
Family History is Associated with Phenotype in Dementia with Lewy Bodies
Abstract
It is currently unknown whether patients with dementia with Lewy bodies (DLB) with relatives with dementia or Parkinson’s disease (familial DLB patients) have a different phenotype than sporadic DLB patients. In this study, we aimed to examine disease onset, rate of cognitive decline, survival, and Alzheimer’s disease (AD) biomarkers in patients with familial DLB (n = 154) and sporadic DLB (n = 137), using linear mixed model analysis and Cox regression analysis, among others. Familial patients had a shorter survival (8.0 years) and more often elevated cerebrospinal fluid AD biomarkers (47%) than sporadic patients (9.0 years; p≤0.001; 30%, p = 0.037). Our findings suggest that genetic factors are important in DLB and that the identification of new genetic factors will probably improve the prediction of prognosis.
INTRODUCTION
Dementia with Lewy bodies (DLB) is one of the most common forms of degenerative dementia in the older population [1]. DLB is diagnosed when dementia is accompanied by at least two of the following four core clinical features: parkinsonism, visual hallucinations, fluctuating cognition, and rapid eye movement (REM) sleep behavior disorders (RBD). DLB can also be diagnosed based on dementia with one core clinical feature, in the presence of reduced dopamine transporter uptake in the basal ganglia, abnormal 123iodine-MIBG myocardial scintigraphy, or polysomnographic confirmation of RBD [2]. Symptoms of DLB are not specific to the disease, but overlap with clinical features of Parkinson’s disease (PD), PD dementia (PDD), and Alzheimer’s disease (AD) [1]. The distinction between DLB and PDD is most challenging, and based on differences in time between onset of dementia and parkinsonism. In PDD, dementia occurs in the context of well-established PD as opposed to DLB, in which dementia occurs before or concurrently with parkinsonism [2]. In addition, pathological and genetic features are also shared between DLB, PD(D), and AD [1, 3, 4]. For example, Lewy bodies containing the α-synuclein protein are the pathological hallmark of DLB, but are also observed in PD(D) [1], and genetic factors, such as the APOE ɛ4 allele and GBA variants, are risk factors for DLB as well as for AD and PD(D), respectively [3, 4]. However, genetic risk factors for AD and PD seem to explain a part of the total phenotypic variance in DLB only [5–7].
Recent studies have indicated that genetic factors play an important role in DLB. The heritable component of DLB has even been estimated at 60% [7]. Families with multiple DLB patients have rarely been described [3, 8]. However, it has been reported that siblings of DLB patients are at higher risk of developing DLB compared to siblings of AD patients [9]. Furthermore, DLB patients more often have a family history of PD or dementia than controls [10, 11]. This finding supports the notion that DLB, PD and dementia share, at least partially, the same genetic factors. This in turn might lead to shared molecular pathways and possibly similar phenotypes.
The APOE ɛ4 allele has been associated with a shorter survival in DLB [12–14] and disease-associated genetic variants in GBA have been associated with an earlier age of onset and death in DLB [15–17]. However, it is currently unknown whether DLB patients with relatives with dementia or PD (familial DLB) have a different phenotype than sporadic DLB patients. The main aim of this study is to examine the role of family history, used as a proxy of genetic factors, in relation to disease onset, rate of cognitive decline, survival, and AD biomarkers. The secondary aim of this study is to explore the aforementioned features in DLB patients with relatives with dementia or PD to examine if their phenotype is more similar to AD or PD.
MATERIALS AND METHODS
Patients and study design
This is a retrospective study in which demographic and clinical data were collected from patients’ medical records. Information on the occurrence of dementia and/or PD in first degree relatives was based on medical records (41%) or a standardized assessment (59%), using a questionnaire or an additional patient/family member interview. A nation-wide registration system containing demographic data about all Dutch citizens was consulted to obtain information about dates of death (collected until December 2018).
A total of 291 patients with probable DLB according to the criteria of Mckeith et al. [18] were enrolled from three hospitals in the Netherlands (Elisabeth-TweeSteden Hospital, Tilburg; Erasmus Medical Center, Rotterdam; Amsterdam University Medical Center, Amsterdam). Patients visited the outpatient clinics of the Neurology departments between 2000 and 2018 and were diagnosed by expert neurologists. Dopamine transporter uptake scans were performed in 128 patients, and were used in the diagnostic process. Familial patients were defined as patients with at least one first degree relative with dementia or PD. Sporadic patients were defined as patients without first degree relatives with dementia or PD. Patients were excluded from the study when no dementia or PD was diagnosed in relatives together with no information on the occurrence of the other disease in relatives (e.g. no relatives with PD and no information of the occurrence of dementia, or vice versa), and when no information on family history was available at all. The distribution of the different groups according to family history is depicted in Supplementary Figure 1.
The study was performed according to the ethical principles of the Declaration of Helsinki and was approved by the local ethics committees (Elisabeth-TweeSteden Hospital: MEC-2016-608, L0318.2016; Erasmus Medical Center: MEC-2015-304, MEC-2016-608; Amsterdam University Medical Center: MEC-2016-061, MEC-2017-2116).
Outcome measures
Several clinical features and AD biomarkers (i.e. the presence of medial temporal lobe atrophy (MTA) [19], a CSF tau/amyloid-β1 - 42 (Aβ42) ratio of >0.52 [20], and ≥1 APOE ɛ4 allele(s) [21]), were explored with respect to family history in DLB.
Age of onset and type of first symptom were based on anamnestic information from the patient or family members. We categorized the type of first symptom into cognitive decline, parkinsonism, and hallucinations. Cognitive decline included descriptions of memory impairment and executive function impairment. Parkinsonism was based on bradykinesia with muscular rigidity, rest tremor, or postural instability [22] or when parkinsonism was noted in the medical files in absence of more specific information. Rate of cognitive decline was based on the available Mini-Mental State Examination (MMSE) scores [23]. Survival was defined as the time between age of onset and death. Magnetic resonance imaging (MRI) scans were evaluated visually and rated according to the MTA scale [19] by radiologists for clinical purposes. CSF was collected by lumbar puncture in polypropylene tubes (Starstedt, Nümbrecht, Germany). Levels of Aβ42, total tau, and p-tau were measured with commercially ELISAs (Innotest®, Fujirebio, Gent, Belgium). APOE genotyping was performed using the LightCycler APOE mutation detection method (Roche Diagnostics GmbH, Mannheim, Germany) after genomic DNA extraction.
Data analysis
Differences in sex, age of onset, type of first symptom, and APOE genotype between familial and sporadic DLB were explored using the χ2 test, Fisher’s exact test, Fisher-Freeman-Halton test, independent Student t test, or Mann-Whitney U test, where appropriate. We analyzed the MTA score and CSF tau/Aβ42 ratio of >0.52 (CSF AD biomarkers) between the groups using linear and logistic regression, respectively, with time between age of onset and date of MRI or lumbar puncture as covariate to correct for possible confounding by disease stage. Linear mixed model analysis was performed to assess changes in MMSE levels over time, while accounting for the correlation between the repeated measurements of each patient. The model included time since first MMSE, family history, and an interaction effect between time and family history to assess differences in the course of cognitive decline between groups. All MMSEs which were administered within 6 months from the previous MMSE were removed, because of possible learning effects. In the random effect structure, covariance type was set on unstructured and a random intercept was included. Differences in survival between familial and sporadic DLB were explored using Kaplan-Meier analysis and Cox regression analysis. We included sex, age of onset, and study center to correct for possible confounding in the linear mixed model analysis and the Cox regression analysis.
In addition, differences in phenotype between DLB patients with a family history of either dementia or PD were explored. Statistical analyses were performed as described above.
Statistical significance for all tests was established at p < 0.05 (two-tailed). The data was analyzed using SPSS software (Version 24).
RESULTS
Differences in phenotype between patients with familial and sporadic DLB
The 154 familial DLB patients and the 137 sporadic DLB patients did not differ in sex, age of onset, type of first symptom (Table 1), or rate of cognitive decline (Table 2). Significantly more familial DLB patients had elevated CSF biomarkers for AD (47%), compared to those with sporadic DLB (30%, p = 0.039). This finding remained significant after correction for time between age of onset and date of lumbar puncture (p = 0.037, Table 1). In addition, a borderline significant result was found concerning the APOE ɛ4 allele, with a higher frequency of this allele in familial patients (65%) compared to sporadic patients (51%, p = 0.069). MTA score was not different between the groups (Table 1).
Table 1
Demographic features, clinical features and biomarkers in the total study group, and group differences
| Total (n = 291) | Familial DLB (n = 154) | Sporadic DLB (n = 137) | p | |
| Study center | 0.011* | |||
| Elisabeth-TweeSteden Hospital | 35 (12%) | 24 (16%) | 11 (8%) | |
| Erasmus Medical Center | 54 (19%) | 20 (13%) | 34 (25%) | |
| Amsterdam University Medical Center | 202 (69%) | 110 (71%) | 92 (67%) | |
| Sex, male | 232 (80%) | 122 (79%) | 110 (80%) | 0.82 |
| Age of onset, y | 66.0 (7.6) | 66.6 (8.4) | 65.3 (6.7) | 0.14 |
| First symptom | ||||
| Cognitive decline | 245 (84%) | 133 (86%) | 112 (82%) | 0.28 |
| Parkinsonism | 29 (10%) | 14 (9%) | 15 (11%) | 0.60 |
| Hallucinations | 17 (6%) | 7 (5%) | 10 (7%) | 0.32 |
| MTA score (average of right and left) (n = 199 [98;101]) | 1 (0.5–1.5) | 1 (0.5–1.5) | 1 (0–1.1) | 0.10a |
| CSF tau/Aβ42 ratio >0.52 (n = 169 [83;86]) | 65 (38%) | 40 (47%) | 25 (30%) | 0.037a, * |
| APOE ɛ4 carrier (n = 160 [76;84]) | 94 (59%) | 55 (65%) | 39 (51%) | 0.069 |
Values are presented as mean (SD), median (IQR) or n (%). DLB, dementia with Lewy bodies; PD, Parkinson’s disease; CSF, cerebrospinal fluid; MTA, medial temporal lobe atrophy, Aβ42, amyloid-β1 - 42, acorrected for time between age of onset and date of MRI or date of lumbar puncture, *p < 0.05.
Table 2
Statistical models regarding rate of cognitive decline and survival in the total study group
| Rate of cognitive decline | β | SE | 95% CI | p | |||
| Uncorrected | Baseline | –0.47 | 0.59 | –1.63–0.70 | 0.43 | ||
| (Linear mixed model analysis) | Change over time | –0.0090 | 0.017 | –0.041–0.024 | 0.59 | ||
| With correction for sex, age of onset, and study center | Baseline | –0.34 | 0.59 | –1.50–0.81 | 0.56 | ||
| (Linear mixed model analysis) | Change over time | –0.0091 | 0.016 | –0.042–0.023 | 0.59 | ||
| Survival | Mediana | SE | 95% CI | p | |||
| Uncorrected | Sporadic DLB | 9.0 | 0.63 | 7.77–10.23 | <0.001* | ||
| (Kaplan-Meier analysis) | Familial DLB | 8.0 | 0.51 | 7.00–9.00 | |||
| B | SE | HR | 95% CI | p | Z-score | ||
| With correction for sex, age of onset, and study center | 0.50 | 0.18 | 1.64 | 1.16–2.31 | 0.005* | 5.71 | |
| (Cox regression analysis) |
aTime between age of onset and death, *p < 0.05.
Differences in survival between patients with familial and sporadic DLB
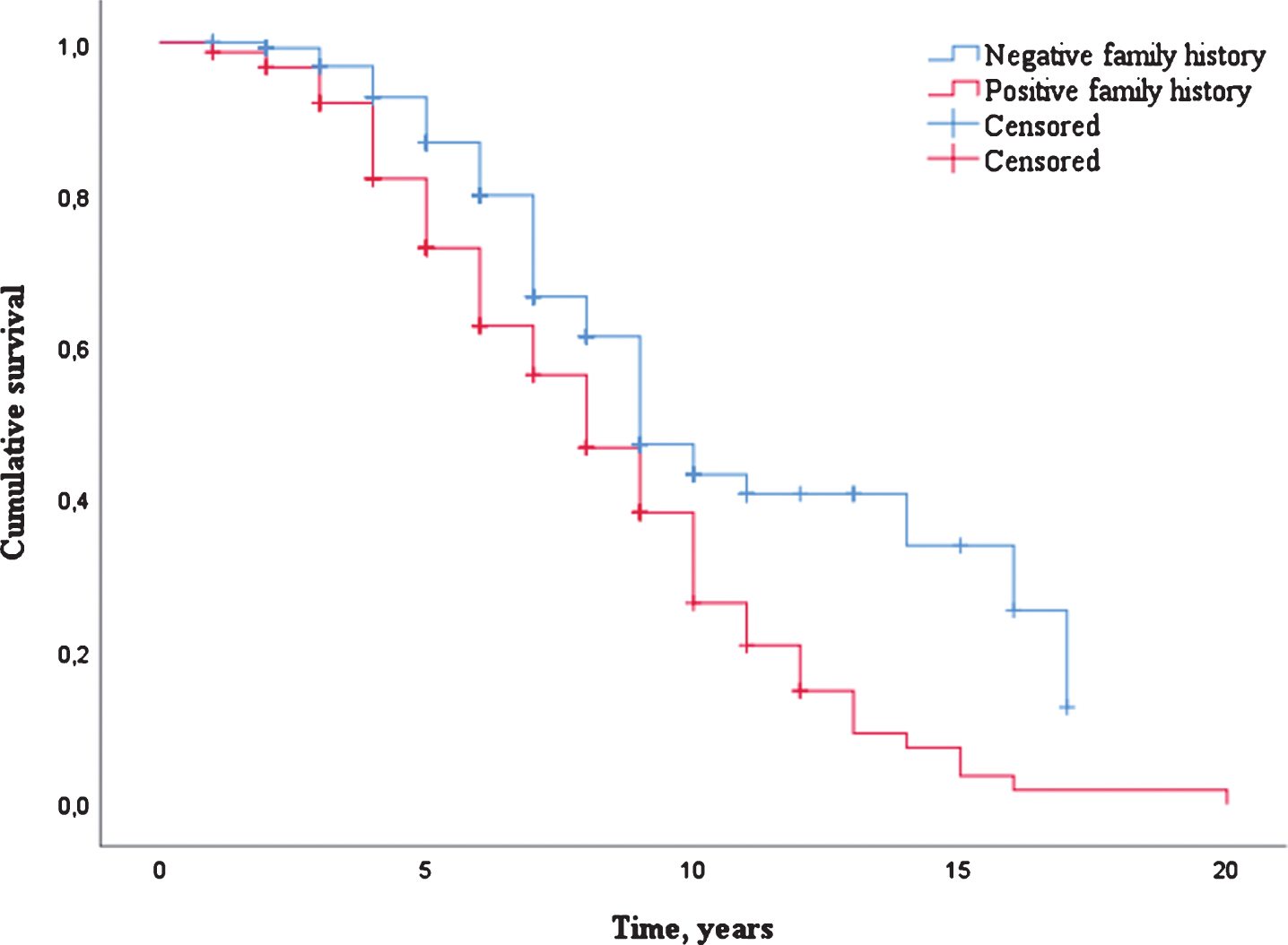
A total of 154 patients (53.1%) were deceased with a median survival of 7.0 years (IQR 5.0–9.0). Uncorrected survival analysis showed that patients with familial DLB had a significantly shorter survival (median 8.0 years, SE 0.51) compared to patients with sporadic DLB (median 9.0 years, SE 0.63, p≤0.001, Table 2 and Fig. 1). This finding remained significant after correction for sex, age of onset and study center (HR 1.64, 95% CI: 1.16–2.31, p = 0.005, Table 2).
Fig.1
Survival distribution (Kaplan-Meier curve) of DLB patients with a negative family history as compared to DLB patients with a positive family history.
Differences in phenotype between DLB patients with a family history of either dementia or PD
A total of 84 patients had a family history of either dementia (n = 69) or PD (n = 15) (Supplementary Figure 1). Parkinsonism was more prevalent as first symptom in DLB patients with a family history of PD (33%) compared to the patients with a family history of dementia (6%, p = 0.008). These groups did not differ in sex, age of onset (Supplementary Table 1), or rate of cognitive decline (Supplementary Table 2).
In addition, there were no significant differences in the presence of the APOE ɛ4 allele, CSF AD biomarkers, and MTA score between the groups (Supplementary Table 1).
Differences in survival between DLB patients with a family history of either dementia or PD
A total of and 43 patients (51.2%) were deceased with a median survival of 8.0 years (IQR 5.0–10.0). There were no differences in survival between DLB patients with relatives with dementia as compared to DLB patients with relatives with PD (Supplementary Table 2).
DISCUSSION
The main finding of this study is that patients with familial DLB have a shorter survival than patients with sporadic DLB. We also found a higher percentage of familial DLB patients with elevated AD biomarkers in their CSF compared to sporadic DLB patients. Several longitudinal studies in DLB showed that concomitant AD pathology is associated with a higher mortality [24–26]. The shorter survival in familial DLB compared to sporadic DLB may be due to concomitant AD pathology in familial DLB, which is reflected in a higher CSF tau/Aβ42 ratio in familial patients compared to sporadic patients. Genetic factors, such as the APOE ɛ4 allele, contribute to the presence of concomitant AD pathology [25, 27]. Interestingly, although only borderline statistically significant, a higher frequency of the APOE ɛ4 allele in familial patients compared to sporadic patients was seen. This suggests that genetic factors are associated with survival, possibly by influencing neuropathology.
Previous studies have shown several possible risk factors for a shorter survival in DLB, such as the presence of a fluctuating cognition, hallucinations at onset and a low MMSE score [14, 26]. Other variables have also been reported to be associated with survival, but are contradictory between different studies (e.g. female and male sex, early and late age of onset) [14, 26]. There were no significant differences in these clinical characteristics between familial and sporadic DLB patients in our study (data not shown for a fluctuating cognition and MMSE score). This might indicate that family history has an effect on survival independent of these clinical characteristics.
In addition, we found parkinsonism to be a more frequent presenting symptom in patients with a positive family history of PD than in patients with a positive family history of dementia. This might be based on more ‘pure’ Lewy body pathology in patients with relatives with PD, and mixed pathology (Lewy body pathology and AD pathology) in patients with relatives with dementia. However, this finding could also be based on recall bias due to familiarity with PD symptoms. Other AD or PD features were equally distributed between patients with a positive family history of dementia or PD, respectively. These results should be interpreted with caution due to the relatively small number of patients in the group with relatives with PD and the presence of non-AD dementias in the group with relatives with dementia.
About half of the DLB patients in this study had at least one first degree relative with dementia or PD. This is in line with previous studies, in which a positive family history for dementia was observed in 39–44% and for PD in 10–24% of patients with DLB [10, 11]. Nonetheless, the percentages that we found could be an overestimation as patients with insufficient information on family history were not taken into account. These patients might be less likely to have relatives with relevant diseases. However, the most important limitation of this study is its retrospective character. Furthermore, information on family history and disease onset was based on anamnestic information, and may have introduced a recall bias. In addition, the patients in this study were predominantly male (80%), which may not be a good representation of the general DLB population [28]. However, our main finding (familial DLB patients have a shorter survival than sporadic DLB patients) stayed significant after correction for sex. At last, the DLB diagnoses in this study were not pathologically confirmed.
Strengths of this study include its large sample size and the enrollment of patients from university medical centers as well as from a general hospital. The latter increases the generalizability of our findings.
In conclusion, we are the first to report that DLB patients with a positive family history of dementia or PD have a shorter survival than DLB patients with a negative family history of these diseases. This suggests that genetic factors contribute to disease course, possibly by influencing the amount of concomitant AD pathology, which is supported by our data. Future studies are necessary to identify which genetic and other contributing factors are accountable for our findings. This knowledge will lead to a better understanding of the pathophysiology of the disease and the overlap with AD and PD(D), and will probably improve the prediction of prognosis.
ACKNOWLEDGMENTS
We would like to thank all patients who participated in this study. This work was supported by ZonMw [grant number 70-73305-98-102] to FJdJ.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0825r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-190825.
REFERENCES
[1] | Walker Z , Possin KL , Boeve BF , Aarsland D ((2015) ) Lewy body dementias. Lancet 386: , 1683–1697. |
[2] | McKeith IG , Boeve BF , Dickson DW , Halliday G , Taylor JP , Weintraub D , Aarsland D , Galvin J , Attems J , Ballard CG , Bayston A , Beach TG , Blanc F , Bohnen N , Bonanni L , Bras J , Brundin P , Burn D , Chen-Plotkin A , Duda JE , El-Agnaf O , Feldman H , Ferman TJ , Ffytche D , Fujishiro H , Galasko D , Goldman JG , Gomperts SN , Graff-Radford NR , Honig LS , Iranzo A , Kantarci K , Kaufer D , Kukull W , Lee VMY , Leverenz JB , Lewis S , Lippa C , Lunde A , Masellis M , Masliah E , McLean P , Mollenhauer B , Montine TJ , Moreno E , Mori E , Murray M , O’Brien JT , Orimo S , Postuma RB , Ramaswamy S , Ross OA , Salmon DP , Singleton A , Taylor A , Thomas A , Tiraboschi P , Toledo JB , Trojanowski JQ , Tsuang D , Walker Z , Yamada M , Kosaka K ((2017) ) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: , 88–100. |
[3] | Vergouw LJM , van Steenoven I , van de Berg WDJ , Teunissen CE , van Swieten JC , Bonifati V , Lemstra AW , de Jong FJ ((2017) ) An update on the genetics of dementia with Lewy bodies. Parkinsonism Relat Disord 43: , 1–8. |
[4] | Orme T , Guerreiro R , Bras J ((2018) ) The genetics of dementia with Lewy bodies: Current understanding and future directions. Curr Neurol Neurosci Rep 18: , 67. |
[5] | Guerreiro R , Escott-Price V , Darwent L , Parkkinen L , Ansorge O , Hernandez DG , Nalls MA , Clark L , Honig L , Marder K , van der Flier W , Holstege H , Louwersheimer E , Lemstra A , Scheltens P , Rogaeva E , St George-Hyslop P , Londos E , Zetterberg H , Ortega-Cubero S , Pastor P , Ferman TJ , Graff-Radford NR , Ross OA , Barber I , Braae A , Brown K , Morgan K , Maetzler W , Berg D , Troakes C , Al-Sarraj S , Lashley T , Compta Y , Revesz T , Lees A , Cairns NJ , Halliday GM , Mann D , Pickering-Brown S , Powell J , Lunnon K , Lupton MK , International Parkinson’s Disease Genomics C, Dickson D , Hardy J , Singleton A , Bras J ((2016) ) Genome-wide analysis of genetic correlation in dementia with Lewy bodies, Parkinson’s and Alzheimer’s diseases. Neurobiol Aging 38: , 214 e217–214 e210. |
[6] | Bras J , Guerreiro R , Darwent L , Parkkinen L , Ansorge O , Escott-Price V , Hernandez DG , Nalls MA , Clark LN , Honig LS , Marder K , Van Der Flier WM , Lemstra A , Scheltens P , Rogaeva E , St George-Hyslop P , Londos E , Zetterberg H , Ortega-Cubero S , Pastor P , Ferman TJ , Graff-Radford NR , Ross OA , Barber I , Braae A , Brown K , Morgan K , Maetzler W , Berg D , Troakes C , Al-Sarraj S , Lashley T , Compta Y , Revesz T , Lees A , Cairns N , Halliday GM , Mann D , Pickering-Brown S , Dickson DW , Singleton A , Hardy J ((2014) ) Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum Mol Genet 23: , 6139–6146. |
[7] | Guerreiro R , Escott-Price V , Hernandez DG , Kun-Rodrigues C , Ross OA , Orme T , Neto JL , Carmona S , Dehghani N , Eicher JD , Shepherd C , Parkkinen L , Darwent L , Heckman MG , Scholz SW , Troncoso JC , Pletnikova O , Dawson T , Rosenthal L , Ansorge O , Clarimon J , Lleo A , Morenas-Rodriguez E , Clark L , Honig LS , Marder K , Lemstra A , Rogaeva E , St George-Hyslop P , Londos E , Zetterberg H , Barber I , Braae A , Brown K , Morgan K , Troakes C , Al-Sarraj S , Lashley T , Holton J , Compta Y , Van Deerlin V , Serrano GE , Beach TG , Lesage S , Galasko D , Masliah E , Santana I , Pastor P , Diez-Fairen M , Aguilar M , Tienari PJ , Myllykangas L , Oinas M , Revesz T , Lees A , Boeve BF , Petersen RC , Ferman TJ , Graff-Radford N , Cairns NJ , Morris JC , Pickering-Brown S , Mann D , Halliday GM , Hardy J , Trojanowski JQ , Dickson DW , Singleton A , International Parkinson’s Disease Genomics Consortium, Stone DJ , Bras J ((2019) ) Heritability and genetic variance of dementia with Lewy bodies. Neurobiol Dis 127: , 492–501. |
[8] | Tsuang DW , DiGiacomo L , Bird TD ((2004) ) Familial occurrence of dementia with Lewy bodies. Am J Geriatr Psychiatry 12: , 179–188. |
[9] | Nervi A , Reitz C , Tang MX , Santana V , Piriz A , Reyes D , Lantigua R , Medrano M , Jimenez-Velazquez IZ , Lee JH , Mayeux R ((2011) ) Familial aggregation of dementia with Lewy bodies. Arch Neurol 68: , 90–93. |
[10] | Boot BP , Orr CF , Ahlskog JE , Ferman TJ , Roberts R , Pankratz VS , Dickson DW , Parisi J , Aakre JA , Geda YE , Knopman DS , Petersen RC , Boeve BF ((2013) ) Risk factors for dementia with Lewy bodies: A case-control study. Neurology 81: , 833–840. |
[11] | Papapetropoulos S , Lieberman A , Gonzalez J , Singer C , Laufer DZ , Mash DC ((2006) ) Family history of dementia: Dementia with Lewy bodies and dementia in Parkinson’s disease. J Neuropsychiatry Clin Neurosci 18: , 113–116. |
[12] | Geiger JT , Ding J , Crain B , Pletnikova O , Letson C , Dawson TM , Rosenthal LS , Pantelyat A , Gibbs JR , Albert MS , Hernandez DG , Hillis AE , Stone DJ , Singleton AB , North American Brain Expression Consortium, Hardy JA , Troncoso JC , Scholz SW ((2016) ) Next-generation sequencing reveals substantial genetic contribution to dementia with Lewy bodies. Neurobiol Dis 94: , 55–62. |
[13] | Keogh MJ , Kurzawa-Akanbi M , Griffin H , Douroudis K , Ayers KL , Hussein RI , Hudson G , Pyle A , Cordell HJ , Attems J , McKeith IG , O’Brien JT , Burn DJ , Morris CM , Thomas AJ , Chinnery PF ((2016) ) Exome sequencing in dementia with Lewy bodies. Transl Psychiatry 6: , e728. |
[14] | Larsson V , Torisson G , Londos E ((2018) ) Relative survival in patients with dementia with Lewy bodies and Parkinson’s disease dementia. PLoS One 13: , e0202044. |
[15] | Shiner T , Mirelman A , Gana Weisz M , Bar-Shira A , Ash E , Cialic R , Nevler N , Gurevich T , Bregman N , Orr-Urtreger A , Giladi N ((2016) ) High frequency of GBA gene mutations in dementia with Lewy bodies among Ashkenazi Jews. JAMA Neurol 73: , 1448–1453. |
[16] | Gamez-Valero A , Prada-Dacasa P , Santos C , Adame-Castillo C , Campdelacreu J , Rene R , Gascon-Bayarri J , Ispierto L , Alvarez R , Ariza A , Beyer K ((2016) ) GBA mutations are associated with earlier onset and male sex in dementia with Lewy bodies. Mov Disord 31: , 1066–1070. |
[17] | Nalls MA , Duran R , Lopez G , Kurzawa-Akanbi M , McKeith IG , Chinnery PF , Morris CM , Theuns J , Crosiers D , Cras P , Engelborghs S , De Deyn PP , Van Broeckhoven C , Mann DM , Snowden J , Pickering-Brown S , Halliwell N , Davidson Y , Gibbons L , Harris J , Sheerin UM , Bras J , Hardy J , Clark L , Marder K , Honig LS , Berg D , Maetzler W , Brockmann K , Gasser T , Novellino F , Quattrone A , Annesi G , De Marco EV , Rogaeva E , Masellis M , Black SE , Bilbao JM , Foroud T , Ghetti B , Nichols WC , Pankratz N , Halliday G , Lesage S , Klebe S , Durr A , Duyckaerts C , Brice A , Giasson BI , Trojanowski JQ , Hurtig HI , Tayebi N , Landazabal C , Knight MA , Keller M , Singleton AB , Wolfsberg TG , Sidransky E ((2013) ) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70: , 727–735. |
[18] | McKeith IG , Dickson DW , Lowe J , Emre M , O’Brien JT , Feldman H , Cummings J , Duda JE , Lippa C , Perry EK , Aarsland D , Arai H , Ballard CG , Boeve B , Burn DJ , Costa D , Del Ser T , Dubois B , Galasko D , Gauthier S , Goetz CG , Gomez-Tortosa E , Halliday G , Hansen LA , Hardy J , Iwatsubo T , Kalaria RN , Kaufer D , Kenny RA , Korczyn A , Kosaka K , Lee VM , Lees A , Litvan I , Londos E , Lopez OL , Minoshima S , Mizuno Y , Molina JA , Mukaetova-Ladinska EB , Pasquier F , Perry RH , Schulz JB , Trojanowski JQ , Yamada M , Consortium on DLB ((2005) ) Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 65: , 1863–1872. |
[19] | Scheltens P , Leys D , Barkhof F , Huglo D , Weinstein HC , Vermersch P , Kuiper M , Steinling M , Wolters EC , Valk J ((1992) ) Atrophy of medial temporal lobes on MRI in “probable” Alzheimer’s disease and normal ageing: Diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry 55: , 967–972. |
[20] | Duits FH , Teunissen CE , Bouwman FH , Visser PJ , Mattsson N , Zetterberg H , Blennow K , Hansson O , Minthon L , Andreasen N , Marcusson J , Wallin A , Rikkert MO , Tsolaki M , Parnetti L , Herukka SK , Hampel H , De Leon MJ , Schroder J , Aarsland D , Blankenstein MA , Scheltens P , van der Flier WM ((2014) ) The cerebrospinal fluid “Alzheimer profile”: Easily said, but what does it mean? Alzheimers Dement 10: , 713–723 e712. |
[21] | Liu CC , Liu CC , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[22] | Gibb WR , Lees AJ ((1988) ) The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 51: , 745–752. |
[23] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[24] | Lemstra AW , de Beer MH , Teunissen CE , Schreuder C , Scheltens P , van der Flier WM , Sikkes SA ((2017) ) Concomitant AD pathology affects clinical manifestation and survival in dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 88: , 113–118. |
[25] | Irwin DJ , Grossman M , Weintraub D , Hurtig HI , Duda JE , Xie SX , Lee EB , Van Deerlin VM , Lopez OL , Kofler JK , Nelson PT , Jicha GA , Woltjer R , Quinn JF , Kaye J , Leverenz JB , Tsuang D , Longfellow K , Yearout D , Kukull W , Keene CD , Montine TJ , Zabetian CP , Trojanowski JQ ((2017) ) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol 16: , 55–65. |
[26] | Jellinger KA , Wenning GK , Seppi K ((2007) ) Predictors of survival in dementia with lewy bodies and Parkinson dementia. Neurodegener Dis 4: , 428–430. |
[27] | Tsuang D , Leverenz JB , Lopez OL , Hamilton RL , Bennett DA , Schneider JA , Buchman AS , Larson EB , Crane PK , Kaye JA , Kramer P , Woltjer R , Trojanowski JQ , Weintraub D , Chen-Plotkin AS , Irwin DJ , Rick J , Schellenberg GD , Watson GS , Kukull W , Nelson PT , Jicha GA , Neltner JH , Galasko D , Masliah E , Quinn JF , Chung KA , Yearout D , Mata IF , Wan JY , Edwards KL , Montine TJ , Zabetian CP ((2013) ) APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 70: , 223–228. |
[28] | Mouton A , Blanc F , Gros A , Manera V , Fabre R , Sauleau E , Gomez-Luporsi I , Tifratene K , Friedman L , Thummler S , Pradier C , Robert PH , David R ((2018) ) Sex ratio in dementia with Lewy bodies balanced between Alzheimer’s disease and Parkinson’s disease dementia: A cross-sectional study. Alzheimers Res Ther 10: , 92. |


