
In Vivo Validation of a Small Molecule Inhibitor of Tau Self-Association in htau Mice
Abstract
Tau oligomers have been shown to transmit tau pathology from diseased neurons to healthy neurons through seeding, tau misfolding, and aggregation that is thought to play an influential role in the progression of Alzheimer’s disease (AD) and related tauopathies. To develop a small molecule therapeutic for AD and related tauopathies, we have developed in vitro and cellular assays to select molecules inhibiting the first step in tau aggregation, the self-association of tau into oligomers. In vivo validation studies of an optimized lead compound were independently performed in the htau mouse model of tauopathy that expresses the human isoforms of tau without inherited tauopathy mutations that are irrelevant to AD. Treated mice did not show any adverse events related to the compound. The lead compound significantly reduced the level of self-associated tau and total and phosphorylated insoluble tau aggregates. The dose response was linear with respect to levels of compound in the brain. A confirmatory study was performed with male htau mice that gave consistent results. The results validated our screening approach by showing that targeting tau self-association can inhibit the entire tau aggregation pathway by using the selected and optimized lead compound whose activity translated from in vitro and cellular assays to an in vivo model of tau aggregation.
INTRODUCTION
The critical unmet need for a disease modifying drug for Alzheimer’s disease (AD) has been highlighted by the failure in the clinic of all late stage drug development programs targeting amyloid-β. Hence, there is an urgent need for alternative approaches for the development of disease-modifying therapeutics (DMT) for AD. Long-term treatment for chronic diseases such as AD requires safe, effective, and economically feasible approaches, especially for early preventive therapeutic interventions.
Tau protein has become an increasingly important target for drug discovery based on advances in understanding its role in AD progression. Initially, the relevance of tau to AD was derived from the discovery that it was the primary component of neurofibrillary tangles (NFT) that were established as a pathological hallmark of AD more than 100 years ago [1]. The prevailing thought was that tau aggregation was merely a consequence of the disease process and not a major contributing factor for the initiation and progression of neurodegenerative disease. Progress in the field has shown that tau can be a causative factor in neurodegenerative diseases and is directly involved in the spread of pathology to neighboring neurons making it an attractive target for the development of DMT for AD and numerous tauopathies [2, 3]. It has also been shown that tau load and distribution correlate with clinical decline [4] and cortical thinning [5] as measured by positron emission tomography (PET).
Tau protein has multiple normal functions including the modulation of microtubule stability, axonal transport, modulation of signaling pathways, and adult neurogenesis [6]. Recent work has shown that toxicity is mediated by a gain of toxic function caused by tau that has become modified by conformational changes accompanied by oligomer formation and aggregation. Tau pathology is associated with the propensity of tau to aggregate and, conversely, the prevention of tau aggregation by mutations preventing the formation of beta-sheet structure [7, 8]. These studies indicate that preventing tau aggregation and subsequent spreading could be an efficacious therapeutic approach for AD-related dementias (ADRD) [9, 10].
Our therapeutic approach is based on the premise that tau oligomers are the acutely toxic aggregated species of tau and their reduction will modify the course of AD. Multiple studies have shown that tau oligomers, not fibrils or tangles, are closely correlated with neuronal loss and memory impairment [11–17]. Tau oligomers also have a causal role in mediating cytotoxicity, disease propagation, and mitochondrial damage [18]. Importantly, tau oligomers cause disruption of neuronal signaling and inhibit the formation of memory in mice. Memory formation was impaired following administration of oligomeric tau to hippocampi, whereas similar treatment with tau monomer did not have an effect. This impairment of memory was also found using oligomers formed from hyperphosphorylated tau purified from human AD brain specimens. Memory-specific mechanisms involved in gene regulation were shown to be disrupted by these extracellular tau oligomers [19, 20]. Tau oligomers have been shown to cause synaptic impairment in other studies as well [21, 22]. Certain forms of tau oligomers are toxic when applied to cultured neurons, whereas tau monomer was not toxic at the same concentrations [23]. The tau oligomer target for the development of therapeutics has been validated in htau mice in studies using treatment with curcumin [24] or by a passive immunotherapeutic approach directed at tau oligomers [25]. The observation that tau pathology progresses to synaptically connected regions of the brain [26, 27] led to the hypothesis that tau can transmit its own pathology from a diseased to a healthy neuron. Recent studies show that tau aggregates and specifically tau oligomers isolated from AD brain may act as templates for the misfolding and aggregation of native tau, thereby seeding the spread of the toxic forms of the protein [28].
These studies indicate that targeting tau oligomers should improve learning and memory and inhibit disease progression in AD and related tauopathies. Tau aggregation inhibitor (TAI) programs have primarily focused on targeting the large and relatively inert fibrils [29–31].
In contrast, our work has focused on the development of a DMT for AD using a small molecule approach targeting tau oligomers by selecting molecules that inhibit tau self-association to prevent the initial step in tau aggregation, thereby reducing the accumulation of all tau aggregates. A lead series of compounds has been optimized for CNS penetration and drug-like properties. A library of compounds with drug-like properties was screened using an assay designed to specifically inhibit the self-association of tau monomers. This differentiated assay used 1) full-length 4R2N tau without mutations, 2) physiologically relevant pH and concentrations of tau, and 3) a proximity-based system for detection of tau self-association within a short incubation time. We selected 57 drug-like molecules predicted to have good CNS penetration from 11 chemical series and 19 singletons. The most active hits of 8 series were chosen as tool compounds and for lead development. Some of these compounds showed good dose response in a cell assay of tau aggregation. The lead compound selected for in vivo studies showed both a good preliminary safety profile using in vitro pharmacology assays, a mini-Ames test, and CNS drug-like properties differentiating it from published TAI [32–35].
The aim of this study was to determine the efficacy of the lead compound in preventing the accumulation of tau aggregates in the htau mouse model best representing tau aggregation in AD. These studies were performed to validate the in vitro and cellular screening assays for selecting compounds and to further advance the lead compound toward preclinical development.
MATERIALS AND METHODS
Animals
The htau mouse model of tauopathy expresses the six CNS isoforms of human tau protein under control of the human tau promoter and in place of endogenous murine tau [36]. There are no mutations in tau in this mouse model making htau an ideal model for studying the development of tau pathology in AD, as there are no mutations in tau associated with AD. For the primary study, 100 male and female mice were used, and in the confirmatory study 45 male htau mice were used. htau mice (Stock No: 005491) were ordered from The Jackson laboratory (Bar Harbor, Maine) and shipped to the Feinstein Institute for Medical Research (FIMR, Northwell Health, Manhasset, NY) where they were housed, treated and analyzed. For controls, tau knockout (KO) and JNPL3 mice were ordered from Taconic Biosciences (Rensselaer, NY), and wild type C57/Bl6 from The Jackson Laboratory (Bar Harbor, Maine). All experiments were in compliance with the FIMR Animal Care and Use Committee.
Antibodies
The antibodies used in these studies were all developed, produced, and formatted for assays in the laboratory of Peter Davies, Ph.D., Director, Litwin-Zucker Center for Alzheimer’s Disease & Memory Disorders, The Feinstein Institute for Medical Research (Manhasset, NY). Pan-tau antibody mAb DA31 epitope spans amino acids 150–190 in 4R2N tau [37]. Additional assays were performed for phospho-tau epitopes pertinent in AD using antibodies PHF-1 (pSer-396/404) [38, 39], CP13 (pSer-202), [40, 41], RZ3 (pThr-231) [42], and MC1 that binds an AD-specific discontinuous epitope of tau, 7EFE9 and 313VDLSKVTSKC322 [43, 44].
Study design
A preventive study was performed treating mice from 2.5 to 6.5 months of age using administration of compound in feed. This enabled stress-free and harm-free administration compared to oral gavage or intraperitoneal injection for large numbers of mice for a relatively long study. One hundred mice were divided into four groups of mixed male and female mice that were treated with feed vehicle or feed formulated to provide a daily dose of 10, 40, or 100 mg/kg mouse. The dose was estimated using an average body weight of 25 g and an average daily consumption of 5 g of feed. The study was performed independently at The Feinstein Institute for Medical Research. The primary endpoint of the study was reduction of insoluble tau aggregates in the brains of the mice with statistical significance. The secondary endpoints were dose-dependent reduction of insoluble tau aggregates, reduction of phosphorylated tau, and reduction of soluble tau. To confirm the findings from the first study in the male mice, a second study was performed using a similar approach in male htau mice. Three groups of male mice (n = 15) were treated for 120 days from three to seven months of age with vehicle, 10 or 40 mg compound/kg mouse dosing in feed. Additionally, the feed formulations were blinded without the use of food coloring for identification.
Feed preparation
The lead compound was administered to mice in the feed. For feed preparation, Open Standard Diet with 20 kcal% Protein, 65 kcal% Carbohydrate, and 15 kcal% Fat (Research Diets, New Brunswick, NJ) was milled without or with the lead compound to produce four lots of feed (0, 50, 200, and 500 mg lead compound per kg diet). Food Drug and Cosmetic Act approved food coloring was milled into the feed to code the lots of feed by color. The compound was formulated to deliver daily doses of 0, 10, 40, or 100 mg/kg/day based on an average consumption of 5 g/day and an average weight of 25 g/mouse. Control mice were fed the same diet without compound (vehicle). The diet used for the confirmatory study was the same as the primary study but it did not contain food dyes. Samples of feed at each dose were randomly selected and assayed in duplicate at the end of treatment to qualify the stability of the compound in the feed. LC-MS/MS analysis of the feed samples was carried out (Quintara Discovery, Hayward, CA) to determine the levels of compound in the feed.
Sample preparation
The methods for preparing and analyzing the specimens used in this study are briefly described as they have been previously published in detail [45–48]. Brain tissue samples were collected in the middle of the wake cycle following the treatment phase of the study. Mice were deeply anesthetized using isoflurane and sacrificed by decapitation using IACUC approved protocols. Mouse brains were divided at the midline; one half was fixed in 4% paraformaldehyde for immunocytochemistry, and the other half was used for biochemical assays of the forebrain and hippocampus. Organs were collected for tissue pathology assessment by first removing the heart and lungs followed by the spleen, liver, and kidneys and transferring to 10% formalin for 24 h for fixation. After organs were documented, they were transferred to a solution of Cryoprotectant (20% glycerol, 2% DMSO, 1x PBS) and stored at 4°C. Serum was prepared from the mice that were bled after sacrifice. Blood was allowed to clot for 30 min at ambient temperature and cleared serum was collected following two rounds centrifugation at 14,000 x g for 10 min at 4°C and stored at –80°C.
Three types of brain preparations were performed for assays to determine the levels of total self-associated tau, Sarkosyl-insoluble tau aggregates, and soluble tau in the heat stable (HS) fractions [46]. For self-associated tau, brain tissue from forebrain or hippocampus was homogenized in 10 volumes of cold Tris-buffered saline, pH 7.4 containing protease and phosphatase inhibitors, cleared by low-speed centrifugation, and stored at –80°C. To prepare Sarkosyl-insoluble tau, the cleared homogenate was incubated for 10 min with 1% Sarkosyl, the insoluble tau was pelleted by ultracentrifugation at 200,000 x g for 30 min and washed with homogenization buffer. The pellet was suspended in 1x Laemmli sample buffer and heated for 10 min at 100°C to dissociate the tau for analysis by ELISA. HS fractions of brain tissue were prepared by homogenization in 10 volumes of ice-cold buffer (50 mM Tris, pH 7.5, 0.8 M NaCl, 5% β-mercaptoethanol), and the cleared homogenate was heated at 100°C for 10 min to precipitate most proteins. Tau has the special characteristic of remaining soluble under these conditions facilitating its purification in the supernatant/HS fraction. Supernatants were cooled and dialyzed against 50 mM Tris pH 7.5 with 1 mM EDTA, 0.1 mM PMSF.
Tau ELISAs
Tau sandwich ELISAs for total tau and phosphorylated tau epitopes were performed with capture antibodies DA31, PHF1, CP13, and RZ3 and all ELISAs used reporter antibody DA9-HRP as described [46]. The tau mono-antibody ELISA was performed using DA9 for both capture antibody and DA9-HRP for reporter antibody [46].
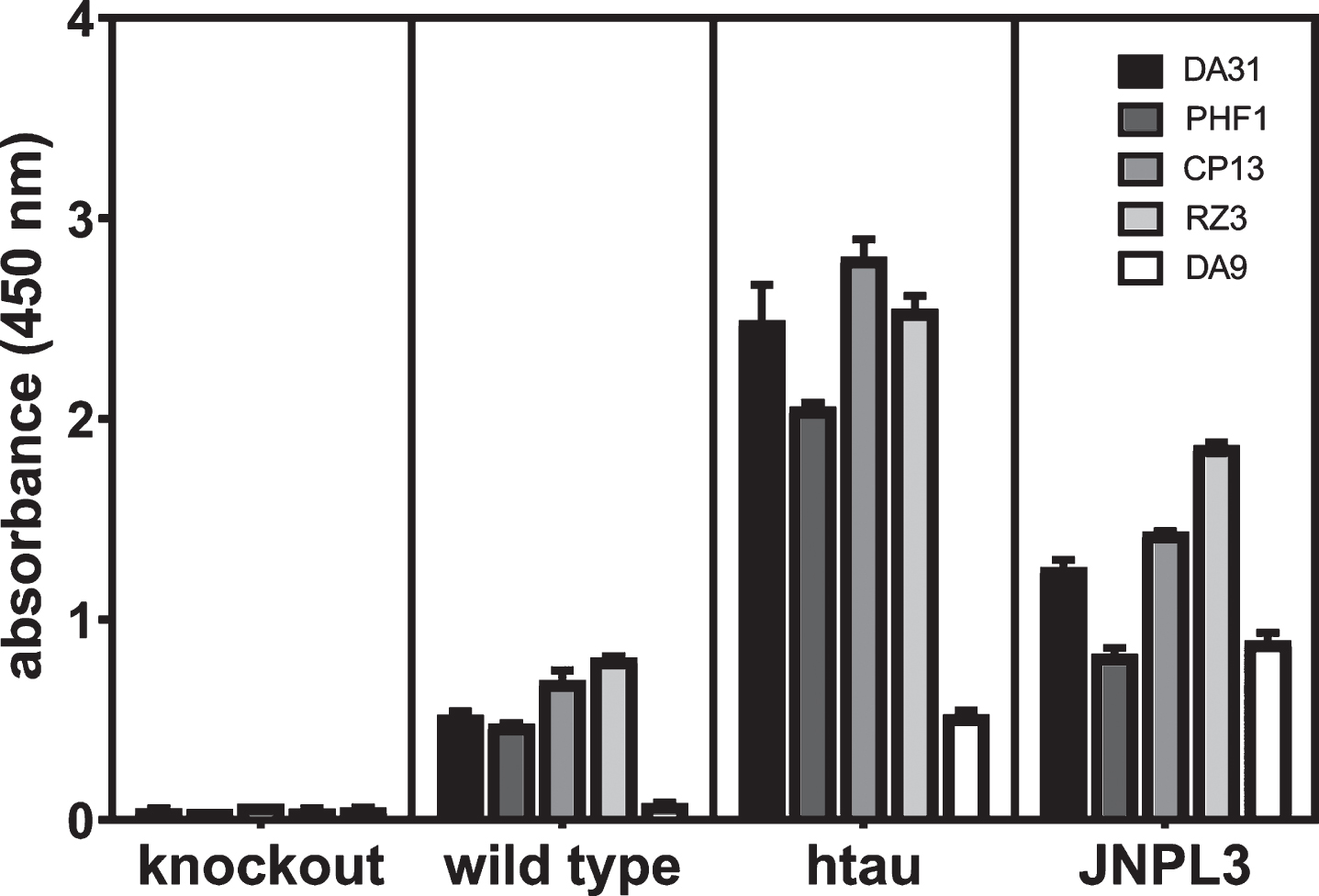
In the cited paper, the wild type mice (C57/Bl6), tau knockout (KO) mice, and normal human brain did not give any signal, whereas brain tissue from htau mice, P301L mice, and AD brain did give a signal. The basis for mono-antibody ELISAs detecting self-associated protein is the use of the same mAb specific for a single epitope in the protein for both the capture and reporter antibodies. Thus, signal is only generated when at least two of the same protein are self-associated [49]. For rigor, the reproducibility of the ELISA formats used in these studies and their performance was evaluated by repeating the assays with control specimens from seven-month-old mice. There was no signal for any of the ELISA formats for total tau, phosphorylated tau, or self-associated tau from the tau KO mice, but there were higher levels of signal in the aged htau and JNPL3 mice (expressing the human tau 4R0N isoform with the P301L mutation) over the wild type mice indicating the method was robust and reproducible (Fig. 1).
Fig.1
Evaluation of the reproducibility and rigor of the ELISAs in these studies. Heat stable fractions from seven-month-old female tau KO, C57/Bl6 wild type, and JNPL3 mice were used for ELISAs measuring total, phosphorylated, and self-associated tau, and total homogenate was used for the assay for self-associated tau as described in the Methods for ELISAs. Serial dilutions of each sample were tested in duplicate at each concentration (4.0, 2.0, 1.0, 0.5, 0.25, and 0.125 ug/ml); the values from the highest sample concentration within the range of the standard curve for each ELISA format are presented.
Immunoblot
Sarkosyl-insoluble preparations from cortex, solubilized in sample buffer were run on 18-well 4-20% polyacrylamide precast Criterion gels (Bio-Rad, Hercules, CA), and protein was transferred to PVDF membrane using the Trans Blot® Turbo™ transfer system (Bio-Rad). The FluorChem R™ (Protein Simple, San Jose, CA) imager was used to capture images, and quantification of signal was performed using AlphaView software (Protein Simple).
Immunocytochemistry
Serial sections were cut with a vibratome from the fixed brain tissue and were immunostained with either mAb MC1 or RZ3 using standard methods [48]. The mAb MC1 recognizes a conformational epitope in misfolded tau [43, 44]. This epitope is present in both soluble forms of tau and in paired helical filaments (PHF) [50, 51] and MC1 reactivity correlates with the severity and progression of AD [52].
Statistical analysis
All statistical analyses were performed using Graph-Pad Prism 7.04 (GraphPad Software, San Diego, CA). Outliers were removed using the ROUT (Q = 1%) method. Unpaired t-tests were used for comparisons of two groups with normal Gaussian distributions; otherwise nonparametric analyses were performed with the Mann Whitney test. One-way analysis of variance (ANOVA) was used for parametric analyses and the Kruskal-Wallis test was used for non-parametric analyses. Error bars indicate standard error of the mean (SEM).
RESULTS
The lead compound was stable and well tolerated by mice at all doses tested
The stability of the compound in the feed over the course of the study was confirmed by determining the level of compound in the lots of feed at the conclusion of the study and comparing them to their original formulations. During the efficacy study the compound was well tolerated at all doses. There were no adverse events or behavioral abnormalities due to the compound, and both male and female control and treated mice exhibited similar weight gain during the study. Prior to the efficacy study the toxicity of the lead compound was assessed by 5-day dosing performed by oral administration (P.O.) with 5 male and 5 female CD-1 mice using 100 mg/kg, the highest dose for the efficacy study. No indication of toxicity or body weight loss was observed during the 5-day dosing period which de-risked the efficacy study for failure due to mouse attrition.
Self-associated tau was reduced in the hippocampus
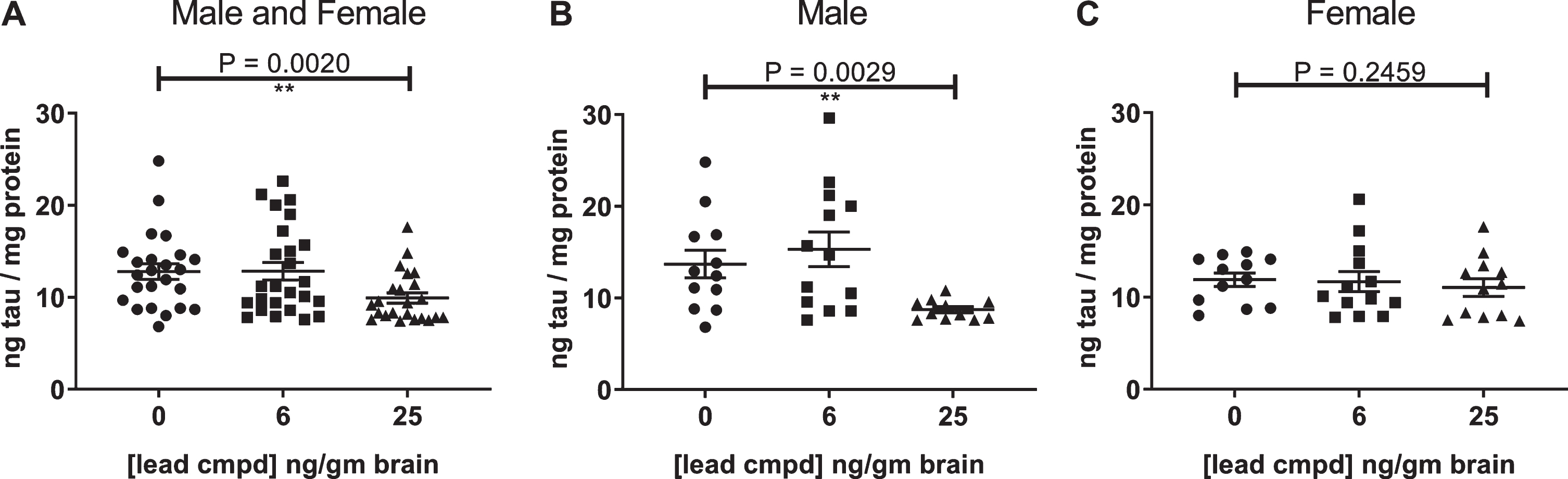
Analysis of the hippocampal homogenates from the male and female htau mice using the DA9 mono-antibody ELISA for tau showed a statistically significant decrease in the level of self-associated tau in the group with the highest level of compound in the brain for the mixed male and female mice or male mice alone, whereas female mice showed a trend in reduction of self-associated tau (Fig. 2).
Fig.2
Self-associated tau in the hippocampus of male and female mice (A), male mice (B), female mice (C) was measured by mono-antibody ELISA formatted with capture antibody DA9 (epitope, tau 102–140) and reporter antibody DA9-HRP. Outliers were removed using the ROUT (Q = 1%) method. p-values for analyses between the control group and the group with 25 ng lead cmpd/gm brain are shown above plots. Analysis between the groups determined statistically significant differences from each other for the male and female mice (Kruskal-Wallis test p = 0.0104) and for the male mice (one-way ANOVA p = 0.0086). **p-value < 0.01.
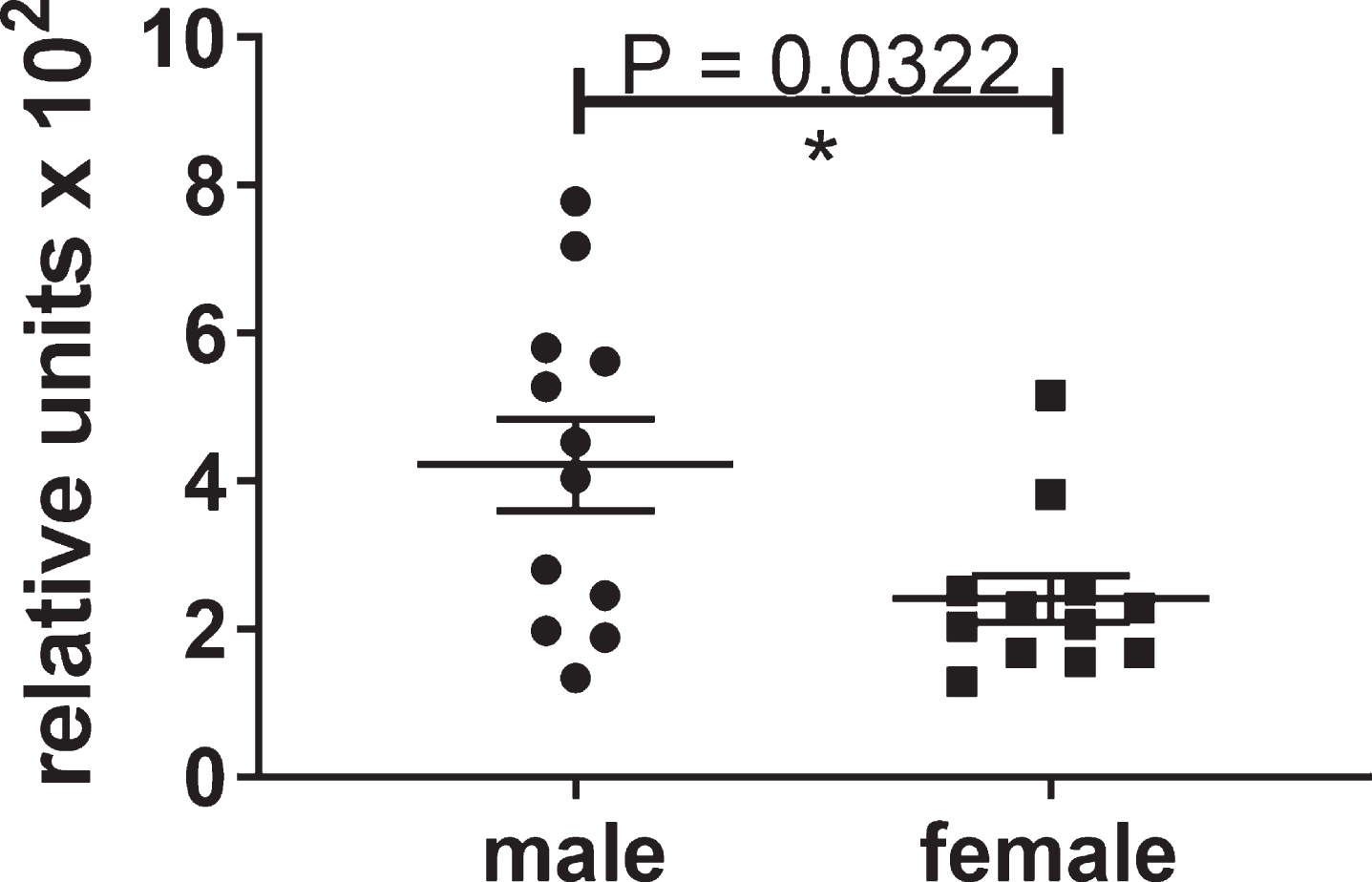
Male mice developed more insoluble tau aggregates than female mice
At the endpoint of the study, the untreated male htau mice had about twice the level of insoluble tau as the untreated female mice at 6.5 months of age (Fig. 3). Therefore, the results for the male htau mice are reported because the lack of insoluble tau in the female mice precluded their use for determining the efficacy of the compound at this age.
Fig.3
Differential levels of insoluble tau aggregates in untreated male and female htau mice at 6.5 months of age. These significant results show that the control group of male htau mice developed about twice the level of insoluble tau aggregates as the female control group at the end of the study. This justified the separate analyses of the male mice which developed enough insoluble tau to determine the efficacy of the lead compound. The levels of Sarkosyl-insoluble tau aggregates were determined by ELISA for total tau formatted with capture antibody DA31 (pan-tau) and reporter Ab DA9 (pan-tau) conjugated to HRP. The values were normalized to total tau in the heat stable fractions of forebrain.
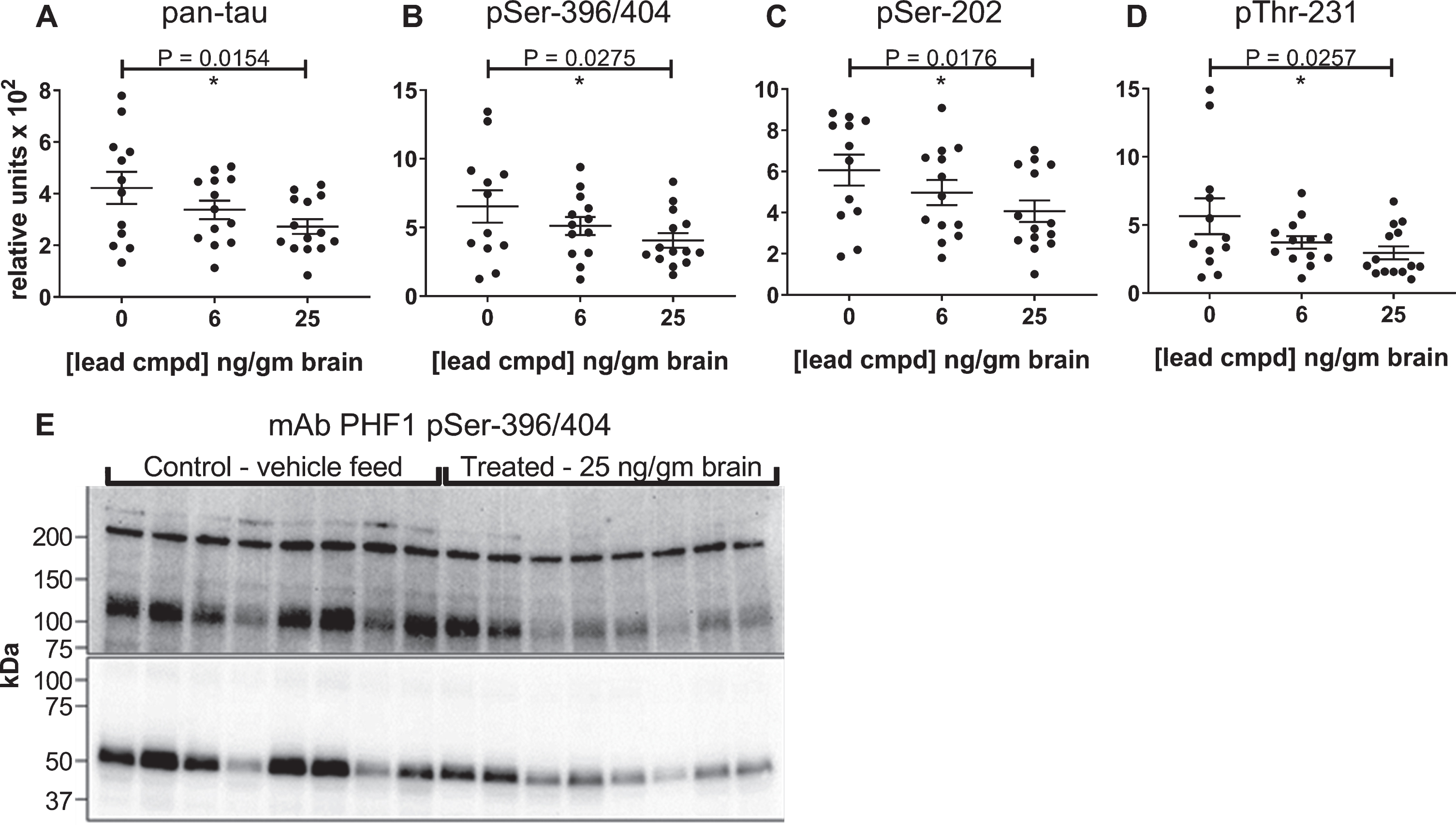
Levels of insoluble tau aggregates were reduced with statistical significance, and the lead compound exhibited a linear dose dependent response with respect to levels of compound in the brain
The biochemical method used to prepare the insoluble protein indicated that the tau in the Sarkosyl-insoluble pellets was associated with aggregated protein, and the ELISA formats provided quantification of the overall levels of total and phosphorylated tau within these pellets. The lead compound caused a statistically significant, linear, dose-dependent reduction of the levels of insoluble tau aggregates and phosphorylated insoluble aggregates in relationship to the levels of compound in the brain (Fig. 4A-D). To further characterize the molecular weights of tau aggregates within the Sarkosyl-insoluble pellets immunoblot analyses were performed. Denaturing methods were required to solubilize the proteins in the pellets (2% SDS, 5% β-mercaptoethanol and heating at 95°C) in order to liberate the tau for quantitation by ELISA and characterization by immunoblot, therefore most of the signal from the immunoblots was from tau monomer species. Within the pellet of aggregated protein, there were additional SDS gel-stable higher molecular weight species of phosphorylated tau that were detected with the PHF1 antibody that were consistent in size with dimer (∼120 kDa), and higher MW oligomers (>200 kDa). The PHF1 immunoblot shows both overall higher levels of phosphorylated tau and aggregated tau in mice from the control group compared to the treated group, consistent with results from the ELISAs (Fig. 4E).
Fig.4
Reduction of insoluble tau levels are dependent on compound concentration in the brain. Sarkosyl-insoluble tau preparations were evaluated by ELISA using pan-tau and p-tau antibodies DA31 (A), PHF1 (B), CP13 (C), and RZ3 (D). Levels of insoluble tau were normalized to total soluble tau in the heat stable fractions of each mouse forebrain and plotted against the average value of compound found in the brains of male C57BL/6 mice. The number of mice in each group: 0 ng/g (n = 12), 6 ng/ng (n = 13), 25 ng/g (n = 14). p-values for pairwise analyses of the control group and the group with 25 ng lead cmpd/gm brain are shown above plots. Immunoblot comparison of vehicle control group with treated group (E). Sarkosyl-insoluble tau preparations from eight mice from each group were run on 4–20% polyacrylamide gradient gel and transferred to a PVDF membrane. Images of short and long capture of chemiluminescent signal are presented in the lower and upper images of the blot, respectively, to show both abundant monomer and less abundant higher molecular weight P-tau species.
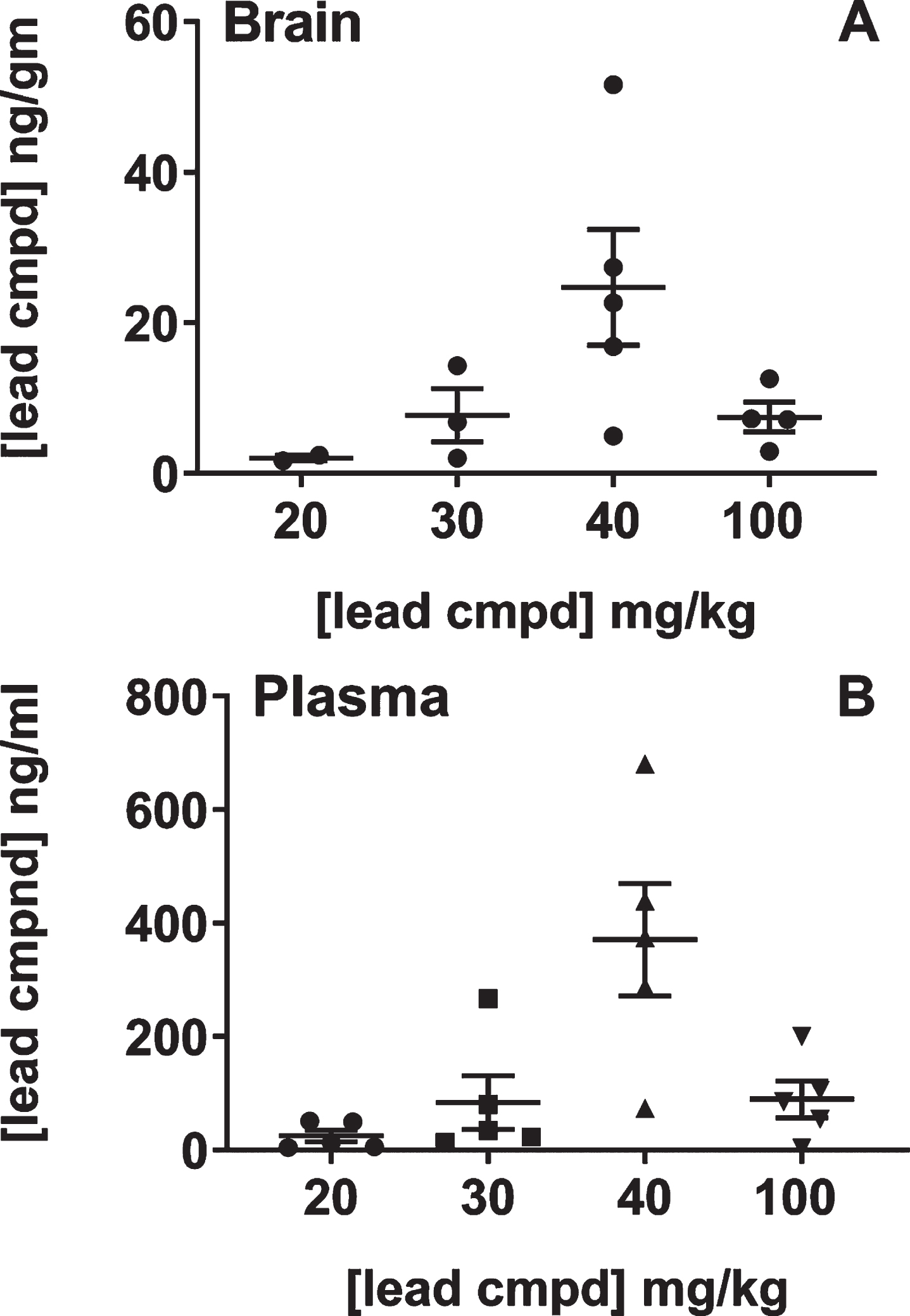
The relationship between the dose in feed and levels of compound in the brain and plasma was studied because of the unexpected result that 40 mg/kg dose was more efficacious than the 100 mg/kg dose. The study was performed with male C57Bl/6 mice as the htau mice have a mixed genetic background that includes C57Bl/6 [37]. Most of the mice in the groups that received the 20 or 30 mg/kg dose in feed had levels of compound in the brain that were below the quantitative limit suggesting that there was a threshold dose of compound in the feed necessary to achieve bioavailability in the brain. The mice treated with 40 or 100 mg/kg dose in feed had an average of 25 or 6 ng/g in brain, respectively. Treatment with the 100 mg/kg dose in the feed led to 3-4-fold lower levels of compound in the brain than the 40 mg/kg treatment group, also indicating an upper limit for dose levels using administration in feed for bioavailibility of the compound in the brain (Fig. 5). Together, these studies showed that there is a linear relationship between efficacy and levels of compound in the brain and explained the non-linear dose response relative to the compound formulations. Additionally, there was a strong positive correlation between plasma and brain compound levels suggesting that plasma levels were driving the concentrations of compound in the brain.
Fig.5
Analysis of brain and plasma levels of lead compound with respect to formulated doses in feed. Groups of male C57BL/6 mice (n = 5) were treated with feed formulated to provide the doses shown on the plot for seven days. Mice were individually caged on the last two days in order to determine the amount of feed each mouse consumed (Murigenics, Vallejo, CA) prior to analyzing compound levels in the brain (A) and plasma (B) by LC-MS/MS (Quintara Discovery, Hayward, CA). Values below quantifiable levels were not included in the groups treated with 20, 30, or 100 mg/kg doses.
Immunocytochemistry with mAb MC1
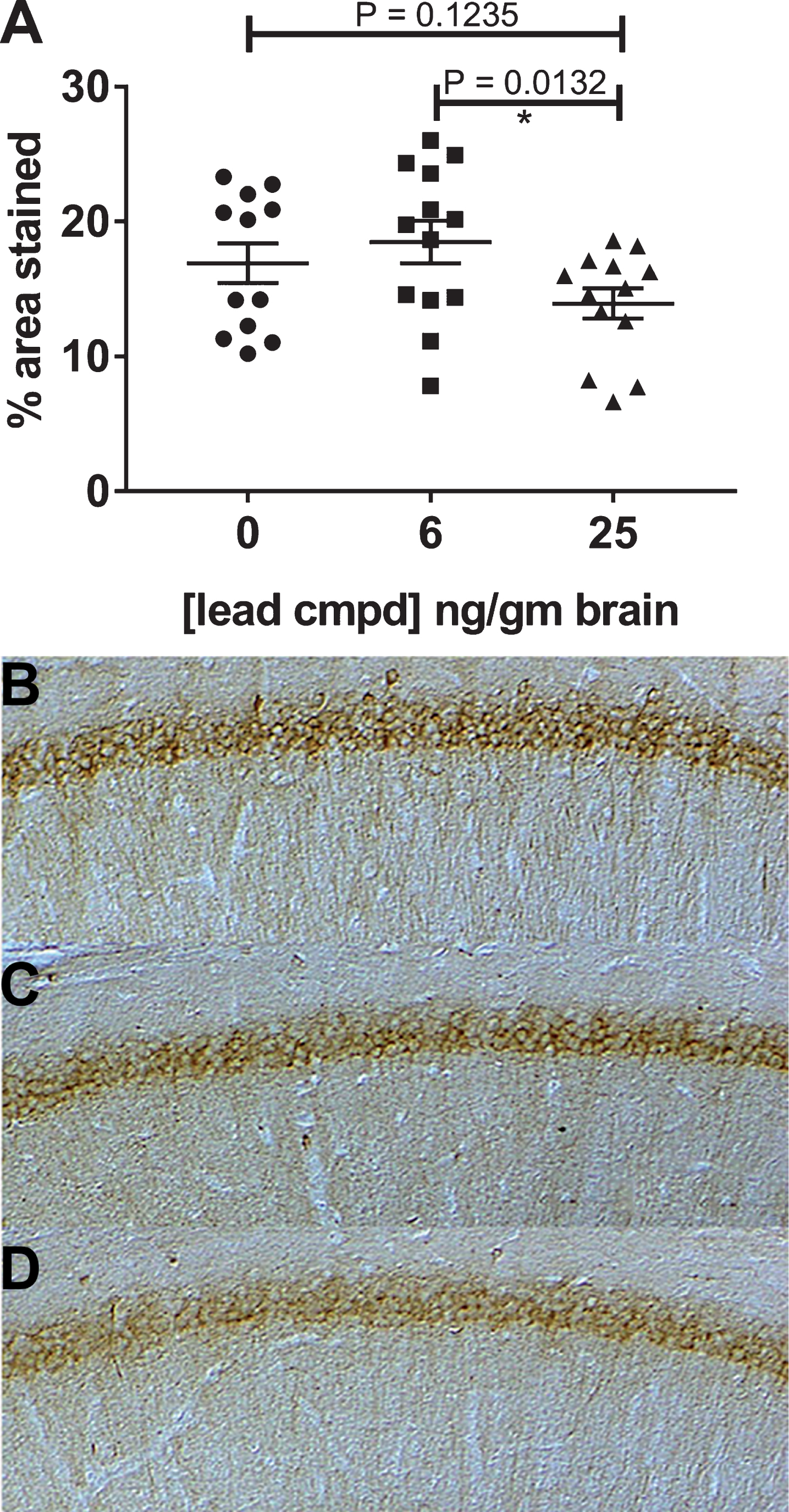
MC1 staining in cortical sections for the treatment groups was similar to the vehicle group. Immunocytochemistry of hippocampal sections with mAb MC1 showed reduction of staining in mice treated with maximally effective dose (Fig. 6). Treatment with the 40 mg/kg dose reduced the accumulation of misfolded tau associated with tau aggregates in the hippocampus and corroborates the data indicating reduction of insoluble tau aggregates and self-associated tau (Figs. 4 and 6) in this treatment group.
Fig.6
Immunohistochemistry of hippocampal slices of male mice. The mAb MC1, recognizing misfolded pathological tau, was used to stain the hippocampal slices from each mouse. Treated mice with 25 ng compound/g brain had significantly reduced levels of MC1 dependent pathological tau compared to treated mice with 6 ng compound/g brain (A). Representative images of the hippocampal region stained with mAb MC1 are shown for the vehicle control (B) and treatment groups 6 and 25 ng/gm brain (C, D).
Analysis of the heat stable fractions from the forebrain and hippocampi of the male mice showed statistical equivalence for levels of total tau and phosphorylation for the four groups except for a 25% increase for phosphorylation at the PHF1 epitope for tau in the forebrain for the group treated with the 40 mg/kg dose compared to the control group (not shown).
Confirmatory study in male htau mice
Compound stability in the feed was tested over the course of the study and was stable confirming that the mice were receiving the estimated dose throughout the study. The groups of mice gained weight at a similar rate during the study.
Exposure evaluated using mice sera
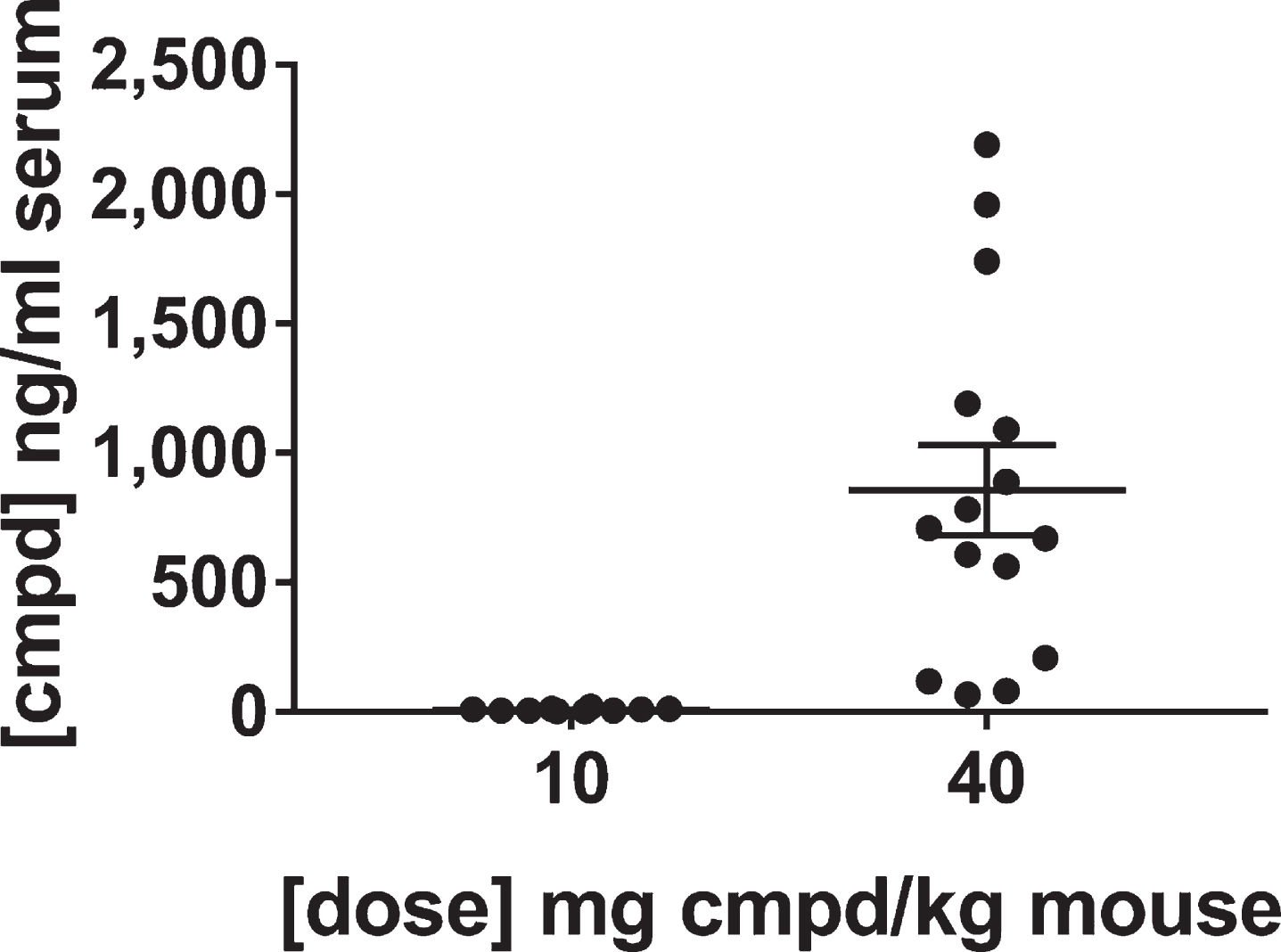
The levels of the lead compound were determined in the sera of the mice at the end of the study. The results confirmed the groups of mice received the appropriate lots of feed. As expected, the feed vehicle did not produce signal, whereas the levels of compound in the group treated with 40 mg/kg dose were on average over 100-fold greater than the group treated with the 10 mg/kg dose. The average value for the compound concentration in the sera was 8.1 ng/ml for the group treated with the 10 mg/kg dose and 857 ng/ml for the group treated with 40 mg/kg dose (Fig. 7). These results are consistent with the dose response bioavailability analysis in brain and plasma (Fig. 4). The variability in exposure between mice at the end of the study is due to the variability in feed consumption over the course of the day, but over the long-term course of treatment the mice should have received equivalent dosing within groups.
Fig.7
Levels of lead compound in the sera of mice in the confirmatory study. Blood and brain specimens were collected at the end of treatment after sacrifice in the middle of the wake cycle and analyzed by LC-MS/MS for levels of the lead compound. Perfusions of the brains were not performed because that would have affected the post-translational modifications of tau. Thus, the brain lysates contained compound from the blood in the brain tissue that precluded their use for determining levels of the compound in the brain.
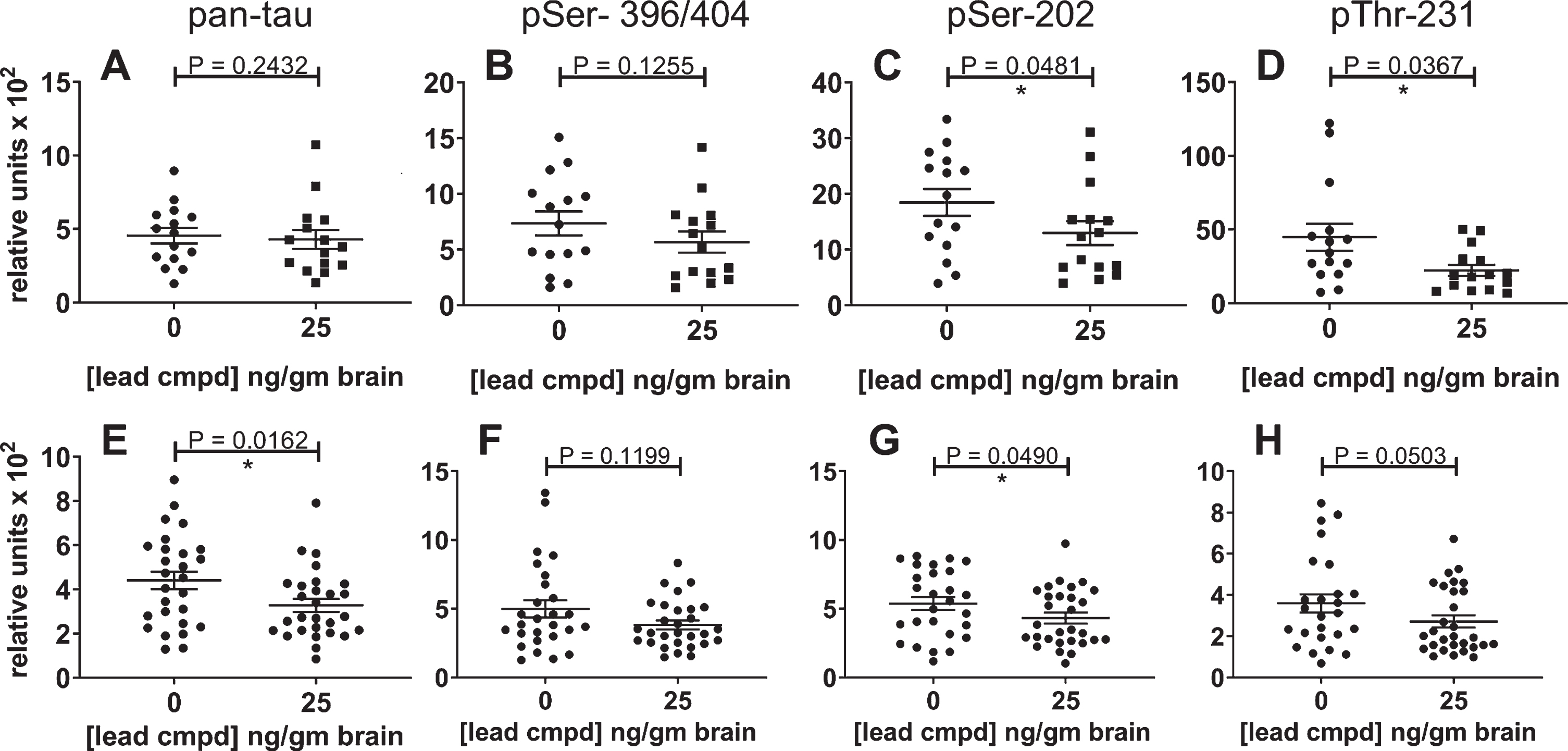
The maximal effective dose in the primary study reduced insoluble tau in the confirmatory study
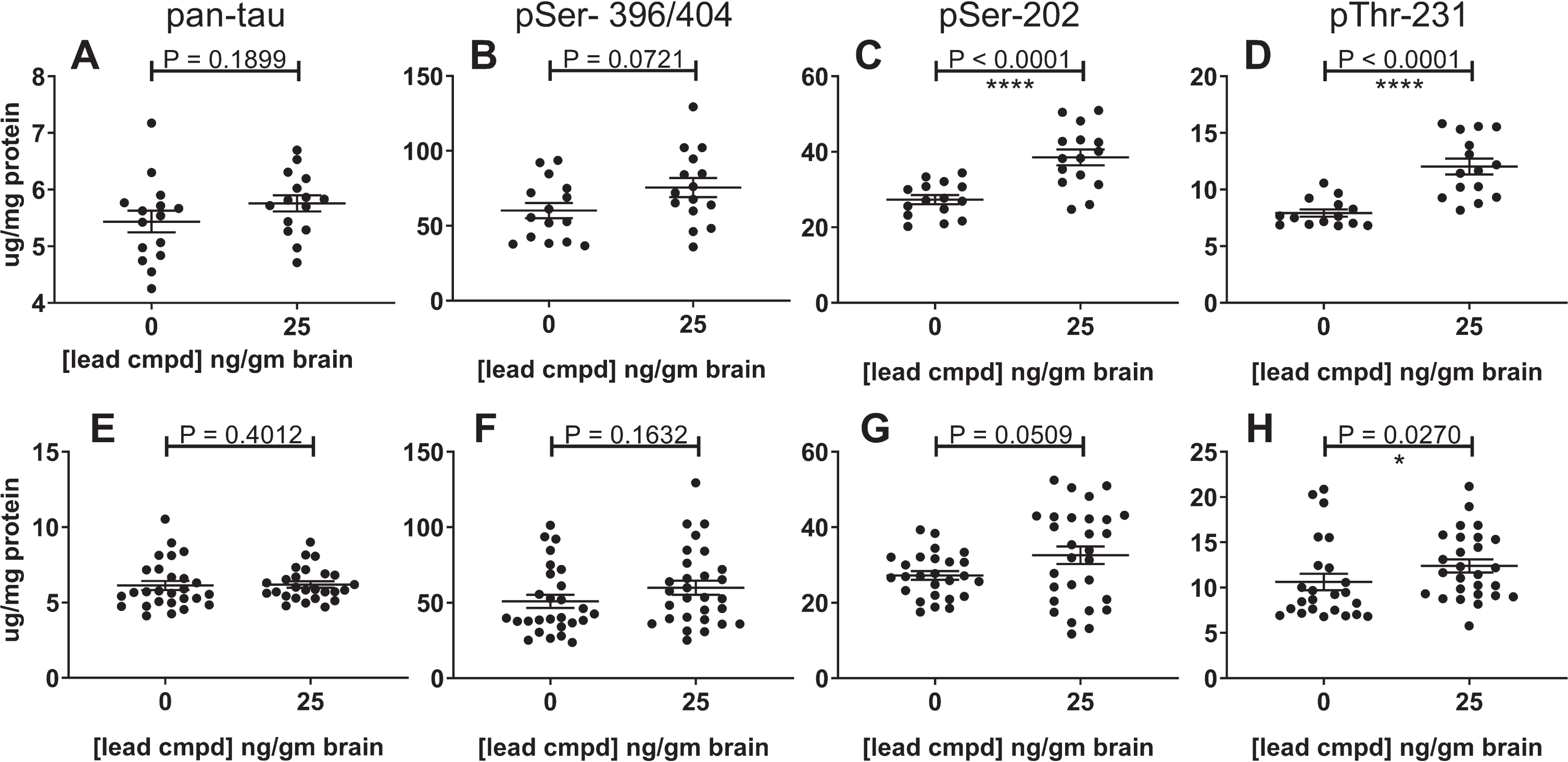
Levels of total insoluble tau and phosphorylated insoluble tau were reduced by 25 ng/g of compound in the brain. There was a trend in reduction for mAbs DA31 and PHF1, and statistically significant reduction with mAbs CP13 and RZ3 (Fig. 8A-D). Data from both studies were combined for the control or treated male mice to increase the group sizes for analysis (Fig. 8E-H). Total insoluble tau was reduced with statistical significance (Fig. 8E), and the levels of phosphorylated forms of insoluble tau were also reduced (Fig. 8F-H).
Fig.8
Reduction of insoluble tau in the confirmatory study and combined study analyses. The results for the confirmatory study are shown independently and in combination with the results of the primary study. The most significant result was found in the combined data analysis of the Sarkosyl-insoluble tau aggregates using the ELISA for total tau (E); it indicates the overall efficacy of lead compound in these studies. The levels of phosphorylated insoluble aggregates in the treated group were also reduced. The number of mice for the confirmatory study groups was 15 (A-E) and for the combined analyses were n = 27 for vehicle control mice and n = 29 for treated mice (E-H). The results for the insoluble phosphorylated tau (PHF1, CP13, or RZ3) were normalized to the respective levels of phosphorylated tau in the heat stable fractions (A-E). Analysis of the combined results for phosphorylated tau were normalized to the results of total tau from the pan-tau ELISA using mAb DA31.
Total and phosphorylated soluble tau levels in the forebrain and hippocampus
The hippocampal heat stable fractions in the confirmatory study showed statistical equivalence for levels of total tau and phosphorylated tau, but in the forebrain there was a highly significant increase of tau phosphorylated at the CP13 and RZ3 epitopes in the heat stable fractions of mice with 25 ng/g brain levels of compound (Fig. 9A-D). The combined data showed a similar result (Fig. 9E-H). It is possible that the reduction in the level of insoluble phosphorylated tau may play a part in the increase of soluble phosphorylated tau. The increase in tau phosphorylation is limited to the forebrain at the PHF1 epitope in the primary study and at the CP13 and RZ3 epitopes in the secondary study suggesting different amounts of kinase and/or phosphatase activity in the forebrain and hippocampus of these mice.
Fig.9
Effect of lead compound on levels of total and phosphorylated soluble tau in the heat-stable fractions from the forebrain. The results from the confirmatory study are shown in panels A-D (n = 15) and from the combination of data from the primary and confirmatory studies in panes E-H (vehicle control group n = 27; treatment group n = 29). The soluble tau from the forebrains of the treated mice showed an increase in phosphorylation at the epitopes indicated. The increase was highly significant in the confirmatory study for pSer-202 (C) and pThr-231 (D), but less significant in combined analyses of the studies (G, H). The results from the P-tau ELISAs using PHF1, CP13, or RZ3 were normalized to the results of total tau from the pan-tau ELISA using mAb DA31.
DISCUSSION
The goal of these studies was to evaluate the efficacy of our lead compound in reducing the level of tau aggregates in a mouse model of tauopathy relevant to AD. The htau mouse model of tauopathy [11] was chosen to evaluate the efficacy of our lead compound because of its relevance to AD. It expresses all six human tau isoforms, without mutation or truncations, under the control of the human tau promoter in the absence of endogenous mouse tau. Six months of age was found to be the earliest age in htau mice at which sufficient tau pathology develops to enable evaluating the efficacy of therapeutics [36]. Here, aggregated tau accumulated in Sarkosyl-insoluble pellet preparations from vehicle control mice that also contained SDS gel-stable higher molecular weight species of tau detected by mAb PHF1 (Fig. 4E) confirming that phosphorylated tau was aggregating in these htau mice by seven months of age. Other mouse models of tauopathy such as rTg4510 [12], P301S [15], and JNPL3 [42] develop tau pathology more rapidly due to the use of tau constructs with mutations found in inherited forms of tauopathies, but not AD, and high levels of expression [12] that make tau more prone to aggregate.
The results show that the primary endpoint of reducing insoluble tau levels with statistical significance was achieved. Similarly, there was a significant reduction in the total levels of self-associated tau which was anticipated as compounds were screened for inhibiting tau self-association to prevent the formation of tau oligomers. Taken together, these results show that the lead compound reduced self-association of soluble tau and inhibited formation of insoluble tau aggregates. The activity translated from in vitro and cellular assays to an in vivo model of tau aggregation. This validated our screening approach and demonstrated that targeting tau self-association can inhibit the entire tau aggregation pathway.
Studies are in progress to further address questions regarding whether the lead compound may have therapeutic efficacy, can ameliorate behavioral deficits, or have benefit for treating inherited forms of tauopathy. Preventive and therapeutic studies are being conducted in the JNPL3 mouse model of tauopathy that expresses the human tau 4R0N isoform with the mutation P301L associated with frontotemporal dementia. These mice develop a motor impairment caused by hind limb weakness [41]. Additionally, therapeutic studies are being carried out using aged htau mice that develop cognitive impairment by 12 months of age [53].
Evidence for treatment efficacy was based on results obtained with the male mice because the female mice did not develop sufficient insoluble tau by seven months of age (Fig. 3). This observation would have been strengthened by the inclusion of a baseline cohort to indicate the level of aggregated and phosphorylated tau at the beginning of treatment at two months of age. The result that the male htau mice developed significantly more tau pathology than the females at the end of the study was unexpected as the initial characterization of the htau model did not indicate sex-based differences [11, 36]. However, more recent studies on the effects of repetitive mild head injury have shown that male htau mice have statistically significant increased levels of phosphorylated tau recognized by mAbs RZ3 and PHF1 at 1- or 15-days post-injury, whereas the female mice did not show these changes [54, 55]. Similarly, in the PS19 mouse model of tauopathy, expressing the human tau isoform 4R1N with the P301S mutation, males developed more pathology than the females [56]. Susceptibility of male mice to the development of aggregated and phosphorylated tau is not universal; female JNPL3 mice, expressing the human tau isoform 4R0N with the P301L mutation, develop insoluble and hyperphosphorylated tau aggregates at a much faster rate than the males [57]. Importantly, a recent study of men and woman on the AD trajectory has shown that women have elevated tau deposits compared to men, as measured with positron emission tomography [58]. Clearly, sex-based differences in the development of tau pathology need to be characterized and addressed to conduct meaningful preclinical and clinical studies.
The differences in the phosphorylation of soluble tau observed in the forebrain and hippocampus of mice with 25 ng/g brain levels of compound needs further investigation. It is possible that the reduction in the level of insoluble phosphorylated tau may play a part in the increase of soluble phosphorylated tau. The increase in tau phosphorylation is limited to the forebrain at the PHF1 epitope in the primary study and at the CP13 and RZ3 epitopes in the secondary study suggesting different amounts of kinase and/or phosphatase activity in the forebrain and hippocampus of these mice at the study endpoint. Differential levels of protein expression for tau isoforms, kinases, and phosphatases has been demonstrated in different regions of the brain that may explain the vulnerability of specific regions of the brain to tau pathology [59]. A recent proteomic analysis of human brain specimens was performed to better understand the vulnerability of specific brain regions to AD. The medial temporal lobe, which includes the hippocampus, was shown to have prominent oxidative phosphorylation impairment compared to the neocortex [60]. The differential effect of the compound in the hippocampus and forebrain of htau mice may partially reflect some of the proteomic differences observed in these published studies. The safest long-term method to treat the large number of mice daily for four months was determined to be administration in feed which was enabled by the stability of the compound in the feed. Administration by feed also provides the mice with prolonged exposure to the compound over the course of the day compared to daily or less frequent administration by oral gavage or intraperitoneal injections. After the efficacy studies were completed dosing studies with administration in feed were performed to better understand why the medium dose gave the best efficacy. The treatment and analytical aspects of the dosing studies were performed blinded at contract research organizations. The dosing study was performed to determine whether the relative levels of compound in the feed were reflected by the exposure levels in the plasma and brains of mice. The feed formulated with the medium dose (200 mg compound/kg feed to provide a daily dose of 40 mg compound/kg mouse) gave an average concentration of 25 ng/g brain, whereas both the lower and high doses (10 and 100 mg/kg) gave brain concentrations that were below quantifiable levels or 6 ng/g, respectively (Fig. 5). Although the dose in feed did not correlate with the concentration in the brain there was, more importantly, a linear correlation between the level of compound in the brain and compound efficacy. In the confirmatory study, the average level of compound in the serum of mice that were treated with the 40 mg/kg dose were over 100-fold greater than the 10 mg/kg treatment group (Fig. 7). At the low dose, non-specific binding may inhibit uptake, and at high levels the compound may form aggregates above a critical concentration range that diminishes its uptake in the intestine and/or its activity. Molecules designed for CNS indications that are optimized for blood-brain-barrier penetration are often hydrophobic and aggregation-prone in aqueous environments. Thus, the dose and formulation of the compound need to be optimized for exposure to select the most efficacious dose [61, 62].
Several therapeutics targeting tau have reached clinical development [2]. There are two small molecule tau aggregation inhibitors in clinical studies; both of these approaches target tau β-sheet, LMTX® (TauRx) [63] and ACI-3024 (AC-Immune). Leuco-Methylthioninium Bis(Hydromethanesulphonate) (LMTX, a stabilized reduced form of methylene blue), failed to meet its clinical endpoints in Phase III clinical studies [64]. The lack of efficacy in the clinical trial may have been due to methylene blue acting by oxidizing the cysteines in tau, a mechanism that is counteracted in vivo by glutathione [65]. More recently, in vitro assays of tau aggregation showed that although methylene blue inhibits tau fibril formation it accelerates tau oligomer formation [66]. Curcumin, a natural compound with pleiotropic effects, and synthetic derivatives such as PE859 [67, 68], Shiga-Y5 [69], and J147 [70] have been used in preclinical or clinical studies targeting protein aggregation and other pathways involved in AD [71]. Because of the low bioavailibility of curcumin, formulation has been a focus for enabling treatment. The posttranslational modification of proteins by O-linked N-acetylglucosamine (O-GlcNAc) inhibits protein aggregation in multiple neurodegenerative diseases [72]. An oral inhibitor of O-GlcNAcase reduces tauopathy in rTg4510 mice [73] and has entered clinical development for progressive supranuclear palsy [74, 75]. It is difficult to evaluate the relative efficacy of the small molecules targeting tau pathways without the performance of simultaneous studies in the same animal models using the same methodology.
Most programs targeting tau are using an immunotherapeutic approach [3, 76–80]. However, there are multiple advantages of using small molecule therapeutics to target the accumulation of tau pathology. Unlike immunotherapeutic approaches targeting tau, small molecules may be selected to be agnostic to specific post-translational modifications, conformational changes or strains of tau aggregates. Orally available, small molecules can be designed to penetrate the blood-brain-barrier, to enter cells where tau is aggregating, and to access extracellular vesicles that may be vectors of tau seeds causing spread of tau pathology, whereas, only 0.1–0.2% of intravenous-administered antibody levels can access the cerebrospinal fluid. An active immunotherapeutic approach may trigger a harmful autoimmune response [76]. The phenomenon of epitope spreading can occur that creates an immune response to other epitopes in the same or similar proteins that precludes limiting the immune response to a single determinant [81]. This mechanism is thought to play a role in multiple autoimmune diseases. Adverse effects due to active immunization are irreversible except for attempting immunosuppression. Furthermore, patients participating in an active immunization trial would be unlikely to be able to participate in another study. A small molecule therapeutic approach is advantageous because it enables cost-effective, patient-centered treatment for the rapidly growing, unserved AD market in the context of significant and growing global healthcare system cost pressures. This is consistent with the increasing global need to deliver value-based care.
In summary, tau load and distribution have been shown to correlate with disease progression and should therefore be an efficacious target for drug discovery. This program is based on the premise that tau oligomers are an acutely toxic species of tau and their material reduction will modify the clinical course of AD. A small molecule approach was taken because an effective chronic drug therapy needs to be safe, effective, conveniently delivered, and economically viable for patients and payers, especially for prevention and early disease intervention. Key advantages versus a biologic approach include greater patient convenience, more predictable efficacy and safety and improved access to the brain, as well as significantly lower patient acquisition costs and improved market access, lower manufacturing costs, simpler scale-up, improved immunogenicity profile, simplified logistics, handling, and storage. This small molecule approach differentiates itself by targeting tau self-association into oligomers, the beginning of the tau aggregation cascade, to inhibit all downstream tau aggregation, as well. Whereas other programs have focused on targeting the formation of tau fibrils or disrupting them. The results presented here have validated this approach in vivo showing a linear dose dependent reduction in both soluble and insoluble tau aggregates with a lead small molecule.
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute on Aging of the National Institutes of Health under Award Numbers R43AG029777, R44AG029777, R43AG033474, R44AG053150, R43AG057325, R43AG062021, and R44AG062021. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank Samantha Decena for her technical assistance and Vanessa Diaz for her administrative assistance with writing the manuscript.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0465r3).
REFERENCES
[1] | Alzheimer A ((1907) ) Über eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und Psychisch-gerichtliche Medizin 64: , 146–148. |
[2] | Medina M ((2018) ) An overview on the clinical development of tau-based therapeutics. Int J Mol Sci 19: , E1160. |
[3] | Jadhav S , Avila J , Schöll M , Kovacs GG , Kövari E , Skrabana R , Evans LD , Kontsekova E , Malawska B , de Silva R , Buee L , Zilka N ((2019) ) A walk through tau therapeutic strategies. Acta Neuropathol Commun 7: , 22. |
[4] | Aschenbrenner AJ , Gordon BA , Benzinger TLS , Morris JC , Hassenstab JJ ((2018) ) Influence of tau PET, amyloid PET, and hippocampal volume on cognition in Alzheimer disease. Neurology 91: , e859–e866. |
[5] | Gordon BA , McCullough A , Mishra S , Blazey TM , Su Y , Christensen J , Dincer A , Jackson K , Hornbeck RC , Morris JC , Ances BM , Benzinger TLS ((2018) ) Cross-sectional and longitudinal atrophy is preferentially associated with tau rather than amyloid β positron emission tomography pathology. Alzheimers Dement (Amst) 10: , 245–252. |
[6] | Tapia-Rojas C , Cabezas-Opazo F , Deaton CA , Vergara EH , JohnsonGVW , Quintanilla RA ((2019) ) It’s all about tau. Prog Neurobiol 175: , 54–76. |
[7] | Sydow A , Van der Jeugd A , Zheng F , Ahmed T , Balschun D , Petrova O , Drexler D , Zhou L , Rune G , Mandelkow E , D’Hooge R , Alzheimer C , Mandelkow EM ((2011) ) Tau-induced defects in synaptic plasticity, learning, and memory are reversible in transgenic mice after switching off the toxic tau mutant. J Neurosci 31: , 2511–2525. |
[8] | Krüger L , Mandelkow EM ((2016) ) Tau neurotoxicity and rescue in animal models of human tauopathies. Curr Opin Neurobiol 36: , 52–58. |
[9] | Holtzman DM , Bateman RJ , Braunstein JB , Budur K , Florian H , Fogelman I , Hu Y , Mendoca N , Verghese PB , West T ((2017) ) Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis 4: , 236–241. |
[10] | Guo T , Noble W , Hanger DP ((2017) ) Roles of tau protein in health and disease. Acta Neuropathol 133: , 665–704. |
[11] | Andorfer C , Acker CM , Kress Y , Hof PR , Duff K , Davies P ((2005) ) Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci 25: , 5446–5454. |
[12] | Santacruz K , Lewis J , Spires T , Paulson J , Kotilinek L , Ingelsson M , Guimaraes A , DeTure M , Ramsden M , McGowan E , Forster C , Yue M , Orne J , Janus C , Mariash A , Kuskowski M , Hyman B , Hutton M , Ashe KH ((2005) ) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309: , 476–481. |
[13] | Spires TL , Orne JD , SantaCruz K , Pitstick R , Carlson GA , Ashe KH , Hyman BT ((2006) ) Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol 168: , 1598–1607. |
[14] | Berger Z , Roder H , Hanna A , Carlson A , Vijayaraghavan R , Yue M , Wszolek Z , Ashe K , Knight J , Dickson D , Andorfer C , Rosenberry TL , Lewis J , Hutton M , Janus C ((2007) ) Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci 27: , 3650–3662. |
[15] | Yoshiyama Y , Higuchi M , Zhang B , Shu-Ming Huang , Iwata N , Saido TC , Maeda J , Suhara T , Trojanowski JQ , Lee V ((2007) ) Synapse loss and microglial activation precede tangles in P301S tauopathy mouse model. Neuron 53: , 337–351. |
[16] | Patterson KR , Remmers C , Fu Y , Brooker S , Kanaan NM , Vana L , Ward S , Reyes JF , Philibert K , Glucksman MJ , Binder LI ((2011) ) Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J Biol Chem 286: , 23063–23076. |
[17] | Lasagna-Reeves CA , Castillo-Carranza DL , Sengupta U , Sarmiento J , Troncoso J , Jackson GR , Kayed R ((2012) ) Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB 26: , 1946–1959. |
[18] | Shafiei SS , Guerrero-Muñoz MJ , Castillo-Carranza DL ((2017) ) Tau oligomers: Cytotoxicity, propagation, and mitochondrial damage. Front Aging Neurosci 9: , 83. |
[19] | Moe JG , Chatterjee I , Davidowitz EJ , Arancio O ((2009) ) Modulation of synaptic function by extracellular tau enriched in oligomers. Alzheimers Dement 5: , P499. |
[20] | Fá M , Puzzo D , Piacentini R , Staniszewski A , Zhang H , Baltrons MA , Li Puma DD , Chatterjee I , Li J , Saeed F , Berman HL , Ripoli C , Gulisano W , Gonzalez J , Tian H , Costa JA , Lopez P , Davidowitz E , Yu WH , Haroutunian V , Brown LM , Palmeri A , Sigurdsson EM , Duff KE , Teich AF , Honig LS , Sierks M , Moe JG , D’Adamio L , Grassi C , Kanaan NM , Fraser PE , Arancio O ((2016) ) Extracellular tau oligomers produce an immediate impairment of LTP and memory. Sci Rep 6: , 19393. |
[21] | Guerrero-Muñoz MJ , Gerson J , Castillo-Carranza DL ((2015) ) Tau oligomers: The toxic player at synapses in Alzheimer’s disease. Front Cell Neurosci 9: , 464. |
[22] | Kaniyappan S , Chandupatla RR , Mandelkow EM , Mandelkow E ((2017) ) Synapse loss and microglial activation precede tangles in P301S tauopathy mouse model. Alzheimers Dement 13: , 1270–1291. |
[23] | Tian H , Davidowitz E , Lopez P , Emadi S , Moe J , Sierks M ((2013) ) Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int J Cell Biol 2013: , 260787. |
[24] | Ma QL , Zuo X , Yang F , Ubeda OJ , Gant DJ , Alaverdyan M , Teng E , Hu S , Chen PP , Maiti P , Teter B , Cole GM , Frautschy SA ((2012) ) Curcumin suppresses soluble tau dimers and corrects molecular chaperone, synaptic, and behavioral deficits in aged human tau transgenic mice. J Biol Chem 288: , 4056–4065. |
[25] | Castillo-Carranza DL , Gerson JE , Sengupta U , Guerroro-Muñoz MJ , Lasanga-Reeves CA , Kayed R ((2014) ) Specific targeting of tau oligomers in Htau mice prevents cognitive impairment and tau toxicity following injection with brain-derived tau oligomeric seeds. J Alzheimers Dis 40: (Suppl 1), S97–S111. |
[26] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[27] | Alafuzoff I , Arzberger T , Al-Sarraj S , Bodi I , Bogdanovic N , Braak H , Bugiani O , Del-Tredici K , Ferrer I , Gelpi E , Giaccone G , Graeber MB , Ince P , Kamphorst W , King A , Korkolopoulou P , Kovács GG , Larionov S , Meyronet D , Monoranu C , Parchi P , Patsouris E , Roggendorf W , Seilhean D , Tagliavini F , Stadelmann C , Streichenberger N , Thal DR , Wharton SB , Kretzschmar H ((2008) ) Staging of neurofibrillary pathology in Alzheimer’s disease: A study of the BrainNet Europe Consortium. Brain Pathol 18: , 484–496. |
[28] | Vaquer-Alicea J , Diamond MI ((2019) ) Propagation of protein aggregation in neurodegenerative diseases. Ann Rev Biochem 88: , 785–810. |
[29] | Ono K , Yoshiike Y , Takashima A , Hasegawa K , Naiki H , Yamada M ((2003) ) Potent anti-amyloidogenic and fibril-destabilizing effects of polyphenols in vitro: Implications for the prevention and therapeutics of Alzheimer’s disease. J Neurochem 87: , 172–181. |
[30] | Bulic B , Pickhardt M , Mandelkow E ((2013) ) Progress and developments in tau aggregation inhibitors for Alzheimer disease. J Med Chem 56: , 4135–4155. |
[31] | Wischik CM , Harrington CR , Storey JM ((2014) ) Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol 88: , 529–539. |
[32] | Moe JG , Krishnamurthy P , Lopez P , Papiani G , Hendrix J, Davidowitz EJ ((2014) ) Drug development of inhibitors of tau oligomer formation for Alzheimer’s disease and tauopathies. Alzheimers Dement 10: , 865–866. |
[33] | Moe JG , Krishnamurthy P , Lopez P , Papiani G , Romero D , Bian H , McDonnel ME , Reitz AB ((2016) ) Small molecule tau oligomerization inhibitors. Alzheimers Dement 12: , 434. |
[34] | Moe JG , Gluchowski C , Krishnamurthy P , Davidowitz EJ ((2017) ) CNS drug-like inhibitors of tau oligomerization. Alzheimers Dement 13: , 1580. |
[35] | Moe JG , Lopez P , Jimenez H , Adrien L , Davies P , Davidowitz EJ ((2018) ) In vivo efficacy of a small molecule inhibitor of tau oligomer formation in htau mice. Alzheimers Dement 14: , 1527–1528. |
[36] | Andorfer C , Kress Y , Espinoza M , de Silva R , Tucker KL , Barde YA , Duff K , Davies P ((2003) ) Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem 86: , 582–590. |
[37] | Davies P ((2000) ) Characterization and use of monoclonal antibodies to tau and paired helical filament tau. Methods Mol Med 32: , 361–373. |
[38] | Greenberg SG , Davies P , Schein JD , Binder I ((1992) ) Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem 267: , 564–569. |
[39] | Otvos L Jr , Feiner L , Lang E , Szendrei GI , Goedert M , Lee VM ((1994) ) Monoclonal antibody PHF-1 recognize tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res 39: , 669–673. |
[40] | Duff K , Knight H , Refolo LM , Sanders S , Yu X , Picciano M , Malester B , Hutton M , Adamson J , Goedert M , Burki K , Davies P ((2000) ) Characterization of pathology in transgenic mice over-expressing human genomic and cDNA tau transgenes. Neurobiol Dis 7: , 87–98. |
[41] | Lewis J , McGowan E , Rockwood J , Melrose H , Nacharaju P , Van Slegtenhorst M , Gwinn-Hardy K , Murphy MP , Baker M , Yu X , Duff K , Hardy J , Corral A , Lin W-L , Yen S-H , Dickon DW , Davies P , HuttonM ((2000) ) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: , 402–405. |
[42] | Vingtdeux V , Davies P , Dickson DW , Marambaud P ((2011) ) AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol 121: , 337–349. |
[43] | Jicha GA , Bowser R , Kazam IG , Davies P ((1997) ) Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res 48: , 128–132. |
[44] | Jicha GA , Lane E , Vincent I , Otvos L , Hoffmann R , Davies P ((1997) ) A conformation- and phosphorylation-dependent antibody recognizing the paired helical filaments of Alzheimer’s disease. J Neurochem 69: , 2087–2095. |
[45] | Acker CM , Forest SK , Zinkowski R , Davies P , d’Abramo C ((2012) ) Sensitive quantitative assays for tau and phospho-tau in transgenic mouse models. Neurobiol Aging 34: , 338–350. |
[46] | Forest SK , Acker CM , d’Abramo C , Davies P ((2013) ) Methods for measuring tau pathology in transgenic mouse models. J Alzheimers Dis 33: , 463–471. |
[47] | d’Abramo C , Acker CM , Jimenez HT , Davies P ((2013) ) Tau passive immunotherapy in mutant P301L mice: Antibody affinity versus specificity. PloS One 8: , e62402. |
[48] | d’Abramo C , Acker CM , Schachter JB , Terracina G , Wang X , Forest SK , Davies P ((2015) ) Detecting tau in serum of transgenic animal models after tau immunotherapy treatment. Neurobiol Aging 37: , 58–65. |
[49] | Vasan S , Mong PY , Grossman A ((2006) ) Interaction of prion protein with small highly structured RNAs: Detection and characterization of Prp-oligomers. Neurochem Res 31: , 629–637. |
[50] | Weaver CL , Espinoza M , Kress Y , Davies P ((2000) ) Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging 21: , 719–727. |
[51] | Fitzpatrick AWP , Falcon B , He S , Murzin AG , Murshudov G , Garringer HJ , Crowther RA , Goedert M , Scheres SHW ((2017) ) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547: , 185–190. |
[52] | Jicha GA , Berenfeld B , Davies P ((1999) ) Sequence requirements for formation of conformational variants of tau similar to those found in Alzheimer’s disease. J Neurosci Res 55: , 713–723. |
[53] | Polydoro M , Acker CM , Duff K , Castillo PE , Davies P ((2009) ) Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology. J Neurosci 29: , 10741–10749. |
[54] | Ferguson SA , Mouzon BC , Lynch C , Lungmus C , Morin A , Crynen G , Carper B , Bieler G , Mufson EJ , Stewart W , Mullan M , Crawford F ((2017) ) Negative impact of female sex on outcomes from repetitive mild traumatic brain injury in hTau mice is age dependent: A chronic effects of neurotrauma consortium study. Front Aging Neurosci 9: , 416. |
[55] | Mouzon B , Bachmeier C , Ojo J , Acker C , Ferguson S , Cryen G , Davies P , Mullan M , Stewart W , Crawford F ((2019) ) Chronic white matter degeneration, but no tau pathology at one-year post-repetitive mild traumatic brain injury in a tau transgenic model. J Neurotrauma 36: , 578–588. |
[56] | Zhang B , Carroll J , Trojanowski JQ , Yao Y , Iba M , Potuzak JS , Hogan AM , Xie SX , Ballatore C , Smith AB 3rd , Lee VM , Brunden KR ((2012) ) The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci 32: , 3601–3611. |
[57] | Sahara N , Murayama M , Higuchi M , Suhara T , Takashima A ((2014) ) Biochemical distribution of tau protein in synaptosomal fraction of transgenic mice expressing human P301L tau. Front Neurol 5: , 26. |
[58] | Buckley RF , Mormino EC , Rabin JS , Hohman TJ , Landau S , Hanseeuw BJ , Jacobs HIL , Papp KV , Amariglio RE , Properzi MJ , Schultz AP , Kirn D , Scott MR , Hedden T , Farrell M , Price J , Chhatwal J , Rentz DM , Villemagne VL , Johnson KA , Sperling RA ((2019) ) Sex differences in the association of global amyloid and regional tau deposition measured by positron emission tomography in clinically normal older adults. JAMA Neurol 76: , 542–551. |
[59] | Hu W , Wu F , Zhang Y , Gong CX , Iqbal K , Liu F ((2017) ) Expression of tau pathology-related proteins in different brain regions: A molecular basis of tau pathogenesis. Front Aging Neurosci 9: , 311. |
[60] | Mendonça CF , Kuras M , Nogueira FCS , Plá I , Hortobágyi T , Csiba L , Palkovits M , Renner É , Döme P , Marko-Varga G , Domont GB , Rezeli M ((2019) ) Proteomic signatures of brain regions affected by tau pathology in early and late stages of Alzheimer’s disease. Neurobiol Dis 130: , 104509. |
[61] | Laplante M , Sabatini DM ((2013) ) Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 126: , 1713–1719. |
[62] | Granath AK , Sigfridsson K ((2016) ) Evaluation of preclinical formulations for a poorly water-soluble compound. Int J Pharm 511: , 630–637. |
[63] | Wilcock GK , Gauthier S , Frisoni GB , Jia J , Hardlund JH , Moebius HJ , Bentham P , Kook KA , Schelter BO , Wischik DJ , Davis CS , Staff RT , Vuksanovic V , Ahearn T , Bracoud L , Shamsi K , Marek K , Seibyl J , Riedel G , Storey JMD , Harrington CR , Wischik CM ((2017) ) Potential of low dose leuco-methylthioninium bis(Hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: Cohort analysis as modified primary outcome in a phase III clinical trial. J Alzheimers Dis 61: , 435–457. |
[64] | Gauthier S , Feldman HH , Schneider LS , Wilcock GK , Frisoni GB , Hardlund JH , Moebius HJ , Bentham P , Kook KA , Wischik DJ , Schelter BO , Davis CS , Staff RT , Bracoud L , Shamsi K , Storey JM , Harrington CR , Wischik CM ((2016) ) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388: , 2873–2884. |
[65] | Crowe A , James MJ , Lee VM , Smith AB 3rd , Trojanowski JQ , Ballatore C , Brunden KR ((2013) ) Aminothienopyridazines and methylene blue affect Tau fibrillization via cysteine oxidation. J Biol Chem 288: , 11024–11037. |
[66] | Soeda Y , Saito M , Maeda S , Ishida K , Nakamura A , Kojima S , Takashima A ((2019) ) Methylene blue inhibits formation of tau fibrils but not of granular tau oligomers: A plausible key to understanding failure of a clinical trial for Alzheimer’s disease. J Alzheimers Dis 68: , 1677–1686. |
[67] | Okuda M , Hijikuro I , Fujita Y , Wu X , Nakayama S , Sakata Y , Noguchi Y , Ogo M , Akasofu S , Ito Y , Soeda Y , Tsuchiya N , Tanaka N , Takahashi T , Sugimoto H ((2015) ) PE859, a novel tau aggregation inhibitor, reduces aggregated tau and prevents onset and progression of neural dysfunction in vivo. PloS One 10: , e0117511. |
[68] | Okuda M , Fugita Y , Hijikuro I , Wada M , Uemura T , Kobayashi Y , Waku T , Tanaka N , Nishimoto T , Izumi Y , Kume T , Akaike A , Takahashi T , Sugimoto H ((2017) ) PE859, a novel curcumin derivative, inhibits amyloid-β and tau aggregation, and ameliorates cognitive dysfunction in senescence-accelerated mouse prone 8. J Alzheimers Dis 59: , 313–328. |
[69] | Yanagisawa D , Hamezah HS , Durani LW , Taguchi H , Tooyama I ((2018) ) Study of tau pathology in male rTg4510 mice fed with a curcumin derivative Shiga-Y5. PloS One 13: , e0208440. |
[70] | Emmanuel IA , Olotu FA , Agoni C , Soliman M ((2019) ) Deciphering the “elixir of life”: Dynamic perspectives into the allosteric modulation of mitochondrial ATP synthase by J147, a novel drug in the treatment of Alzheimer’s disease. Chem Biodivers 16: , e1900085. |
[71] | Chen M , Du ZY , Zheng X , Li DL , Zhou RP , Zhang K ((2018) ) Use of curcumin in diagnosis, prevention, and treatment of Alzheimer’s disease. Neural Regen Res 13: , 742–752. |
[72] | Ryan P , Xu M , Davey AK , Danon JJ , Mellick GD , Kassiou M , Rudrawar S ((2019) ) The O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem Neurosci 10: , 2209–2221. |
[73] | Hastings NB , Wang X , Song L , Butts BD , Grotz D , Hargreaves R , Fred Hess J , Hong KK , Huang CR , Hyde L , Laverty M , Lee J , Levitan D , Lu SX , Maguire M , Mahadomrongkul V , McEachern EJ , Ouyang X , Rosahl TW , Selnick H , Stanton M , Terracina G , Vocadlo DJ , Wang G , Duffy JL , Parker EM , Zhang L ((2017) ) Inhibition of O-GlcNAcase leads to elevation of O-GlcNAc tau and reduction of tauopathy and cerebrospinal fluid tau in rTg4510 mice. Mol Neurodegener 12: , 39. |
[74] | Smith R , Schain M , Nilsson C , Strandberg O , Olsson T , Hägerström D , Jögi J , Borroni E , Schöll M , Honer M , Hansson OO ((2016) ) Increased basal ganglia binding of 18 F-AV-1451 in patients with progressive supranuclear palsy. Mov Disord 32: , 108–114. |
[75] | Selnick H , Hess JF , Tang C , Liu K , Schachter J , Ballard JE , Marcus JN , Klein DJ , Wang X , Pearson M , Savage MJ , Kaul R , Li T , Vocadlo DJ , Zhou Y , Zhu Y , Mu C , Wang Y , Wei Z , Bai C , Duffy JL , McEachern EJ ((2019) ) Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for tauopathies. J Med Chem, doi: 10.1021/acs.jmedchem.9b01090 |
[76] | Davies P ((2016) ) Passive immunotherapy for tau pathology. In Developing Therapeutics for Alzheimer’s Disease, WolfeMS, ed. Elsevier Inc., pp. 371–379. |
[77] | Bittar A , Sengupta U , Kayed R ((2018) ) Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies. NPJ Vaccines 3: , 9. |
[78] | Sigurdsson EM ((2018) ) Tau immunotherapies for Alzheimer’s disease and related tauopathies: Progress and potential pitfalls. J Alzheimers Dis 64: , S555–S565. |
[79] | Novak P , Kontsekova E , Zilka N , Novak M ((2018) ) Ten years of tau-targeted immunotherapy: The path walked and the roads ahead. Front Neurosci 12: , 798. |
[80] | Novak P , Zilka N , Zilkova M , Kovacech B , Skrabana R , Ondrus M , Fialova L , Kontsekova E , Otto M , Novak M ((2019) ) AADvac1, an active immunotherapy for Alzheimer’s disease and non-Alzheimer tauopathies: An overview of preclinical and clinical development. J Prev Alzheimers Dis 6: , 63–69. |
[81] | Vanderlugt CL , Miller SD ((2002) ) Epitope spreading in immune-mediated diseases: Implications for immunotherapy. Nat Rev Immunol 2: , 85–95. |


