
Oral Immunization with Soybean Storage Protein Containing Amyloid-β 4–10 Prevents Spatial Learning Decline
Abstract
Amyloid-β (Aβ) plays a central role in the pathogenesis of Alzheimer’s disease (AD). Because AD pathologies begin two decades before the onset of dementia, prevention of Aβ amyloidosis has been proposed as a mean to block the pathological cascade. Here, we generate a transgenic plant-based vaccine, a soybean storage protein containing Aβ4–10, named Aβ+, for oral Aβ immunization. One mg of Aβ+ or control protein (Aβ–) was administered to TgCRND8 mice once a week from 9 weeks up to 58 weeks. Aβ+ immunization raised both anti-Aβ antibodies and cellular immune responses. Spatial learning decline was prevented in the Aβ+ immunized group in an extended reference memory version of Morris water maze test from 21 to 57 weeks. In Tris-buffered saline (TBS), sodium dodecyl sulfate (SDS), and formic acid (FA) serial extractions, all sets of Aβ species from Aβ monomer, low to high molecular weight Aβ oligomers, and Aβ smears had different solubility in TgCRND8 brains. Aβ oligomers decreased in TBS fractions, corresponding to an increase in high molecular weight Aβ oligomers in SDS extracts and Aβ smears in FA fraction of the Aβ+ treated group. There was significant inhibition of histological Aβ burden, especially in diffuse plaques, and suppression of microglial inflammation. Processing of amyloid-β protein precursor was not different between Aβ+ and Aβ– groups. No evidence of amyloid-related inflammatory angiopathy was observed. Thus, Aβ+ oral immunization could be a promising, cheap, and long-term safe disease-modifying therapy to prevent the pathological process in AD.
INTRODUCTION
Based on the amyloid cascade hypothesis in Alzheimer’s disease (AD) [1, 2], many disease-modifying therapies (DMTs) are now being developed. However, none have succeeded in phase III clinical trials. Although the first clinical trial with an amyloid-β (Aβ) vaccine, AN1792, was stopped because of meningoencephalitis [3], subsequent studies revealed that AN1792 induced anti-Aβ antibodies, removed Aβ accumulations, and slowed the progression rate of cognitive dysfunction [4, 5]. Since then, many trials have attempted to improve the safety of Aβ immunotherapies by avoiding T-cell autoimmune responses [6–8]. In a phase Ib randomized trial, aducanumab, an antibody against aggregated forms of Aβ, reduced Aβ burden accompanied by a slowing of cognitive impairment in prodromal and mild AD patients [9]. A phase II study of Aβ vaccine CAD106 against Aβ1–6 evoked a strong serological response and demonstrated acceptable safety and tolerability [10]. A phase IIa trial of Aβ vaccine UB-311 against Aβ1–14 has been started based on favorable phase I trial results [11]. Case studies, the Alzheimer Disease Neuroimaging Initiative (ADNI), and the Dominantly Inherited Alzheimer’s Network (DIAN) have shown that AD pathology begins more than 20 years before the onset of dementia [12–14]. For this reason, DMT trials aimed at preventing the onset of AD, such as the DIAN-Trials Unit [15] and Alzheimer’s Prevention Initiative (API) [16], are now ongoing.
Mucosal vaccination is the ideal immunization for good accessibility, needle-free delivery, and protective immune responses in both mucosal and systemic immune compartments [17]. This method induces regulatory T cells, leading to a decrease in the systemic T-cell response and increased secretion of immune-inhibitory cytokines. Plants are advantageous platforms for recombinant vaccines because of their low cost, industrial scale production, and the absence of contamination from toxins and pathogens that are produced in bacterial and yeast systems [18]. Although plant-based Aβ vaccines using potatoes, tomatoes, green pepper leaves, rice, and tobacco have been reported [19–21], their effect on amyloid deposition and learning was examined only in the study with transgenic rice [21]. Booster injections of Aβ peptide were necessary in their procedure, and most transgenic plants did not produce sufficient amounts of Aβ. We have developed an approach based upon an innovative transgenic soybean that produces 870 mg/g of transgenic soybean seed storage protein containing Aβ4–10 (Aβ+), which is sufficient sequence of Th2 epitope [22] for safe oral immunization without T-cell responses and additional Aβ peptide booster injections [23]. Here, we validate the efficacy of Aβ+ oral vaccines using an AD mouse model, TgCRND8 [24, 25].
MATERIALS AND METHODS
Preparation of transgenic soybean protein Aβ+
Three tandem repeats of the Th2 epitope portion of Aβ4–10 (FRHDSGY) [22] were inserted into three portions of the flexible disordered regions II-IV of soybean glycinin A1aB1b, a carrier subunit protein of 11S globulins [26] (Fig. 1A). The seed specific glycinin promoter, cDNA for A1aB1b containing Aβ4–10 sequences or a wild type A1aB1b cassette for controls, and glycinin 1 terminator constructs to plasmids were transformed into soybean immature embryos. A1aB1b with Aβ4–10 and wild type A1aB1b were expressed in protein storage vacuoles [23]. Purified A1aB1b containing Aβ4–10, referred to as Aβ+, and wild type control A1aB1b, referred to as Aβ– were used.
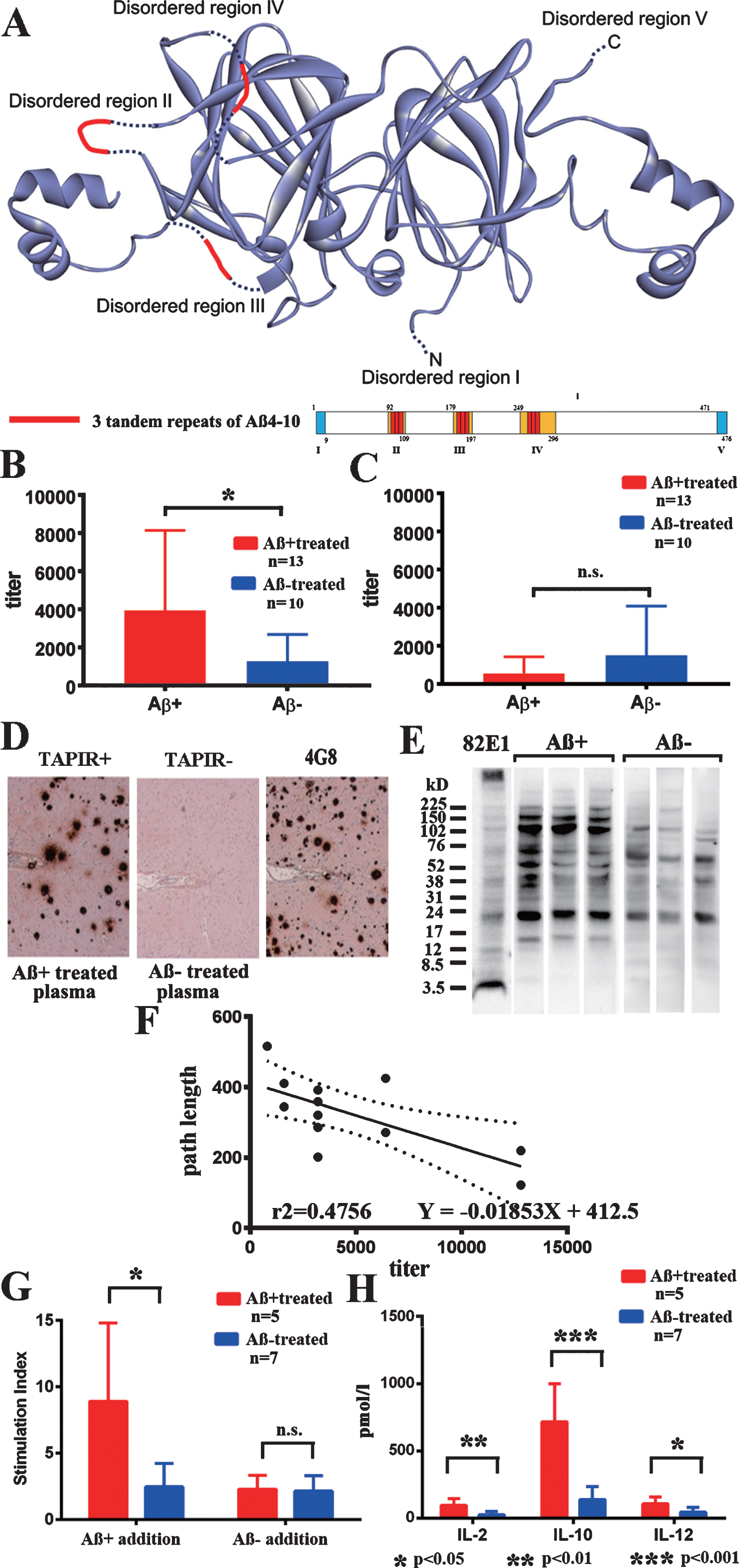
Fig.1
Transgenic plant protein A1aB1b and immune responses. A) The structure of transgenic plant protein A1aB1b containing Aβ4–10 (Aβ+). Three tandem repeats of Aβ4–10 were inserted into the three disordered regions of soybean 11S globulin, A1aB1b, that are marked in red. B) IgG antibody titer against Aβ+ in plasma from Aβ+ immunized mice (red, n = 12) significantly increased compared with Aβ– immunized mice (blue, n = 10, p < 0.05). C) IgG antibody titer against Aβ– in plasma was much less than that against Aβ+ and did not differ between Aβ+ (red, n = 12) and Aβ– (blue, n = 10) immunized mice. D) Aβ+ immunized mouse plasma diluted with blocking solution (1:2,000) labeled senile plaque Aβ amyloid in an AD brain (TAPIR+; left). Control stain of the AD brain using Aβ– treated mouse plasma (1:2,000, TAPIR-; middle) and anti-Aβ antibody (4G8; right). The figures show representative staining from Aβ+ and Aβ– treated mice. E) The SDS fraction of non-oral vaccine treated TgCRND8 mice was stained with plasma (1:400) from Aβ+ and Aβ– treated mice. Plasma from Aβ+ treated mice showed more staining compared with that from Aβ– treated mice in bands at 16, 40, 56, 102, 150, and 200 kD. F) The antibody titer against Aβ+ and average path length to reach the hidden platform during 1–10 days of the last Morris water maze test showed a significant linear regression correlation. Determination coefficients (r2 = 0.4756) and regression equations (Y = –0.01853X + 412.5) are shown (n = 12; n = 6 for 43 weeks, and n = 6 for 59 weeks). G) Thymidine uptake by stimulation of Aβ+ was significantly increased in Aβ+ treated mouse splenocytes (red, n = 5) compared with those in the Aβ– treated group (blue, n = 7, p < 0.05). No increase in thymidine uptake by stimulation of Aβ– was shown in both Aβ+ and Aβ– treated groups (p = 0.628). H) Stimulation of Aβ+ significantly increased the amounts of released cytokines, IL–2 (p < 0.01), IL–10 (p < 0.001), and IL–12 (p < 0.05) in Aβ+ treated mouse splenocytes (red, n = 5) compared with those in Aβ– treated mice (blue, n = 7).
Oral immunization
TgCRND8 expresses a mutant (K670N/M671L and V717F) human amyloid-β protein precursor (AβPP) 695 transgene under the regulation of the Syrian hamster prion promoter on a C3H/B6 strain background [24, 25]. TgCRND8 mice show spatial learning deterioration at 3 months of age that are accompanied by both increasing levels of Aβ and increasing numbers of amyloid plaques in the brain [24]. One mg of purified Aβ+ or control Aβ– with 10 μg cholera toxin subunit B (Crucell, Leiden, Netherlands) was administered into the guts via a catheter every week from 9 weeks old until 22∼58 weeks old. All animal experiments followed the ARRIVE guidelines, and were approved by the Ethics Committee of Hirosaki University (approval number M13007-1).
Morris water maze (MWM) test
Memory was evaluated by a spatial reference memory version of the MWM test every 4 weeks, as previously described [24, 25, 27]. Tests began on the first day of 13 weeks old and continued for 9 more consecutive days just 4 weeks after the first oral administration at 9 weeks old. These consecutive 10-day tests were repeated every 4 weeks until 21, 41, and 57 weeks old. The swim path of a mouse during each trial was recorded by a video camera connected to a video tracking system (Noldus EthoVision XT, Noldus Information Technology, Wageningen, Netherlands). The mouse was given 4 consecutive 60 s training trials for 10 days. The location of a hidden escape platform was in the center of one of the pool’s quadrants and was left in the same position during 10 consecutive days. Probe trial was administered 24 h after the 10th day of training. During the probe trial, the escape platform was removed from the pool, and the mice were allowed to search the pool uninterrupted for 60 s [24, 27].
Brain preparation
Under anesthesia with halothane, brains and cerebrospinal fluid (CSF) were collected at 23 weeks (Aβ+ n = 9, male 5, female 4, Aβ– n = 11, male 4, female 7), 43 weeks (Aβ+ n = 7, male 4, female 3, Aβ– n = 6, male 4, female 2), and 59 weeks (Aβ+ n = 6, male 4, female 2, Aβ– n = 7, male 5, female 2) after the last MWM test. Brains were removed and cut into sagittal sections along the midline. One hemisphere was fixed in 4% paraformaldehyde with 0.1 M phosphate-buffered saline (PBS, pH 7.6) for 8 h, and embedded in paraffin. The other half of the brain was fractionated by three sequential extraction steps using Tris-buffered saline (TBS) with protease inhibitors (Complete®, Roche Diagnostics, Basel, Switzerland), 2% sodium dodecyl sulfate (SDS) in water with the same protease inhibitors, and then 70% formic acid (FA) in water for biochemical analysis of Aβ species [28, 29].
Immune response to oral administration of Aβ+ and Aβ-
Microplates (MICROLON, Greiner bio-one, Austria) were coated overnight at 4°C with Aβ+, Aβ–, or Aβ1–42 peptides (0.5 μg/well) with 0.01 M PBS (pH 7.4), washed with PBS, and blocked with Blocker Casein in PBS (Thermo Fisher, Waltham, MA). After incubating with plasma samples in each well for 45 min at room temperature and washing, samples were reacted with anti-mouse IgG- or IgA-conjugated horseradish peroxidase (Thermo Fisher) in Blocker Casein PBS at 37°C for 30 min, and color development using 100 μl of tetramethylbenzidine for 15 min was performed. H2SO4 was added to stop the reaction, and signals were measured at 450 nm using an ELISA reader.
Splenocytes from 59-week-old mice were isolated, cultured, and restimulated, as previously described [30]. Aβ+, Aβ–, or Aβ1–42 was added to splenocytes at final concentrations of 100 μg/ml in triplicated wells, and 1 μCi of [3H]-thymidine was added to cells at 72 h. Cells were harvested after 18 h and thymidine incorporation was measured using a 1450 Microbeta liquid scintillation counter (Perkin Elmer, Waltham, MA). The stimulation index (SI) was calculated using the following formula: counts per minute (CPM) of the well with antigen per CPM with no antigen. An SI index >3 indicates a proliferative cellular immune response of the splenocytes. Supernatants were collected just before the addition of [3H]-thymidine and stored at –80°C for cytokine assays. Released cytokines were measured using Mouse Pro-Inflammatory TH1/TH2 9-plex (MesoScale Discovery, Rockville, MD) according to the manufacturer’s protocol. The Multi-Spot ELISA plates were precoated with antibodies specific for the following cytokines: interferon-γ, Interleukin (IL)-1β, IL-10, IL-12 total, IL-2, IL-4, IL-5, keratinocyte chemoattractant/human growth-regulated oncogene (KC/GRO), and tumor necrosis factor-α (TNF-α), and detected with SULFO-TAG detection antibodies. Light emitted upon electrochemical stimulation was read using a SECTOR Imager 2400A (Meso Scale Discovery).
Antibodies for western blot and immunostaining
The following antibodies to Aβ were used for western blots and immunostaining. Monoclonal antibodies: 82E1 (anti-Aβ1–16, IBL, Fujioka, Gunma, Japan), BA-27 (anti-Aβ1–40) [31], BC-05 (anti-Aβ35–43) [31], 4G8 (anti-Aβ18–22, Signet Lab), and 6E10 (anti-Aβ3–8, Covance Research Products Inc); Polyclonal antibodies: Aβ-N (anti-Aβ1–5, IBL), Ab9204 [32], anti-Aβ40 (Cat# 44–348, Thermo Fisher), and anti-Aβ42 (Cat#44–344, Thermo Fisher). A monoclonal antibody against Aβ4–10, named PEP3, was newly produced. Other antibodies included Iba1 for microglial markers (Wako Cat# 019-19471), anti-GFAP (Dako Cat# N1506), anti-CD5 (Cat# 550522 Clone 53-7.3, BD Pharrmingen, Franklin Lakes, NJ), anti-AβPP antibody Saeko (anti-C-terminal 30 amino acids of AβPP [29]), anti-mouse tau antibody, TAU-5 (Thermo Fisher Cat# MA1-26600), and PHF-1 against phosphorylated tau (pTau) at serine 396/serine 404 (gift from Davies P).
ELISA for levels of Aβ40, Aβ42, AβOs, and sAβPP
Human β Amyloid ELISA Kits for Aβx - 40 and Aβx - 42 (294-64701 for Aβx - 40; 290-62601 for Aβx - 42; Wako), Human Amyloid β oligomers (82E1-specific) Assay Kit-IBL (#27725, IBL [33]), a Human sAPPα (highly sensitive) Assay Kit (#27734, IBL), and a Human sAPPβ-sw (highly sensitive) Assay Kit (#27733, IBL).
Western blot analysis
All prepared samples were boiled at 70°C for 10 min in SDS sample buffer, separated on a 4–12% NuPAGE Bis–Tris Gel (Cat# NP0321, Thermo Fisher), and electrotransferred to an Immobilon P (MerckMillipore, Burlington, MA) membrane at 100 V for 1.5 h. The signal intensities of proteins labeled using Supersignal (Cat#34076, Thermo Fisher) were quantified using a luminoimage analyzer (LAS 1000-mini, Fuji Film, Tokyo, Japan). Aβ42 peptides (Cat#A9810, Sigma-Aldrich, St. Louis, MO) were used as control Aβ peptides.
Pathological analysis
Five-μm-thick sections were immersed in 0.5% periodic acid to block intrinsic peroxidase, and then treated with 99% FA for Aβ and tau staining for 3 min. After blocking with 5% normal goat or horse serum in 50 mM PBS (pH 7.4) containing 0.05% Tween 20 and 4% Block Ace (Cat# UK-B80, DS Pharma Biomedical, Suita, Osaka, Japan), sections were incubated overnight with the primary antibodies. Specific labeling was visualized using a Vectastain Elite ABC kit (Vector, Burlingame, CA). Tissue sections were counterstained with hematoxylin. Immunostaining areas of Aβ or Iba1 in 10 randomly selected ROIs (872 μm×671 μm) in the frontal, temporal, and parietal cortex of 3 serial slides were measured in total using Image Pro Plus ver.4.5 (Media Cybernetics, Rockville, MD) after adjustment for artifact staining. The presence of hemorrhage was examined using Berlin blue staining. For the tissue amyloid plaque immunoreactivity (TAPIR) to identify antibody raised against Aβ+, paraffin sections of brains from AD patients and controls were stained with plasma from Aβ+ and Aβ– treated mice diluted with blocking solution (1:2,000). Congo red stain was used to stain core plaques.
Statistical analysis
Data were expressed as the mean±standard deviation (SD) except for Fig. 2. Two-way analysis of variance (ANOVA), with post hoc tests (Bonferroni’s multiple comparisons test), was used for analyzing longitudinal alteration. In Fig. 2, data are expressed as the mean±standard error (SE), and two-way repeated ANOVA was used. Mann–Whitney U test was applied for comparison between two groups. Prism 7 software (GraphPad, La Jolla, CA) and IBM SPSS Statistics 25 (IBM, Armonk, NY) were used for statistical analyses. A value of p < 0.05 was considered to be significant for all statistical tests.
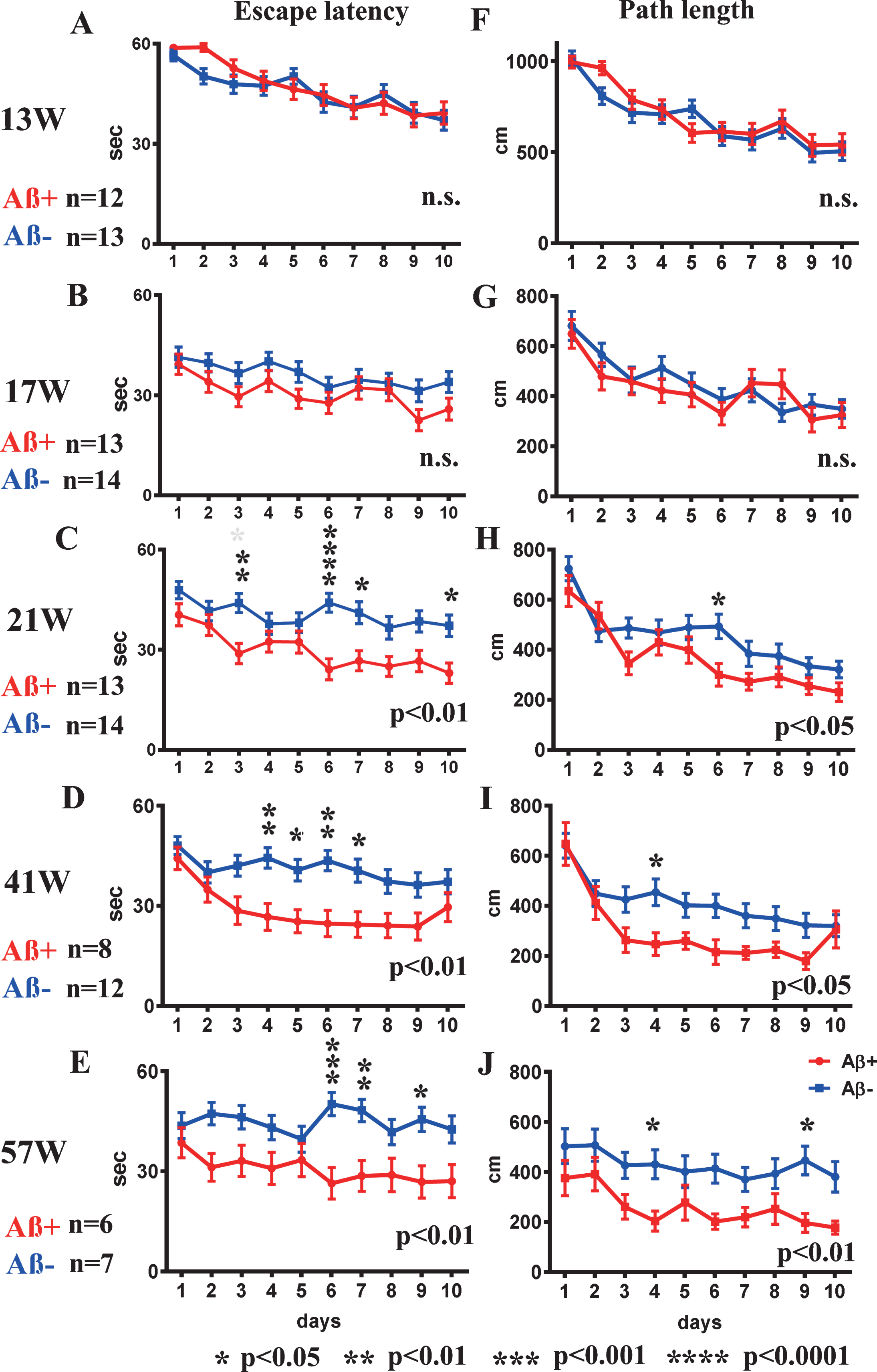
Fig.2
Spatial reference memory version of the MWM test from 13 to 57 weeks old. A-E) escape latency, and F-J) path-length at 13, 17, 21, 41, and 57 weeks. Escape latency and path-length analyses showed significant improvement in Aβ+ treated mice (red line) from 21 weeks old (C, H) compared with Aβ– treated mice (blue line) (escape latency p < 0.01, path-length p < 0.05). Aβ+ treated mice continued to show significantly better performances in escape latency and path-length than those of Aβ– treated mice until 57 weeks old (p < 0.05 to 0.01; D, E, I, J). Statistical significance by two-way repeated ANOVA was shown in lower right of each line graph. The asterisk shows the result of post hoc analysis at each day. Mann–Whitney U test was applied for comparison between two groups. Analyzed mice numbers are n = 12 for Aβ+, n = 13 for Aβ– at 13 weeks; n = 13 for Aβ+, n = 14 for Aβ– at 17, 21 weeks; n = 8 for Aβ+, n = 12 for Aβ– at 41 weeks; and n = 6 for Aβ+, n = 7 for Aβ– at 57 weeks.
RESULTS
Oral immunization raised adapted immune responses
Significant positive rates of IgG antibodies against Aβ+ were found in the Aβ+ treated group (Fig. 1B; p < 0.05). IgG antibodies against Aβ– were much lower than those against Aβ+, and there was no difference between the Aβ+ and Aβ– treated groups (Fig. 1C). IgG antibodies against Aβ1–42 peptides, and IgA antibodies against Aβ+, Aβ–, and Aβ1–42 were not detected. Plasma (1:2,000) from 59-week-old Aβ+ immunized mice labeled Aβ amyloid plaques in the human AD brain (TAPIR+; Fig. 1D). The SDS fraction of non-oral vaccine treated TgCRND8 mice was stained with plasma (1:400) from Aβ+ and Aβ– treated mice. Plasma from Aβ+ treated mice showed more staining compared with that from Aβ– treated mice in bands at 16, 40, 56, 102, 150, and 200 kD (Fig. 1E). The antibody titer against Aβ+and average path length to reach the hidden platform during 1–10 days of the last Morris water maze test showed a significant linear regression correlation (Fig. 1F; p < 0.05). This finding suggested a close correlation between evoked anti-Aβ oligomers antibody titers and preservation of special learning ability, as shown in the Fig. 2.
Splenocytes from the Aβ+ treated group showed significant proliferation against Aβ+ addition compared with those of the Aβ– treated group (Fig. 1G; p < 0.05). Aβ– treated splenocytes did not react with both Aβ+ and Aβ– addition. Significantly increased levels of IL-2, IL-10, and IL-12 were revealed in the media from the Aβ+ treated group compared with the Aβ– treated group (p < 0.01 for IL-2, p < 0.001 for IL-10, and p < 0.05 for IL-12). The levels of interferon-γ, IL-1β, IL-4, IL-5, KC/GRO, and TNF-α were not different between the groups. IL-10 levels were markedly increased compared with those of IL-12, suggesting inhibition of proinflammatory cytokines and the predominance of a Th2 response (Fig. 1H).
Aβ+ prevented spatial learning decline
In the first trial at 13 weeks, there were no differences between Aβ+ and Aβ– treated mice in escape latency or path-length to reach the hidden platform (Fig. 2A and F). For escape latency and path-length analysis, improvements due to learning effects were recognized in the second trial in both groups (Fig. 2B, G). The escape latency and path-length in Aβ+ treated mice were significant shorter than those of the Aβ– treated group at 21 weeks old (escape latency Fig. 2C; p < 0.01, path-length Fig. 2H; p < 0.05) suggesting improved learning in Aβ+ treated mice. Aβ+ treated mice continued to show significantly better performances than Aβ– treated mice until 57 weeks old (Fig. 2D, E, I, and J; p < 0.05–0.01). There were no significant differences in the probe trials.
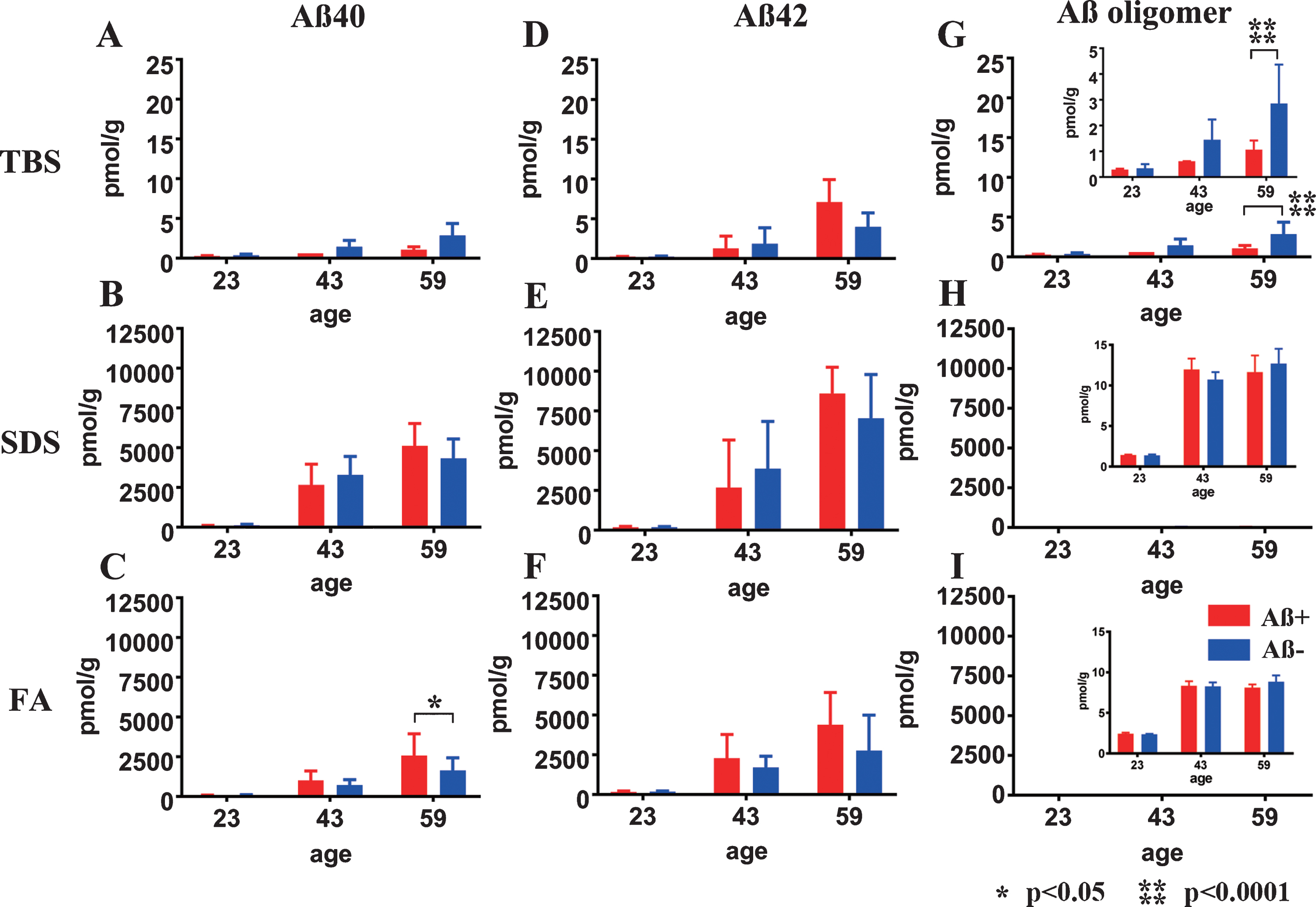
Decreased soluble AβOs by ELISA
Almost all Aβ40 and Aβ42 monomers accumulated in the SDS and FA fractions (Fig. 3B, C, E, F). In the TBS, SDS, and FA soluble fractions, the amount of Aβ40 and Aβ42 did not differ between the Aβ+ and Aβ– groups at any of the ages tested (Fig. 3A, B, D–F), except for increased Aβ40 in the FA fraction of Aβ+ group (Fig. 3C; p < 0.05). AβOs measured by 82E1/82E1 ELISA longitudinally showed that the amount of AβOs in the TBS fraction was significantly decreased in the Aβ+ treated group compared with the Aβ– treated group (Fig. 3G; p < 0.0001). AβOs ELISA showed trace amounts of soluble AβOs in both the SDS and FA extracted fractions compared with the amounts of Aβ40 and Aβ42 in the Aβ+ and Aβ– groups, and no difference between the Aβ+ and Aβ– treated groups (Fig. 3H, I).
Fig.3
Longitudinal change in Aβ40, Aβ42, and AβOs in TBS, SDS, and FA fractions from Aβ+ or Aβ– treated mouse brains. Amounts of Aβx - 40 (A–C), Aβx - 42 (D–F), and AβOs (G–I) in TBS, SDS, and FA fractions of Aβ+ (red) and Aβ– (blue) immunized mouse brains measured using ELISA at 23, 43, and 59 weeks old. There were no significant differences in the levels of Aβx - 40 (A, B) and Aβx - 42 (D–F) between Aβ+ and Aβ– treated groups at any time points or in any fractions, except for the Aβx - 40 in the FA fraction at 59 weeks old (C: *p<0.05). Significant suppression of AβOs in TBS soluble fractions was revealed in Aβ+ treated mice compared with Aβ– treated mice (G and enlarged illustration, ****p < 0.0001). Amounts of AβOs in SDS and FA fractions were very low compared with Aβ40 and Aβ42 in the same fraction (H, I). Mice at 23 weeks (n = 9 for Aβ+, n = 11 for Aβ–), 43 weeks (n = 7 for Aβ+, n = 6 for Aβ–), and 59 weeks (n = 6 for Aβ+, n = 7 for Aβ–) were measured by ELISA.
Longitudinal appearance of AβOs species in TgCRND8 without oral immunization trial
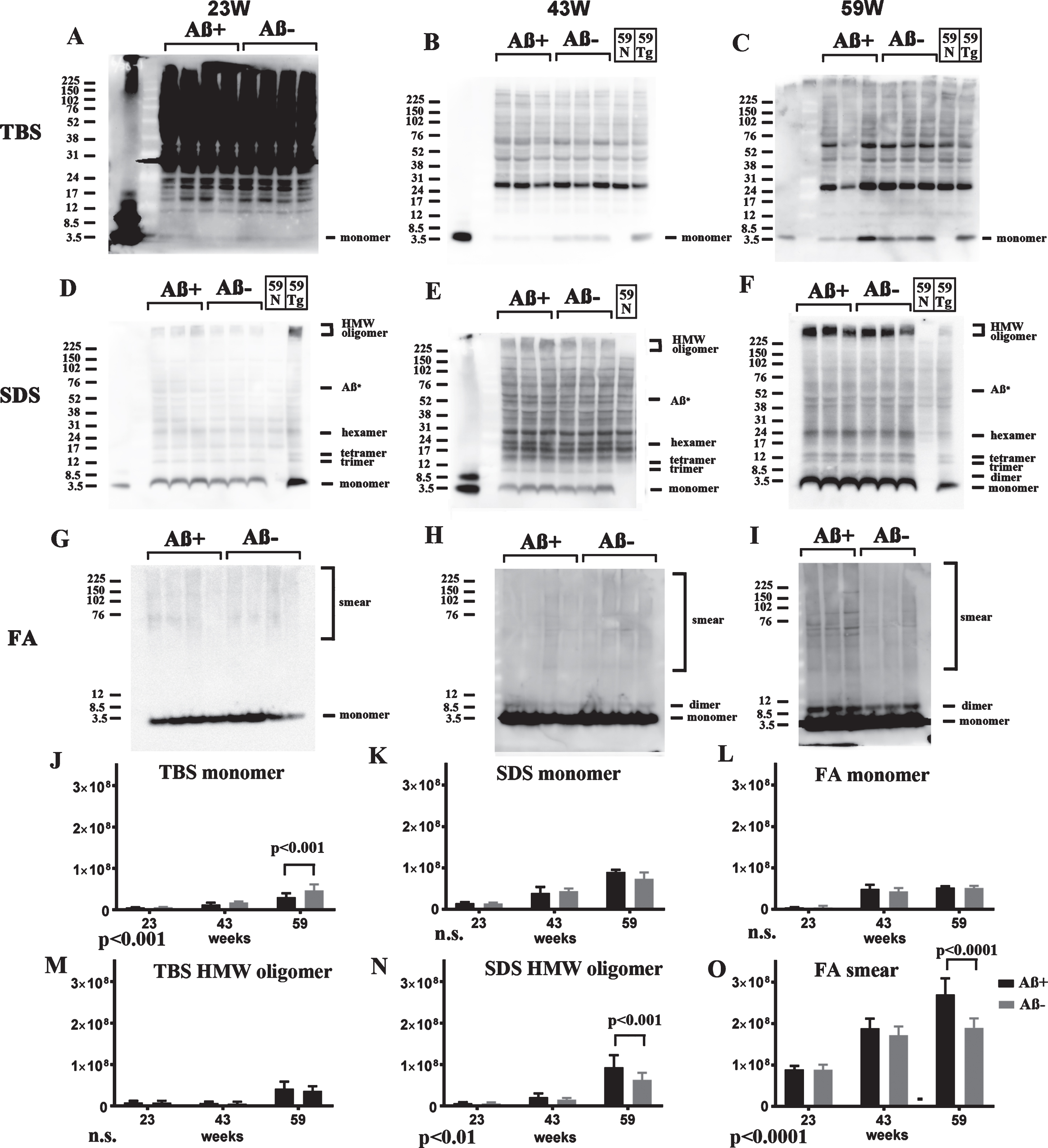
Since 82E1/82E1 ELISA could only measure some AβOs species, the basic age-dependent presence of all Aβ species were directly examined by western blots in 3-step brain extracts from TgCRND8 at 13, 23, 43, and 59 weeks of age. In TBS fractions, Aβ monomers were labeled by 82E1 from 13 weeks, and the amount increased with age (Fig. 4A, D). AβOs except high molecular weight (HMW) AβOs larger than 200 kDa were difficult to detect in this fraction because of the existence of mouse IgGs, as shown in lane 59N from a non-transgenic TgCRND8 littermate at 59 weeks old (Fig. 4A, G). In SDS fractions, however, there were marked Aβ monomers, low molecular weight (LMW) AβOs (di-, tri-, tetra-, and Aβ*56 [34], and HMW AβOs larger than 200 kDa. Respective AβOs species increased with age (Fig. 4B, E, H). In FA fractions, Aβ monomers, Aβ dimers and diffuse smear patterns were observed (Fig. 4C, F, I). Many Aβ species with different solubility and aggregation properties were accumulated from the early period in TgCRND8 brains, suggesting that 82E1/82E1 ELISA detected only some of the accumulated AβOs. Aβ monomers increased with age in the TBS, SDS, and FA fractions, HMW oligomers increased with age in the TBS and SDS fractions, and Aβ smear increased in the FA fractions, although not significantly with the small sample size.
Fig.4
Age-dependent increase of Aβ monomer and Aβ oligomers in brains of TgCRND8 without an oral immunization trial (A–L), antibody epitope mapping of aggregated synthetic Aβ1–42 (M), and immunostaining of TgCRND8 brains without an oral immunization trial (N). D–I are quantification of Aβ monomer in each fraction (D–F), HMW oligomers in TBS and SDS fraction (G, H), and Aβ smear in the FA fraction (I). 59N indicates nontransgenic littermates at 59 weeks. A) In TBS fractions, Aβ monomers were detected by 82E1 from 13 weeks and the amount increased with age (D). Other molecular weight oligomers except HMW oligomers (G) were difficult to detect because of the existence of mouse IgGs. B) In SDS fractions, Aβ monomers and AβOs, including di-, tri-, tetra-, and Aβ*56, and HMW AβOs, were visualized from 13 weeks. Respective species increased with age (E, H). C) In FA fractions, Aβ monomers from 13 weeks, Aβ dimers from 43 weeks, and diffuse smear patterns from 43 weeks were found (F, I). J–L) 4G8, anti-Aβ40, and anti-Aβ42 weakly detected LMW AβOs, but could not detect HMW AβOs. M) HMW AβOs were detected by antibodies against the N-terminus (82E1, Ab9204, Aβ-N, and PEP3), and were weakly detected by antibodies against the mid portion of Aβ (6E10 and 4G8), but were not detected by anti-Aβ42 and BC-05. Anti-C-terminus to Aβ40 (anti-Aβ40 and BA-27) did not detect Aβ1–42. N) Immunostaining of TgCRND8 brains using Aβ-N (1:1000), anti-Aβ40 (1:1000), and anti-Aβ42 (1:400) showed age-dependent Aβ deposition. Aβ burden labeled by anti-Aβ40 and anti-Aβ42 were weaker than those by Aβ-N. Bar represents 100 μm. One mouse at 13 weeks, 2 mice at 23 weeks, 43 weeks, and 59 weeks were used for analysis.
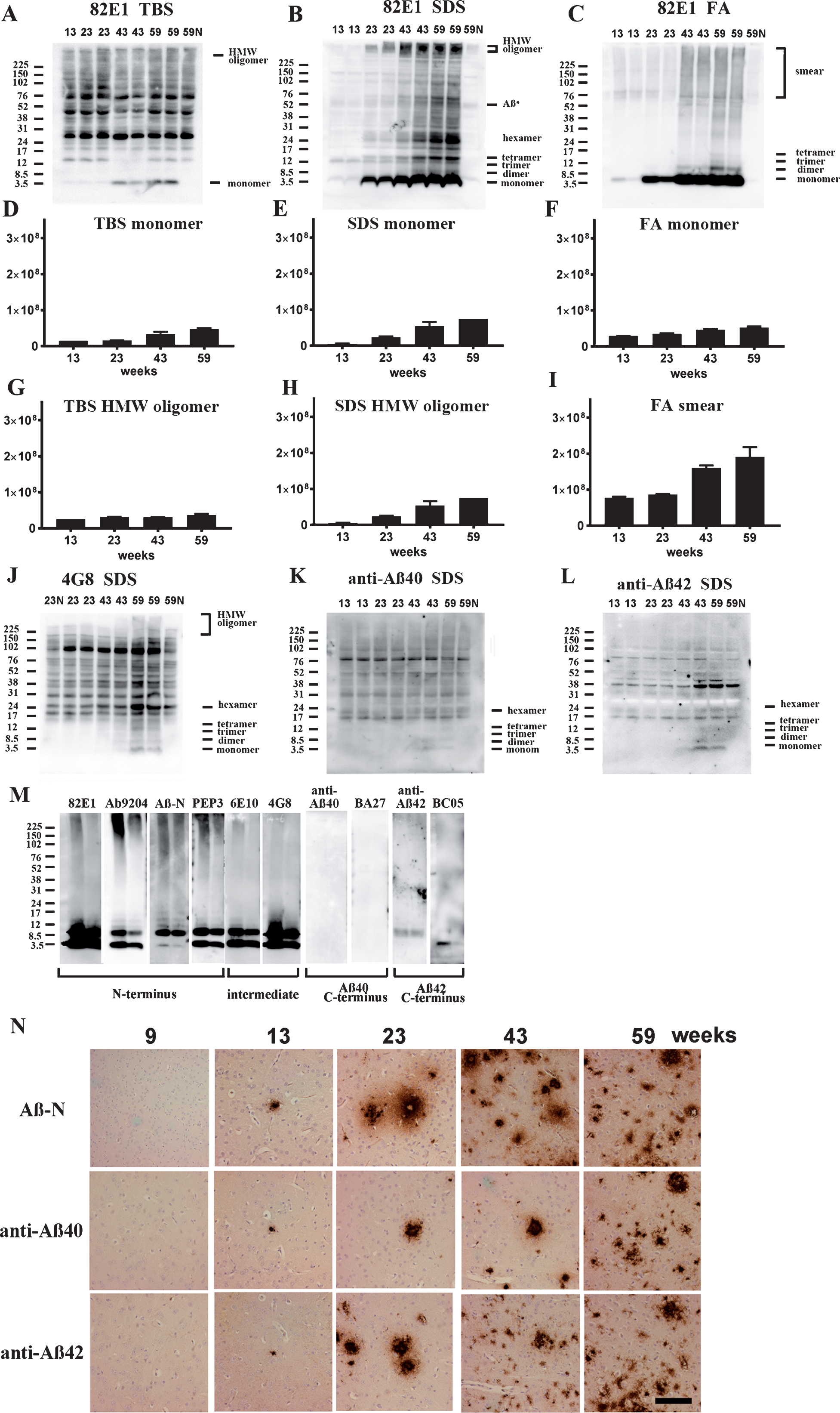
4G8, anti-Aβ40, and anti-Aβ42 weakly detected LMW AβOs, but HMW AβOs could not be detected (Fig. 4J–L). These findings suggest that the C-terminal site of Aβ caused conformational changes in Aβ when incorporated into HMW large assemblies, leading to C-terminal epitope blockade of Aβ species including Aβ40 and Aβ42. In support of this, western blots of aggregated synthetic Aβ42 showed that N-terminus antibodies (Ab9204, Aβ-N, and PEP3) clearly detected HMW AβOs; however, antibodies against the mid portion (6E10 and 4G8) and C-terminus 42 (anti-Aβ42 and BC-05) of Aβ could not detect HMW AβOs (Fig. 4M). Antibodies against C-terminus 40 (anti-Aβ40, BA-27) did not detect synthetic Aβ42. The C-terminal epitopes of HMW AβOs were blocked resulting no staining by C-terminus-specific antibodies. Immunostaining of TgCRND8 brains showed decreased immunostaining by anti-Aβ40 or anti-Aβ42 compared with that by Aβ-N (Fig. 4N). C-terminal epitope blockade of HMW AβOs is one reason for the decrease of Aβ burden labeled by anti-C terminal antibodies.
Western blotting of Aβ+ treated brains
Based on these analyses, Aβ species in three fractions of both Aβ+ and Aβ– groups were analyzed by western blots using 82E1 at 23, 43, and 59 weeks of age. In TBS fractions, soluble Aβ monomers in the Aβ+ treated group were decreased at 23, 43, and 59 weeks. Trace amounts of HMW AβOs were detected at 59 weeks; however, there was no difference between the Aβ+ and Aβ– groups (Fig. 5A–C). In SDS fractions, Aβ monomers and LMW AβOs were detected equally at 23, 43, and 59 weeks. However, accumulation of HMW AβOs was slightly increased in the Aβ+ treated group at 23, 43, and 59 weeks (Fig. 5D–F). In FA fractions, monomers and dimers of Aβ increased with age equally in both groups (Fig. 5G–I). Smear patterns of Aβ were markedly observed in the Aβ+ group at 59 weeks of age (Fig. 5I). Using quantification of the bands, TBS soluble Aβ monomer decreased significantly in Aβ+ treated groups by two-way ANOVA (p < 0.001, Fig. 5J). Using post-hoc analysis, significance was detected at 59 weeks (p < 0.001, Fig. 5J). SDS soluble HMW oligomers and Aβ smear in the FA fraction increased significantly in Aβ+ treated groups by two-way ANOVA (Fig. 5N, p < 0.01 for SDS soluble HMW oligomers, and Fig. 5O, p < 0.0001 for Aβ smear). By post-hoc analysis significance was detected at 59 weeks (p < 0.001 for SDS soluble HMW oligomers, Fig. 5N, and p < 0.0001 for Aβ smear, Fig. 5O). HMW oligomers in the TBS fraction and Aβ monomer in the SDS and FA fractions did not differ between the Aβ+ and Aβ– groups (Fig. 5M,K,L). Since TBS soluble AβOs detected by 82E1/82E1 ELISA seem to be recovered as Aβ monomers in western blots using SDS sample buffer, these results correspond to AβOs ELISA result that Aβ+ immunization suppressed soluble AβOs. Furthermore, it was suggested that Aβ+ immunization accelerated the conversion of LMW AβOs to HMW AβOs in SDS fractions, and finally increased smear Aβ accumulation in most insoluble FA fractions.
Fig.5
Longitudinal comparison of Aβ species (23, 43, and 59 weeks) in TBS (A–C), SDS (D–F), and FA (G–I) extracts from Aβ+ and Aβ– treated mice in western blots using 82E1. For controls, a nontransgenic mouse at 59 weeks (59N) and a TgCRND8 mouse at 59 weeks without treatment (59Tg) were used. Aβ monomer in each fraction (J–L), HMW oligomers in the TBS fraction (M), and SDS fraction (N), and Aβ smear in the FA fraction (O) were quantified. The results of two-way ANOVA are shown in the lower left of each figure (J–O). In TBS fractions, soluble Aβ monomers in Aβ+ treated mice were decreased compared with those in Aβ– treated mice at 23, 43, and 59 weeks (A–C, J). To detect Aβ monomers clearly at 23 weeks, the membrane was exposed for longer than the other blots (A). Small amounts of HMW AβOs were detected only at 59 weeks, but they did not differ between the Aβ+ and Aβ– groups (A–C, M). In SDS fractions, Aβ monomers and LMW AβOs were detected equally at 23, 43, and 59 weeks (D–F, K). Accumulation of HMW AβOs was inversely increased in the Aβ+ treated group at 23, 43, and 59 weeks (D–F, N). In FA fractions, monomers and dimers of Aβ increased with age equally in both groups (G–I, L). However, smear patterns of Aβ were markedly visualized in the Aβ+ group at 59 weeks (G–I, O). Mice at 23 weeks (n = 9 for Aβ+, n = 11 for Aβ–), 43 weeks (n = 7 for Aβ+, n = 6 for Aβ–), and 59 weeks (n = 6 for Aβ+, n = 7 for Aβ–) were used.
Suppressed Aβ-immunoreactive load and adverse reaction
Age-related Aβ-immunoreactive load measured using the total area of Aβ-N immunostaining was significantly suppressed in the Aβ+ treated group compared with that of the Aβ– group (Fig. 6A, C; p < 0.0001). Congo red staining showed that most Aβ deposition was Congo red-negative diffuse plaques (Fig. 6A). Separate evaluation of core plaques (Fig. 6D; p < 0.0001) and diffuse plaques (Fig. 6E; p < 0.0001) revealed that a large part of the age-related increase in the area of amyloid burden and also decrease by immunization consisted of diffuse plaques. Immunostaining by anti-Aβ40 or anti-Aβ42 also showed significant suppression of amyloid burden in the Aβ+ group compared with the Aβ– group (Fig. 6F; p < 0.001, 6G p < 0.0001).
Fig.6
Staining of Aβ+ or Aβ– treated brains with Aβ-N and Congo red (A), microglial marker Iba1 (B) in Aβ+ or Aβ– treated mouse brains, quantification of Aβ burden (C–G), Iba1 (H), western blot of AβPP and CTFs (I), ELISA of sAβPPα (J), sAβPPβ (K), western blotting of total tau (L), phosphorylated tau (M), CSF Aβ40 (N), and Aβ42 (O) measured by ELISA. A) Aβ-immunoreactive load using Aβ-N was suppressed in the Aβ+ treated group compared with that of the Aβ– group. Most Aβ staining showed diffuse plaques that are not stained with Congo red. B) Microgliosis was weaker in the Aβ+ treated group than that in the Aβ– treated group. Bar represents 100 μm in A and 200 μm in B. C) The area labeled by Aβ-N was significantly suppressed in the Aβ+ treated group compared with that in the Aβ– treated group (p < 0.0001). D, E) The area occupied by diffuse plaques was more than 3-fold of that by core plaques. Both plaques were suppressed by Aβ+ treatment (D, p < 0.0001; E, p < 0.0001). Large part of the Aβ decrease by immunization consisted of diffuse plaques. Aβ burdens detected by anti-Aβ40 (F) and anti-Aβ42 (G) were significantly decreased in the Aβ+ treated group (F, p < 0.001; G, p < 0.0001). H) The area of Iba1 staining in Aβ+ treated mice was smaller than that in Aβ– treated mice (p < 0.0001). The numbers of mice analyzed: 23 weeks (n = 9 for Aβ+, n = 11 for Aβ–), 43 weeks (n = 7 for Aβ+, n = 6 for Aβ–), and 59 weeks (n = 6 for Aβ+, n = 7 for Aβ–). I) AβPP, CTFβ, and CTFα, and other CTFs in SDS fractions did not differ between the Aβ+ and Aβ– treated groups from 23 to 59 weeks. (J, K) The amount of sAβPPα and sAβPPβ in the TBS fractions detected by ELISA did not differ between Aβ+ (Red) and Aβ– (Blue) treated groups from 23 to 59 weeks. L, M) The amount of total tau (TAU-5) and phosphorylated tau (pTau) (PHF-1) did not differ between Aβ+ and Aβ– treated mice from 23 to 59 weeks. N, O) Aβ40 and Aβ42 in CSF decreased with aging. They were decreased in Aβ+ treated mice compared to Aβ– treated mice, although there was no significance difference between Aβ+ and Aβ– mice (Aβ+ n– =– 7 for 23 weeks, n = 4 for 43 weeks and n = 6 for 59 weeks; Aβ– n = 9 for 23 weeks, n = 2 for 43 weeks, and n = 4 for 59 weeks).
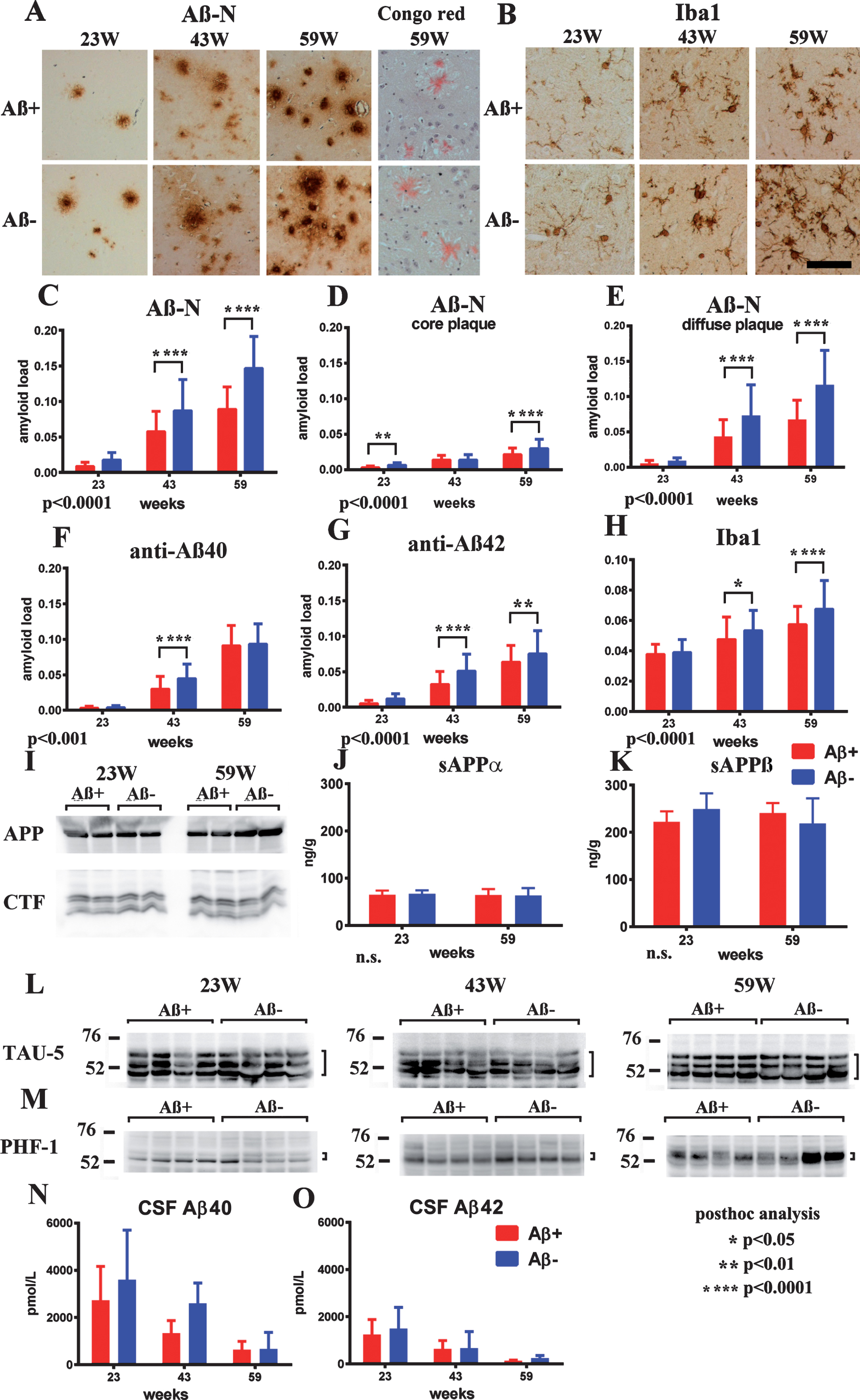
Microglial burden based on Iba1 labeling was significantly decreased in the Aβ+ treated group compared with the Aβ– treated group (Fig. 6B, H; p < 0.0001). Astrocytosis detected using an anti-GFAP antibody was not different between both groups of mice at any age. Both infiltration of CD5-positive lymphocytes or macrophages, and microhemorrhage by Berlin blue staining, were not detected in both Aβ+ and Aβ– treated mice in any examined brain sections (data not shown).
No alteration in AβPP processing and tau
Full length AβPP, C-terminal fragments of AβPP (CTFs) showed no differences between the Aβ+ and Aβ– groups from 23 to 59 weeks (Fig. 6I). The amount of α- and β-cleaved soluble AβPP (sAβPPα, sAβPPβ) detected by ELISA also showed no differences in both groups during 23–59 weeks (Fig. 6J, K). Levels of the total tau and pTau amount (Fig. 6L, M) did not differ between the Aβ+ and Aβ– treated groups at all ages. Additionally, there were almost no bands of pTau in western blots of FA extracts of TgCRND8 brains, corresponding with the lack of neurofibrillary tangles in TgCRND8 brains by Gallyas silver staining (data not shown).
CSF Aβ40 and Aβ42
Aβ40 and Aβ42 in CSF decreased with aging. They were decreased in Aβ+ treated mice compared with Aβ– treated mice, although there was no significant difference between Aβ+ and Aβ– mice with the small sample size (Aβ+ n = 7 for 23 weeks, n = 4 for 43 weeks and n = 6 for 59 weeks; Aβ– n = 9 for 23 weeks, n = 2 for 43 weeks, and n = 4 for 59 weeks) because of the difficulty in drawing CSF from mice (Fig. 6N,O).
DISCUSSION
To evaluate the potential utility of Aβ+ oral immunization to prevent cognitive decline in TgCRND8, we adopted an extended reference memory version of the MWM test, requiring hippocampus-dependent spatial working memory, with a longitudinal design that mimicked human clinical trials [24]. At 21 weeks, escape latencies and path-lengths became worse in the Aβ– group, whereas the Aβ+ group remained in the same levels. The performances of the Aβ+ group were significantly better during 21–59 weeks than those in the Aβ– group. Our extended MWM test study showed the preventative efficacy of Aβ+ oral immunization on Aβ-related learning impairment. As previously indicated [35], probe tests were inadequate for extended MWM test which consisted of extensive long-time and repeated behavioral evaluation. The same repeated trials in every 4 weeks in Aβ immunization trial of TgCRND8 mice also failed to show significant difference in probe trials [24]. Extensive overtraining by trials for a long duration may achieve the saturated level of memory, and may make it difficult to detect subtle changes in probe trials even among hippocampal damaged animals [36, 37]. Recent cohort observation studies have confirmed that Aβ amyloidosis caused impairment of episodic memory in the preclinical stage, and executive and global functions in the symptomatic stage before onset of dementia [38–40]. Rigorous and detailed cognitive assessments to repeatedly evaluate subtle cognitive impairments over a long duration are necessary for preclinical prevention trials such as DIAN, API, or A4 studies [15, 16, 41]. Based on these findings, our extended reference memory version of the MWM test is a competent way to develop preventive DMTs in basic level of model animal experiments.
A previous Aβ42 immunization study reported a reduction in behavior impairment and plaques in TgCRND8 mice; however, Aβ amounts measured by ELISA did not decrease [24]. Administration of anti-Aβ antibody m266 to PDAPP mice and BAM-10 to Tg2576 mice rapidly reversed the memory deficits without altering brain Aβ burden [42, 43]. The same findings were observed in our experiments, which revealed reduced cognitive impairment and Aβ burden, but no differences in the total amounts of Aβ40 and Aβ42 measured by ELISA between Aβ+ and Aβ– groups except for increased Aβ40 in the FA fraction. Decreased TBS-soluble AβOs were the significantly suppressed Aβ species. This observation supported the notion that these soluble AβOs species contribute to neuronal/synaptic injury and cognitive impairment.
Because the ELISA-based Aβ assay could not recover the whole amount of Aβ species, we next intensely surveyed Aβ species using western blotting of the same 3-step extracts to avoid overlooking other cardinal Aβ molecules. Analyses of untreated TgCRND8 mice revealed the age-related accumulation of all Aβ species: Aβ monomers in the TBS fraction; a large part of all sets of AβOs with LMW to HMW in the SDS fraction [28]; and age-related Aβ monomers, dimers, and smears in the FA fraction. These different LMW to HMW AβOs species were not measured by AβOs ELISA, and the C-terminus of HMW AβOs species were blocked [44]. Histological examination also confirmed increased plaque immunoreactivity by N-terminal antibodies compared to those by anti-Aβ40 and Aβ42. Thus, diverse Aβ species with conformational changes, modifications, and different lengths and solubilities were accumulated in the brains. For this reason, we carefully analyzed Aβ accumulation using not only ELISA, but also conventional western blots to decide which is the cardinal Aβ molecule that is toxic for the nervous system and causes cognitive dysfunction.
In a comparison study between Aβ+ and Aβ– treated groups, visualized TBS soluble Aβ monomers were decreased by Aβ+ treatment during weeks 23–59. Because AβOs in the TBS fraction were easily degraded into monomers using SDS sample buffer in western blotting experiments, these findings may partially correspond with the decreased amount of AβOs detected by ELISA in the Aβ+ treated group. Consistent with ELISA findings, almost all insoluble aggregated Aβ species in the SDS and FA fractions did not show obvious differences between the Aβ+ and Aβ– treated groups. Conversely, HMW AβOs in the SDS fraction and smear Aβ in the FA fraction were increased in the Aβ+ treated group compared with the Aβ– treated group. These findings suggested that toxic soluble AβOs in Aβ+ treated brains may be sequestered into highly insoluble and aggregated AβOs and Aβ fibrils. The same findings have been suggested previously, because the amyloid fibril is a protective structure in AD pathology [45–47]. CSF Aβ40 and Aβ42 levels were decreased in Aβ+ treated mice compared with Aβ– treated mice, although there was no significant difference between Aβ+ and Aβ– mice because of the small sample size. Previous immunization studies showed increased Aβ in CSF by increased clearance of Aβ [48]. Decreased amounts of CSF Aβ are thought to be due to the deposition of Aβ in the brain, because CSF Aβ decreases correlate with Aβ plaques [29, 49]. Decreased amounts of Aβ in Aβ+ treated mice suggested that the effect of immunization is not by enhanced clearance of Aβ but sequestration of Aβ in an insoluble fraction in the brain.
Histological evaluation showed that reduction of diffuse plaques was more prominent than the reduction of core plaques by Aβ+ immunization. Diffuse plaques were composed of scant Aβ amyloid fibrils. No difference of Aβ levels by ELISA may imply a lower decrease in core plaques. Together with biochemical analyses, Aβ+ immunization may facilitate the sequestration of toxic soluble AβOs into insoluble and HMW AβOs within the compact aggregated Aβ amyloid core. Liu et al. separated AβOs into neurotoxic type 1 AβOs and non-neurotoxic type 2 AβOs. Type 2 AβOs, which occupy the majority of AβOs sequestrated around dense core plaques (∼95% in 21-month-old Tg2576 mice), accounted for less than 15% of the cortex [46]. Hong et al. showed that diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in AD brain [50]. It is still unclear what types of AβOs exert the main neurotoxicity [33, 42–54]. We have previously shown that accumulation of Aβ dimers in lipid rafts is the earliest event corresponding to behavioral deficits in Tg2576 mice [55]. Aβ dimers are also shown to be cardinal molecules for synaptic dysfunction in Aβ amyloid cascades in AD pathogenesis [56, 57]. Injection of LMW oligomers into the brain induced rapid and persistent impairment of memory, associated with decreased hippocampal synaptophysin [58]. AβOs neurotoxicity and their induction of tauopathy are canceled by AβOs-specific monoclonal antibodies [59]. The recent success of a phase Ib randomized trial of aducanumab also showed that antibodies against aggregated forms of Aβ are clinically useful [9]. Together with these findings, our study suggested that soluble AβOs are the cardinal Aβ species responsible for neurotoxicity and are valid targets for DMTs.
Inverse increases in HMW Aβ oligomers and Aβ smear have not been reported in previous Aβ immunization studies. There are some studies on decreased Aβ neurotoxicity through the accelerated conversion of Aβ oligomers into Aβ fibrils. Using chaperone protein HspB1, Aβ oligomers were converted into large nontoxic aggregates, and their toxicity was sequestrated [60]. Peptides that enhance the formation of amorphous aggregates of Aβ attenuated the paralysis of transgenic Caenorhabditis elegans [61]. Meanwhile, activated microglia are shown to take up Aβ, cluster Aβ inside, and release aggregated Aβ, which contributed to plaque growth [62]. Based on these reports, we speculated the presence of sequestration mechanisms that convert toxic Aβ oligomers into HMW Aβ aggregates for reduction of Aβ toxicity.
Both ELISA and TAPIR showed the presence of specific IgG antibodies against Aβ4–10 structural epitopes within A1aB1b in Aβ+ immunized mice. In western blotting, antibodies were shown to detect several Aβ oligomers. Splenocytic proliferation and cytokine release against Aβ+ stimulation also implied the presence of adaptive cellular immune responses. We found no histological meningoencephalitis or bleeding in mouse brains. Our mucosal Aβ+ immunization significantly decreased microgliosis, indicating that it suppressed glial-mediated inflammatory responses. These findings suggested that our Aβ+ oral immunization could safely raise moderate and continuous innate and humoral immune responses.
Our Aβ+ immunization did not alter basic AβPP processing or the signaling process of the AβPP intracellular C-terminal domain [63]. Accumulation of pTau was only detected in dystrophic neurites associated with core plaques. Pathological conversion and spreading of aggregated tau was facilitated in the dystrophic neurites around core plaques [64]. Because numbers of core plaques were not so different, the amount of pTau was not significantly different.
Preclinical initiation and long-term maintenance of DMT against Aβ amyloidosis have been proposed to halt the progression of cognitive impairment and the onset of dementia. This strategy, which will likely necessitate weekly or monthly injections of antibodies, carries two risks. First, it may enhance risks of side effects. Second, the cost of long-term treatment with antibodies is likely to be prohibitively high when applied on national scales to large numbers of people. The use of oral plant vaccines offers advantages of extremely low cost and better safety compared with synthetic compounds or antibodies. For these reasons, our plant-based soybean Aβ+ oral vaccine strategy is cheap, safe and as effective as others.
In conclusion, our results revealed that Aβ+ oral immunization suppressed soluble AβOs production and prevented cognitive impairment without obvious adverse reactions. Oral immunization by Aβ+ could be a promising DMT for prevention of the pathological processes of AD.
ACKNOWLEDGMENTS
We thank Eiki Tsushima for statistical analysis, Kaori Haga for research assistance, Takaomi C. Saido for Ab9201, and Peter Davies for PHF-1. Research reported in this publication was supported by the Longevity Science Committee of the Ministry of Health and Welfare of Japan; Scientific Research (C) (18K07385 MS and 19K07989 TK) from the Ministry of Education, Science, and Culture of Japan; Study of prevention for neurodegenerative diseases by new antiaging methods in Hirosaki University Institutional Research Grant; Development of Fundamental Technologies for the Production of High-value Materials Using Transgenic Plants by the Ministry of Economy, Trade, and Industry of Japan.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0023r1).
REFERENCES
[1] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[2] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[3] | Orgogozo JM , Gilman S , Dartigues JF , Laurent B , Puel M , Kirby LC , Jouanny P , Dubois B , Eisner L , Flitman S , Michel BF , Boada M , Frank A , Hock C ((2003) ) Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology 61: , 46–54. |
[4] | Gilman S , Koller M , Black RS , Jenkins L , Griffith SG , Fox NC , Eisner L , Kirby L , Rovira MB , Forette F , Orgogozo JM ; AN1792(QS-21)-201 Study Team ((2005) ) Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64: , 1553–1562. |
[5] | Serrano-Pozo A , William CM , Ferrer I , Uro-Coste E , Delisle MB , Maurage CA , Hock C , Nitsch RM , Masliah E , Growdon JH , Frosch MP , Hyman BT ((2010) ) Beneficial effect of human anti-amyloid-β active immunization on neurite morphology and tau pathology. Brain 133: , 1312–1327. |
[6] | Bard F , Barbour R , Cannon C , Carretto R , Fox M , Games D , Guido T , Hoenow K , Hu K , Johnson-Wood K , Khan K , Kholodenko D , Lee C , Lee M , Motter R , Nguyen M , Reed A , Schenk D , Tang P , Vasquez N , Seubert P , Yednock T ((2003) ) Epitope and isotype specificities of antibodies to β-amyloid peptide for protection against Alzheimer’s disease-like neuropathology. Proc Natl Acad Sci U S A 101: , 2023–2028. Erratum in Proc Natl Acad Sci U S A (2004) 101, 11526. |
[7] | Lemere CA , Masliah E ((2010) ) Can Alzheimer disease be prevented by amyloid-β immunotherapy? Nat Rev Neurol 6: , 108–119. |
[8] | Wisniewski T , Goñi F ((2015) ) Immunotherapeutic approaches for Alzheimer’s disease. Neuron 85: , 1162–1176. |
[9] | Sevigny J , Chiao P , Bussière T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[10] | Vandenberghe R , Riviere ME , Caputo A , Sovago J , Maguire RP , Farlow M , Marotta G , Sanchez-Valle R , Scheltens P , Ryan JM , Graf A ((2016) ) Active Aβ immunotherapy CAD106 in Alzheimer’s disease: A phase 2b study. Alzheimers Dement (N Y) 3: , 10–22. |
[11] | Wang CY , Wang PN , Chiu MJ , Finstad CL , Lin F , Lynn S , Tai YH , De Fang X , Zhao K , Hung CH , Tseng Y , Peng WJ , Wang J , Yu CC , Kuo BS , Frohna PA ((2017) ) UB-311, a novel UBITh® amyloid β peptide vaccine for mild Alzheimer’s disease. Alzheimers Dement (N Y) 3: , 262–272. |
[12] | Lippa CF , Nee LE , Mori H , St George-Hyslop P ((1998) ) Abeta-42 deposition precedes other changes in PS-1 Alzheimer’s disease. Lancet 352: , 1117–1118. |
[13] | Weiner MW , Veitch DP , Aisen PS , Beckett LA , Cairns NJ , Cedarbaum J , Green RC , Harvey D , Jack CR , Jagust W , Luthman J , Morris JC , Petersen RC , Saykin AJ , Shaw L , Shen L , Schwarz A , Toga AW , Trojanowski JQ ; Alzheimer’s Disease Neuroimaging Initiative ((2015) ) 2014 Update of the Alzheimer’s Disease Neuroimaging Initiative: A review of papers published since its inception. Alzheimers Dement 11: , e1–120. |
[14] | Bateman RJ , Xiong C , Benzinger TL , Fagan AM , Goate A , Fox NC , Marcus DS , Cairns NJ , Xie X , Blazey TM , Holtzman DM , Santacruz A , Buckles V , Oliver A , Moulder K , Aisen PS , Ghetti B , Klunk WE , McDade E , Martins RN , Masters CL , Mayeux R , Ringman JM , Rossor MN , Schofield PR , Sperling RA , Salloway S , Morris JC ; Dominantly Inherited Alzheimer Network ((2012) ) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367: , 795–804. |
[15] | Mills SM , Mallmann J , Santacruz AM , Fuqua A , Carril M , Aisen PS , Althage MC , Belyew S , Benzinger TL , Brooks WS , Buckles VD , Cairns NJ , Clifford D , Danek A , Fagan AM , Farlow M , Fox N , Ghetti B , Goate AM , Heinrichs D , Hornbeck R , Jack C , Jucker M , Klunk WE , Marcus DS , Martins RN , Masters CM , Mayeux R , McDade E , Morris JC , Oliver A , Ringman JM , Rossor MN , Salloway S , Schofield PR , Snider J , Snyder P , Sperling RA , Stewart C , Thomas RG , Xiong C , Bateman RJ ((2013) ) Preclinical trials in autosomal dominant AD: Implementation of the DIAN-TU trial. Rev Neurol (Paris) 169: , 737–743. |
[16] | Reiman EM , Langbaum JB , Fleisher AS , Caselli RJ , Chen K , Ayutyanont N , Quiroz YT , Kosik KS , Lopera F , Tariot PN ((2011) ) Alzheimer’s Prevention Initiative: A plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis Suppl 3: , 321–329. |
[17] | Shahid N , Daniell H ((2016) ) Plant-based oral vaccines against zoonotic and non-zoonotic diseases. Plant Biotechnol J 14: , 2079–2099. |
[18] | Kwon KC , Verma D , Singh ND , Herzog R , Daniell H ((2013) ) Oral delivery of human biopharmaceuticals, autoantigens and vaccine antigens bioencapsulated in plant cells. Adv Drug Deliv Rev 65: , 782–799. |
[19] | Rosales-Mendoza S , Rubio-Infante N , Zarazúa S , Govea-Alonso DO , Martel-Gallegos G , Moreno-Fierros L ((2014) ) Plant-based vaccines for Alzheimer’s disease: An overview. Expert Rev Vaccines 13: , 429–441. |
[20] | Gonzalez-Castro R , Acero Galindo G , García Salcedo Y , Uribe Campero L , Vazquez Perez V , Carrillo-Tripp M , Gevorkian G , Gomez Lim MA ((2018) ) Plant-based chimeric HPV-virus-like particles bearing amyloid-β epitopes elicit antibodies able to recognize amyloid plaques in APP-tg mouse and Alzheimer’s disease brains. Inflammopharmacology 26: , 817–827. |
[21] | Nojima J , Maeda A , Aoki S , Suo S , Yanagihara D , Watanabe Y , Yoshida T , Ishiura S ((2011) ) Effect of rice-expressed amyloid β in the Tg2576 Alzheimer’s disease transgenic mouse model. Vaccine 29: , 6252–6258. |
[22] | McLaurin J , Cecal R , Kierstead ME , Tian X , Phinney AL , Manea M , French JE , Lambermon MH , Darabie AA , Brown ME , Janus C , Chishti MA , Horne P , Westaway D , Fraser PE , Mount HT , Przybylski M , St George-Hyslop P ((2002) ) Therapeutically effective antibodies against amyloid-β peptide target amyloid-β residues 4-10 and inhibit cytotoxicity and fibrillogenesis. Nat Med 8: , 1263–1269. |
[23] | Maruyama N , Fujiwara K , Yokoyama K , Cabanos C , Hasegawa H , Takagi K , Nishizawa K , Uki Y , Kawarabayashi T , Shouji M , Ishimoto M , Terakawa T ((2014) ) Stable accumulation of seed storage proteins containing vaccine peptides in transgenic soybean seeds. J Biosci Bioeng 118: , 441–447. |
[24] | Janus C , Pearson J , McLaurin J , Mathews PM , Jiang Y , Schmidt SD , Chishti MA , Horne P , Heslin D , French J , Mount HT , Nixon RA , Mercken M , Bergeron C , Fraser PE , St George-Hyslop P , Westaway D ((2000) ) Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature 408: , 979–982. |
[25] | Chishti MA , Yang DS , Janus C , Phinney AL , Horne P , Pearson J , Strome R , Zuker N , Loukides J , French J , Turner S , Lozza G , Grilli M , Kunicki S , Morissette C , Paquette J , Gervais F , Bergeron C , Fraser PE , Carlson GA , George-Hyslop PS , Westaway D ((2001) ) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276: , 21562–21570. |
[26] | Adachi M , Takenaka Y , Gidamis AB , Mikami B , Utsumi S ((2001) ) Crystal structure of soybean proglycinin A1aB1b homotrimer. J Mol Biol 305: , 291–305. |
[27] | Janus C ((2004) ) Search strategies used by APP transgenic mice during spatial navigation in the Morris water maze. Learn Mem 11: , 337–346. |
[28] | Harigaya Y , Shoji M , Kawarabayashi T , Kanai M , Nakamura T , Iizuka T , Igeta Y , Saido TC , Sahara N , Mori H , Hirai S ((1995) ) Modified amyloid β protein ending at 42 or 40 with different solubility accumulates in the brain of Alzheimer’s disease. Biochem Biophys Res Commun 211: , 1015–1022. |
[29] | Kawarabayashi T , Younkin LH , Saido TC , Shoji M , Ashe KH , Younkin SG ((2001) ) Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci 21: , 372–381. |
[30] | Maier M , Seabrook TJ , Lemere CA ((2005) ) Modulation of the humoral and cellular immune response in Aβ immunotherapy by the adjuvants monophosphoryl lipid A (MPL), cholera toxin B subunit (CTB) and E. coli enterotoxin LT(R192G). Vaccine 23: , 149–159. |
[31] | Suzuki N , Cheung TT , Cai XD , Odaka A , Otvos L Jr , Eckman C , Golde TE , Younkin SG ((1994) ) An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science 264: , 1336–1340. |
[32] | Saido TC , Iwatsubo T , Mann DM , Shimada H , Ihara Y , Kawashima S ((1995) ) Dominant and differential deposition of distinct β-amyloid peptide species, AβN3(pE), in senile plaques. Neuron 14: , 457–466. |
[33] | Xia W , Yang T , Shankar G , Smith IM , Shen Y , Walsh DM , Selkoe DJ ((2009) ) A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol 66: , 190–199. |
[34] | Lesné S , Koh MT , Kotilinek L , Kayed R , Glabe CG , Yang A , Gallagher M , Ashe KH ((2006) ) A specific amyloid-β protein assembly in the brain impairs memory. Nature 440: , 352–357. |
[35] | Markowska AL , Long JM , Johnson CT , Olton DS ((1993) ) Variable-interval probe test as a tool for repeated measurements of spatial memory in the water maze. Behav Neurosci 107: , 627–632. |
[36] | Morris RGM , Schenk F , Tweedie F , Jarrard LE ((1990) ) Ibotenate lesions of hippocampus and/or subiculum: Dissociating components of allocentric spatial learning. Eur J Neurosci 2: , 1016–1028. |
[37] | Boksa P , Krishnamurthy A , Brooks W ((1995) ) Effects of a period of asphyxia during birth on spatial learning in the rat. Pediatr Res 37: , 489–96. |
[38] | Hedden T , Oh H , Younger AP , Patel TA ((2013) ) Meta-analysis of amyloid-cognition relations in cognitively normal older adults. Neurology 80: , 1341–1348. |
[39] | Insel PS , Mattsson N , Mackin RS , Schöll M , Nosheny RL , Tosun D , Donohue MC , Aisen PS , Jagust WJ , Weiner MW ; Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Accelerating rates of cognitive decline and imaging markers associated with β-amyloid pathology. Neurology 86: , 1887–1896. |
[40] | Petersen RC , Wiste HJ , Weigand SD , Rocca WA , Roberts RO , Mielke MM , Lowe VJ , Knopman DS , Pankratz VS , Machulda MM , Geda YE , Jack CR Jr ((2016) ) Association of elevated amyloid levels with cognition and biomarkers in cognitively normal people from the community. JAMA Neurol 73: , 85–92. |
[41] | Sperling RA , Rentz DM , Johnson KA , Karlawish J , Donohue M , Salmon DP , Aisen P ((2014) ) The A4 study: Stopping AD before symptoms begin? Sci Transl Med 6: , 228fs13. |
[42] | Dodart JC , Bales KR , Gannon KS , Greene SJ , DeMattos RB , Mathis C , DeLong CA , Wu S , Wu X , Holtzman DM , Paul SM ((2002) ) Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci 5: , 452–457. |
[43] | Kotilinek LA , Bacskai B , Westerman M , Kawarabayashi T , Younkin L , Hyman BT , Younkin S , Ashe KH ((2002) ) Reversible memory loss in a mouse transgenic model of Alzheimer’s disease. J Neurosci 22: , 6331–6335. |
[44] | Ahmed M , Davis J , Aucoin D , Sato T , Ahuja S , Aimoto S , Elliott JI , Van Nostrand WE , Smith SO ((2010) ) Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils. Nat Struct Mol Biol 17: , 561–567. |
[45] | Cheng IH , Scearce-Levie K , Legleiter J , Palop JJ , Gerstein H , Bien-Ly N , Puoliväli J , Lesné S , Ashe KH , Muchowski PJ , Mucke L ((2007) ) Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem 282: , 23818–23828. |
[46] | Liu P , Reed MN , Kotilinek LA , Grant MK , Forster CL , Qiang W , Shapiro SL , Reichl JH , Chiang AC , Jankowsky JL , Wilmot CM , Cleary JP , Zahs KR , Ashe KH ((2015) ) Quaternary structure defines a large class of amyloid-β oligomers neutralized by sequestration. Cell Rep 11: , 1760–1771. |
[47] | Ryan TM , Roberts BR , McColl G , Hare DJ , Doble PA , Li QX , Lind M , Roberts AM , Mertens HD , Kirby N , Pham CL , Hinds MG , Adlard PA , Barnham KJ , Curtain CC , Masters CL ((2015) ) Stabilization of nontoxic Aβ-oligomers: Insights into the mechanism of action of hydroxyquinolines in Alzheimer’s disease. J Neurosci 35: , 2871–2884. |
[48] | Siemers ER , Sundell KL , Carlson C , Case M , Sethuraman G , Liu-Seifert H , Dowsett SA , Pontecorvo MJ , Dean RA , Demattos R ((2016) ) Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 12: , 110–120. |
[49] | Strozyk D , Blennow K , White LR , Launer LJ ((2003) ) CSF Aβ42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology 60: , 652–656. |
[50] | Hong W , Wang Z , Liu W , O’Malley TT , Jin M , Willem M , Haass C , Frosch MP , Walsh DM ((2018) ) Diffusible, highly bioactive oligomers represent a critical minority of soluble Aβ in Alzheimer’s disease brain. Acta Neuropathol 136: , 19–40. |
[51] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation. Nature 416: , 535–539. |
[52] | Lacor PN , Buniel MC , Chang L , Fernandez SJ , Gong Y , Viola KL , Lambert MP , Velasco PT , Bigio EH , Finch CE , Krafft GA , Klein WL ((2004) ) Synaptic targeting by Alzheimer’s-related amyloid β oligomers. J Neurosci 24: , 10191–10200. |
[53] | Cleary JP , Walsh DM , Hofmeister JJ , Shankar GM , Kuskowski MA , Selkoe DJ , Ashe KH ((2005) ) Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci 8: , 79–84. |
[54] | Tomiyama T , Matsuyama S , Iso H , Umeda T , Takuma H , Ohnishi K , Ishibashi K , Teraoka R , Sakama N , Yamashita T , Nishitsuji K , Ito K , Shimada H , Lambert MP , Klein WL , Mori H ((2010) ) A mouse model of amyloid β oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss. J Neurosci 30: , 4845–4856. |
[55] | Kawarabayashi T , Shoji M , Younkin LH , Wen-Lang L , Dickson DW , Murakami T , Matsubara E , Abe K , Ashe KH , Younkin SG ((2004) ) Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J Neurosci 24: , 801–809. |
[56] | Shankar GM , Li S , Mehta TH , Garcia-Munoz A , Shepardson NE , Smith I , Brett FM , Farrell MA , Rowan MJ , Lemere CA , Regan CM , Walsh DM , Sabatini BL , Selkoe DJ ((2008) ) Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14: , 837–842. |
[57] | Müller-Schiffmann A , Herring A , Abdel-Hafiz L , Chepkova AN , Schäble S , Wedel D , Horn AH , Sticht H , de Souza Silva MA , Gottmann K , Sergeeva OA , Huston JP , Keyvani K , Korth C ((2016) ) Amyloid-β dimers in the absence of plaque pathology impair learning and synaptic plasticity. Brain 139: , 509–525. |
[58] | Figueiredo CP , Clarke JR , Ledo JH , Ribeiro FC , Costa CV , Melo HM , Mota-Sales AP , Saraiva LM , Klein WL , Sebollela A , De Felice FG , Ferreira ST ((2013) ) Memantine rescues transient cognitive impairment caused by high-molecular-weight Aβ oligomers but not the persistent impairment induced by low-molecular-weight oligomers. J Neurosci 33: , 9626–9634. |
[59] | Takamura A , Okamoto Y , Kawarabayashi T , Yokoseki T , Shibata M , Mouri A , Nabeshima T , Sun H , Abe K , Urisu T , Yamamoto N , Shoji M , Yanagisawa K , Michikawa M , Matsubara E ((2011) ) Extracellular and intraneuronal HMW-AbetaOs represent a molecular basis of memory loss in Alzheimer’s disease model mouse. Mol Neurodegener 6: , 20. |
[60] | Ojha J , Masilamoni G , Dunlap D , Udoff RA , Cashikar AG ((2011) ) Sequestration of toxic oligomers by HspB1 as a cytoprotective mechanism. Mol Cell Biol 31: , 3146–3157. |
[61] | Yang A , Wang C , Song B , Zhang W , Guo Y , Yang R , Nie G , Yang Y , Wang C ((2017) ) Attenuation of β-amyloid toxicityandby accelerated aggregation. Neurosci Bull 33: , 405–412. |
[62] | Baik SH , Kang S , Son SM , Mook-Jung I ((2016) ) Microglia contributes to plaque growth by cell death due to uptake of amyloid β in the brain of Alzheimer’s disease mouse model. Glia 64: , 2274–2290. |
[63] | Kimberly WT , Zheng JB , Guénette SY , Selkoe DJ ((2001) ) The intracellular domain of the β-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem 276: , 40288–40292. |
[64] | Li T , Braunstein KE , Zhang J , Lau A , Sibener L , Deeble C , Wong PC ((2016) ) The neuritic plaque facilitates pathological conversion of tau in an Alzheimer’s disease mouse model. Nat Commun 7: , 12082. |


