
Telomere Length and the Risk of Alzheimer’s Disease: The Rotterdam Study
Abstract
There is a wide interest in biomarkers that capture the burden of detrimental factors as these accumulate with the passage of time, i.e., increasing age. Telomere length has received considerable attention as such a marker, because it is easily quantified and it may aid in disentangling the etiology of dementia or serve as predictive marker. We determined the association of telomere length with risk of Alzheimer’s disease and all-cause dementia in a population-based setting. Within the Rotterdam Study, we performed quantitative PCR to measure mean leukocyte telomere length in blood. We determined the association of telomere length with risk of Alzheimer’s disease until 2016, using Cox regression models. Of 1,961 participants (mean age 71.4±9.3 years, 57.1% women) with a median follow-up of 8.3 years, 237 individuals were diagnosed with Alzheimer’s disease. We found a U-shaped association between telomere length and risk of Alzheimer’s disease: compared to the middle tertile the adjusted hazard ratio was 1.59 (95% confidence interval (CI), 1.13–2.23) for the lowest tertile and 1.47 (1.03–2.10) for the highest tertile. Results were similarly U-shaped but slightly attenuated for all-cause dementia. In conclusion, shorter and longer telomere length are both associated with an increased risk of Alzheimer’s disease in the general population.
INTRODUCTION
Dementia is a devastating disease affecting millions of people worldwide. While advances over the past decades have unraveled several pathological mechanisms underlying dementia, chronological age remains the most important predictor for dementia onset [1]. There is, therefore, a wide interest in biomarkers that capture the burden of detrimental factors as these accumulate with the passage of time, i.e., increasing age. Such markers, that are preferably easily quantified, may aid in disentangling the etiology of dementia or ultimately serve as predictive markers. Against this background, telomere length has received considerable attention [2].
Telomeres are repetitive base pair sequences at the end of chromosomes and facilitate complete chromosome replication [3]. Since the replication machinery is unable to copy the ends of DNA, telomeres will shorten with each cell division. A change in telomere length resulting in shortened telomeres induces a DNA damage response that leads to a growth arrest during which cells attempt to repair the damage and if DNA damage is irreparable, replicative senescence or cell death is triggered [4]. Very short telomeres have relatively low cancer risk (due to cell senescence blocking cell division), but could be associated with increased DNA damage [5]. Given the tight link with cell death, short telomere length has been linked to poorer survival [6, 7] and other age-related diseases, including dementia as has been shown in a recent meta-analysis [8]. Conversely, there is evidence that over-elongated telomeres represent pathological cell function with decreased DNA repair and increased cancer risk [9], which has also been shown in human embryonic stem cells [10], while “normally” long telomeres are protective, including against DNA damage. With respect to Alzheimer’s disease, there is also evidence suggesting an increased risk of mild cognitive impairment related to both short and long telomeres [11]. Additionally, hippocampal cells of Alzheimer’s disease brains are shown to have longer telomeres compared to control samples [12]. It is, thus, conceivable that not only shorter but also longer telomere length may be detrimental for dementia risk. However, the temporal relation between longer telomere length and dementia has so far not been investigated. Therefore, the aim of this study is to investigate the association between blood cell telomere length and risk of Alzheimer’s disease and to explore non-linearity within this association.
METHODS
Design and study population
This study was embedded within the Rotterdam Study, a prospective population-based cohort study among middle-aged and elderly individuals in the Netherlands. Three cohorts were established in 1990, 2000, and 2006, respectively. From each cohort, participants were invited every 4–5 years to undergo follow-up examinations. We refer the reader to the Rotterdam Study methods paper for further information [13]. For this study, a total of 2,140 participants were randomly selected from the first (1990–2004, N = 7983) and third cohorts (2006–2008, N = 3932) for telomere length measurement. From these participants, 1,961 were dementia-free at baseline visit and had complete telomere length data available at baseline (Fig. 1).
Fig.1
Flowchart of study population.
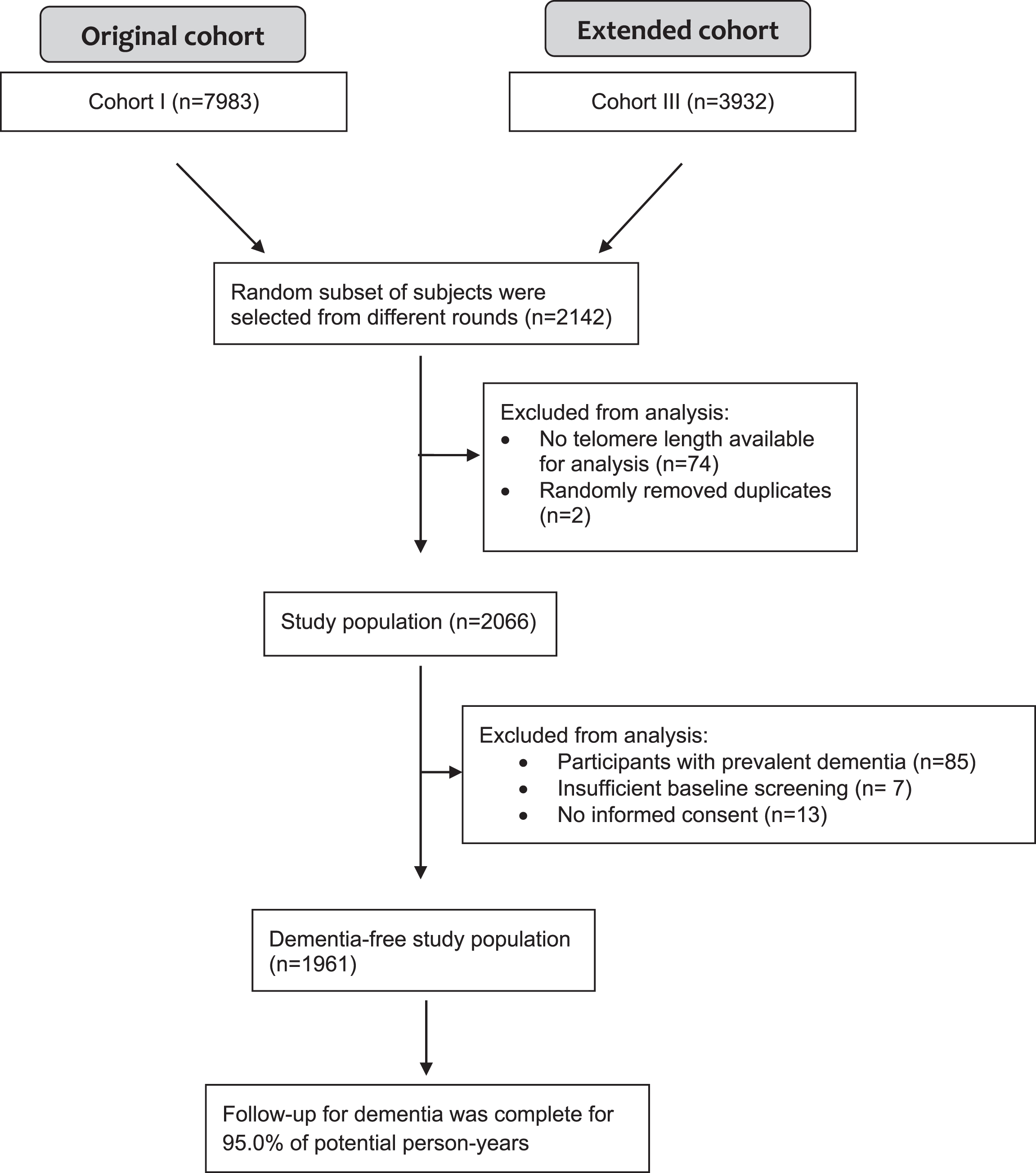
The Rotterdam Study has been approved by the Medical Ethics Committee of the Erasmus MC and by the Ministry of Health, Welfare and Sport of the Netherlands, implementing the Wet Bevolkingsonderzoek: ERGO (Population Studies Act: Rotterdam Study). All participants provided written informed consent to participate in the study and to obtain information from their treating physicians.
Assessment of telomere length
Blood for all samples was collected in EDTA tubes and stored at –20°C until DNA isolation. DNA isolation was done with a method based on salting out, with different lysis buffers for the different cohorts. The OD260/280 and OD260/230 for both isolation methods did not differ significantly between the two cohorts. The average OD260/280 ratio was 1.90 with a standard deviation of 0.10 and the average OD260/230 ratio was 1.61 with a standard deviation of 0.46 in a random sample of 168. Telomere length was measured using a qPCR assay based on the method described by Cawthon [14] with minor modifications. The SDS software of the 7900HT qPCR machine is not able to analyze the qPCR data produced at two different temperature points. Therefore, we chose the singleplex assay for this study. For each sample, the telomere and 36B4 assay were run in separate wells but in the same 384 wells PCR plate. Each reaction contained 5 ng DNA, 1μM of each of the telomere primers (tel1b-forward: G.2ptG.2ptT.2ptT.2ptT.2ptG.2ptT.2ptT.2ptT.2ptG.2ptG.2ptG.2ptT.2ptT.2ptT.2ptGGGTTTGGGTTTGGGTTTGGGTT, tel2b-reverse: GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT) or 250 nM of the 34B4 primers (36B4u-forward: CAGCAAGTGGGAAGGTGTAATCC, 36B4d-reverse: CCCATTCTATCATCAACGGGTACAA) and 1× Quantifast SYBR green PCR Mastermix (Qiagen). The reactions for both assays were performed in duplicate for each sample in a 7900HT machine (Applied Biosystems). Ct values and PCR efficiencies were calculated per plate using the MINER algorithm [15]. Duplicate Ct values that had a coefficient of variance (CV) of more than 1% were excluded from further analysis. Using the average Ct value per sample and the average PCR efficiency per plate the samples were quantified using the formula Q = 1/(1 + PCR eff)∧Ct. The relative telomere length was calculated by dividing the Q of the telomere assay by the Q of the 34B4 assay. To validate the assay, 96 random samples were run twice and the CV of that experiment was 4.5%. Telomere length was measured in 37 batches.
Ascertainment of incident dementia
Baseline and follow-up ascertainment methods for dementia have previously been described in detail [16]. In brief, participants were screened for dementia at baseline and subsequent center visits. In addition, the entire cohort was continuously under surveillance for dementia through electronic linkage of the study database with medical records. A consensus panel led by a consultant neurologist established the final diagnosis according to standard criteria for dementia (DSM-III-R) and Alzheimer’s disease (NINCDS–ADRDA). Within this period, participants were censored at date of dementia diagnosis, death, loss to follow-up, or 1 January 2016, whichever came first.
APOE genotyping
APOE genotype was determined using polymerase chain reaction on coded DNA samples in the baseline cohort [17] and with a bi-allelic Tacqman assay (rs7412 and rs429358) in the extensions of the Rotterdam Study [13]. APOE ɛ4 carrier status was defined carriers versus non-carriers of the ɛ4 allele.
Other covariates
Potential confounding factors were chosen on the basis of previous literature [16, 18]. All covariates were measured at baseline. Educational attainment was categorized as low (primary education or lower vocational education), intermediate (secondary education or intermediate vocational education), and high educational level (higher vocational education or university). Smoking habits were categorized as current, former and never smoking. Body mass index was calculated as weight in kilograms per height in meters squared. Blood pressure was measured twice at the right brachial artery with the participant in sitting position, out of which the mean was used. Hypertension was defined as a resting blood pressure exceeding 140/90 mmHg or the use of blood pressure lowering medication. Hypercholesterolemia was defined as a total cholesterol of ≥6.2 mmol/L or the use of serum lipid-reducing agents. White blood cell counts were assessed with a Coulter AcT diff2 Hematology Analyzer at the research center in Ommoord. On 14 February 2005, a new device of the same brand and type was installed. Prevalent stroke was assessed at baseline during interview and we verified these data with medical records. After study entry, we continuously monitored participants after enrollment for incident stroke through linkage of the study database with files from general practitioners. Files of nursing home physicians and files from general practitioners of participants who moved out of the district were also checked. We obtained additional information from hospital records. When potential strokes were found, they were reviewed by research physicians and verified by an experienced neurologist.
Statistical analysis
Missing covariate data (maximum 2.2%) were imputed using 5-fold multiple imputation based on determinant, outcome, and included covariates. In all models, we adjusted for age continuously, sex and center visit since data was randomly selected from 4 center visits from 2 subcohorts. We additionally adjusted for educational attainment, APOE ɛ4 carrier status, smoking, body mass index, hypertension, hypercholesterolemia, and white blood cell count in a second model, all at time of blood draw. We first explored linearity of telomere length with Alzheimer’s disease and dementia risk by entering T/S ratio as restricted cubic spline with 3 knots in Cox regression models. In order to further quantify potential non-linearity, T/S ratio was subsequently divided into tertiles, defined as T/S ratio of 0.31–0.87, 0.87–1.02, and 1.02–1.79, respectively. To provide a parsimonious description of the telomere-dementia relationship and facilitate communication of the results, we assessed the association between telomere length in tertiles, comparing lowest and highest tertiles that represent the shortest and longest telomere lengths, respectively, to the middle tertile, with risk of Alzheimer’s disease and dementia. We repeated the analysis for Alzheimer’s disease, by adjusting not only for age at blood draw, but also for age at dementia diagnosis or date of last follow-up as an attempt to assess survival bias. We further verified that age was robustly controlled for by comparing results across models using five different ways of age-adjustment. These included no adjustment for age, entering age and age2 in the model, entering age as cubic splines, repeating the analyses with age rather than follow-up time as the time scale and finally regressing the effect of age out of T/S ratio before creating T/S ratio tertiles. We performed an additional adjustment for plate number to rule out batch effects. The proportional hazards assumption was assessed using Schoenfeld residuals. We next repeated the analyses for Alzheimer’s disease, but excluding all participants with prior clinical stroke at baseline and censoring for incident clinical stroke during follow-up using Cox models.
We also assessed interaction by age, sex, education, or APOE ɛ4 carrier status by stratification and testing for multiplicative interaction by including the product of the interacting factor and T/S ratio continuously as a restricted cubic spline (4 knots) to take into account the non-linear relationship between T/S ratio and Alzheimer’s disease. We stratified for age by 3 groups, namely 45–65 years, 65–85 years, and 85–105 years.
All analyses were performed using RStudio version 0.99.903 (R version 3.4.3 [19], RStudio, Inc., Boston, MA). We used the following R packages for our analyses: mice, survival, rms, and scales.
RESULTS
Baseline characteristics of the study population are presented in Table 1. The mean age of the participants was 71.4±9.3 years with 57.1% women. During a median follow-up of 7.3 (interquartile range 4.9–11.7) years (26,025 person-years), 305 individuals were diagnosed with dementia of whom 237 were Alzheimer’s disease.
Table 1
Baseline characteristics of the dementia-free study population
| Characteristics | Dementia-free cohort (N = 1961) |
| Women | 1120 (57.1%) |
| Age, y | 71.4±9.3 |
| Cohort | |
| First | 1543 (78.7%) |
| Third | 418 (21.3%) |
| Education | |
| Primary education | 366 (18.9%) |
| Lower/intermediate general education | 806 (41.5%) |
| Intermediate vocational education | 535 (27.6%) |
| Higher vocational education | 233 (12.0%) |
| Apolipoprotein E ɛ4 carriership | |
| Non-carrier (ɛ2/ɛ2, ɛ2/ɛ3, or ɛ3/ɛ3) | 1411 (72.0%) |
| Carrier (ɛ4/ɛ4, ɛ3/ɛ4, or ɛ2/ɛ4) | 550 (28.0%) |
| Smoking | |
| Current | 405 (21.1%) |
| Former | 921 (48.0%) |
| Never | 592 (30.9%) |
| Body mass index, kg/m2 | 27.2±4.1 |
| Hypertension | 700 (36.5%) |
| Hypercholesterolemia | 949 (48.8%) |
| White blood cell count | 6.9±2.3 |
| Telomere length, T/S ratio | 0.95±0.18 |
N, number of participants included in study. Data presented as mean (standard deviation) for continuous variables and number (percentages) for categorical variables. T/S ratio is relative telomere to single copy gene ratio. Data represent original data without imputed values. Number of missing values are 43 (2.2%) for smoking, 21 (1.1%) for education, 27 (1.4%) for hypertension, 17 (0.9%) for hypercholesterolemia, 29 (1.5%) for body mass index, and 157 (8.0%) for white blood cell count. Data for apolipoprotein E ɛ4 carriership was complete.
There was evidence of non-linearity in the association between telomere length and Alzheimer’s disease (Supplementary Figure 1). Compared to the middle tertile of telomere length, we found an adjusted hazard ratio (aHR) of 1.59; 95% confidence interval (CI), 1.13–2.23 for lowest tertile of telomere length and 1.47; 95% CI, 1.03–2.10 for highest tertile of telomere length for Alzheimer’s disease, with similar but lower estimates for dementia (Table 2). When additionally adjusting for age at dementia diagnosis or date of last follow-up, we found an aHR of 1.52; 95% CI, 1.07–2.18 for lowest tertile of telomere length and 1.75; 95% CI, 1.21–2.54 for highest tertile of telomere length for Alzheimer’s disease, compared to the middle tertile. Other approaches of age-adjustment yielded similar results (Table 3). There were no batch effects. The proportional hazards assumption was met for all models. Results were slightly attenuated but remained U-shaped after excluding participants with prior clinical stroke at baseline and censoring for incident clinical stroke: compared to middle tertile aHR: 1.44, 1.02–2.05 for lowest tertile of telomere length and aHR 1.30, 95% CI 0.90–1.89 for highest tertile of telomere length.
Table 2
Telomere length and the risk of Alzheimer’s disease and all-cause dementia
| n/N | Alzheimer’s disease | n/N | All-Cause dementia | |||
| Model I HR, 95% CI | Model II HR, 95% CI | Model I HR, 95% CI | Model II HR, 95% CI | |||
| Telomere length | ||||||
| Tertile 1 (T/S ratio 0.31–0.87) | 106/654 | 1.54, 1.10–2.15 | 1.59, 1.13–2.23 | 129/654 | 1.25, 0.94–1.66 | 1.27, 0.96–1.70 |
| Tertile 2 (T/S ratio 0.87–1.02) | 54/654 | 1 (reference) | 1 (reference) | 80/654 | 1 (reference) | 1 (reference) |
| Tertile 3 (T/S ratio 1.02–1.79) | 77/653 | 1.41, 0.99–2.00 | 1.47, 1.03–2.10 | 96/653 | 1.19, 0.88–1.60 | 1.25, 0.92–1.69 |
n, number of cases; N, number of persons at risk; HR, hazard ratio; CI, confidence interval; T/S ratio, relative telomere to single copy gene ratio. Cox regression model I: Adjusted for age, sex, and visit. Cox regression model II: Adjusted for age, sex, visit, education, APOE ɛ4 carrier status, smoking, body mass index, hypertension, hypercholesterolemia, and white blood cell count.
Table 3
Association between telomere length and Alzheimer’s disease using different modes of age adjustment
| Adjustment | Alzheimer’s disease | |||||
| No age adjustment | Age + age2 | Cubic splines for age† | Age as time-scale | Regressing out the effect of age of T/S ratio* | ||
| n/N | HR, 95% CI | HR, 95% CI | HR, 95% CI | HR, 95% CI | HR, 95% CI | |
| Telomere length | ||||||
| Tertile 1 (shortest) | 106/654 | 2.08, 1.49–2.91 | 1.64, 1.17–2.30 | 1.64, 1.17–2.30 | 1.63, 1.16–2.29 | 1.33, 0.96–1.86 |
| Tertile 2 (middle) | 54/654 | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) | 1 (reference) |
| Tertile 3 (longest) | 77/653 | 1.25, 0.88–1.77 | 1.45, 1.01–2.06 | 1.45, 1.02–2.06 | 1.44, 1.01–2.06 | 1.22, 0.87–1.69 |
n, number of cases; N, number of persons at risk; HR, hazard ratio; CI, confidence interval; T/S ratio, relative telomere to single copy gene ratio. †Cubic splines with 3 knots, similar results for higher degree polynomials. Models adjusted for age, sex, visit, education, APOE ɛ4 carrier status, smoking, body mass index, hypertension, hypercholesterolemia, and white blood cell count. *Regressing the effect of age out of T/S ratio before creating T/S ratio tertiles; no additional adjustment for age in the model; similar results for additionally regressing out the effect of sex.
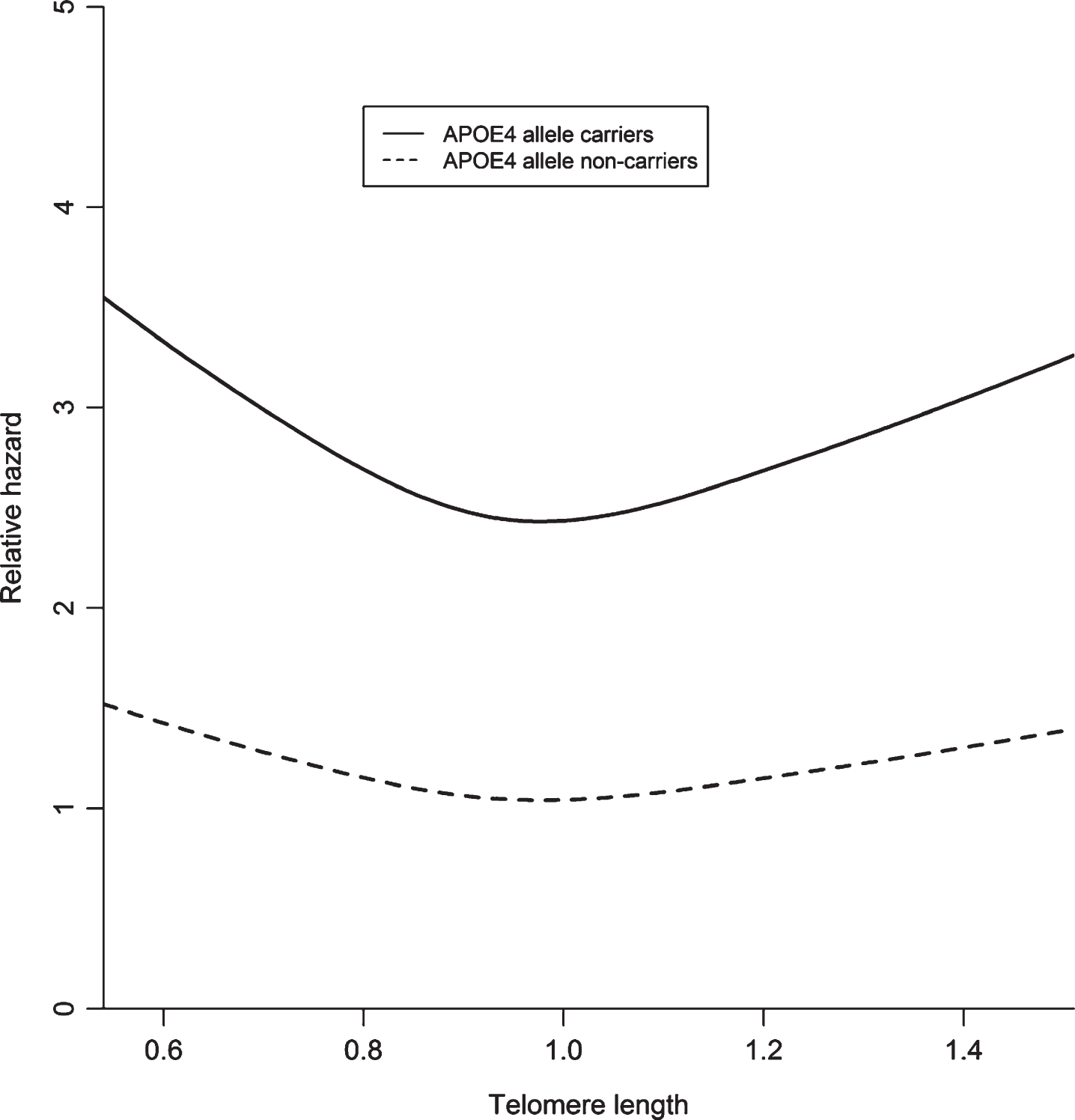
In the stratified analyses, the association between shorter and longer telomeres and Alzheimer’s disease was stronger in APOE ɛ4 carriers compared to non-carriers (Fig. 2) with the multiplicative interaction term yielding p = 0.087. Stratification for age, sex, and education did not provide different risk estimates (p-values for interaction: p = 0.554, p = 0.600, and p = 0.123, respectively).
Fig.2
A visual representation of telomere length and the risk of Alzheimer’s disease stratified by APOE ɛ4 carriership with restricted cubic splines in Cox model II, adjusted for age, sex, study visit, education, APOE ɛ4 carrier status, smoking, body mass index, hypertension, hypercholesterolemia and white blood cell count. Number of Alzheimer’s disease cases in APOE ɛ4 carrier group: 102 with 550 persons at risk; number of cases in the APOE ɛ4 non-carrier group: 135 with 1411 persons at risk.
DISCUSSION
In this study, shorter as well as longer telomere length were significantly associated with increased risk of dementia, specifically Alzheimer’s disease.
Thus far, focus in the dementia field has been on shorter telomere length with studies finding observationally as well as genetically associations between shorter telomere length and increased risk of Alzheimer’s disease [20–23]. Moreover, a recent meta-analysis which included 13 primary studies demonstrated significantly shorter telomere length in 860 Alzheimer’s disease patients compared to 2,022 controls [8]. Interestingly, we found that in addition to shorter telomere length also longer telomere length was associated with an increased dementia risk. This finding is in line with previous studies [24, 25], particularly the study by Roberts and others [11] showing that short and long telomeres increase risk of amnestic mild cognitive impairment. Yet, we emphasize that further replication is necessary to confirm the robustness of our findings with dementia.
An explanation for our observations with longer telomere length might be survival bias, where presumably participants with the longest telomere length live long enough to develop dementia. This is, however, less likely since associations remained after additional adjustment for age at dementia diagnosis or end of follow-up. Alternatively, it is plausible that a disturbed equilibrium of telomere length toward either direction is harmful. Indeed, over-elongated telomeres become fragile and accumulate DNA damage, as has been shown by an experimental study in human embryonic stem cells, and very tight control of telomere length homeostasis is regulated by not only telomerase-dependent elongation, but also by telomere trimming events [10]. Another study has shown that telomerase RNA component (TERC) knockout mice with Alzheimer’s disease, which present with telomere shortening, slows down the progression of Aβ pathology, showing a more protective effect of short telomere length [26], thereby supporting a detrimental effect of longer telomere length. The potential biological link between over-elongated telomeres and higher Alzheimer’s disease risk remains to be further elucidated. In contrast with longer telomere length, more is known about the potential biological link between shorter telomeres and increased Alzheimer’s disease risk. Neurons lacking telomerase reverse transcriptase (TERT) in knockout mice and thus have shorter telomeres, display an increased production of oxidative species and an increase in cellular oxidative damage in response to tau, demonstrating a harmful effect of shorter telomeres [27]. Therefore, telomerase may play different roles in the tau and amyloid pathology via multiple mechanisms [28]. In addition, microglial cellular senescence may also be an important mechanism in the development of Alzheimer’s disease, which is suggested to be exacerbated by the presence of amyloid [29, 30]. Further studies are warranted to differentiate between a true biological association and survival bias. If a true biological association exists between longer telomere length and risk of dementia, telomerase therapies, which have been successful in mice [31, 32], would need further fine-tuning to prevent over-elongation of telomere length.
Interestingly, we found stronger effects for Alzheimer’s disease compared to all-cause dementia. This might be reflective of the fact that Alzheimer’s disease is a more homogeneous entity than all-cause dementia, which includes a plethora of etiologies.
Certain limitations must be taken into account. First, we measured telomere length in leukocytes in blood. This may not be representative of telomere length in the brain, in particular glial cells. However, a previous study has shown a direct correlation between telomeres measured from peripheral blood and cerebellum [33], suggesting blood cell telomere length to be a valid measurement. Another limitation of measuring telomere length in leukocytes is that lymphocytes are the only peripheral blood cells expressing telomerase activity and therefore are able to maintain telomere length despite proliferation, while granulocytes lack the ability for further cell division [34]. Therefore, it is possible that the older individuals without dementia have a greater proportion of lymphocytes (fewer granulocytes) [35], and thus longer telomere length. Unfortunately, we were not able to adjust for the percentage of lymphocytes to overcome this issue, since this data was not available for all center visits. Another issue is that leukocytes divide peripherally in response to various stresses (e.g., infection) and the telomere length may therefore change unexpectedly. Also, research centers may vary in reliability in measuring leukocyte telomere length. Second, we measure mean telomere length, while cell senescence is suggested to be related to and modulated by the shortest telomere length per cell [36]. Third, the external validity of the results may be limited due to the single center nature of this study. Finally, despite using different approaches yielding similar results, the effect of age may not have been taken into account entirely. Nevertheless, given the link of older age with shorter telomere length and older age with Alzheimer’s disease, controlling for age would if anything strengthen the association between longer telomere length and risk of Alzheimer’s disease.
In conclusion, shorter and longer telomere length are associated with an increased risk of Alzheimer’s disease in a sample of elders in The Netherlands. Further studies are warranted to confirm our findings, in particular studies with imaging or fluid biomarkers available for dementia diagnosis, and to unravel any underlying biological pathway from possible methodological bias.
ACKNOWLEDGMENTS
We gratefully acknowledge the study participants of the Ommoord district and their general practitioners and pharmacists for their devotion in contributing to the Rotterdam Study. We also thank all staff that facilitated assessment of participants in the Rotterdam Study throughout the years. Special thanks to Jeremy Labrecque for the statistical advice.
This work was supported by the European Union’s Horizon 2020 research and innovation programme [grant number 667375] (“CoSTREAM”); the Erasmus Medical Center and Erasmus University Rotterdam; the Netherlands Organization for Scientific Research (NWO) [grant numbers 948-00-010, 918-46-615]; the Netherlands Organization for Health Research and Development (ZonMw); The Research Institute for Diseases in the Elderly (RIDE); the Ministry of Education, Culture and Science; the Ministry of Health, Welfare and Sports; the European Commission (DG XII); and the Municipality of Rotterdam.
The funding source had no role in study design, collection, analysis, interpretation of data, writing of the report or decision to submit the article for publication.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0759r2).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-190759.
REFERENCES
[1] | GBD 2016 Dementia Collaborators ((2019) ) Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18: , 88–106. |
[2] | Blackburn EH , Epel ES , Lin J ((2015) ) Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 350: , 1193–1198. |
[3] | Martinez P , Blasco MA ((2015) ) Replicating through telomeres: a means to an end. Trends Biochem Sci 40: , 504–515. |
[4] | Martinez P , Blasco MA ((2018) ) Heart-breaking telomeres. Circ Res 123: , 787–802. |
[5] | Min J , Wright WE , Shay JW ((2017) ) Alternative lengthening of telomeres can be maintained by preferential elongation of lagging strands. Nucleic Acids Res 45: , 2615–2628. |
[6] | Wang Q , Zhan Y , Pedersen NL , Fang F , Hagg S ((2018) ) Telomere length and all-cause mortality: a meta-analysis. Ageing Res Rev 48: , 11–20. |
[7] | Fasching CL ((2018) ) Telomere length measurement as a clinical biomarker of aging and disease. Crit Rev Clin Lab Sci 55: , 443–465. |
[8] | Forero DA , Gonzalez-Giraldo Y , Lopez-Quintero C , Castro-Vega LJ , Barreto GE , Perry G ((2016) ) Meta-analysis of telomere length in Alzheimer’s disease. J Gerontol A Biol Sci Med Sci 71: , 1069–1073. |
[9] | Zhang X , Zhao Q , Zhu W , Liu T , Xie SH , Zhong LX , Cai YY , Li XN , Liang M , Chen W , Hu QS , Zhang B ((2017) ) The association of telomere length in peripheral blood cells with cancer risk: a systematic review and meta-analysis of prospective studies. Cancer Epidemiol Biomarkers Prev 26: , 1381–1390. |
[10] | Rivera T , Haggblom C , Cosconati S , Karlseder J ((2017) ) A balance between elongation and trimming regulates telomere stability in stem cells. Nat Struct Mol Biol 24: , 30–39. |
[11] | Roberts RO , Boardman LA , Cha RH , Pankratz VS , Johnson RA , Druliner BR , Christianson TJ , Roberts LR , Petersen RC ((2014) ) Short and long telomeres increase risk of amnestic mild cognitive impairment. Mech Ageing Dev 141-142: , 64–69. |
[12] | Thomas P , NJ OC , Fenech M ((2008) ) Telomere length in white blood cells, buccal cells and brain tissue and its variation with ageing and Alzheimer’s disease. Mech Ageing Dev 129: , 183–190. |
[13] | Ikram MA , Brusselle GGO , Murad SD , van Duijn CM , Franco OH , Goedegebure A , Klaver CCW , Nijsten TEC , Peeters RP , Stricker BH , Tiemeier H , Uitterlinden AG , Vernooij MW , Hofman A ((2017) ) The Rotterdam Study: 2018 update on objectives, design and main results. Eur J Epidemiol 32: , 807–850. |
[14] | Cawthon RM ((2002) ) Telomere measurement by quantitative PCR. Nucleic Acids Res 30: , e47. |
[15] | Codd V , Mangino M , van der Harst P , Braund PS , Kaiser M , Beveridge AJ , Rafelt S , Moore J , Nelson C , Soranzo N , Zhai G , Valdes AM , Blackburn H , Leach IM , de Boer RA , Wellcome Trust Case Control C, Goodall AH , Ouwehand W , van Veldhuisen DJ , van Gilst WH , Navis G , Burton PR , Tobin MD , Hall AS , Thompson JR , Spector T , Samani NJ ((2010) ) Variants near TERC are associated with mean telomere length. Nat Genet 42: , 197–199. |
[16] | de Bruijn RF , Bos MJ , Portegies ML , Hofman A , Franco OH , Koudstaal PJ , Ikram MA ((2015) ) The potential for prevention of dementia across two decades: the prospective, population-based Rotterdam Study. BMC Med 13: , 132. |
[17] | Slooter AJ , Cruts M , Kalmijn S , Hofman A , Breteler MM , Van Broeckhoven C , van Duijn CM ((1998) ) Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Arch Neurol 55: , 964–968. |
[18] | Nilsonne G , Tamm S , Månsson KNT , Åkerstedt T , Lekander M ((2015) ) Leukocyte telomere length and hippocampus volume: a meta-analysis. F1000Res 4: , 1073. |
[19] | R Core Team ((2017) ) R: A language and environment for statistical computing. R Foundation for Statistical Computing. Vienna, Austria. |
[20] | Honig LS , Kang MS , Schupf N , Lee JH , Mayeux R ((2012) ) Association of shorter leukocyte telomere repeat length with dementia and mortality. Arch Neurol 69: , 1332–1339. |
[21] | Zhan Y , Song C , Karlsson R , Tillander A , Reynolds CA , Pedersen NL , Hagg S ((2015) ) Telomere length shortening and Alzheimer disease–a Mendelian randomization study. JAMA Neurol 72: , 1202–1203. |
[22] | Scheller Madrid A , Rasmussen KL , Rode L , Frikke-Schmidt R , Nordestgaard BG , Bojesen SE ((2019) ) Observational and genetic studies of short telomeres and Alzheimer’s disease in 67,000 and 152,000 individuals: a Mendelian randomization study. Eur J Epidemiol, doi: 10.1007/s10654-019-00563-w. |
[23] | Guo Y , Yu H ((2019) ) Leukocyte telomere length shortening and Alzheimer’s disease etiology. J Alzheimers Dis 69: , 881–885. |
[24] | Wikgren M , Karlsson T , Lind J , Nilbrink T , Hultdin J , Sleegers K , Van Broeckhoven C , Roos G , Nilsson LG , Nyberg L , Adolfsson R , Norrback KF ((2012) ) Longer leukocyte telomere length is associated with smaller hippocampal volume among non-demented APOE epsilon3/epsilon3 subjects. PLoS One 7: , e34292. |
[25] | Wikgren M , Karlsson T , Nilbrink T , Nordfjall K , Hultdin J , Sleegers K , Van Broeckhoven C , Nyberg L , Roos G , Nilsson LG , Adolfsson R , Norrback KF ((2012) ) APOE epsilon4 is associated with longer telomeres, and longer telomeres among epsilon4 carriers predicts worse episodic memory. Neurobiol Aging 33: , 335–344. |
[26] | Rolyan H , Scheffold A , Heinrich A , Begus-Nahrmann Y , Langkopf BH , Holter SM , Vogt-Weisenhorn DM , Liss B , Wurst W , Lie DC , Thal DR , Biber K , Rudolph KL ((2011) ) Telomere shortening reduces Alzheimer’s disease amyloid pathology in mice. Brain 134: , 2044–2056. |
[27] | Spilsbury A , Miwa S , Attems J , Saretzki G ((2015) ) The role of telomerase protein TERT in Alzheimer’s disease and in tau-related pathology in vitro. J Neurosci 35: , 1659–1674. |
[28] | Liu MY , Nemes A , Zhou QG ((2018) ) The emerging roles for telomerase in the central nervous system. Front Mol Neurosci 11: , 160. |
[29] | Flanary B ((2005) ) The role of microglial cellular senescence in the aging and Alzheimer diseased brain. Rejuvenation Res 8: , 82–85. |
[30] | Flanary BE , Sammons NW , Nguyen C , Walker D , Streit WJ ((2007) ) Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 10: , 61–74. |
[31] | Jaskelioff M , Muller FL , Paik JH , Thomas E , Jiang S , Adams AC , Sahin E , Kost-Alimova M , Protopopov A , Cadinanos J , Horner JW , Maratos-Flier E , Depinho RA ((2011) ) Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature 469: , 102–106. |
[32] | Bernardes de Jesus B , Vera E , Schneeberger K , Tejera AM , Ayuso E , Bosch F , Blasco MA ((2012) ) Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol Med 4: , 691–704. |
[33] | Lukens JN , Van Deerlin V , Clark CM , Xie SX , Johnson FB ((2009) ) Comparisons of telomere lengths in peripheral blood and cerebellum in Alzheimer’s disease. Alzheimers Dement 5: , 463–469. |
[34] | Akbar AN , Vukmanovic-Stejic M ((2007) ) Telomerase in T lymphocytes: use it and lose it? J Immunol 178: , 6689–6694. |
[35] | van der Willik KD , Fani L , Rizopoulos D , Licher S , Fest J , Schagen SB , Ikram MK , Ikram MA ((2019) ) Balance between innate versus adaptive immune system and the risk of dementia: a population-based cohort study. J Neuroinflammation 16: , 68. |
[36] | Hemann MT , Strong MA , Hao LY , Greider CW ((2001) ) The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 107: , 67–77. |


