated neurons is pervasive [39, 40]. This kind of neurodegeneration results in a continual release of neuronal debris into the brain interstitial space, which subsequently makes its way into the cerebrospinal fluid and, finally, the peripheral circulation. We believe that the presence of this debris in the peripheral circulation triggers increased production of debris-specific autoantibodies, including those directed against key, abundant cell surface targets that are critical for proper neuronal function [41]. Further, we have proposed that, in the context of BBB compromise, some autoantibodies with cognate targets on neuronal surfaces can bind to these surfaces and elicit a sequence of events that contributes to AD pathology and plays a role in neuronal Aβ42 deposition and perhaps the downstream formation of amyloid plaques.
In the present study, we used differentiated SH-SY5Y neuroblastoma cells as a model system to test the above scenario. Surprisingly, IgG autoantibodies from AD serum showed a similar level of binding to cells as did serum from a non-demented, age-matched control subject and a younger subject. This finding, in parallel with our previous studies, raises the possibility that all human sera contain brain-reactive (including neuron-binding) autoantibodies, regardless of age or the presence of disease, and that these autoantibodies are present in the general circulation prior to initiation of AD-related pathology. Our previous studies that have used human protein microarrays to examine autoantibody profiles across a wide range of ages support this idea and have shown that complex autoantibody profiles that include thousands of autoantibodies are commonly (perhaps ubiquitously) expressed in the blood in individuals of all ages [26]. It also suggests that increased BBB permeability may be an important disease trigger that elicits a cascade of events beginning with the binding of autoantibodies to their neuronal cell surface targets. We also found that binding of serum autoantibodies to SH-SY5Y cells increased the rate and extent of internalization of exogenous soluble Aβ42 added to the culture medium in a physiologically relevant dose (100 nM), as determined previously [3]. Aβ42 was initially observed within small, uniformly sized granules in neurites and in the perinuclear cytoplasm, but these later were concentrated within larger juxtanuclear clusters. Interestingly, as was seen for IgG binding, quantitative comparison of the effects of sera from an AD patient, an age-matched non-demented subject, and a younger non-demented control individual revealed no significant differences in the extent of Aβ42 accumulation at 72 h. This finding suggests that neuron-binding autoantibodies are present in the blood regardless of the presence or absence of disease. Furthermore, this effect was largely attributed to the IgG fraction of the blood since cells treated with IgG purified from AD serum showed levels of intracellular Aβ42 accumulation comparable to the cells treated with whole serum, and depletion of IgG from serum markedly reduced Aβ42 accumulation. It was interesting to see that the amount of Aβ42 accumulation was higher in cells treated with FITC-Aβ42 alone than in cells treated with the same concentration of FITC-Aβ42 plus serum that was depleted of IgG. We attribute this reduction in FITC-Aβ42 internalization in the group treated with IgG-depleted AD serum to the fact that FITC-Aβ42 has to compete with an increased number of non-specific, transitory cell surface binding events due to the higher level of serum proteins and other components which are absent in the FITC-Aβ42 only group.
Neuronal internalization of exogenous Aβ42 is enhanced by autoantibodies directed against a single abundant neuronal surface protein
Previous studies have suggested that Aβ42 binds to α7nAChR with picomolar affinity [42] and that this binding on the surfaces of α7nAChR-expressing neurons in AD brains leads to internalization of the resulting Aβ42-α7nAChR complexes and their accumulation within the lysosomal compartment as confirmed using immunohistochemistry [9, 12, 13]. Here, we show that IgG autoantibodies present in the blood can augment this process. The selectivity of IgG autoantibodies for certain types of neurons, especially pyramidal neurons, is evident and reflected by the differential pattern of immunoreactivity amongst neurons with virtually identical exposures to the same nearby leaking vessel. For example, immunohistochemistry frequently shows intensely immunoreactive neurons interspersed among completely immuno-negative neurons in the same sections. This observation, which has been documented in human, rodent, and pig brains as well as in organotypic brain slice cultures derived from adult mice, is difficult to explain based on a Fc-mediated IgG binding mechanism via Fc receptors [23, 31, 33]. Similarly, our observations on the rescue of crosslinking of Fab fragments with anti-Fab antibodies also argues against an Fc-receptor binding mechanism. In this case, Fc fragments of the anti-Fab antibodies bound to Fab fragments would be expected to be oriented away from the plasma membrane of these cells.
In the present study, we also tested the possibility that an IgG autoantibody directed against a single abundant neuronal surface protein, in this case the α7nAChR, can influence the rate and extent of exogenous Aβ42 internalization in neurons. Results showed that treatment of cells with anti-α7nAChR antibodies significantly increased Aβ42 accumulation over that of cells exposed to Aβ42 alone. In addition, Aβ42 and α7nAChR were internalized and trafficked together to the lysosomal compartment as in neurons of AD brains. This shows that autoantibodies directed against a single, relatively abundant cell surface protein are capable of driving Aβ42 accumulation, with the expectation that the potency of this effect would be dependent on the titer of the specific neuron-binding autoantibody as well as the relative abundance of the receptor or target protein on the cell surface. Of course, the presence of multiple autoantibodies to cell surface proteins would, accordingly, be expected to magnify this effect, thereby increasing the rate of Aβ42 deposition as well as precipitating synaptic and neuronal loss.
Our results also provide explanation for selective vulnerability of cholinergic and cholinoceptive neurons to Aβ42 deposition in AD brains. The strong binding affinity of Aβ42 to α7nAChR in neurons that express this receptor may selectively enhance Aβ42 accumulation in these cells, which helps to explain why cholinergic tone is diminished in the early phases of AD and, similarly, why anticholinesterases provide modest and short-lived benefits only at this mild stage. The accumulation of Aβ42 may trigger various neurodegenerative changes, including interfering with the ability of neurons to support their extensive dendrite trees and axonal projections, and may ultimately contribute to more widespread neuronal loss and aberrant circuitry and connectivity patterns as the disease progresses. Of course, many different autoantibodies may be able to bind to neurons with and without α7nAChRs. However, the reduced affinity of Aβ42 for the surfaces of cells lacking this receptor suggests that these cells would experience little or no Aβ42 accumulation at this concentration compared to cells that express α7nAChR. It is reasonable to suggest, however, that at later stages of the disease, when amyloid-burdened cells and BBB damage are more rampant and widespread, Aβ42 levels are high enough to accumulate appreciably in other cell types throughout the brain. Moreover, we would like to emphasize that the chronic interaction between other autoantibodies and neuronal surface proteins may initiate a more general loss of these proteins due to receptor-mediated endocytosis, a loss that could lead to cognitive changes as a cumulative result of neuronal functional deficits that expand into networks that span cortical and subcortical regions.
Autoantibody-mediated augmentation of Aβ42 internalization requires an antibody cross-linking step
We have proposed that extensive cross-linking of antigen targets present on neuronal surfaces by bivalent brain-reactive autoantibodies triggers endocytosis and internalization of autoantibody-target protein complexes. Chronic receptor stripping via endocytosis could contribute to intraneuronal accumulation of exogenous Aβ42 and depletion of key proteins and receptors from cell surface membranes. To test this, we compared the effects of bivalent and purified monovalent autoantibodies, both derived from human serum, on the rate and extent of Aβ42 internalization. We found that differentiated SH-SY5Y cells treated with monovalent F(ab) fragments exhibited reduced exogenous Aβ42 internalization and accumulation. We were able to rescue the native cross-linking function by addition of anti-F(ab) antibodies to medium containing monovalent F(ab) fragments, which restored the rate and extent of intracellular Aβ42 accumulation to levels comparable to that of cells treated with AD patient serum and IgG purified from AD patient serum. This phenomenon has been linked to the pathophysiology of other antibody-mediated neuropsychiatric disorders, including N-methyl D-aspartate receptor encephalitis [43]. Although IgG was the only antibody species under investigation in the present study, there is a growing literature to suggest that other less abundant isotypes, including IgM and IgA, may also share a role in the pathophysiology in question, which is an active area of research across neuropsychiatry and psychoneuroimmunology. IgG may not be unique in its ability to induce cross-linking and Aβ42 internalization and, with future studies, it may be revealed that all autoantibodies are capable of similarly contributing to the amyloidogenic burden of neurons. Furthermore, we propose that this mechanism of neurodegeneration, initiated by increased local BBB permeability, may be applicable to other parts of the brain, with the type of symptoms dictated by the brain region and connectivity affected [43, 44]. It is important to note, however, that other mechanisms of cellular uptake are likely at play in Aβ42 internalization, including caveolin-, clathrin-, and phagocytosis-mediated mechanisms, which were not explored in the present study.
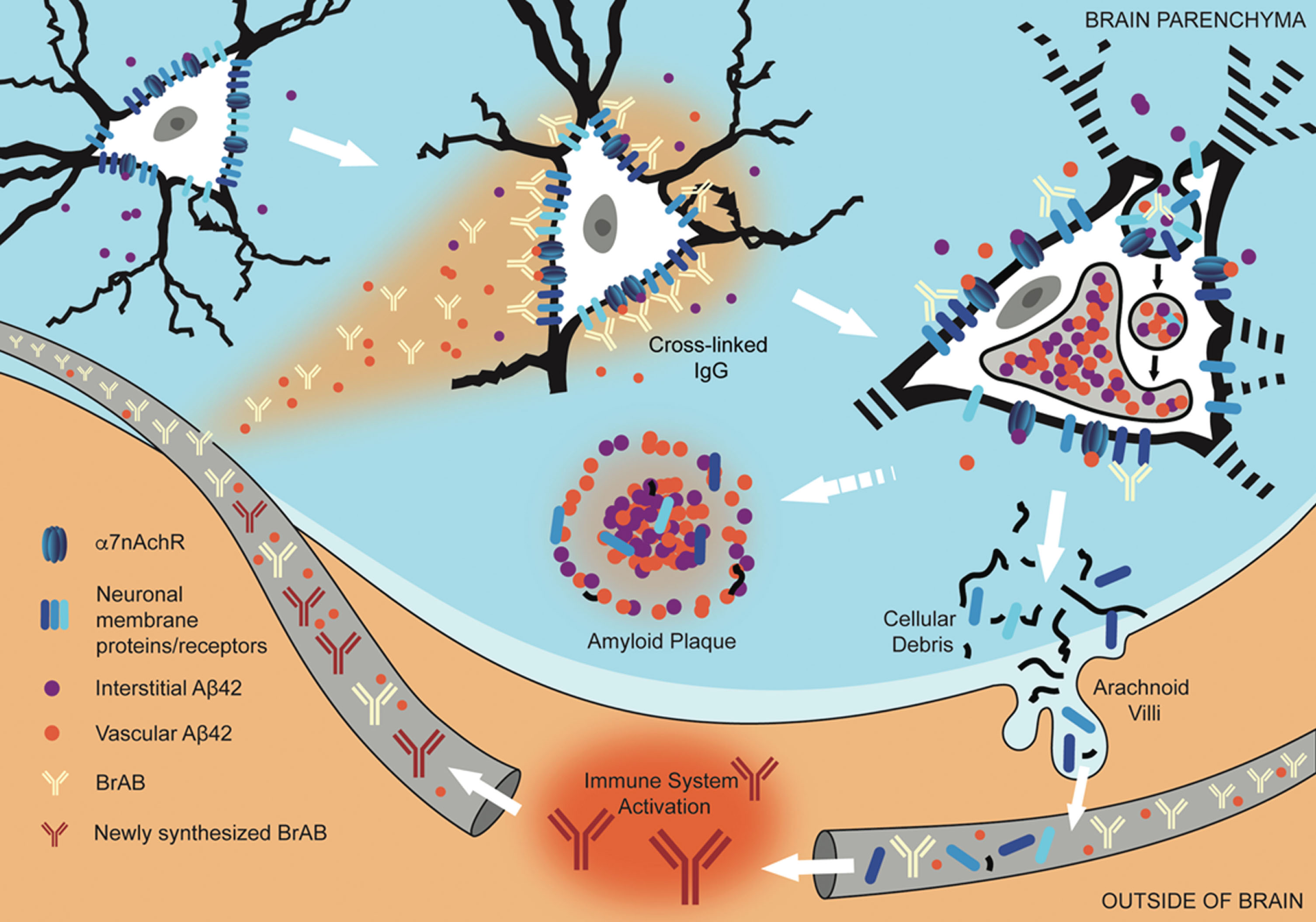
Fig.8
Interaction of extravasated BrABs with NSPs triggers endocytosis and results in intraneuronal accumulation of Aβ42 and depletion of NSPs from cell surfaces. Under conditions of BBB compromise, blood-borne Aβ42 and brain-reactive autoantibodies flood into the brain parenchyma. Some interact with cognate neuronal surface proteins (including receptors; NSPs), while Aβ42 binds to α7nAChRs with picomolar affinity. Bivalent autoantibodies cross-link their targets on neuronal surfaces, which can trigger endocytosis and internalization of cross-linked NSPs and surface-bound Aβ42-α7nAChR complexes. Within the acidic environment of endosomes and lysosomes, Aβ42 forms aggregates which are non-degradable and tend to accumulate. Over time, chronic leak of autoantibodies into the brain tissue results in continual internalization of Aβ42 within neurons, which interferes with their proper functioning and leads to failure to maintain their dendritic trees and associated synapses. Death and lysis of these Aβ42-overburdened neurons contributes to the formation of amyloid plaques (APs). This cell death is also a source of neuronal debris that finds its way into cerebrospinal fluid and, via arachnoid villi, into the general circulation, where it triggers an immune response leading to production of autoantibodies directed against such neuronal debris. Under conditions of increased BBB permeability, these are able to access cell surface-bound targets in the brain and thus perpetuate the engine that drives the observed pathology.
Conclusion
In conclusion, these results are consistent with the notion that, in the context of increased BBB permeability, chronic binding of brain-reactive autoantibodies to abundant cell surface protein targets and the resulting antibody-mediated endocytosis may be a major pathway for early pathogenic neuronal deposition of soluble Aβ42 originating from the blood in AD brains. This mechanism could also account for the depletion of key cell surface proteins leading to the observed neuronal dysfunction and loss of synapses associated with early AD-related neuropathology.
ACKNOWLEDGMENTS
The authors wish to express their gratitude to Natasha L. Hesketh, B.S., RowanSOM, GSBS, for lending her artistic talent in the production of Fig. 8.
The authors wish to thank the Osteopathic Heritage Foundation and the Dean’s endowment for primary care research for support of this project.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0962r3).
REFERENCES
[1] | Alzheimer’s Association ((2011) ) 2011 Alzheimer’s disease facts and figures. Alzheimers Dement 7: , 208–244. |
[2] | Dickson DW ((1997) ) Neuropathological diagnosis of Alzheimer’s disease: A perspective from longitudinal clinicopathological studies. Neurobiol Aging 18: , S21–26. |
[3] | Clifford PM , Zarrabi S , Siu G , Kinsler KJ , Kosciuk MC , Venkataraman V , D’Andrea MR , Dinsmore S , Nagele RG ((2007) ) Abeta peptides can enter the brain through a defective blood-brain barrier and bind selectively to neurons. Brain Res 1142: , 223–236. |
[4] | Di Domenico F , Sultana R , Barone E , Perluigi M , Cini C , Mancuso C , Cai J , Pierce WM , Butterfield DA ((2011) ) Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer’s disease subjects. J Proteomics 74: , 1091–1103. |
[5] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging and the Alzheimer’s Association workgroup. Alzheimers Dement 7: , 280–292. |
[6] | Morris JC ((2005) ) Early-stage and preclinical Alzheimer disease. Alzheimer Dis Assoc Disord 19: , 163–165. |
[7] | Omtri RS , Thompson KJ , Tang X , Gali CC , Panzenboeck U , Davidson MW , Kalari KR , Kandimalla KK ((2018) ) Differential effects of Alzheimer’s disease Abeta40 and 42 on endocytosis and intraneuronal trafficking. Neuroscience 373: , 159–168. |
[8] | Wesen E , Jeffries GDM , Matson Dzebo M , Esbjorner EK ((2017) ) Endocytic uptake of monomeric amyloid-beta peptides is clathrin- and dynamin-independent and results in selective accumulation of Abeta(1-42) compared to Abeta(1-40). Sci Rep 7: , 2021. |
[9] | D’Andrea MR , Nagele RG , Wang H-Y , Peterson PA , Lee DH ((2001) ) Evidence that neurons accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer’s disease. Histopathology 38: , 120–134. |
[10] | Hu X , Crick SL , Bu G , Frieden C , Pappu RV , Lee J-M ((2009) ) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc Natl Acad Sci U S A 106: , 20324. |
[11] | Hung SY , Huang WP , Liou HC , Fu WM ((2015) ) LC3 overexpression reduces Abeta neurotoxicity through increasing alpha7nAchR expression and autophagic activity in neurons and mice. Neuropharmacology 93: , 243–251. |
[12] | Nagele RG , D’Andrea MR , Anderson WJ , Wang HY ((2002) ) Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110: , 199–211. |
[13] | Gouras GK , Almeida CG , Takahashi RH ((2005) ) Intraneuronal Abeta accumulation and origin of plaques in Alzheimer’s disease. Neurobiol Aging 26: , 1235–1244. |
[14] | Wirths O ((2017) ) Altered neurogenesis in mouse models of Alzheimer disease. Neurogenesis (Austin) 4: , e1327002. |
[15] | Medeiros R , Castello NA , Cheng D , Kitazawa M , Baglietto-Vargas D , Green KN , Esbenshade TA , Bitner RS , Decker MW , LaFerla FM ((2014) ) alpha7 Nicotinic receptor agonist enhances cognition in aged 3xTg-AD mice with robust plaques and tangles. Am J Pathol 184: , 520–529. |
[16] | Oakley H , Cole SL , Logan S , Maus E , Shao P , Craft J , Guillozet-Bongaarts A , Ohno M , Disterhoft J , Van Eldik L , Berry R , Vassar R ((2006) ) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation. J Neurosci 26: , 10129–10140. |
[17] | Umeda T , Tomiyama T , Kitajima E , Idomoto T , Nomura S , Lambert MP , Klein WL , Mori H ((2012) ) Hypercholesterolemia accelerates intraneuronal accumulation of Abeta oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci 91: , 1169–1176. |
[18] | Friedrich RP , Tepper K , Rönicke R , Soom M , Westermann M , Reymann K , Kaether C , Fändrich M ((2010) ) Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc Natl Acad Sci U S A 107: , 1942–1947. |
[19] | Gouras GK , Tsai J , Naslund J , Vincent B , Edgar M , Checler F , Greenfield JP , Haroutunian V , Buxbaum JD , Xu H , Greengard P , Relkin NR ((2000) ) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: , 15–20. |
[20] | Wang HY , Stucky A , Liu J , Shen C , Trocme-Thibierge C , Morain P ((2009) ) Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s disease brain. J Neurosci 29: , 10961–10973. |
[21] | Wang HY , Lee DH , D’Andrea MR , Peterson PA , Shank RP , Reitz AB ((2000) ) beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology. J Biol Chem 275: , 5626–5632. |
[22] | D’Andrea MR , Nagele RG , Wang HY , Lee DH ((2002) ) Consistent immunohistochemical detection of intracellular beta-amyloid42 in pyramidal neurons of Alzheimer’s disease entorhinal cortex. Neurosci Lett 333: , 163–166. |
[23] | Nagele RG , Clifford PM , Siu G , Levin EC , Acharya NK , Han M , Kosciuk MC , Venkataraman V , Zavareh S , Zarrabi S , Kinsler K , Thaker NG , Nagele EP , Dash J , Wang HY , Levitas A ((2011) ) Brain-reactive autoantibodies prevalent in human sera increase intraneuronal amyloid-beta(1-42) deposition. J Alzheimers Dis 25: , 605–622. |
[24] | Sedeyn JC , Wu H , Hobbs RD , Levin EC , Nagele RG , Venkataraman V ((2015) ) Histamine induces Alzheimer’s disease-like blood brain barrier breach and local cellular responses in mouse brain organotypic cultures. Biomed Res Int 2015: , 937148. |
[25] | Hyman BT , Trojanowski JQ ((1997) ) Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol 56: , 1095–1097. |
[26] | Nagele EP , Han M , Acharya NK , DeMarshall C , Kosciuk MC , Nagele RG ((2013) ) Natural IgG autoantibodies are abundant and ubiquitous in human sera, and their number is influenced by age, gender, and disease. PLoS One 8: , e60726. |
[27] | DeMarshall C , Goldwaser EL , Sarkar A , Godsey GA , Acharya NK , Thayasivam U , Belinka BA , Nagele RG ((2017) ) Autoantibodies as diagnostic biomarkers for the detection and subtyping of multiple sclerosis. J Neuroimmunol 309: , 51–57. |
[28] | DeMarshall CA , Nagele EP , Sarkar A , Acharya NK , Godsey G , Goldwaser EL , Kosciuk M , Thayasivam U , Han M , Belinka B , Nagele RG ((2016) ) Detection of Alzheimer’s disease at mild cognitive impairment and disease progression using autoantibodies as blood-based biomarkers. Alzheimers Dement (Amst) 3: , 51–62. |
[29] | Han M , Nagele E , DeMarshall C , Acharya N , Nagele R ((2012) ) Diagnosis of Parkinson’s disease based on disease-specific autoantibody profiles in human sera. PLoS One 7: , e32383. |
[30] | Nagele E , Han M , Demarshall C , Belinka B , Nagele R ((2011) ) Diagnosis of Alzheimer’s disease based on disease-specific autoantibody profiles in human sera. PLoS One 6: , e23112. |
[31] | Levin EC , Acharya NK , Han M , Zavareh SB , Sedeyn JC , Venkataraman V , Nagele RG ((2010) ) Brain-reactive autoantibodies are nearly ubiquitous in human sera and may be linked to pathology in the context of blood-brain barrier breakdown. Brain Res 1345: , 221–232. |
[32] | Buckley K , Kelly RB ((1985) ) Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol 100: , 1284–1294. |
[33] | Acharya NK , Levin EC , Clifford PM , Han M , Tourtellotte R , Chamberlain D , Pollaro M , Coretti NJ , Kosciuk MC , Nagele EP , Demarshall C , Freeman T , Shi Y , Guan C , Macphee CH , Wilensky RL , Nagele RG ((2013) ) Diabetes and hypercholesterolemia increase blood-brain barrier permeability and brain amyloid deposition: Beneficial effects of the LpPLA2 inhibitor darapladib. J Alzheimers Dis 35: , 179–198. |
[34] | Baker-Nigh A , Vahedi S , Davis EG , Weintraub S , Bigio EH , Klein WL , Geula C ((2015) ) Neuronal amyloid-beta accumulation within cholinergic basal forebrain in ageing and Alzheimer’s disease. Brain 138: , 1722–1737. |
[35] | Lana E , Khanbolouki M , Degavre C , Samuelsson EB , Akesson E , Winblad B , Alici E , Lithner CU , Behbahani H ((2017) ) Perforin promotes amyloid beta internalisation in neurons. Mol Neurobiol 54: , 874–887. |
[36] | Lorenzen A , Samosh J , Vandewark K , Anborgh PH , Seah C , Magalhaes AC , Cregan SP , Ferguson SS , Pasternak SH ((2010) ) Rapid and direct transport of cell surface APP to the lysosome defines a novel selective pathway. Mol Brain 3: , 11. |
[37] | Dahm L , Ott C , Steiner J , Stepniak B , Teegen B , Saschenbrecker S , Hammer C , Borowski K , Begemann M , Lemke S , Rentzsch K , Probst C , Martens H , Wienands J , Spalletta G , Weissenborn K , Stocker W , Ehrenreich H ((2014) ) Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol 76: , 82–94. |
[38] | DeMarshall CA , Han M , Nagele EP , Sarkar A , Acharya NK , Godsey G , Goldwaser EL , Kosciuk M , Thayasivam U , Belinka B , Nagele RG ((2015) ) Potential utility of autoantibodies as blood-based biomarkers for early detection and diagnosis of Parkinson’s disease. Immunol Lett 168: , 80–88. |
[39] | Hamos JE , DeGennaro LJ , Drachman DA ((1989) ) Synaptic loss in Alzheimer’s disease and other dementias. Neurology 39: , 355–361. |
[40] | Bertoni-Freddari C , Fattoretti P , Solazzi M , Giorgetti B , Di Stefano G , Casoli T , Meier-Ruge W ((2003) ) Neuronal death versus synaptic pathology in Alzheimer’s disease. Ann N Y Acad Sci 1010: , 635–638. |
[41] | Snyder HM , Carrillo MC , Grodstein F , Henriksen K , Jeromin A , Lovestone S , Mielke MM , O’Bryant S , Sarasa M , Sjogren M , Soares H , Teeling J , Trushina E , Ward M , West T , Bain LJ , Shineman DW , Weiner M , Fillit HM ((2014) ) Developing novel blood-based biomarkers for Alzheimer’s disease. Alzheimers Dement 10: , 109–114. |
[42] | Wang HY , Lee DHS , Davis CB , Shank RP ((2000) ) Amyloid peptide 1-42 binds selectively and with picomolar affinity to a7 nicotinic acetylcholine receptors. J Neurochem 75: , 1155–1161. |
[43] | Moscato EH , Peng X , Jain A , Parsons TD , Dalmau J , Balice-Gordon RJ ((2014) ) Acute mechanisms underlying antibody effects in anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol 76: , 108–119. |
[44] | Goldwaser EL , Acharya NK , Nagele RG ((2015) ) Cerebrovascular and blood-brain barrier compromise: A mechanistic link between vascular disease and Alzheimer’s disease subtypes of neurocognitive disorders. J Parkinsons Dis Alzheimer Dis 2: , 10. |


